

Abstract
The mechanistic target of rapamycin (mTOR) pathway coordinates the metabolic activity of eukaryotic cells through environmental signals, including nutrients, energy, growth factors, and oxygen. In the nervous system, the mTOR pathway regulates fundamental biological processes associated with neural development and neurodegeneration. Intriguingly, genes that constitute the mTOR pathway have been found to be germline and somatic mutation from patients with various epileptic disorders. Hyperactivation of the mTOR pathway due to said mutations has garnered increasing attention as culprits of these conditions : somatic mutations, in particular, in epileptic foci have recently been identified as a major genetic cause of intractable focal epilepsy, such as focal cortical dysplasia. Meanwhile, epilepsy models with aberrant activation of the mTOR pathway have helped elucidate the role of the mTOR pathway in epileptogenesis, and evidence from epilepsy models of human mutations recapitulating the features of epileptic patients has indicated that mTOR inhibitors may be of use in treating epilepsy associated with mutations in mTOR pathway genes. Here, we review recent advances in the molecular and genetic understanding of mTOR signaling in epileptic disorders. In particular, we focus on the development of and limitations to therapies targeting the mTOR pathway to treat epileptic seizures. We also discuss future perspectives on mTOR inhibition therapies and special diagnostic methods for intractable epilepsies caused by brain somatic mutations.
Keywords: mTORC1, mTORC2, Epilepsy, Malformation of cortical development
INTRODUCTION
The discovery of the mechanistic target of rapamycin (mTOR) began with the identification of new antimicrobial agents in soil samples from Rapa Nui (also known as Easter Island) [93]. This new antimicrobial agent, called rapamycin (clinically called sirolimus), was found to exhibit immunosuppressive and anti-cancer potential and to be of use as an antiepileptic drug [43,47,144]. For two decades, endeavors to define the function of mTOR revealed that mTOR coordinates environmental signals and metabolic activity by forming two distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [130]. mTOR was first found in brain lysates, suggesting the importance of the brain-specific function of mTOR [125]. Indeed, the mTOR pathway has been found to regulate a variety of functions in the brain from brain development to degeneration [85].
Human genetic studies of epileptic patients and epilepsy models of aberrant activation of the mTOR pathway have demonstrated the importance of activating mutations in the mTOR pathway in neurodevelopmental disorders with epilepsy [66]. With the discovery of mutations activating the mTOR pathway as genetic causes of medically intractable epilepsy [4], mTOR inhibitors, such as sirolimus and everolimus, have emerged as being of potential medical use in treating intractable epilepsy patients [33]. However, with the limited efficacy and significant drawbacks of clinically available mTOR inhibitors, new drugs that are more effective and tolerable based on the understanding of pathogenic mechanisms are required. Key discoveries in research on the role of the mTOR pathway in epilepsy are summarized in Supplementary Box 1.
THE MTOR PATHWAY
mTOR is a serine/threonine protein kinase that forms the core subunit of two functionally distinct protein complexes : mTORC1 and mTORC2 [130] (Fig. 1). mTORC1 comprises three core components : mTOR, Raptor (regulatory protein associated with mTOR), and mLST8 (mammalian lethal with Sec13 protein 8, also known as GßL) [130]. In response to environmental signals, mTORC1 acts to control the balance between anabolism and catabolism [130]. mTORC2 controls proliferation, survival, and cytoskeleton organization [130]. In a nutrient-rich environment, cells convert energy sources into macromolecules, such as proteins, lipids, and nucleotides. In nutrientstarved environments, however, cells downregulate the production of macromolecules and rely on catabolic pathways, such as autophagy, for energy.
Fig. 1.
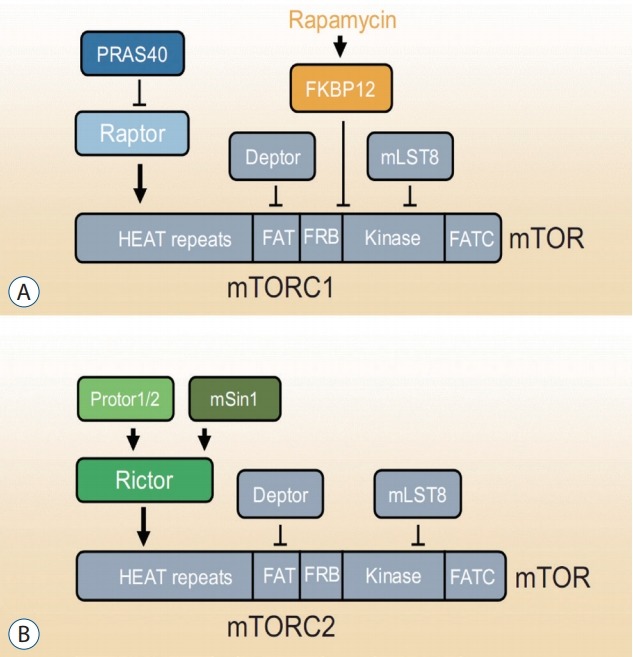
mTORC1 and mTORC2. A : The components of mTORC1 and respective binding site on mTOR. B : The components of mTORC2 and respective binding site on mTOR. PRAS40 : proline-rich Akt substrate of 40 kDa, Raptor : regulatory protein associated with mTOR, FKBP12 : FK506 binding protein 12, Deptor : DEP domain-containing mTORinteracting protein, mLST8 : mammalian lethal with Sec13 protein 8, mTOR : mechanistic target of rapamycin, mTORC1 : mTOR complex 1, mSin1 : mammalian stress-activated protein kinase-interacting protein, Rictor : rapamycin-insensitive companion of mammalian target of rapamycin, mTORC2 : mTOR complex 2.
The mTOR pathway receives various inputs from upstream signaling pathways in response to growth factors, amino acids, energy, oxygen, and stress [130], and the upstream pathways of mTOR can be divided as energy-sensing PI3K-PTEN-AKT-TSC and amino acid-sensing GATOR2-GATOR1-Rag GTPase pathways (Fig. 2) : phosphatidylinositol-3-kinase (PI3K) is critical to integrating insulin signaling for growth and survival [130]. PTEN antagonizes the action of PI3K. Akt is activated by PI3K and is a positive regulator of mTORC1 via inhibition of Tuberous Sclerosis Complex (TSC). TSC is a heterotrimeric complex comprising TSC1, TSC2, and TBC1D7 [38]. TSC inhibits mTORC1 by acting as a GTPase activating protein for Ras homolog enriched in brain (Rheb) [67]. Rheb is a small GTPase that activates mTORC1 by directly binding to mTORC1 on the surface of lysosomes [88]. Meanwhile, Rag GTPase, a component of the amino acid sensing pathway [127], activates mTORC1 by promoting translocation of mTORC1 to the lysosomal surface. Upstream regulators of Rag GTPase in amino acid signaling are the GATOR1 and GATOR2 complexes [10]. The GATOR1 complex, consisting of DEPDC5, Nprl2, and Nprl3, inhibits the mTORC1 pathway by acting as a guanine exchange factor for Rag GTPase. The GATOR2 complex, consisting of Mios, WDR24, WDR59, Seh1L, and Sec13, is a positive regulator of the mTORC1 pathway by inhibiting GATOR1. KICSTOR, which is composed of four proteins, KPTN, ITFG2, C12orf66, and SZT2, recruits GATOR1 to the lysosome to inhibit Rag GTPase [150]. Leucyl-tRNA synthetase, which is another amino acid sensor, functions as a GTPase activating protein for Rag GTPase [57]. mTORC1 senses amino acids in an intra-lysosome fashion. Lysosomal amino acid regulates Rag GTPase via v-ATPase, which increases the guanine exchange factor activity of Ragulator towards Rag GTPase [159]. SLC38A9 is a sensor of lysosomal arginine and activates mTORC1 [69]. Additional novel mTOR upstream regulators, including a methionine sensor, have recently been discovered [1,55].
Fig. 2.

Upstream and downstream of mTORC1 and mTORC2. The signaling network of mTORC1 and mTORC2. Positive regulators of mTORC1 signaling are shown in blue to green. Negative regulators of mTORC1 signaling are shown in red to yellow.
For macromolecule metabolism, mTORC1 regulates translation through inhibitory eukaryotic initiation factor 4E (eIF4E)-binding protein 1/2/3 (4E-BPs) and the S6 kinases (S6Ks) [21,50]. Translational control occurs predominantly at the initiation step, which commences with the binding of the eukaryotic translation initiation factor 4F (eIF4F) complex to the 5’cap [52,135]. As the limiting component of the eIF4F complex, eIF4E is considered to be a critical determinant in translation of mRNA [37]. Facilitating eIF4F formation and the progression of translation, mTORC1 phosphorylates (inactivates) the 4E-BPs, leading to their dissociation from eIF4E [51,60]. The S6Ks activate the eukaryotic translation initiation factor 4B (eIF4B), which is an activator of the eukaryotic translation initiation factor 4A, leading to an increase in the helicase activity of eIF4A and the initiation of translation [39,61].
Stimulating lipid synthesis, mTORC1 interacts with the sterol responsive element binding proteins transcription factors [116]. For sufficient supply of nucleotides during growth, mTORC1 promotes purine and pyrimidine nucleotide biosynthesis through MTHFD2 and the carbamoyl-phosphate synthetase [17]. Through increased translation of the HIF1α transcription factor that drives the expression of glycolytic enzymes, mTORC1 further promotes growth by changing glucose metabolism from oxidative phosphorylation to glycolysis [130]. The mTORC1 pathway also activates the transcriptional coactivator PGC1α for increased mitochondrial biosynthesis [34]. Meanwhile, mTORC1 inhibits autophagy, which plays an important role in scavenging damaged and harmful cellular structures and sustains energy homeostasis, through ULK1 [118]. Recently, it was also demonstrated that mTORC1 regulates ribosomal protein degradation through NUFIP1 [151].
mTORC2 largely functions as a primary effector of the insulin/PI3K signaling [130] (Fig. 2). Generally, the role of the mTORC2 pathway is thought to overlap with the insulin signaling pathway, considering that phenotypes caused by perturbation of the mTORC2 pathway are very similar to those elicited by perturbation of insulin signaling [105]. Upon phosphorylation by mTORC2 [128], Akt promotes cell survival and proliferation. mTORC2 further regulates cellular proliferation and survival through the AGC (PKA/PKG/PKC) family of protein kinases.
PHYSIOLOGICAL ROLES OF THE MTOR PATHWAY IN THE BRAIN
The mTOR pathway plays important roles in development and aging of the central nervous system (CNS) : the mTORC1 pathway is a core regulator of neural development, cortical architecture, neuronal morphology, circuit formation, synaptic plasticity, and neurodegeneration [85]. Perturbation of the mTOR pathway has been found to disrupt several developmental processes. Defective telencephalic development was reported in mTOR mutant rats, called “flat-top” mutants [59]. The loss of function of tuberous sclerosis complex 2 (Tsc2), which is an upstream inhibitor of mTOR, was found to disrupt neuroepithelial growth [119]. Altogether, these studies demonstrated that the mTOR pathway is important for brain development by regulating neural stem cells. Meanwhile, activation of the mTOR pathway via conditional deletion of Tsc2 was found to lead to the disruption of cortical layering [147]. Additionally, increased activity of the mTOR pathway due to mTOR mutations, loss of Tsc1/2, or Pten, was described as eliciting hypertrophy of soma, fewer dendritic spine, increased axon length, and increased dendritic complexity in cortical neurons [53,77,78,137,138].
Perturbation of the mTOR pathway has also been found to adversely affect neural circuit formation. The mTOR pathway regulates axon length [53] and axon guidance in response to environmental signals [156]. Among neurons, local protein synthesis in synapses distant from the soma is mediated by mTOR and is critical for the formation of the neural circuit. The expression levels of synaptic proteins, such as the Arc and Synapsin, are increased by activation of mTOR [81,117]. Hyperactivation of the mTOR pathway by loss of Pten has been shown to increase glutamatergic and GABAergic signals [148]. In addition to neurons, the mTOR pathway has been found to regulate glial cells during neural circuit formation. Deletion of the core component of the mTORC1 or the mTORC2, Raptor or Rictor, was reported to result in defective myelination and oligodendrocyte maturation [18,146]. Conditional Tsc1 knockout in the astrocyte reportedly disrupted electrical signaling in the mouse brain by abrogating neural circuitry [141]. Interestingly, researchers have discovered crosstalk between neurons and glial cells as evidenced by a loss of myelination upon loss of TSC1 in neurons [96]. In TSC patients, the critical role of the mTOR pathway in neural circuit formation was echoed by reductions in white matter and cortical connectivity [112].
Neuronal synapses are crucial to storing memories. Observations of impaired memory formation and synaptic function in response to rapamycin treatment suggest the important role of the mTOR pathway in memory formation [25]. Indeed, the mTOR pathway has been found to play a crucial role in both long-term potentiation (LTP) and long-term depression (LTD) : activation of the mTOR pathway via Tsc1+/-, Tsc2+/-, or 4E-BP2 knockout (KO), results in a decrease in LTP threshold [8,41,145]. Meanwhile, reduced mTOR pathway activity disrupts LTD through metabotropic glutamate receptors (mGluRs) that disturb local protein synthesis [62,64].
ACTIVATION OF MTOR PATHWAY IN NEURODEVELOPMENTAL DISORDERS WITH EPILEPSY
Neurodevelopmental disorders, such as malformations of cortical development (MCD), Cowden syndrome (CS), PIK-3CA-related overgrowth spectrum (PROS), and hamartoma tumor syndromes, commonly present cortical abnormalities and are highly associated with epilepsy, developmental delay, and autism-spectrum disorders [4,12,70,80,97,106,133]. These neurodevelopmental disorders with epilepsy are a common cause of drug-resistant epilepsy in children requiring surgery for treatment [4,12]. However, about 50% of these individuals continue to experience seizures after surgical resection of the epileptic focus [94]. In addition to clinical intractability, refractory epileptic seizures also pose tremendous socioeconomic burden, based on poor quality of life, to patients and their caregivers [28].
Aberrant activation of the mTOR pathway has been identified in brain lesions from patients with neurodevelopmental disorders with epilepsy, especially in MCD [15,86,100]. With these results, researchers have suspected genetic mutations as culprits of the observed mTOR pathway activation in neurodevelopmental disorders [32]. Indeed, a number of neurodevelopmental disorders with epilepsy has been shown to be caused by germline or somatic mutations in the mTOR pathway : these mutations can be categorized as those affecting the energy-sensing PI3K-PTEN-AKT-TSC pathway and those affecting the amino acid-sensing GATOR2-GATOR1-Rag GTPase pathway (Table 1).
Table 1.
Mutations in the mTOR pathway in the neurodevelopmental disorders with epilepsy
| Gene | Mutation type | Effect of the mutation on the mTOR pathway | Disease |
|---|---|---|---|
| PIK3CA | Germline | Activating | CS, PROS |
| Somatic | Activating | PROS, MEG, HME, MCAP, MPPH, FCD | |
| PTEN | Germline | Inactivating | CS, BRRS, LDD, Proteus syndrome, Proteus-like syndrome |
| AKT3 | Germline | Activating | MEG, HME, MEG with polymicrogyria, MEG with PNH |
| Somatic | Activating | HME, FCD | |
| TSC1/2 | Germline | Inactivating | TSC |
| Inherited – autosomal dominant | Inactivating | TSC | |
| Somatic | Inactivating | FCD | |
| TBC1D7 | Inherited – autosomal recessive | Inactivating | MEG |
| RHEB | Somatic | Activating | HME |
| STRADA | Inherited – autosomal recessive | Inactivating | PMSE |
| MTOR | Germline | Activating | SKS |
| Somatic | Activating | SKS, FCD, HME | |
| DEPDC5 | Inherited – autosomal dominant | Inactivating | FFEVF, ADNFLE, FMTLE |
| Germline | Inactivating | MCD, epileptic spasm, FCD | |
| Somatic | Inactivating | FCD, pachygyria, MCD | |
| NPRL2 | Germline | Inactivating | TLE, MCD |
| NPRL3 | Germline | Inactivating | TLE, MCD, FCD |
| KPTN | Inherited – autosomal recessive | Inactivating | Familial intellectual disability-macrocephaly syndrome, Macrocephaly |
| SZT2 | Inherited – autosomal recessive | Inactivating | Infantile encephalopathy with epilepsy |
| C12ORF66 | Germline | Inactivating | Macrocephaly |
mTOR : mechanistic target of rapamycin, CS : cowden syndrome, PROS : PIK3CA-related overgrowth spectrum, MEG : megalencephaly, HME : hemimegalencephaly, MCAP : megalencephaly-capillary malformation, MPPH : megalencephaly-polydactylyl-polymicrogyria-hydrocephalus, FCD : focal cortical dysplasia, PTEN : phosphatase and tensin homolog, BRRS : Bannayan-Riley-Ruvalcaba syndrome, LDD : Lhermitte-Duclos disease, PNH : periventricular nodular heterotopia, TSC : tuberous sclerosis, RHEB : Ras homolog enriched in brain, STRADA : STE20-related kinase adaptor α, PMSE : polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome, MTOR : mechanistic target of rapamycin, SKS : Smith-Kingsmore syndrome, FFEVF : familial focal epilepsy with variable foci, ADNFLE : autosomal dominant nocturnal epilepsy, FMTLE : familial mesial temporal lobe epilepsy, TLE : temporal lobe epilepsy
MUTATIONS IN THE ENERGY-SENSING PI3K-PTEN-AKT-TSC PATHWAY
Germline mutations in the PIK3CA gene, which activates the mTOR pathway, have been reported in CS [106]. Characterized by hamartomatous overgrowth of tissues, CS is also accompanied by epileptic seizure in a subset of patients [98,113]. There are also other brain and body overgrowth disorders with epilepsy that are caused by post-zygotic mutations in the PIK3CA pathway, which are termed PROS [70]. Additionally, mosaic or brain somatic mutations in PIK3CA have been reported in several other neurodevelopmental disorders with epilepsy, including the megalencephaly (MEG), hemimegalencephaly (HME), megalencephaly-capillary malformation (MCAP), megalencephaly-polydactyly-polymicrogyria-hydrocephalus syndromes, and focal cortical dysplasia (FCD) type IIa/IIb [36,68,79,99,122,132].
Classically, germline PTEN mutation has been reported in hamartoma tumor syndromes, including CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like conditions that share the pathological phenotypes of macrocephaly and megalencephaly [97]. Epileptic seizures have been reported in patients with germline mutation in PTEN [29,30,90].
Brain somatic mutations in AKT3 have primarily been detected in MCD, including the HME and FCD. Brain somatic gain of function mutations in AKT3 were first demonstrated to cause HME [79], followed by identification of somatic AKT3 mutations in FCD [68]. Germline AKT3 mutations were also reported in MEG [104]. To date, germline AKT3 mutations have been reported in HME, MEG, MEG with polymicrogyria, and MEG with periventricular nodular heterotopia [2].
TSC1 and TSC2 germline mutations have been identified in TSC patients [142]. Brain somatic mutations in TSC1 and TSC2 have also been discovered in FCD type IIb [82]. Homozygous TBC1D7 loss of function mutation have been identified in MEG patients [3,22]. More recently, brain somatic mutation in RHEB were identified in an HME patient [126].
STE20-related kinase adaptor α (STRADA) is a negative regulator of mTOR, by activating AMPK [107]. A loss-of-function mutation in STRADA was reported in autosomal recessive disease polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome, also called Pretzel syndrome (PMSE) [111].
Germline or mosaic mutation in MTOR has been described in the Smith-Kingsmore syndrome (SKS) OMIM #616638, which is characterized by epileptic seizure and neurodevelopmental defect [54]. SKS is a rare disease for which only 23 patients have been reported as of 2018 [54,101,102]. Brain somatic mutations in MTOR have also been described in FCD and HME, with varying degrees of mutational burden [91]. All of these mutations have been found to be missense gain-of-function mutations that activate the mTOR pathway.
MUTATIONS IN THE AMINO ACID-SENSING GATOR2-GATOR1-RAG GTPASE PATHWAY
Amino acid-sensing pathway is mediated by the GATOR2-GATOR1-Rag GTPase pathway. Since GATOR1 is a negative regulator of the mTORC1 pathway, researchers proposed that loss-of-function of GATOR1 would lead to neurodevelopmental disorders with epilepsy via hyperactivation of the mTORC1 pathway. Germline mutations in DEPDC5, NPRL2, and NPRL3 have been reported in familial focal epilepsy with variable foci, autosomal dominant nocturnal epilepsy, temporal lobe epilepsy, and familial mesial temporal lobe epilepsy [13,121]. In addition, germline mutations in DEPDC5, NPRL2, and NPRL3 have also been discovered in MCD and epileptic spasms patients [24,131,134]. Additionally, mosaicism and brain somatic mutation in GATOR1 have been reported in MCD [36], pachygyria [26] and familial focal epilepsy with focal cortical dysplasia [14]. Interestingly, it was recently demonstrated that epilepsy patients with focal cortical dysplasia carry a germline mutation in one allele of DEPDC5 and a brain somatic mutation in the other allele, suggesting a two-hit hypothesis as a disease mechanism [14,120]. Autosomal recessive mutations in the KICSTOR complex, including KPTN [9,108], SZT2 [11,143], and the genomic locus that contains C12orf66 [95], have been identified in neurodevelopmental disorders with epilepsy. Until now, mutations in GATOR2 have not been identified in epilepsy patients.
EPILEPSY ANIMAL MODELS OF HYPERACTIVATION OF THE MTOR PATHWAY
For extensive research into epilepsy, genetic animal models of mutations activating the mTOR pathway in epilepsy patients have been developed (Table 2). The first genetic model of epilepsy was the Pten KO mouse reported in 2001 [6,78]. Although Eker rats with germline mutations in the Tsc2 gene were the first genetic animal model of mTOR hyperactivation, the model was not suitable for analyzing epileptic seizures due to early phase lethality [155]. In 2002, the first TSC model with epilepsy, which was the astrocyte-specific Tsc1 KO model, was reported [141]; neuron-specific KO of Tsc1 was also found to lead to spontaneous seizure in the mouse model [96]. After these, the Tsc2 GFAP KO mouse model was developed and shown to lead to epilepsy [157]. A mouse model expressing human PI3KCA mutation in developing neural progenitors was generated to recapitulate pathological features of PROS syndrome, including epilepsy [123]. A knock-in mouse model with gain-of-function mutation in Akt3 has also been found to elicit spontaneous seizure [140].
Table 2.
Epilepsy animal models with genetic mTOR pathway hyperactivation
| Gene | Role and function | Genotype | Reference |
|---|---|---|---|
| PI3K | PI3K is a critical signaling pathway which integrates insulin signaling to the growth and survival | Hyperactive Pik3ca Nestin KI | [22] |
| PTEN | PTEN is a phosphatase and a negative regulator of the PI3K-PTEN-AKT-TSC pathway | Pten KO | [1,14] |
| Pten GFAP KO | [13,17,23] | ||
| Pten NSE KO | [33] | ||
| Inducible Pten KO | [145] | ||
| AKT | AKT is a positive regulator of the mTORCl via inhibition of TSC and the mTORC2 | Akt3 KI | [25] |
| Hyperactive Akt3 KI via in utero electroporation at the SVZ | [2] | ||
| TSC1/2 | TSC1/2 is a negative regulator of the mTORCl via inhibiting the action of RHEB | Tsc1 GFAP KO | [26] |
| Tsc1 SynI KO | [37] | ||
| Tsc1 Nestin KO | [8] | ||
| Tsc1 Emxl KO | [4] | ||
| Tsc1 KO via in utero electroporation at the SVZ | [15] | ||
| Tsc2 GFAP KO | [27,44] | ||
| Tsc2 KO via in utero electroporation at the SVZ | [15] | ||
| mTOR | mTOR is a serine/threonine kinase regulating various downstream targets which coordinate metabolism | Hyperactive MTOR KI via in utero electroporation at the SVZ | [16] |
| RHEB | RHEB activate the mTORC1 on the lysosomal membrane | Constitutively active Rheb KI via in utero electroporation at the SVZ | [9] |
| DEPDC5 | DEPDC5 comprise the amino acid-sensing pathway and is a negative regulator of the mTORCl | Depdc5 KO via in utero electroporation at the SVZ in Depec5 +/- mouse | [21] |
mTOR : mechanistic target of rapamycin, PI3K : phosphatidylinositol-3-kinase, KI : knock-in, PTEN : phosphatase and tensin homologon chromosome 10, GFAP : glial fibrillary acidic protein, KO : knock-out, NSE : neuron specific enonlase, SVZ : subventricular zone, TSC : tuberous sclerosis, SynI : synapsin I, RHEB : Ras homologue enriched in brain
Recently, epilepsy models of brain somatic mutations, which recapitulate focal malformations of cortical development (FMCD), have been reported. A mouse model of FMCD was first generated by in utero electroporation, thereby providing a small portion of neurons to express known mTOR mutations in human FMCD patients [7,83,110]; these models developed spontaneous seizures. Additionally, FMCD models of brain somatic mutation in mTOR pathway genes, such as RHEB or TSC, have consistently recapitulated spontaneous seizures [63,82]. Heterozygote GATOR1 gene (Depdc5, Nprl2, and Nprl3) KO animals have not been found to recapitulate epileptic seizures observed in patients with germline mutations in the GATOR1 genes [40,65,72,92]. Interestingly, FMCD mouse model caused by a biallelic two-hit mutation (brain somatic and germline) in Depdc5 was shown to lead to spontaneous seizure [120]. Homozygous KO of Szt2, a component of the KICSTOR complex, has been reported as leading to low seizure threshold in mice [46].
MTOR PATHWAY ACTIVATION IN EPILEPTOGENESIS
Epileptogenesis refers to the process leading to the first spontaneous seizure after pro-epileptogenic insult (e.g., genetic defect, brain injury, status epilepticus, or cancer). Epileptogenesis is a brain-wide circuit rewiring process comprising reorganization of microcircuits to long-range circuits, as well as gliosis, blood-brain barrier damage, inflammation, and neurodegeneration [114].
Since epilepsy is the consequence of electrical changes, electrophysiological characteristics in animal models and epilepsy patients have been analyzed in an attempt to understand the epileptogenic mechanism. Studies have indicated that highfrequency oscillations on electroencephalogram are characteristic of FCD [20]. Organotypic brain slice analysis of resected FCD lesions has revealed an electrographic firing pattern of ictal-like discharges after 4-aminopyridine treatment [5], which decreased with treatment with GABAA receptor antagonist.
In brain slice cultures of hyperactive mTOR signaling, researchers have found both the frequency and amplitude of the miniature excitatory postsynaptic current to be increased in glutamatergic synaptic transmission of the hippocampus [89,138]. In GABAergic neurons, hyperactive mTOR signaling increased evoked synaptic responses in the hippocampus [148]. In a subset of auditory cortical neurons of Pten conditional KO, synaptic inputs from the long-range connections, including the contralateral auditory cortex and thalamus, and local connections were increased [152].
In an epilepsy animal model of brain somatic mutations in the mTOR pathway, mutation-carrying cortical neurons showed an increased capacitance, increased cell size, reduced input resistance indicative of a higher current needed to reach the voltage threshold of the action potential, and increased gain of firing frequency [120]. Also, spontaneous excitatory postsynaptic current frequency was decreased in the mTOR pathway mutation-carrying cortical neurons [84,120].
There have been tremendous advances in the identification of the contribution of mTOR downstream functions to neuronal hypertrophy, dendritic branching, axon length, and neuronal migration. Although numerous epilepsy animal models have been utilized to unravel the path to epileptogenesis, the downstream mechanism of mTOR signaling that accounts for epileptogenesis remains unclear. The complexity of the downstream output of the mTOR pathway has hampered attempts to understand the epileptogenic mechanism. Using genetic manipulation techniques, normalizing major downstream outputs of hyperactive mTOR signaling, including translation via overexpressing constitutively active 4E-BP or knockdown of S6Ks [84] and autophagy via knockdown of OFD1 [110], have failed to prevent or even reduce epileptogenesis. Further studies defining the downstream pathway of mTOR in epileptogenesis will be necessary.
INHIBITION OF THE MTOR PATHWAY IN TREATMENT OF EPILEPTIC DISORDERS
Researchers have hypothesized that epilepsy resulting from mTOR pathway activation could be treated by mTOR inhibition, and mTOR inhibition therapies have been evaluated in several clinical trials for epilepsy (Table 3). Rapalogs, including sirolimus and everolimus, have been reported as promising new anti-epileptics because they can penetrate the blood brain-barrier [71]. Rapalogs have been extensively tested and have gained the US Food and Drug Administration (FDA) approval as anti-epileptics in TSC [27,47,73].
Table 3.
Clinical studies with the mTOR inhibitors for the anti-epileptic effects
| Type of study | Disease | Drug and dose | Number and age of patient(s) | Duration of treatment | Anti-epileptic effect | Refs |
|---|---|---|---|---|---|---|
| Prospective, open-label, phase I/II clinical trial | TSC | Everolimus | 16 patients; 3 year-old or older | Median duration : 21.5 months (range, 4.7–34.4) | Reduction in seizure frequency in 9/16 patients | [11] |
| 4.7–5.6 mg/m2/day | ||||||
| Case report | TSC | Everolimus | 1 patient; 10-year-old man | 12 months | Cessation of seizure | [20] |
| 4.5 mg/m2/day | ||||||
| Prospective, multicenter, openlabel, phase I/II clinical trial | TSC | Everolimus | 20 patients; median age : 8 years (age range, 2–21) | 12 weeks | Reduction in seizure frequency in 17/20 patients, 4 ofthese patients were seizure-free at 12 weeks | [24] |
| 5 mg/m2/day | ||||||
| Prospective, double-blind, parallel-group, placebo-controlled, multicenter phase III | TSC | Everolimus | 8 patients, children under the age of 3 | 35 months (range, 33–38) | Cessation of seizures in 1 patient, significant (at least a 50%) reduction in the number of seizures in 2 patients | [10] |
| 4.5 mg/m2/day | ||||||
| Case study series | TSC | Everolimus | 6 patients; median age : 5 years (age range, 2–12) | 36 weeks | Reduction in seizure frequency in 4/6 patients | [29] |
| 5–7 mg/day | ||||||
| Open-label, single center case series | TSC | Everolimus | 1 patients; 14-year-old female | 18 months | 25-50% seizure reduction | [3] |
| 5 mg/day | ||||||
| Case study | TSC | Everolimus | 1 patient; 13.5-year-old female | 12 days | Seizure aggravation | [30] |
| 5 mg/m2/day | ||||||
| Case study | TSC | Everolimus | 1 patient; 13-year-old female | 1.5 year | Reduction in seizure frequency | [28] |
| 5 mg/day | ||||||
| Prospective, open-label, phase I/II clinical trial | TSC | Everolimus | 14 patients; median age : 8 years (age range, 2.0–21.3) | 48 months | Reduction in seizure frequency in 13/14 patients (over 50% seizure reduction) | [12] |
| 5 mg/m2/day | ||||||
| Core phase, phase III, randomized, double-blind, placebo-controlled | TSC | Everolimus | 366 patients (including placebo group); median age; 10.1 years (age range, 2.2–56.3) | 18 weeks | Response rate; 15.1% with placebo 28.2% with low-exposure everolimus 40.0% with high-exposure everolimus | [7] |
| Low exposure group; 5.2 mg/m2/day (range, 1.3–14.5) | ||||||
| High exposure group; 7.5 mg/m2/day (range, 1.4–24.4) | ||||||
| Extension phase, phase III, randomized, double-blind, placebo-controlled | TSC | Everolimus | 294 patients; median age : 8.7 years (age range, 2.2–18.0) | 1 year | Sustained seizure reduction in 48.05%; Median percentage reduction in seizure frequency: 48.2% | [5] |
| 5–9 mg/m2/day | ||||||
| Case study series | TSC | Sirolimus | 3 patients; 15 years (age range, 5.5–21 years) | Median 4 months (range, 3–5) | Reduction in seizure frequency in 2/3 patients | [6] |
| 1.5 mg/kg/day | ||||||
| Case report | TSC | Sirolimus | 1 patient; 9-year-old female | 10 months | Reduction in seizure frequency | [18] |
| 0.15 mg/kg/day | ||||||
| Case study series | PMSE | Sirolimus | 6 patients; median age : 3 years (age range, 5 months–5 year) | 6 months | Reduction in seizure frequency | [19] |
| 1-5 mg/m2/day | ||||||
| Open-label, single center case | TSC | Sirolimus | 6 patients; median age : 6 years (age range, 3–17) | Median duration : 18 months (range, 6–36) | Over 50% reduction in seizure frequency in 5/6 patients | [3] |
| 1 mg/m2/day | ||||||
| Case report | HME | Sirolimus | 1 patient; 3-month-old man | 3 months | Seizure reduction (over 50%) | [31] |
| 1 mg/m2/day |
mTOR : mechanistic target of rapamycin, TSC : tuberous sclerosis, PMSE : polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome, HME : hemimegalencephaly
The first study to show the beneficial effect of the rapalogs on epilepsy did so in a mouse model of TSC [158]. In this study, early treatment with sirolimus prevented the development of epilepsy. The anti-epileptic effect of rapalogs has also been demonstrated in other genetic models of epilepsy with mTOR pathway hyperactivation, including Pten [87] KO and Strada KO [111]. Recently, intractable epilepsy mouse models with brain somatic mutation in the mTOR pathway were found to be cured by rapalogs [63,82,83]. Interestingly, epileptic seizure was almost completely suppressed by mTOR inhibitors in mouse models of TSC KO or mTOR activating mutation, but only partially suppressed in Pten KO mouse models [87]. These results suggest that an independent mechanism of epileptogenesis distinct from that in TSC KO or mTOR mutation is present in the Pten KO model. In nongenetic seizure models, including the pilocarpine-induced epilepsy model, kainic acid-induced epilepsy model, and absence epilepsy model, rapalogs partially reduced seizures. Citraro et al. [31] reported a representative studies of the anti-epileptic effect of rapalogs in an animal model of intractable epilepsy.
Everolimus have been approved by the FDA for treating subependymal giant astrocytoma, kidney tumors, and partial epilepsy in TSC patients. The first trial to test the efficacy of everolimus to treat epileptic seizure was reported in 2010 for TSC patients [74]. In this study, seizure frequency was reduced in nine of 16 patients with everolimus. Following studies supported the anti-epileptic effects of everolimus or sirolimus (Table 3).
The prospective, randomized, multicenter, placebo-controlled study testing the seizure suppression effect of everolimus on seizure frequency in TSC patients with drug-resistant epilepsy reported positive results [49]. Recently, long-term results of the prospective, open-label, non-randomized study of everolimus for treating epileptic seizure in TSC indicated that reduction in seizure frequency is sustained for up to 4 years [75]. This effect has been confirmed in the long-term follow-up of the EXIST3 trial [35]. Large-scale studies on the anti-epileptic effect of everolimus have described response rates for everolimus and a median seizure frequency reduction in respondents of about 40% [35,75]. In 2017 and 2018, respectively, both the European Medicinal Agency in Europe and the US FDA approved everolimus as an adjunctive therapy in partial-onset seizures in TSC patients aged 2 years and older. However, there are reports of non-responders, seizure aggravation, or an withdrawal after everolimus administration [35,149]. Meanwhile, clinical trials for treating epileptic seizure in FCD type II with everolimus have recently started (clinicaltrials.gov identifier NCT03198949).
The first report of the anti-epileptic effect of sirolimus in TSC patients, which appears to be similar to that of everolimus [23], was in 2009 [103]. In HME patients with brain somatic mutation in mTOR, sirolimus administration reduced seizures [153]. Interestingly, sirolimus has been shown to prevent epilepsy in PMSE patients [111].
Animal and human treatment studies reported that withdrawal of rapalogs provokes seizure recurrence [73]. In electrophysiological analysis of brain slices of resected epileptic foci, everolimus was found to reduce spontaneous excitatory postsynaptic activity, burst discharges, and epileptiform activity in TSC, FCD, and HME cases; effects were subtle in epilepsy patients without mTOR mutations [27].
While rapalogs have been approved for use in various human diseases, concerns have been raised for their long-term use and their adverse effect profile, including potentially serious adverse effects. The adverse effects of rapalogs include immunosuppression, mucositis, hyperlipidemia, hyperglycemia, diabetes-like syndrome, and fatal pneumonitis [42,76,109,139]. In a phase III clinical trial with everolimus, over 90% of 111 patients experienced an adverse effects of any grade [48]. With everolimus treatment for over 1 year, grade 3 or 4 adverse events, which are severe adverse events, were reported in 45% of younger patients and 38% of older patients among 150 patients [35]. We should stress that developmental delay caused by rapalogs [63] should be a concern, considering that the onset of intractable epilepsy with mTOR pathway mutation is usually before adolescence [4]. Additionally, treatment with rapalogs has been found to induce adverse nervous system-specific effects altering sociability [129], learning and memory [16,25], and anxiety [56] in mouse models.
For epilepsy caused by brain somatic mutations, therapeutic regimens must seek to avoid perturbations outside of the CNS, and in this regard, antisense oligonucleotide (ASO) drugs appear to be promising therapeutic options. ASO is a complementary oligonucleotide sequence of sense mRNA sequences that hampers normal gene expression processes, including splicing, transcription, and translation [136]. The characteristics of the bloodbrain barrier prevent ASO from permeating outside of the CNS when administered into the cerebrospinal fluid via intrathecal injection. Moreover, the half-life of ASO drugs is more than 3 months [19]. Recently, ASO drugs have been approved by the FDA for treating various types of neurodegenerative disorders [45]. We suspect that intrathecal injection of ASO drugs targeting mTOR itself, mTOR mutation, or downstream targets of mTOR will prove effective in treating intractable epilepsies while avoiding the adverse effects of rapalogs outside of the CNS.
CONCLUSION
Over the last two decades, extensive research to outline the roles of the mTOR pathway, to identify mutations in the mTOR pathway in human epilepsy patients, and to develop mTOR inhibitors have led to the discovery of novel strategies for diagnosing and treating intractable epilepsy. The overarching goal of this clinical research is attaining seizure-free status with few to no adverse effects in epilepsy patients. While rapalogs have proven to be effective in controlling seizures, complete seizure cessation has not been recorded in most of patients [35,75]. Also, about half of all TSC patients fail to respond to rapalogs. These discrepancies suggest that there might be an unknown biological mechanism at play. Considering about 40% of patients treated with rapalogs experience a severe adverse effect, it will be necessary to develop more potent and tolerable therapies.
While FMCD patients with drug-resistant epilepsy typically undergo surgical resection of the epileptic focus for treating the seizure, up to 50% of patients do not respond to therapy. Based on preclinical and clinical studies of TSC with rapalogs, it may no longer be necessary to perform highly invasive surgical resection for seizure treatment. However, there is a problem in that diagnosing FMCD patients with low-frequency brain somatic mutation in the mTOR pathway requires analyzing DNA from brain tissue after surgical resection. To alleviate this issue, minimally invasive diagnosis through biomarkers in patient cerebrospinal fluid would prove invaluable.
Acknowledgments
This work was supported by grants from the Suh Kyungbae Foundation (to J.H.L.), Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (H15C3143 and H16C0415 to J.H.L.), and Cheong-Am Science Fellow supported by POSCO Cheong-Am Foundation (to J.K.K).
Footnotes
J.H.L is a co-founder of SoVarGen, Inc. that develops new diagnostics and therapeutics for brain disorders. The remaining authors declare no competing financial interests.
INFORMED CONSENT
Informed consent was obtained from all individual participants included in this study.
Supplementary Materials
The online-only data supplement is available with this article at https://doi.org/10.3340/jkns.2019.0027.
Key discoveries in research on the role of the mTOR pathway in epilepsy.
References
- 1.Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science. 2017;358:807–813. doi: 10.1126/science.aan6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alcantara D, Timms AE, Gripp K, Baker L, Park K, Collins S, et al. Mutations of AKT3 are associated with a wide spectrum of developmental disorders including extreme megalencephaly. Brain. 2017;140:2610–2622. doi: 10.1093/brain/awx203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alfaiz AA, Micale L, Mandriani B, Augello B, Pellico MT, Chrast J, et al. TBC1D7 mutations are associated with intellectual disability, macrocrania, patellar dislocation, and celiac disease. Hum Mutat. 2014;35:447–451. doi: 10.1002/humu.22529. [DOI] [PubMed] [Google Scholar]
- 4.Aronica E, Becker AJ, Spreafico R. Malformations of cortical development. Brain Pathol. 2012;22:380–401. doi: 10.1111/j.1750-3639.2012.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avoli M, Bernasconi A, Mattia D, Olivier A, Hwa GG. Epileptiform discharges in the human dysplastic neocortex: in vitro physiology and pharmacology. Ann Neurol. 1999;46:816–826. doi: 10.1002/1531-8249(199912)46:6<816::aid-ana3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 6.Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- 7.Baek ST, Copeland B, Yun EJ, Kwon SK, Guemez-Gamboa A, Schaffer AE, et al. An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development. Nat Med. 2015;21:1445–1454. doi: 10.1038/nm.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baple EL, Maroofian R, Chioza BA, Izadi M, Cross HE, Al-Turki S, et al. Mutations in KPTN cause macrocephaly, neurodevelopmental delay, and seizures. Am J Hum Genet. 2014;94:87–94. doi: 10.1016/j.ajhg.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012;150:1196–1208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basel-Vanagaite L, Hershkovitz T, Heyman E, Raspall-Chaure M, Kakar N, Smirin-Yosef P, et al. Biallelic SZT2 mutations cause infantile encephalopathy with epilepsy and dysmorphic corpus callosum. Am J Hum Genet. 2013;93:524–529. doi: 10.1016/j.ajhg.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bast T, Ramantani G, Seitz A, Rating D. Focal cortical dysplasia: prevalence, clinical presentation and epilepsy in children and adults. Acta Neurol Scand. 2006;113:72–81. doi: 10.1111/j.1600-0404.2005.00555.x. [DOI] [PubMed] [Google Scholar]
- 13.Baulac S. MTOR signaling pathway genes in focal epilepsies. In: Rossignol E, Carmant L, Lacaille JC, editors. Progress in Brain Research. Vol. 226. Amsterdam: Elsevier; 2016. pp. 61–79. [DOI] [PubMed] [Google Scholar]
- 14.Baulac S, Ishida S, Marsan E, Miquel C, Biraben A, Nguyen DK, et al. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann Neurol. 2015;77:675–683. doi: 10.1002/ana.24368. [DOI] [PubMed] [Google Scholar]
- 15.Baybis M, Yu J, Lee A, Golden JA, Weiner H, McKhann G 2nd, et al. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol. 2004;56:478–487. doi: 10.1002/ana.20211. [DOI] [PubMed] [Google Scholar]
- 16.Beaumont V, Zhong N, Fletcher R, Froemke RC, Zucker RS. Phosphorylation and local presynaptic protein synthesis in calcium- and calcineurindependent induction of crayfish long-term facilitation. Neuron. 2001;32:489–501. doi: 10.1016/s0896-6273(01)00483-4. [DOI] [PubMed] [Google Scholar]
- 17.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323–1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bercury KK, Dai J, Sachs HH, Ahrendsen JT, Wood TL, Macklin WB. Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. J Neurosci. 2014;34:4466–4480. doi: 10.1523/JNEUROSCI.4314-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bishop KM. Progress and promise of antisense oligonucleotide therapeutics for central nervous system diseases. Neuropharmacology. 2017;120:56–62. doi: 10.1016/j.neuropharm.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 20.Blümcke I. Neuropathology of focal epilepsies: a critical review. Epilepsy Behav. 2009;15:34–39. doi: 10.1016/j.yebeh.2009.02.033. [DOI] [PubMed] [Google Scholar]
- 21.Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL. Control of p70 S6 kinase by kinase activity of FRAP in vivo. Nature. 1995;377:441–446. doi: 10.1038/377441a0. [DOI] [PubMed] [Google Scholar]
- 22.Capo-Chichi JM, Tcherkezian J, Hamdan FF, Décarie JC, Dobrzeniecka S, Patry L, et al. Disruption of TBC1D7, a subunit of the TSC1-TSC2 protein complex, in intellectual disability and megalencephaly. J Med Genet. 2013;50:740–744. doi: 10.1136/jmedgenet-2013-101680. [DOI] [PubMed] [Google Scholar]
- 23.Cardamone M, Flanagan D, Mowat D, Kennedy SE, Chopra M, Lawson JA. Mammalian target of rapamycin inhibitors for intractable epilepsy and subependymal giant cell astrocytomas in tuberous sclerosis complex. J Pediatr. 2014;164:1195–1200. doi: 10.1016/j.jpeds.2013.12.053. [DOI] [PubMed] [Google Scholar]
- 24.Carvill GL, Crompton DE, Regan BM, McMahon JM, Saykally J, Zemel M, et al. Epileptic spasms are a feature of DEPDC5 mTORopathy. Neurol Genet. 2015;1:e17. doi: 10.1212/NXG.0000000000000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casadio A, Martin KC, Giustetto M, Zhu H, Chen M, Bartsch D, et al. A transient, neuron-wide form of creb-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell. 1999;99:221–237. doi: 10.1016/s0092-8674(00)81653-0. [DOI] [PubMed] [Google Scholar]
- 26.Cen Z, Guo Y, Lou Y, Jiang B, Wang J, Feng J. De novo mutation in DEPDC5 associated with unilateral pachygyria and intractable epilepsy. Seizure. 2017;50:1–3. doi: 10.1016/j.seizure.2017.03.014. [DOI] [PubMed] [Google Scholar]
- 27.Cepeda C, Levinson S, Yazon VW, Barry J, Mathern GW, Fallah A, et al. Cellular antiseizure mechanisms of everolimus in pediatric tuberous sclerosis complex, cortical dysplasia, and non-mTOR-mediated etiologies. Epilepsia Open. 2018;3(Suppl Suppl 2):180–190. doi: 10.1002/epi4.12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen HH, Chen C, Hung SC, Liang SY, Lin SC, Hsu TR, et al. Cognitive and epilepsy outcomes after epilepsy surgery caused by focal cortical dysplasia in children: early intervention maybe better. Child’s Nerv Syst. 2014;30:1885–1895. doi: 10.1007/s00381-014-2463-y. [DOI] [PubMed] [Google Scholar]
- 29.Cheung KM, Lam CW, Chan YK, Siu WK, Yong L. Atypical focal cortical dysplasia in a patient with Cowden syndrome. Hong Kong Med J. 2014;20:165–167. doi: 10.12809/hkmj133863. [DOI] [PubMed] [Google Scholar]
- 30.Child ND, Cascino GD. Mystery case: Cowden syndrome presenting with partial epilepsy related to focal cortical dysplasia. Neurology. 2013;81:e98–e99. doi: 10.1212/WNL.0b013e3182a55ef0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Citraro R, Leo A, Constanti A, Russo E, De Sarro G. mTOR pathway inhibition as a new therapeutic strategy in epilepsy and epileptogenesis. Pharmacol Res. 2016;107:333–343. doi: 10.1016/j.phrs.2016.03.039. [DOI] [PubMed] [Google Scholar]
- 32.Crino PB. Focal brain malformations: a spectrum of disorders along the mTOR cascade. Novartis Found Smyp. 2007;288:260–272. doi: 10.1002/9780470994030.ch18. discussion 272-281. [DOI] [PubMed] [Google Scholar]
- 33.Crino PB. The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol. 2016;12:379–392. doi: 10.1038/nrneurol.2016.81. [DOI] [PubMed] [Google Scholar]
- 34.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1a transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 35.Curatolo P, Franz DN, Lawson JA, Yapici Z, Ikeda H, Polster T, et al. Adjunctive everolimus for children and adolescents with treatmentrefractory seizures associated with tuberous sclerosis complex: post-hoc analysis of the phase 3 EXIST-3 trial. Lancet Child Adolesc Health. 2018;2:495–504. doi: 10.1016/S2352-4642(18)30099-3. [DOI] [PubMed] [Google Scholar]
- 36.D’Gama AM, Geng Y, Couto JA, Martin B, Boyle EA, LaCoursiere CM, et al. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol. 2015;77:720–725. doi: 10.1002/ana.24357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Benedetti A, Joshi-Barve S, Rinker-Schaeffer C, Rhoads RE. Expression of antisense RNA against initiation factor eIF-4E mRNA in HeLa cells results in lengthened cell division times, diminished translation rates, and reduced levels of both eIF-4E and the p220 component of eIF-4F. Mol Cell Biol. 1991;11:5435–5445. doi: 10.1128/mcb.11.11.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell. 2012;47:535–546. doi: 10.1016/j.molcel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and ßTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 40.Dutchak PA, Laxman S, Estill SJ, Wang C, Wang Y, Wang Y, et al. Regulation of hematopoiesis and methionine homeostasis by mTORC1 inhibitor NPRL2. Cell Rep. 2015;12:371–379. doi: 10.1016/j.celrep.2015.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, et al. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eisen T, Sternberg CN, Robert C, Mulders P, Pyle L, Zbinden S, et al. Targeted therapies for renal cell carcinoma: review of adverse event management strategies. J Natl Cancer Inst. 2012;104:93–113. doi: 10.1093/jnci/djr511. [DOI] [PubMed] [Google Scholar]
- 43.Eng CP, Sehgal SN, Vézina C. Activity of rapamycin (AY-22,989) against transplanted tumors. J Antibiot (Tokyo) 1984;37:1231–1237. doi: 10.7164/antibiotics.37.1231. [DOI] [PubMed] [Google Scholar]
- 44.European Chromosome 16 Tuberous Sclerosis Consortium Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305–15. doi: 10.1016/0092-8674(93)90618-z. [DOI] [PubMed] [Google Scholar]
- 45.Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;337:1723–1732. doi: 10.1056/NEJMoa1702752. [DOI] [PubMed] [Google Scholar]
- 46.Frankel WN, Yang Y, Mahaffey CL, Beyer BJ, O’Brien TP. Szt2, a novel gene for seizure threshold in mice. Genes Brain Behav. 2009;8:568–576. doi: 10.1111/j.1601-183X.2009.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franz DN, Agricola K, Mays M, Tudor C, Care MM, Holland-Bouley K, et al. Everolimus for subependymal giant cell astrocytoma: 5-year final analysis. Ann Neurol. 2015;78:929–938. doi: 10.1002/ana.24523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, et al. Everolimus for subependymal giant cell astrocytoma in patients with tuberous sclerosis complex: 2-year open-label extension of the randomised EXIST-1 study. Lancet Oncol. 2014;15:1513–1520. doi: 10.1016/S1470-2045(14)70489-9. [DOI] [PubMed] [Google Scholar]
- 49.French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. 2016;388:2153–2163. doi: 10.1016/S0140-6736(16)31419-2. [DOI] [PubMed] [Google Scholar]
- 50.Gingras A, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated by the Akt(PKB) signaling pathway. Genes Dev. 1998;12:502–513. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, et al. Regulation of 4E-BP1 phosphorylation : a novel two-step mechanism. Genes Dev. 1999;13:1422–1437. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors : effectors of mRNA recruitment of translation. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 53.Gong X, Zhang L, Huang T, Lin T V., Miyares L, Wen J, et al. Activating the translational repressor 4E-BP or reducing S6K-GSK3ß activity prevents accelerated axon growth induced by hyperactive mTOR in vivo. Hum Mol Genet. 2015;24:5746–5758. doi: 10.1093/hmg/ddv295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gordo G, Tenorio J, Arias P, Santos-Simarro F, García-Miñaur S, Moreno JC, et al. mTOR mutations in Smith-Kingsmore syndrome: four additional patients and a review. Clin Genet. 2018;93:762–775. doi: 10.1111/cge.13135. [DOI] [PubMed] [Google Scholar]
- 55.Gu X, Orozco JM, Saxton RA, Condon KJ, Liu GY, Krawczyk PA, et al. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science. 2017;358:813–818. doi: 10.1126/science.aao3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hadamitzky M, Herring A, Kirchhof J, Bendix I, Haight MJ, Keyvani K, et al. Repeated systemic treatment with rapamycin affects behavior and amygdala protein expression in rats. Int J Neuropsychopharmacol. 2018;21:592–602. doi: 10.1093/ijnp/pyy017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, et al. LeucyltRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012;149:410–424. doi: 10.1016/j.cell.2012.02.044. [DOI] [PubMed] [Google Scholar]
- 58.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 59.Hentges K, Thompson K, Peterson A. The flat-top gene is required for the expansion and regionalization of the telencephalic primordium. Development. 1999;126:1601–9. doi: 10.1242/dev.126.8.1601. [DOI] [PubMed] [Google Scholar]
- 60.Hiremath LS, Webb NR, Rhoads RE. Immunological detection of the messenger RNA cap-binding protein. J Biol Chem. 1985;260:7843–7849. [PubMed] [Google Scholar]
- 61.Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 62.Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hsieh LS, Wen JH, Claycomb K, Huang Y, Harrsch FA, Naegele JR, et al. Convulsive seizures from experimental focal cortical dysplasia occur independently of cell misplacement. Nat Commun. 2016;7:11753. doi: 10.1038/ncomms11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- 65.Hughes J, Dawson R, Tea M, McAninch D, Piltz S, Jackson D, et al. Knockout of the epilepsy gene Depdc5 in mice causes severe embryonic dysmorphology with hyperactivity of mTORC1 signalling. Sci Rep. 2017;7:12618. doi: 10.1038/s41598-017-12574-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iffland PH, 2nd, Crino PB. Focal cortical dysplasia: gene mutations, cell signaling, and therapeutic implications. Annu Rev Pathol. 2017;12:547–571. doi: 10.1146/annurev-pathol-052016-100138. [DOI] [PubMed] [Google Scholar]
- 67.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jansen LA, Mirzaa GM, Ishak GE, O’Roak BJ, Hiatt JB, Roden WH, et al. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain. 2015;138(Pt 6):1613–1628. doi: 10.1093/brain/awv045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jung J, Genau HM, Behrends C. Amino acid-dependent mTORC1 regulation by the lysosomal membrane protein SLC38A9. Mol Cell Biol. 2015;35:2479–2494. doi: 10.1128/MCB.00125-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Keppler-Noreuil KM, Parker VE, Darling TN, Martinez-Agosto JA. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am J Med Genet C Semin Med Genet. 2016;172:402–421. doi: 10.1002/ajmg.c.31531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klawitter J, Gottschalk S, Hainz C, Leibfritz D, Christians U, Serkova NJ. Immunosuppressant neurotoxicity in rat brain models: oxidative stress and cellular metabolism. Chem Res Toxicol. 2010;23:608–619. doi: 10.1021/tx900351q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kowalczyk MS, Hughes JR, Babbs C, Sanchez-Pulido L, Szumska D, Sharpe JA, et al. Nprl3 is required for normal development of the cardiovascular system. Mamm Genome. 2012;23:404–415. doi: 10.1007/s00335-012-9398-y. [DOI] [PubMed] [Google Scholar]
- 73.Krueger DA, Care MM, Agricola K, Tudor C, Mays M, Franz DN. Everolimus long-term safety and efficacy in subependymal giant cell astrocytoma. Neurology. 2013;80:574–580. doi: 10.1212/WNL.0b013e3182815428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;369:1801–1811. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 75.Krueger DA, Wilfong AA, Mays M, Talley CM, Agricola K, Tudor C, et al. Long-term treatment of epilepsy with everolimus in tuberous sclerosis. Neurology. 2016;87:2408–2415. doi: 10.1212/WNL.0000000000003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kuegler PB, Zimmer B, Waldmann T, Baudis B, Ilmjärv S, Hescheler J, et al. Markers of murine embryonic and neural stem cells, neurons and astrocytes: reference points for developmental neurotoxicity testing. ALTEX. 2010;27:17–42. doi: 10.14573/altex.2010.1.16. [DOI] [PubMed] [Google Scholar]
- 77.Kumar V, Zhang MX, Swank MW, Kunz J, Wu G. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, et al. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- 79.Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44:941–945. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee SK, Kim DW. Focal cortical dysplasia and epilepsy surgery. J Epilepsy Res. 2013;3:43–47. doi: 10.14581/jer.13009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTORdependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lim JS, Gopalappa R, Kim SH, Ramakrishna S, Lee M, Kim WI, et al. Somatic mutations in TSC1 and TSC2 cause focal cortical dysplasia. Am J Hum Genet. 2017;100:454–472. doi: 10.1016/j.ajhg.2017.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lim JS, Kim WI, Kang HC, Kim SH, Park AH, Park EK, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. 2015;21:395–400. doi: 10.1038/nm.3824. [DOI] [PubMed] [Google Scholar]
- 84.Lin TV, Hsieh L, Kimura T, Malone TJ, Bordey A. Normalizing translation through 4E-BP prevents mTOR-driven cortical mislamination and ameliorates aberrant neuron integration. Proc Natl Acad Sci U S A. 2016;113:11330–11335. doi: 10.1073/pnas.1605740113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lipton JO, Sahin M. The neurology of mTOR. Neuron. 2014;84:275–291. doi: 10.1016/j.neuron.2014.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ljungberg MC, Bhattacharjee MB, Lu Y, Armstrong DL, Yoshor D, Swann JW, et al. Activation of mammalian target of rapamycin in cytomegalic neurons of human cortical dysplasia. Ann Neurol. 2006;60:420–429. doi: 10.1002/ana.20949. [DOI] [PubMed] [Google Scholar]
- 87.Ljungberg MC, Sunnen CN, Lugo JN, Anderson AE, D’Arcangelo G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis Model Mech. 2009;2:389–398. doi: 10.1242/dmm.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 89.Luikart BW, Schnell E, Washburn EK, Bensen AL, Tovar KR, Westbrook GL. Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J Neurosci. 2011;31:4345–4354. doi: 10.1523/JNEUROSCI.0061-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marchese M, Conti V, Valvo G, Moro F, Muratori F, Tancredi R, et al. Autism-epilepsy phenotype with macrocephaly suggests PTEN, but not GLIALCAM, genetic screening. BMC Med Genet. 2014;15:26. doi: 10.1186/1471-2350-15-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marsan E, Baulac S. Review: mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol Appl Neurobiol. 2018;44:6–17. doi: 10.1111/nan.12463. [DOI] [PubMed] [Google Scholar]
- 92.Marsan E, Ishida S, Schramm A, Weckhuysen S, Muraca G, Lecas S, et al. Depdc5 knockout rat: a novel model of mTORopathy. Neurobiol Dis. 2016;89:180–189. doi: 10.1016/j.nbd.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 93.Martel RR, Klicius J, Galet S. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can J Physiol Pharmacol. 1977;55:48–51. doi: 10.1139/y77-007. [DOI] [PubMed] [Google Scholar]
- 94.Martinez-Lizana E, Fauser S, Brandt A, Schuler E, Wiegand G, Doostkam S, et al. Long-term seizure outcome in pediatric patients with focal cortical dysplasia undergoing tailored and standard surgical resections. Seizure. 2018;62:66–73. doi: 10.1016/j.seizure.2018.09.021. [DOI] [PubMed] [Google Scholar]
- 95.Mc Cormack A, Sharpe C, Gregersen N, Smith W, Hayes I, George AM, et al. 12q14 microdeletions: additional case series with confirmation of a macrocephaly region. Case Rep Genet. 2015;2015:192071. doi: 10.1155/2015/192071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mester J, Eng C. When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet. 2013;163C:114–121. doi: 10.1002/ajmg.c.31364. [DOI] [PubMed] [Google Scholar]
- 98.Millichap J. Cowden syndrome with cortical malformation and epilepsy. Pediatr Neurol Briefs. 2013;34:7. [Google Scholar]
- 99.Mirzaa GM, Conway RL, Gripp KW, Lerman-Sagie T, Siegel DH, deVries LS, et al. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A. 2012;158A:269–291. doi: 10.1002/ajmg.a.34402. [DOI] [PubMed] [Google Scholar]
- 100.Miyata H, Chiang AC, Vinters HV. Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol. 2004;56:510–9. doi: 10.1002/ana.20234. [DOI] [PubMed] [Google Scholar]
- 101.Moosa S, Böhrer-Rabel H, Altmüller J, Beleggia F, Nürnberg P, Li Y, et al. Smith-Kingsmore syndrome: a third family with the MTOR mutation c.5395G>A p.(Glu1799Lys) and evidence for paternal gonadal mosaicism. Am J Med Genet A. 2017;173:264–267. doi: 10.1002/ajmg.a.37999. [DOI] [PubMed] [Google Scholar]
- 102.Mroske C, Rasmussen K, Shinde DN, Huether R, Powis Z, Lu HM, et al. Germline activating MTOR mutation arising through gonadal mosaicism in two brothers with megalencephaly and neurodevelopmental abnormalities. BMC Med Genet. 2015;16:102. doi: 10.1186/s12881-015-0240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Muncy J, Butler IJ, Koenig KM. Rapamycin reduces seizure frequency in tuberous sclerosis complex. J Child Neurol. 2009;24:477. doi: 10.1177/0883073808324535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nellist M, Schot R, Hoogeveen-Westerveld M, Neuteboom RF, van der Louw EJ, Lequin MH, et al. Germline activating AKT3 mutation associated with megalencephaly, polymicrogyria, epilepsy and hypoglycemia. Mol Genet Metab. 2015;114:467–473. doi: 10.1016/j.ymgme.2014.11.018. [DOI] [PubMed] [Google Scholar]
- 105.Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle. 2011;10:2305–2316. doi: 10.4161/cc.10.14.16586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Orloff MS, He X, Peterson C, Chen F, Chen JL, Mester JL, et al. Germline PIK3CA and AKT1 mutations in cowden and cowden-like syndromes. Am J Hum Genet. 2013;92:76–80. doi: 10.1016/j.ajhg.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Orlova KA, Parker WE, Heuer GG, Tsai V, Yoon J, Baybis M, et al. STRADa deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J Clin Invest. 2010;120:1591–1602. doi: 10.1172/JCI41592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pajusalu S, Reimand T, Õunap K. Novel homozygous mutation in KPTN gene causing a familial intellectual disability-macrocephaly syndrome. Am J Med Genet A. 2015;167A:1913–1915. doi: 10.1002/ajmg.a.37105. [DOI] [PubMed] [Google Scholar]
- 109.Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf. 2013;12:177–186. doi: 10.1517/14740338.2013.752814. [DOI] [PubMed] [Google Scholar]
- 110.Park SM, Lim JS, Ramakrishina S, Kim SH, Kim WK, Lee J, et al. Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination. Neuron. 2018;99:83–97. doi: 10.1016/j.neuron.2018.05.039. e7. [DOI] [PubMed] [Google Scholar]
- 111.Parker WE, Orlova KA, Parker WH, Birnbaum JF, Krymskaya VP, Goncharov DA, et al. Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci Transl Med. 2013;5:182ra53. doi: 10.1126/scitranslmed.3005271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Peters JM, Taquet M, Prohl AK, Scherrer B, van Eeghen AM, Prabhu SP, et al. Diffusion tensor imaging and related techniques in tuberous sclerosis complex: review and future directions. Future Neurol. 2013;8:583–597. doi: 10.2217/fnl.13.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pilarski R. Cowden syndrome: a critical review of the clinical literature. J Genet Couns. 2009;18:13–27. doi: 10.1007/s10897-008-9187-7. [DOI] [PubMed] [Google Scholar]
- 114.Pitkänen A, Lukasiuk K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 2011;10:173–186. doi: 10.1016/S1474-4422(10)70310-0. [DOI] [PubMed] [Google Scholar]
- 115.Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–48. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Raab-Graham KF, Haddick PC, Jan YN, Jan LY. Activity- and mTORdependent suppression of Kv1.1 channel mRNA translation in dendrites. Science. 2006;314:144–148. doi: 10.1126/science.1131693. [DOI] [PubMed] [Google Scholar]
- 118.Rabanal-Ruiz Y, Otten EG, Korolchuk VI. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017;61:565–584. doi: 10.1042/EBC20170027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rennebeck G, Kleymenova EV, Anderson R, Yeung RS, Artzt K, Walker CL. Loss of function of the tuberous sclerosis 2 tumor suppressor gene results in embryonic lethality characterized by disrupted neuroepithelial growth and development. Proc Natl Acad Sci U S A. 1998;95:15629–15634. doi: 10.1073/pnas.95.26.15629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ribierre T, Deleuze C, Bacq A, Baldassari S, Marsan E, Chipaux M, et al. Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J Clin Investig. 2018;128:2452–2458. doi: 10.1172/JCI99384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ricos MG, Hodgson BL, Pippucci T, Saidin A, Ong YS, Heron SE, et al. Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann Neurol. 2016;79:120–131. doi: 10.1002/ana.24547. [DOI] [PubMed] [Google Scholar]
- 122.Rivière JB, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, Conway RL, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Roy A, Skibo J, Kalume F, Ni J, Rankin S, Lu Y, et al. Mouse models of human PIK3CA-related brain overgrowth have acutely treatable epilepsy. Elife. 2015;4:e12703. doi: 10.7554/eLife.12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sabatini DM. Twenty-five years of mTOR: uncovering the link from nutrients to growth. Proc Natl Acad Sci U S A. 2017;114:11818–11825. doi: 10.1073/pnas.1716173114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 126.Salinas V, Vega P, Piccirilli MV, Chicco C, Ciraolo C, Christiansen S, et al. Identification of a somatic mutation in the RHEB gene through high depth and ultra-high depth next generation sequencing in a patient with Hemimegalencephaly and drug resistant Epilepsy. Eur J Med Genet. 2018 doi: 10.1016/j.ejmg.2018.11.005. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 127.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 129.Saré RM, Song A, Loutaev I, Cook A, Maita I, Lemons A, et al. Negative effects of chronic rapamycin treatment on behavior in a mouse model of fragile X syndrome. Front Mol Neurosci. 2018;10:452. doi: 10.3389/fnmol.2017.00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Scerri T, Riseley JR, Gillies G, Pope K, Burgess R, Mandelstam SA, et al. Familial cortical dysplasia type IIA caused by a germline mutation in DEPDC5. Ann Clin Transl Neurol. 2015;2:575–580. doi: 10.1002/acn3.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schick V, Majores M, Engels G, Spitoni S, Koch A, Elger CE, et al. Activation of Akt independent of PTEN and CTMP tumor-suppressor gene mutations in epilepsy-associated Taylor-type focal cortical dysplasias. Acta Neuropathol. 2006;112:715–725. doi: 10.1007/s00401-006-0128-y. [DOI] [PubMed] [Google Scholar]
- 133.Schwartzkroin PA, Walsh CA. Cortical malformations and epilepsy. Ment Retard Dev Disabil Res Rev. 2000;6:268–280. doi: 10.1002/1098-2779(2000)6:4<268::AID-MRDD6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 134.Sim JC, Scerri T, Fanjul-Fernández M, Riseley JR, Gillies G, Pope K, et al. Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann Neurol. 2016;79:132–137. doi: 10.1002/ana.24502. [DOI] [PubMed] [Google Scholar]
- 135.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Southwell AL, Skotte NH, Bennett CF, Hayden MR. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol Med. 2012;18:634–643. doi: 10.1016/j.molmed.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 137.Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. 2014;83:1131–1143. doi: 10.1016/j.neuron.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- 139.Teutonico A, Schena PF, Di Paolo S. Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol. 2005;16:3128–3135. doi: 10.1681/ASN.2005050487. [DOI] [PubMed] [Google Scholar]
- 140.Tokuda S, Mahaffey CL, Monks B, Faulkner CR, Birnbaum MJ, Danzer SC, et al. A novel Akt3 mutation associated with enhanced kinase activity and seizure susceptibility in mice. Hum Mol Genet. 2011;20:988–999. doi: 10.1093/hmg/ddq544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, et al. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52:285–296. doi: 10.1002/ana.10283. [DOI] [PubMed] [Google Scholar]
- 142.van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805–808. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- 143.Venkatesan C, Angle B, Millichap JJ. Early-life epileptic encephalopathy secondary to SZT2 pathogenic recessive variants. Epileptic Disord. 2016;18:195–200. doi: 10.1684/epd.2016.0828. [DOI] [PubMed] [Google Scholar]
- 144.Vézina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic I. Taxonomy of the producing Streptomycete and isolation of the active principle. J Antibiot (Tokyo) 1975;28:721–726. doi: 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- 145.von der Brelie C, Waltereit R, Zhang L, Beck H, Kirschstein T. Impaired synaptic plasticity in a rat model of tuberous sclerosis. Eur J Neurosci. 2006;23:686–692. doi: 10.1111/j.1460-9568.2006.04594.x. [DOI] [PubMed] [Google Scholar]
- 146.Wahl SE, McLane LE, Bercury KK, Macklin WB, Wood TL. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J Neurosci. 2014;34:4453–4465. doi: 10.1523/JNEUROSCI.4311-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Way SW, Mckenna J 3rd, Mietzsch U, Reith RM, Wu HC, Gambello MJ. Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum Mol Genet. 2009;18:1252–1265. doi: 10.1093/hmg/ddp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Weston MC, Chen H, Swann JW. Multiple roles for mammalian target of rapamycin signaling in both glutamatergic and GABAergic synaptic transmission. J Neurosci. 2012;32:11441–11452. doi: 10.1523/JNEUROSCI.1283-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wiemer-Kruel A, Woerle H, Strobl K, Bast T. Everolimus for the treatment of subependymal giant cell astrocytoma probably causing seizure aggravation in a child with tuberous sclerosis complex: a case report. Neuropediatrics. 2014;45:129–131. doi: 10.1055/s-0033-1360481. [DOI] [PubMed] [Google Scholar]
- 150.Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature. 2017;543:438–442. doi: 10.1038/nature21423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wyant GA, Abu-Remaileh M, Frenkel EM, Laqtom NN, Dharamdasani V, Lewis CA, et al. Nufip1 is a ribosome receptor for starvation-induced ribophagy. Science. 2018;360:751–758. doi: 10.1126/science.aar2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xiong Q, Oviedo HV, Trotman LC, Zador AM. PTEN regulation of local and long-range connections in mouse auditory cortex. J Neurosci. 2012;32:1643–1652. doi: 10.1523/JNEUROSCI.4480-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Xu Q, Uliel-Sibony S, Dunham C, Sarnat H, Flores-Sarnat L, Brunga L, et al. mTOR inhibitors as a new therapeutic strategy in treatment resistant epilepsy in hemimegalencephaly: a case report. J Child Neurol. 2019;34:132–138. doi: 10.1177/0883073818813238. [DOI] [PubMed] [Google Scholar]
- 154.Yeung RS, Katsetos CD, Klein-szanto A. Subependymal astrocytic hamartomas in the Eker rat model of tuberous sclerosis. Am J Pathol. 1997;151:1477–1486. [PMC free article] [PubMed] [Google Scholar]
- 155.Yeung RS, Xiao GH, Jin F, Lee WC, Testa JR, Knudson AG. Predisposition to renal carcinoma in the Eker rat is determined by germ-line mutation of the tuberous sclerosis 2 (TSC2) gene. Proc Natl Acad Sci U S A. 1994;91:11413–11416. doi: 10.1073/pnas.91.24.11413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yoon BC, Zivraj KH, Holt CE. Local translation and mRNA trafficking in axon pathfinding. Results Probl Cell Differ. 2009;48:269–288. doi: 10.1007/400_2009_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zeng LH, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum Mol Genet. 2011;20:445–454. doi: 10.1093/hmg/ddq491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zoncu R, Efeyan A, Sabatini DM. MTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Key discoveries in research on the role of the mTOR pathway in epilepsy.