

Abstract
Human pluripotent stem cells (hPSCs) offer many potential applications for drug screening and ‘disease in a dish’ assay capabilities. However, a more ambitious goal is to develop cell therapeutics using hPSCs to generate and replace somatic cells that are lost as a result of disease or injury. This Spotlight article will describe the state of progress of some of the hPSC-derived therapeutics that offer the most promise for clinical use. Lessons from developmental biology have been instrumental in identifying signaling molecules that can guide these differentiation processes in vitro, and will be described in the context of these cell therapy programs.
KEY WORDS: Human Pluripotent Stem Cell, Clinical medicine, HPSC-cardiomyocytes
Summary: This opinion piece considers the progress in the use of human PSCs in regenerative medicine – fuelled by advances in developmental biology – in five areas that offer great promise for clinical applications.
Introduction
Human embryonic stem cells (hESCs) were first reported in 1998 by Dr Jamie Thomson's group, and their reprogrammed cousins, human induced pluripotent stem cells (hiPSCs), were described in 2007 (Yu et al., 2007; Takahashi et al., 2007). Scientists, medical professionals and the lay public were quick to realize the potential of these cells, and research moved quickly in several directions. As a result, there have been major advances in human developmental biology, drug development, toxicology, human genetics, tissue engineering and regenerative medicine. Within these broad areas, the premise of replacing somatic cells with differentiated hPSCs for regenerative purposes is arguably the most challenging, for several reasons (see Box 1): first and foremost is the difficulty of developing in vitro methods that generate the target cell type at sufficient purity and with the appropriate cellular functions. Differentiation protocols commonly yield heterogeneous cell populations, and it is crucial to define the threshold level of each non-target cell type that presents no medical risk to patients receiving the therapy. Residual undifferentiated hPSCs are a particular concern and must be reduced to a level that is demonstrably safe and minimizes the risk of teratoma and teratocarcinoma. Cell graft survival and integration are another important aspect of achieving therapeutic benefit. In this context, the phenotypic maturity of cells differentiated from hPSCs can significantly affect these parameters. Similarly, the format in which cells are delivered can impact their survival, integration and, ultimately, their functional benefit. The extent to which these challenges are being addressed by the therapeutic programs described below will probably affect their success in the clinic.
Box 1. The regulatory path from the lab to the clinic
Advancing a PSC-derived cell therapy from the laboratory to a Phase 1 clinical trial requires demonstrating to the FDA or other regulatory body that the production process is well controlled and the product is safe and efficacious in animal models. In the case of an allogeneic cell therapy, it also requires establishing and characterizing cell banks of undifferentiated PSCs. A crucial characteristic is a normal karyotype, to minimize the risk of transplanting transformed cells. The same demonstration of normal karyotype is required for iPSCs intended for autologous cell therapy. Although the PSC differentiation process can be developed in a research lab, ultimately, the production process must be adapted to current Good Manufacturing Practices (cGMP) conditions to generate clinical material. This requires the development and execution of Standard Operating Procedures (SOPs) for every step of the process to ensure reproducibility and tight control. In addition, the cells generated by this process must meet strict product specifications. These specifications are established through an iterative process in which production runs are assayed and then tested for efficacy and safety. Specifications for hPSC-derived therapeutics typically include purity of the target cell type, as well as quantitation of contaminating cell types in the final product. In addition to efficacy testing, hPSC-derived cell therapies need to be evaluated for tumorigenicity and biodistribution in animal models, as well as standardized assays for sterility and adventitious agents, before they can be used in a clinical trial.
This Spotlight article focuses on the use of human pluripotent stem cells (hPSCs) in regenerative medicine. We describe five areas that offer great promise for clinical applications: spinal cord injury, retinal blindness, heart failure, diabetes and Parkinson's disease (Fig. 1), and we conclude with a few thoughts about the current state of the field and speculate on its immediate future. Space limitations dictate that we focus on clinical or near-clinical data, so we apologize to colleagues whose more fundamental studies are not described. In this regard, it is worth noting that early clinical data are not often reported in peer-reviewed journals, and when they are, the publications lag significantly behind completion of the studies. Therefore, we have included data from less traditional sources as a way to inform the reader of the most current progress, and noted the source of that information in the accompanying text.
Fig. 1.
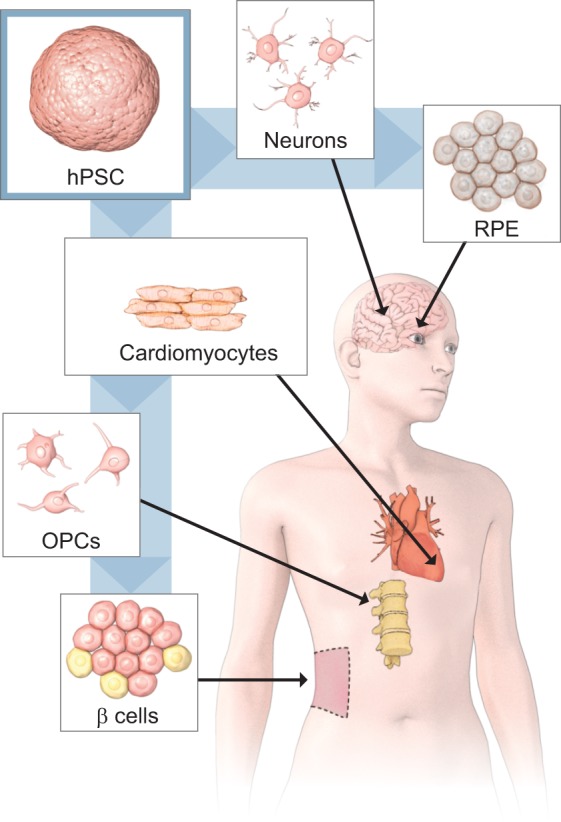
hPSC-derived cell therapeutics advancing to clinical testing. hPSC-derived cell therapeutics advancing to clinical testing include retinal pigment epithelium (RPE) for retinal degenerative diseases, dopaminergic neurons (Neurons) for Parkinson's disease, cardiomyocytes for heart disease, oligodendrocyte progenitor cells (OPCs) for spinal cord injury and β-islet cells (β cells) for diabetes.
Spinal cord injury
Traumatic injury to the spinal cord can result in the permanent loss of neural conduction through descending motor tracts and ascending sensory tracts. Although the death of neurons and breaks in axonal structures are the primary cause of the functional deficit, a neuronal replacement strategy for spinal cord injury is exceedingly challenging. However, it has been reported that spinal contusion injury is characterized by axons that remain intact in the corticospinal tract early after the injury but that are demyelinated (Bresnahan et al., 1976; Blight, 1983; Totoiu and Keirstead, 2005). Without appropriate myelination, nerve impulse conduction velocity is severely compromised (Nashmi and Fehlings, 2001), and axons are susceptible to degeneration (Irvine and Blakemore, 2008). Therefore, numerous studies have been conducted in animal models of spinal injury, in which remyelinating cells were transplanted with the expectation that they differentiate into mature myelinating oligodendrocytes in vivo and restore axonal conduction. This therapeutic approach has demonstrated improved motor function in animal models (Keirstead et al., 2005; Bambakidis and Miller, 2004; Cao et al., 2005; Lee et al., 2005, Biernaskie et al., 2007; Yasuda et al., 2011; Hawryluk et al., 2014; All et al., 2015). However, it is worth noting that the beneficial effects of trophic factors secreted by transplanted cells might also contribute to functional efficacy (Zhang et al., 2006).
Based on these findings, the Geron Corporation developed an hESC-derived oligodendrocyte progenitor cell (OPC), termed GRN-OPC1, to treat spinal cord injury in humans (Nistor et al., 2005; Keirstead et al., 2005). GRN-OPC1 cells were obtained by exposing the H1 hESC line (Thomson et al., 1998) to a staged 42-day differentiation protocol (Nistor et al., 2005). In this method, differentiation was initiated by culturing the cells in suspension using a chemically defined medium containing epidermal growth factor (EGF) and fibroblast growth factor (FGF) 2 (Nistor et al., 2005), which closely resembles the culture conditions developed for isolating neural progenitor cells from the embryonic rodent brain (Vescovi et al., 1993). Retinoic acid was also added early in the differentiation process, presumably because it is known to promote neural ectoderm differentiation in hESCs (Levenberg et al., 2003).
The safety and therapeutic efficacy of GRN-OPC1 cells transplanted in acute thoracic spinal cord injury patients was assessed in an open-label Phase 1 clinical trial initiated in 2010, the first trial ever to use a pluripotent stem cell-derived therapeutic. At the time, there was some controversy in the field as to whether it was too soon for clinical testing of hPSC-derived therapies and whether the GRN-OPC1 cells were sufficiently committed to the oligodendrocyte phenotype for optimal remyelination. As described in an interview with Geron's Chief Scientific Officer, Dr Jane Lebkowski (Lebkowski, 2011), the trial was designed as a dose-escalation study, in which the first cohort of transiently immunosuppressed patients receive a low dose of 2×106 cells delivered as a suspension by direct injection into the injury site. As is typical of a dose-escalation study for a first clinical trial, the initial dose is for evaluating safety and is not expected to show efficacy. Based on the 2.5×105 cell dose of GRN-OPC1 that improved motor function in 200-g rats, an efficacious dose for humans scaled to body weight would be at least 2×107 cells. Geron discontinued all of its stem cell programs in late 2011 for business reasons, and the GRN-OPC1 trial was halted with four patients enrolled.
Two years after Geron terminated its stem cell programs, the technologies and assets were acquired by Asterias Biotherapeutics, and GRN-OPC1 cells were renamed AST-OPC1. At the time of a recent oral presentation at the American Society of Gene & Cell Therapy annual meeting (Lebkowski, 2014), five patients had been treated in the first cohort of the AST-OPC1 trial. As reported in the presentation, although some minor (grade 1-3) adverse events were associated with the transplanted cells, no serious adverse events (grade 4 or 5) were observed in serial MRI analyses from one week to one year post-transplant. Importantly, there was no evidence of expansive tumors or cysts or of antibody- or cell-mediated rejection. Annual neurological evaluations of motor and sensory function for up to three years did not reveal any major improvements in these five patients, as expected for the low dose of cells administered in this first phase of the trial. The long-term follow-up will extend to 15 years from the time of treatment, as a rigorous test of safety. Improvements of one or two vertebral segments in the level of thoracic spinal cord injury would not necessarily be expected to impact function and quality of life. By contrast, repair of one or two segments in cervical injuries could have profound benefits, particularly regarding upper limb function and dependence on ventilator-assisted breathing. Under the sponsorship of Asterias Biotherapeutics, a Phase 1/2a trial has been approved by the FDA for evaluating dose escalation of AST-OPC1 in patients with functionally complete cervical injuries, and enrollment has begun. In the absence of any FDA-approved therapeutics for acute spinal cord injury, beneficial effects of AST-OPC1s would be an important finding. Even if this therapy does not ultimately improve function in spinal cord injury, it might still benefit other therapeutic indications in which axonal demyelination is a contributing factor.
Retinal degeneration
A major clinical focus of hPSC-derived cell therapy has been the replacement of damaged retinal pigment epithelium (RPE) in the eye. The RPE serves many functions: it absorbs scattered light and supports photoreceptors through phagocytosis of photoreceptor outer segment membranes; it also supplies nutrients and trophic factors, and buffers extracellular ionic changes (Sparrrow et al., 2010). RPE deterioration or malfunction is associated with several retinal dystrophies, including age-related macular degeneration (AMD) and Stargardt's macular dystrophy (Sunness et al., 1999; Armstrong et al., 1998). In these retinal dystrophies, loss of RPE is associated with loss of photoreceptors as a result of the crucial support provided by the RPE (Sarks et al., 1988; Weng et al., 1999). Based on these findings, there are compelling reasons to think that an hPSC-RPE therapy would have utility for treating these diseases, assuming that sufficient photoreceptors remain at the time of treatment.
Several clinical trials have been advanced to evaluate hPSC-derived RPE in AMD and Stargardt's disease. The first were initiated in 2011 and sponsored by Advanced Cell Technology/Ocata Therapeutics. Two open-label Phase 1/2 trials in the United States, one for Stargardt's disease and one for the more common dry form of AMD, are using a dose-escalation strategy to evaluate safety and efficacy. In parallel, Advanced Cell Technology/Ocata Therapeutics started a Phase 1/2 trial for Stargardt's disease in the United Kingdom testing the same hPSC-RPE therapeutic.
For the US trials, a surprisingly simple cell preparation protocol was used: RPE cells were generated from the MA09 hESC line using an embryoid body-based spontaneous differentiation approach (Schwartz et al., 2012). Clusters of presumptive RPE were identified by the appearance of darkly pigmented polygonal cells and manually isolated from plated embryoid bodies for expansion (Klimanskaya et al., 2004). The finished product, designated MA09-hRPE, was characterized for purity by quantitative immunocytochemical staining with the RPE markers bestrophin, PAX6, MITF and ZO1. Phagocytosis was functionally assayed by flow cytometry for cells containing fluorescent bioparticles after a 16-24 h exposure (Schwartz et al., 2012).
For clinical testing, a cell suspension of MA09-hRPE was injected into the subretinal space, in the pericentral macula, after vitrectomy in immunosuppressed patients. Note that this region is likely to contain more viable photoreceptors that the central macula in these retinal dystrophies and, as a result, should be more amenable to functional repair. However, delivery of a cell suspension does not assure uniform and appropriate distribution of the grafted cells.
A report from the two US trials described that, aside from adverse events associated with the surgical procedure and the immune-suppression treatment, none of the 18 patients (nine suffering from Stargardt's disease and nine from AMD) experienced serious adverse effects from the transplanted cells (Schwartz et al., 2015), and no evidence of teratoma formation or adverse proliferation was observed. Although the small number of patients did not allow a reliable statistical analysis of efficacy, six of the nine AMD patients showed some improvement in visual acuity in the treated eye at 6 months post-transplantation. Among the eight Stargardt's disease patients tested at 6 months, three showed some improvement and one showed a modest decline. However, there is some controversy on whether the reported increases in visual acuity reflect functional improvement mediated by the grafted RPE or an adaptation over time by which the patient learns to use less damaged parts of the retina to see (Sunness, 2015). Based on the safety of MA09-hRPE in these 18 patients, the FDA approved an amendment to the protocol, allowing enrollment of AMD and Stargardt's disease patients with better baseline visual acuity (Ocata Therapeutics, Form 10-K Annual Report for 2013; http://ir.ocata.com/sec-filings?page=11#document-11944-0001019687-14-001176). No data have been reported yet for the MA09-hRPE Stargardt's trial in the UK.
The use of MA09-hRPE cells has also been approved for a US Phase 1/2 trial in myopic macular degeneration, sponsored by the Jules Stein Eye Institute at UCLA, as well as a Korean Phase 1/2 trial in Stargardt's disease and AMD sponsored by Cha Biotech, a licensee of Ocata's technology. The latter trial recently reported 12-month follow-up data from four patients that show no adverse effects and improved visual acuity following RPE transplantation, supporting the findings of Schwartz et al., 2015 (Song et al., 2015).
There are several other groups pursuing hESC-RPE therapies at earlier stages of clinical development. Cell Cure Neurosciences recently opened a Phase 1/2a dose-escalation study using hESC-RPE, designated OpRegen, to treat dry AMD. The process for generating OpRegen cells relies on enhancing RPE differentiation by treating hESCs in suspension with nicotinamide and Activin A (Idelson et al., 2009). Whereas the beneficial effect of nicotinamide is ascribed to reducing neural cell death in the differentiating cultures, Activin A had previously been reported to promote RPE differentiation (Fuhrmann et al., 2000). Notably, OpRegen cells were developed without the use of xenobiotic reagents, a regulatory benefit for this particular therapy.
A Phase 1 trial for hESC-derived RPE in wet AMD was sponsored by Pfizer in collaboration with the University College London/UK in 2012, but that study has not yet enrolled any patients. In contrast to the previous studies which used cell suspensions of MA09-hRPE and OpRegen cells, the Pfizer trial is based on transplanting a polyester membrane patch seeded with a monolayer of hESC-RPE cells. This preparation more closely resembles the polarized monolayer structure of the healthy RPE in vivo, and might be advantageous for patients in which native RPE and underlying Bruch's membrane are more severely disrupted. This cell/scaffold type of device is also being developed by Regenerative Patch Technologies. In this case, the device, coated with vitronectin and seeded with hESC-RPE cells, is a mesh frame supporting ultrathin membranes of parylene designed to replicate the permeability properties of Bruch's membrane (Lu et al., 2012; Clegg, 2014). However, Regenerative Patch Technologies has not yet received FDA approval for clinical testing.
Retinal disease is also the target of the first iPSC-derived therapy to enter the clinic. An open-label study was approved in Japan to treat wet-type AMD with autologous iPSC-RPE. The RIKEN CDB website (http://www.cdb.riken.jp/en/news/2014/researchs/0915_3047.html) reported that autologous skin-derived iPSCs were differentiated into RPE and, in September 2014, transplanted as a scaffold-free cell sheet in a patient with AMD. Details of the cell preparation were not available at the time of this Review, but the protocol used is presumably a variation of the one recently published by the Takahashi team (Kamao et al., 2014). According to a recent blog posting, Dr Masayo Takahashi, the Principal Investigator for this study, confirmed that this trial was temporarily suspended due to changes in Japanese regulations that limit iPSC-related research and because mutations were found in the second patient's iPSCs (http://www.ipscell.com/2015/07/firstipscstop/). In a recent interview, Dr Takahashi indicated that the second patient will receive allogeneic rather than autologous iPSC-RPE (http://msemporda.blogspot.com.es/p/isscr-2015.html).
In summary, preliminary clinical data for the use of hPSC-RPE in retinal dystrophies appears favorable with respect to safety. Improvements in visual acuity were observed, but linking these improvements to therapeutic efficacy can be challenging under some circumstances, given that spontaneously improved visual acuity in the face of worsening macular degeneration has been described (Sunness et al., 2000, 2014).
Heart failure
Heart failure is caused by a variety of conditions that damage or overwork the heart, but cardiac ischemia and myocardial infarction are most highly associated with the disease (Mosterd and Hoes, 2007). The goal of treating heart failure patients is to improve cardiac contractility, and hPSC-derived cell therapies offer two possible mechanisms of action to achieve this. First, cardiomyocytes derived from hPSCs could integrate into the host myocardium and directly contribute to contractile function. Second, hPSC-derived cells could induce regenerative processes by virtue of paracrine factors released from the grafted cells. The latter mechanism is the basis for an investigator-sponsored early-stage trial initiated by the group of Dr Philippe Menasché at the Assistance Publique-Hospitaux de Paris to evaluate the therapeutic value of hESC-derived cardiac progenitors (CPs). For this trial, the I6 hESC line is expanded on a feeder layer of clinical-grade human fibroblasts and differentiated using bone morphogenetic protein (BMP)-2 and the FGF receptor kinase inhibitor SU-5402 for 4 days (Menasché et al., 2014). BMP-2 and the closely related TGF-β superfamily member BMP-4 are important regulators of mesoderm formation and cardiogenesis (Winnier et al., 1995; Schultheiss et al., 1997; Song et al., 2014; Luo et al., 2015). Inhibition of FGF signaling was observed to improve the efficiency of cardiac differentiation mediated by BMP-2 (Tomescot et al., 2007). Such a brief 4-day differentiation period raises the potential for residual undifferentiated cells. To reduce the number of undifferentiated cells remaining before therapeutic administration, the differentiating cells were immunomagnetically collected, based on their expression of the SSEA1 antigen. SSEA1 expression is reportedly induced by ESC differentiation (Leschik et al., 2008), and this sorting step achieved over 95% SSEA1+ cell purity. Importantly, the subcutaneous injection of sorted cells at such high purity failed to elicit teratoma formation in immunocompromised mice (Menasché et al., 2014). Although the RNA expression level of the CP marker Isl-1 was elevated in the resultant cell population relative to undifferentiated I6 hESCs, it is difficult to know the actual percentage of CPs in the clinical cell preparation.
For the Phase 1 trial, hESC-derived CPs embedded in a fibrin patch are attached to the epicardial surface of the infarct, secured with a flap made of autologous pericardium during open-chest surgical procedures for coronary artery bypass, mitral valve repair or mitral valve replacement, and patients are immunosuppressed for 2 months after transplantation. It is important to note that this cell patch is unlikely to integrate electrically with the host myocardium and contribute directly to contractile strength. Rather, its ability to improve heart function is assumed to rely on paracrine factors released from the grafted cells, which elicit endogenous repair mechanisms.
The first procedure using such technique was recently described (Menasché et al., 2015). The patient was a 68-year-old diabetic female with chronic heart failure due to infarction, receiving coronary artery bypass surgery to revascularize part of her damaged heart. The CP patch was grafted to a different area of damaged myocardium, which was not suitable for a bypass graft. Three months after the patch graft, although the patient developed donor-specific anti-MHC antibodies, indicating an allogeneic immune response, no arrhythmias were detected from her implanted defibrillator's monitor, and no cardiac masses were observed by echocardiography. Further, the patient's clinical status was improved (reduction in clinical stage of heart failure, improvement in ejection fraction and walking ability, increased wall motion in the region receiving the patch). Even though these results are encouraging, it is not possible to know how much benefit was derived from the bypass graft compared with that derived from the hESC-CP seeded patch.
In parallel, two academic groups have planned to evaluate the therapeutic value of hESC-derived cardiomyocytes in early-stage clinical trials. The group led by Dr Joseph Wu at Stanford University, with support from the California Institute for Regenerative Medicine, has recently described a method for differentiating hPSCs to cardiomyocytes with a chemically defined culture system to treat end-stage heart failure (Burridge et al., 2014). This protocol used sequential stimulation and inhibition of the canonical Wnt pathway by small molecules, as previously reported (Lian et al., 2012). Their planned Phase 1 safety trial will target heart failure patients receiving a left ventricular assist device as a bridge to heart transplantation (Joseph Wu, personal communication). In contrast to the program at Stanford that targets end-stage heart failure patients, our group, at the University of Washington, is pursuing a preclinical program to use hESC-derived cardiomyocytes to treat subacute myocardial infarction. Our strategy is to re-muscularize the injured heart early after infarction to prevent heart failure from developing. Our first nonhuman primate preclinical study using hESC-derived cardiomyocytes differentiated with Activin A and BMP-4 demonstrated robust engraftment of the cardiomyocytes and electrical coupling to the host myocardium (Chong et al., 2014). However, transient arrhythmias were observed in the treated animals and the small infarct size provided an insufficient therapeutic window for demonstrating efficacy. We are currently addressing these issues with additional studies in large animals and are planning for a Phase 1/2 clinical trial in acute myocardial infarct patients with severe cardiac injury.
Preclinical programs aiming to integrate hESC-derived cardiomyocytes into the damaged heart will clearly need to address the risk of autonomous pacemaker activity and arrhythmogenesis to generate advances in these therapies. In addition, finding methods to avoid graft rejection and to ensure the long-term contractile contribution of the grafted cells will be a challenge. This could be addressed with a life-long immunosuppressive drug regimen or by genetically engineering the cells to be less immunoreactive.
Diabetes
Type 1 diabetes results from the auto-immune destruction of β-cells, the insulin-secreting cells of the pancreas, making patients dependent on an exogenous supply of insulin to regulate their glycemia. However, the long-term complications arising from imprecise blood sugar management in patients using insulin can be serious (Gerich, 1986). Therefore, cell replacement therapies such as pancreas and islet transplants provide a better alternative than insulin therapy for glycemic control (Truong et al., 2005), but the availability of donor tissue is limited. In response to this need, efforts are currently being focused on the production of hPSC-derived insulin-secreting cells for cell replacement therapy.
Viacyte is developing hESC-derived pancreatic endodermal cells to treat Type 1 diabetes. Their cell production process involves a stepwise protocol utilizing growth factor signaling pathways involved in the normal embryonic development of PDX1+ pancreatic endoderm, which gives rise to β-cells (D'Amour et al., 2005, 2006; Kroon et al., 2008; Schulz et al., 2012). First, a high concentration of Activin A was used to differentiate CyT49 hESCs into definitive endoderm. Indeed, previous studies in zebrafish and Xenopus had demonstrated that Activin A and Nodal initiate the formation of the precursor of the definitive endoderm, the mesendoderm (Gamer and Wright, 1995; Rodaway et al., 1999; Osada and Wright, 1999; Lee et al., 2011; Rosa et al., 2014). Signaling through FGFRIIIB stimulates pancreatic progenitor proliferation (Bhushan et al., 2001; Elghazi et al., 2002). Thus, the resultant endodermal cells were expanded with Keratinocyte growth factor/FGF7 and then exposed to modulators of signaling pathways involved in pancreas development (Stafford and Prince, 2002; Hebrok et al., 1998; Kim and Melton, 1998; Rossi et al., 2001), to promote their differentiation to PDX+ endodermal cells: TTNPB, a retinoid analogue; cyclopamine, a Hedgehog inhibitor; and Noggin, a BMP antagonist.
Whereas initial studies grew cells as adherent cultures, Viacyte has modified the differentiation to a suspension culture method, allowing them to scale production for clinical use (Schulz et al., 2012). This is an important criterion for successful commercialization. Note that the PDX1+ pancreatic progenitor cells derived with this protocol are not fully differentiated and require a maturation step in vivo to reach the definitive β-cell state in which they secrete insulin in response to increasing extra-cellular glucose. Relying on the in vivo environment for the last stage of differentiation could introduce variability based on patient differences. Conversely, when differentiation to the terminal cell type is executed completely in vitro, consistent processes can be used throughout to ensure reproducible yield and quality of β-cells, and these parameters can be measured before the therapeutic is administered. Recently described methods for differentiation of hPSCs to functional β-islet-like cells in vitro (Pagliuca et al., 2014; Rezania et al., 2014) are being commercially developed by Semma Therapeutics and Betalogics, and might offer advantages for future cell therapy development.
As Type 1 diabetes is an auto-immune disease that targets β-cells, it is essential that the grafted cells be immunologically protected. Thus, Viacyte is developing a therapy designated VC-01 that combines pancreatic endodermal cells and a proprietary encapsulation device that prevents immune cells from contacting the engrafted cells. As described in an oral presentation by Viacyte at the Stem Cell Meeting on the Mesa (Laikind, 2014), the encapsulation device is freely permeable to glucose and insulin but impermeable to bigger molecules and cells. Furthermore, neovascularization around the device will presumably facilitate glucose sensing and insulin release systemically. This type of encapsulation should effectively prevent a cell-mediated immune response to the allogeneic cargo. Ideally, islet-reactive antibodies characteristic of Type I diabetes will also be prevented from entering the device and mediating humoral rejection of the encapsulated cells, but optimizing the device so that the desired permeability characteristics are maintained even if a fibrotic response occurs might be a challenge. However, this will be most crucial for achieving the same kinetics of glucose sensing and insulin release than those occurring in a normal physiologic setting. According to the company's website (http://viacyte.com/products/vc-01-diabetes-therapy/), pre-clinical studies performed in mice suggest that the VC-01 therapy effectively prevents hyperglycemia when the mouse β-cells are ablated, although these studies do not appear to be published.
A Phase 1/2 open-label dose-escalation trial will evaluate the safety and efficacy of VC-01 to treat Type 1 diabetic patients. As described at the aforementioned meeting (Laikind, 2014), the first cohort of patients (n=3) will receive a sub-therapeutic dose of encapsulated cells and the second cohort (n=36) will receive the full therapeutic dose. In addition, four small ‘sentinel’ devices containing cells will be implanted. These sentinel implants will be removed sequentially during the course of the trial for evaluation of cell viability, vascularization and cell differentiation. The primary endpoints for the trial are safety and efficacy, as measured by circulating C-peptide levels, a marker of endogenous insulin production. According to the company's website (http://viacyte.com/news-events-2/in-the-news/), the first patient in the trial was enrolled in October 2014.
Parkinson's disease
Parkinson's disease is a neurodegenerative disorder that mainly affects neurons that secrete dopamine in the substantia nigra, a region of the midbrain that controls movement. Dopaminergic neuronal loss leads to severe motor deficits, and currently available drug treatments do not alleviate the symptoms durably (Giugni and Okun, 2014). Deep brain stimulation can reduce motor symptoms, but serious adverse events are often associated with the surgical procedure (Deuschl et al., 2006; Weaver et al., 2009). Cell replacement therapy remains a potentially transformative treatment for this degenerative disease.
The development of PSC-derived dopaminergic neurons for treating Parkinson's disease was predicated on a series of early open-label studies indicating that fetal ventral mesencephalic tissue containing dopaminergic neurons from the developing substantia nigra imparted clinical benefit after transplantation into the striatum, the innervation target for nigral neurons (Lindvall et al., 1992, 1994; Freed et al., 1992). However, two subsequent randomized, double-blind studies using ventral mesencephalic tissue failed to demonstrate robust efficacy (Freed et al., 2001; Olanow et al., 2003). Moreover, these studies revealed graft-associated dyskinesia in a subset of patients, probably due to heterogeneous distribution of dopaminergic neurons in the striatum, and excessive contamination of grafts by serotonergic neurons (Carlsson et al., 2006; Politis et al., 2010). Building on these observations and refining patient selection, the TRANSEURO trial has been initiated to develop fetal midbrain tissue transplants further (Moore et al., 2014).
Alternatively, protocols for differentiating hPSCs into dopaminergic neurons have been pursued by a number of laboratories. Three of these groups have plans to evaluate clinically hPSC-derived dopaminergic neurons for Parkinson's disease in the near future, and will be briefly described below. Although these three groups have not yet begun clinical trials, we have included their work in this Review based on our belief that Parkinson's disease is a logical target for hPSC-based therapy, and that multiple independent programs increase the likelihood of seeing these studies brought to the clinic.
The group led by Dr Lorenz Studer at the Memorial Sloan Kettering Institute, with support from the New York Stem Cell Foundation, heads a consortium to produce clinical-grade human dopaminergic neural progenitor cells for a Phase 1 trial. This group described a protocol for differentiating hESCs into immature dopaminergic neurons, which incorporated simultaneous inhibition of BMP, shown to induce neural tissue in Xenopus and zebrafish embryos (Lamb et al., 1993; Londin et al., 2005; Wawersik et al., 2005), and TGF-β for neural induction (Chambers et al., 2009). The neuralized cells were subsequently differentiated into FOXA2+ floor-plate cells using a combination of recombinant SHH and the small-molecule agonist purmorphamine, which both activate the Hedgehog pathway, required for floor-plate formation (Chiang et al., 1996). As canonical Wnt signaling induces dopaminergic neurons specifically from the midbrain floor plate (Joksimovic et al., 2009), this protocol also used the GSK3B inhibitor CHIR99021 to promote differentiation towards the FOXA2+/LMX1A+ midbrain floor-plate phenotype further. Finally, a cocktail of BDNF, GDNF, ascorbic acid, dibutyryl cAMP, TGF-β3 and DAPT, a Notch pathway inhibitor, was used to promote differentiation to an immature dopaminergic neuronal phenotype expressing tyrosine hydroxylase, the rate-limiting enzyme for dopamine synthesis (Kriks et al., 2011; Steinbeck et al., 2015). Using this approach, the authors were able to produce and engraft dopamine neurons in the striata of Parkinsonian mice, rats and monkeys (Kriks et al., 2011).
Studer's team recently performed a key efficacy study in which Parkinsonism had been induced in mice by chemical ablation of dopaminergic neurons in the right striatum (Steinbeck et al., 2015). As previously observed (Kriks et al., 2011), transplantation of hESC-derived dopamine neurons corrected the motor deficit. In this study, however, the authors engineered the hESCs to express a light-induced chloride channel, HALO, allowing them to silence the grafts by delivering light through a fiber-optic cable. Photo-activation of the chloride channel induced an immediate return of the Parkinsonian symptoms, and switching off the light restored normal motor function. This is strong evidence for a direct effect of the graft cells in the neural circuit rather than for a paracrine, trophic effect.
Dr Jeanne Loring's lab at The Scripps Research Institute is leading an effort to advance autologous hiPSC-derived dopaminergic neurons for clinical testing. As described in a presentation by Dr Loring at the Rejuvenation Biotechnology Conference (Loring, 2014), her lab has created iPSCs from eight Parkinson's disease patients, and is using a modification of the differentiation protocol developed by the Studer lab (Kriks et al., 2011) to differentiate the cells to immature dopaminergic neurons for transplant (Loring, 2014). Similarly, the laboratory of Dr Jun Takahashi, of Kyoto University, is developing dopaminergic neurons from hiPSCs with the intent to test these cells in Parkinson's disease patients using a method similar to the aforementioned protocol (Kriks et al., 2011), but with the addition of a cell-sorting step to enrich for immature dopaminergic neurons (Doi et al., 2014).
Although these therapeutic programs are probably a few years away from clinical evaluation, they represent a significant potential to impact the motor deficits in Parkinson's disease. The issue of contaminating serotonergic neurons and heterogeneous cell preparations that might be associated with primary fetal cells can be better controlled for with an hPSC-derived therapeutic approach. It will be interesting to see whether autologous iPSC-derived dopaminergic neurons impart sustained functional recovery in Parkinson patients, given their derivation from afflicted individuals.
Closing thoughts
Seventeen years have passed since hPSCs were first described (Thomson et al., 1998). At the time of their discovery, many saw the potential for these cells to impact clinical medicine. However, some have criticized the slow progress of their clinical application, suggesting that remaining challenges are insurmountable and that hPSCs will not fulfil their promises (Sanganalmath and Bolli, 2013; Anderson et al., 2014). We consider this criticism unfair. Scaling cell production, controlling differentiation, achieving successful engraftment and demonstrating efficacy are challenging undertakings. Although we undoubtedly underestimated how challenging this would be, the last 15 years have seen significant progress in the translational research of hPSC-derived therapeutics. Researchers can now produce clinical-scale and clinical-grade stem cell derivatives which show good efficacy in the best animal models. Clearly, there is still room for improving the survival of transplanted cells and developing delivery methods which optimize their integration into host tissues, and these parameters are under active investigation in a number of labs.
The first in-human trials for spinal cord injury, retinal degeneration, diabetes and heart repair have begun, and the race is on for Parkinson's disease. These clinical studies and subsequent late-stage trials will address additional challenges for bringing these therapies to the market. The duration of the beneficial effects will be an important criterion for widespread use and will factor into reimbursement by payers, ultimately determining the accessibility of the treatment to patients. When immunosuppressive drugs are required, these clinical trials will determine whether immunosuppression is an acceptable supplement to the cell therapy. Although challenges remain, the early trials we have described may provide a glimpse of the full potential for this therapeutic platform, and along the way inspire enthusiasm for a new era in regenerative medicine.
Footnotes
Competing interests
C.E.M. is a scientific co-founder and equity holder in BEAT Biotherapeutics.
Funding
The authors gratefully acknowledge financial support from the UW Medicine Heart Regeneration Program and research grants from the National Institutes of Health[R01 HL084642, P01 HL094374, P01 GM081619, U01 HL100405] and the Fondation Leducq Transatlantic Network of Excellence.
References
- All A. H., Gharibani P., Gupta S., Bazley F. A., Pashai N., Chou B.-K., Shah S., Resar L. M., Cheng L., Gearheart J. D. et al. (2015). Early intervention for spinal cord injury with human induced pluripotent stem cells oligodendrocyte progenitors. PLoS ONE 10, e116933 10.1371/journal.pone.0116933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson M. E., Goldhaber J., Houser S. R., Puceat M. and Sussman M. A. (2014). Embryonic stem cell-derived cardiac myocytes are not ready for human trials. Circ. Res. 115, 335-338. 10.1161/CIRCRESAHA.114.304616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong J. D., Meyer D., Xu S. and Elfervig J. L. (1998). Long-term follow-up of Stargardt's disease and fundus flavimaculatus. Ophthalmology 105, 448-457. 10.1016/S0161-6420(98)93026-3 [DOI] [PubMed] [Google Scholar]
- Bambakidis N. C. and Miller R. H. (2004). Transplantation of oligodendrocyte precursors and sonic hedgehog results in improved function and white matter sparing in the spinal cords of adult rats after contusion. Spine J. 4, 16-26. 10.1016/j.spinee.2003.07.004 [DOI] [PubMed] [Google Scholar]
- Biernaskie J., Sparling J. S., Liu J., Shannon C. P., Plemel J. R., Xie Y., Miller F. D. and Tetzlaff W. (2007). Skin-derived precursors generate myelinating Schwann cells that promote remyelination and functional recovery after contusion spinal cord injury. J. Neurosci. 27, 9545-9559. 10.1523/JNEUROSCI.1930-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan A., Itoh N., Kato S., Thiery J. P., Czernichow P., Bellusci S. and Scharfmann R. (2001). FGF10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development 128, 5109-5117. [DOI] [PubMed] [Google Scholar]
- Blight A. R. (1983). Cellular morphology of chronic spinal cord injury in the cat: analysis of myelinated axons by line-sampling. Neuroscience 10, 521-543. 10.1016/0306-4522(83)90150-1 [DOI] [PubMed] [Google Scholar]
- Bresnahan J. C., King J. S., Martin G. F. and Yashon D. (1976). A neuroanatomical analysis of spinal cord injury in the rhesus monkey (Macaca mulatta). J. Neurol. Sci. 28, 521-542. 10.1016/0022-510X(76)90122-2 [DOI] [PubMed] [Google Scholar]
- Burridge P. W., Matsa E., Shukla P., Lin Z. C., Churko J. M., Ebert A. D., Lan F., Diecke S., Huber B., Mordwinkin N. M. et al. (2014). Chemically defined generation of human cardiomyocytes. Nat. Methods 11, 855-860. 10.1038/nmeth.2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q., Xu X.-M., DeVries W. H., Enzmann G. U., Ping P., Tsoulfas P., Wood P. M., Bunge M. B. and Whittemore S. R. (2005). Functional recovery in traumatic spinal cord injury after transplantation of multineurotrophin-expressing glial-restricted precursor cells. J. Neurosci. 25, 6947-6957. 10.1523/JNEUROSCI.1065-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson T., Winkler C., Lundblad M., Cenci M. A., Björklund A. and Kirik D. (2006). Graft placement and uneven pattern of reinnervation in the striatum is important for development of graft-induced dyskinesia. Neurobiol. Dis. 21, 657-668. 10.1016/j.nbd.2005.09.008 [DOI] [PubMed] [Google Scholar]
- Chambers S. M., Fasano C. A., Papapetrou E. P., Tomishima M., Sadelain M. and Studer L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotech. 27, 275-280. 10.1038/nbt.1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C., Litingtung Y., Lee E., Young K. E., Corden J. L., Westphal H. and Beachy P. A. (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Science 383, 407-413. 10.1038/383407a0 [DOI] [PubMed] [Google Scholar]
- Chong J. J. H., Yang X., Don C. W., Minami E., Liu Y.-W., Weyers J. J., Mahoney W. M., Van Biber B., Cook S. M., Palpant N. J. et al. (2014). Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 510, 273-277. 10.1038/nature13233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg D. (2014). Stem Cell Meeting on the Mesa , Oral presentation, Oct 7th, 2014. https://www.youtube.com/watch?v=pDQ2SD9aLt0 [Google Scholar]
- D'Amour K. A., Agulnick A. D., Eliazer S., Kelly O. G., Kroon E. and Baetge E. E. (2005). Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotech. 23, 1534-1541. 10.1038/nbt1163 [DOI] [PubMed] [Google Scholar]
- D'Amour K. A., Bang A. G., Eliazer S., Kelly O. G., Agulnick A. D., Smart N. G., Moorman M. A., Kroon E., Carpenter M. K. and Baetge E. E. (2006). Production of pancreatic hormone–expressing endocrine cells from human embryonic stem cells. Nat. Biotech. 24, 1392-1401. 10.1038/nbt1259 [DOI] [PubMed] [Google Scholar]
- Deuschl G., Schade-Brittinger C., Krack P., Volkmann J., Schäfer H., Bötzel K., Daniels C., Deutschländer A., Dillman U., Eisner W. et al. (2006). A randomized trial of deep-brain stimulation for Parkinson's disease. New Engl. J. Med. 355, 896-908. 10.1056/NEJMoa060281 [DOI] [PubMed] [Google Scholar]
- Doi D., Samata B., Katsukawa M., Kikuchi T., Morizane A., Ono Y., Sekiguchi K., Nakagawa M., Parmar M. and Takahashi J. (2014). Isolation of human induced pluripotent stem cell-derived dopaminergic progenitors by cell sorting for successful transplantation. Stem Cell Rep. 2, 337-350. 10.1016/j.stemcr.2014.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elghazi L., Cras-Meneur C., Czernichow P. and Scharfmann R. (2002). Role for FGFR2IIIB-mediated signals in controlling pancreatic endocrine progenitor cell proliferation. Proc. Natl. Acad. Sci. USA 99, 3884-3889. 10.1073/pnas.062321799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed C. R., Breeze R. E., Rosenberg N. L., Schneck S. A., Kriek E., Qi J.-X., Lone T., Zhang Y.-b., Snyder J. A., Wells T. H. et al. (1992). Survival of implanted fetal dopamine cells and neurologic improvement 12 to 46 months after transplantation for Parkinson's disease. N. Engl. J. Med. 327, 1549-1555. 10.1056/NEJM199211263272202 [DOI] [PubMed] [Google Scholar]
- Freed C. R., Greene P. E., Breeze R. E., Tsai W.-Y., DuMouchel W., Kao R., Dillon S., Winfield H., Culver S., Trojanowski J. Q. et al. (2001). Transplantation of embryonic dopamine neurons for severe Parkinson's disease. N. Engl. J. Med. 344, 710-719. 10.1056/NEJM200103083441002 [DOI] [PubMed] [Google Scholar]
- Fuhrmann S., Levine E. M. and Reh T. A. (2000). Extraocular mesenchyme patterns the optic vesicle during early eye development in the embryonic chick. Development 127, 4599-4609. [DOI] [PubMed] [Google Scholar]
- Gamer L. W. and Wright C. V. E. (1995). Autonomous endodermal determination in Xenopus: regulation of expression of the pancreatic gene XlHbox 8. Dev. Biol. 171, 240-251. 10.1006/dbio.1995.1275 [DOI] [PubMed] [Google Scholar]
- Gerich J. E. (1986). Insulin-dependent diabetes mellitus: pathophysiology. Mayo Clin. Proc. 61, 787-791. 10.1016/S0025-6196(12)64818-6 [DOI] [PubMed] [Google Scholar]
- Giugni J. C. and Okun M. S. (2014). Treatment of advanced Parkinson's disease. Curr. Opin. Neurol. 27, 450-460. 10.1097/WCO.0000000000000118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawryluk G. W. J., Spano S., Chew D., Wang S., Erwin M., Chamankhah M., Forgione N. and Fehlings M. G. (2014). An examination of the mechanisms by which neural precursors augment recovery following spinal cord injury: a key role for remyelination. Cell Transplant. 23, 365-380. 10.3727/096368912X662408 [DOI] [PubMed] [Google Scholar]
- Hebrok M., Kim S. K. and Melton D. A. (1998). Notochord repression of endodermal sonic hedgehog permits pancreas development. Genes Dev. 12, 1705-1713. 10.1101/gad.12.11.1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idelson M., Alper R., Obolensky A., Ben-Shushan E., Hemo I., Yachimovich-Cohen N., Khaner H., Smith Y., Wiser O., Gropp M. et al. (2009). Directed differentiation of human embryonic stem cells into functional retinal pigment epithelium cells. Cell Stem Cell 5, 396-408. 10.1016/j.stem.2009.07.002 [DOI] [PubMed] [Google Scholar]
- Irvine K. A. and Blakemore W. F. (2008). Remyelination protects axons from demyelination-associated axon degeneration. Brain 131, 1464-1477. 10.1093/brain/awn080 [DOI] [PubMed] [Google Scholar]
- Joksimovic M., Yun B. A., Kittappa R., Anderegg A. M., Chang W. W., Taketo M. M., McKay R. D. G. and Awatramani R. B. (2009). Wnt antagonism of SHH facilitates midbrain floor plate neurogenesis. Nat. Neurosci. 12, 125-131. 10.1038/nn.2243 [DOI] [PubMed] [Google Scholar]
- Kamao H., Mandai M., Okamoto S., Sakai N., Suga A., Sugita S., Kiryu J. and Takahashi M. (2014). Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Rep. 2, 205-218. 10.1016/j.stemcr.2013.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keirstead H. S., Nistor G., Bernal G., Totoiu M., Cloutier F., Sharp K. and Steward O. (2005). Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J. Neurosci. 25, 4694-4705. 10.1523/JNEUROSCI.0311-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. K. and Melton D. A. (1998). Pancreas development is promoted by cyclopamine, a hedgehog signaling inhibitor. Proc. Natl. Acad. Sci. USA 95, 13036-13041. 10.1073/pnas.95.22.13036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimanskaya I., Hipp J., Rezai K. A., West M., Atala A. and Lanza R. (2004). Derivation and comparative assessment of retinal pigment epithelium from human embryonic stem cells using transcriptomics. Cloning Stem Cells 6, 217-245. 10.1089/clo.2004.6.217 [DOI] [PubMed] [Google Scholar]
- Kriks S., Shim J.-W., Piao J., Ganat Y. M., Wakeman D. R., Xie Z., Carrillo-Reid L., Auyeung G., Antonacci C., Buch A. et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature 480, 547-551. 10.1038/nature10648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon E., Martinson L. A., Kadoya K., Bang A. G., Kelly O. G., Eliazer S., Young H., Richardson M., Smart N. G., Cunningham J. et al. (2008). Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotech. 26, 443-452. 10.1038/nbt1393 [DOI] [PubMed] [Google Scholar]
- Laikind P. (2014). Stem Cell Meeting on the Mesa, Oral presentation, Oct 7th, 2014 https://www.youtube.com/watch?v=dg1yQETjCmQ.
- Lamb T. M., Knecht A. K., Smith W. C., Stachel S. E., Economides A. N., Stahl N., Yancopolous G. D. and Harland R. M. (1993). Neural induction by the secreted polypeptide noggin. Science 262, 713-718. 10.1126/science.8235591 [DOI] [PubMed] [Google Scholar]
- Lebkowski J. (2011). GRNOPC1: the world's first embryonic stem cell-derived therapy. Regen. Med. 6(6 Suppl.), 11-13. 10.2217/rme.11.77 [DOI] [PubMed] [Google Scholar]
- Lebkowski J. (2014). Phase 1 clinical trial of human embryonic stem cell-derived oligodendrocyte progenitors inpatients with neurologically complete thoracic spinal cord injury: Results and next steps. American Society of Gene & Cell Therapy annual meeting presentation https://www.youtube.com/watch?v=8rG6bzd2mz4. [Google Scholar]
- Lee K. H., Yoon D. H., Park Y. G. and Lee B. H. (2005). Effects of glial transplantation on functional recovery following acute spinal cord injury. J. Neurotrauma 22, 575-589. 10.1089/neu.2005.22.575 [DOI] [PubMed] [Google Scholar]
- Lee K. L., Lim S. K., Orlov Y. L., Yit L. Y., Yang H., Ang L. T., Poellinger L. and Lim B. (2011). Graded Nodal/Activin signaling titrates conversion of quantitative phospho-Smad2 levels into qualitative embryonic stem cell fate decisions. PLoS Genet. 7, e1002130 10.1371/journal.pgen.1002130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leschik J., Stefanovic S., Brinon B. and Pucéat M. (2008). Cardiac commitment of primate embryonic stem cells. Nat. Protoc. 3, 1381-1387. 10.1038/nprot.2008.116 [DOI] [PubMed] [Google Scholar]
- Levenberg S., Huang N. F., Lavik E., Rogers A. B., Itskovitz-Eldor J. and Langer R. (2003). Differentiation of human embryonic stem cells on three-dimensional polymer scaffolds. Proc. Natl. Acad. Sci. USA 100, 12741-12746. 10.1073/pnas.1735463100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X., Hsiao C., Wilson G., Zhu K., Hazeltine L. B., Azarin S. M., Raval K. K., Zhang J., Kamp T. J. and Palecek S. P. (2012). Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 109, E1848-E1857. 10.1073/pnas.1200250109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindvall O., Widner H., Rehncrona S., Brundin P., Odin P., Gustavii B., Frackowiak R., Leenders K. L., Sawle G., Rothwell J. C. et al. (1992). Transplantation of fetal dopamine neurons in Parkinson's disease: one-year clinical and neurophysiological observations in two patients with putaminal implants. Ann. Neurol. 31, 155-165. 10.1002/ana.410310206 [DOI] [PubMed] [Google Scholar]
- Lindvall O., Sawle G., Widner H., Rothwell J. C., Björklund A., Brooks D., Brundin P., Frackowiak R., Marsden C. D., Odin P. et al. (1994). Evidence for long-term survival and function of dopaminergic grafts in progressive Parkinson's disease. Ann. Neurol. 35, 172-180. 10.1002/ana.410350208 [DOI] [PubMed] [Google Scholar]
- Londin E. R., Niemiec J. and Sirotkin H. I. (2005). Chordin, FGF signaling, and mesodermal factors cooperate in zebrafish neural induction. Dev. Biol. 279, 1-19. 10.1016/j.ydbio.2004.11.016 [DOI] [PubMed] [Google Scholar]
- Loring J. F. (2014). Rejuvenation Biotechnology Conference, Oral presentation, August, 2014 and https://www.youtube.com/watch?v=LWrdtFLFc9s. [Google Scholar]
- Lu B., Zhu D., Hinton D., Humayun M. S. and Tai Y.-C. (2012). Mesh-supported submicron parylene-C membranes for culturing retinal pigment epithelial cells. Biomed. Microdevices 14, 659-667. 10.1007/s10544-012-9645-8 [DOI] [PubMed] [Google Scholar]
- Luo W., Zhao X., Jin H., Tao L., Zhu J., Wang H., Hemmings B. A. and Yang Z. (2015). Akt1 signaling coordinates BMP signaling and β-catenin activity to regulate second heart field progenitor development. Development 142, 732-742. 10.1242/dev.119016 [DOI] [PubMed] [Google Scholar]
- Menasché P., Vanneaux V., Fabreguettes J. R., Bel A., Tosca L., Garcia S., Bellamy V., Farouz Y., Pouly J., Damour O. et al. (2014). Towards a clinical use of human embryonic stem cell-derived cardiac progenitors: a translational experience. Eur. Heart J. 36, 743-750. 10.1093/eurheartj/ehu192 [DOI] [PubMed] [Google Scholar]
- Menasché P., Vanneaux V., Hagège A., Bel A., Cholley B., Cacciapuoti I., Parouchev A., Benhamouda N., Tachdjian G., Tosca L. et al. (2015). Human embryonic stem cell-derived cardiac progenitors for severe heart failure treatment: first clinical case report . Eur. Heart J. 36, 2011-2017. 10.1093/eurheartj/ehv189 [DOI] [PubMed] [Google Scholar]
- Moore S. F., Guzman N. V., Mason S. L., Williams-Gray C. H. and Barker R. A. (2014). Which patients with Parkinson's disease participate in clinical trials? One centre's experiences with a new cell based therapy trial (TRANSEURO). J. Parkinsons Dis. 4, 671-676. 10.3233/JPD-140432 [DOI] [PubMed] [Google Scholar]
- Mosterd A. and Hoes A. W. (2007). Clinical epidemiology of heart failure. Heart 93, 1137-1146. 10.1136/hrt.2003.025270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashmi R. and Fehlings M. G. (2001). Mechanisms of axonal dysfunction after spinal cord injury: with an emphasis on the role of voltage-gated potassium channels. Brain Res. Rev. 38, 165-191. 10.1016/S0165-0173(01)00134-5 [DOI] [PubMed] [Google Scholar]
- Nistor G. I., Totoiu M. O., Haque N., Carpenter M. K. and Keirstead H. S. (2005). Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia 49, 385-396. 10.1002/glia.20127 [DOI] [PubMed] [Google Scholar]
- Olanow C. W., Goetz C. G., Kordower J. H., Stoessl A. J., Sossi V., Brin M. F., Shannon K. M., Nauert G. M., Perl D. P., Godbold J. et al. (2003). A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson's disease. Ann. Neurol. 54, 403-414. 10.1002/ana.10720 [DOI] [PubMed] [Google Scholar]
- Osada S. I. and Wright C. V. E. (1999). Xenopus nodal-related signaling is essential for mesendodermal patterning during early embryogenesis. Development 126, 3229-3240. [DOI] [PubMed] [Google Scholar]
- Pagliuca F. C., Millman J. R., Gürtler M., Segel M., Van Dervort A., Ryu J. H., Peterson Q. P., Greiner D. and Melton D. A. (2014). Generation of functional human pancreatic β cells in vitro. Cell 159, 428-439. 10.1016/j.cell.2014.09.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politis M., Wu K., Loane C., Quinn N. P., Brooks D. J., Rehncrona S., Bjorklund A., Lindvall O. and Piccini P. (2010). Serotonergic neurons mediate dyskinesia side effects in Parkinson's patients with neural transplants. Sci. Transl. Med. 2,38ra46 10.1126/scitranslmed.3000976 [DOI] [PubMed] [Google Scholar]
- Rezania A., Bruin J. E., Arora P., Rubin A., Batushansky I., Asadi A., O'Dwyer S., Quiskamp N., Mojibian M., Albrecht T. et al. (2014). Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotech. 32, 1121-1133. 10.1038/nbt.3033 [DOI] [PubMed] [Google Scholar]
- Rodaway A., Takeda H., Koshida S., Broadbent J., Price B., Smith J. C., Patient R. and Holder N. (1999). Induction of the mesendoderm in the zebrafish germ ring by yolk cell-derived TGF-β family signals and discrimination of mesoderm and endoderm by FGF. Development 126, 3067-3078. [DOI] [PubMed] [Google Scholar]
- Rosa A., Papaioannou M. D., Krzyspiak J. E. and Brivanlou A. H. (2014). miR-373 is regulated by TGFβ signaling and promotes mesendoderm differentiation in human embryonic stem cells. Dev. Biol. 391, 81-88. 10.1016/j.ydbio.2014.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi J. M., Dunn N. R., Hogan B. L. M. and Zaret K. S. (2001). Distinct mesodermal signals, including BMPs from the septum transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev. 15, 1998-2009. 10.1101/gad.904601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanganalmath S. K. and Bolli R. (2013). Cell therapy for heart failure: a comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circ. Res. 113, 810-834. 10.1161/CIRCRESAHA.113.300219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarks J. P., Sarks S. H. and Killingsworth M. C. (1988). Evolution of geographic atrophy of the retinal pigment epithelium. Eye 2, 552-577. 10.1038/eye.1988.106 [DOI] [PubMed] [Google Scholar]
- Schultheiss T. M., Burch J. B. and Lassar A. B. (1997). A role for bone morphogenetic proteins in the induction of cardiac myogenesis. Genes Dev. 11, 451-462. 10.1101/gad.11.4.451 [DOI] [PubMed] [Google Scholar]
- Schulz T. C., Young H. Y., Agulnick A. D., Babin M. J., Baetge E. E., Bang A. G., Bhoumik A., Cepa I., Cesario R. M., Haakmeester C. et al. (2012). A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PLoS ONE 7, e37004 10.1371/journal.pone.0037004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S. D., Hubschman J.-P., Heilwell G., Franco-Cardenas V., Pan C. K., Ostrick R. M., Mickunas E., Gay R., Klimanskaya I. and Lanza R. (2012). Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet 379, 713-720. 10.1016/S0140-6736(12)60028-2 [DOI] [PubMed] [Google Scholar]
- Schwartz S. D., Regillo C. D., Lam B. L., Eliott D., Rosenfeld P. J., Gregori N. Z., Hubschman J.-P., Davis J. L., Heilwell G., Spirn M. et al. (2015). Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt's macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet 385, 509-516. 10.1016/S0140-6736(14)61376-3 [DOI] [PubMed] [Google Scholar]
- Song J., McColl J., Camp E., Kennerley N., Mok G. F., McCormick D., Grocott T., Wheeler G. N. and Munsterberg A. E. (2014). Smad1 transcription factor integrates BMP2 and Wnt3a signals in migrating cardiac progenitor cells. Proc. Natl. Acad. Sci. USA 111, 7337-7342. 10.1073/pnas.1321764111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W. K., Park K.-M., Kim H.-J., Lee J. H., Choi J., Ching S. Y., Shim S. H., Del Priore L. V. and Lanza R. (2015). Treatment of macular degeneration using embryonic stem cell-derived retinal pigment epithelium: preliminary results in Asian patients. Stem Cell Rep. 4, 1-13. 10.1016/j.stemcr.2015.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrrow J. R., Hicks D. and Hamel C. P. (2010). The retinal pigment epithelium in health and disease. Curr. Mol. Med. 10, 802-823. 10.2174/156652410793937813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford D. and Prince V. E. (2002). Retinoic acid signaling is required for a critical early step in zebrafish pancreatic development. Curr. Biol. 12, 1215-1220. 10.1016/S0960-9822(02)00929-6 [DOI] [PubMed] [Google Scholar]
- Steinbeck J. A., Choi S. J., Mrejeru A., Ganat Y., Deisseroth K., Sulzer D., Mosharov E. V. and Studer L. (2015). Optogenetics enables functional analysis of human embryonic stem cell–derived grafts in a Parkinson's disease model. Nat. Biotech. 33, 204-209. 10.1038/nbt.3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunness J. S. (2014). Spontaneous improvement in visual acuity in age-related geographic atrophy of the macula. JAMA Ophthamol. 132, 356-357. 10.1001/jamaophthalmol.2014.21 [DOI] [PubMed] [Google Scholar]
- Sunness J. S. (2015). Stem cells in age-related macular degeneration and Stargardt's macular dystrophy. Lancet 386, 29 10.1016/S0140-6736(15)61201-6 [DOI] [PubMed] [Google Scholar]
- Sunness J. S., Gonzalez-Baron J., Applegate C. A., Bressler N. M., Tian Y., Hawkins B., Barron Y. and Bergman A. (1999). Enlargement of atrophy and visual acuity loss in the geographic atrophy form of age-related macular degeneration. Ophthalmology 106, 1768-1779. 10.1016/S0161-6420(99)90340-8 [DOI] [PubMed] [Google Scholar]
- Sunness J. S., Applegate C. A. and Gonzalez-Baron J. (2000). Improvement of visual acuity over time in patients with bilateral geographic atrophy from age-related macular degeneration. Retina 20, 162-169. 10.1097/00006982-200002000-00009 [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K. and Yamanaka S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861-872. 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]
- Thomson J. A., Itskovitz-Eldor J., Shapiro S. S., Waknitz M. A., Swiergiel J. J., Marshall V. S. and Jones J. M. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145-1147. 10.1126/science.282.5391.1145 [DOI] [PubMed] [Google Scholar]
- Tomescot A., Leschik J., Bellamy V., Dubois G., Messas E., Bruneval P., Desnos M., Hagège A. A., Amit M., Itskovitz J. et al. (2007). Differentiation in vivo of cardiac committed human embryonic stem cells in postmyocardial infarcted rats. Stem Cells 25, 2200-2205. 10.1634/stemcells.2007-0133 [DOI] [PubMed] [Google Scholar]
- Totoiu M. O. and Keirstead H. S. (2005). Spinal cord injury is accompanied by chronic progressive demyelination. J. Comp. Neurol. 486, 373-383. 10.1002/cne.20517 [DOI] [PubMed] [Google Scholar]
- Truong W., Lakey J. R., Ryan E. A. and Shapiro A. M. (2005). Clinical islet transplantation at the University of Alberta—the Edmonton experience. Clin. Transpl. 153-172. [PubMed] [Google Scholar]
- Vescovi A. L., Reynolds B. A., Fraser D. D. and Weiss S. (1993). bFGF regulates the proliferative fate of unipotent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells. Neuron 11, 951-966. 10.1016/0896-6273(93)90124-A [DOI] [PubMed] [Google Scholar]
- Wawersik S., Evola C. and Whitman M. (2005). Conditional BMP inhibition in Xenopus reveals stage-specific roles for BMPs in neural and neural crest induction. Dev. Biol. 277, 425-442. 10.1016/j.ydbio.2004.10.002 [DOI] [PubMed] [Google Scholar]
- Weaver F. M., Follett K., Stern M., Hur K., Harris C., Marks W. J., Rothlind J., Sagher O., Reda D., Moy C. S. et al. (2009). Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA 301, 63-73. 10.1001/jama.2008.929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J., Mata N. L., Azarian S. M., Tzekov R. T., Birch D. G. and Travis G. H. (1999). Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell 98, 13-23. 10.1016/S0092-8674(00)80602-9 [DOI] [PubMed] [Google Scholar]
- Winnier G., Blessing M., Labosky P. A. and Hogan B. L. (1995). Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 9, 2105-2116. 10.1101/gad.9.17.2105 [DOI] [PubMed] [Google Scholar]
- Yasuda A., Tsuji O., Shibata S., Nori S., Takano M., Kobayashi Y., Takahashi Y., Fujiyoshi K., Hara C. M., Miyawaki A. et al. (2011). Significance of remyelination by neural stem/progenitor cells transplanted into the injured spinal cord. Stem Cells 29, 1983-1994. 10.1002/stem.767 [DOI] [PubMed] [Google Scholar]
- Yu J., Vodyanik M. A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J. L., Tian S., Nie J., Jonsdottir G. A., Ruotti V., Stewart R. et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917-1920. 10.1126/science.1151526 [DOI] [PubMed] [Google Scholar]
- Zhang Y. W., Denham J. and Thies R. S. (2006). Oligodendrocyte progenitor cells derived from human embryonic stem cells express neurotrophic factors. Stem Cells Dev. 15, 943-952. 10.1089/scd.2006.15.943 [DOI] [PubMed] [Google Scholar]