

Summary
HIV‐2 is thought to have entered the human population in the 1930s through cross‐species transmission of SIV from sooty mangabeys in West Africa. Unlike HIV‐1, HIV‐2 has not led to a global pandemic, and recent data suggest that HIV‐2 prevalence is declining in some West African states where it was formerly endemic. Although many early isolates of HIV‐2 were derived from patients presenting with AIDS‐defining illnesses, it was noted that a much larger proportion of HIV‐2‐infected subjects behaved as long‐term non‐progressors (LTNP) than their HIV‐1‐infected counterparts. Many HIV‐2‐infected adults are asymptomatic, maintaining an undetectable viral load for over a decade. However, despite lower viral loads, HIV‐2 progresses to clinical AIDS without therapeutic intervention in most patients. In addition, successful treatment with anti‐retroviral therapy (ART) is more challenging than for HIV‐1. HIV‐2 is significantly more sensitive to restriction by host restriction factor tripartite motif TRIM5α than HIV‐1, and this difference in sensitivity is linked to differences in capsid structure. In this review we discuss the determinants of HIV‐2 disease progression and focus on the important interactions between TRIM5α and HIV‐2 capsid in long‐term viral control.
Keywords: capsid, HIV‐2, long term non‐progression, TRIM5
Introduction
HIV‐2 was first isolated in 1986 from healthy commercial sex workers in Senegal and named HTLV‐IV. Shortly afterwards a similar virus (named LAV‐II) was isolated from two West African patients with AIDS and renamed HIV‐2 1. Subsequent molecular characterization showed that HIV‐2 was related to HIV‐1, but was closer to simian immunodeficiency virus (SIV) derived from macaques displaying an AIDS‐like syndrome 1. It has since been established that HIV‐2 entered the human population in approximately 1938 from a virus infecting sooty mangabeys (SIVmm) in West Africa 2.
An estimate of HIV‐2 prevalence of 1–2 million infected people worldwide is widely cited 3. However, as HIV‐2 is not regularly included in national testing strategies and requires specialist laboratory facilities for accurate diagnosis 4 the current prevalence is unknown. The recent falls in national HIV‐2 prevalence in some West African countries 5 have led to predictions that the epidemic will reach extinction in approximately 2068 3, 6.
In contrast to untreated HIV‐1 infection, longitudinal follow‐up of a rural community cohort with HIV‐2 infection demonstrated that 30–40% of infected people exhibit low or undetectable viral loads with AIDS‐free survival for up to 10 years 7. These HIV‐2 long‐term non‐progressors (LTNPs) had mortality rates equivalent to the uninfected population, although most people infected with HIV‐2 have moderately higher mortality rates than the HIV‐negative population 7, 8. After 8 years of follow‐up, the mortality rate for HIV‐1 infection is approximately double that of HIV‐2 infection without treatment 7, 9. This distinction between HIV‐1 and HIV‐2 mortality was highlighted in a recent report from a cohort in Bissau with an unusually high proportion of subjects with known dates of infection, in which the median survival time for HIV‐1 was 8·2 years and for HIV‐2 it was 15·6 years 10. In this study it was also noted that HIV‐2‐infected people developed clinical AIDS with a higher mean CD4+ T cell percentage than seen for HIV‐1. The risk of HIV‐2 disease progression is dependent on viral load – people with viral loads > 10 000 copies/ml are likely to progress to AIDS at the same rate as those with HIV‐1 7. The differential effect of plasma viral load on mortality in HIV‐2 infection is illustrated in Fig. 1. Furthermore, when matched for low CD4+ counts (<100 cells/ml), mortality in HIV‐2 infection is equivalent to that of HIV‐1 8. Spontaneous undetectable viraemia and an indolent disease course in HIV‐1 is rare 11, whereas this is a common feature of HIV‐2 infection.
Figure 1.
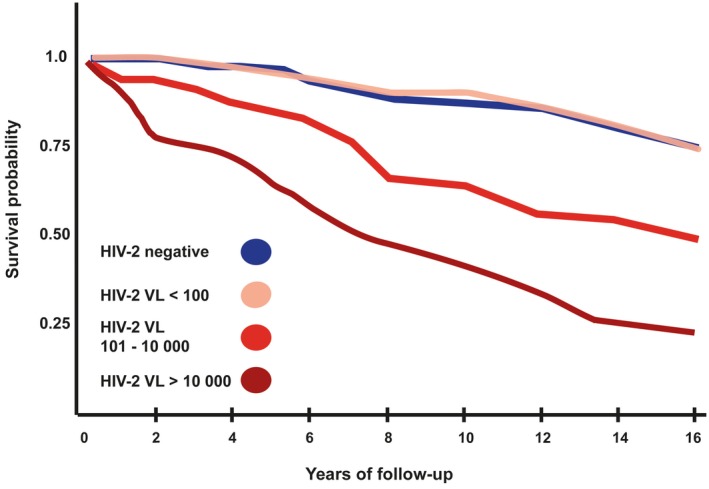
Mortality after years of follow‐up based on HIV‐2 viral load. HIV‐2 VL = HIV‐2 viral load measured in copies/ml. These survival curves are adapted from Kaplan–Meier survival curves for HIV‐2 when stratified for viral load 12. Increasing viral load level has a strong association with decreasing survival probability 12.
In this review we discuss the relative contributions to HIV‐2 disease progression of viral, immunological and host factors. We focus on the potential importance of the interaction between HIV‐2 capsid and human tripartite motif containing protein TRIM5α, and discuss how these viral and host factors may contribute to delayed disease progression in HIV‐2 infection.
TRIM5α and HIV‐2
TRIM5α is a host restriction factor, one of a group of intracellular anti‐viral proteins which act as the effector arm of the interferon (IFN) response to disrupt the HIV life cycle at multiple points 13. The structure of TRIM proteins is characterized by a conserved modular tripartite motif consisting of a Really Interesting New Gene (RING) domain followed by one or two zinc binding areas named B‐box, a coiled coil (CC) region and in some cases a PRY‐SPRY domain 14. TRIM5 is transcribed into five isoforms and its anti‐viral activity is mediated by isoform TRIM5α. The other isoforms result in truncated proteins lacking a PRY‐SPRY domain and inhibit TRIM5α activity, therefore the proportion of TRIM5α among total TRIM5 transcripts may influence HIV restriction 15. Although TRIM proteins are structurally similar, their functions are diverse: here we will focus on the anti‐retroviral mechanisms of TRIM5α.
Host restriction factors play a key role in the prevention of cross‐species movement of lentiviruses: for example, the orthologue TRIM5α derived from rhesus macaques potently restricts HIV‐1, whereas the human equivalent does not 16. TRIM5 is one of the fastest‐evolving genes in the human genome 17. TRIM5 has evolved under positive selection pressure, due probably to various pathogens exerting selection pressure at different periods of human evolution 18. Positive selection on residues in the PRY‐SPRY domain is prominent in TRIM5, particularly in an amino acid motif, which determines viral capsid specificity and hence viral restriction 18. TRIM5α monomers bind at the interface of three capsid hexamers. This binding site was shown for rhesus TRIM5α and HIV‐1 and is associated with high‐affinity binding and determines species‐specific restriction of TRIM5α orthologues 19.
TRIM5α’s ability to restrict HIV‐1 in old world monkey (OWM) cells by targeting viral capsid was reported in 2004 20. The sensitivity of HIV‐2 to TRIM5α is determined by a region of the capsid which corresponds to the cyclophilin binding loop in HIV‐1 21. Depletion of cyclophilin A enhances TRIM5α‐mediated restriction and reduces infectivity of HIV‐2 22.The anti‐retroviral mechanism of TRIM5α acts at the pre‐integration phase of the HIV life‐cycle and is summarized in Fig. 2. TRIM5α in rhesus macaques is also capable of late viral restriction by incorporation into nascent HIV‐2 virions, thereby decreasing the production of functional new virus particles. This late restriction is a saturable process, as increased virion production may overwhelm it 23. In feline cell lines permissive to HIV‐2 infection, experimentally induced expression of human TRIM5α restricts viral replication and TRIM5α‐specific short interfering RNA rescues infectivity in resistant cells 21. Additional anti‐retroviral mechanisms include that TRIM5 binding to the incoming capsid prevents nuclear entry via nucleoporin channels 24 and also stimulates transforming growth factor (TGF) beta‐activated kinase 1 (TAK‐1) phosphorylation, which activates the nuclear factor kappa B (NF‐κB) transcription pathway 25. This capsid‐specific recognition then stimulates further downstream transcription of inflammatory cytokines, which enhances the anti‐viral response against HIV.
Figure 2.
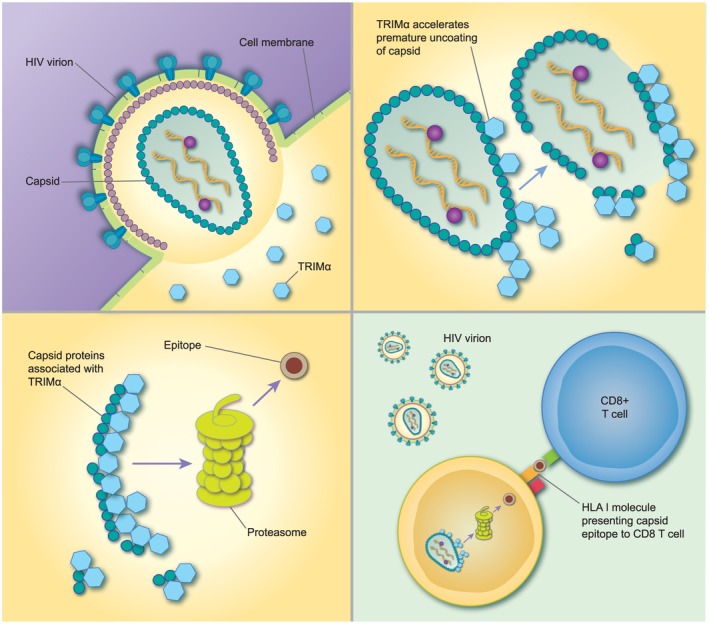
Mechanism of retroviral restriction by tripartite motif (TRIM)5α: (a) after viral entry TRIM5α monomers binds to the capsid and oligomerize with additional TRIM5α molecules. (b) These are then poly‐ubiquitinated via TRIM’s E3 ligase activity. This prematurely uncoats the capsid, disrupting the reverse transcription/capsid complex (RTC). (c) The RTC is then recruited to the cellular proteasome for degradation. (d) We hypothesize that enhanced proteasomal processing selects for epitopes which are associated with protective gag‐specific CD8+ T cell responses presented on human leucocyte antigen (HLA) class I molecules 32, 33.
Despite high TRIM diversity, TRIM5 genotype has minimal impact on HIV‐1 disease progression 26. Moreover, TRIM5α does not appear to drive evolutionary change in HIV‐1 capsid sequences from clade B 27. HIV‐infected patients who are homozygous for the H43Y substitution develop AIDS more quickly than heterozygotes or H43 homozygotes 28 and this substitution modestly impairs the restrictive capacity of TRIM5α 28. Furthermore, the appearance of putative TRIM5α viral escape variants has been found to precede rapid immunological decline in HIV‐1 infection 29. Increased expression of TRIM5α in peripheral blood mononuclear cells is associated with a reduced risk of incident HIV‐1 infection 30. The impact of TRIM5 genotype and expression on HIV‐2 disease outcomes has not been reported. A primate model of human HIV‐2 infection using SIVsm‐infected Indian macaques showed that certain TRIM5 variants were associated with increased memory CD4+ cells and longer AIDS‐free survival. This longitudinal study also demonstrated the development of escape mutants to TRIM5α, which could accelerate immunological decline, resulting in simian AIDS in the test subjects 31.
Viral factors associated with HIV‐2 progression
Contribution to disease progression by viral replicative capacity, diversity and fitness
The HIV genome consists of several genes in an open reading frame, represented in Fig. 3. Differences between the HIV‐1 and HIV‐2 genomes are shown in Fig. 3. Viral replicative capacity, genetic diversity and intrahost evolution rates have all been linked to differences in disease outcomes in HIV infection and are discussed in turn.
Figure 3.
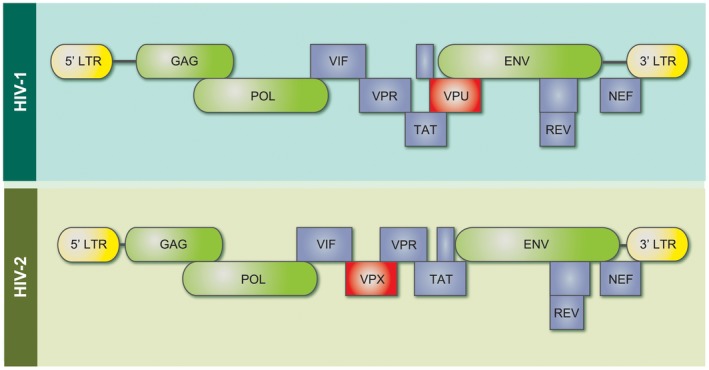
Differences in the HIV‐1 and HIV‐2 genomes. Gag genes display approximately 60% protein sequence similarity between HIV‐1 and 2, whereas env has approximately 40% similarity 34. HIV‐1 possesses the accessory protein Vpu, which degrades both surface CD4 receptors and tetherin, thereby enhancing virion release. HIV‐2 contains vpx: this protein counteracts restriction factor SAM domain and HD domain‐containing protein 1 (SAMHD1), which is present in myeloid cell lines (adapted from 36).
Measuring virion production in cell cultures with a radio‐labelled reverse transcriptase assay, it has been shown in HIV‐1‐infected patients that a greater viral replicative capacity (RC) in the transmitted/founder (T/f) strain is associated with greater immune activation, higher viral load set‐points and faster disease progression 37. The RC of Gag‐MJ4 chimeric viruses correlates closely with that of full‐length T/f molecular clones. This suggests that the gag sequences of the T/f strains influence replicative capacity. In an epidemiologically linked transmission cohort, cytotoxic T lymphocyte (CTL) pressure on gag in transmitted viruses negatively influenced viral RC 38. Residue substitutions in all gag regions influenced RC, although only a few mutations occurred in capsid, which is a highly conserved region 38. Further studies did not support the sole importance of gag and have suggested that multiple genes interact to determine RC 39.
An alternative approach to measuring replicative capacity is to use competition assays with different viral strains. Using this method, Arien et al. showed that most HIV‐2 isolates had lower RC than HIV‐1 40 and additional studies demonstrated that clinical HIV‐2 isolates from aviraemic patients had a lower RC than those from viraemic patients 41. This indicates that viral RC is probably also an important contributor to disease progression in HIV‐2 infection.
HIV‐1 diversity shows a strong correlation with viral load, fitness and disease progression, but less is known about HIV‐2 diversity and divergence influence disease outcomes 42. Skar et al. reported that HIV‐2 had a greater evolutionary rate than HIV‐1, specifically in the gp125 region that contains the V3 loop 43. This region binds to CD4+ receptors and thereby exposes the V3 loop to neutralizing antibodies. Mutations were more likely to be synonymous and therefore due to viral factors (purifying selection) as opposed to immunological (positive selection) pressure – escape mutants to neutralizing antibodies were notably rare. In contrast, findings from a Senegalese cohort showed limited divergence of env 44. One possible reason for the disparity in the results of these studies is that the two patient profiles were different. The Senegalese cohort were ART‐naive, with limited disease progression, and had a long follow‐up time. Skar et al. reported on env divergence in patients with mostly progressive HIV‐2 infection. Many of these patients displayed X4 tropism, with high viral loads and low CD4+ counts. In addition, slow disease progression has been associated with lower env diversity and vice versa 45. A recent study stratified HIV‐2‐infected patients into fast and slow progressors and showed that HIV‐2 env evolutionary rates in fast progressors are approximately double that of slow progressors 46. Furthermore, stronger positive selection pressure on conserved residues in the envelope was associated with slower disease progression. The following trends emerge: individuals with controlled HIV‐2 infection have viruses with a low replicative rate, low viral loads and diversity. Those that develop progressive disease have higher viral loads and greater diversity, with prominent negative selection pressure.
A study of env and gag sequences showed that viral loads in phylogenetically linked HIV‐2 infection clusters were discordant 47. Therefore, viral genetic factors, at least in gag and env, probably do not account for the full spectrum of disease progression in HIV‐2 infection. This finding strengthens the hypothesis that interaction between viral and immunological factors are collective determinants of disease outcomes in HIV‐2.
The importance of gag
The viral gag gene encodes structural proteins capsid (CA/p26), nucleocapsid (NC) and matrix (MA). After an HIV virion enters a cell the capsid houses the viral RNA and facilitates reverse transcription during transit to the nucleus. HIV‐1 capsids prevent both innate immune sensing of viral cDNA by the cGAS‐STING [cyclic guanosine monophosphate–adenosine monophosphate (GMP–AMP) synthase and stimulator of IFN genes] pathway as well as cDNA degradation and sensing by endogenous DNase enzymes such as TREX1 (three prime repair exonuclease 1) 48, 49. In contrast to HIV‐1, HIV‐2 is able to replicate in dendritic cells and stimulates innate immune sensing via its capsid in a cyclophilin A‐dependent manner 35. Capsid structures are highly conserved among retroviruses, and mutations in their sequence may either reduce viral infectivity or increase the susceptibility of the virus to host immune responses 50, 51. It was recently reported that HIV‐2 capsid binds more strongly than HIV‐1 to the non‐POU domain‐containing octamer‐binding protein (NONO) protein in the nucleus, promoting DNA sensing by cGAS and stimulating a potent IFN response 52. The capsid residues involved in NONO binding are highly conserved in primate lentiviruses 52. Capsid sequences also determine the pathway for nuclear import and influence the choice of integration sites for viral DNA 53. The choice of integration site can then influence viral load, depending on how transcriptionally active the site is.
Polymorphisms in HIV‐2 capsid sequences have been implicated in multiple aspects of disease progression. Capsid sequences differ between patients with low and high HIV‐2 viral loads, a key determinant of immune activation and disease progression. Specifically, patients with low viral loads often displayed prolines in capsid at positions 119, 159 and 178. Prolines in these positions had an additive effect on decreasing viraemia 54. In addition, prolines in these positions are associated with enhanced proteasomal processing of a CD8+ T cell gag epitope 165‐DRFYKSLRA‐173. This epitope is associated with greater gag‐specific T cell responses as determined by enzyme‐linked immunospot (ELISPOT) and is a frequent target for T cell responses in patients with low viral loads 55. Therefore, it is possible that capsid sequences directly influence adaptive immune responses. HIV‐2 capsid sequences with a proline at position 119 are more sensitive to restriction by the anti‐viral protein TRIM5α (rhesus macaque) than sequences with alternative amino acids at this position. This enhanced sensitivity to TRIM5α may be linked to conformational changes in the capsid protein 56, 57. HIV‐1 NL4‐3 chimeras which contain HIV‐2 capsid sequences are more susceptible to restriction by human TRIM5α than the wild‐type virus, although proline substitutions at position 119 do not correlate with TRIM5α restriction 58. Furthermore, HIV‐2 CA sequences derived from rapid progressors and LTNP do not differ in their sensitivity to TRIM5α – although they are all more sensitive than HIV‐1 22.
Despite the increased sensitivity of HIV‐2 capsid to TRIM5α restriction, as well as the strong cytotoxic T cell response directed towards gag, there is no evidence of positive selection pressure on gag divergence in a cross‐sectional analysis of viral sequences from the Caió community cohort 59. Purifying selection pressure predominates in interhost evolution, and this reflects the constrained evolution of this gene.
HLA/KIR and other host genetic associations with HIV‐2 disease outcomes
The most robust immunogenetic associations with LTNP in HIV‐1 are found in HLA alleles and CCR5 polymorphisms 60. Few studies have reported on the impact of human leucocyte antigen (HLA) genotypes on HIV‐2 infection outcomes. This reflects the paucity of well‐characterized cohorts in West Africa, compounded by the variability of HLA repertoires present in different ethnic groups 61.
Yindom et al. noted that HLA class I associations were strongest for HLA B*1503, which correlated with high viral loads and low CD4+ counts. Surprisingly, HLA B*5703 did not correlate with low viral load, despite its strong association with LTNP in HIV‐1 infection 62. In Senegalese female sex workers HLA‐B35, together with the HLA‐B35‐Cw4 and HLA‐A23‐C7 haplotypes, were associated with increased risk of disease progression 63. Three additional HLA types have been associated with lower odds of having a detectable viral load in HIV‐2 infection, HLA‐B*58:01, HLA‐DPB1*10:01 and HLA‐DRB1*11:01 64.
Many associations between compound HLA/killer cell immunoglobulin‐like receptors (KIR) genotypes and outcomes in HIV‐1 infection have been reported, but in the Caió cohort there were no significant associations between individual KIR alleles or HLA/KIR genotypes and either the risk of infection with HIV‐2 or HIV‐2 disease progression 62.
An additional study from the Caió cohort reported that a haplotype for polymorphisms rs11575097‐rs10849523 in the CD4 gene was associated with high viral loads in HIV‐2 65. In the same study, no correlations were found between polymorphisms in the CD209 gene encoding the dendritic cell‐specific intercellular adhesion molecule (ICAM)‐3‐grabbing nonintegrin, and HIV‐2 susceptibility or progression.
There is clear potential for further genetic studies to shed light on the mechanisms of HIV‐2 disease progression.
CD4+ T cell responses and HIV‐2 disease outcomes
One of the hallmarks of HIV‐1 infection is the progressive loss of CD4+ T helper cells. Strikingly, the subset of HIV‐1‐specific CD4+ cells is particularly susceptible to HIV‐1 infection 66. However, despite the fact that the HIV‐2‐specific CD4+ cell response is relatively well‐preserved in most patients, HIV‐2‐specific CD4+ T cells are as vulnerable to HIV‐2 infection as their HIV‐1 specific counterparts are to HIV‐1 infection 66, 67. The relative proportion of infected virus‐specific CD4+ T cells between HIV‐2 controllers and progressors has not been investigated.
CD4+ cells from aviraemic HIV‐2‐infected patients show preserved proliferative and cytokine‐releasing functions, IFN‐γ and interleukin (IL)‐2 being the dominant cytokines released 67, 68. Aviraemic patients’ CD4+ cells have a broader cytokine profile; they release IFN‐γ, IL‐2, tumour necrosis factor (TNF)‐α, macrophage inflammatory protein (MIP)‐1β and CD107 69, whereas in HIV‐2 viraemic progressors the functional profiles of HIV‐specific CD4+ T cells are similar to those seen in HIV‐1 infection. This could suggest that HIV‐2 disease progression is associated with greater infection of HIV‐2‐specific CD4+ populations, leading to reduced T helper cell activity. A greater CD4+ response against homologous gag proteins correlates positively with immune activation and negatively with HIV‐2 proviral load, suggesting that the host responses promoting immune activation limits HIV‐2 viral replication 70. Expanded T regulatory populations coupled with increasing programmed cell death 1–programmed cell death ligand 1 (PD1–PDL1) expression on T cells have been observed in patients with advanced HIV‐2 disease and low CD4+ counts, but these findings did not correlate with viraemia. This observation may indicate that aberrant immune tolerance mechanisms in the face of long‐standing immune activation can drive HIV‐2 disease progression 71, 72, 73.
Cytotoxic T cell responses and disease outcomes
Many studies have implicated the HIV‐1‐specific CD8+ CTL response as the major factor controlling viral replication from acute infection through the asymptomatic stage of infection 74. In HIV‐1 the CTL response tends to be broad, targeting a wide variety of epitopes, but is largely undermined by escape mutants that evade CTL recognition 75. Despite the importance of the CTL response in HIV‐1 control, progressors cannot be distinguished from non‐progressors based on the magnitude or breadth of their HIV‐1‐specific CTL response. There are characteristics of CTL found in non‐progressors, including restriction by ‘protective’ HLA molecules such as HLA‐B27 or B57, polyfunctionality, proliferative capacity, levels of cytolytic molecules and, potently, the ability to suppress HIV replication in vitro 76. In addition, greater targeting of HIV‐1 gag is associated with lower viral loads 77.
The CTL response in HIV‐2 is characterized as being narrow, consistently targeting a smaller number of epitopes than HIV‐1 78. CTL from HIV‐2‐infected subjects produce a wider range of cytokines, as well as greater quantities of individual cytokines per cell, compared to those from HIV‐1‐infected individuals 69. This is linked with the high functional avidity reported for some HIV‐2‐specific CTL responses 78. Using ELISPOT to measure CTL responses to gag peptides, aviraemic patients showed higher‐magnitude T cell responses than viraemic patients 79. Gag‐specific CTL responses were shown to correlate strongly and inversely with viral load in HIV‐2‐infected subjects, with most viraemic patients lacking gag‐specific responses (which would be rare in HIV‐1 infection) 80. CD8+ cells from aviraemic patients can potently suppress viral replication in CD4+ cells; these CD8+ cells have an early differentiation phenotype and are potent effectors 81. Therefore, there are both quantitative and qualitative differences between CTL responses in HIV‐2‐infected subjects with and without detectable viraemia, suggesting that the generation of a potent and effective CTL response is central to limiting HIV‐2 replication. Robust gag‐specific CD8+ T cell responses are common in subjects with undetectable viral load, raising the question of how these CTL populations are maintained in the absence of detectable viraemia. HIV‐2 replication is detectable in gut mucosa in patients who are otherwise aviraemic, and this may offer an explanation if CTL are stimulated through persistent HIV‐2 replication in gut‐associated lymphoid tissue 82.
It is unclear why some HIV‐2‐infected patients do not generate effective gag‐specific CTL responses, although we hypothesize that this may be related to a relatively weak interaction between TRIM5α and HIV‐2 capsid (see below).
Immune activation
Many of the characteristic features of HIV‐1 disease progression are thought to be a consequence of prolonged immune activation. Immune activation also appears to be driving HIV‐2 disease progression: the soluble activation marker beta‐2 microglobulin predicts HIV‐2 disease progression as accurately as plasma viral load in untreated subjects 83. T cell activation markers closely correlate with plasma viral load in viraemic patients – however, there is a subgroup of patients with undetectable viraemia who exhibit elevated T cell activation 78. There has been speculation that the lower levels of immune activation in the many aviraemic HIV‐2‐infected patients constitute the key factor explaining their slower rates of disease progression 84. Levels of biomarkers associated with enhanced immune activation are equivalent in HIV‐1 and HIV‐2 infection, and when viral load is taken into account HIV‐2 appears to elicit greater immune activation per unit of viral load than does HIV‐1 85. Moreover, immune activation in patients with HIV‐1 and HIV‐2 infection is equivalent when matched for CD4+ count 86.
A key driver of immune activation in HIV‐1‐infected subjects is the increased translocation into the blood of microbial products from the gut, including lipopolysaccharide (LPS), which promotes macrophage activation 87. LPS levels were elevated in an African cohort with HIV‐2 infection and correlated with disease progression 88. However, this was not observed in a European cohort, even though there was evidence of significant T cell and monocyte activation 89. In marked contrast to HIV‐1 infection, mucosal integrity and gut‐associated lymphocyte numbers are preserved in aviraemic HIV‐2 infection (with or without ART), despite the presence of replicating virus in the gut mucosa 82. These findings suggest that there is a significant difference in HIV‐associated gut mucosal pathology between HIV‐1 and HIV‐2 infection, although how this might link with HIV‐2 progression is not entirely clear.
Other host factors associated with HIV‐2 progression
Disease progression in HIV reflects a complex interplay between social factors and biological factors which influence how rapidly a person will develop AIDS after HIV infection. Age at infection and sex are important determinants in this dynamic process.
Being male is an important determining factor in disease outcomes for both HIV‐1 and HIV‐2 90. These findings may be explained, in part, by the social behaviour of men resulting in poor adherence to ART and health‐seeking behaviour later in infection 91. In addition, baseline mortality rates due to non‐infectious causes are higher in males, and this contributes to the observed higher mortality rates in HIV‐infected men 92.
Social factors do not fully account for the discrepancy in disease progression rates between males and females. Plasmacytoid dendritic cells from females produce higher transcription levels of several IFN‐stimulated genes in response to IFN‐α than those from males 93. This enhanced response may be beneficial in acute infection, limiting viral set point and initial CD4+ count decline 94. Females, on average, have lower viral load set points coupled with an enhanced type I IFN response and greater CD8+ T cell activation; this may favour rapid CD4+ recovery and improved outcomes after ART initiation 95, 96. Statistical modelling of transmitted viral sequences indicates that transmission bottlenecks in heterosexuals exist, and that they favour the transmission of viruses with greater fitness from females to males 97. It has been postulated that because women have a large population of cells in the vaginal mucosa which are susceptible to HIV infection, they therefore have a lower barrier to infection than men. This bottleneck selects viruses infecting men with greater predicted replicative capacity based on the presence of specific amino acid residues. The viruses which infect men may therefore predispose them to accelerated disease progression.
HIV‐1‐infected males in Cameroon had an elevated risk of virological failure after ART initiation which was not linked to pre‐existing viral resistance or poor adherence. Furthermore, females in this study showed greater CD4+ count recovery after 24 months of ART 98. Most African HIV‐2 cohorts contain more women than men, and here male sex has also been associated with higher mortality 99.
In HIV‐1, advanced age at infection is a powerful co‐factor for accelerated disease progression – this is probably due to immunological senescence 100. Males are often older than females when they initiate ART, which may therefore explain their diminished immunological reconstitution 101. HIV‐2 prevalence in Guinea‐Bissau was highest among females in the 45–60‐year age group 102 and in the pre‐ART era older HIV‐2‐infected adults had mortality rates equivalent to the HIV‐negative population, suggesting that age was not a risk factor for progression in HIV‐2 infection 103. In contrast, data from the ANRS cohort showed that age greater than 40 years was a significant risk factor for HIV‐2 disease progression after ART initiation 104.
Conclusion: possible causal factors which promote HIV‐2 disease progression
We believe that greater priority should be given to HIV‐2 research because of the unique insights this human model can provide about the pathophysiology of HIV‐1, particularly a better understanding of host and viral factors associated with long‐term viral control. The interaction between the viral capsid, host restriction factors and the cellular immune response may be central to maintaining durable control of viral replication in HIV‐2 infection. Multiple lines of evidence indicate that TRIM5α may, soon after initiating viral uncoating, target the viral capsid for proteasomal degradation 32. During this process, HIV‐2 capsids enriched in proline residues may favour the efficient processing of CTL epitopes that are associated with a long‐lasting, protective gag‐specific CTL response. The strength of the capsid/TRIM5α interaction is also related to the extent to which the type I IFN responses are triggered 105; combinations of capsid and TRIM5α that bind with high affinity could lead to a potent CTL response targeting gag epitopes. In aviraemic patients this CTL response may potentially be maintained by ongoing, low‐level mucosal replication in the gut. HIV‐2‐specific CTL may be able to control viral replication and maintain a low level of immune activation for many years, without the emergence of viral escape mutants 59. A caveat to the search for a functional cure is that therapies which maintain a low viral load may not always result in a reversal of immunodeficiency if immune activation remains high 106.
Given HIV’s unparalleled ability to adapt to its host, future therapies which aim to induce a state of prolonged clinical latency in the absence of regular therapeutic intervention (either ART or infusions of monoclonal antibodies) will need to target multiple aspects of viral replication. TRIM5α restriction acts at the pre‐integration phase of the life‐cycle, displays cross‐talk with the adaptive immune response and may negatively affect viral RC. There is evidence to support this proposition, as HIV‐1 CTL escape variants display increased sensitivity to restriction by TRIM5α 107. The RC of these escape variants may be further reduced, given the functional constraints on capsid structure 108.
TRIM5 has been successfully edited using the CRISPR‐Cas 9 system to effect single nucleotide polymorphisms which augment HIV‐1 restriction 109 and recent advances in CRISPR technology may allow for multiple specific edits to the human genome with greater accuracy and lower risk of off‐site effects 110. It is possible to envisage ex‐vivo editing of TRIM5 to generate a patient‐specific pool of HIV‐resistant CD4+ T cells in the future. Understanding how HIV‐2 replication is durably controlled by TRIM5α may provide valuable insights into working towards this future scenario.
Disclosures
We report no conflicts of interest.
Author contributions
M. B. drafted the final manuscript. S. R. J. contributed substantially to the writing and editing of this manuscript.
Acknowledgements
M. B. is funded via a Commonwealth Scholarship. The SR‐J laboratory receives grant support from the Research Council of Norway, the Rosetrees Foundation, the British HIV Association and the Centre for AIDS research, University of Kumamoto. Ashley Jacobs is thanked for graciously assisting M. B. with critical feedback to previous drafts.
References
- 1. Clavel F, Guétard D, Brun‐Vézinet F et al Isolation of a new human retrovirus from West African patients with AIDS. Science 1986; 233:343–6. Available at: http://www.ncbi.nlm.nih.gov/pubmed/2425430 (accessed 13 Novermber 2017). [DOI] [PubMed] [Google Scholar]
- 2. Faria NR, Hodges‐Mameletzis I, Silva JC et al Phylogeographical footprint of colonial history in the global dispersal of human immunodeficiency virus type 2 group A. J Gen Virol 2012; 93(Pt 4):889–99. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22190015 (accessed 14 December 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Visseaux B, Damond F, Matheron S, Descamps D, Charpentier C. HIV‐2 molecular epidemiology. Infect Genet Evol 2016; 46:233–40. Available at: https://linkinghub.elsevier.com/retrieve/pii/S1567134816303471 (accessed 20 November 2016). [DOI] [PubMed] [Google Scholar]
- 4. Forbi JC, Esona MD, Iperepolu HO, Adoga MP, Agwale SM. Absence of routine molecular testing and prevalence of HIV‐2 infection in regions hardest‐hit by HIV infection. J Infect Dev Ctries 2012; 6:854–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23276739 (accessed 31 October 2018). [DOI] [PubMed] [Google Scholar]
- 5. Olesen JS, Jespersen S, da Silva ZJ et al HIV‐2 continues to decrease, whereas HIV‐1 is stabilizing in Guinea‐Bissau. AIDS 2018; 32:1193–8. Available at: http://insights.ovid.com/crossref?an=00002030-201806010-00013 (accessed 31 October 2018). [DOI] [PubMed] [Google Scholar]
- 6. Fryer HR, Van Tienen C, Van Der Loeff MS et al Predicting the extinction of HIV‐2 in rural Guinea‐Bissau. AIDS 2015; 29:2479–86. Available at: http://www.ncbi.nlm.nih.gov/pubmed/26485121 (accessed 19 January 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van der Loeff M, Larke N, Kaye S et al Undetectable plasma viral load predicts normal survival in HIV‐2‐infected people in a West African village. Retrovirology 2010; 7:46 Available at: http://www.ncbi.nlm.nih.gov/pubmed/20482865 (accessed 31 October 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tchounga B, Ekouevi DK, Balestre E, Dabis F. Mortality and survival patterns of people living with HIV‐2. Curr Opin HIV AIDS 2016; 1 Available at: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=01222929-900000000-99453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nyamweya S, Hegedus A, Jaye A, Rowland‐Jones S, Flanagan KL, Macallan DC. Comparing HIV‐1 and HIV‐2 infection: lessons for viral immunopathogenesis. Rev Med Virol 2013; 23:221–40. Available at: http://doi.wiley.com/10.1002/rmv.1739 (accessed 22 January 2017). [DOI] [PubMed] [Google Scholar]
- 10. Esbjörnsson J, Månsson F, Kvist A et al Long‐term follow‐up of HIV‐2‐related AIDS and mortality in Guinea‐Bissau: a prospective open cohort study. Lancet HIV 2018; Available at: https://linkinghub.elsevier.com/retrieve/pii/S2352301818302546 (accessed 18 December 2018). [DOI] [PubMed] [Google Scholar]
- 11. Okulicz JF, Marconi VC, Landrum ML et al Clinical outcomes of elite controllers, viremic controllers, and long‐term nonprogressors in the US department of defense HIV natural history study. J Infect Dis 2009; 200:1714–23. Available at: https://academic.oup.com/jid/article-lookup/doi/10.1086/646609 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 12. Schim van der Loeff MF, Jaffar S, Aveika AA et al Mortality of HIV‐1, HIV‐2 and HIV‐1/HIV‐2 dually infected patients in a clinic‐based cohort in The Gambia. AIDS 2002; 16:1775–83. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12218389. [DOI] [PubMed] [Google Scholar]
- 13. Doyle T, Goujon C, Malim MH. HIV‐1 and interferons: who’s interfering with whom? Nat Rev Microbiol 2015; 13:403–13. Available at: http://www.nature.com/doifinder/10.1038/nrmicro3449 (accessed 28 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uchil PD, Quinlan BD, Chan W‐T, Luna JM, Mothes WTRIM. E3 ligases interfere with early and late stages of the retroviral life cycle. PLOS Pathog 2008; 4:e16 Available at: http://www.ncbi.nlm.nih.gov/pubmed/18248090 (accessed 30 October 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Battivelli E, Migraine J, Lecossier D et al Modulation of TRIM5alpha activity in human cells by alternatively spliced TRIM5 isoforms. J Virol 2011; 85:7828–35. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21632761 (accessed 25 October 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilson SJ, Webb BLJ, Maplanka C et al Rhesus macaque TRIM5 alleles have divergent antiretroviral specificities. J Virol 2008; 82:7243–7. Available at: http://jvi.asm.org/cgi/doi/10.1128/JVI.00307-08 (accessed 29 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meyerson NR, Sawyer SL. Two‐stepping through time: mammals and viruses. Trends Microbiol 2011; 19:286–94. Available at: https://www.sciencedirect.com/science/article/pii/S0966842X11000540 (accessed 25 Dec 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sawyer SL, Emerman M, Malik HS, Finlay M, Barton G. Discordant evolution of the adjacent antiretroviral genes TRIM22 and TRIM5 in mammals. PLOS Pathog 2007; 3:e197 Available at: http://dx.plos.org/10.1371/journal.ppat.0030197 (accessed 4 August 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morger D, Zosel F, Bühlmann M et al The three‐fold axis of the HIV‐1 capsid lattice is the species‐specific binding interface for TRIM5α. J Virol 2017:JVI.01541‐17 Available at: http://www.ncbi.nlm.nih.gov/pubmed/29237846%0Ahttp://jvi.asm.org/lookup/doi/10.1128/JVI.01541-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5α restricts HIV‐1 infection in Old World monkeys. Nature 2004; 427:848–53. Available at: http://www.nature.com/doifinder/10.1038/nature02343 (accessed 28 November 2016). [DOI] [PubMed] [Google Scholar]
- 21. Ylinen LMJ, Keckesova Z, Wilson SJ, Ranasinghe S, Towers GJ. Differential restriction of human immunodeficiency virus type 2 and simian immunodeficiency virus SIVmac by TRIM5 alleles. J Virol 2005; 79:11580–7. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16140735 (accessed 23 January 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mamede JI, Damond F, de Bernardo A et al Cyclophilins and nucleoporins are required for infection mediated by capsids from circulating HIV‐2 primary isolates. Sci Rep 2017; 7:45214 Available at: http://www.nature.com/articles/srep45214 (accessed 4 January 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ohmine S, Sakuma R, Sakuma T, Thatava T, Takeuchi H, Ikeda Y. The antiviral spectra of TRIM5α orthologues and human TRIM family proteins against lentiviral production. PLOS ONE 2011; 6(:e16121 Available at: http://www.ncbi.nlm.nih.gov/pubmed/21264255 (accessed 19 September 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Veillette M, Bichel K, Pawlica P et al The V86M mutation in HIV‐1 capsid confers resistance to TRIM5α by abrogation of cyclophilin a‐dependent restriction and enhancement of viral nuclear import. Retrovirology 2013; 10:25 Available at: http://retrovirology.biomedcentral.com/articles/10.1186/1742-4690-10-25 (accessed 1 November 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pertel T, Hausmann S, Morger D et al TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011; 472:361–5. Available at: http://www.nature.com/nature/journal/v472/n7343/pdf/nature09976.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goldschmidt V, Bleiber G, May M et al Role of common human TRIM5alpha variants in HIV‐1 disease progression. Retrovirology 2006; 3:54 Available at: http://www.ncbi.nlm.nih.gov/pubmed/16925802 (accessed 17 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rahm N, Gfeller D, Snoeck J et al Susceptibility and adaptation to human TRIM5α alleles at positive selected sites in HIV‐1 capsid. Virology 2013; 441:162–70. Available at: https://www.sciencedirect.com/science/article/pii/S0042682213001748 (accessed 3 November 2018). [DOI] [PubMed] [Google Scholar]
- 28. van Manen D, Rits MAN, Beugeling C et al The Effect of Trim5 polymorphisms on the clinical course of HIV‐1 infection. PLOS Pathog 2008; 4:e18 Available at: http://dx.plos.org/10.1371/journal.ppat.0040018 (accessed 17 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kootstra NA, Navis M, Beugeling C, vanDort KA, Schuitemaker H. The presence of the Trim5α escape mutation H87Q in the capsid of late stage HIV‐1 variants is preceded by a prolonged asymptomatic infection phase. AIDS 2007; 21:2015–23. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17885291 (accessed 6 August 2017). [DOI] [PubMed] [Google Scholar]
- 30. Sewram S, Singh R, Kormuth E et al Human TRIM5α expression levels and reduced susceptibility to HIV‐1 infection. J Infect Dis 2009; 199:1657–63. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19388851 (accessed 23 August 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu F, Ourmanov I, Riddick N et al TRIM5α restriction affects clinical outcome and disease progression in simian immunodeficiency virus‐infected rhesus macaques. J Virol 2015; 89:2233–40. Available at: http://www.ncbi.nlm.nih.gov/pubmed/25473059 (accessed 23 January 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jimenez‐Moyano E, Ruiz A, Kløverpris HN et al Nonhuman TRIM5 variants enhance recognition of HIV‐1‐infected cells by CD8+ T cells. J Virol 2016; 90:8552–62. Available at: http://jvi.asm.org/lookup/doi/10.1128/JVI.00819-16 (accessed 28 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakayama EE, Shioda T. Impact of TRIM5α in vivo . AIDS 2015; 29:1733–43. Available at: http://www.ncbi.nlm.nih.gov/pubmed/26372380 (accessed 20 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guyader M, Emerman M, Sonigo P, Clavel F, Montagnier L, Alizon M. Genome organization and transactivation of the human immunodeficiency virus type 2. Nature 1987; 326:662–9. [DOI] [PubMed] [Google Scholar]
- 35. Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, Littman DR. A cryptic sensor for HIV‐1 activates antiviral innate immunity in dendritic cells. Nature 2010; 467:214–7. Available at: http://www.ncbi.nlm.nih.gov/pubmed/20829794 (accessed 24 January 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beer BE, Bailes E, Sharp P, Hirsch V. Diversity and evolution of primate lentiviruses. LANL HIV sequence database; Available at: https://www.hiv.lanl.gov/content/sequence/HIV/REVIEWS/Beer99/Beer.html (accessed 3 December 2017). [Google Scholar]
- 37. Claiborne DT, Prince JL, Scully E et al Replicative fitness of transmitted HIV‐1 drives acute immune activation, proviral load in memory CD4+ T cells, and disease progression. Proc Natl Acad Sci USA 2015; 112:E1480–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/25730868 (accessed 27 March 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prince JL, Claiborne DT, Carlson JM et al Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV‐1 pathogenesis. PLOS Pathog 2012; 8:e1003041 Available at: http://www.ncbi.nlm.nih.gov/pubmed/23209412 (accessed 27 March 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Selhorst P, Combrinck C, Ndabambi N et al Replication capacity of viruses from acute infection drives HIV‐1 disease progression. J Virol 2017; 91:e01806–16. Available at: http://www.ncbi.nlm.nih.gov/pubmed/28148791 (accessed 18 November 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ariën KK, Abraha A, Quiñones‐Mateu ME, Kestens L, Vanham G, Arts EJ. The replicative fitness of primary human immunodeficiency virus type 1 (HIV‐1) group M, HIV‐1 group O, and HIV‐2 isolates. J Virol internet] 2005. Jul 15 [cited 2017 Nov 13]; 79:8979–90. Available at: http://jvi.asm.org/cgi/doi/10.1128/JVI.79.14.8979-8990.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blaak H, van derEnde ME, Boers PHM, Schuitemaker H, Osterhaus ADME. In vitro replication capacity of HIV‐2 variants from long‐term aviremic individuals. Virology 2006; 353:144–54. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0042682206003606 (accessed 13 November 2017). [DOI] [PubMed] [Google Scholar]
- 42. Esbjörnsson J, Månsson F, Kvist A et al Inhibition of HIV‐1 disease progression by contemporaneous HIV‐2 infection. N Engl J Med 2012; 367:224–32. Available at: http://www.nejm.org/doi/abs/10.1056/NEJMoa1113244 (accessed 13 November 2017). [DOI] [PubMed] [Google Scholar]
- 43. Skar H, Borrego P, Wallstrom TC et al HIV‐2 genetic evolution in patients with advanced disease is faster than that in matched HIV‐1 patients. J Virol 2010; 84:7412–5. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2898231&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. MacNeil A, Sankalé J, Meloni ST, Sarr AD, Mboup S, Kanki P. Long‐term intrapatient viral evolution during HIV‐2 infection. J Infect Dis 2007; 195:726–33. Available at: http://jid.oxfordjournals.org/lookup/doi/10.1086/511308 [DOI] [PubMed] [Google Scholar]
- 45. Lemey P, Kosakovsky Pond SL, Drummond AJ et al Synonymous substitution rates predict HIV disease progression as a result of underlying replication dynamics. PLOS Comput Biol 2007; 3:0282–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Palm AA, Lemey P, Jansson M et al Low postseroconversion CD4 + T‐cell level is associated with faster disease progression and higher viral evolutionary rate in HIV‐2 infection. MBio 2019; 10 Available at: http://www.ncbi.nlm.nih.gov/pubmed/30622192 (accessed 15 January 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. de Silva TI, van Tienen C, Onyango C et al Population dynamics of HIV‐2 in rural West Africa: comparison with HIV‐1 and ongoing transmission at the heart of the epidemic. AIDS 2013; 27:125–34. Available at: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00002030-201301020-00015 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 48. Lahaye X, Satoh T, Gentili M et al The capsids of HIV‐1 and HIV‐2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic Cells. Immunity 2013; 39:1132–42. Available at: http://linkinghub.elsevier.com/retrieve/pii/S1074761313005013 (accessed 9 November 2016). [DOI] [PubMed] [Google Scholar]
- 49. Kumar S, Morrison JH, Dingli D, Poeschla E. HIV‐1 activation of innate immunity depends strongly on the intracellular level of TREX1 and sensing of incomplete reverse transcription products. J Virol 2018; 92:e00001–18. Available at: http://www.ncbi.nlm.nih.gov/pubmed/29769349 (accessed 2 January 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Forshey BM, vonSchwedler U, Sundquist WI, Aiken C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol 2002; 76:5667–77. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11991995 (accessed 23 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schommers P, Martrus G, Matschl U et al Changes in HIV‐1 capsid stability induced by common cytotoxic‐T‐lymphocyte‐driven viral sequence mutations. J Virol 2016; 90:7579–86. Available at: http://www.ncbi.nlm.nih.gov/pubmed/27279617 (accessed 23 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lahaye X, Gentili M, Silvin A et al NONO detects the nuclear HIV capsid to promote cGAS‐mediated innate immune activation. Cell 2018; 175:488–501.e22. [DOI] [PubMed] [Google Scholar]
- 53. Schaller T, Ocwieja KE, Rasaiyaah J et al HIV‐1 capsid–cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLOS Pathog 2011; 7:e1002439 Available at: http://dx.plos.org/10.1371/journal.ppat.1002439 (accessed 4 March 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Onyango CO, Leligdowicz A, Yokoyama M et al HIV‐2 capsids distinguish high and low virus load patients in a West African community cohort. Vaccine 2010; 28:B60–7. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0264410X09012389 (accessed 21 November 2016). [DOI] [PubMed] [Google Scholar]
- 55. Jallow S, Leligdowicz A, Kramer HB et al The presence of prolines in the flanking region of an immunodominant HIV‐2 gag epitope influences the quality and quantity of the epitope generated. Eur J Immunol 2015; 45:2232–42. Available at: http://doi.wiley.com/10.1002/eji.201545451 (accessed 21 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Song H, Nakayama EE, Yokoyama M, Sato H, Levy JA, Shioda T. A single amino acid of the human immunodeficiency virus type 2 capsid affects its replication in the presence of cynomolgus monkey and human TRIM5 s. J Virol 2007; 81:7280–5. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17475650 (accessed 23 January 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miyamoto T, Yokoyama M, Kono K, Shioda T, Sato H, Nakayama EE. A single amino acid of human immunodeficiency virus type 2 capsid protein affects conformation of two external loops and viral sensitivity to TRIM5α. PLOS ONE 2011; 6:e22779 Available at: http://dx.plos.org/10.1371/journal.pone.0022779 (accessed 21 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Takeuchi JS, Perche B, Migraine J et al High level of susceptibility to human TRIM5α conferred by HIV‐2 capsid sequences. Retrovirology 2013; 10:50 Available at: http://retrovirology.biomedcentral.com/articles/10.1186/1742-4690-10-50 (accessed 29 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. de Silva TI, Leligdowicz A, Carlson J et al HLA‐associated polymorphisms in the HIV‐2 capsid highlight key differences between HIV‐1 and HIV‐2 immune adaptation. AIDS 2018; 1 Available at: http://insights.ovid.com/crossref?an=00002030-900000000-97305 (accessed 26 February 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Naranbhai V, Carrington M. Host genetic variation and HIV disease: from mapping to mechanism. Immunogenetics 2017; 69:489–98. Available at: http://link.springer.com/10.1007/s00251-017-1000-z (accessed 25 July 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Allsopp CE, Harding RM, Taylor C et al Interethnic genetic differentiation in Africa: HLA class I antigens in The Gambia. Am J Hum Genet 1992; 50:411–21. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1734720 (accessed 8 December 2017). [PMC free article] [PubMed] [Google Scholar]
- 62. Yindom L‐M, Leligdowicz A, Martin MP et al Influence of HLA class I and HLA‐KIR compound genotypes on HIV‐2 infection and markers of disease progression in a Manjako community in West Africa. J Virol 2010; 84:8202–8. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2916551&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Diouf K, Sarr AD, Eisen G, Popper S, Mboup S, Kanki P. Associations between MHC class I and susceptibility to HIV‐2 disease progression. J Hum Virol; 5:1–7. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12352262 (accessed 8 December 2017). [PubMed] [Google Scholar]
- 64. Thomsen D, Erikstrup C, Jespersen S et al The influence of human leukocyte antigen‐types on disease progression among HIV‐2 infected patients in Guinea‐Bissau. AIDS 2018; 32:721–8. Available at: http://www.ncbi.nlm.nih.gov/pubmed/29369163 (accessed 5 September 2018). [DOI] [PubMed] [Google Scholar]
- 65. Hennig BJ, Velez‐Edwards DR, Schim van der Loeff MF et al CD4 intragenic SNPs associate with HIV‐2 plasma viral load and CD4 count in a community‐based study from Guinea‐Bissau, West Africa. J Acquir Immune Defic Syndr 2011; 56:1–8. Available at: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00126334-201101010-00001 (accessed 8 December 2017). [DOI] [PubMed] [Google Scholar]
- 66. Douek DC, Brenchley JM, Betts MR et al HIV preferentially infects HIV‐specific CD4+ T cells. Nature 2002; 417:95–8. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11986671 (accessed 15 November 2017). [DOI] [PubMed] [Google Scholar]
- 67. Duvall MG, Jaye A, Dong T et al Maintenance of HIV‐specific CD4+ T cell help distinguishes HIV‐2 from HIV‐1 infection. J Immunol 2006; 176:6973–81. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16709858 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 68. Alatrakchi N, Damond F, Matheron S et al Proliferative, IFNgamma and IL‐2‐producing T‐cell responses to HIV‐2 in untreated HIV‐2 infection. AIDS 2006; 20:29–34. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16327316 (accessed 8 December 2017). [DOI] [PubMed] [Google Scholar]
- 69. Duvall MG, Precopio ML, Ambrozak DA et al Polyfunctional T cell responses are a hallmark of HIV‐2 infection. Eur J Immunol 2008; 38:350–63. Available at: http://doi.wiley.com/10.1002/eji.200737768 (accessed 10 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Foxall RB, Cortesão CS, Albuquerque AS, Soares RS, Victorino RMM, Sousa AE. Gag‐specific CD4+ T‐cell frequency is inversely correlated with proviral load and directly correlated with immune activation in infection with human immunodeficiency virus type 2 (HIV‐2) but not HIV‐1. J Virol 2008; 82:9795–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18653457 (accessed 17 February 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Francisco LM, Sage PT, Sharpe AH. The PD‐1 pathway in tolerance and autoimmunity. Immunol Rev 2010; 236:219–42. Available at: http://www.ncbi.nlm.nih.gov/pubmed/20636820 (accessed 14 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Soares RS, Tendeiro R, Foxall RB et al Cell‐associated viral burden provides evidence of ongoing viral replication in aviremic HIV‐2‐infected patients. J Virol 2011; 85:2429–38. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21159859 (accessed 8 January 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tendeiro R, Foxall RB, Baptista AP et al PD‐1 and its ligand PD‐L1 are progressively up‐regulated on CD4 and CD8 T‐cells in HIV‐2 infection irrespective of the presence of viremia. AIDS 2012; 26:1065–71. Available at: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00002030-201206010-00002 (accessed 10 November 2016). [DOI] [PubMed] [Google Scholar]
- 74. McMichael AJ, Rowland‐Jones SL. Cellular immune responses to HIV. Nature 2001; 410:980–7. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11309628 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 75. Phillips RE, Rowland‐Jones S, Nixon DF et al Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 1991; 354:453–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1721107 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 76. O’Connell KA, Bailey JR, Blankson JN. Elucidating the elite: mechanisms of control in HIV‐1 infection. Trends Pharmacol Sci 2009; 30:631–7. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0165614709001539 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 77. Kiepiela P, Ngumbela K, Thobakgale C et al CD8+ T‐cell responses to different HIV proteins have discordant associations with viral load. Nat Med 2007; 13:46–53. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17173051 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 78. Leligdowicz A, Onyango C, Yindom L‐M et al Highly avid, oligoclonal, early‐differentiated antigen‐specific CD8+ T cells in chronic HIV‐2 infection. Eur J Immunol 2010; 40:1963–72. Available at: http://doi.wiley.com/10.1002/eji.200940295 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 79. Leligdowicz A, Yindom L‐M, Onyango C et al Robust Gag‐specific T cell responses characterize viremia control in HIV‐2 infection. J Clin Invest 2007; 117:3067–74. Available at: http://www.jci.org/cgi/doi/10.1172/JCI32380 (accessed 18 November 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. de Silva TI, Peng Y, Leligdowicz A et al Correlates of T‐cell‐mediated viral control and phenotype of CD8+ T cells in HIV‐2, a naturally contained human retroviral infection. Blood 2013; 121:4330–9. Available at: http://www.bloodjournal.org/cgi/doi/10.1182/blood-2012-12-472787 (accessed 18 November 2016). [DOI] [PubMed] [Google Scholar]
- 81. Angin M, Wong G, Papagno L et al Preservation of lymphopoietic potential and virus suppressive capacity by CD8+ T cells in HIV‐2‐infected controllers. J Immunol 2016;1600693 Available at: http://www.jimmunol.org/cgi/doi/10.4049/jimmunol.1600693. [DOI] [PubMed] [Google Scholar]
- 82. Fernandes SM, Pires AR, Matoso P et al HIV‐2 infection is associated with preserved GALT homeostasis and epithelial integrity despite ongoing mucosal viral replication. Mucosal Immunol 2017; mi201744 Available at: http://www.nature.com/doifinder/10.1038/mi.2017.44 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 83. Jaffar S, Grant AD, Whitworth J, Smith PG, Whittle H. The natural history of HIV‐1 and HIV‐2 infections in adults in Africa: a literature review. Bull World Health Organ 2004; 82:462–9. [PMC free article] [PubMed] [Google Scholar]
- 84. Grossman Z, Meier‐Schellersheim M, Sousa AE, Victorino RMM, Paul WE. CD4+ T‐cell depletion in HIV infection: are we closer to understanding the cause? Nat Med 2002; 8:319–23. Available at: http://www.nature.com/doifinder/10.1038/nm0402-319 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 85. Nyamweya S, Townend J, Zaman A et al Are plasma biomarkers of immune activation predictive of HIV progression: a longitudinal comparison and analyses in HIV‐1 and HIV‐2 infections? PLOS ONE 2012; 7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sousa AE, Carneiro J, Meier‐Schellersheim M, Grossman Z, Victorino RMM. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV‐1 and HIV‐2 but only indirectly to the viral load. J Immunol 2002; 169:3400–6. [DOI] [PubMed] [Google Scholar]
- 87. Brenchley JM, Price DA, Schacker TW et al Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 2006; 12:1365–71. Available at: http://www.nature.com/doifinder/10.1038/nm1511 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 88. Nowroozalizadeh S, Månsson F, da Silva Z et al Microbial translocation correlates with the severity of both HIV‐1 and HIV‐2 infections. J Infect Dis 2010; 201:1150–4. Available at: https://academic.oup.com/cid/article-lookup/doi/10.1086/651430 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 89. Cavaleiro R, Tendeiro R, Foxall RB et al Monocyte and myeloid dendritic cell activation occurs throughout HIV type 2 infection, an attenuated form of HIV disease. J Infect Dis 2013; 207:1730–42. Available at: https://academic.oup.com/jid/article-lookup/doi/10.1093/infdis/jit085 (accessed 18 November 2017). [DOI] [PubMed] [Google Scholar]
- 90. Jespersen S, Hønge BL, Esbjörnsson J et al Differential effects of sex in a West African cohort of HIV‐1, HIV‐2 and HIV‐1/2 dually infected patients: men are worse off. Trop Med Int Heal 2016; 21:253–62. Available at: http://www.ncbi.nlm.nih.gov/pubmed/26616349 (accessed 13 November 2017). [DOI] [PubMed] [Google Scholar]
- 91. Fonsah JY, Njamnshi AK, Kouanfack C et al Adherence to antiretroviral therapy (ART) in Yaoundé‐Cameroon: association with opportunistic infections, depression, art regimen and side effects. PLOS ONE 2017; 12:e0170893 Available at: http://www.ncbi.nlm.nih.gov/pubmed/28141867 (accessed 4 April 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cornell M, Schomaker M, Garone DB et al Gender differences in survival among adult patients starting antiretroviral therapy in South Africa: a multicentre cohort study. PLOS Med 2012;9:e1001304 Available at: http://www.ncbi.nlm.nih.gov/pubmed/22973181 (accessed 4 April 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chang JJ, Woods M, Lindsay RJ et al Higher expression of several interferon‐stimulated genes in HIV‐1‐infected females after adjusting for the level of viral replication. J Infect Dis 2013; 208:830–8. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23757341 (accessed 3 November 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Addo MM, Altfeld M. Sex‐based differences in HIV type 1 pathogenesis. J Infect Dis 2014; 209(Suppl 3):S86–92. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24966195 (accessed November 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sterling TR, Vlahov D, Astemborski J. Initial plasma HIV‐1 RNA levels and progression to AIDS in women and men. N Engl J Med 2001; 344:720–5. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11236775 (accessed 11 February 2018). [DOI] [PubMed] [Google Scholar]
- 96. Meier A, Chang JJ, Chan ES et al Sex differences in the Toll‐like receptor‐mediated response of plasmacytoid dendritic cells to HIV‐1. Nat Med 2009; 15:955–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19597505 (accessed 11 February 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Carlson JM, Schaefer M, Batorsky R et al Selection bias at the heterosexual HIV‐1 transmission bottleneck. Science 2014; 345:1254031-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Boullé C, Kouanfack C, Laborde‐Balen G et al Gender differences in adherence and response to antiretroviral treatment in the Stratall trial in rural district hospitals in Cameroon. JAIDS J Acquir Immune Defic Syndr 2015; 69:355–64. Available at: http://www.ncbi.nlm.nih.gov/pubmed/26181708 (accessed 4 April 2017). [DOI] [PubMed] [Google Scholar]
- 99. Peterson I, Togun O, de Silva T et al Mortality and immunovirological outcomes on antiretroviral therapy in HIV‐1 and HIV‐2‐infected individuals in The Gambia. AIDS 2011; 25:2167–75. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21881480 (accessed 4 March 2018). [DOI] [PubMed] [Google Scholar]
- 100. Darby SC, Ewart DW, Giangrande PL, Spooner RJ, Rizza CR. Importance of age at infection with HIV‐1 for survival and development of AIDS in UK haemophilia population. UK Haemophilia Centre Directors’ Organisation. Lancet 1996; 347:1573–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/8667864 (accessed 13 November 2017). [DOI] [PubMed] [Google Scholar]
- 101. Balestre E, Eholié SP, Lokossue A et al Effect of age on immunological response in the first year of antiretroviral therapy in HIV‐1‐infected adults in West Africa. AIDS 2012; 26:951–7. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22382142 (accessed 18 November 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Aaby P, Ariyoshi K, Buckner M et al Age of wife as a major determinant of male‐to‐female transmission of HIV‐2 infection: a community study from rural West Africa. AIDS 1996; 10:1585–90. Available at: http://www.ncbi.nlm.nih.gov/pubmed/8931796 (accessed 13 November 2017). [DOI] [PubMed] [Google Scholar]
- 103. Poulsen AG, Aaby P, Larsen O et al 9‐year HIV‐2‐associated mortality in an urban community in Bissau, west Africa. Lancet 1997; 349:911–4. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0140673696044029 (accessed 13 November 2017). [DOI] [PubMed] [Google Scholar]
- 104. Matheron S, Pueyo S, Damond F et al Factors associated with clinical progression in HIV‐2 infected‐patients: the French ANRS cohort. AIDS 2003; 17:2593–601. Available at http://www.ncbi.nlm.nih.gov/pubmed/14685053. [DOI] [PubMed] [Google Scholar]
- 105. Pertel T, Hausmann S, Morger D et al TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011; 472:361–5. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21512573 (accessed 30 November 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hunt PW, Brenchley J, Sinclair E et al Relationship between T cell activation and CD4+ t cell count in HIV‐seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis 2008; 197:126–33. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18171295 (accessed 24 January 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Merindol N, El‐Far M, Sylla M et al HIV‐1 capsids from B27/B57+ elite controllers escape Mx2 but are targeted by TRIM5α, leading to the induction of an antiviral state. PLOS Pathog 2018; 14:e1007398 Available at: http://dx.plos.org/10.1371/journal.ppat.1007398 (accessed 20 December 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Rihn SJ, Wilson SJ, Loman NJ et al Extreme genetic fragility of the HIV‐1 capsid. PLOS Pathog 2013; 9:e1003461 Available at: http://www.ncbi.nlm.nih.gov/pubmed/23818857 (accessed 15 March 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Dufour C, Claudel A, Joubarne N et al Editing of the human TRIM5 gene to introduce mutations with the potential to inhibit HIV‐1. PLOS ONE 2018; 13:e0191709 Available at: http://dx.plos.org/10.1371/journal.pone.0191709 (accessed 1 February 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hu JH, Miller SM, Geurts MH et al Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018; Available at: http://www.nature.com/doifinder/10.1038/nature26155 (accessed 4 March 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]