

ABSTRACT
Hypertrophic cardiomyopathy is the most frequently occurring inherited cardiovascular disease, with a prevalence of more than one in 500 individuals worldwide. Genetically acquired dilated cardiomyopathy is a related disease that is less prevalent. Both are caused by mutations in the genes encoding the fundamental force-generating protein machinery of the cardiac muscle sarcomere, including human β-cardiac myosin, the motor protein that powers ventricular contraction. Despite numerous studies, most performed with non-human or non-cardiac myosin, there is no clear consensus about the mechanism of action of these mutations on the function of human β-cardiac myosin. We are using a recombinantly expressed human β-cardiac myosin motor domain along with conventional and new methodologies to characterize the forces and velocities of the mutant myosins compared with wild type. Our studies are extending beyond myosin interactions with pure actin filaments to include the interaction of myosin with regulated actin filaments containing tropomyosin and troponin, the roles of regulatory light chain phosphorylation on the functions of the system, and the possible roles of myosin binding protein-C and titin, important regulatory components of both cardiac and skeletal muscles.
KEY WORDS: Cardiac myosin, Cardiomyopathy mutations, Force–velocity curves
Summary: The underlying molecular basis of genetic-based cardiomyopathy diseases is largely unknown. This review describes recent molecular studies that have used human cardiac proteins to begin to elucidate the mechanisms whereby mutations cause disease.
Historical perspective
Dozens of laboratories, working over the course of more than a century and using a wide variety of approaches, have contributed importantly to our understanding of muscle contraction (Rall, 2014). The muscle sarcomere may well be the most studied protein complex in cell biology, partially due to its extraordinarily high degree of structural organization.
I (J.A.S.) was asked to preface my talk at the 2015 Journal of Experimental Biology Symposium on Muscle: Molecules to Motion with a brief historical perspective of my laboratory's specific contributions to research on actin and myosin, and how those contributions led to our current focus on understanding the molecular basis of hypertrophic and dilated cardiomyopathy. My laboratory, over the last several decades, has been devoted to developing both in vivo and in vitro systems to understand the molecular basis of energy transduction by the myosin family of molecular motors (Spudich, 2011, 2012). Our interests have included the roles of myosins in non-muscle cells and the molecular basis of muscle contraction. Essential for our research was the development of in vitro motility assays for purified actin and myosin in the 1980s (Kron and Spudich, 1986; Spudich et al., 1985). We used these assays to show that the globular head of myosin, known as subfragment-1 (S1), is the motor domain of the myosin molecule (Toyoshima et al., 1987), and, together with molecular genetic approaches, we provided early strong functional evidence that the light-chain binding region of the S1 acts as a swinging lever arm during chemo-mechanical coupling (e.g. Shih et al., 2000; Uyeda et al., 1996). Our study on changing the lever arm length and even removing the light-chain binding region altogether was an early work that showed that the heavy chain, truncated after the converter domain (Fig. 1, myosin head structure on the right), is the ‘motor domain’ of the molecule, and the lever arm amplifies allosteric movements in the motor domain to obtain a larger stroke size – the distance myosin moves an actin filament per ATP hydrolyzed (Uyeda et al., 1996). Using the physics of laser trapping, we simplified the motility assay to the single molecule level, which allowed the measurement of the stroke size (∼10 nm) as well as the intrinsic force produced by the motor (a few piconewtons) (Finer et al., 1994). With these tools in hand, and after studying a variety of myosin motor types over several decades, my laboratory is now fully focused on one of the most clinically important members of the myosin family of molecular motors, human β-cardiac myosin.
Fig. 1.

Sliding velocity as a function of lever arm length. The Dictyosteium discoideum myosin II gene was genetically engineered to produce four forms of S1 with different lever arm lengths (shown on the right), expressed in D. discoideum, and purified as described previously (Uyeda et al., 1996). The normal D. discoideum myosin S1 has two light chains. Constructs containing zero light chains and one light chain were expressed, as well as one containing three light chains. The latter was created by inserting an additional essential light chain (magenta) binding domain into the myosin II heavy chain. The fulcrum was inferred from the intercept of the straight line with the x-axis. vo, unloaded sliding velocity; d, stroke size; ts, strongly bound state time. Data are from Uyeda et al. (1996).
Human cardiomyopathy mutations are a leading cause of cardiac death
The human β-cardiac myosin motor is a major site in the sarcomere for hundreds of missense mutations that result in a spectrum of heart diseases called cardiomyopathies. Genetically based hypertrophic cardiomyopathy (HCM), an autosomal dominant inherited disease (Geisterfer-Lowrance et al., 1990; Seidman and Seidman, 2000, 2001), is clinically characterized by thickening of the ventricular walls with a resultant decrease in ventricular chamber size that occurs without a predisposing cause. Systolic performance of the heart is preserved or even increased, but relaxation capacity is diminished. HCM affects more than one in 500 individuals (Harvey and Leinwand, 2011; Maron, 2011; Maron et al., 1995; Semsarian et al., 2015). Dilated cardiomyopathy (DCM), in contrast, is characterized by thinning of the heart walls, enlargement of the ventricular chambers, and weakness of the cardiac muscle, resulting in an inability to raise the cardiac output to meet the demands of even modest exercise (Hershberger et al., 2013).
There are reported to be more than 300 pathogenic mutations in β-cardiac myosin (Buvoli et al., 2008; Colegrave and Peckham, 2014; Seidman and Seidman, 2001; Walsh et al., 2010). Most of these are HCM mutations in the globular head domain, myosin S1 (Colegrave and Peckham, 2014). The other major site in the sarcomere for HCM-causing mutations is a regulatory protein known as myosin binding protein-C (MyBP-C). MyBP-C is thought to put a break on the contraction of the muscle, which can be regulated by its phosphorylation (Chow et al., 2014; Kensler et al., 2011; Seidman and Seidman, 2000; van Dijk et al., 2014). A smaller but significant set of mutations are found in the tropomyosin–troponin complex. Tropomyosin (Tm) and the three troponin subunits, troponin T (TnT), troponin I (TnI), and troponin C (TnC), form the Ca2+-regulatory complex of the sarcomere. The actin, myosin, tropomyosin, and the three troponins constitute a six-component system that is considered to be the fundamental regulated contractile complex of the sarcomere (Fig. 2). MyBP-C, which is in the vicinity of the myosin heads in the sarcomere, should be added as a seventh component of the fundamental regulated complex. Furthermore, the giant protein titin is also in the vicinity of the myosin heads in the sarcomere and has been shown to carry DCM mutations (Gerull et al., 2002; Herman et al., 2012; LeWinter and Granzier, 2013, 2014).
List of symbols and abbreviations.
- d
distance
- DCM
dilated cardiomyopathy
- ELC
essential light chain
- f
intrinsic force
- Fensemble
ensemble force
- HCM
hypertrophic cardiomyopathy
- MyBP-C
myosin binding protein-C
- Nt
total number of functionally available heads
- RLC
regulatory light chain
- S1
subfragment-1
- sS1
short subfragment-1
- tc
total cycle time
- Tm
tropomyosin
- TnC
troponin C
- TnI
troponin I
- TnT
troponin T
- ts
strongly bound state time
- v
velocity
- WT
wild type
Fig. 2.
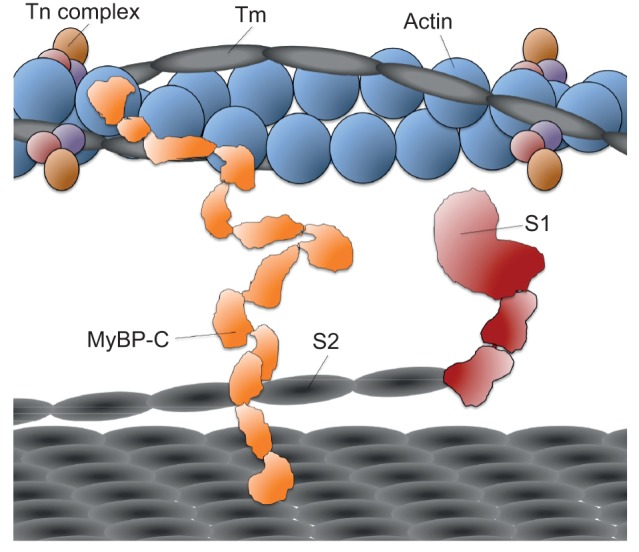
Schematic drawing to scale of essential components of the sarcomere. Tropomyosin (Tm) and the three subunits of troponin (Tn complex) decorate the actin filament. The domains of myosin are the S1 head (only one of two are shown for clarity), the coiled-coil S2, and the light meromyosin that forms the thick filament (bottom grey platform). The N-terminal domains of myosin binding protein-C (MyBP-C) bind to the thin actin filament and the C-terminal domains bind to the thick myosin filament. Titin (not shown) traverses in between the thin and thick filaments, closely associated with the thick filament backbone. Interactions among titin, MyBP-C, S1, and S2 are possible and perhaps regulatory, but have not been fully explored.
Much more is known about the role of the Tm.Tn regulatory complex in cardiac contraction than that of the regulatory proteins MyBP-C and titin. There is good evidence that domains of MyBP-C near the N terminus bind to the regulated actin filament while its C terminus binds to the myosin thick filament backbone (Fig. 2), thus playing a regulatory role in muscle contraction (Chow et al., 2014; Kensler et al., 2011; van Dijk et al., 2014). Studies also suggest that MyBP-C binds to myosin S2, the coiled-coil domain just C-terminal to the globular S1 head, as well as to the regulatory light chain (RLC) of the myosin S1 (Harris et al., 2004; Kampourakis et al., 2014; Pfuhl and Gautel, 2012; Ratti et al., 2011). Titin has been thought of as an element involved in passive tension in the sarcomere (Granzier and Irving, 1995), but as discussed below, MyBP-C and titin may both contribute importantly to the regulation of ensemble force production by the myosin.
The force–velocity curve is fundamental to muscle contraction
As described above, clinically HCM mutations are associated with hypercontractility, whereas DCM mutations are associated with hypocontractility. This has led to the general hypothesis that HCM mutations are hypercontractile and DCM mutations are hypocontractile at the fundamental molecular level. Many important studies have contributed to our current understanding of the effects of some HCM and DCM mutations on various functions of the purified actomyosin complex, usually its ATPase activity or its velocity in the in vitro motility assay (for review, see Moore et al., 2012; Sivaramakrishnan et al., 2009). Only a few studies have attempted to explore the effects of the mutations on the power output generated by a reconstituted actin–myosin complex (Aksel et al., 2015; Debold et al., 2007; Greenberg et al., 2010; Sommese et al., 2013b). Ultimately, the molecular basis of changes in power output is what one needs to understand.
Power (P) output comes directly from the force–velocity (F–V) curve of muscle contraction, as P=F×v (Fig. 3). The force axis on this curve is related to the load on the muscle; the force the muscle produces must overcome the load. Ventricular function is influenced by the two types of loads that are applied to cardiac muscle. One is preload, the load applied by the volume of blood filling the left ventricle that results in stretching of the myofibrils prior to the initiation of ejection. The other is afterload, the load due to pressure in the systemic circulation during contraction when the aortic valve is open. The afterload is the primary load that the heart needs to overcome to eject blood out of the left ventricle.
Fig. 3.
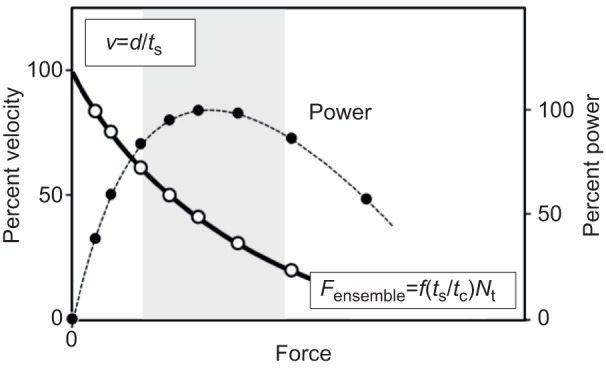
Schematic plot of a force–velocity (F–V) curve for heart muscle. The power curve is generated by multiplying the force times the velocity at each point along the F–V curve. The grey zone is the region of the power curve thought to dominate systolic contraction.
A somewhat oversimplified but useful view of velocity (v) and ensemble force (Fensemble) breaks these down into five parameters, f, d, ts, tc, and Nt (Fig. 3). The velocity at any load is a reflection of d/ts, the distance moved per strongly-bound state time of the motor to actin. Fensemble is the intrinsic force f of each motor domain multiplied by the total number of myosin heads in a force-producing state at any moment. The latter is the total number of heads functionally available (Nt) times the duty ratio. The duty ratio for those heads available to interact with the actin is ts/tc, where ts is the strongly bound state time of myosin on the actin at a particular load and tc is the total cycle time of the actin-activated myosin chemomechanical cycle. The weakly bound states of the motor are the ATP- and ADP-Pi-bound states, while the strongly bound states are the ADP-bound states (Fig. 4). Thus the ensemble force in the sarcomere is Fensemble=f(ts/tc)Nt.
Fig. 4.
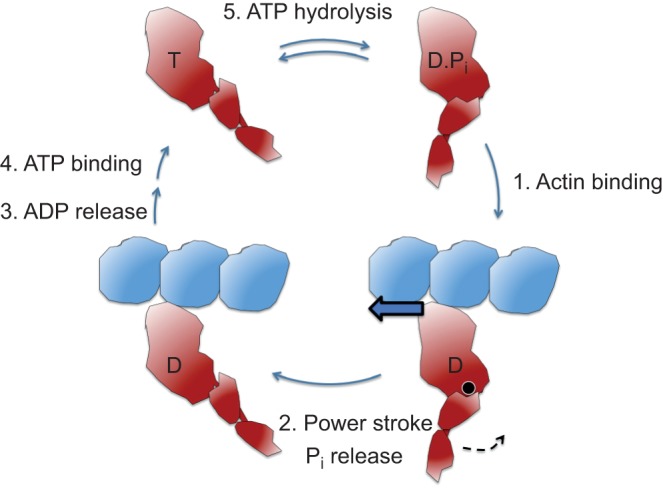
Schematic figure of the key steps of the actin-activated myosin chemomechanical cycle. The total cycle involves five key steps. The pre-stroke states of S1 are shown on the right and the post-stroke states on the left. Step 1: The pre-stroke S1 with bound ADP (D) and phosphate (Pi) undergoes a weak to strong transition in binding to actin. This is the rate-limiting step in the cycle and determines the total cycle time (tc), which is 1/kcat, where kcat is the turnover number, or the number of substrate molecules each enzyme site converts to product per unit time. Step 2: While tightly bound to actin, the lever arm swings about its fulcrum point (black circle) to the right to its post-stroke position (black arrow), moving the actin filament to the left (bold blue arrow) with respect to the myosin thick filament. Step 3: ADP release frees the active site for binding of ATP (T). This step is related to the velocity of the actin filament sliding as the filament cannot move any faster than the myosin heads can let go. Step 4: ATP binding weakens the interaction of the S1 to actin. Although the ATP-bound heads still maintain a weak affinity for the actin, this state is typically drawn as dissociated from the actin filament. Step 5: ATP hydrolysis results in cocking of the head into the pre-stroke state (Shih et al., 2000).
We have been examining these individual parameters for several myosin HCM and DCM mutations studied in a recombinant form of human β-cardiac myosin (short S1 or sS1) that consists of residues 1–808 (the motor domain elongated by the essential light chain [ELC] binding domain) co-expressed with the human cardiac ventricular ELC (MYL3) (Fig. 1, second myosin head image).
Changes in intrinsic force, cycle time and strongly bound state time are generally small for HCM and DCM mutations
Although several-fold changes in intrinsic force, cycle time or strongly bound state time have been reported in studies of HCM and DCM using non-human myosins compared with wild type (WT), we have found only small (10–50%) changes in any of these parameters for the HCM mutant forms we have studied using the human β-cardiac sS1 (R403Q, R453C, R719W, R723G, and R663H). In general, these relatively small changes are not surprising, as most patients with HCM do not begin to exhibit symptoms until at least the third decade of life (and often much later).
The DCM mutants (I201T, A223T, R237W, S532P, and F764L) showed somewhat larger changes compared with WT than the HCM mutants. M531R, a left ventricular non-compaction mutant, showed the largest change, the tc of which was decreased 1.7-fold compared with WT (Aksel et al., 2015).
In general, we are finding that the DCM mutations all behave similarly; for example, they all increase tc. In contrast, the HCM mutations are variable, some appearing more hypocontractile with regard to these parameters and others slightly hypercontractile. These variabilities led us to develop a metric that relates to Fensemble more directly, using a loaded in vitro motility assay.
An ensemble force metric derived from a loaded in vitro motility assay
One way to measure actin gliding velocities at different loads is using a loaded in vitro motility assay in which increasing concentrations of an actin binding protein is utilized as the load-bearing molecule (Bing et al., 2000; Greenberg and Moore, 2010; Haeberle and Hemric, 1995; Warshaw et al., 1990). This assay has been used to compare power output and maximal force generation between myosin variants (Greenberg et al., 2010; Haeberle, 1994; Malmqvist et al., 2004) and between different solution conditions (Greenberg and Moore, 2010; Janson et al., 1992). The load-bearing molecule often used is α-actinin.
The α-actinin assay does not work well, however, with the small motor domain sS1, presumably because of the small size of this domain compared with that of two-headed myosin constructs (Aksel et al., 2015). To obtain information about the relative ensemble forces of mutant and WT human β-cardiac sS1, we developed an utrophin-based loaded in vitro motility assay (Aksel et al., 2015) (Fig. 5A). The actin-binding protein utrophin is used to put a load on the actin filaments that the myosin must overcome. To achieve proper control of myosin and load molecule ratios, a truncated utrophin construct of similar size to sS1 is engineered to anchor on a glass surface via the same attachment used for sS1. Both the velocity of the filaments and the percentage of mobile filaments decrease as utrophin concentration is increased. These two parameters can be combined into what we call the percent time mobile (Fig. 5B). This parameter is a proxy for the ensemble force generated by the motor molecule (Aksel et al., 2015). Importantly, to facilitate these experiments, we also developed a rapid, objective filament-tracking software called FAST (Aksel et al., 2015). (For details of the FAST classes and routines, see the FAST package available at http://spudlab.stanford.edu/fast-for-automatic-motility-measurements/. FAST is currently available for both Linux [Ubuntu 14.04] and Mac operating systems. Academic license for FAST is available for free on http://spudlab.stanford.edu/fast-for-automatic-motility-measurements/.)
Fig. 5.
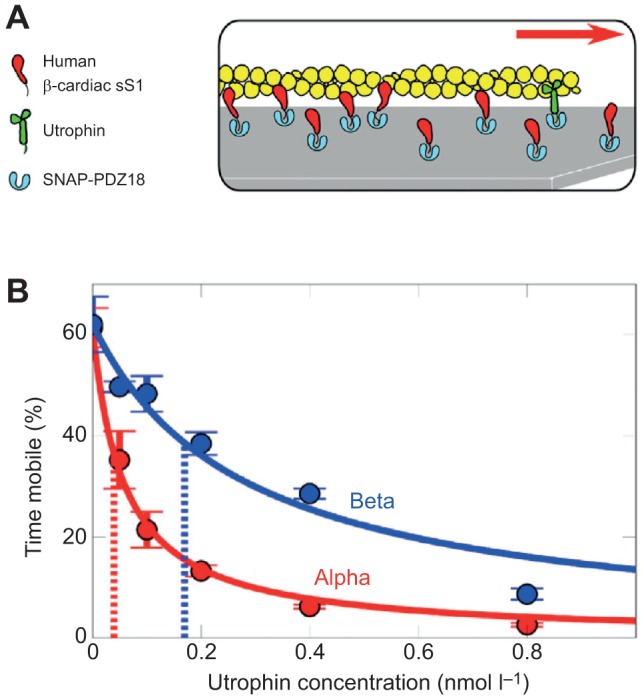
Cartoon of the loaded in vitro motility assay and data obtained for human α- and β-cardiac sS1. (A) Myosin sS1 (red) and utrophin (green) are anchored to a microscope cover slip on the surface via the same attachment system involving binding of a C-terminal eight-residue peptide that specifically binds to SNAP-PDZ18 (light blue), which coats the surface. The utrophin puts a load on the actin filament and slows its gliding velocity. (B) Loaded in vitro motility percent time mobile data for α- (red) and β- (blue) human cardiac sS1. Solid lines show the best stop-model fit (Aksel et al., 2015) to the percent time mobile data. Dashed lines mark values determined from the fit that reflect the relative force generation by the two motors; a lower x-intercept means weaker force. Data are from Aksel et al., (2015).
We have used the utrophin loaded motility assay and FAST to characterize human α- and β-cardiac sS1 (Aksel et al., 2015). As seen in Fig. 5B, the amount of utrophin needed to overcome the motility of human α-cardiac sS1 is considerably less than that needed to overcome the human β-cardiac sS1, consistent with α-cardiac sS1 being a weaker motor (producing less Fensemble) than β-cardiac sS1 (Malmqvist et al., 2004). We have also applied this approach to mutants that show opposite mechanical and enzymatic phenotypes with respect to each other, the hypercontractile left ventricular non-compaction mutant M531R and the hypocontractile DCM mutant S532P (Aksel et al., 2015). By this test, M531R is a stronger motor than WT, while S532P is a weaker motor. We also showed that omecamtiv mecarbil, a previously discovered cardiac specific myosin activator (Malik et al., 2011), increases β-cardiac sS1 Fensemble generation as judged by this loaded in vitro motility assay (Aksel et al., 2015). We are now applying this approach to more than 15 cardiomyopathy mutant forms of human β-cardiac sS1.
HCM and DCM mutations in the tropomyosin–troponin complex are Ca2+-sensitizing and Ca2+-desensitizing, respectively
The tropomyosin–troponin complex is the Ca2+ sensor in the muscle and is composed of tropomyosin and three troponin subunits: (1) TnC, which binds Ca2+; (2) TnI, which binds actin and tropomyosin and is the inhibitory subunit; and (3) TnT, which binds tropomyosin and communicates the Ca2+ signal from TnC to tropomyosin (Brown and Cohen, 2005; Kobayashi and Solaro, 2005). In the absence of Ca2+, tropomyosin is in a position along the actin filament that blocks the binding of myosin. Upon Ca2+ binding to TnC, the inhibitory region of TnI dissociates from tropomyosin, allowing the tropomyosin to move azimuthally on the actin filament, revealing the myosin binding sites on actin. Mutations in these proteins have been studied using various methods, including: animal models, skinned fibers into which mutant proteins have been exchanged, and reconstituted in vitro molecular studies using endogenous and/or recombinant proteins such as ATPase, fluorescence, and motility assays (e.g. Miller et al., 2001; Mirza et al., 2005).
Extensive studies by many investigators have yielded important advances in understanding the effects of HCM- and DCM-causing mutations on a reconstituted six-component system consisting of actin, myosin, tropomyosin, and the three troponin subunits (for reviews, see Hitchcock-DeGregori, 2008; Kobayashi and Solaro, 2005; Moss et al., 2004; Tardiff, 2011; Willott et al., 2010). Most biochemical thin filament studies have been performed using non-human cardiac myosin (e.g. Szczesna et al., 2000; Venkatraman et al., 2005, 2003) or skeletal myosin (e.g. Kobayashi et al., 2013; Lin et al., 1996; Mirza et al., 2005; Tobacman et al., 1999). More recent studies have used human β-cardiac myosin, where some specific differences in biochemical parameters were found compared with earlier non-human myosin studies (Bloemink et al., 2014; Deacon et al., 2012; Gupte et al., 2015; Sommese et al., 2013b). Regardless of the source of the mammalian myosin, however, the general paradigm holds true that HCM mutations in tropomyosin or troponin are sensitizers to Ca2+ (e.g. Fig. 6 shows HCM mutations for three TnI mutations), while DCM mutations are desensitizers (Willott et al., 2010).
Fig. 6.
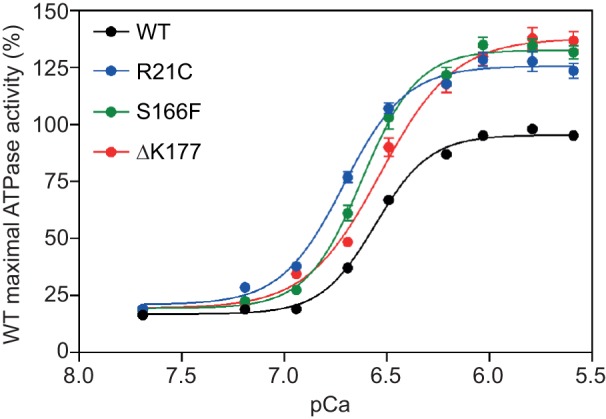
Effects of TnI mutations on Ca2+ regulation of actin-activated myosin S1 ATPase. Percent of the maximal wild-type (WT) ATPase activity (100%) is plotted as a function of the negative logarithm of the Ca2+ concentration. WT (black) is compared with R21C (blue), S166F (green) and ΔK177 (red). Maximum WT activity was set to 100% and all other activities were normalized to this. Mean activities±standard error are plotted. Myosin S1 was prepared by proteolytic cleavage of bovine cardiac myosin.
The myosin mesa and a possible role for MyBP-C and titin in regulating the number of functionally available myosin heads in the sarcomere
Besides β-cardiac myosin, the other major site in the sarcomere for HCM mutations is the regulatory protein MyBP-C (Buvoli et al., 2008; Seidman and Seidman, 2011; Walsh et al., 2010). MyBP-C is thought to put a break on the contraction of the muscle, which can be regulated by its phosphorylation (Chow et al., 2014; Kensler et al., 2011; Seidman and Seidman, 2000; van Dijk et al., 2014). There is good evidence that domains of MyBP-C near the N terminus bind to the regulated actin filament, whereas its C terminus binds to the myosin thick filament backbone (Fig. 2), playing a regulatory role in muscle contraction (Chow et al., 2014; Kensler et al., 2011; van Dijk et al., 2014). MyBP-C is in the A-band region of the sarcomere in close proximity to the myosin heads (Fig. 2). Another regulator, the giant molecule titin (Granzier and Irving, 1995; LeWinter and Granzier, 2013, 2014), extends through the A-band region, running from the Z-disc, to which actin filaments are bound, to the M-line, the center of the sarcomere. Thus, each half sarcomere has titin molecules running its entire length, where the titin plays an important role in passive tension of the sarcomere (Granzier and Irving, 1995) as well as other roles (LeWinter and Granzier, 2014).
A recent report made the suggestion that MyBP-C and/or other proteins in the vicinity of the myosin heads may play a role in sequestering some of the heads and taking them out of the functionally available pool, reducing Nt and thus Fensemble (Spudich, 2015). The concept of MyBP-C removing myosin heads from the functioning pool of heads in the sarcomere has been suggested previously, with evidence for binding of MyBP-C to S2 and the RLC (for reviews, see Kampourakis et al., 2014; Moss et al., 2015). But there may be a site on the motor domain of S1, which has previously been overlooked, that participates in such sequestration of myosin heads. This site has been called the myosin mesa, which is highly conserved among the cardiac myosins, from mouse α-cardiac to human β-cardiac myosin (Spudich, 2015). The mesa has many more arginine residues on its surface than lysine residues, the opposite of usual protein surfaces (Warwicker et al., 2014). Arginines are known to play a role in transient interactions between different proteins (Jones et al., 2000). Thus, the cluster of eight arginine residues on the mesa, R169, R204, R249, R403, R442, R453, R652, and R663, could be involved in transient interactions between the mesa and another protein or protein domain (e.g. MyBP-C, titin, myosin S2) (Fig. 7, blue-labeled amino acids). Strikingly, all eight of these arginines are sites of HCM mutations (Alfares et al., 2015). A number of other non-arginine cardiomyopathy mutations are also on the mesa in the vicinity of these arginines (Fig. 7). Besides a potential MyBP-C interaction with the myosin mesa, titin could be another player in such an interaction. Indeed, Granzier and colleagues reported that certain titin domains in the A-band of the sarcomere have conserved surface patterns and show weak interaction with myosin S1 (Muhle-Goll et al., 2001).
Fig. 7.

Locations of 17 hypertrophic cardiomyopathy mutations in the myosin catalytic domain. (A) Cartoon showing two groups of HCM mutations. Four mutations are in the converter domain (dark green), and 13 are on or very near the myosin mesa (bright green). Sixteen of the HCM mutations were chosen because they have been documented to be the cause of HCM in families carrying these mutations (Alfares et al., 2015). The 17th is M531R, which, while only documented in one family, is a left ventricular non-compaction mutant myosin that appears to be hypercontractile in our studies. (B) The top view of the mesa showing 13 mutations on or near the mesa surface. The residues labeled in blue denote the positively charged amino acids. (C) The same view as in B except the surface charge distribution is shown. I263T, N444S, and M531R are slightly below the surface in acidic pockets, while the remainder of the mutations are on or very near the surface and most are arginine residues, seven of which form a particularly large domain of positive charge on the right half of the mesa as viewed.
An intriguing hypothesis is that the myosin mesa HCM mutations reduce the affinity of S1's putative binding partner(s) (e.g. MyBP-C, titin, myosin S2), releasing those heads to now be involved in the contractile process. This would result in hypercontractility of the muscle, which is characteristic of HCM clinically. Because the myosin motor domain is highly allosteric and there may be other binding sites for the putative interacting partner, this molecular mechanism for HCM hypercontractility may extend to other HCM mutations as well, and this mechanism could be a unifying hypothesis as to why HCM mutations are hypercontractile clinically.
Conclusion and future perspectives
Our laboratory is focused on changes in biochemical and biomechanical parameters in HCM and DCM mutant forms of human β-cardiac myosin. Our studies use both the two-component human system of actin and myosin (Aksel et al., 2015; Sommese et al., 2013b) and the six-component reconstituted human system: actin, myosin, Tm, TnC, TnI, and TnT (Gupte et al., 2015; Sommese et al., 2013a). From the considerations described here and elsewhere (Spudich, 2015), it is possible that many of the HCM mutations in the motor domain of myosin alter the effects of a putative MyBP-C, titin or myosin S2 interaction with the mesa surface. Thus, reconstitution of the eight-component system is likely to be necessary to fully understand the effects of the HCM mutations. This may equally apply to the DCM mutations.
Similarly, the effects of small molecule activators and inhibitors on the human cardiac ventricular system (e.g. Malik et al., 2011) will likely have to be understood at the eight-component level in molecular studies to correlate effects seen in the reconstituted system with those observed in skinned fibers, cardiomyocytes, the whole organ, and animals.
Acknowledgements
We thank Allan Borrayo for technical assistance in maintaining cell cultures and virus preparations.
Footnotes
Competing interests
J.A.S. is a founder of and owns shares in Cytokinetics, Inc., and MyoKardia, Inc., biotech companies that are developing therapeutics that target the sarcomere.
Author contributions
J.A.S. and K.M.R. developed the general concepts and approaches of the research program. T.A. developed the loaded in vitro motility assay. J.S. built the laser trap for single molecule analysis. S.N., M.K., A.A., S.S.S., T.A., J.S., R.F.S., S.R.B., E.C.Y., S.S., R.T., C.C., C.L., D.T., and K.M.R. performed experiments and, together with J.A.S., analyzed the data. S.R.B. carried out the experiments shown in Fig. 6. J.A.S. prepared the manuscript, which S.N., M.K., A.A., S.S.S., T.A., J.S., R.F.S., S.R.B., E.C.Y., S.S., R.T., C.C., C.L., D.T., and K.M.R. edited prior to submission.
Funding
This work was supported by the National Institutes of Health [GM33289 and HL117138 to J.A.S.]; the Human Frontiers Science Program [to J.A.S.]; a National Institutes of Health Cellular, Biochemical, and Molecular Sciences Training Grant [to S.R.B. and C.L.]; a Stanford Interdisciplinary Graduate Fellowship [to R.F.S.]; a Stanford Dean's Postdoctoral Fellowship [to S.N.]; the National Science Foundation Graduate Research Fellowship Program [to S.R.B.]; and the Lucile Packard Child Health Research Institute Postdoctoral Grant [to A.S.A.]. Deposited in PMC for release after 12 months.
References
- Aksel T., Choe Yu E., Sutton S., Ruppel K. M. and Spudich J. A. (2015). Ensemble force changes that result from human cardiac myosin mutations and a small-molecule effector. Cell Rep. 11, 910-920. 10.1016/j.celrep.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfares A. A., Kelly M. A., McDermott G., Funke B. H., Lebo M. S., Baxter S. B., Shen J., McLaughlin H. M., Clark E. H., Babb L. J. et al. (2015). Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet. Med 17, 880-888. 10.1038/gim.2014.205 [DOI] [PubMed] [Google Scholar]
- Bing W., Knott A. and Marston S. B. (2000). A simple method for measuring the relative force exerted by myosin on actin filaments in the in vitro motility assay: evidence that tropomyosin and troponin increase force in single thin filaments. Biochem. J. 350, 693-699. 10.1042/bj3500693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemink M., Deacon J., Langer S., Vera C., Combs A., Leinwand L. and Geeves M. A. (2014). The hypertrophic cardiomyopathy myosin mutation R453C alters ATP binding and hydrolysis of human cardiac beta-myosin. J. Biol. Chem. 289, 5158-5167. 10.1074/jbc.M113.511204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. H. and Cohen C. (2005). Regulation of muscle contraction by tropomyosin and troponin: how structure illuminates function. Adv. Protein Chem. 71, 121-159. 10.1016/S0065-3233(04)71004-9 [DOI] [PubMed] [Google Scholar]
- Buvoli M., Hamady M., Leinwand L. A. and Knight R. (2008). Bioinformatics assessment of β-myosin mutations reveals myosin's high sensitivity to mutations. Trends Cardiovasc. Med. 18, 141-149. 10.1016/j.tcm.2008.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow M. L., Shaffer J. F., Harris S. P. and Dawson J. F. (2014). Altered interactions between cardiac myosin binding protein-C and alpha-cardiac actin variants associated with cardiomyopathies. Arch. Biochem. Biophys. 550-551, 28-32. 10.1016/j.abb.2014.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrave M. and Peckham M. (2014). Structural implications of beta-cardiac myosin heavy chain mutations in human disease. Anat. Rec. 297, 1670-1680. 10.1002/ar.22973 [DOI] [PubMed] [Google Scholar]
- Deacon J. C., Bloemink M. J., Rezavandi H., Geeves M. A. and Leinwand L. (2012). Identification of functional differences between recombinant human α and β cardiac myosin motors. Cell. Mol. Life Sci. 69, 2261-2277. 10.1007/s00018-012-0927-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debold E. P., Schmitt J. P., Patlak J. B., Beck S. E., Moore J. R., Seidman J. G., Seidman C. and Warshaw D. M. (2007). Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am. J. Physiol. Heart Circ. Physiol. 293, H284-H291. 10.1152/ajpheart.00128.2007 [DOI] [PubMed] [Google Scholar]
- Finer J. T., Simmons R. M. and Spudich J. A. (1994). Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature 368, 113-119. 10.1038/368113a0 [DOI] [PubMed] [Google Scholar]
- Geisterfer-Lowrance A. A. T., Kass S., Tanigawa G., Vosberg H.-P., McKenna W., Seidman C. E. and Seidman J. G. (1990). A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 62, 999-1006. 10.1016/0092-8674(90)90274-I [DOI] [PubMed] [Google Scholar]
- Gerull B., Gramlich M., Atherton J., McNabb M., Trombitás K., Sasse-Klaassen S., Seidman J. G., Seidman C., Granzier H., Labeit S. et al. (2002). Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 30, 201-204. 10.1038/ng815 [DOI] [PubMed] [Google Scholar]
- Granzier H. L. and Irving T. C. (1995). Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys. J. 68, 1027-1044. 10.1016/S0006-3495(95)80278-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg M. J. and Moore J. R. (2010). The molecular basis of frictional loads in the in vitro motility assay with applications to the study of the loaded mechanochemistry of molecular motors. Cytoskeleton (Hoboken) 67, 273-285. 10.1002/cm.20441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg M. J., Kazmierczak K., Szczesna-Cordary D. and Moore J. R. (2010). Cardiomyopathy-linked myosin regulatory light chain mutations disrupt myosin strain-dependent biochemistry. Proc. Natl. Acad. Sci. USA 107, 17403-17408. 10.1073/pnas.1009619107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte T. M., Haque F., Gangadharan B., Sunitha M. S., Mukherjee S., Anandhan S., Rani D. S., Mukundan N., Jambekar A., Thangaraj K. et al. (2015). Mechanistic heterogeneity in contractile properties of α-tropomyosin (TPM1) mutants associated with inherited cardiomyopathies. J. Biol. Chem. 290, 7003-7015. 10.1074/jbc.M114.596676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeberle J. R. (1994). Calponin decreases the rate of cross-bridge cycling and increases maximum force production by smooth muscle myosin in an in vitro motility assay. J. Biol. Chem. 269, 12424-12431. [PubMed] [Google Scholar]
- Haeberle J. R. and Hemric M. E. (1995). Are actin filaments moving under unloaded conditions in the in vitro motility assay? Biophys. J. 68, 306S-310S; discussion 310S-311S. [PMC free article] [PubMed] [Google Scholar]
- Harris S. P., Rostkova E., Gautel M. and Moss R. L. (2004). Binding of myosin binding protein-C to myosin subfragment S2 affects contractility independent of a tether mechanism. Circ. Res. 95, 930-936. 10.1161/01.RES.0000147312.02673.56 [DOI] [PubMed] [Google Scholar]
- Harvey P. A. and Leinwand L. A. (2011). The cell biology of disease: cellular mechanisms of cardiomyopathy. J. Cell Biol. 194, 355-365. 10.1083/jcb.201101100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman D. S., Lam L., Taylor M. R. G., Wang L., Teekakirikul P., Christodoulou D., Conner L., DePalma S. R., McDonough B., Sparks E. et al. (2012). Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 366, 619-628. 10.1056/NEJMoa1110186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger R. E., Hedges D. J. and Morales A. (2013). Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531-547. 10.1038/nrcardio.2013.105 [DOI] [PubMed] [Google Scholar]
- Hitchcock-DeGregori S. E. (2008). Tropomyosin: function follows structure. Adv. Exp. Med. Biol. 644, 60-72. 10.1007/978-0-387-85766-4_5 [DOI] [PubMed] [Google Scholar]
- Janson L. W., Sellers J. R. and Taylor D. L. (1992). Actin-binding proteins regulate the work performed by myosin II motors on single actin filaments. Cell Motil. Cytoskeleton 22, 274-280. 10.1002/cm.970220407 [DOI] [PubMed] [Google Scholar]
- Jones S., Marin A. and Thornton J. M. (2000). Protein domain interfaces: characterization and comparison with oligomeric protein interfaces. Protein Eng. 13, 77-82. 10.1093/protein/13.2.77 [DOI] [PubMed] [Google Scholar]
- Kampourakis T., Yan Z., Gautel M., Sun Y.-B. and Irving M. (2014). Myosin binding protein-C activates thin filaments and inhibits thick filaments in heart muscle cells. Proc. Natl. Acad. Sci. USA 111, 18763-18768. 10.1073/pnas.1413922112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler R. W., Shaffer J. F. and Harris S. P. (2011). Binding of the N-terminal fragment of C0-C2 of cardiac MyBP-C to cardiac F-actin. J. Struct. Biol. 174, 44-51. 10.1016/j.jsb.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T. and Solaro R. J. (2005). Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu. Rev. Physiol. 67, 39-67. 10.1146/annurev.physiol.67.040403.114025 [DOI] [PubMed] [Google Scholar]
- Kobayashi M., Debold E. P., Turner M. A. and Kobayashi T. (2013). Cardiac muscle activation blunted by a mutation to the regulatory component, troponin T. J. Biol. Chem. 288, 26335-26349. 10.1074/jbc.M113.494096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron S. J. and Spudich J. A. (1986). Fluorescent actin filaments move on myosin fixed to a glass surface. Proc. Natl. Acad. Sci. USA 83, 6272-6276. 10.1073/pnas.83.17.6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeWinter M. M. and Granzier H. L. (2013). Titin is a major human disease gene. Circulation 127, 938-944. 10.1161/CIRCULATIONAHA.112.139717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeWinter M. M. and Granzier H. L. (2014). Cardiac titin and heart disease. J. Cardiovasc. Pharmacol. 63, 207-212. 10.1097/FJC.0000000000000007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D., Bobkova A., Homsher E. and Tobacman L. S. (1996). Altered cardiac troponin T in vitro function in the presence of a mutation implicated in familial hypertrophic cardiomyopathy. J. Clin. Invest. 97, 2842-2848. 10.1172/JCI118740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik F. I., Hartman J. J., Elias K. A., Morgan B. P., Rodriguez H., Brejc K., Anderson R. L., Sueoka S. H., Lee K. H., Finer J. T. et al. (2011). Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science 331, 1439-1443. 10.1126/science.1200113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmqvist U. P., Aronshtam A. and Lowey S. (2004). Cardiac myosin isoforms from different species have unique enzymatic and mechanical properties. Biochemistry 43, 15058-15065. 10.1021/bi0495329 [DOI] [PubMed] [Google Scholar]
- Maron B. J. (2011). Hypertrophic cardiomyopathy. In Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine, 9th edn (ed. R. O. Bonow, D. L. Mann, D. P. Zipes and P. Libby). St Louis, MO: WB Saunders. [Google Scholar]
- Maron B. J., Gardin J. M., Flack J. M., Gidding S. S., Kurosaki T. T. and Bild D. E. (1995). Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation 92, 785-789. 10.1161/01.CIR.92.4.785 [DOI] [PubMed] [Google Scholar]
- Miller T., Szczesna D., Housmans P. R., Zhao J., de Freitas F., Gomes A. V., Culbreath L., McCue J., Wang Y., Xu Y. et al. (2001). Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy-linked troponin T (I79N) mutation. J. Biol. Chem. 276, 3743-3755. 10.1074/jbc.M006746200 [DOI] [PubMed] [Google Scholar]
- Mirza M., Marston S., Willott R., Ashley C., Mogensen J., McKenna W., Robinson P., Redwood C. and Watkins H. (2005). Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J. Biol. Chem. 280, 28498-28506. 10.1074/jbc.M412281200 [DOI] [PubMed] [Google Scholar]
- Moore J. R., Leinwand L. and Warshaw D. M. (2012). Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ. Res. 111, 375-385. 10.1161/CIRCRESAHA.110.223842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss R. L., Razumova M. and Fitzsimons D. P. (2004). Myosin crossbridge activation of cardiac thin filaments: implications for myocardial function in health and disease. Circ. Res. 94, 1290-1300. 10.1161/01.RES.0000127125.61647.4F [DOI] [PubMed] [Google Scholar]
- Moss R. L., Fitzsimons D. P. and Ralphe J. C. (2015). Cardiac MyBP-C regulates the rate and force of contraction in mammalian myocardium. Circ. Res. 116, 183-192. 10.1161/CIRCRESAHA.116.300561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhle-Goll C., Habeck M., Cazorla O., Nilges M., Labeit S. and Granzier H. (2001). Structural and functional studies of titin's fn3 modules reveal conserved surface patterns and binding to myosin S1: a possible role in the Frank-Starling mechanism of the heart. J. Mol. Biol. 313, 431-447. 10.1006/jmbi.2001.5017 [DOI] [PubMed] [Google Scholar]
- Pfuhl M. and Gautel M. (2012). Structure, interactions and function of the N-terminus of cardiac myosin binding protein C (MyBP-C): who does what, with what, and to whom? J. Muscle Res. Cell Motil. 33, 83-94. 10.1007/s10974-012-9291-z [DOI] [PubMed] [Google Scholar]
- Rall J. A. (2014). Mechanism of Muscular Contraction. New York, NY: Springer. [Google Scholar]
- Ratti J., Rostkova E., Gautel M. and Pfuhl M. (2011). Structure and interactions of myosin-binding protein C domain C0: cardiac-specific regulation of myosin at its neck? J. Biol. Chem. 286, 12650-12658. 10.1074/jbc.M110.156646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman C. E. and Seidman J. G. (2000). Hypertrophic cardiomyopathy. In The Metabolic and Molecular Bases of Inherited Disease (ed. Scriver C. R., Beaudet A. L., Valle D., Sly W. S., Childs K. W. and Vogelstein B.), pp. 5532-5452 New York: McGraw-Hill. [Google Scholar]
- Seidman J. G. and Seidman C. E. (2001). The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 104, 557-567. 10.1016/S0092-8674(01)00242-2 [DOI] [PubMed] [Google Scholar]
- Seidman C. E. and Seidman J. G. (2011). Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ. Res. 108, 743-750. 10.1161/CIRCRESAHA.110.223834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semsarian C., Ingles J., Maron M. S. and Maron B. J. (2015). New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 65, 1249-1254. 10.1016/j.jacc.2015.01.019 [DOI] [PubMed] [Google Scholar]
- Shih W. M., Gryczynski Z., Lakowicz J. R. and Spudich J. A. (2000). A FRET-based sensor reveals large ATP hydrolysis-induced conformational changes and three distinct states of the molecular motor myosin. Cell 102, 683-694. 10.1016/S0092-8674(00)00090-8 [DOI] [PubMed] [Google Scholar]
- Sivaramakrishnan S., Ashley E., Leinwand L. and Spudich J. (2009). Insights into human β-cardiac myosin function from single molecule and single cell studies. J. Cardiovasc. Transl. Res. 2, 426-440. 10.1007/s12265-009-9129-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommese R. F., Nag S., Sutton S., Miller S. M., Spudich J. A. and Ruppel K. M. (2013a). Effects of troponin T cardiomyopathy mutations on the calcium sensitivity of the regulated thin filament and the actomyosin cross-bridge kinetics of human beta-cardiac myosin. PLoS ONE 8, e83403 10.1371/journal.pone.0083403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommese R. F., Sung J., Nag S., Sutton S., Deacon J. C., Choe E., Leinwand L. A., Ruppel K. and Spudich J. A. (2013b). Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proc. Natl. Acad. Sci. USA 110, 12607-12612. 10.1073/pnas.1309493110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich J. A. (2011). Molecular motors: forty years of interdisciplinary research. Mol. Biol. Cell 22, 3936-3939. 10.1091/mbc.E11-05-0447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich J. A. (2012). One path to understanding energy transduction in biological systems. Nat. Med. 18, 1478-1482. 10.1038/nm.2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich J. A. (2015). The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem. Soc. Trans. 43, 64-72. 10.1042/BST20140324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich J. A., Kron S. J. and Sheetz M. P. (1985). Movement of myosin-coated beads on oriented filaments reconstituted from purified actin. Nature 315, 584-586. 10.1038/315584a0 [DOI] [PubMed] [Google Scholar]
- Szczesna D., Zhang R., Zhao J., Jones M., Guzman G. and Potter J. D. (2000). Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. J. Biol. Chem. 275, 624-630. 10.1074/jbc.275.1.624 [DOI] [PubMed] [Google Scholar]
- Tardiff J. C. (2011). Thin filament mutations: developing an integrative approach to a complex disorder. Circ. Res. 108, 765-782. 10.1161/CIRCRESAHA.110.224170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobacman L. S., Lin D., Butters C., Landis C., Back N., Pavlov D. and Homsher E. (1999). Functional consequences of troponin T mutations found in hypertrophic cardiomyopathy. J. Biol. Chem. 274, 28363-28370. 10.1074/jbc.274.40.28363 [DOI] [PubMed] [Google Scholar]
- Toyoshima Y. Y., Kron S. J., McNally E. M., Niebling K. R., Toyoshima C. and Spudich J. A. (1987). Myosin subfragment-1 is sufficient to move actin filaments in vitro. Nature 328, 536-539. 10.1038/328536a0 [DOI] [PubMed] [Google Scholar]
- Uyeda T. Q., Abramson P. D. and Spudich J. A. (1996). The neck region of the myosin motor domain acts as a lever arm to generate movement. Proc. Natl. Acad. Sci. USA 93, 4459-4464. 10.1073/pnas.93.9.4459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk S. J., Bezold K. L. and Harris S. P. (2014). Earning stripes: myosin binding protein-C interactions with actin. Pflugers Arch. 466, 445-450. 10.1007/s00424-013-1432-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman G., Harada K., Gomes A. V., Kerrick W. G. L. and Potter J. D. (2003). Different functional properties of troponin T mutants that cause dilated cardiomyopathy. J. Biol. Chem. 278, 41670-41676. 10.1074/jbc.M302148200 [DOI] [PubMed] [Google Scholar]
- Venkatraman G., Gomes A. V., Kerrick W. G. L. and Potter J. D. (2005). Characterization of troponin T dilated cardiomyopathy mutations in the fetal troponin isoform. J. Biol. Chem. 280, 17584-17592. 10.1074/jbc.M409337200 [DOI] [PubMed] [Google Scholar]
- Walsh R., Rutland C., Thomas R. and Loughna S. (2010). Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 115, 49-60. 10.1159/000252808 [DOI] [PubMed] [Google Scholar]
- Warshaw D. M., Desrosiers J. M., Work S. S. and Trybus K. M. (1990). Smooth muscle myosin cross-bridge interactions modulate actin filament sliding velocity in vitro. J. Cell Biol. 111, 453-463. 10.1083/jcb.111.2.453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warwicker J., Charonis S. and Curtis R. A. (2014). Lysine and arginine content of proteins: computational analysis suggests a new tool for solubility design. Mol. Pharm. 11, 294-303. 10.1021/mp4004749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willott R. H., Gomes A. V., Chang A. N., Parvatiyar M. S., Pinto J. R. and Potter J. D. (2010). Mutations in troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J. Mol. Cell. Cardiol. 48, 882-892. 10.1016/j.yjmcc.2009.10.031 [DOI] [PubMed] [Google Scholar]