

Abstract
Autoimmune pancreatitis (AIP) is a rare condition classified in 2 subtypes. Their distinction relies on a combination of clinical, serological, morphological and histological features. Type 1 is a pancreatic manifestation of IgG4-related disease characterized by multiorgan infiltration by IgG4 plasmocytes. In this condition, hepatobiliary infiltration is frequent and often mimics cholangiocarcinoma or primary sclerosing cholangitis. On the other hand, type 2 is commonly limited to the pancreas. Herein, we describe the case of a patient who presented a type 2 AIP associated with cholangiopathy, a condition not described in the established criteria. He first developed a pancreatitis identified as type 2 by the typical histopathological features and lack of IgG4 in the serum and tissue. Despite a good clinical response to steroids, cholestasis persisted, identified by MR cholangiography as a stricture of the left hepatic duct with dilatation of the intrahepatic bile duct in segments 2 and 3. Biliary cytology was negative. Evolution was favorable but after steroid tapering a few months later, the patient suffered from recurrence of the pancreatitis as well as progression of biliary attempt, suspicious for cholangiocarcinoma. As the investigations again ruled out neoplastic infiltration or primary sclerosing cholangitis, azathioprine was initiated with resolution of both pancreatic and biliary attempts.
Keywords: Autoimmune pancreatitis, Biliary involvement
Introduction
Autoimmune pancreatitis (AIP) is a rare form of pancreatitis. Two separate subtypes have been identified. Many classifications have been proposed for their diagnosis and, recently, an international consensus (International Consensus Diagnostic Criteria from the International Association of Pancreatology) [1].
Type 1 is more common in middle-aged to elderly Asian men and is a pancreatic manifestation of a systemic immunoglobulin G4 (IgG4)-related disease. The hepatobiliary system is involved in 60–80% of patients [2]. It is characterized by a lymphoplasmacytic infiltrate rich in IgG4 plasma cells, storiform fibrosis and obliterative venulitis. IgG4 serum levels are highly elevated and IgG4 tissue infiltration causes multi-organ damage.
Type 2 has been described mainly in Europe and the USA in younger people. The histological pattern, different from type 1 PAI, shows the presence of periductal granulocyte epithelial lesion [1, 3]. Definitive diagnosis of type 2 AIP requires histology, as serum immunoglobulin levels are normal and extrapancreatic involvement is not described, except for inflammatory bowel disease in 20–30% of cases [4].
Even though international guidelines facilitate AIP diagnosis, it remains challenging because of the heterogeneous presentation that can especially mimic pancreas or bile duct cancer [5]. Diagnosis relies on clinical, serological, morphological and histological arguments as well as rapid response to steroid therapy [1, 6, 7]. The distinction between both subtypes is important to predict the evolution of the disease, extrapancreatic involvement and the rate of relapse.
We present a case of a type 2 AIP, with biliopancreatic manifestation and relapse during steroid tapering.
Case Report
A 62-year-old man of Eritrean origin was admitted to our hospital for a 1-month epigastric transfixing pain and a 6% weight loss. An abdominal CT scan revealed an enlargement of the pancreas which appeared hypoperfused (mainly on the body and tail), suggesting an acute pancreatitis. There were no bile duct dilatations or pancreatic calcifications. He reported no relevant medical history except for smoking. Laboratory analysis showed moderate inflammation with C-reactive protein 3 times the upper normal limits (UNL), significant anicteric cholestasis (AP 2 times UNL, GGT 10 times UNL, normal total bilirubin) and a slightly elevated lipase 1.5 times UNL. Calcium and lipid levels were normal: CA19-9 was 230 kU/l (n < 37), and total IgG was 9 g/L with no IgG4 subclass. Thus, anamnestic and clinical investigations as well as blood tests were nonsuggestive of an etiology.
An endoscopic ultrasonography was performed: bile ducts were normal and tissue from the enlarged hypoechoic pancreas was obtained through core biopsies with a 22-gauge needle (SharkCore Medtronic Covidien AG, Neuhausen am Rheinfall, Switzerland). The histological results identified abundant neutrophilic infiltration of the pancreatic gland with fibrosis and rare plasma cells negative for IgG4 immunostaining, suggesting the diagnosis of type 2 AIP. Cytology was not conclusive because of the absence of cells. A thoracic CT scan eliminated extrapancreatic lesions and a colonoscopy ruled out an associated inflammatory bowel disease.
The patient was initially treated with oral steroids at a dose of 40 mg per day. Despite a good clinical response with pain resolution, elevated liver biochemistry persisted after 2 weeks of treatment. An MR cholangiopancreatography (MRCP) was thereby performed in February 2017 and showed persisting pancreatitis as well as a stricture of the left hepatic duct and intrahepatic bile duct dilatation in segments 2 and 3. There was no peribiliary infiltration or left liver atrophy. However, as the biliary attempt persisted 1 month later on a control MRCP (March 23, 2017) despite resolution of the pancreatitis, a cholangiocarcinoma had to be excluded. The patient underwent percutaneous transhepatic cholangiography with sampling of the stricture while an internal-external drain was maintained. Biliary cytology revealed clusters of glandular cells with enlarged nuclei, without arguments for dysplasia and fibro-inflammatory tissue.
After 1 month, steroids were tapered by 5 mg per week until June 2017. Liver tests remained stable (AP 1.5 times UNL, GGT 8 times UNL, normal total bilirubin) as well as CA19-9 (200 kU/L). A cholangiography through the drain was obtained and, as the intrahepatic dilatation and the stricture regressed, it was removed after a clamping test.
A follow-up MRCP was planned in August 2017, 2 months after stopping steroids. When presenting for this exam, the patient complained of sudden jaundice, pruritus and fatigue. Biological test results were as follows: AP 3 times UNL, GGT 10 times UNL and total bilirubin 10 times UNL. The imagery revealed recurrence of the left hepatic duct stricture associated with dilated intrahepatic biliary tree in segments 2, 3 and 4, surrounding left liver infiltration and a second stricture of the main bile duct near the convergence with slightly dilated right intrahepatic bile ducts as well as a tumefaction of the pancreas, suspicious of an acute recurrent pancreatitis.
Percutaneous cholangiography was repeated in September 2017 and sampling of the main bile duct stricture was obtained through brushing, while two internal-external drains were maintained in the anterior and posterior biliary sector of the right liver. Cytology demonstrated benign bile duct cells with polymorphonuclear granulocytes.
An ultrasound-guided percutaneous biopsy of the infiltrated liver was achieved and showed fibrous tissue associated with a dense polymorphic inflammation, suggesting an inflammatory pseudotumor. Surrounding tissue was also obtained. It revealed mild portal fibrosis, edema and intrahepatic and canalicular cholestasis, secondary to bile ducts obstruction. Antinuclear antibodies were negative and a PET CT confirmed the absence of other organ damage (see Fig. 1, 2, 3, 4).
Fig. 1.
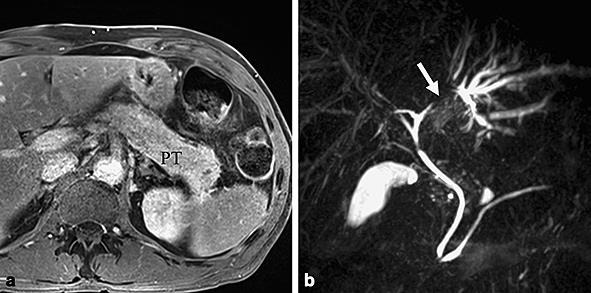
Baseline MR imaging. a Axial gadolinium-enhanced T1 image showing signs of acute pancreatitis with an enlargement of the pancreatic tail and a mild stranding of the peripancreatic spaces. b MR cholangiopancreatography showing a stricture of the left hepatic duct with upstream dilatation of the bile ducts of the left hepatic lobe.
Fig. 2.
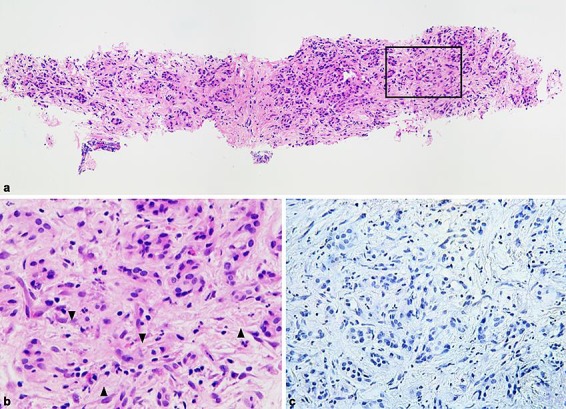
a Overview of one representative SharkCore biopsy showing a slight nonstoriform interlobular fibrosis of the pancreatic tissue, exhibiting a heterogeneous inflammatory infiltrate with predominant neutrophilic accumulation within the interlobular spaces (inset). b Detailed view of the inset in a showing neutrophilic infiltrates (arrowhead) with inconspicuous periacinar lymphoplasmacytic and histiocytic accumulation. c Detailed view of the immunostaining results showing no IgG4-positive plasma cells. This combination of histological and immunostaining findings is highly suggestive for type 2 autoimmune pancreatitis. H&E staining. Original magnification, a ×10, b ×60. IgG4 stain (Cell marque©, 1: 50). Original magnification, c ×40.
Fig. 3.
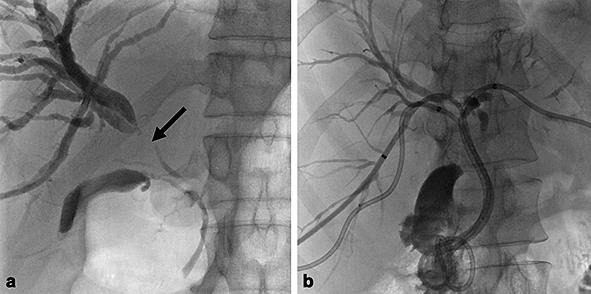
a The second percutaneous transhepatic cholangiography shows a main stricture (arrow) of the right hepatic duct with upstream dilatation of the bile ducts of the right liver. b Three internal external drainage tube are placed in the right anterior and posterior segments of the right liver and in the left hepatic lobe.
Fig. 4.
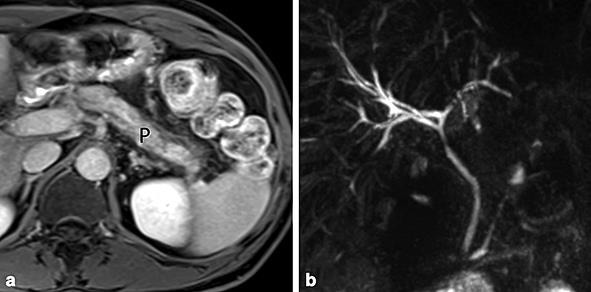
Control MR imaging. a Axial gadolinium-enhanced T1 image showing diffuse hypotrophy of the pancreas, but without signs of acute pancreatitis. b MR cholangiopancreatography showing a mild dilatation of the intrahepatic bile ducts, but without residual strictures of the main bile ducts.
The patient developed cholangitis treated by additional percutaneous drainage of the left biliary tree and antibiotic course. After resolution of the infection, drains were sequentially removed and steroids were re-started in October 2017, with early adjunction of azathioprine at a dose of 2 mg/kg per day. Under this therapy, normalization of bilirubin and improvement of cholestasis were rapidly obtained (within 2 weeks), with AP 1.5 times UNL and GGT 3 times UNL. Steroids were progressively tapered until discontinuation. After 5 months of follow-up under azathioprine, the patient remains asymptomatic, cholestasis is still improving and the last MRCP, performed in February 2018, shows a clear regression of the biliary and pancreatic attempts.
Discussion
Diagnosis of AIP, which is a group of heterogeneous diseases, is challenging, especially for type 2, which is less frequent and is studied to a lesser extent than type 1. In small case reports and series, the association of biliary involvement and pancreatitis [8] is usually suggestive of type 1 AIP, as IgG4-related biliary disease [9]. To our knowledge, very few cases of biliary involvement have been described with type 2 AIP. In 2012, a case was reported of a child with granulocytic epithelial lesion, a sclerosing cholangitis. The authors also reviewed 245 liver biopsies (103 children and 142 adults) of patients with a diagnosis of autoimmune or primary sclerosing cholangitis and identified 5 cases of neutrophilic bile duct lesions, and only 1 had concomitant pancreatitis. All of these patients were in remission after steroid course and no recurrence was observed over 6–16 years of follow-up [10]. One case was also described in 2013 in a child, with a similar evolution. Although this entity is not really known, it has been suggested that this form of bile duct lesion may account for a subgroup of juvenile sclerosing cholangitis.
Most of the time, autoimmune cholangitis appears as a stricture and dilatation of bile ducts, hard to distinguish from cholangiocarcinoma or primary sclerosing cholangitis [11]. Making the right diagnosis is crucial because treatment and outcomes are not the same. Tissue samplings, mandatory to exclude cancer, were obtained repetitively by endoscopic ultrasound, endoscopic retrograde cholangiopancreatography and targeted liver biopsy, with no evidence of malignant cells, and regression of the pancreatobiliary manifestation is an argument to rule out cholangiocarcinoma. The rapid development of biliary lesions as well as response to immunosuppressor therapy were also against the diagnosis of primary sclerosing cholangitis. Furthermore, liver histology did not exhibit the classical chronic nonsuppurative lymphocytic cholangitis. Finally, most of the primary sclerosing cholangitis cases are associated with inflammatory bowel disease [4]. The absence of high IgG4 serum levels, pancreatobiliary histology and exclusion of extrapancreatic manifestation ruled out type 1 AIP. Ultimately, secondary cholangitis was also eliminated: HIV testing was negative, and abdominal ultrasound and CT scan eliminated extra- or intrahepatic lithiasis as well as hepatic artery thrombosis. We therefore concluded that this was a case of an autoimmune cholangiopathy related to type 2 AIP.
As the pancreatobiliary attempt worsened under the steroid course, we started a second-line treatment with azathioprine, with an effective outcome.
Statement of Ethics
This case report was conducted ethically in accordance with the World Medical Association Declaration of Helsinki. The patient gave written informed consent to publish his case (including publication of images).
Disclosure Statement
The authors have no affiliation with any organization with a direct or indirect financial interest in the subject matter discussed in the manuscript. This manuscript has not been submitted to, nor is under review at, another journal or other publishing venue.
Author Contributions
All authors participated in drafting the article and revising it critically for important intellectual content, and all authors approved the final version.
References
- 1.Shimosegawa T, Chari ST, Frulloni L, Kamisawa T, Kawa S, Mino-Kenudson M, et al. International Association of Pancreatology International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas. 2011 Apr;40((3)):352–8. doi: 10.1097/MPA.0b013e3182142fd2. [DOI] [PubMed] [Google Scholar]
- 2.Okazaki K, Chari ST, Frulloni L, Lerch MM, Kamisawa T, Kawa S, et al. International consensus for the treatment of autoimmune pancreatitis. Pancreatology. 2017 Jan-Feb;17((1)):1–6. doi: 10.1016/j.pan.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Sureka B, Rastogi A. Autoimmune Pancreatitis. Pol J Radiol. 2017 Feb;82:233–9. doi: 10.12659/PJR.900899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorenzo D, Maire F, Stefanescu C, Gornet JM, Seksik P, Serrero M, et al. Features of Autoimmune Pancreatitis Associated with Inflammatory Bowel Diseases. Clin Gastroenterol Hepatol. 2017 doi: 10.1016/j.cgh.2017.07.033. [DOI] [PubMed] [Google Scholar]
- 5.Geary K, Yazici C, Seibold A, Guzman G. IgG4-Related Cholangiopathy and Its Mimickers: A Case Report and Review Highlighting the Importance of Early Diagnosis. Int J Surg Pathol. 2018 Apr;26((2)):165–73. doi: 10.1177/1066896917730902. [DOI] [PubMed] [Google Scholar]
- 6.Sah RP, Chari ST. Autoimmune pancreatitis: an update on classification, diagnosis, natural history and management. Curr Gastroenterol Rep. 2012 Apr;14((2)):95–105. doi: 10.1007/s11894-012-0246-8. [DOI] [PubMed] [Google Scholar]
- 7.Kamisawa T, Chari ST, Lerch MM, Kim MH, Gress TM, Shimosegawa T. Recent advances in autoimmune pancreatitis: type 1 and type 2. Gut. 2013 Sep;62((9)):1373–80. doi: 10.1136/gutjnl-2012-304224. [DOI] [PubMed] [Google Scholar]
- 8.Hart PA, Zen Y, Chari ST. Recent Advances in Autoimmune Pancreatitis. Gastroenterology. 2015 Jul;149((1)):39–51. doi: 10.1053/j.gastro.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 9.Takuma K, Kamisawa T, Igarashi Y. Autoimmune pancreatitis and IgG4-related sclerosing cholangitis. Curr Opin Rheumatol. 2011 Jan;23((1)):80–7. doi: 10.1097/BOR.0b013e3283412f60. [DOI] [PubMed] [Google Scholar]
- 10.Kawa S, Hamano H. Clinical features of autoimmune pancreatitis. J Gastroenterol. 2007 May;42((S18 Suppl 18)):9–14. doi: 10.1007/s00535-007-2044-x. [DOI] [PubMed] [Google Scholar]
- 11.Nakazawa T, Ohara H, Sano H, Ando T, Aoki S, Kobayashi S, et al. Clinical differences between primary sclerosing cholangitis and sclerosing cholangitis with autoimmune pancreatitis. Pancreas. 2005 Jan;30((1)):20–5. [PubMed] [Google Scholar]