

Abstract
The evolutionarily conserved Escherichia coli translesion DNA polymerase IV (DinB) is one of three enzymes that can bypass potentially deadly DNA lesions on the template strand during DNA replication. Remarkably, however, DinB is the only known translesion DNA polymerase active in RecA-mediated strand exchange during error-prone double-strand break repair. In this process, a single-stranded DNA (ssDNA)–RecA nucleoprotein filament invades homologous dsDNA, pairing the ssDNA with the complementary strand in the dsDNA. When exchange reaches the 3′ end of the ssDNA, a DNA polymerase can add nucleotides onto the end, using one strand of dsDNA as a template and displacing the other. It is unknown what makes DinB uniquely capable of participating in this reaction. To explore this topic, we performed molecular modeling of DinB's interactions with the RecA filament during strand exchange, identifying key contacts made with residues in the DinB fingers domain. These residues are highly conserved in DinB, but not in other translesion DNA polymerases. Using a novel FRET-based assay, we found that DinB variants with mutations in these conserved residues are less effective at stabilizing RecA-mediated strand exchange than native DinB. Furthermore, these variants are specifically deficient in strand displacement in the absence of RecA filament. We propose that the amino acid patch of highly conserved residues in DinB-like proteins provides a mechanistic explanation for DinB's function in strand exchange and improves our understanding of recombination by providing evidence that RecA plays a role in facilitating DinB's activity during strand exchange.
Keywords: DNA polymerase, DNA synthesis, DNA damage, DNA repair, homologous recombination, DinB, DNA polymerase IV, RecA
Introduction
Bacterial genomes accumulate lesions due to a variety of endogenous and exogenous DNA-damaging agents, which result in replication fork stalling and ultimately mutagenesis (1, 2). Genomic stability is critical for cell survival and bacteria use high-fidelity repair pathways, such as homologous recombination (3, 4), to ensure this stability.
Homologous recombination's main function is to repair double-strand breaks (DSBs)4 resulting from DNA damage and replication fork stalling (homologous recombination reviewed in Ref. 4). In this pathway, the RecBCD complex binds both ends of a DSB and degrades the double-stranded DNA (dsDNA) until it encounters a Chi site (4–6), a hotspot for homologous recombination that consists of an 8-bp sequence repeated across the genome (7, 8). Upon encountering a Chi site, the RecBCD complex changes function and begins to degrade only one strand of DNA in the 5′–to–3′ direction, creating a long 3′ overhang (4, 5, 9), which is in turn coated by RecA giving rise to the RecA nucleoprotein filament (10, 11). This filament performs the search for homologous dsDNA (4). The next step is formation of a displacement loop (D-loop), in which single-stranded DNA (ssDNA) in the RecA nucleoprotein filament pairs with homologous dsDNA. In the D-loop the ssDNA displaces a strand of the dsDNA, known as the displaced strand. The stability of the D-loop depends on homology, which provides the system with the potential for reversibility in the case of a low-homology match (12–15). When a D-loop with enough homology is formed, a DNA polymerase binds and extends the D-loop using the ssDNA as primer and the complementary strand as template (4). The strand-exchange products remain reversible until there is no longer ssDNA available (12–14, 16–18), but the probability of reversal is potentially lowered with each nucleotide added.
High-fidelity pathways are overwhelmed when DNA damage is severe, and replicative DNA polymerases encounter lesions in the template DNA, which causes potentially lethal replication fork stalling (1, 19). Under these conditions, cells resort to low fidelity DNA damage tolerance pathways (e.g. translesion synthesis) to survive. Translesion synthesis DNA polymerases synthesize across from lesions on the template strand that would otherwise stall replicative DNA polymerases (e.g. DNA polymerase III) and cause cell death. DNA polymerase IV (DinB), a highly conserved translesion DNA polymerase, bypasses N2-deoxyguanosine adducts (20–23) and alkylation lesions (20, 24–26). DinB's active site, which is wider than that of higher-fidelity DNA polymerases, allows it to accommodate and bypass these lesions (27, 28). Conversely, DinB's active site does not allow for the same geometric error checking that is observed in high-fidelity DNA polymerases (27, 28), which causes DinB to misincorporate nucleotides and create −1 frameshift mutations (29, 30).
Stressful environmental conditions can trigger a switch from high-fidelity DSB repair, which uses DNA polymerase III to extend the D-loop, to error-prone DSB repair, which uses DinB (31–34). Interestingly, the two other highly-conserved translesion DNA polymerases expressed in Escherichia coli, DNA polymerases II and V, do not participate in RecA-mediated strand exchange during error-prone double-strand break repair (35). In fact, these polymerases have been found to inhibit D-loop extension in vivo and in vitro (33, 36–39). Although the mechanistic details of DinB's role in RecA-mediated strand exchange are unknown, previous work suggests that extension of the initiating strand by DinB stabilizes D-loops and that DinB's unusually high intracellular concentration (2500 nm under DNA damaging conditions (29)) helps it to access the site of strand exchange (35). This is consistent with new work indicating that most DinB molecules carry out DNA synthesis outside replisomes (35, 40).
To gain insights into the mechanism of DinB synthesis during strand exchange, here we identify a patch of DinB residues located in the DinB fingers domain that are predominantly conserved in DinB-like proteins. We performed structural modeling, which predicts that these residues are likely positioned between the dsDNA template and displaced strands following D-loop formation, possibly contributing to or stabilizing displacement of the displaced strand.
Among these conserved residues are DinB cysteine 66 (Cys-66) and arginine 38 (Arg-38). We have previously shown that Cys-66 is important for DinB's interaction with RecA (25, 26). Structural modeling predicts that Cys-66 is located near the displaced strand, whereas Arg-38 is near the template strand during strand exchange. We report here that both Cys-66 and Arg-38 are important for DinB to perform strand displacement. The evidence shown in this report provides both mechanistic insights into the role of DinB in RecA-mediated strand exchange and a possible explanation for DinB's function as the only known translesion DNA polymerase to promote this process.
Results
Molecular modeling of DinB in RecA-mediated strand exchange indicates the DinB fingers domain separates dsDNA
To further the understanding of how DinB facilitates strand exchange during DSB repair, we performed molecular modeling of DinB interacting with a RecA nucleoprotein filament and dsDNA (Fig. 1A). In this model, the RecA–DNA portion represents the D-loop formation stage of homologous recombination (i.e. when the 3′ end of the incoming nucleoprotein filament anneals to the template strand, thereby separating the dsDNA). Our model of DinB interaction at this stage reveals how DinB likely interacts with the RecA filament and dsDNA before DinB extends the D-loop (Fig. 1A). The model suggests that upon DinB binding, the template and displaced strands of the heteroduplex are separated by DinB's fingers domain (highlighted in yellow, Fig. 1B). Here, we investigate whether this separation of the dsDNA is part of how DinB facilitates strand displacement through DNA synthesis during strand exchange.
Figure 1.
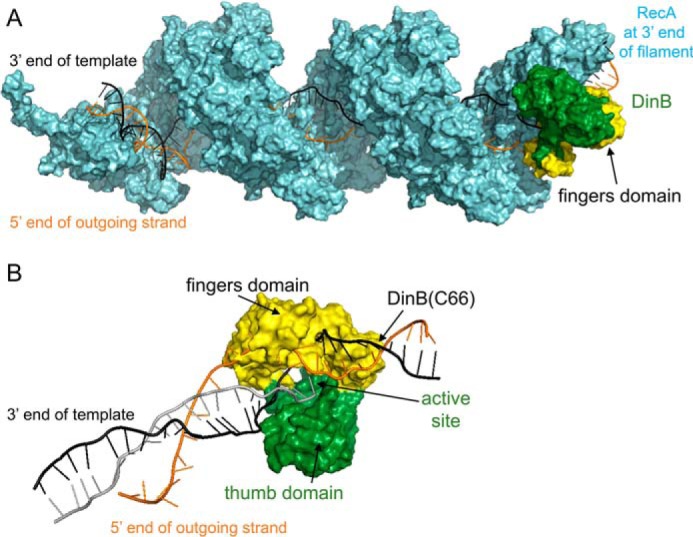
Molecular modeling of DinB in RecA-mediated strand exchange indicates the DinB fingers domain separates dsDNA. A, this model of RecA–DNA–DinB interaction represents the homologous recombination stage where strand exchange has taken place up to the 3′ extremity of the incoming strand. Interactions were modeled using structures and models as follows. The ssDNA (gray, largely obscured by RecA), template strand of dsDNA (black), displaced strand of dsDNA (orange), and RecA (cyan) were modeled using the PDB structure 3CMW (51) that had been extended by three turns (52) in a previous model (53). DinB (green and yellow) was modeled using the PDB structure 4IRC (43). The RecA nucleoprotein filament and DinB are shown interacting with the RecA molecule that lies at the 3′ end of the nucleoprotein filament. DinB and the RecA nucleoprotein filament were surface-rendered with PyMOL (42). The fingers domain of DinB is highlighted in yellow. B, DinB and DNA are shown without RecA to permit visualization of DinB's interaction with three DNA strands during strand exchange. Structures have been turned ∼180° about the horizontal axis with respect to A. DinB's fingers domains (yellow) appears to separate the template (black) and displaced (orange) strands of the dsDNA.
Several residues predominantly conserved in DinB are predicted to be near the template or displaced strands during strand exchange
Because DinB is the only known translesion DNA polymerase that can extend strand-exchange products, we used multiple sequence alignments to investigate the conservation of the DinB fingers domain. Several residues in this area of DinB are highly conserved in DinB-like proteins (magenta and blue residues in Fig. 2A) but are not conserved in translesion DNA polymerases V (Fig. 2B) or II (Fig. 2C). Our structural modeling predicts several of these conserved residues to interact with either the displaced (Val-53, Met-57, Cys-66, and Pro-67) or the template strand (Arg-38 and Ala-62) during RecA-mediated strand exchange (magenta and blue residues in Fig. 3). The notion that these residues, which appear to separate the dsDNA during strand exchange, are only found in DinB-like proteins suggests an explanation for why DinB is the only known translesion DNA polymerase that extends strand-exchange products.
Figure 2.
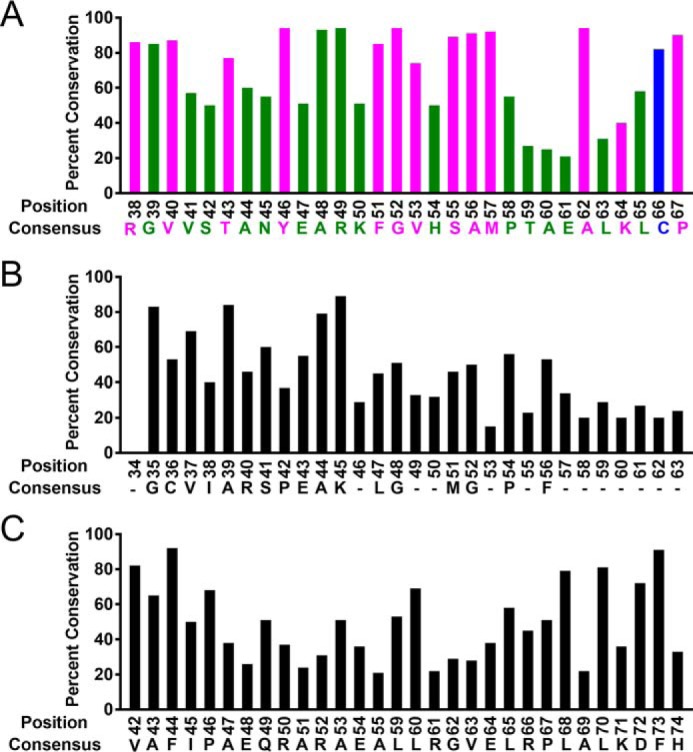
Several residues of the DinB fingers domain are highly conserved only in DinB-like proteins. Multiple sequence alignments show that an amino acid patch, depicted here in magenta and blue (A) located in the fingers domain of DinB, is highly conserved in DinB-like proteins but not in DNA polymerase V (B) or in DNA polymerase II (C) homologs. Percent conservation values for A–C are reported in Table S1.
Figure 3.
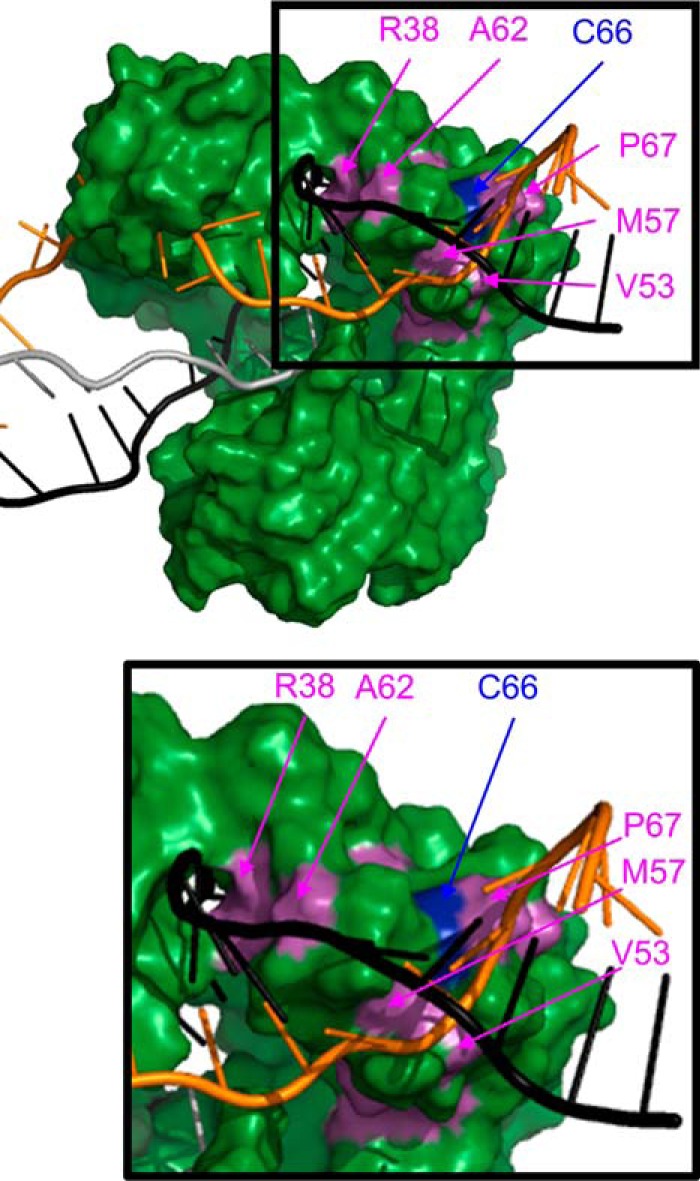
Modeling suggests that several conserved DinB residues are located near the template (black) and displaced (orange) strands during strand exchange. Depiction of DinB from surface-rendered model from Fig. 1 shows that cysteine 66 (blue) and other predominantly conserved residues only in DinB-like proteins (magenta) are located near the template (Arg-38 and Ala-62) and displaced (Val-53, Met-57, Cys-66, and Pro-67) strands of dsDNA during strand exchange. Enlargement of the boxed area is provided to better visualize the positions of the highlighted residues. Residues in green, magenta, and blue color correspond to residues of the same color in Fig. 2.
Notably, the conserved Cys-66 in DinB is predicted to be adjacent to the displaced strand during strand exchange (blue residue in Figs. 2A and 3). We have previously reported that this residue is important for DinB interaction with RecA during translesion DNA synthesis (26). The DinB(C66A) variant maintains comparable DNA synthesis activity to the native enzyme (Fig. S1) (25); however, it demonstrates increased interaction with RecA and decreased fidelity on damaged DNA templates (25, 26). In addition, Cys-66 is hypothesized to be important for the structure of the DinB fingers domain (25). Mutating this residue in silico to a smaller amino acid appears to eliminate an interaction between Cys-66 and Phe-51 in the fingers domain (Fig. S2). This may cause the flexibility previously observed in the DinB(C66A) variant, which does not appear to significantly affect the DNA synthesis activity of the enzyme (Fig. S1) (25).
Because of Cys-66's previously identified importance to the DinB–RecA interaction (25), its predicted proximity to the displaced strand during strand exchange (Fig. 3), and its potential importance for the structural stability of the fingers domain (Fig. S2) (25), we investigated Cys-66's role during RecA-mediated strand exchange.
Residue Cys-66 is important for strand-exchange activity
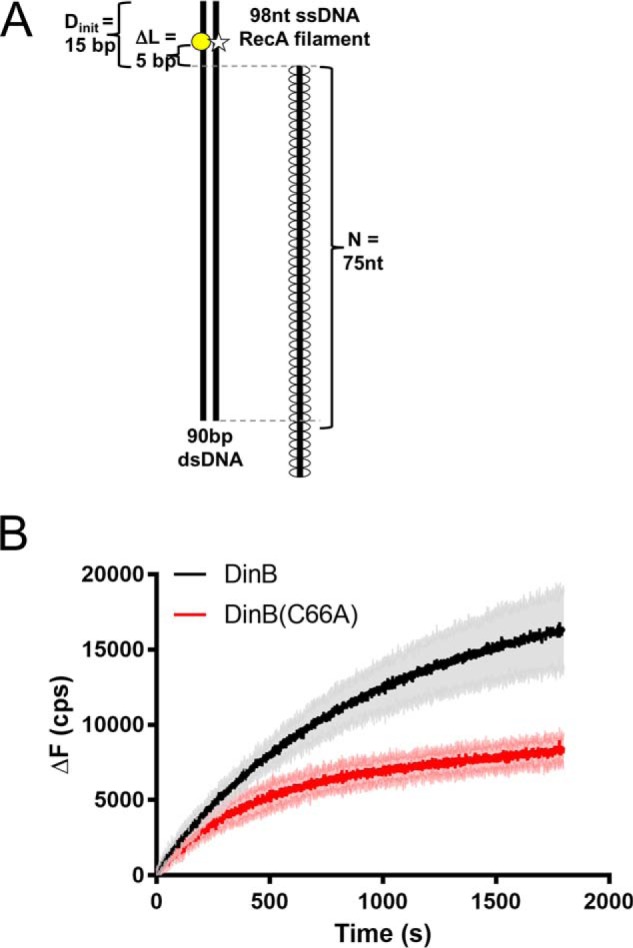
To measure strand exchange, a RecA–ssDNA filament was mixed with labeled dsDNA (Fig. 4A). The number of homologous bases between the 3′ end of the ssDNA filament and the dsDNA was varied from 20 to 75 bp (Fig. 4A). The template strand of dsDNA was labeled with rhodamine (yellow circle, Fig. 4A) at a location 5 bp beyond the end of the region of homology. When annealed to the displaced strand, this rhodamine quenched a fluorescein label located nearby on the displaced strand of the dsDNA (white star, Fig. 4A). As the RecA–ssDNA filament invaded the dsDNA, DinB used the ssDNA as a primer and the complementary strand in the dsDNA as a template for extension. As the template and displaced strands were separated by the polymerase, fluorescence quenching became less efficient, and the fluorescence signal increased. An increase in fluorescence above the baseline indicated the following: 1) that the initial strand-exchange product was stable enough to allow DinB time to bind, and 2) that DinB could synthesize enough nucleotides to disrupt the dsDNA at the location of the fluorophores. In addition, as DinB synthesized beyond five nucleotides, each nucleotide insertion may have acted to stabilize the strand-exchange product, as the region of homology between the ssDNA and the template strand increased.
Figure 4.

Highly conserved fingers domain residue is important for DinB's activity during strand exchange. A, schematic shows experimental setup for RecA-dependent strand-exchange experiments. Fluorescently-labeled dsDNA (left) is mixed with each of a set of ssDNA RecA filaments (right). Each in the set of filaments contains a different length of homology (N) to the labeled dsDNA (signified by gray, green, magenta, and blue lines). In the dsDNA, the template strand is labeled with rhodamine (yellow circle), and the displaced strand is labeled with fluorescein (white star). These fluorophores are located 5 bp away from area of homology between dsDNA and ssDNA RecA filament (ΔL = 5 bp). The proximity of the rhodamine quenches fluorescein fluorescence until template and displaced strands are separated. Fluorescence increases when the ssDNA RecA filament invades the dsDNA, and DinB synthesizes DNA using the ssDNA filament as a primer. Five nucleotide insertions are needed to separate the displaced strand at the location of the fluorescent labels and relieve quenching. B, when only RecA or only DinB is mixed with the highest homology ssDNA RecA filament (N = 75, blue filament in A), the fluorescent labels are not efficiently separated, indicating that the dsDNA is still annealed at the location of the labels. When both proteins are present in the absence of dCTP, dGTP, and dTTP (dATP is present for nucleoprotein filament assembly), baseline fluorescence is observed. A RecA filament with full homology to the dsDNA (N = 90) is used to determine maximum possible fluorescence in the assay. C, DinB stabilizes strand exchange in a homology-dependent manner. As homology increases between the ssDNA RecA filament and the fluorescently-labeled dsDNA, DinB efficiently separates the dsDNA at the location of the fluorescent labels. This indicates that increased homology allows DinB to more efficiently stabilize strand-exchange products. D, DinB(C66A) stabilizes strand-exchange products, but it does so with less efficiency than the native enzyme. N indicates the length of homology between dsDNA and ssDNA filament; ΔL indicates the distance between region of homology on dsDNA and fluorescent label; Dinit indicates the distance between fluorescent label and closet end of dsDNA; ΔF indicates the change in fluorescence measured in counts/s with respect to the fluorescence at 0 s. Experiments were performed in triplicate with similar results. Representative data are shown.
Disruption of dsDNA depended on the presence of RecA as there was little observable change in fluorescence (from time 0; ΔF) when RecA was absent (Fig. 4B, DinB only). We observed a nonzero baseline of fluorescence with only RecA, suggesting that RecA altered the dsDNA at least 5 bp beyond the 3′ end of the filament (Fig. 4B, RecA only N = 75). This may be caused by an alteration in the topology of the DNA that occurs as RecA forms the D-loop. As expected, addition of DinB and no dNTPs did not significantly alter the RecA baseline fluorescence due to the lack of DNA synthesis (Fig. 4B, RecA, DinB, No dNTPs). In addition, a maximum ΔF for this assay was measured using a ssDNA filament that contained a full 90 bp of homology to the dsDNA (Fig. 4B, RecA only N = 90).
RecA, dNTPs, and DinB synthesis disrupted the dsDNA, which in turn relieved quenching and increased fluorescence in experiments with different lengths of homology (see increasing fluorescence in Fig. 4C). The increase in fluorescence due to the presence of DinB depended on the homology between the ssDNA filament and the dsDNA. We note that 75 bp of homology was required to observe a fluorescence increase due to DinB synthesis (compare Fig. 4C to RecA, DinB, no dNTPs control in Fig. 4B). This indicates that under the conditions of the experiment, 75 bp of homology was required for the RecA-mediated strand-exchange product to be stable enough for DinB to alleviate quenching at the fluorophores in the dsDNA. Lower homology (<75 bp) does not allow for DinB binding and synthesis.
Although DinB(C66A) primer extension was comparable with the native enzyme's (Fig. S1) (25, 26), it was not as proficient in the strand-exchange assays (Fig. 4D). Although disruption of dsDNA depended on the length of homology between the ssDNA RecA filament and the dsDNA, synthesis by DinB(C66A) with 75 bp of homology disrupted the dsDNA far less than that of DinB (compare blue lines in Fig. 4, C and D). This is indicative of lower DNA synthesis during strand exchange by DinB(C66A) than the native enzyme.
In these experiments, the initial rate is a measure of the rate at which the enzyme can disrupt dsDNA and cause a change in fluorescence. The maximum ΔF is indicative of the stability of the strand-exchange product at the location of the fluorescent labels, as it is a point of equilibrium between separated and annealed dsDNA.
A direct comparison of the reactions using 75 bp of homology (Fig. 5A) is highlighted in Fig. 5B. In this assay DinB(C66A) had a slightly (∼20%) lower initial rate (ΔF/s) than DinB (Fig. 5B; p < 0.0001; enlargement of Fig. 5B from 0 to 200 s is provided in Fig. S3), indicating that the rate at which the dsDNA was initially separated was approximately only 20% slower than for DinB. This difference is possibly due in part to minor differences in the enzymes' rate of primer extension (Fig. S1). However, the maximum fluorescence reached by DinB(C66A) was ∼50% lower than that reached by DinB (Fig. 5B; p < 0.01). This indicates that the stability of the resulting strand-exchange products was poorer due to less DNA synthesis and likely reversed more often than those created by DinB. These data suggest that although DinB(C66A) initially separated the dsDNA 20% more slowly, its ability for DNA synthesis at some point beyond the fluorophores either slowed down further or stopped prematurely to result in a less stable strand-exchange product. The observed slow down cannot be accounted for simply by the slight differences in the enzyme's primer-extension activity.
Figure 5.
DinB stabilizes strand-exchange products significantly better than DinB(C66A). A, as in Fig. 4, RecA-dependent strand-exchange experiments utilize an ssDNA RecA filament and dsDNA that is fluorescently labeled on either strand to cause quenching. These fluorophores are located 5 bp away from the region of homology between the dsDNA and the ssDNA RecA filament (ΔL = 5 bp). Shown here is the ssDNA RecA filament with the highest homology, 75 nt. Experiments were performed using all four dNTPs. B, shown is the direct comparison between the fluorescence obtained with DinB and DinB(C66A) in experiments with 75 bp of homology between the RecA ssDNA filament and the dsDNA (directly compares blue lines from Fig. 4, C and D). Both DinB and DinB(C66A) stabilize strand-exchange products and separate the dsDNA at the location of the labels, but DinB does this better than the variant. The DinB reaction has a significantly higher initial rate (18.59 ± 0.3395 cps) than the DinB(C66A) reaction (14.34 ± 0.2784 cps, p value < 0.0001). The DinB reaction also reaches a significantly higher maximum ΔF (16,987 ± 2519 cps) than the DinB(C66A) reaction (8893 ± 913; p value < 0.01). N indicates the length of homology between dsDNA and ssDNA filament; ΔL indicates the distance between region of homology on dsDNA and fluorescent label; Dinit indicates the distance between fluorescent label and closet end of dsDNA; ΔF indicates the change in fluorescence measured in counts/s with respect to the fluorescence at 0 s. Experiments were performed in triplicate. Mean ± S.D. (shaded region surrounding curves) is shown.
DinB(C66A) can insert a single nucleotide during strand exchange as well as DinB
To further investigate the mechanisms underlying DinB's strand-exchange activity, we used dsDNA containing a fluorescent label located only a single bp outside of the homologous region (Fig. 6A). In this experiment, DinB must insert only a single nucleotide to separate the rhodamine (on the template strand) from the fluorescein (on the displaced strand) and increase fluorescence. When only a single nucleotide insertion was required (Fig. 6B), DinB(C66A) performed similarly to native DinB (compare Figs. 5B and 6B). The initial rate of the DinB curve was not significantly different from that of DinB(C66A) (enlargement of Fig. 6B from 0 to 50 s is provided in Fig. S3). Notably, the DinB curve did reach a significantly higher maximum ΔF (p < 0.01). This again suggests that although there is little to no difference in the initial rates of dsDNA separation, the final strand-exchange products made by DinB(C66A) are less stable (i.e. they reverse more often) and therefore likely had lower final homology.
Figure 6.

DinB(C66A) can insert a single nucleotide during strand exchange as well as DinB. A, RecA-dependent strand-exchange experiments were performed similarly to Fig. 5, except fluorophores are located 1 bp away from the region of homology between the dsDNA and the ssDNA RecA filament (ΔL = 1 bp). Only a single nucleotide insertion is required to separate fluorophores and relieve quenching. B, similar to Fig. 5, experiments used all four dNTPs. Initial rates of DinB (68.48 ± 3.769 cps) and DinB(C66A) (61.86 ± 2.306) reactions were not significantly different. The DinB reaction reaches a significantly higher maximum fluorescence (17,163 ± 913 cps) than DinB(C66A) (13,859 ± 580 cps; p value < 0.01). Experiments were performed in triplicate. Mean ± S.D. (shaded region surrounding curves) is shown. C, experiments were performed as in B, except that only dATP, the nucleotide required for a single insertion, was included. The DinB reaction has a significantly lower initial rate (56.31 ± 0.8425 cps) than the DinB(C66A) reaction (58.57 ± 0.6877 cps, p value < 0.05). The maximum ΔF values reached by DinB (9756 ± 1267 cps) and DinB(C66A) (11,679 ± 180 cps) were not significantly different. Experiments were performed in triplicate. Mean ± S.D. (shaded region surrounding curves) is shown. N indicates the length of homology between dsDNA and ssDNA filament; ΔL indicates the distance between region of homology on dsDNA and fluorescent label; Dinit indicates the distance between fluorescent label and closest end of dsDNA; ΔF indicates the change in fluorescence measured in counts/s with respect to the fluorescence at 0 s. Experiments were performed in triplicate. Mean ± S.D. (shaded region surrounding curves) is shown.
Although only a single nucleotide insertion is required to observe fluorescence in this assay, the presence of all dNTPs allows for more than one insertion. Each nucleotide insertion, in turn, may increase the stability of the strand-exchange product, thereby preventing a decrease in fluorescence that is caused by the displaced strand reannealing the template. Because initial rates of the curves indicate that DinB and DinB(C66A) insert the first nucleotide at a similar rate, we hypothesized that the difference in maximum ΔF in Fig. 6B was due to difference in DNA synthesis after the first insertion.
To observe only a single nucleotide insertion, the experiment was repeated using only the dNTP required for the first insertion dATP. Interestingly, we find that DinB(C66A) performed the first nucleotide insertion at a slightly higher rate (p < 0.05; Fig. 6C, enlargement of Fig. 6C from 0 to 100 s is provided in Fig. S3) than DinB. This is consistent with previous work, which indicates that DinB(C66A) inserts dATP with greater efficiency than the native enzyme (26). In addition, DinB(C66A) was previously discovered to bind more efficiently to RecA (25), which may facilitate the DinB(C66A) binding to the intricate DNA substrate in this assay. These data suggest that DinB(C66A) can insert the first nucleotide during strand exchange with at least the same efficiency as DinB.
In addition, here we eliminate the potential for differences in DNA synthesis beyond the fluorophores and find no significant difference in the maximum ΔF reached by the two enzymes (Fig. 6C). This supports our hypothesis that the difference in maximum ΔF in previous assays was due to a difference in DNA synthesis beyond the fluorophores.
To rule out previously observed differences between DinB and DinB(C66A) fidelity (26) as the cause of differences in activity, we tested the fidelity of both proteins using each single dNTP under the experimental conditions shown in Fig. 6A. In these experiments, the first nucleotide insertion by DinB should be a dATP across from a dTTP on the template strand. We find that DinB inserted dATP well; the increase in fluorescence for the dATP insertion was comparable when all dNTPs are present (Fig. S4). When an incorrect dNTP for the insertion was provided (Fig. S4), the fluorescence was comparable with the RecA only (no DinB) fluorescence (Fig. S4) and with the RecA only (no DinB, no dNTPs) fluorescence (Fig. S4). These experiments were also performed in conditions requiring five nucleotide insertions (Fig. S5), and comparable results were observed, indicating that differences in fidelity between the enzymes is not detectable in the fluorescence readings of these assays.
These data show that the DinB(C66A) variant can perform the first nucleotide insertion during strand exchange at least as efficiently as the native enzyme.
DinB(C66A) variant is deficient in strand displacement
To understand the difference between DinB(C66A)'s high efficiency in inserting a single nucleotide and low efficiency in separating dsDNA 5 bp beyond the ssDNA filament, we consulted our model of DinB interactions during strand exchange (Fig. 1). Our modeling indicates that upon DinB binding, the template and displaced strands of the heteroduplex are separated by DinB's fingers domain (Figs. 1B and 3). In this manner, 1–2 nucleotides (nt) of the template strand that lie directly beyond the 3′ end of the invading RecA nucleoprotein filament are “free” (Fig. 3) and prevented from annealing to the displaced strand (orange strand in Figs. 1B and 3). This information suggests a possible explanation for the DinB(C66A)'s observed activities. We hypothesized that DinB(C66A) can efficiently synthesize using a free template (Fig. S1) (24, 25); however, it would not be able to separate the displaced strand during strand exchange. To test this, we performed strand displacement experiments in the absence of RecA. In these experiments, DinB must displace the strand that is annealed to the template, without the help of RecA, before it can synthesize DNA.
To our knowledge, DinB's innate ability to perform DNA synthesis with strand displacement in the absence of RecA has not been previously investigated. To do this experiment, an ssDNA template (90-nt bottom black line in Fig. 7A) was annealed to an ssDNA primer (2-nt gray line in Fig. 7A) and a 75-nt ssDNA that mimics a displaced strand (75-nt top black line in Fig. 7A). Only 61 nt of the 75-nt ssDNA were complementary to the template strand. The other noncomplementary 14 nt formed a short flap of unannealed ssDNA on the 5′ end of the 75-nt ssDNA (Fig. 7A). This strand was labeled with fluorescein at the nucleotide directly following the 3′ end of the unannealed flap (Fig. 7A). As primer extension occurs, the displaced strand is separated from the template strand; therefore, fluorescence of the fluorescein label changes as the displaced strand moves away from the template strand.
Figure 7.

DinB(C66A) variant is deficient in RecA-independent strand displacement. A, graphic depiction of the DNA substrate used in these experiments. A 29-nt ssDNA primer (gray line) was annealed to a 90-nt template (bottom black line) as well as to a 75-nt fluorescently-labeled oligonucleotide (top black line with the fluorophore represented by the star). The 75-nt oligonucleotide displaced strand is composed of a 61-nt complementary to the 90-bp template and of a 14-nt unannealed flap located at the 5′ end. The fluorescein label (depicted by the star) on the displaced strand was located on the first nucleotide of the 75-bp complementary region. If displaced and template strands are separated, fluorescence is altered. DinB must insert a single nucleotide onto the end of the primer to displace the labeled nucleotide. B, experiments with all dNTPs added show that DinB stabilizes strand displacement after a short lag (highlighted by the enlargement in C) with greater efficiency than the DinB(C66A) variant. Initial velocities for both proteins from 0 to 50 s are not significantly different from zero. The rate of the DinB reaction from 100 to 200 s (46.40 ± 1.188 cps) is significantly higher than the velocity of the DinB(C66A) reaction at the same time point (11.80 ± 1.140 cps; p value < 0.0001). The DinB reaction also reaches a significantly higher maximum ΔF (28,053 ± 6272 cps) than the DinB(C66A) reaction (9798 ± 1290 cps; p value < 0.01). Maximum ΔF was measured using a measure of the fluorescence caused by the outgoing strand alone at about 37,000 cps. Change in fluorescence was measured in counts/s with respect to the fluorescence at 0 s. Experiments were performed in triplicate. Mean ± S.D. (shaded region surrounding curves) is shown in B. Only the mean is shown in C for better visualization of lag. D, E. coli strain containing a system described by Ponder et al. (31) was used to examine mutagenesis in response to an induced DSB (left). The endonuclease I–SceI created a DSB 11.4 kb on the E. coli chromosome downstream of the lac gene. The lac gene in this strain contains a mutation preventing cell growth when the only carbon source is lactose represented here by lacmut. The Lac− to Lac+ reversion is measured as the number of Lac+ colonies observed when the test strain is grown on lactose-only containing minimal medium divided by the total number of colonies observed when the strain is grown on glucose-only containing minimal medium (right). The (C66A) mutation in DinB (red) significantly reduces the rate of Lac+ reversion in the presence of a DSB. For comparison purposes, we used ΔdinB and dinB+ strains shown in gray and black, respectively. Data shown is mean of 12 replicates ± S.E. * indicates a p value < 0.05 when comparing the dinB+ and dinB(C66A) strains by two-tailed t test.
When the fluorescein is located directly beside the end of the primer (Fig. 7A), DinB must insert only a single nucleotide to separate the fluorescein-containing nucleotide on the displaced strand. In this case, DinB showed a short lag before efficiently separating the displaced strand (black line in Fig. 7B, an enlargement is provided in Fig. 7C to highlight the lag). During the first 50 s, there was no significant fluorescence increase. This lag likely coincides with DinB binding to the DNA before DNA synthesis. Notably, this lag was absent in all RecA-dependent experiments, possibly because the RecA–DinB interaction facilitates DinB binding to the intricate DNA template.
DinB(C66A) demonstrated a similar lag as DinB (Fig. 7B, enlargement provided in Fig. 7C; the rate from 0 to 50 s is not significantly different from zero). Although DinB(C66A) has shown similar activity to native DinB in primer-extension assays (25, 26), DinB(C66A) was significantly less proficient at DNA synthesis during strand displacement. The rate from 100 to 200 s was about 75% lower than the same interval in DinB's reaction (p < 0.0001), and the DinB(C66A) reaction reached a maximum ΔF that is about 65% lower than DinB (p < 0.05).
Together, these data indicate that DinB can synthesize from a primer in templates with a topology that mimics strand displacement in recombination intermediates. Remarkably, this ability is independent of RecA. In addition, residue Cys-66, which is highly conserved in only DinB-like proteins, is critical to DinB's ability to perform strand displacement.
DinB(C66A)-containing strain induces significantly less mutagenesis during DSB repair
DinB activity during error-prone DSB repair has been shown to cause mutagenesis (31–33, 35). Error-prone DSB repair-induced mutagenesis is significantly reduced in a ΔdinB strain (31). Ponder et al. (31) have previously described a system with which to assess this DinB-dependent mutagenesis. A chromosomal reporter was constructed that encodes an arabinose-inducible I–SceI endonuclease (31), which creates a DSB at an 18-bp I–SceI recognition site located ∼11.4 kb downstream from a lac gene containing the LacI33 allele. Cells with this allele are Lac− (i.e. unable to use lactose as a sole carbon source) because of a +1 frameshift in the lac gene (31). The reversion from Lac− to Lac+ is measured by counting Lac+ colonies on lactose-containing minimal medium (31). Ponder et al. (31) showed that deletion of the dinB gene significantly reduced the number of Lac+ reversions. Here, we confirm these results and find that the test E. coli strain with a chromosomal dinB(C66A) allele at the dinB locus produces significantly fewer Lac+ revertants than the dinB+ strain (Fig. 7D; 5–10-fold decrease, p < 0.05 at all time points). These data support our model that DinB(C66A) is not as proficient as DinB during error-prone DSB repair in vivo. This experiment also provides a proof of principle that the obtained in vitro data correlates well with the in vivo behavior of the enzyme.
Template-binding DinB residue Arg-38 is also important for strand displacement
To further our understanding of the role of the fingers domain in DinB's synthesis strand exchange, we investigated another of the highly conserved residues that we identified by modeling. Like Cys-66, Arg-38 is predominantly conserved in DinB-like proteins. Although our structural modeling predicts Cys-66 to be located near the displaced strand in strand exchange, Arg-38 is predicted to be located near the template strand. Arginine's positive charge makes it an excellent candidate for interaction with the negatively charged DNA strand. To test the importance of this residue to DinB's strand-exchange activity, we constructed a variant in which only this residue was mutated to an uncharged alanine (DinB(R38A)). We found that the DNA synthesis activity using an undamaged DNA template is comparable with that of the native enzyme (about 20% lower than native DinB activity) (Fig. S1). Like DinB(C66A), the DinB(R38A) derivative stabilized strand-exchange products but did so with reduced efficiency compared with native DinB (compare Figs. 8A with 4C).
Figure 8.

DinB(R38A) variant is deficient in strand displacement. A, reactions performed as in Fig. 4. DinB(R38A) stabilizes strand-exchange products, but it does so with less efficiency than the native enzyme. N indicates is the length of homology between dsDNA and ssDNA filament. Experiments were performed in triplicate. Mean is shown. B, reactions were performed as in Fig. 6A with all four dNTPs. DinB has a significantly higher initial rate (68.48 ± 3.769 cps) than DinB(R38A) (55.85 ± 1.855 cps; p value < 0.0001) in this assay. The DinB reaction reaches a significantly higher maximum fluorescence (17,163 ± 913 cps) than DinB(R38A) (9949 ± 1742 cps; p value < 0.01). Experiments were performed in triplicate. Mean ± S.D. (shaded region surrounding curves) is shown. C, experiments in which only a single insertion is required were performed as in Fig. 6A using only dATP, the nucleotide required for the single insertion. DinB had a significantly higher initial rate (56.31 ± 0.8425 cps) than DinB(R38A) (40.88 ± 1.561 cps; p value < 0.0001) in this assay. The maximum ΔF values of DinB (9756 ± 1267 cps) and DinB(R38A) (7539 ± 1470 cps) were not significantly different. Experiments were performed in triplicate. Mean ± S.D. (shaded region surrounding curves) is shown. ΔF indicates the change in fluorescence measured in counts/s with respect to the fluorescence at 0 s. D, ability of the enzyme for strand displacement was tested as in Fig. 7A with all four dNTPs. DinB(R38A) has little to no activity in this assay. Maximum ΔF was measured using a measure of the fluorescence caused by the outgoing strand alone as about 37,000 cps. Experiments were performed in triplicate. Mean ± S.D. (shaded region surrounding curves) is shown.
When only one nucleotide insertion was required to separate fluorophores and all dNTPs were provided (as in Fig. 6B), DinB(R38A) had an 18% lower initial rate and a 40% lower maximum ΔF than native DinB (Fig. 8B; p < 0.0001 and p < 0.01).
When only the dATP was provided, the difference in the initial rate between the two enzymes remained similar (about 25% Fig. 8C, p < 0.0001), whereas the maximum ΔF values were not significantly different (Fig. 8C). These data suggest that Arg-38 is important for DNA synthesis during strand exchange. Again, we hypothesized that although DinB(R38A) can insert nucleotides across from a free template, it cannot efficiently separate a displaced strand to insert nucleotides.
Notably, we find that DinB(R38A) was fully deficient in strand displacement activity in the absence of RecA (Fig. 8B; experimental setup shown in Fig. 7A). Because our modeling (Fig. 3) and the crystal structure of DinB during primer extension (43) indicate that this residue is located near the template strand during strand exchange and primer extension, respectively, it is likely that DinB(Arg-38) aids strand displacement by stabilizing the template strand during DNA synthesis.
In addition, these data indicate an additional active role for RecA in strand exchange beyond simply forming a D-loop, because DinB(R38A) can perform some DNA synthesis during RecA-dependent strand exchange, but it is not able to perform any detectable DNA synthesis during RecA-independent strand displacement.
Discussion
Stressful environmental conditions can trigger a switch from high-fidelity DSB repair, which uses DNA polymerase III to extend the D-loop, to error-prone DSB repair, which uses DinB for this function (31–34). DinB is the only of three E. coli translesion DNA polymerases known to promote RecA-mediated DSB repair (35). In fact, the other E. coli translesion DNA polymerases II and V actually inhibit this process (33, 36–39). In addition, DinB significantly improves the efficiency of recombination events during transduction, a horizontal gene transfer process, whereas translesion DNA polymerase V appears to inhibit these events (25).
Our model of DinB's interaction with DNA during RecA-mediated strand exchange predicts that the DinB fingers domain lies directly between the template and displaced strands during strand exchange. Interestingly, several residues in this domain are highly conserved in DinB, but not in the translesion DNA polymerases that inhibit strand exchange (Fig. 2). Notably, our molecular modeling indicates that several of the conserved residues in the fingers domain of DinB are located near the template strand (Arg-38 and Ala-62, Fig. 3) or the displaced strand (Val-53, Met-57, Cys-66, and Pro-67, Fig. 3) during strand exchange. The conservation and location of these residues suggests a structural explanation for DinB to be the only E. coli translesion DNA polymerase with a known role in RecA-mediated strand exchange.
Our group previously showed the Cys-66 residue is highly conserved among DinB-like proteins (25). We also showed that it is important to the RecA-dependent increase in fidelity during translesion synthesis (25, 26), and in DinB's ability to promote recombination during transduction. Furthermore, we show here that Cys-66 is central to DinB's ability to perform strand displacement during RecA-mediated strand exchange. Notably, we have previously predicted that DinB was likely to interact with the RecA nucleoprotein filament (44), and not just with RecA alone, and here we showed that to be the case.
We have found that the DinB(C66A) variant is more flexible in the fingers domain than the native enzyme; however, it does not appear to be degraded in vivo, and the catalytic core of the enzyme is not significantly affected (25). Analysis of DinB's crystal structure (43) reveals that Cys-66 appears to interact with Phe-51 (Fig. S2). The orientation of the atoms of Cys-66 in relation to the benzene ring of Phe-51 suggests that these two residues may form an aromatic thiol π-type hydrogen bond (45), which would be disrupted in the DinB(C66A) variant. The loss of this bonding may explain the structural flexibility that was previously observed (25). It is likely that Cys-66 is important to DinB's ability to perform strand displacement, because of its stabilization of the structure of the fingers domain and not just through direct interaction between Cys-66 and dsDNA. The importance of Cys-66 in the structure of the fingers domain may also explain its importance to the many DinB functions outlined above.
The crystal structures of DinB bound to a primer and an undamaged (43) or lesion-containing (46) template indicate that Arg-38 interacts with the template strand. Our structural model predicts Arg-38 to interact with the template strand during strand exchange as well (Fig. 3). In addition, a previous study characterizing the DinB(R38A) variant determined that Arg-38 is responsible for suppressing misincorporations during bypass of N2-deoxyguanosine adducts (47). Given the complete lack of DinB(R38A) strand displacement activity in vitro, it is probable that this highly conserved residue is involved in template DNA binding during strand displacement.
We observed here that native DinB experiences a significant lag before starting DNA synthesis during strand displacement in the absence of RecA (Fig. 7C). Presumably, this lag is caused by a lower affinity of DinB for this intricate DNA substrate. Interestingly, this lag is absent when RecA is available to form the D-loop before synthesis (Fig. 6B). Additionally, the DinB(R38A) mutant can perform RecA-dependent strand exchange, although it is completely devoid of the strand displacement function in the absence of RecA. These data suggest that RecA facilitates DinB's activity during strand exchange. One possible mechanism for this would be that RecA recruits DinB to the site of strand exchange by the protein–protein interaction that we have previously observed (25, 26, 44). Here, we observe that RecA affects the annealing of dsDNA at a location that is 5 bp beyond where the 3′ end of the nucleoprotein filament binds (Fig. 4B). This suggests that RecA alters the topology of the dsDNA once the D-loop has formed, which may facilitate DinB's binding to this intricate DNA substrate and the progression of strand displacement required for synthesis.
Our observations provide mechanistic insights that support DinB's hypothesized role as the DNA polymerase that initially performs primer extension at the D-loop during certain replication fork stalling events (35). The Cys-66, Arg-38, and likely the surrounding amino acid patch that are predominantly conserved (Fig. 2) in DinB and DinB-like proteins are important for strand displacement (Fig. 7). Taken together, the data outlined here suggest a likely role for this conserved amino acid patch in strand displacement by separating the template and displaced strands of dsDNA.
Experimental procedures
Protein purification
DinB was purified from both the TMCΔT strain (BL21-AI ΔdinB, ΔumuDC, ΔrecA (48)) by ion-exchange chromatography and hydrophobic interaction chromatography as described previously (25, 48–50). DinB(C66A) was purified from the TMCΔD strain (BL21-AI ΔdinB, ΔumuDC (26)) using an identical methodology.
Strand-exchange assay
RecA nucleoprotein filament was formed by adding ssDNA (0.06 μm; Tables 1 and 2), RecA (2 μm; New England Biolabs, Ipswich, MA), dATP (1 mm), pyruvate kinase (10 units/ml), phosphoenolpyruvate (3 mm), and single-stranded binding protein (0.2 μm, Epicenter, Madison, WI) in RecA buffer (70 mm Tris-HCl, 10 mm MgCl2, and 5 mm DTT, pH 7.6) at 37 °C for 10 min.
Table 1.
Oligonucleotides used in RecA-dependent strand-exchange assays in Figs. 4 and 5
F* indicates fluorescein-labeled nucleotide; Rho* indicates rhodamine-labeled nucleotide. All oligonucleotides purchased from Integrated DNA Technologies (Coralville, IA).

Table 2.
Oligonucleotides used in RecA-dependent strand-exchange assays in Fig. 6
F* indicates fluorescein-labeled nucleotide; Rho* indicates rhodamine-labeled nucleotide. All oligonucleotides purchased from Integrated DNA Technologies (Coralville, IA).

The dsDNA constructs (Tables 1 and 2) were prepared by annealing the complementary oligonucleotides at temperatures from 90 to 40 °C with 1 °C steps equilibrated for 1 min in a thermocycler.
For the strand-exchange reactions, a 0.06 μm 98-nt ssDNA/RecA filament was mixed with 0.06 μm rhodamine-fluorescein–labeled dsDNA and E. coli DinB. The mixture was rapidly transferred to a quartz cuvette. For DinB measurements, the RecA buffer contained 0.1 mg/ml BSA, 2 mm dATP, and 1.0 mm dNTPs (or 1 mm of each required dNTP). The final polymerase concentration was 100 nm in all experiments.
FRET experiments were performed with a Fluoromax 4C spectrofluorometer (Horiba, Edison, NJ) by following the emission of the fluorescein label at 518 nm every 1 s for 30 min and 493-nm excitation. The integration was 0.5 s and the bandwidth 2 nm. Samples were kept at 37 °C.
Molecular modeling
The DinB–RecA–DNA complex was assembled as follows. The RecA–DNA part represents the homologous recombination stage where strand exchange has taken place up to the 3′ extremity of the incoming strand. The heteroduplex product of strand exchange is bound to the filament DNA-binding site I, and the displaced strand is in the filament-binding site II. The DNA in site I was directly taken from the crystal structure with PDB code 3CMW (51), which has been extended to three filament turns using the PTools library (52). The structure of the displaced strand was taken from former modeling work where we simulated the early stage of strand exchange (53). The geometry of the junction between the heteroduplex 3′ extremity and the double-stranded B-form DNA tail (D-loop junction), formed by the complementary strand and the displaced strand, was also taken from that former work, such as it resulted from spontaneous strand exchange. The B-form tail interacts with the filament groove, notably with the C-terminal domain of the RecA monomer at the 3′ extremity of the filament. The three-turn filament with three DNA strands included was stable during the 100-ns molecular dynamics simulation in solvated environment at 300 K, performed with NAMD 2.10 (54) and the CHARMM 27 force field (55). The geometry of DinB in complex with the RecA filament was obtained by superposing the terminal 4 bp of the DNA in the crystal structure of the DinB–DNA complex with PDB code 4IRC (43) on the four terminal heteroduplex bp. Despite the heteroduplex being strongly unwound and stretched with respect to B-form DNA, the superposition was made possible by the fact that the heteroduplex is structured as a succession of bp triplets in the B-form, separated by intercalation sites that concentrate the stretching/unwinding deformations. Steric overlaps that resulted from the brute superposition of DinB–DNA on the heteroduplex, notably between DinB and the displaced strand, the DinB thumb and a region of the last RecA monomer close to loop L1, the little finger and the L2 loops of the last two RecA monomers, were removed using interactive and flexible simulations with the BioSpring device and associated software (56). These simulations did not use any knowledge-based restraint. When adjusting the complementary and outgoing strand regions situated 3′ of the D-loop junction, we were careful to minimally displace the strands from their initial relaxed position in the RecA filament. Adaptation of the complementary strand to the presence of DinB necessitated its backbone displacement over a 6-base region, thus locally following the direction taken by the template strand in the DinB complex; this displacement was compatible with conserving most of the Watson-Crick interactions with the outgoing strand, which itself varied little from its geometry in the DinB-free RecA filament.
RecA-independent strand displacement synthesis
Experiments were performed in RecA buffer at 37 °C. A solution containing DNA template (200 nm), dNTPs (1 mm), BSA (0.1 mg/ml), and E. coli DinB or DinB(C66A) (100 nm) was transferred to a quartz cuvette. Measurements of fluorescein emission at 37 °C were performed at 518 nm (excitation at 493 nm). Oligonucleotides used in these experiments are described in Table 3.
Table 3.
Oligonucleotides used in RecA-independent strand displacement assays in Fig. 7
F* indicates fluorescein-labeled nucleotide; Rho* indicates rhodamine-labeled nucleotide. All oligonucleotides purchased from Integrated DNA Technologies (Coralville, IA).

Multiple sequence alignments
Multiple sequence alignments (MSAs) of DinB, DNA polymerase V, and DNA polymerase II sequences were performed using CLC Bio (Qiagen, Redwood City, CA). DinB and DNA polymerase V sequences were obtained from NCBI and hand curated using previously described criteria (25). The DinB MSA utilized 472 sequences from over 100 bacterial species; the DNA polymerase V MSA utilized 2694 sequences from over 600 bacterial species. The DNA polymerase II sequences were obtained from NCBI and hand curated to the following criteria: were annotated as a DNA polymerase II and were at least 600 amino acids long and were shorter than 900 amino acids long. The DNA polymerase II MSA utilized 124 sequences from over 100 bacterial species.
In vivo DSB-induced mutagenesis assay
The DSB-induced mutagenesis assay was performed as described previously (31). The dinB(C66A) allele was mobilized by P1 transduction (41) from MG1655 dinB(C66A) (25) into the assay strain (31). Strains to be tested were streaked from frozen stock on M9 medium supplemented with vitamin B1 with glucose as the sole carbon source and grown for 2 days at 37 °C. Twelve single colonies per strain were inoculated into M9 liquid medium supplemented with vitamin B1 and with glucose as the sole carbon source and grown for 48 h at 37 °C until saturation. Cultures were then deposited on the surface of M9 medium plates supplemented as above but with lactose as the sole carbon source to measure Lac+ revertant colonies. Viable cell measurements were performed on the same kind of M9 medium plates with glucose as the sole carbon source. Revertant colony counts were performed 3–5 days after plating.
Author contributions
T. F. T., C. D., M. P., and V. G. G. conceptualization; T. F. T. formal analysis; T. F. T., C. P., and V. G. G. funding acquisition; T. F. T., C. D., A.-E. M., B. H. N., and C. P. investigation; T. F. T., A.-E. M., and C. P. visualization; T. F. T., C. D., A.-E. M., C. P., M. P., and V. G. G. methodology; T. F. T. writing-original draft; T. F. T., C. D., B. H. N., C. P., M. P., and V. G. G. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Susan Rosenberg from Baylor College of Medicine for the generous gift of the strains required for the in vivo DSB-induced mutagenesis assay. T. F. T. and V. G. G. thank the members of the Godoy and Chai laboratories for their helpful conversations. Simulations were performed using HPC resources from GENCI-CINES Grant 2017-A0040707438 (to C. P.).
This work was supported in part by National Institutes of Health NIGMS Grant RO1GM088230 (to V. G. G.) and Northeastern University. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5, Table S1, and supporting Refs. 1–4.
- DSB
- double-strand break
- DinB
- DNA polymerase IV
- D-loop
- displacement loop
- ssDNA
- single-stranded DNA
- Cys-66
- DinB cysteine 66
- Arg-38
- DinB arginine 38
- DinB(C66A)
- DinB variant with single amino acid mutation cysteine 66 to alanine
- DinB(R38A)
- DinB variant with single amino acid mutation arginine 38 to alanine
- nt
- nucleotide
- MSA
- multiple sequence alignment
- PDB
- Protein Data Bank.
References
- 1. Friedberg E. C., Walker G. C., Siede W., Wood R. D., Schultz R. A., and Ellenburger T. (2006) DNA Repair and Mutagenesis, ASM Press, Washington, D. C. [Google Scholar]
- 2. Sutton M. D., Smith B. T., Godoy V. G., and Walker G. C. (2000) The SOS response: recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu. Rev. Genet. 34, 479–497 10.1146/annurev.genet.34.1.479 [DOI] [PubMed] [Google Scholar]
- 3. Cox M. M. (2007) in Topics in Current Genetics. Molecular Genetics of Recombination. (Aguilera A., and Rothstein R., eds) pp. 53–94, Springer-Verlag, Berlin, Heidelberg, Germany [Google Scholar]
- 4. Kowalczykowski S. C. (2015) An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 7, a016410 10.1101/cshperspect.a016410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith G. R. (2012) How RecBCD enzyme and chi promote DNA break repair and recombination: a molecular biologist's view. Microbiol. Mol. Biol. Rev. 76, 217–228 10.1128/MMBR.05026-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dillingham M. S., and Kowalczykowski S. C. (2008) RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. 72, 642–671 10.1128/MMBR.00020-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lam S. T., Stahl M. M., McMilin K. D., and Stahl F. W. (1974) Rec mediated recombinational hot spot activity in bacteriophage λ. II. A mutation which causes hot spot activity. Genetics 77, 425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malone R. E., Chattoraj D. K., Faulds D. H., Stahl M. M., and Stahl F. W. (1978) Hotspots for generalized recombination in the Escherichia coli chromosome. J. Mol. Biol. 121, 473–491 10.1016/0022-2836(78)90395-9 [DOI] [PubMed] [Google Scholar]
- 9. Anderson D. G., and Kowalczykowski S. C. (1997) The recombination hot spot χ is a regulatory element that switches the polarity of DNA degradation by the RecBCD enzyme. Genes Dev. 11, 571–581 10.1101/gad.11.5.571 [DOI] [PubMed] [Google Scholar]
- 10. Register J. C. 3rd., and Griffith J. (1985) The direction of RecA protein assembly onto single strand DNA is the same as the direction of strand assimilation during strand exchange. J. Biol. Chem. 260, 12308–12312 [PubMed] [Google Scholar]
- 11. Anderson D. G., and Kowalczykowski S. C. (1997) The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a χ-regulated manner. Cell 90, 77–86 10.1016/S0092-8674(00)80315-3 [DOI] [PubMed] [Google Scholar]
- 12. Lee J. Y., Qi Z., and Greene E. C. (2016) ATP hydrolysis promotes duplex DNA release by the RecA presynaptic complex. J. Biol. Chem. 291, 22218–22230 10.1074/jbc.M116.740563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsieh P., Camerini-Otero C. S., and Camerini-Otero R. D. (1992) The synapsis event in the homologous pairing of DNAs: RecA recognizes and pairs less than one helical repeat of DNA. Proc. Natl. Acad. Sci. U.S.A. 89, 6492–6496 10.1073/pnas.89.14.6492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Danilowicz C., Yang D., Kelley C., Prévost C., and Prentiss M. (2015) The poor homology stringency in the heteroduplex allows strand exchange to incorporate desirable mismatches without sacrificing recognition in vivo. Nucleic Acids Res. 43, 6473–6485 10.1093/nar/gkv610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bazemore L. R., Folta-Stogniew E., Takahashi M., and Radding C. M. (1997) RecA tests homology at both pairing and strand exchange. Proc. Natl. Acad. Sci. U.S.A. 94, 11863–11868 10.1073/pnas.94.22.11863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Greene E. C. (2016) DNA sequence alignment during homologous recombination. J. Biol. Chem. 291, 11572–11580 10.1074/jbc.R116.724807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rosselli W., and Stasiak A. (1991) The ATPase activity of RecA is needed to push the DNA strand exchange through heterologous regions. EMBO J. 10, 4391–4396 10.1002/j.1460-2075.1991.tb05017.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu J. (2011) in DNA Recombination, Methods and Protocols (Tsubouchi H., ed), pp. 363–383, Humana Press Inc., Totowa, NJ [Google Scholar]
- 19. Goodman M. F. (2002) Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 71, 17–50 10.1146/annurev.biochem.71.083101.124707 [DOI] [PubMed] [Google Scholar]
- 20. Benson R. W., Norton M. D., Lin I., Du Comb W. S., and Godoy V. G. (2011) An active site aromatic triad in Escherichia coli DNA pol IV coordinates cell survival and mutagenesis in different DNA-damaging agents. PLoS ONE 6, e19944 10.1371/journal.pone.0019944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jarosz D. F., Godoy V. G., Delaney J. C., Essigmann J. M., and Walker G. C. (2006) A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature 439, 225–228 10.1038/nature04318 [DOI] [PubMed] [Google Scholar]
- 22. Jarosz D. F., Godoy V. G., and Walker G. C. (2007) Proficient and accurate bypass of persistent DNA lesions by DinB DNA polymerases. Cell Cycle 6, 817–822 10.4161/cc.6.7.4065 [DOI] [PubMed] [Google Scholar]
- 23. Yuan B., Cao H., Jiang Y., Hong H., and Wang Y. (2008) Efficient and accurate bypass of N2-(1-carboxyethyl)-2′-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 8679–8684 10.1073/pnas.0711546105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bjedov I., Dasgupta C. N., Slade D., Le Blastier S., Selva M., and Matic I. (2007) Involvement of Escherichia coli DNA polymerase IV in tolerance of cytotoxic alkylating DNA lesions in vivo. Genetics 176, 1431–1440 10.1534/genetics.107.072405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cafarelli T. M., Rands T. J., Benson R. W., Rudnicki P. A., Lin I., and Godoy V. G. (2013) A single residue unique to dinb-like proteins limits formation of the polymerase IV multiprotein complex in Escherichia coli. J. Bacteriol. 195, 1179–1193 10.1128/JB.01349-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cafarelli T. M., Rands T. J., and Godoy V. G. (2014) The DinB*RecA complex of Escherichia coli mediates an efficient and high-fidelity response to ubiquitous alkylation lesions. Environ. Mol. Mutagen. 55, 92–102 10.1002/em.21826 [DOI] [PubMed] [Google Scholar]
- 27. Friedberg E. C., Fischhaber P. L., and Kisker C. (2001) Error-prone DNA polymerases: novel structures and the benefits of infidelity. Cell 107, 9–12 10.1016/S0092-8674(01)00509-8 [DOI] [PubMed] [Google Scholar]
- 28. Yang W. (2003) Damage repair DNA polymerases Y. Curr. Opin. Struct. Biol. 13, 23–30 10.1016/S0959-440X(02)00003-9 [DOI] [PubMed] [Google Scholar]
- 29. Kim S. R., Matsui K., Yamada M., Gruz P., and Nohmi T. (2001) Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol. Genet. Genomics 266, 207–215 10.1007/s004380100541 [DOI] [PubMed] [Google Scholar]
- 30. Nohmi T. (2006) Environmental stress and lesion-bypass DNA polymerases. Annu. Rev. Microbiol. 60, 231–253 10.1146/annurev.micro.60.080805.142238 [DOI] [PubMed] [Google Scholar]
- 31. Ponder R. G., Fonville N. C., and Rosenberg S. M. (2005) A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol. Cell 19, 791–804 10.1016/j.molcel.2005.07.025 [DOI] [PubMed] [Google Scholar]
- 32. Shee C., Gibson J. L., Darrow M. C., Gonzalez C., and Rosenberg S. M. (2011) Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 108, 13659–13664 10.1073/pnas.1104681108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pomerantz R. T., Kurth I., Goodman M. F., and O'Donnell M. E. (2013) Preferential D-loop extension by a translesion DNA polymerase underlies error-prone recombination. Nat. Struct. Mol. Biol. 20, 748–755 10.1038/nsmb.2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mallik S., Popodi E. M., Hanson A. J., and Foster P. L. (2015) Interactions and localization of Escherichia coli error-prone DNA polymerase IV after DNA damage. J. Bacteriol. 197, 2792–2809 10.1128/JB.00101-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lovett S. T. (2006) Replication arrest-stimulated recombination: dependence on the RecA paralog, RadA/Sms and translesion polymerase, DinB. DNA Repair 5, 1421–1427 10.1016/j.dnarep.2006.06.008 [DOI] [PubMed] [Google Scholar]
- 36. Sommer S., Bailone A., and Devoret R. (1993) The appearance of the UmuD'C protein complex in Escherichia coli switches repair from homologous recombination to SOS mutagenesis. Mol. Microbiol. 10, 963–971 10.1111/j.1365-2958.1993.tb00968.x [DOI] [PubMed] [Google Scholar]
- 37. Boudsocq F., Campbell M., Devoret R., and Bailone A. (1997) Quantitation of the inhibition of Hfr x F-recombination by the mutagenesis complex UmuD'C. J. Mol. Biol. 270, 201–211 10.1006/jmbi.1997.1098 [DOI] [PubMed] [Google Scholar]
- 38. Berdichevsky A., Izhar L., and Livneh Z. (2002) Error-free recombinational repair predominates over mutagenic translesion replication in E. coli. Mol. Cell 10, 917–924 10.1016/S1097-2765(02)00679-2 [DOI] [PubMed] [Google Scholar]
- 39. Rehrauer W. M., Bruck I., Woodgate R., Goodman M. F., and Kowalczykowski S. C. (1998) Modulation of RecA nucleoprotein function by the mutagenic UmuD'C protein complex. J. Biol. Chem. 273, 32384–32387 10.1074/jbc.273.49.32384 [DOI] [PubMed] [Google Scholar]
- 40. Henrikus S. S., Wood E. A., McDonald J. P., Cox M. M., Woodgate R., Goodman M. F., van Oijen A. M., and Robinson A. (2018) DNA polymerase IV primarily operates outside of DNA replication forks in Escherichia coli. PLoS Genet. 14, e1007161 10.1371/journal.pgen.1007161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thomason L. C., Costantino N., and Court D. L. (2007) E. coli genome manipulation by P1 transduction. Curr. Protoc. Mol. Biol. 2007, Chapter 1, Unit 1.17 10.1002/0471142727.mb0117s79 [DOI] [PubMed] [Google Scholar]
- 42. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 2.0, Schrödinger, LLC [Google Scholar]
- 43. Sharma A., Kottur J., Narayanan N., and Nair D. T. (2013) A strategically located serine residue is critical for the mutator activity of DNA polymerase IV from Escherichia coli. Nucleic Acids Res. 41, 5104–5114 10.1093/nar/gkt146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Godoy V. G., Jarosz D. F., Simon S. M., Abyzov A., Ilyin V., and Walker G. C. (2007) UmuD and RecA directly modulate the mutagenic potential of the Y family DNA polymerase DinB. Mol. Cell 28, 1058–1070 10.1016/j.molcel.2007.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Duan G., Smith V. H., and Weaver D. F. (2001) Characterization of aromatic-thiol π-type hydrogen bonding and phenylalanine-cysteine side chain interactions through ab initio calculations and protein database analyses. Mol. Phys. 99, 1689–1699 10.1080/00268970110063917 [DOI] [Google Scholar]
- 46. Kottur J., Sharma A., Gore K. R., Narayanan N., Samanta B., Pradeepkumar P. I., and Nair D. T. (2015) Unique structural features in DNA polymerase IV enable efficient bypass of the N2 adduct induced by the nitrofurazone antibiotic. Structure 23, 56–67 10.1016/j.str.2014.10.019 [DOI] [PubMed] [Google Scholar]
- 47. Sholder G., Creech A., and Loechler E. L. (2015) How Y-Family DNA polymerase IV is more accurate than Dpo4 at dCTP insertion opposite an N2-dG adduct of benzo[a]pyrene. DNA Repair 35, 144–153 10.1016/j.dnarep.2015.09.020 [DOI] [PubMed] [Google Scholar]
- 48. Tashjian T. F., Lin I., Belt V., Cafarelli T. M., and Godoy V. G. (2017) RNA primer extension hinders DNA synthesis by Escherichia coli mutagenic DNA polymerase IV. Front. Microbiol. 8, 288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Studier F. W. (2005) Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
- 50. Beuning P. J., Simon S. M., Godoy V. G., Jarosz D. F., and Walker G. C. (2006) Characterization of Escherichia coli translesion synthesis polymerases and their accessory factors. Methods Enzymol. 408, 318–340 10.1016/S0076-6879(06)08020-7 [DOI] [PubMed] [Google Scholar]
- 51. Chen Z., Yang H., and Pavletich N. P. (2008) Mechanism of homologous recombination from the RecA–ssDNA/dsDNA structures. Nature 453, 489–494 10.1038/nature06971 [DOI] [PubMed] [Google Scholar]
- 52. Boyer B., Ezelin J., Poulain P., Saladin A., Zacharias M., Robert C. H., and Prévost C. (2015) An integrative approach to the study of filamentous oligomeric assemblies, with application to RecA. PLoS ONE 10, e0116414 10.1371/journal.pone.0116414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang D., Boyer B., Prévost C., Danilowicz C., and Prentiss M. (2015) Integrating multi-scale data on homologous recombination into a new recognition mechanism based on simulations of the RecA–ssDNA/dsDNA structure. Nucleic Acids Res. 43, 10251–10263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mackerell A. D. Jr., Feig M., and Brooks C. L. 3rd. (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulation. J. Comput. Chem. 25, 1400–1415 10.1002/jcc.20065 [DOI] [PubMed] [Google Scholar]
- 56. Molza A. E., Férey N., Czjzek M., Le Rumeur E., Hubert J. F., Tek A., Laurent B., Baaden M., and Delalande O. (2014) Innovative interactive flexible docking method for multi-scale reconstruction elucidates dystrophin molecular assembly. Faraday Discuss. 169, 45–62 10.1039/C3FD00134B [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.