

ABSTRACT
Neural tube closure relies on the apical constriction of neuroepithelial cells. Research in frog and fly embryos has found links between the levels of intracellular calcium, actomyosin dynamics and apical constriction. However, genetic evidence for a role of calcium in apical constriction during mammalian neurulation is still lacking. Secretory pathway calcium ATPase (SPCA1) regulates calcium homeostasis by pumping cytosolic calcium into the Golgi apparatus. Loss of function in Spca1 causes cranial exencephaly and spinal cord defects in mice, phenotypes previously ascribed to apoptosis. However, our characterization of a novel allele of Spca1 revealed that neurulation defects in Spca1 mutants are not due to cell death, but rather to a failure of neuroepithelial cells to apically constrict. We show that SPCA1 influences cell contractility by regulating myosin II localization. Furthermore, we found that loss of Spca1 disrupts actin dynamics and the localization of the actin remodeling protein cofilin 1. Taken together, our results provide evidence that SPCA1 promotes neurulation by regulating the cytoskeletal dynamics that promote apical constriction and identify cofilin 1 as a downstream effector of SPCA1 function.
KEY WORDS: Atp2c1, Neural tube closure, Apical constriction, Ca2+, Cofilin 1, Non-muscle myosin II, Mouse
Highlighted Article: The Golgi-localized calcium pump SPCA1 promotes mouse neurulation by regulating the cytoskeletal dynamics that promote apical constriction, influencing the subcellular localization of myosin II and the actin-remodeling protein cofilin 1.
INTRODUCTION
The mammalian brain and spinal cord develop from the neural tube, an embryonic structure that starts out as a flat epithelial sheet and undergoes extensive morphogenetic rearrangements collectively known as neurulation. During mammalian neurulation, the neural plate epithelia first bends at the ventral midline, creating two neural folds that elevate, further bend and eventually fuse at their dorsal aspect to form the hollow neural tube (Jacobson and Tam, 1982). Perturbation of this process in humans results in neural tube defects, a class of birth disorders that are found in 1/2000 births in developed countries and as frequently as 27.8/2000 births in developing areas (Wallingford et al., 2013).
An important morphogenetic mechanism driving neural tube closure is apical constriction (Haigo et al., 2003; Hildebrand and Soriano, 1999). Apical constriction comprises a reduction in the apical surface of a cell, causing an overall change in cell shape that can ultimately result in the bending of epithelial sheets, the delamination of mesenchymal cells, or wound healing, depending on the particular setting (reviewed by Martin and Goldstein, 2014). Research in cell culture, as well as in fly and vertebrate embryos, has identified a common molecular machinery implicated in apical constriction. This machinery involves the binding of non-muscle myosin II motors to apically localized actin filaments and their subsequent contraction, which is regulated through the phosphorylation of the myosin light chain (pMLC) (Hildebrand, 2005; Martin et al., 2009; Mason et al., 2013). Links between actomyosin dynamics, apical constriction and neural tube closure are also supported by pharmacological interventions that interfere with MLC phosphorylation and actomyosin remodeling (Escuin et al., 2015; Kinoshita et al., 2008; Nishimura et al., 2012).
Evidence from studies in Drosophila supports a role for calcium in the control of apical constriction in different settings. In wound-healing assays, wounding of the cuticle epithelia elicits apical constriction through a wave of actin enrichment and myosin contractility that correlates with elevated levels of calcium near the wound site (Antunes et al., 2013). Additionally, manipulation of calcium levels during fly embryogenesis disrupts apical myosin contractility and dorsal closure (Hunter et al., 2014). In vertebrates, several lines of evidence support roles for calcium during neurulation. First, rat and chicken embryos cultured in calcium-free media, or in the presence of drugs that block calcium uptake, develop neural tube defects (Ferreira and Hilfer, 1993; Lee and Nagele, 1986; O'Shea, 1982; Smedley and Stanisstreet, 1985). Conversely, treating chicken embryos with ionophores to increase intracellular calcium causes an initial precocious bending of neural folds (Ferreira and Hilfer, 1993) that later resolves into neural tube closure arrest (Lee et al., 1978). Although these studies reveal that intracellular calcium levels are crucial for neural tube closure, a direct link between calcium and apical constriction during neurulation has only recently been uncovered by two recent studies using live imaging of frog embryos. These studies revealed that apical constriction during neurulation involves cell-autonomous and asynchronous pulsed apical contractions that are preceded by flashes of calcium (Christodoulou and Skourides, 2015; Suzuki et al., 2017). These calcium pulses also correlate with accumulation of F-actin in the medial region of neuroepithelial cells, suggesting that calcium regulates apical constriction by influencing actin dynamics and/or myosin contractility. Treating frog embryos with thapsigargin, a drug that increases intracellular calcium, disrupts calcium pulses, as well as the dynamics and spatial distribution of constriction events, ultimately blocking neural tube closure (Christodoulou and Skourides, 2015). This evidence indicates that neuroepithelial cells must cycle through periods of high and low calcium to enable pulsatile contractions and complete neural tube closure. However, how calcium regulates apical constriction and the mechanisms that control calcium homeostasis in the neural tube remain unclear.
Genetic screens and targeted mutagenesis in mice have led to the identification of more than 200 genes that are essential for neural tube closure. The study of these mouse mutants has provided insight into the processes that promote neurulation, revealing important roles for apoptosis, proliferation, cytoskeletal dynamics, cell signaling, planar cell polarity and dorsoventral neural tube patterning (Harris and Juriloff, 2010). In contrast, genetic evidence for a role of calcium in neural tube closure is limited. Secretory pathway calcium ATPase 1 (SPCA1, also known as ATP2C1) is a P-type Ca2+ ATPase localized to the trans-Golgi network, where it serves as the primary pump responsible for pumping calcium into the Golgi apparatus (Dode et al., 2005; Lissandron et al., 2010; Van Baelen et al., 2003; Wootton et al., 2004). Targeted deletion of Spca1 in mice results in exencephaly and embryonic lethality around embryonic day (E) 11.5. Both neural tube defects and embryonic lethality in Spca1 mutants have been attributed to widespread apoptosis due to Golgi stress (Okunade et al., 2007). Therefore, although Spca1 embryos provide support for a role of calcium during neurulation, a link between calcium and apical constriction has not been established in mammals.
Here, we describe the characterization of a novel N-ethyl-N-nitrosourea (ENU)-induced mutation in Spca1. We show that the neural tube defects of Spca1 mutants precede the onset of apoptosis, demonstrating that cell death is not the primary cause for these malformations. In-depth analysis of Spca1 embryos using molecular markers revealed that loss of Spca1 disrupts apical constriction in the neural tube. Similar to mutations in the actin-binding proteins shroom family member 3 (SHROOM3) and cofilin 1 (CFL1), loss of SPCA1 function disrupts the organization of the apical actomyosin cytoskeleton. We demonstrate that SPCA1 is required to regulate myosin II localization, as well as actin dynamics, and identify CFL1 as a downstream effector of SPCA1 function. Together, these data provide the first genetic evidence that perturbation of calcium homeostasis impairs apical constriction during neural tube closure and provide mechanistic insight into the role of SPCA1 in regulating the cytoskeletal dynamics that execute apical constriction.
RESULTS
A mouse forward genetic screen identifies a novel allele of Spca1
The 1D mutation was found during an ENU mutagenesis screen to identify recessive lethal mutations affecting mouse embryonic morphogenesis. Homozygote 1D mutants showed a characteristic exencephalic phenotype, with an open neural tube extending from the midbrain to the anterior spinal cord (Fig. 1A,B). This fully penetrant phenotype was observed as early as E8.75 and was the only discernible defect in 1D embryos until their developmental arrest and embryonic lethality between E10.5 and E12.5. Analysis of transverse embryonic sections at E9.5 revealed that the medial hinge point at the floorplate of the neuroepithelia was similar in 1D mutants and wild-type littermates (Fig. 1C-F, black arrowheads). However, the neural folds failed to bend dorsolaterally in 1D embryos, causing the neuroepithelia at the cranial region to remain open, with an exposed convex surface (Fig. 1C,D). At the spinal cord, the neural tube of 1D mutants also showed a convex surface, but the non-neural epithelium was able to bridge over the dorsal aspect of the neuroepithelia, avoiding the exposure of the neural folds (Fig. 1E,F, unfilled arrowhead). In a small subset of 1D embryos, the neural tube was fully exposed throughout the anterior-posterior axis (craniorachischisis; 2/83 embryos). Together, these phenotypes support the conclusion that the gene disrupted by the 1D mutation is required for proper dorsolateral bending of the neural tube throughout the anterior-posterior embryonic axis.
Fig. 1.
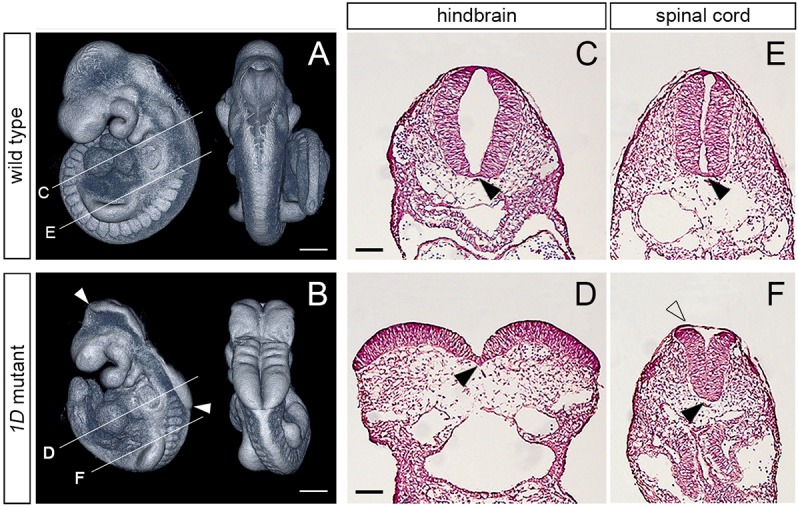
The 1D mutation causes exencephaly and spinal cord defects. (A,B) Lateral (left) and dorsal (right) views of E9.5 wild-type and 1D mutant embryos obtained from micro computed tomography (microCT) scans. Arrowheads in B demarcate the extent of exencephaly in the 1D mutant. Lines indicate the level of cross-sections shown in C-F. (C-F) Cross sections through the hindbrain (C,D) and spinal cord (E,F) regions in E9.5 wild type and 1D mutants. Black arrowheads point to the medial hinge point. Unfilled arrowhead in F points to the surface epithelia covering the abnormally shaped neuroepithelia in 1D embryos. Scale bars: 300 µm (A,B); 100 µm (C-F).
To identify the mutation responsible for the neural tube defects in 1D mutants, we first used PCR amplification of short tandem repeat polymorphisms to map the 1D mutation to the 2.65 Mbp region on chromosome 9 defined by markers D9Mit24 and D9Mit310. Of the 23 genes in this region, the gene encoding secretory pathway Ca2+ ATPase 1 (Spca1) stood out as a good candidate because of the similar exencephalic phenotype of 1D embryos and Spca1 null mutants (Okunade et al., 2007). Sequencing of the Spca1 cDNA from 1D embryos identified a single T-to-A transversion in the coding region, which is predicted to result in a non-conservative V183E amino acid substitution in SPCA1 (Fig. 2A,B). Alignment of the mouse SPCA1 sequence with related Ca2+ ATPase pumps revealed that the non-polar nature of V183 is conserved across species (Fig. 2C), as well as within other mouse Ca2+ ATPase family members (Fig. 2D), suggesting that this residue is important for SPCA1 function. To determine whether the predicted V183E mutation in 1D mutants represents a loss of function in Spca1, we performed a genetic complementation test between the 1D mutation and a null allele of Spca1 obtained from the KOMP repository (Spca1KO; clone EPD0858_4_C07; Skarnes et al., 2011). We found that transheterozygote embryos carrying the 1D and Spca1 KO alleles showed similar exencephaly and lethality to both homozygote 1D mutants and Spca1KO embryos (Fig. 2E-H). Consequently, we interpret the results from our genetic complementation test as genetic evidence that the 1D mutation is a novel allele of Spca1, and that the excencephalic phenotype in 1D embryos is due to loss of SPCA1 function.
Fig. 2.
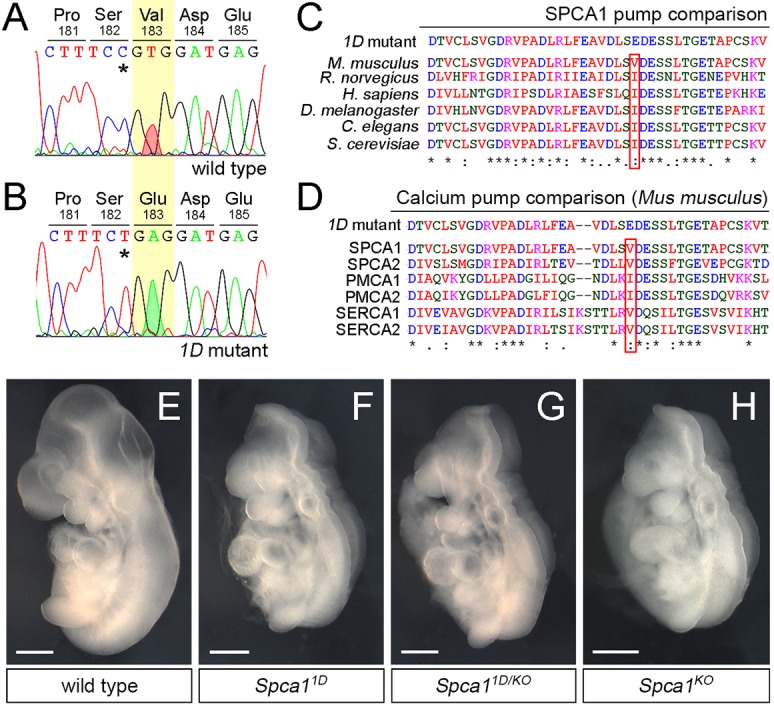
1D mutants contain a non-conservative mutation in SPCA1. (A,B) Sanger sequencing traces for the Spca1 coding region in wild type and 1D mutants. 1D embryos contain a T-to-A nucleotide difference (red and green shaded peaks), which is predicted to cause a Val-to-Glu amino acid change at position 183 (highlighted in yellow). Residue numbers are based on NP_778190 sequence. Note that the Spca1 sequence shown contains a known silent polymorphism between the FvB (wild-type) and C57J/B6 (1D mutant) strains (C-T SNP, asterisks). (C,D) Protein sequence alignments showing the conserved nature of the mouse SPCA1 V183 residue across species (C) and in comparison with other mouse Ca2+ ATPases (D). Conservation is indicated for identical residues (asterisks), for residues with strong similar properties (double dot) and for residues with weakly similar properties (dot). SPCA2, PMCA1, PMCA2, SERCA1 and SERCA2 are also known as ATP2C2, ATP2B1, ATP2B2, ATP2A1 and ATP2A2, respectively. (E-H) Lateral views of E9.5 wild-type (E), Spca11D homozygotes (F), Spca11D/KO trans heterozygote (G) and homozygote Spca1KO (H) embryos. Scale bars: 500 µm.
Calcium homeostasis is disrupted in Spca11D mutants
We next investigated how the 1D mutation disrupts SPCA1. The 1D mutation did not disrupt the localization of SPCA1 to the Golgi apparatus, as shown by immunofluorescence staining of wild-type and Spca1 mutant mouse embryonic fibroblasts (MEFs) (Fig. 3A-F) and neuroepithelial cells (Fig. S1). Based on the structural analysis of the related sarco/endoplasmic reticulum Ca2+ ATPase pump (SERCA; Toyoshima et al., 2000) and a homology-modeled protein structure of SPCA1 (obtained through SWISS-MODEL; Schwede et al., 2003), the 1D V183E mutation in SPCA1 is predicted to disrupt a β-sheet in the actuator domain, a region that undergoes dramatic conformational changes essential for the calcium pumping cycle (Toyoshima et al., 2000). Consequently, we hypothesized that the 1D V183E mutation disrupts the ability of SPCA1 to pump calcium. To test this hypothesis, we performed calcium clearance assays in MEFs using the fluorescence-based cytosolic calcium reporter GCaMP2 (Lissandron et al., 2010; Wang et al., 2008). MEFs obtained from wild-type, Spca11D and Spca1KO embryos were transfected with the cytosolic GCaMP2 reporter, then fluorescence was monitored by live imaging in resting conditions (Ca2+-free media) and upon addition of 2 mM Ca2+. We found that the initial intracellular calcium levels in resting conditions were elevated in Spca11D and Spca1KO cells compared with wild-type MEFs (Fig. 3G, asterisk), a result consistent with a role for SPCA1 in maintaining low cytosolic calcium levels. The addition of calcium to the culture media triggered a sudden increase in cytosolic calcium in all MEFs, regardless of their genotype (Fig. 3G,I). Although clearance of these high calcium levels was initiated in both wild-type and Spca1 mutant cells, Spca11D MEFs failed to clear cytosolic calcium as efficiently as wild-type cells, resulting in higher resting levels of cytosolic calcium at the end of the experiment (Fig. 3G,H). Notably, calcium clearance in Spca11D mutant cells followed a similar curve as in null Spca1KO MEFs, demonstrating that the V183E mutation in 1D mutants severely impairs the ability of SPCA1 to pump calcium. Together with the similarities between the exencephalic phenotypes of Spca11D and Spca1KO embryos, these results indicate that the 1D allele represents a strong loss-of-function allele of Spca1.
Fig. 3.

Effects of the 1D mutation on the localization and function of SPCA1. (A-F) Immunohistochemistry with SPCA1 (A,C,E, green) and GM130 (GOLGA2) (B,D,F, magenta) antibodies in wild-type (A,B), Spca1KO (C,D) and Spca11D (E,F) MEFs. Scale bars: 20 µm. (G-J) Calcium clearance assays in wild-type (black, n=9), Spca1KO (blue, n=6) and Spca11D (yellow, n=9) MEFs in the absence (G,H) or presence (I,J) of drug inhibitors. Drug treatments were as follows: wild-type MEFs treated with 100 nM thapsigargin (Th; SERCA inhibitor, orange, n=8), Spca11D MEFs treated with 100 nM thapsigargin (green, n=7) and Spca11D MEFs treated with sodium orthovanadate (violet, n=4). At time 0, cells were spiked with 2 mM Ca2+ (black dashed lines). Resting cytosolic calcium levels 10 min after introduction of calcium (gray dashed lines) are quantified in H and J. Asterisks in G and I indicate measurements in resting conditions before the addition of Ca2+. Error bars indicate s.e.m. ns, not significant; *P<0.05, **P<0.01.
Although Spca11D and Spca1KO MEFs were not able to clear cytosolic calcium as efficiently as wild-type cells in our assays, they still showed a remarkable ability to regulate calcium homeostasis, an effect that we hypothesized was due to the activity of other calcium pumps involved in calcium clearance. To understand further the relative contribution of SPCA1 to calcium homeostasis, we performed calcium clearance assays as described above, but using drugs known to inhibit specific Ca2+ ATPases. In these experiments, fluorescence from the cytosolic GCaMP2 reporter in wild-type and Spca11D MEFs was measured in the absence or presence of 100 nM thapsigargin, a selective inhibitor of the endoplasmic reticulum-localized SERCA Ca2+ pumps (Lytton et al., 1991), and 100 µM orthovanadate, which has been described to inhibit all Ca2+ ATPases (Barrabin et al., 1980). We found that inhibiting multiple Ca2+ ATPases increased the severity of calcium clearance defects (Fig. 3I,J), consistent with a collaborative role for all Ca2+ ATPases in maintaining a low concentration of cytosolic calcium. Notably, inhibition of SERCA pumps alone did not increase the resting levels of cytosolic calcium to the levels seen in Spca1 mutants (Fig. 3I, orange and yellow lines), suggesting that SPCA1 is particularly important in maintaining low cytosolic calcium concentrations. These findings, together with the fact that SPCA1 is one of the predominant Ca2+ ATPases in neuroepithelial cells (Fig. S2; Brunskill et al., 2014), suggest that calcium homeostasis during neural tube closure relies on Spca1 function.
Neural tube defects precede apoptosis in Spca1 mutants
A previous study attributed the neural tube defects of Spca1 knockout embryos to widespread apoptosis (Okunade et al., 2007). However, this study quantified apoptosis at a developmental stage near the onset of embryonic lethality in Spca1 embryos, making it unclear whether or not apoptosis was the primary cause of the neural tube defects. To establish whether apoptosis precedes the appearance of an open neural tube in Spca1 embryos, we first performed dissections at different embryonic stages to determine when morphological defects in the Spca1 mutant neuroepithelia can be first observed. We found that at E8.75, wild-type embryos showed neural folds that were in close apposition (Fig. 4A). In contrast, the neural tube was open in E8.75 Spca11D embryos, with the dorsal-most aspects of the neuroepithelia remaining separate from each other throughout the cranial region (Fig. 4B). At this early developmental stage, the characteristic convex surface of the Spca11D mutant neuroepithelia was observed in the hindbrain (Fig. 4B, unfilled arrowhead), but not in the midbrain (Fig. 4B, white arrowhead). However, by E9.5 the neuroepithelia surface was convex at both cranial regions in all Spca11D embryos. Together, these results indicate that neural tube defects in Spca1 mutants develop between E8.5 and E9.5.
Fig. 4.

Apoptosis in Spca11D mutants. (A,B) MicroCT scans of E8.75 wild-type and Spca11D embryos (dorsal views). White arrowhead indicates midbrain area, unfilled arrowhead hindbrain area. (C-H) Immunodetection of activated caspase 3 (αCasp3) in wild-type and Spca11D embryos at E8.75 (C,D), E9.5 (E,F) and E10.5 (G,H). (I) Plot represents average apoptotic index (activated caspase 3 foci/total number of nuclei) in wild-type and Spca11D embryos at E8.75, E9.5 and E10.5 (n=10 sections from three biological replicates for each stage and genotype). HM, head mesenchyme; NT, neural tube. Error bars indicate s.e.m. Scale bars: 300 µm (A,B); 100 µm (C-H).
To determine whether apoptosis correlates with the appearance of neural tube defects in Spca11D embryos, we used immunofluorescence with activated caspase 3 (αCasp3) antibodies, as well as terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays (Mirkes et al., 2001). The number of apoptotic cells was quantified in transverse sections of wild type and Spca11D mutants at three developmental stages: E8.75, E9.5 and E10.5. Consistent with the onset of embryonic lethality in Spca11D mutants, we found that the number of αCasp3-positive cells was elevated in both the neural tube and surrounding head mesenchyme of E10.5 Spca11D embryos (Fig. 4G-I). However, at E8.75 and E9.5, when neural defects become morphologically evident in Spca11D mutants, we found no significant difference in apoptosis with respect to wild-type littermates (Fig. 4C-F,I). Experiments using TUNEL assays confirmed these results (not shown). Therefore, although our analysis showed that there is widespread apoptosis in Spca11D embryos as they reach their stage of embryonic lethality, our results demonstrate that neural tube defects in Spca11D mutants precede the generalized apoptosis observed in Spca11D mutants, indicating that the exencephalic phenotype of Spca11D embryos is not caused by cell death.
SPCA1 is required for proper apical constriction in the neuroepithelia
To investigate further the mechanisms underlying neural tube defects in Spca1 mutants, we sought to establish parallels between the phenotypes of Spca1 embryos and other mouse mutants with neural tube defects. Spca1 embryos were similar to Shroom3 and cofilin 1 (Cfl1) mouse mutants in several aspects, including a predominantly cranial exencephaly, a closed, but convex-shaped spinal cord, and low-penetrance craniorachischisis (Grego-Bessa et al., 2015; Gurniak et al., 2005; Hildebrand and Soriano, 1999). Because loss of function of Shroom3 and Cfl1 disrupts apical constriction, we wondered whether SPCA1 also influences this morphogenetic process. To explore whether neuroepithelial cells in Spca1 embryos fail to apically constrict, we examined en face views of E9.5 wild-type and Spca11D neuroepithelia at the hindbrain region, using phalloidin staining to reveal F-actin enrichment at the cell cortex. Images were digitally analyzed with ImageJ (Eliceiri, 2017) to quantify the apical cell area of individual cells (Fig. 5). This analysis revealed that the average apical cell area was increased in Spca11D mutants compared with wild-type littermates (Fig. 5E) and that the number of constricted cells in the Spca1 mutant neuroepithelia was dramatically reduced compared with wild-type controls (Fig. 5C,D, maximally constricted cells shown in deep blue and purple; see Fig. 5A′,B′ for representative examples of wild-type and Spca11D embryos). These results demonstrate that apical constriction is disrupted in Spca1 mutants. We also investigated whether loss of SPCA1 function interferes with other known morphogenetic processes involved in the closure of the neural tube. However, our data indicates that loss of Spca1 does not affect either proliferation (Fig. S3), Shh signaling and dorsoventral neural tube identity (Fig. S4), planar cell polarity (Fig. S5) or the polarity of the neuroepithelia (Fig. S6). Together, these findings identify a specific requirement for SPCA1 in the apical constriction of neuroepithelial cells during neural tube closure.
Fig. 5.

Analysis of apical constriction in Spca11D mutants. (A,B) Phalloidin staining of F-actin in the anterior spinal cord region of whole-mount wild-type and Spca11D embryos (en face view). (A′,B′) Semi-automated cell outlines generated in ImageJ and color coded based on size of apical area (see C,D). Scale bar: 20 µm. (C,D) Distribution of cell apical areas in wild-type and Spca11D neuroepithelia. (E) Box plot showing the apical area of cells in wild-type and Spca11D neuroepithelia. Data in C-E corresponds to 834 cells from four wild-type embryos and 966 cells from four Spca11D mutant embryos. The line represents the median, the box indicates the distance between the first and third quartile (interquartile range; IQR), whiskers represent ±1.5*IQR and points represent all individual measurements. *P<0.001.
Spca1 is required for proper localization of myosin II motors to actin filaments
Given the similarities between the phenotypes of Spca1 and Shroom3 mutants, we investigated whether the lack of apical constriction in these embryos has a common molecular basis. Because SHROOM3 regulates apical constriction by influencing actomyosin dynamics (Hildebrand, 2005; McGreevy et al., 2015), we first analyzed the organization of actin and myosin II in Spca11D mutants. To this end, we examined en face views of E9.5 wild-type and Spca11D neuroepithelia at the hindbrain region using non-muscle myosin heavy chain IIB (NMHCIIB; also known as MYH10) antibodies and phalloidin staining (Fig. 6). Myosin II and F-actin were enriched at apical cell junctions in wild-type E9.5 neuroepithelia (Fig. 6A,A′). However, in Spca11D embryos, we found that NMHCIIB lost its sharp junctional localization and was enriched in the cytoplasm of neuroepithelial cells (Fig. 6B,B′). To corroborate this cytoplasmic enrichment, we used ImageJ to quantify the amount of medial and junctional NMHCIIB accumulation in cells from wild-type and Spca11D embryos. This analysis confirmed that the junctional-to-medial ratio of NMHCIIB was reduced in Spca11D samples compared with wild-type littermates (Fig. 6C, n>1000 cells from four embryos per genotype, P<0.001). Western blot quantification of NMHCIIB in embryonic extracts showed that Spca11D mutants have levels of NMHCIIB similar to those of wild-type embryos (Fig. S7A,B), indicating that the cytoplasmic enrichment in Spca1 mutants is not due to an upregulation in the levels of NMHCIIB. Because the localization of NMHCIIB at the junctional domain is crucial for the generation of the contractile forces that promote apical constriction (Hildebrand, 2005), our results suggest that Spca1 regulates neural tube closure by regulating NMHCIIB localization in neuroepithelial cells.
Fig. 6.
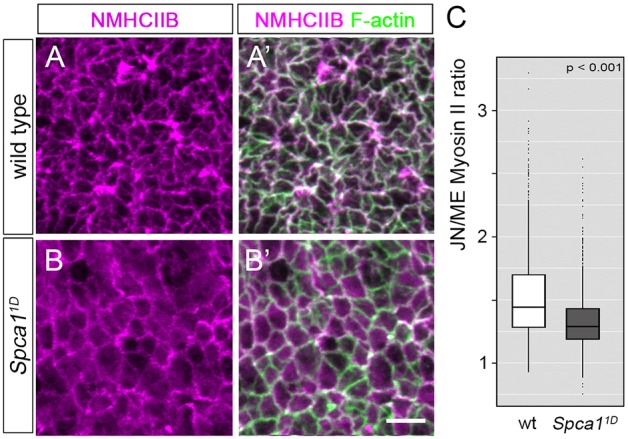
Myosin localization and phosphorylation in Spca11D mutants. (A-B′) Immunodetection of NMHCIIB and F-actin in E9.5 wild-type (A,A′) and Spca11D (B,B′) embryos. Scale bar: 5 µm. (C) Quantification of junctional (JN) and medial (ME) NMHCIIB in E9.5 wild-type (white) and Spca11D (gray) embryos (n>1000 cells from four embryos for each genotype). The line represents the median, the box indicates the distance between the first and third quartile (interquartile range; IQR), whiskers represent ±1.5 *IQR and points indicate outliers.
The effect of Spca1 loss of function on NMHCIIB junctional localization is similar to that previously described for Shroom3 embryos (McGreevy et al., 2015), providing support for the hypothesis that neural tube defects in these mutants might originate through common molecular mechanisms. However, despite the similarities between Spca1 and Shroom3 mutants, our results also revealed important discrepancies. First, Shroom3 deficiency results in diffuse and discontinuous junctional F-actin, with a concomitant increase in cytoplasmic F-actin (McGreevy et al., 2015). In contrast, en face views of the Spca11D neuroepithelia failed to detect any visible alteration in F-actin localization (Figs 5A,B, 6A-B′, 7C′,D′ and Fig. S8A,B) and quantification of medial versus junctional actin did not reveal any significant difference between Spca11D mutants and wild-type littermates (Fig. S8F). Second, loss of Shroom3 has been shown to cause a decrease in the levels of phosphorylated myosin light chain 2 (Grego-Bessa et al., 2015; Nishimura and Takeichi, 2008). However, quantification of pMLC2 using western blotting indicated that pMLC2 levels were not significantly altered between Spca11D mutants and littermate controls (Fig. S7A,B, P=0.38). Furthermore, the levels of myosin light chain kinase (MLCK1; also known as MYLK) and myosin targeting phosphatase subunit 1 (MYPT1; PPP1R12A), which influence MLC2 phosphorylation (Somlyo and Somlyo, 2003) were unaffected by loss of Spca1 (Fig. S7A,B). In addition to these differences, we found that SHROOM3 was properly localized at the apical domain of neuroepithelial cells in Spca11D mutants (Fig. S9). Therefore, taken together, these results indicate that although SHROOM3 and SPCA1 have some common effects on the cytoskeletal organization of the neuroepithelia, it is unlikely that SPCA1 regulates neurulation by influencing SHROOM3 function. Instead, our data suggest that SPCA1 controls apical constriction by directly influencing NMHCIIB localization.
Fig. 7.

Cofilin 1 localization and actin dynamics in Spca11D mutants. (A-B′) Immunodetection of CFL1 (magenta) and F-actin (green) in transverse sections of the E9.5 wild-type and Spca11D neuroepithelia. Scale bar: 20 µm. (A″-B‴) Magnified views of CFL1 and F-actin localization in the boxed areas in A′ and B′. Scale bar: 5 µm. (C-D′) En face views of CFL1 (magenta) and F-actin (green) localization in E9.5 wild-type and Spca11D embryos. Scale bar: 5 µm. (E) Western blot of F- and G-actin fractions isolated from wild-type and Spca11D embryos (right) and quantification of F/G actin ratios (left, n=5). Error bars indicate s.e.m. *P<0.05.
SPCA1 regulates CFL1 localization and actin dynamics
We also investigated the molecular basis for the similarities between the neurulation and apical constriction defects of Spca1 and Cfl1 embryos. Interestingly, CFL1 has been shown to bind SPCA1 and to influence its calcium-pumping activity (von Blume et al., 2011; Kienzle et al., 2014), raising the possibility that loss of SPCA1 function contributes to the neurulation defects of CFL1 mutants. We wondered whether SPCA1 could reciprocally influence CFL1 function. To test this possibility, we analyzed CFL1 localization and F-actin dynamics in Spca1 embryos.
As previously described (Grego-Bessa et al., 2015), we found that CFL1 localized to both the apical and basal domains of the neuroepithelia in wild-type E9.5 embryos (Fig. 7A). Our analysis of Spca11D transverse sections did not detect any defect in CFL1 localization at the basal domain (Fig. 7A,B). However, we found that CFL1 failed to colocalize with apical F-actin filaments in Spca11D mutants and, instead, accumulated in cytoplasmic blebs at the apical surface of the neuroepithelium (Fig. 7A″-B‴). We were able to confirm the effects of Spca1 loss of function on apical CFL1 localization through en face analysis of the neuroepithelia. In wild-type embryos, CFL1 was localized to punctate structures distributed both at the medial cytoplasmic domain, as well as at cell junctions, where it colocalized with actin (Fig. 7C,C′). In Spca11D mutants, however, CFL1 localized exclusively at the medial domain and was excluded from the junctional actin cytoskeleton (Fig. 7D,D′). Together, these results provide evidence that SPCA1 activity is required for the proper subcellular localization of CFL1 in the apical domain of the neuroepithelia.
Because CFL1 regulates actin dynamics by binding to actin filaments (Lappalainen et al., 1997), we hypothesized that SPCA1 might control neural tube closure by regulating actin dynamics through CFL1. CFL1 controls actin dynamics by severing actin filaments, thereby producing free barbed ends that can serve to enhance actin polymerization or depolymerization depending on the local concentration of several regulating factors (Bravo-Cordero et al., 2013). Our analysis of Spca11D embryos did not reveal visible alterations in the distribution of F-actin compared with wild-type controls, in either transverse neural tube sections or through en face analysis of the neuroepithelia (Fig. S8A-D). However, quantification of pixel intensity from confocal images revealed that apical F-actin was decreased in Spca11D mutants compared with wild-type controls (Fig. S8G,H). Additionally, analysis of F-actin and G-actin fractions from wild-type and Spca11D embryos revealed a decrease in the F-actin/G-actin ratio in Spca11D mutants (Fig. 7E), providing evidence that loss of SPCA1 function disrupts actin dynamics. Taken together with our previous findings, our results suggest that SPCA1 regulates apical constriction by influencing CFL1 localization and activity.
DISCUSSION
In this study, we identify a new allele of Spca1 and demonstrate that, contrary to previous reports, the neurulation defects caused by Spca1 loss of function are not due to widespread apoptosis. We show that loss of Spca1 impairs the apical constriction of neuroepithelial cells, but does not interfere with dorsoventral neural tube patterning, nor with the integrity or polarity of the neural epithelia. We provide evidence that SPCA1 is required to maintain low cytoplasmic calcium levels. This finding, together with the fact that SPCA1 is a predominant calcium ATPase in neural tissues, indicates that SPCA1 has an important role regulating calcium dynamics in neuroepithelial cells. Together, results from our characterization of Spca1 mutants demonstrate that SPCA1 is crucial for the apical constriction of neuroepithelial cells during mammalian neurulation, suggesting that clearance of cytosolic calcium levels is important for this process. Furthermore, our findings demonstrate that SPCA1 is required to regulate the actomyosin dynamics that drive apical constriction and identify CFL1 as an important downstream effector of SPCA1 function during neural tube closure.
Molecular effectors of SPCA1 function
Spca1 embryos are remarkably similar to mutants in Shroom3 and Cfl1, suggesting that apical constriction defects in these mutants stem from a common molecular mechanism. We show that Spca1 embryos have defects in the localization of myosin II motors to junctional actin filaments and the stability of the actin cytoskeleton. Interestingly, the abnormal localization of myosin II to the medial domain is similar in Spca1 and Shroom3 mutants, raising the possibility that SHROOM3 may function as a downstream effector of SPCA1. However, our results show that loss of SPCA1 has different effects than loss of SHROOM3 function on F-actin localization (Fig. S8; McGreevy et al., 2015) and MLC2 phosphorylation (Fig. S7; Grego-Bessa et al., 2015; Nishimura and Takeichi, 2008), as well as on the stage of embryonic lethality (E18.5 for Shroom3 and E11.5 for Spca1; Hildebrand and Soriano, 1999; Okunade et al., 2007). These differences, together with the fact that we did not observe any abnormal localization of SHROOM3 in the Spca1 neuroepithelia (Fig. S9), all suggest that the neurulation and apical constriction defects in Spca1 mutants are not due to a loss of SHROOM3 function.
Our analysis of Spca1 embryos also revealed similarities and differences with respect to Cfl1 mutants. One of the most striking differences is in the effects of SPCA1 and CFL1 loss of function on the basal domain of the neuroepithelia. CFL1 localizes at both the apical and basal domains of neuroepithelial cells, and these different domains have been proposed to have separate roles in regulating cytoskeletal organization. At the basal domain, Cfl1 mutants show ectopic localization of F-actin, MHCIIB and pMLC2, as well as disruptions of the basement membrane that give the Cfl1 neuroepithelia a multilayered appearance (Grego-Bessa et al., 2015). Our analysis of Spca1 mutants did not detect any of the basal phenotypes characteristic of Cfl1 mutants, nor any abnormality in the localization of the basal CFL1 pool. However, we found that CFL1 failed to localize to actin filaments in the apical domain, where CFL1 is required to regulate actomyosin dynamics (Grego-Bessa et al., 2015). These findings, together with the fact that SPCA1 localizes to the apical aspect of neuroepithelial cells, suggest that SPCA1 regulates the apical, but not the basal, pool of CFL1. Results from the analysis of embryos carrying Spca1 and Cfl1 mutant alleles are consistent with SPCA1 and CFL1 having cooperative effects during neural tube closure and embryonic development (Fig. S11).
CFL1 localization and activity are regulated by phosphorylation. In its unphosphorylated state, CFL1 can bind actin and promote its polymerization/depolymerization, but phosphorylation at Ser3 renders CFL1 unable to bind and remodel actin filaments (reviewed by Bravo-Cordero et al., 2013). The levels of CFL1 phosphorylation are regulated by the action of the LIM-domain protein kinase (LIMK), which phosphorylates CFL1 upon being activated by ROCK (Maekawa et al., 1999; Scott et al., 2010), as well as by the slingshot protein phosphatase (Niwa et al., 2002). Interestingly, SSH activity is regulated by the calcium-dependent proteins calcineurin (Wang et al., 2005) and Ca2+/calmodulin-dependent protein kinase II (Zhao et al., 2012), suggesting that the levels of intracellular calcium are crucial for CFL1 activity. It will be interesting to address whether SPCA1 influences CFL1 localization by regulating its phosphorylation, but western blot experiments using embryonic extracts were unable to resolve whether the levels of CFL1 phosphorylation and its regulators are significantly disrupted in Spca1 mutants (Fig. S10). One possibility is that CFL1 localization is regulated by SPCA1 through mechanisms other than protein phosphorylation. Alternatively, loss of Spca1 activity might cause significant local differences in pCFL1 levels that, although difficult to detect by western blotting, could ultimately be responsible for the abnormal CFL1 localization we observed in Spca1 mutants. As tools become available, it will be important to investigate further whether the local activity of CFL1 and its regulators is disrupted at the neural tube epithelia of Spca1 mutants.
The effects of Spca1 loss of function on CFL1 localization and actin dynamics (Fig. 7) suggest that CFL1 is an important downstream effector of SPCA1. However, complete loss of CFL1 leads to an expansion of the apical F-actin domain, as well as to increased F-actin/G-actin ratios (Grego-Bessa et al., 2015). In contrast, we observed a small decrease in F-actin accumulation at the apical domain of the Spca1 neuroepithelia (Fig. S8G,H), as well as a decrease in F-actin/G-actin ratios in Spca1 embryos compared with wild-type littermate controls (Fig. 7E). Furthermore, loss of Cfl1 causes a decrease in pMLC2 levels (Grego-Bessa et al., 2015), whereas the levels of pMLC2 were unaffected in Spca1 embryos. Therefore, although abnormal localization of apical CFL1 to F-actin in Spca1 mutants strongly suggests a role for CFL1 as a downstream effector of SPCA1, the differences between Spca1 and Cfl1 mutants argue that apical constriction defects in the Spca1 neuroepithelia cannot be exclusively explained by a loss of CFL1 function. We speculate that CFL1 might not be the only downstream effector of SPCA1, and that the different effects of SPCA1 and CFL1 loss of function on actin dynamics and MLC2 phosphorylation could be due to collateral effects of SPCA1 activity on additional actin remodelers, as well as other regulators of myosin contractility. In the future, it will be interesting to explore further the molecular basis for the links between calcium homeostasis, CFL1 and actin dynamics in Spca1 mutants.
Evidence for a conserved role for calcium in the control of apical constriction
Research in cell culture, fly and vertebrate embryos has identified actin filaments and myosin II motors as a common machinery required for apical constriction. However, the organization of these cytoskeletal components differs between organisms and developmental contexts (Martin and Goldstein, 2014). For instance, studies during Drosophila gastrulation and frog neurulation indicate that the forces that promote apical constriction depend on both medial and junctional actomyosin networks (Coravos and Martin, 2016; Martin et al., 2009; Mason et al., 2013; Solon et al., 2009). In contrast, the analysis of chicken and mouse embryos (Nishimura and Takeichi, 2008; Schroeder, 1970), as well as our own observations, indicate that actomyosin complexes are localized at cell junctions during neural tube closure. These differences question whether contractile forces for apical constriction are regulated through similar mechanisms across different organisms and developmental contexts, and whether findings in one model system can be extrapolated to other settings. In this respect, our finding that SPCA1 is required for apical constriction and neural tube closure in mice adds to the growing body of evidence from flies and frogs providing links between calcium and apical constriction.
During frog neurulation, calcium flashes and actin remodeling precede contractile activity (Christodoulou and Skourides, 2015). Therefore, it is tempting to speculate that, despite differences in cytoskeletal organization between frog and mice neuroepithelia, calcium pulses might also take place in mammals and that the duration and/or frequency of these pulses could be controlled by SPCA1. Additional studies will be required to uncover whether this is the case. Nonetheless, by providing genetic evidence that SPCA1 is required to control apical constriction during mammalian neurulation, our results not only indicate that a role for calcium in apical constriction has been conserved through evolution, but also implicate SPCA1 as an important player in the control of intracellular calcium levels during neural tube closure.
Biological oscillations often involve a delayed negative-feedback mechanism (Cao et al., 2016). In this respect, our findings, together with data from the literature, support a mutual regulatory relationship between SPCA1 and CFL1. CFL1 has been shown to be regulated by calcium levels through the activities of Ca2+/calmodulin-dependent kinase II (CaMKII) and Ca2+/calmodulin dependent phosphatase (calcineurin) (Wang et al., 2005; Zhao et al., 2012). Once active, cofilin remodels actin, influences myosin contractility (Wiggan et al., 2012) and binds to SPCA1, stimulating its calcium-pumping activity (von Blume et al., 2011; Kienzle et al., 2014). We show that SPCA1 activity influences myosin II contractility and CFL1 localization to actin filaments. Thus, it is tempting to speculate that SPCA1 and CFL1 could be part of a feedback loop mechanism, whereby a pulse of cytosolic calcium initiates a cascade of events leading to CFL1 activation, actomyosin remodeling and contractility, as well as calcium clearance by SPCA1, preparing the cell for another pulse of cytosolic calcium and subsequent contractile cycle. Although further experiments will be required to test this model, by identifying CFL1 as an important effector of SPCA1 during mouse neurulation, our results provide important insight into the regulatory interactions that could contribute to the coordination of calcium pulses and actomyosin dynamics during neural tube closure.
Calcium and neural tube defects
Neural tube malformations continue to represent a large proportion of birth defects in humans, in part due to their complex etiology, involving both genetic and environmental factors (Wallingford et al., 2013). Our results from the analysis of Spca1 mouse mutants raise the question of whether environmental calcium may play a role in the etiology of human neural tube defects. Epidemiological studies have indicated that the incidence of neural tube defects is lower in areas with high levels of calcium in the drinking water (hard water) and higher in areas with little or no calcium (soft water) (Lowe et al., 1971). Additionally, calcium levels were found to be significantly lower in the amniotic fluid of fetuses with neural defects compared with non-affected individuals (Dawson et al., 1999). Intake of compounds known to influence calcium homeostasis has also been linked to the development of neural tube closure defects. For instance, caffeine, the neuro-stimulating component in many popular beverages, stimulates the release of calcium from the Golgi apparatus by binding to ryanodine receptors (Lissandron et al., 2010) and has been shown to disrupt neural tube closure in rat, mouse and chicken embryos (Ma et al., 2012; Marret et al., 1997; Wilkinson and Pollard, 1994). Additionally, valproic acid, a drug used to treat depression and other mood disorders, affects calcium homeostasis (Ghodke-Puranik et al., 2013) and increases the incidence of neural tube defects (Hughes et al., 2018). Our results from the analysis of mouse Spca1 mutants, together with the growing body of evidence supporting a role for calcium in neurulation, urges additional epidemiological and clinical studies to examine how calcium homeostasis influences neural tube closure in humans, as well as whether counteracting dietary deficiencies in elemental calcium during early pregnancy could reduce the incidence of neural tube defects.
MATERIALS AND METHODS
ENU screen, mouse strains and genotyping
ENU mutagenesis was performed as previously described (Horner and Caspary, 2011) on Mus musculus C57BL/6J male mice (Jackson Laboratory stock #000664). FVB/NJ females (Jackson Laboratory stock #001800) were used to outcross mutagenized males and their progeny for more than ten generations. All experiments involving mice were performed according to standard operating procedures approved by Cornell's Institutional Animal Care and Use Committee. Animals and embryos were genotyped by PCR using polymorphic microsatellite markers (see Table S1) on DNA from proteinase K-treated tissue samples. Spca1 knockout mice were generated from Knock Out Mouse Project clone EPD0858_4_C07 (Skarnes et al., 2011). Chimeras with germline transmission were bred to Sox2-Cre mice (Hayashi et al., 2002) to generate the Spca1 knockout allele (Spca1KO), which was then maintained into a C57BL/6NJ background. Cfl1 knockout mice (Gurniak et al., 2005) were obtained from Kathryn Anderson (Sloan Kettering Institute, NY, USA) and raised in an FVB/NJ background. See Table S1 for Spca1KO genotyping primers.
Linkage analysis and positional cloning
ENU mutants were mapped using a genome-wide selection of microsatellite markers polymorphic between FVB/NJ and C57BL6/J (see Table S2 in the supplementary material). The Spca1 coding region was PCR amplified and sequenced from a cDNA library obtained from embryonic RNA (RNA-STAT 60, Amsbio) using SuperScript III First-Strand (Invitrogen 18080051). See Table S1 for primer sequences.
Mouse embryo analysis
Matings were set up and monitored for vaginal plugs. Embryos of the desired stage were dissected in cold PBS-BSA and fixed overnight in 4% paraformaldehyde (PFA) at 4°C. After fixation, embryos were processed for whole-mount in situ hybridization, whole-mount immunofluorescence (en face views) or for immunofluorescence on 10 µm cryosections (embedded in OCT compound). Whole-mount RNA in situ hybridization was performed as previously described (Garcia-Garcia et al., 2008). Immunofluorescence was performed following standard protocols (Nagy et al., 2003) using the antibodies indicated in Table S1. Samples were mounted in Fluoroshield Mounting Medium containing DAPI (Abcam, ab104139) and imaged with a Zeiss LSM 710 Confocal using Plan-Apochromat 63×/1.4 Oil DIC M27 or C-Apochromat 40×/1.2 W Corr M27 objectives (Cornell University BRC Imaging Facility). Gender did not affect any of the embryonic phenotypes described.
For micro-computed tomography (microCT) imaging, E9.5 and E8.75 embryos were fixed in PFA, rinsed in PBS, dehydrated in methanol, then stained in 1% iodine (I2)/100% methanol for 3 days at room temperature. Samples were scanned at 2.7 µm resolution using a Zeiss Xradia Versa 520 XRM (Cornell University BRC Imaging Facility). 3D reconstructions of the data were generated using OsiriX software (Pixmeo SARL).
Cell culture
Primary MEFs were obtained from whole E9.5 wild-type and mutant embryos. All experiments were performed on primary MEFs maintained in DMEM/10% fetal bovine serum for fewer than six passages. Immunohistochemistry was performed following standard procedures using the antibodies indicated in Table S1. For calcium imaging, MEFs were grown on 35-mm glass-bottom dishes (Greiner Bio-One, 627860) and transfected with pCMV-GCaMP2 (provided by Michael Kotlikoff, Cornell University, NY, USA) using Lipofectamine 2000 (Thermo Fisher Scientific, 11668027). Fluorescence levels upon calcium stimulation were quantified 36-48 h after transfection as previously described (McCombs and Palmer, 2008). Cells were equilibrated in Ca2+-free Kreb's buffer 30-60 min prior to imaging. Confocal images were acquired every 5 s for 10 min on a Zeiss LSM710 confocal microscope equipped with a humidified chamber at 37°C using a C-Apochromat 40×/1.2 W Corr M27 objective and definite autofocus (Cornell University BRC Imaging Facility). Relative fluorescence intensity for each time point was calculated as fluorescence intensity/(Rmax−Rmin)*100.
Immunoblotting and measurement of F/G-actin ratios
A pool of E9.5 wild-type embryos or individual E9.5 mutant embryos were lysed in RIPA buffer containing protease and phosphatase inhibitors (Thermo Fisher Scientific, A32961). Protein concentrations were measured using a BCA protein assay (Thermo Fisher Scientific, 23227) and equal amounts of wild-type and mutant protein were loaded in adjacent lanes. Western blots were performed using standard protocols using HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, 115-035-044/87768) and Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, 32106). Band intensity was quantified using the ImageJ Gel Analyzer tool. GAPDH and αTubulin were used as loading controls. F/G-actin fractions were quantified as previously described (Rasmussen et al., 2010). Analysis of pCFL1 to CFL1 ratios was carried out by western blotting of protein isolates from cranial neural tube extracts of E9.5 embryos that were run on 12.5% SuperSep Phos-Tag gels (Wako Pure Chemical Industries, 195-17991).
Fluorescence quantification and image processing
Apoptosis
ImageJ was used to draw regions of interest (ROIs) around the neuroepithelium and head mesenchyme in confocal images of caspase 3- and DAPI-stained embryo sections. The total number of nuclei (DAPI channel) was quantified using the ‘Image-based Tool for Counting Nuclei’ (ITCN) ImageJ plugin. Caspase 3 foci were manually counted in each image. The apoptotic index was calculated as the number of apoptotic cells divided by the total number of cells in the ROI.
Apical area
Maximum intensity projections of the apical region of phalloidin-stained en face images were obtained. The ‘Tissue Cell Geometry Stats’ plugin (with manual correction) in ImageJ was used to outline individual cells and measure their apical area.
Myosin II localization
Maximum intensity projections of the apical region of en face actin stained images were obtained. The ‘Tissue Cell Geometry Stats’ ImageJ plugin was used to outline individual cells in the F-actin channel. Cell ROIs were copied to the NMHCIIB channel. A custom script was used to expand each cell ROI by 3 pixels/measure the fluorescence intensity (Total Intensity) and then contract the ROI by 5 pixels/measure intensity (Medial Intensity). Junctional intensity was calculated as total intensity minus medial intensity. Data are reported as junctional/medial intensity ratio.
Planar cell polarity quantification
Quantification of the planar distribution of NMHCIIB was performed using a method similar to that described by McGreevy et al. (2015). Using ImageJ, segmented ROIs were drawn around individual junctions and the mean fluorescence intensity was obtained for each junction. The images were thresholded to consider only the top 50% of pixels. The fluorescence intensity of each junction was normalized to the average fluorescence intensity of all junctions in the same sample. The orientation of the junction relative to the mediolateral axis was computed by measuring the angle of the major axis of an ellipse fitted to each ROI.
Statistical analysis
Statistical analysis was performed using Excel or R software. P-values were calculated using two-tailed unpaired Student's t-tests unless otherwise indicated.
Acknowledgements
We thank Katherine Alexander and Kathryn Anderson for helpful discussions and comments on the manuscript; Dr Tim Reinhardt and Dr Michael Kotlikoff for reagents; Cornell's CARE staff for mice husbandry and care; and personnel at the Transgenic, Confocal and Electron microscopy facilities for help with experiments. The Cornell Stem Cell and Transgenic Core Facility was supported by the New York State Department of Health (C029155); Cornell Center for Materials Research facilities were supported by the National Science Foundation (DMR-1120296) and the Cornell Biotechnology Resource Center Imaging Facility was supported by the National Institutes of Health (S10RR025502 and S10OD012287).
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: M.J.G.-G., J.M.B.; Validation: M.J.G.-G., J.M.B.; Formal analysis: J.M.B.; Investigation: J.M.B.; Writing-original draft: M.J.G.-G., J.M.B.; Writing-review & editing: M.J.G.-G.; Visualization: M.J.G.-G., J.M.B.; Supervision: M.J.G.-G.; Funding acquisition: M.J.G.-G.
Funding
Work in the García-García lab was supported by the National Science Foundation (IOS-1452543). J.M.B. was funded by the Cornell Center for Vertebrate Genomics.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.170019.supplemental
References
- Antunes M., Pereira T., Cordeiro J. V., Almeida L. and Jacinto A. (2013). Coordinated waves of actomyosin flow and apical cell constriction immediately after wounding. J. Cell Biol. 202, 365-379. 10.1083/jcb.201211039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrabin H., Garrahan P. J. and Rega A. F. (1980). Vanadate inhibition of the Ca2+-ATPase from human red cell membranes. Biochim. Biophys. Acta 600, 796-804. 10.1016/0005-2736(80)90482-4 [DOI] [PubMed] [Google Scholar]
- Bravo-Cordero J. J., Magalhaes M. A. O., Eddy R. J., Hodgson L. and Condeelis J. (2013). Functions of cofilin in cell locomotion and invasion. Nature Publishing Group 14, 405-415. 10.1038/nrm3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill E. W., Potter A. S., Distasio A., Dexheimer P., Plassard A., Aronow B. J. and Potter S. S. (2014). A gene expression atlas of early craniofacial development. Dev. Biol. 391, 133-146. 10.1016/j.ydbio.2014.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Lopatkin A. and You L. (2016). Elements of biological oscillations in time and space. Nat. Struct. Mol. Biol. 23, 1030-1034. 10.1038/nsmb.3320 [DOI] [PubMed] [Google Scholar]
- Chen Z. F. and Behringer R. R. (1995). twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 9, 686-699. 10.1101/gad.9.6.686 [DOI] [PubMed] [Google Scholar]
- Christodoulou N. and Skourides P. A. (2015). Cell-autonomous Ca2+ flashes elicit pulsed contractions of an apical actin network to drive apical constriction during neural tube closure. 13, 2189-2202. 10.1016/j.celrep.2015.11.017 [DOI] [PubMed] [Google Scholar]
- Coravos J. S. and Martin A. C. (2016). Apical sarcomere-like actomyosin contracts nonmuscle drosophila epithelial cells. Dev. Cell 39, 346-358. 10.1016/j.devcel.2016.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson E. B., Evans D. R., Harris W. A. and Van Hook J. W. (1999). Amniotic fluid B12, calcium, and lead levels associated with neural tube defects. Am. J. Perinatol. 16, 373-378. 10.1055/s-2007-993887 [DOI] [PubMed] [Google Scholar]
- Dode L., Andersen J. P., Raeymaekers L., Missiaen L., Vilsen B. and Wuytack F. (2005). Functional comparison between secretory pathway Ca2+/Mn2+-ATPase (SPCA) 1 and sarcoplasmic reticulum Ca2+-ATPase (SERCA) 1 isoforms by steady-state and transient kinetic analyses. J. Biol. Chem. 280, 39124-39134. 10.1074/jbc.M506181200 [DOI] [PubMed] [Google Scholar]
- Eliceiri K. (2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529 10.1186/s12859-017-1934-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escuin S., Vernay B., Savery D., Gurniak C. B., Witke W., Greene N. D. E. and Copp A. J. (2015). Rho-kinase-dependent actin turnover and actomyosin disassembly are necessary for mouse spinal neural tube closure. J. Cell Sci. 128, 2468-2481. 10.1242/jcs.164574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira M. C. and Hilfer S. R. (1993). Calcium regulation of neural fold formation: visualization of the actin cytoskeleton in living chick embryos. Dev. Biol. 159, 427-440. 10.1006/dbio.1993.1253 [DOI] [PubMed] [Google Scholar]
- Garcia-Garcia M. J., Shibata M. and Anderson K. V. (2008). Chato, a KRAB zinc-finger protein, regulates convergent extension in the mouse embryo. Development 135, 3053-3062. 10.1242/dev.022897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodke-Puranik Y., Thorn C. F., Lamba J. K., Leeder J. S., Song W., Birnbaum A. K., Altman R. B. and Klein T. E. (2013). Valproic acid pathway. Pharmacogenet. Genomics 23, 236-241. 10.1097/FPC.0b013e32835ea0b2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grego-Bessa J., Hildebrand J. and Anderson K. V. (2015). Morphogenesis of the mouse neural plate depends on distinct roles of cofilin 1 in apical and basal epithelial domains. Development 142, 1305-1314. 10.1242/dev.115493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurniak C. B., Perlas E. and Witke W. (2005). The actin depolymerizing factor n-cofilin is essential for neural tube morphogenesis and neural crest cell migration. Dev. Biol. 278, 231-241. 10.1016/j.ydbio.2004.11.010 [DOI] [PubMed] [Google Scholar]
- Haigo S. L., Hildebrand J. D., Harland R. M. and Wallingford J. B. (2003). Shroom induces apical constriction and is required for hingepoint formation during neural tube closure. Curr. Biol. 13, 2125-2137. 10.1016/j.cub.2003.11.054 [DOI] [PubMed] [Google Scholar]
- Harris M. J. and Juriloff D. M. (2010). An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res. Part A Clin. Mol. Teratol. 88, 653-669. 10.1002/bdra.20676 [DOI] [PubMed] [Google Scholar]
- Hayashi S., Lewis P., Pevny L. and McMahon A. P. (2002). Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev. 119 Suppl. 1, S97-S101. 10.1016/S0925-4773(03)00099-6 [DOI] [PubMed] [Google Scholar]
- Hildebrand J. D. (2005). Shroom regulates epithelial cell shape via the apical positioning of an actomyosin network. J. Cell Sci. 118, 5191-5203. 10.1242/jcs.02626 [DOI] [PubMed] [Google Scholar]
- Hildebrand J. D. and Soriano P. (1999). Shroom, a PDZ domain-containing actin-binding protein, is required for neural tube morphogenesis in mice. Cell 99, 485-497. 10.1016/S0092-8674(00)81537-8 [DOI] [PubMed] [Google Scholar]
- Horner V. L. and Caspary T. (2011). Creating a ‘hopeful monster’: mouse forward genetic screens. Methods Mol. Biol. 770, 313-336. 10.1007/978-1-61779-210-6_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A., Greene N. D. E., Copp A. J. and Galea G. L. (2018). Valproic acid disrupts the biomechanics of late spinal neural tube closure in mouse embryos. Mech. Dev. 149, 20-26. 10.1016/j.mod.2017.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter G. L., Crawford J. M., Genkins J. Z. and Kiehart D. P. (2014). Ion channels contribute to the regulation of cell sheet forces during Drosophila dorsal closure. Development 141, 325-334. 10.1242/dev.097097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson A. G. and Tam P. P. L. (1982). Cephalic neurulation in the mouse embryo analyzed by SEM and morphometry. Anat. Rec. 203, 375-396. 10.1002/ar.1092030308 [DOI] [PubMed] [Google Scholar]
- Kienzle C., Basnet N., Crevenna A. H., Beck G., Habermann B., Mizuno N. and, von Blume J. (2014). Cofilin recruits F-actin to SPCA1 and promotes Ca2+-mediated secretory cargo sorting. J. Cell Biol. 206, 635-654. 10.1083/jcb.201311052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita N., Sasai N., Misaki K. and Yonemura S. (2008). Apical accumulation of Rho in the neural plate is important for neural plate cell shape change and neural tube formation. Mol. Biol. Cell 19, 2289-2299. 10.1091/mbc.e07-12-1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen P., Fedorov E. V., Fedorov A. A., Almo S. C. and Drubin D. G. (1997). Essential functions and actin-binding surfaces of yeast cofilin revealed by systematic mutagenesis. EMBO J. 16, 5520-5530. 10.1093/emboj/16.18.5520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. and Nagele R. G. (1986). Toxic and teratologic effects of verapamil on early chick embryos: evidence for the involvement of calcium in neural tube closure. Teratology 33, 203-211. 10.1002/tera.1420330207 [DOI] [PubMed] [Google Scholar]
- Lee H., Nagele R. and Karasanyi N. (1978). Inhibition of neural tube closure by ionophore A23187 in chick embryos. Experientia 34, 518-520. 10.1007/BF01935965 [DOI] [PubMed] [Google Scholar]
- Lissandron V., Podini P., Pizzo P. and Pozzan T. (2010). Unique characteristics of Ca2+ homeostasis of the trans-Golgi compartment. Proc. Natl Acad. Sci. USA 107, 9198-9203. 10.1073/pnas.1004702107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe C. R., Roberts C. J. and Lloyd S. (1971). Malformations of central nervous system and softness of local water supplies. Br. Med. J. 2, 357-361. 10.1136/bmj.2.5758.357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton J., Westlin M. and Hanley M. R. (1991). Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 266, 17067-17071. [PubMed] [Google Scholar]
- Ma Z.-L., Qin Y., Wang G., Li X.-D., He R.-R., Chuai M., Kurihara H. and Yang X. (2012). Exploring the caffeine-induced teratogenicity on neurodevelopment using early chick embryo. PLoS ONE 7, e34278 10.1371/journal.pone.0034278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M., Ishizaki T., Boku S., Watanabe N., Fujita A., Iwamatsu A., Obinata T., Ohashi K., Mizuno K. and Narumiya S. (1999). Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285, 895-898. 10.1126/science.285.5429.895 [DOI] [PubMed] [Google Scholar]
- Marret S., Gressens P., Van-Maele-Fabry G., Picard J. and Evrard P. (1997). Caffeine-induced disturbances of early neurogenesis in whole mouse embryo cultures. Brain Res. 773, 213-216. 10.1016/S0006-8993(97)00938-4 [DOI] [PubMed] [Google Scholar]
- Martin A. C. and Goldstein B. (2014). Apical constriction: themes and variations on a cellular mechanism driving morphogenesis. Development 141, 1987-1998. 10.1242/dev.102228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A. C., Kaschube M. and Wieschaus E. F. (2009). Pulsed contractions of an actin-myosin network drive apical constriction. Nature 457, 495-499. 10.1038/nature07522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason F. M., Tworoger M. and Martin A. C. (2013). Apical domain polarization localizes actin-myosin activity to drive ratchet-like apical constriction. Nat. Cell Biol. 15, 926-936. 10.1038/ncb2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCombs J. E. and Palmer A. E. (2008). Measuring calcium dynamics in living cells with genetically encodable calcium indicators. Methods 46, 152-159. 10.1016/j.ymeth.2008.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGreevy E. M., Vijayraghavan D., Davidson L. A. and Hildebrand J. D. (2015). Shroom3 functions downstream of planar cell polarity to regulate myosin II distribution and cellular organization during neural tube closure. Biol. Open 4, 186-196. 10.1242/bio.20149589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkes P. E., Little S. A. and Umpierre C. C. (2001). Co-localization of active caspase-3 and DNA fragmentation (TUNEL) in normal and hyperthermia-induced abnormal mouse development. Teratology 63, 134-143. 10.1002/tera.1024 [DOI] [PubMed] [Google Scholar]
- Moury J. D. and Schoenwolf G. C. (1995). Cooperative model of epithelial shaping and bending during avian neurulation: autonomous movements of the neural plate, autonomous movements of the epidermis, and interactions in the neural plate/epidermis transition zone. Dev. Dyn. 204, 323-337. 10.1002/aja.1002040310 [DOI] [PubMed] [Google Scholar]
- Nagy A., Behringer R. R., Gertsenstein M. Vintersten K. (2003). Manipulating the Mouse Embryo: A Laboratory Manual, 3rd edn Cold Spring Harbor Laboratory Press. [Google Scholar]
- Nishimura T. and Takeichi M. (2008). Shroom3-mediated recruitment of Rho kinases to the apical cell junctions regulates epithelial and neuroepithelial planar remodeling. Development 135, 1493-1502. 10.1242/dev.019646 [DOI] [PubMed] [Google Scholar]
- Nishimura T., Honda H. and Takeichi M. (2012). Planar cell polarity links axes of spatial dynamics in neural-tube closure. Cell 149, 1084-1097. 10.1016/j.cell.2012.04.021 [DOI] [PubMed] [Google Scholar]
- Niwa R., Nagata-Ohashi K., Takeichi M., Mizuno K. and Uemura T. (2002). Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108, 233-246. 10.1016/S0092-8674(01)00638-9 [DOI] [PubMed] [Google Scholar]
- Okunade G. W., Miller M. L., Azhar M., Andringa A., Sanford L. P., Doetschman T., Prasad V. and Shull G. E. (2007). Loss of the Atp2c1 secretory pathway Ca2+-ATPase (SPCA1) in mice causes golgi stress, apoptosis, and midgestational death in homozygous embryos and squamous cell tumors in adult heterozygotes. J. Biol. Chem. 282, 26517-26527. 10.1074/jbc.M703029200 [DOI] [PubMed] [Google Scholar]
- O'Shea K. S. (1982). Calcium and neural tube closure defects: an in vitro study. Birth Defects Orig. Artic. Ser. 18, 95-106. [PubMed] [Google Scholar]
- Rasmussen I., Pedersen L. H., Byg L., Suzuki K., Sumimoto H. and Vilhardt F. (2010). Effects of F/G-actin ratio and actin turn-over rate on NADPH oxidase activity in microglia. BMC Immunol. 11, 44 10.1186/1471-2172-11-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder T. E. (1970). Neurulation in Xenopus laevis. An analysis and model based upon light and electron microscopy. J. Embryol. Exp. Morphol. 23, 427-462. [PubMed] [Google Scholar]
- Schwede T., Kopp J., Guex N. and Peitsch M. C. (2003). SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31, 3381-3385. 10.1093/nar/gkg520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott R. W., Hooper S., Crighton D., Li A., König I., Munro J., Trivier E., Wickman G., Morin P., Croft D. R. et al. (2010). LIM kinases are required for invasive path generation by tumor and tumor-associated stromal cells. J. Cell Biol. 191, 169-185. 10.1083/jcb.201002041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes W. C., Rosen B., West A. P., Koutsourakis M., Bushell W., Iyer V., Mujica A. O., Thomas M., Harrow J., Cox T. et al. (2011). A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337-342. 10.1038/nature10163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley M. J. and Stanisstreet M. (1985). Calcium and neurulation in mammalian embryos. J. Embryol. Exp. Morphol. 89, 1-14. [PubMed] [Google Scholar]
- Solon J., Kaya-Çopur A., Colombelli J. and Brunner D. (2009). Pulsed forces timed by a ratchet-like mechanism drive directed tissue movement during dorsal closure. Cell 137, 1331-1342. 10.1016/j.cell.2009.03.050 [DOI] [PubMed] [Google Scholar]
- Somlyo A. P. and Somlyo A. V. (2003). Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325-1358. 10.1152/physrev.00023.2003 [DOI] [PubMed] [Google Scholar]
- Suzuki M., Sato M., Koyama H., Hara Y., Hayashi K., Yasue N., Imamura H., Fujimori T., Nagai T., Campbell R. E. et al. (2017). Distinct intracellular Ca(2+) dynamics regulate apical constriction and differentially contribute to neural tube closure. Development 144, 1307-1316. 10.1242/dev.141952 [DOI] [PubMed] [Google Scholar]
- Toyoshima C., Nakasako M., Nomura H. and Ogawa H. (2000). Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature 405, 647-655. 10.1038/35015017 [DOI] [PubMed] [Google Scholar]
- Van Baelen K., Vanoevelen J., Callewaert G., Parys J. B., De Smedt H., Raeymaekers L., Rizzuto R., Missiaen L. and Wuytack F. (2003). The contribution of the SPCA1 Ca2+ pump to the Ca2+ accumulation in the Golgi apparatus of HeLa cells assessed via RNA-mediated interference. Biochem. Biophys. Res. Commun. 306, 430-436. 10.1016/S0006-291X(03)00977-X [DOI] [PubMed] [Google Scholar]
- von Blume J., Alleaume A.-M., Cantero-Recasens G., Curwin A., Carreras-Sureda A., Zimmermann T., van Galen J., Wakana Y., Valverde M. A. and Malhotra V. (2011). ADF/cofilin regulates secretory cargo sorting at the TGN via the Ca2+ ATPase SPCA1. Dev. Cell 20, 652-662. 10.1016/j.devcel.2011.03.014 [DOI] [PubMed] [Google Scholar]
- Wallingford J. B., Niswander L. A., Shaw G. M. and Finnell R. H. (2013). The continuing challenge of understanding, preventing, and treating neural tube defects. Science 339, 1222002-1222002 10.1126/science.1222002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Shibasaki F. and Mizuno K. (2005). Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J. Biol. Chem. 280, 12683-12689. 10.1074/jbc.M411494200 [DOI] [PubMed] [Google Scholar]
- Wang Q., Shui B., Kotlikoff M. I. and Sondermann H. (2008). Structural basis for calcium sensing by GCaMP2. Structure 16, 1817-1827. 10.1016/j.str.2008.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggan O., Shaw A. E., DeLuca J. G. and Bamburg J. R. (2012). ADF/cofilin regulates actomyosin assembly through competitive inhibitionof myosin II binding to F-actin. Dev. Cell 22, 530-543. 10.1016/j.devcel.2011.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson J. M. and Pollard I. (1994). In utero exposure to caffeine causes delayed neural tube closure in rat embryos. Teratog. Carcinog. Mutagen. 14, 205-211. 10.1002/tcm.1770140502 [DOI] [PubMed] [Google Scholar]
- Wootton L. L., Argent C. C. H., Wheatley M. and Michelangeli F. (2004). The expression, activity and localisation of the secretory pathway Ca2+ -ATPase (SPCA1) in different mammalian tissues. Biochim. Biophys. Acta 1664, 189-197. 10.1016/j.bbamem.2004.05.009 [DOI] [PubMed] [Google Scholar]
- Zhao J.-W., Gao Z.-L., Ji Q.-Y., Wang H., Zhang H.-Y., Yang Y.-D., Xing F.-J., Meng L.-J. and Wang Y. (2012). Regulation of cofilin activity by CaMKII and calcineurin. Am. J. Med. Sci. 344, 462-472. 10.1097/MAJ.0b013e318244745b [DOI] [PubMed] [Google Scholar]