

ABSTRACT
Mitochondria oxidize substrates to generate the ATP that fuels muscle contraction and locomotion. This review focuses on three steps in oxidative phosphorylation that have independent roles in setting the overall mitochondrial ATP flux and thereby have direct impact on locomotion. The first is the electron transport chain, which sets the pace for oxidation. New studies indicate that the electron transport chain capacity per mitochondria declines with age and disease, but can be revived by both acute and chronic treatments. The resulting higher ATP production is reflected in improved muscle power output and locomotory performance. The second step is the coupling of ATP supply from O2 uptake (mitochondrial coupling efficiency). Treatments that elevate mitochondrial coupling raise both exercise efficiency and the capacity for sustained exercise in both young and old muscle. The final step is ATP synthesis itself, which is under dynamic control at multiple sites to provide the 50-fold range of ATP flux between resting muscle and exercise at the mitochondrial capacity. Thus, malleability at sites in these subsystems of oxidative phosphorylation has an impact on ATP flux, with direct effects on exercise performance. Interventions are emerging that target these three independent subsystems to provide many paths to improve ATP flux and elevate the muscle performance lost to inactivity, age or disease.
KEY WORDS: Magnetic resonance spectroscopy, Muscle energetics, Mitochondrial coupling, P/O, Exercise efficiency, Exercise capacity
Summary: This review focuses on three steps in oxidative phosphorylation that have independent roles in setting the overall mitochondrial ATP flux and thereby have direct impacts on exercise performance.
Introduction
Mitochondria are the powerhouses of biological tissues. In muscles, they link oxidation of substrates to phosphorylation that generates ATP. The contractile fibers then use ATP to generate force and motion. The role of mitochondria as the terminal sink for O2 in the respiratory system that sets the limit to maximum O2 uptake at the muscle level is well established (Weibel et al., 1991). A causal pathway linking this oxidation to the phosphorylation that generates ATP to fuel muscle force production and exercise performance is also clear. However, less clear is the direct role that mitochondria play in setting the limits to exercise performance. A few studies have made this connection in human subjects using exercise-training experiments, which are well known for raising the capacity for ATP supply (Jubrias et al., 2001) and mitochondrial volume density (Hoppeler et al., 1985). These studies show that a direct increase in muscle performance results from the greater energy supply capacity after endurance training. Thus mitochondria provide the bridge between the pathways that delivery oxygen and the synthesis of ATP that fuels muscle contraction and exercise performance.
One path to elevate ATP supply to raise exercise performance is to increase the mitochondrial content of muscle, as is typically found in endurance training of young subjects (Hoppeler et al., 1985). A second path is to improve the capacity for ATP generation per mitochondrion by targeting the processes underlying ATP supply. One such mechanism is the coupling of oxidation to phosphorylation (mitochondrial coupling efficiency), which can be improved to elevate ATP generation per O2 uptake. An example of this improvement is the acute effect of dietary nitrate on mitochondrial coupling efficiency, with direct effects on exercise efficiency in humans (Jones, 2014; Larsen et al., 2007). Free nitrate is released in the muscle cell by drinking beetroot juice, and acts via the nitrous oxide pathway to elevate mitochondrial ATP synthesis per O2 uptake (often expressed divided by 2 to yield the biochemical term of P/O). After drinking beetroot juice, the human subjects showing the largest increase in P/O had a correspondingly elevated exercise efficiency (leg power output per O2 uptake) on a cycle ergometer (Larsen et al., 2007). This link between mitochondrial and exercise efficiencies is the predicted response based on the thermodynamic connection between these processes (Whipp and Wasserman, 1969). Thus it is possible to adjust the underlying processes in oxidative phosphorylation to improve exercise performance.
A second example of the malleability of oxidative phosphorylation comes from the rapid effect of an antioxidant targeted to the mitochondrion, SS-31, on the capacity of mitochondria to generate ATP. A 1 h infusion of SS-31 in old mice raised P/O by 50% but doubled the phosphorylation capacity (ATPmax) in vivo in hindlimb muscles (Siegel et al., 2013). This greater rise in ATPmax than in P/O implies not only improvement in mitochondrial coupling efficiency but also a rapid rise in the capacity for electron transport chain (ETC) flux (O2 uptake capacity). The mechanism for this increase in the ETC flux capacity is thought to be stabilization of cardiolipin on the inner mitochondrial membrane, thereby restoring a key bridge in electron flow through the ETC (Birk et al., 2014; Szeto, 2014). These treatments demonstrate that there are many sites in oxidative phosphorylation that are potential targets for treatment to improve mitochondrial function. What is remarkable about beetroot juice and SS-31 is their speed of action in raising ATP supply capacity and exercise performance (1 h!). In contrast, many months of exercise training are needed to achieve the same goal (Jubrias et al., 2001). Thus multiple sites in oxidative phosphorylation have the potential to elevate energy supply and exercise performance – very rapidly with some treatments – without increasing the mitochondrial pool.
In this review I evaluate the three major steps in oxidative phosphorylation for malleability (Fig. 1) (Nicholls and Ferguson, 2002) that can improve exercise performance. The first step is the ETC, which oxidizes NADH to pump H+ to generate a proton motive force across the inner mitochondrial membrane. The second step uses the proton motive force to drive phosphorylation via the F1F0-ATP synthase. The third step involves short-circuiting the H+ gradient via a number of processes that leak H+ through the inner mitochondrial membrane, thereby circumventing phosphorylation. My goal is to evaluate how each step contributes to ATP production and thereby has impact on exercise performance. The exciting implication of these insights is that mitochondria have multiple sites of malleability that provide targets for optimizing function to improve ATP flux and elevate muscle exercise performance in both healthy and diseased states.
List of symbols and abbreviations.
- ANT
adenine nucleotide transporter
- ATPmax
mitochondrial phosphorylation capacity
- CV
cardiovascular
- ETC
electron transport chain
- Km
substrate affinity in a Michaelis–Menten reaction
- NA(c,f)
capillary density
- NADH
reduced nicotinamide adenine dinucleotide
- P/O
coupling coefficient for oxidative phosphorylation (ATP/O2÷2)
- Pmax
power output by the legs at the aerobic limit
- ROS
reactive oxygen species
- V̇O2,max
whole-body capacity for oxygen uptake
- V̇V(mt,f)
mitochondrial volume density
Fig. 1.

Diagram of the inner mitochondrial membrane, showing three processes that underlie oxidative phosphorylation. (1) The electron transport chain, which involves NADH oxidation; (2) the uncoupling of oxidation from phosphorylation by leaking H+ across the inner mitochondrial membrane (Leak); and (3) phosphorylation to generate ATP (ATP synthesis). F0-F1 denotes the ATP synthase, UCP is uncoupling protein and ANT is the adenine nucleotide transporter.
Electron transport chain
The ETC and oxidative flux
Setting the pace for oxidation and providing the H+ gradient for mitochondrial phosphorylation is the ETC. Isolated mitochondria are found to have a similar density of ETC complexes on the inner membrane (cristae) of mitochondria among different skeletal muscles (Schwerzmann et al., 1989). The respiratory capacity of isolated mitochondria is very near to the maximal oxygen uptake rate of mitochondria in vivo, as estimated in intact muscles of a wide variety of animals (i.e. 5 ml O2 ml−1 mitochondria per minute) (Hoppeler, 1990; Hoppeler and Lindstedt, 1985). Thus the structure and function of the ETC appears to be highly conserved, providing a biochemical basis for a uniform maximum oxidation capacity of mitochondria among mammalian skeletal muscles.
Linking oxidative flux to performance
The constant maximal O2 uptake rate of mitochondria in vivo is derived from the close association of maximal O2 uptake and the mitochondrial content of the whole-body musculature across mammalian species. From mice to nearly elephant-sized mammals, exercise elicits a maximum O2 flux (V̇O2,max) that matches the size of the mitochondrial pool in the musculature providing the sink for O2 uptake (Weibel et al., 1991). The same correlation holds for humans exercising on an ergometer. The V̇O2,max elicited by exercise is directly proportional to the mitochondrial content of the vastus lateralis muscle (Hoppeler et al., 1973). This association makes sense because the vastus lateralis consumes nearly 40% of the oxygen during cycling exercise (see Conley et al., 2000a). The next step to muscle power output also holds: mitochondrial content varies directly with the maximum sustained power output by legs during cycling (Hoppeler et al., 1985). Thus the mitochondrial capacity for using O2 is linked to the sustained capacity for generating muscle power. This example illustrates that the steps in the pathway from O2 uptake to aerobic performance by the legs go through the mitochondria that use O2 to generate ATP.
Testing the ETC–muscle performance link
Exercise training provides a test of the connections between oxidation, mitochondrial content and exercise performance. Eight weeks of endurance training raised in equal proportions the muscle mitochondrial content and leg power output (Hoppeler et al., 1985). Fig. 2A shows this similar relative increase in mitochondrial volume density [V̇V(mt,f)] and maximum sustained leg power output on an ergometer. Paradoxically, a smaller relative change in V̇O2,max is apparent. However, Fig. 2B resolves this paradox by showing that it is the absolute rise in V̇O2,max that agrees with the predicted rise in O2 uptake by the muscle to generate this leg power output (based on the oxidative efficiency of power generation by muscle, 2.99 ml O2 min−1=1 watt). This finding that the predicted rise in O2 uptake at the muscle level was close to the measured increase in V̇O2,max at the whole-body level suggests that training on an ergometer results in adaptation primarily in the muscles used in cycling (e.g. the quadriceps and, to a lesser extent, the hamstrings). For example, leg power output – and therefore O2 uptake – increased by 35%, while whole-body V̇O2,max increased by only 18%. Thus a large increase occurred in the leg muscles generating power, while non-locomotory muscles associated with O2 delivery, such as the heart and diaphragm, most likely had smaller increases more in accord with the 18% ΔV̇O2,max. These endurance-training results illustrate the direct connections from mitochondria to O2 uptake to muscle exercise performance.
Fig. 2.
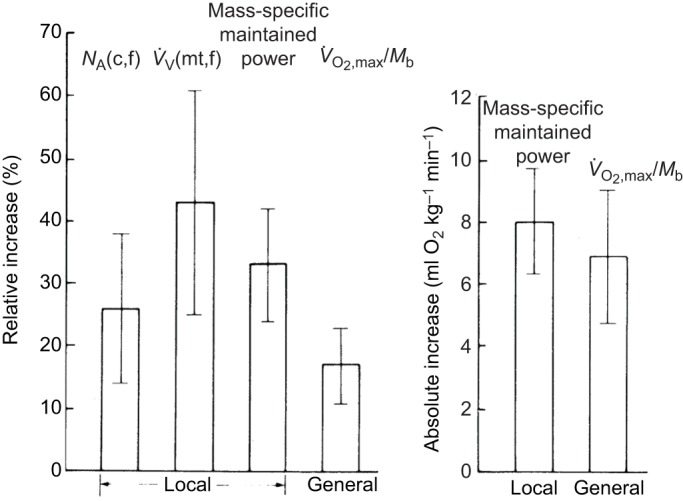
Muscle ultrastructure, leg performance and whole-body adaptations to endurance training in adults. (A) Comparison of changes in capillary density [NA(c,f)], volume density of mitochondria [V̇V(mt,f)], mass-specific maintained power, and mass-specific maximal O2 consumption (V̇O2,max/Mb). (B) Comparison of the absolute increases in mass-specific maintained power and V̇O2,max/Mb. Error bars indicate ±95% confidence intervals. From Hoppeler et al. (1985), reprinted with permission from the American Physiological Society.
ETC flux and muscle power output
The central role in muscle performance specifically played by the ETC in mitochondria is apparent in the many studies that have altered O2 delivery. Breathing hypoxic air results in myoglobin saturation reaching the critical threshold for limiting mitochondrial oxidation at lower work rates than under normoxic conditions (Richardson et al., 1995a,b, 2001). A separate study used blood doping via infusion of erythrocytes to have the opposite effect – to raise the limit to oxidation. The elevated hemoglobin levels resulting from red cell re-infusion raised both V̇O2,max and leg power output (Turner et al., 1993). However, in neither case – raising or lowering the cardiovascular (CV) O2 delivery – was a proportional change found in V̇O2 or exercise performance (leg power output). Instead, a smaller change was found in both cases, which is consistent with the shared role of CV O2 delivery and mitochondrial oxidation in determining V̇O2 (Lindstedt and Conley, 2001; Kascar and Burns, 1973). This shared control emphasizes the crucial role of both the CV delivery and the O2 sink in mitochondria – the ETC – in exercise performance. Ironically, much effort has been made to increase CV O2 delivery in athletes, with rather small effects on V̇O2,max (Lindstedt and Conley, 2001). Nonetheless, the small increments that are achieved by altering one of many factors that contribute to the limits of exercise performance in a world-class competition may still be enough to make the athlete a winner.
Lower function in aging mitochondria
A disconnection between mitochondria and ATPmax with impact on exercise performance is evident in old muscle (Conley et al., 2007a,b,c). This disconnection was evident from a greater loss in ATPmax than expected based on the reduction in mitochondrial content in muscle of elderly subjects (68 years old) versus adults (38 years old) (Conley et al., 2000b). The result was an ATPmax per mitochondrial content that was approximately half that found in adult subjects. The lower muscle ATPmax was paralleled by reduced leg power output per muscle volume at the aerobic limit (Conley et al., 2013b,c). Thus, reduced mitochondrial content and lower function per content combine to limit ATPmax in old muscle, which was reflected in depressed exercise performance in elderly muscle.
Lower ETC flux in old mitochondria?
Mouse muscle shows many of the same mitochondrial energetic changes with aging as with humans (Marcinek et al., 2005; Siegel et al., 2013, 2012). Mitochondrial uncoupling, reduced ATPmax and lower exercise function are all found in the hindlimb muscles of old versus young mice (Siegel et al., 2013). A unique finding in mice is that ETC content actually increases with age in hindlimb muscle (Siegel et al., 2012), as compared with the net loss in human muscle that is expected from the reduction in mitochondrial content with age (Conley et al., 2000b). This rise in ETC content in the face of a drop in ATPmax suggests that there is a lower capacity for flux through the ETC in mouse muscle with age. Thus, two parallel pathways likely contribute to the lower ATPmax of mitochondria with age in mice. The first pathway is a lower efficiency of converting O2 uptake into ATP synthesis (low P/O) due to H+ leak that does not lead to phosphorylation. The second appears to be a lower capacity for O2 flux to generate ATP resulting from a reduced oxidative capacity per ETC complex with age.
Elevating ETC flux in old mitochondria
Reversal of these changes with age is found with interventions in both old mice (described above) and elderly humans. A 6-mo endurance training (ET) program in elderly human subjects raised both mitochondrial capacity and exercise performance (Conley et al., 2013b; Jubrias et al., 2001). These two changes are shown in Fig. 3 as elevations in ATPmax of the quadriceps (ΔATPmax, 32%) and the power output by the legs at V̇O2,max (ΔPmax, 17%). The smaller rise in Pmax versus ATPmax (Δ17%/Δ32%=0.53) is exactly what is expected from the 50% contraction efficiency of converting ATP into muscle work in humans (Nelson et al., 2011).
Fig. 3.
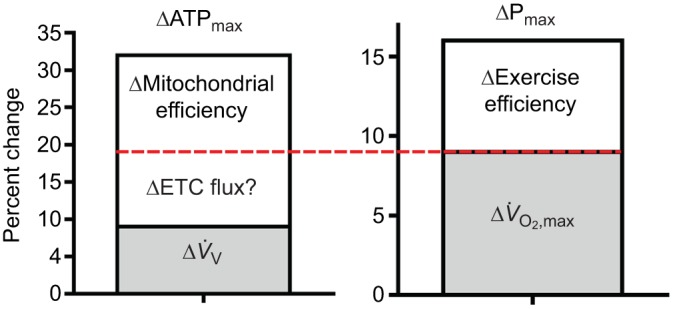
Mitochondrial ATP production capacity and leg performance improvements with endurance training in the elderly. Average increases in mitochondrial ATP production capacity (ΔATPmax) of the quadriceps muscles and in leg power output at the aerobic capacity (ΔPmax) after a 6-mo endurance-training (ET) program. The red horizontal line separates the relative contribution of ΔV̇O2,max versus Δexercise efficiency to ΔPmax. This line also provides estimates of the contribution of ΔETC flux (ΔO2 uptake) versus Δmitochondrial coupling efficiency to ΔATPmax. The Δexercise efficiency at the whole-body level is likely due to Δmitochondrial efficiency because muscle fiber types were unchanged with ET. In addition, only half of the change in ΔV̇O2,max is accounted for by Δmitochondrial volume density [ΔV̇V(mt,f)] in muscle, which points to increased ΔETC flux per mitochondrion (ΔETC flux?) to account for the other half of the increased O2 flux. The question marks (?) indicate likely changes at the mitochondrial level based on the measured changes in whole-body and leg performance.
Identifying mechanisms
We (Conley et al., 2013b) used a combination of mitochondrial and exercise performance changes to reveal that a decrease in mitochondrial coupling is a key factor in the lower performance of elderly versus adult subjects. A similar approach is taken in Fig. 3 to identify the mitochondrial mechanisms underlying the improved ATPmax with ET in the elderly. This figure shows that the rise in Pmax was the result of increases in whole-body O2 flux (ΔV̇O2,max) and better exercise efficiency, as distinguished by the red horizontal line. We showed previously that reduced mitochondrial coupling efficiency can be determined from the quotient of exercise and contractile efficiency (Conley et al., 2013b), based on a thermodynamic analysis (Whipp and Wasserman, 1969). Because fiber type composition did not change with ET in these subjects (Conley et al., 2013b; Jubrias et al., 2001), the contractile efficiency is likely also to be unchanged. This leaves the improvement in mitochondrial coupling efficiency as the predominant determinant of the greater exercise efficiency. The horizontal red line in Fig. 3 indicates that this estimated mitochondrial efficiency rise represents approximately one-third of the improvement in ATPmax.
Another one-third of the rise in ATPmax is due to greater mitochondrial volume density [ΔV̇V(mt,f)]. However, the red line indicates that there appears to be more O2 uptake at the whole-body level (ΔV̇O2,max) than indicated by ΔV̇V(mt,f). This disparity suggests that an increased O2 flux (ΔETC flux capacity) per mitochondrial content also contributes to the rise in ATPmax. Connecting whole-body O2 uptake to mitochondrial O2 flux makes sense given that ΔV̇O2,max appears to be largely due to O2 uptake at the muscle level, as shown in Fig. 2. The elevation in ETC flux capacity of mitochondria implied by the results shown in Fig. 3 is consistent with a restoration of the ETC capacity that is lost with age in both mice and human muscle. It is also consistent with the rapid mitochondrial functional improvements resulting from treatment by the targeted antioxidant SS-31 in old mice (Siegel et al., 2013). This treatment doubled ATPmax but activated only a 50% rise in mitochondrial efficiency (ΔP/O). This discrepancy points to ΔETC flux capacity of mitochondria as responsible for the other half of the elevation in ATPmax in these old muscles. These results argue that low flux per ETC content is an important part of the loss of exercise performance with age and that the interventions – ET and SS-31 – help to restore lost mitochondrial capacity and exercise performance with age. Thus a comparison of changes in mitochondrial and exercise performance in humans and direct measures of mitochondrial function in vivo in mice suggests that two mechanisms underlie ΔATPmax after these interventions: improved mitochondrial coupling efficiency (P/O) and greater capacity for O2 flux per mitochondrial content (ΔETC flux capacity).
An important implication of reversing functional loss per mitochondria is that the long-held belief that this age-related dysfunction is irreversible can be rejected (Harman, 1956). Instead, there are many processes that are malleable (e.g. low P/O) and some damage that may be treatable (e.g. cardiolipin oxidation; Birk et al., 2014; Szeto, 2014) to at least partially restore function of mitochondria to improve exercise function of old animals, including elderly humans.
Mitochondrial coupling
The malleability of ATP generation per O2 uptake is well illustrated by the impact of dietary nitrate. The short-circuiting of phosphorylation underlying low ATP generated per O2 results from a number of processes (Fig. 1) and directly reduces exercise efficiency (Conley et al., 2013b; Larsen et al., 2007; Whipp and Wasserman, 1969). In addition, the low P/O can also limit the mitochondrial capacity (ATPmax) to reduce the leg power output at the aerobic maximum (Conley et al., 2013c). Thus, mechanisms that bypass phosphorylation represent factors independent of the ETC that influence both the efficiency and capacity for exercise.
Short-circuiting phosphorylation
Many mechanisms uncouple oxidative phosphorylation by dissipating the H+ gradient without generating ATP. Such non-phosphorylating H+ movement across the inner mitochondrial membrane can be facilitated by chemical agents, such as dinitrophenol (Marcinek et al., 2004), a high proton motive force (high membrane potential) that increases passive leak (Brand, 2000), or channels that pass H+ through the membrane (Harper et al., 2004). Hibernators take advantage of the heat dissipation that accompanies this leak to warm up to normothermic levels after a winter of low metabolism and depressed body temperature. This is accomplished by active mechanisms of uncoupling using hormonal stimulation of H+ flux via uncoupling channel (UCP1) in brown fat, which raises oxidation without phosphorylation (Nicholls and Locke, 1984).
Uncoupling in muscle
Less dramatic uncoupling is found in skeletal muscle, but the lower mitochondrial efficiency nonetheless impacts a number of human functions. A higher H+ leak in muscle has been suggested to be an important factor in energy expenditure and body weight regulation. Higher mitochondrial leak rates have been found in mitochondria isolated from muscle from individuals that lose weight more quickly than cohorts with stable weight (Thrush et al., 2014). Supporting these findings are in vivo measurements that reveal a higher level of mitochondrial oxidation in individuals exhibiting mitochondrial uncoupling in human vastus lateralis (Conley et al., 2013a). This uncoupling was found in sedentary young subjects (P/O=1.4) relative to active individuals (2.1) of the same age. Independent approaches have confirmed greater coupling in university students (P/O=2.1; Cettolo et al., 2007) and active adults (P/O=2.4; Conley et al., 2013b) that approach the theoretical maximum (2.3–2.5; Brand, 2005). Isolated mitochondria studies also found improved coupling in athletic individuals (e.g. Zoll et al., 2002). High coupling levels near the theoretical maximum indicate a small role for uncoupling mechanisms in these physically active individuals, and suggests that part of the high exercise efficiency of athletes (Coyle, 2005) lies not only in more efficient fiber types but also inmitochondria working at peak efficiency, as predicted by Whipp and Wasserman (1969).
Uncoupling factors in muscle
Independent studies have found mitochondrial uncoupling in humans in muscles with higher type I fibers and lipid content (e.g. tibialis anterior, P/O=2.0; Amara et al., 2007). This is true even in athletes (soleus muscle; Befroy et al., 2008), compared with muscles of predominantly type II fiber composition and low lipid contents (first dorsal interosseous muscle, P/O=2.7; Amara et al., 2007). This association of uncoupling with high intracellular lipid content may reflect the role of fatty acid oxidation and accompanying reactive oxygen species (ROS) generation in H+ leak (Toime and Brand, 2010) that, in turn, results in exercise inefficiency. Both fatty acids and ROS have been implicated in elevated H+ leak that is regulated by UCP3 (Costford et al., 2007; Toime and Brand, 2010). Recently, a connection between ROS defense via glutathione redox state has been suggested as a mechanism regulating UCP3 activity in H+ leak (Mailloux et al., 2013). Reversible glutathionation of UCP3 in response to ROS and redox state provides a dynamic post-translational mechanism for regulating mitochondrial uncoupling. It also provides insight into findings from several studies that show that the UCP3 level is inversely related to exercise efficiency in humans and other animals (Mogensen et al., 2006; Schaeffer et al., 2005; Schrauwen et al., 1999).
Impact of uncoupling on performance
Both uncoupling in muscle (Amara et al., 2007) and a higher metabolic cost of locomotion have been repeatedly found among the elderly (Hortobagyi et al., 2011; Mian et al., 2006; Ortega and Farley, 2007, 2015). A mitochondrial basis for this exercise inefficiency is apparent in two studies using independent methods. In a comparison of adult and elderly subjects, a lower mitochondrial coupling efficiency was quantitatively linked to the reduced cycling efficiency with age (Conley et al., 2013b). This direct connection of mitochondria to exercise efficiency was apparent because the other contributing efficiency – contractile coupling – was not different between the groups. A study of walking speed in elderly subjects found that mitochondrial efficiency was an important contributor to reduced exercise capacity (Coen et al., 2013). In addition, the lower mitochondrial capacity (ATPmax) that resulted from uncoupling was associated with greater fatigability of older subjects, which is an important factor limiting the mobility of the aged (Santanasto et al., 2015). Thus, the impact of mitochondrial uncoupling on exercise efficiency and capacity affects the mobility of the elderly in two ways: it limits the speed and the sustainability of walking.
Improving uncoupling and elevating performance
Treatments that reverse mitochondrial uncoupling provide a direct test of the link to exercise performance. The effect of dietary nitrate is an excellent example of how improvement in mitochondrial coupling elevates exercise efficiency with an acute treatment (Jones, 2014; Larsen et al., 2007). This direct example and the indirect examples in human endurance training and SS-31 treatment in mice provide further evidence that raising ATP flux per O2 uptake represents a path to elevated exercise performance. Thus, reversing mitochondrial uncoupling is an independent and parallel pathway from increased ETC flux for elevating exercise performance.
Phosphorylation
The final step in oxidative phosphorylation – the control of ATP synthesis itself – is also malleable. The need for a flexible control of phosphorylation lies in the failure of a simple regulation mechanism to balance mitochondrial ATP synthesis with contractile ATP demand. This classical feedback mechanism involves the product of ATP use – ADP – as the signal to activate phosphorylation in proportion to ATP need (Chance and Williams, 1956). Underlying this simple feedback are two sophisticated systems that set the ADP level. The first is the creatine kinase equilibrium (see Conley et al., 2001), which determines the ADP level based on metabolites that protect the ATP in the cell (e.g. the high-energy buffer, PCr). The second is a three-protein complex that bridges the inner and outer mitochondrial membranes to communicate this ADP to the mitochondrial matrix (see Perry et al., 2012). In this classic feedback mechanism, ADP rises with increased ATP demand to activate greater ATP supply by the mitochondria. However, a striking example of the failure of this simple system is the finding of large changes in cardiac oxygen uptake and work without an apparent change in ADP, as revealed by non-invasive magnetic resonance studies of intact heart (Balaban et al., 1986). Skeletal muscle also demonstrates the inadequacy of ADP levels alone to account for mitochondrial flux (Conley et al., 2001; Jeneson et al., 2009; Perry et al., 2011). These results indicate that despite an elegant system that underlies simple feedback control, more dynamic control is needed to regulate phosphorylation in proportion to contractile ATP demand.
Additional control mechanisms
A novel regulatory mechanism that fits this large dynamic range between resting and maximal flux is an allosteric regulation (i.e. second-order regulation) of the adenine nucleotide transporter (ANT) (Jeneson et al., 2009). This model of amplified sensitivity of mitochondria acts through cooperative binding of ADP that is analogous to the binding of O2 to hemoglobin. The sensitivity to ADP is reduced in resting muscle with low ATP demands but rises sharply with ADP level due to cooperative binding. The result is an activation system for ATP supply that can meet the low ATP demand of the resting state as well as the high flux in contracting muscle. Thus an ADP sensitivity that varies with concentration is one mechanism that accounts for the high dynamic range of oxidative phosphorylation between resting and active muscle.
Malleability of ADP affinity (Km)
The large dynamic range of fluxes can also be accommodated by a shift in the sensitivity to ADP (i.e. a variable Km), which has been found with exercise (Perry et al., 2012), in athletic individuals (Zoll et al., 2002) and with dietary interventions (Herbst et al., 2014). The high Km for ADP activation of phosphorylation under resting conditions has been found to be greatly reduced with exercise and training, indicating an adjustable ADP sensitivity. The site for this change in regulation is suggested to be the transport protein complex involving three proteins that together bridge the inner mitochondrial membrane (ANT1), membrane space [mitochondrial creatine kinase (miCK)] and outer membrane [voltage-dependent anion channel (VDAC)] (see fig. 4 in Perry et al., 2012). This protein complex is responsible for exporting ATP from the matrix in exchange for ADP import and is driven by the H+ gradient. The suggestion is that modifying the affinity of miCK results in a shift of the apparent Km for ADP activation of phosphorylation. The retention of these changes in Km in permeabilized fibers originating from a muscle biopsy suggests that covalent post-translational mechanisms are at play (e.g. phosphorylation, acetylation and glutathionation; Perry et al., 2012). Translational mechanisms also achieve a shift in the ADP Km that improves sensitivity to ADP regulation of phosphorylation, as shown by the effect of a 3-week dietary Omega-3 supplementation on isolated fiber preparations from human vastus lateralis (Herbst et al., 2014). Upregulation of ANT appears to be the basis of the greater sensitivity of the ADP Km that improves control of phosphorylation. There are also reports of direct post-translational effects on the F1F0 ATP synthase that modulate phosphorylation in cardiac tissue (Wang et al., 2013). These results demonstrate a highly dynamic regulation of phosphorylation with roles for classic allosteric factors (Jeneson et al., 2009) and translational modifications (Herbst et al., 2014), as well as emerging post-translational mechanisms (Wang et al., 2013). Together, their impact on exercise performance is to provide a sensitive regulation of ATP supply between rest and exercise, at different exercise levels and perhaps priming after repeated exercise bouts.
Conclusions
These examples demonstrate that the steps in oxidative phosphorylation, which transduce substrates into fuel, are not fixed but remarkably dynamic. Here I have presented evidence of malleability of the factors determining flux at each stage of oxidative phosphorylation. These separate sites of regulation mean that parallel pathways can be targeted to modulate ATP flux. Improving the coupling of oxidative phosphorylation raises the efficiency of ATP production, while elevating flux through the ETC can separately raise the capacity for ATP production. These flux increases have corresponding effects on the efficiency and capacity of muscle power production. The importance of these separate regulatory sites is that parallel pathways can be targeted to modulate ATP flux with impact on the efficiency and capacity for exercise. Remarkably, agents that target these sites can improve exercise performance within an hour of treatment. These treatments reveal sites that can be manipulated to improve exercise performance even in healthy young subjects but are likely already optimized to maximize performance in athletes. Importantly, these treatment approaches show promise in reversing deficits in function with age and disease. Thus mitochondrial function can be modulated at multiple sites with independent affects on ATP flux that have separate contributions to exercise performance.
Acknowledgements
Thanks go to Sharon Jubrias, David Marcinek and Eric Shankland for insights and discussions.
Footnotes
Competing interests
The author declares no competing or financial interests.
Funding
Support for the work described in this review came from: US National Institutes of Health grants R01 AGAR10853, RC2 AG036606, R01 AR41928 and R01 AR45184, as well as the CHDI Foundation, University of Washington Royalty Research fund, and Seattle Children's Mitochondrial Guild. Deposited in PMC for release after 12 months.
References
- Amara C. E., Shankland E. G., Jubrias S. A., Marcinek D. J., Kushmerick M. J. and Conley K. E. (2007). Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc. Natl. Acad. Sci. USA 104, 1057-1062. 10.1073/pnas.0610131104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban R. S., Kantor H. L., Katz L. A. and Briggs R. W. (1986). Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science 232, 1121-1123. 10.1126/science.3704638 [DOI] [PubMed] [Google Scholar]
- Befroy D. E., Petersen K. F., Dufour S., Mason G. F., Rothman D. L. and Shulman G. I. (2008). Increased substrate oxidation and mitochondrial uncoupling in skeletal muscle of endurance-trained individuals. Proc. Natl. Acad. Sci. USA 105, 16701-16706. 10.1073/pnas.0808889105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk A. V., Chao W. M., Bracken C., Warren J. D. and Szeto H. H. (2014). Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 171, 2017-2028. 10.1111/bph.12468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M. D. (2000). Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp. Gerontol. 35, 811-820. 10.1016/S0531-5565(00)00135-2 [DOI] [PubMed] [Google Scholar]
- Brand M. D. (2005). The efficiency and plasticity of mitochondrial energy transduction. Biochem. Soc. Trans. 33, 897-904. 10.1042/BST20050897 [DOI] [PubMed] [Google Scholar]
- Cettolo V., Cautero M., Tam E. and Francescato M. P. (2007). Mitochondrial coupling in humans: assessment of the P/O2 ratio at the onset of calf exercise. Eur. J. Appl. Physiol. 99, 593-604. 10.1007/s00421-006-0382-7 [DOI] [PubMed] [Google Scholar]
- Chance B. and Williams G. R. (1956). The respiratory chain and oxidative phosphorylation. Adv. Enzymol. 17, 65-134. 10.1002/9780470122624.ch2 [DOI] [PubMed] [Google Scholar]
- Coen P. M., Jubrias S. A., Distefano G., Amati F., Mackey D. C., Glynn N. W., Manini T. M., Wohlgemuth S. E., Leeuwenburgh C., Cummings S. R. et al. (2013). Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 68, 447-455. 10.1093/gerona/gls196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley K. E., Esselman P. E., Jubrias S. A., Cress M. E., Inglin B., Mogadam C. and Schoene R. B. (2000a). Ageing, muscle properties and maximal O2 uptake rate in humans. J. Physiol. 526, 211-217. 10.1111/j.1469-7793.2000.00211.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley K. E., Jubrias S. A. and Esselman P. C. (2000b). Oxidative capacity and ageing in human muscle. J. Physiol. 526, 203-210. 10.1111/j.1469-7793.2000.t01-1-00203.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley K. E., Kemper W. F. and Crowther G. J. (2001). Limits to sustainable muscle performance: interaction of glycolysis and respiration. J. Exp. Biol. 204, 3189-3194. [DOI] [PubMed] [Google Scholar]
- Conley K. E., Amara C. E., Jubrias S. A. and Marcinek D. J. (2007a). Mitochondrial function, fibre types and ageing: new insights from human muscle in vivo. Exp. Physiol. 92, 333-339. 10.1113/expphysiol.2006.034330 [DOI] [PubMed] [Google Scholar]
- Conley K. E., Jubrias S. A., Amara C. E. and Marcinek D. J. (2007b). Mitochondrial dysfunction: impact on exercise performance and cellular aging. Exerc. Sport Sci. Rev. 35, 43-49. 10.1249/JES.0b013e31803e88e9 [DOI] [PubMed] [Google Scholar]
- Conley K. E., Marcinek D. J. and Villarin J. (2007c). Mitochondrial dysfunction and age. Curr. Opin. Clin. Nutr. Metab. Care 10, 688-692. 10.1097/MCO.0b013e3282f0dbfb [DOI] [PubMed] [Google Scholar]
- Conley K. E., Amara C. E., Bajpeyi S., Costford S. R., Murray K., Jubrias S. A., Arakaki L., Marcinek D. J. and Smith S. R. (2013a). Higher mitochondrial respiration and uncoupling with reduced electron transport chain content in vivo in muscle of sedentary versus active subjects. J. Clin. Endocrinol. Metab. 98, 129-136. 10.1210/jc.2012-2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley K. E., Jubrias S. A., Cress M. E. and Esselman P. (2013b). Exercise efficiency is reduced by mitochondrial uncoupling in the elderly. Exp. Physiol. 98, 768-777. 10.1113/expphysiol.2012.067314 [DOI] [PubMed] [Google Scholar]
- Conley K. E., Jubrias S. A., Cress M. E. and Esselman P. C. (2013c). Elevated energy coupling and aerobic capacity improves exercise performance in endurance-trained elderly subjects. Exp. Physiol. 98, 899-907. 10.1113/expphysiol.2012.069633 [DOI] [PubMed] [Google Scholar]
- Costford S. R., Seifert E. L., Bézaire V., Gerrits M. F., Bevilacqua L., Gowing A. and Harper M.-E. (2007). The energetic implications of uncoupling protein-3 in skeletal muscle. Appl. Physiol. Nutr. Metab. 32, 884-894. 10.1139/H07-063 [DOI] [PubMed] [Google Scholar]
- Coyle E. F. (2005). Improved muscular efficiency displayed as Tour de France champion matures. J. Appl. Physiol. (1985) 98, 2191-2196. 10.1152/japplphysiol.00216.2005 [DOI] [PubMed] [Google Scholar]
- Harman D. (1956). Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298-300. 10.1093/geronj/11.3.298 [DOI] [PubMed] [Google Scholar]
- Harper M.-E., Bevilacqua L., Hagopian K., Weindruch R. and Ramsey J. J. (2004). Ageing, oxidative stress, and mitochondrial uncoupling. Acta Physiol. Scand. 182, 321-331. 10.1111/j.1365-201X.2004.01370.x [DOI] [PubMed] [Google Scholar]
- Herbst E. A. F., Paglialunga S., Gerling C., Whitfield J., Mukai K., Chabowski A., Heigenhauser G. J. F., Spriet L. L. and Holloway G. P. (2014). Omega-3 supplementation alters mitochondrial membrane composition and respiration kinetics in human skeletal muscle. J. Physiol. 592, 1341-1352. 10.1113/jphysiol.2013.267336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppeler H. (1990). The different relationship of VO2max to muscle mitochondria in humans and quadrupedal animals. Respir. Physiol. 80, 137-145. 10.1016/0034-5687(90)90077-C [DOI] [PubMed] [Google Scholar]
- Hoppeler H. and Lindstedt S. L. (1985). Malleability of skeletal muscle in overcoming limitations: structural elements. J. Exp. Biol. 115, 355-364. [DOI] [PubMed] [Google Scholar]
- Hoppeler H., Lüthi P., Claassen H., Weibel E. and Howald H. (1973). The ultrastructure of the normal human skeletal muscle. Pflügers Arch. 344, 217-232. 10.1007/bf00588462 [DOI] [PubMed] [Google Scholar]
- Hoppeler H., Howald H., Conley K. E., Lindstedt S. L., Claassen H., Vock P. and Weibel E. R. (1985). Endurance training in humans: aerobic capacity and structure of skeletal muscle. J. Appl. Physiol. 59, 320-327. [DOI] [PubMed] [Google Scholar]
- Hortobagyi T., Finch A., Solnik S., Rider P. and DeVita P. (2011). Association between muscle activation and metabolic cost of walking in young and old adults. J. Gerontol. A Biol. Sci. Med. Sci. 66A, 541-547. 10.1093/gerona/glr008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeneson J. A. L., Schmitz J. P. J., van den Broek N. M. A., van Riel N. A. W., Hilbers P. A. J., Nicolay K. and Prompers J. J. (2009). Magnitude and control of mitochondrial sensitivity to ADP. Am. J. Physiol. Endocrinol. Metab. 297, E774-E784. 10.1152/ajpendo.00370.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A. M. (2014). Influence of dietary nitrate on the physiological determinants of exercise performance: a critical review. Appl. Physiol. Nutr. Metab. 39, 1019-1028. 10.1139/apnm-2014-0036 [DOI] [PubMed] [Google Scholar]
- Jubrias S. A., Esselman P. C., Price L. B., Cress M. E. and Conley K. E. (2001). Large energetic adaptations of elderly muscle to resistance and endurance training. J. Appl. Physiol. 90, 1663-1670. [DOI] [PubMed] [Google Scholar]
- Kascar H. and Burns J. (1973). The control of flux. Soc. Exp. Biol. 27, 65-104. [PubMed] [Google Scholar]
- Larsen F. J., Weitzberg E., Lundberg J. O. and Ekblom B. (2007). Effects of dietary nitrate on oxygen cost during exercise. Acta Physiol. (Oxf.) 191, 59-66. 10.1111/j.1748-1716.2007.01713.x [DOI] [PubMed] [Google Scholar]
- Lindstedt S. L. and Conley K. E. (2001). Human aerobic performance: too much ado about limits to VO2. J. Exp. Biol. 204, 3195-3199. [DOI] [PubMed] [Google Scholar]
- Mailloux R. J., McBride S. L. and Harper M.-E. (2013). Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem. Sci. 38, 592-602. 10.1016/j.tibs.2013.09.001 [DOI] [PubMed] [Google Scholar]
- Marcinek D. J., Schenkman K. A., Ciesielski W. A. and Conley K. E. (2004). Mitochondrial coupling in vivo in mouse skeletal muscle. Am. J. Physiol. Cell Physiol. 286, C457-C463. 10.1152/ajpcell.00237.2003 [DOI] [PubMed] [Google Scholar]
- Marcinek D. J., Schenkman K. A., Ciesielski W. A., Lee D. and Conley K. E. (2005). Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J. Physiol. 569, 467-473. 10.1113/jphysiol.2005.097782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian O. S., Thom J. M., Ardigo L. P., Narici M. V. and Minetti A. E. (2006). Metabolic cost, mechanical work, and efficiency during walking in young and older men. Acta Physiol. (Oxf.) 186, 127-139. 10.1111/j.1748-1716.2006.01522.x [DOI] [PubMed] [Google Scholar]
- Mogensen M., Bagger M., Pedersen P. K., Fernström M. and Sahlin K. (2006). Cycling efficiency in humans is related to low UCP3 content and to type I fibres but not to mitochondrial efficiency. J. Physiol. 571, 669-681. 10.1113/jphysiol.2005.101691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson F. E., Ortega J. D., Jubrias S. A., Conley K. E. and Kushmerick M. J. (2011). High efficiency in human muscle: an anomaly and an opportunity? J. Exp. Biol. 214, 2649-2653. 10.1242/jeb.052985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D. and Ferguson S. (2002). Bioenergetics 3. London: Academic Press. [Google Scholar]
- Nicholls D. G. and Locke R. M. (1984). Thermogenic mechanisms in brown fat. Physiol. Rev. 64, 1-64. [DOI] [PubMed] [Google Scholar]
- Ortega J. D. and Farley C. T. (2007). Individual limb work does not explain the greater metabolic cost of walking in elderly adults. J. Appl. Physiol. 102, 2266-2273. 10.1152/japplphysiol.00583.2006 [DOI] [PubMed] [Google Scholar]
- Ortega J. D. and Farley C. T. (2015). Effects of aging on mechanical efficiency and muscle activation during level and uphill walking. J. Electromyogr. Kinesiol. 25, 193-198. 10.1016/j.jelekin.2014.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry C. G. R., Kane D. A., Lin C.-T., Kozy R., Cathey B. L., Lark D. S., Kane C. L., Brophy P. M., Gavin T. P., Anderson E. J. et al. (2011). Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem. J. 437, 215-222. 10.1042/BJ20110366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry C. G. R., Kane D. A., Herbst E. A. F., Mukai K., Lark D. S., Wright D. C., Heigenhauser G. J. F., Neufer P. D., Spriet L. L. and Holloway G. P. (2012). Mitochondrial creatine kinase activity and phosphate shuttling are acutely regulated by exercise in human skeletal muscle. J. Physiol. 590, 5475-5486. 10.1113/jphysiol.2012.234682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson R., Noyszewski E., Kendrick K., Leigh J. and Wagner P. (1995a). Hypoxia proportionately reduces muscle VO2max and myoglobin-PO2: evidence that VO2max is dependent on O2 supply. FASEB J. 9, A351, 2035. [Google Scholar]
- Richardson R. S., Noyszewski E. A., Kendrick K. F., Leigh J. S. and Wagner P. D. (1995b). Myoglobin O2 desaturation during exercise. Evidence of limited O2 transport. J. Clin. Invest. 96, 1916-1926. 10.1172/JCI118237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson R. S., Newcomer S. C. and Noyszewski E. A. (2001). Skeletal muscle intracellular PO2 assessed by myoglobin desaturation: response to graded exercise. J. Appl. Physiol. 91, 2679-2685. [DOI] [PubMed] [Google Scholar]
- Santanasto A. J., Glynn N. W., Jubrias S. A., Conley K. E., Boudreau R. M., Amati F., Mackey D. C., Simonsick E. M., Strotmeyer E. S., Coen P. M. et al. (2015). Skeletal muscle mitochondrial function and fatigability in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1379-1385. 10.1093/gerona/glu134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer P. J., Villarin J. J., Pierotti D. J., Kelly D. P. and Lindstedt S. L. (2005). Cost of transport is increased after cold exposure in Monodelphis domestica: training for inefficiency. J. Exp. Biol. 208, 3159-3167. 10.1242/jeb.01703 [DOI] [PubMed] [Google Scholar]
- Schrauwen P., Troost F. J., Xia J., Ravussin E. and Saris W. H. M. (1999). Skeletal muscle UCP2 and UCP3 expression in trained and untrained male subjects. Int. J. Obes. 23, 966-972. 10.1038/sj.ijo.0801026 [DOI] [PubMed] [Google Scholar]
- Schwerzmann K., Hoppeler H., Kayar S. R. and Weibel E. R. (1989). Oxidative capacity of muscle and mitochondria: correlation of physiological, biochemical, and morphometric characteristics. Proc. Natl. Acad. Sci. USA 86, 1583-1587. 10.1073/pnas.86.5.1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel M. P., Wilbur T., Mathis M., Shankland E. G., Trieu A., Harper M.-E. and Marcinek D. J. (2012). Impaired adaptability of in vivo mitochondrial energetics to acute oxidative insult in aged skeletal muscle. Mech. Ageing Dev. 133, 620-628. 10.1016/j.mad.2012.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel M. P., Kruse S. E., Percival J. M., Goh J., White C. C., Hopkins H. C., Kavanagh T. J., Szeto H. H., Rabinovitch P. S. and Marcinek D. J. (2013). Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 12, 763-771. 10.1111/acel.12102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto H. H. (2014). First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 171, 2029-2050. 10.1111/bph.12461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrush A. B., Zhang R., Chen W., Seifert E. L., Quizi J. K., McPherson R., Dent R. and Harper M.-E. (2014). Lower mitochondrial proton leak and decreased glutathione redox in primary muscle cells of obese diet-resistant versus diet-sensitive humans. J. Clin. Endocrinol. Metab. 99, 4223-4230. 10.1210/jc.2014-1726 [DOI] [PubMed] [Google Scholar]
- Toime L. J. and Brand M. D. (2010). Uncoupling protein-3 lowers reactive oxygen species production in isolated mitochondria. Free Radic. Biol. Med. 49, 606-611. 10.1016/j.freeradbiomed.2010.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner D. L., Hoppeler H., Noti C., Gurtner H.-P., Gerber H., Schena F., Kayser B. and Ferretti G. (1993). Limitations to VO2,max in humans after blood retransfusion. Respir. Physiol. 92, 329-341. 10.1016/0034-5687(93)90017-5 [DOI] [PubMed] [Google Scholar]
- Wang S.-B., Murray C. I., Chung H. S. and Van Eyk J. E. (2013). Redox regulation of mitochondrial ATP synthase. Trends Cardiovasc. Med. 23, 14-18. 10.1016/j.tcm.2012.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel E. R., Taylor C. R. and Hoppeler H. (1991). The concept of symmorphosis: a testable hypothesis of structure-function relationship. Proc. Natl. Acad. Sci. USA 88, 10357-10361. 10.1073/pnas.88.22.10357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whipp B. J. and Wasserman K. (1969). Efficiency of muscular work. J. Appl. Physiol. 26, 644-648. [DOI] [PubMed] [Google Scholar]
- Zoll J., Sanchez H., N'Guessan B., Ribera F., Lampert E., Bigard X., Serrurier B., Fortin D., Geny B., Veksler V. et al. (2002). Physical activity changes the regulation of mitochondrial respiration in human skeletal muscle. J. Physiol. 543, 191-200. 10.1113/jphysiol.2002.019661 [DOI] [PMC free article] [PubMed] [Google Scholar]