

Abstract
endo-β-1,2-Glucanase (SGL) is an enzyme that hydrolyzes β-1,2-glucans, which play important physiological roles in some bacteria as a cyclic form. To date, no eukaryotic SGL has been identified. We purified an SGL from Talaromyces funiculosus (TfSGL), a soil fungus, to homogeneity and then cloned the complementary DNA encoding the enzyme. TfSGL shows no significant sequence similarity to any known glycoside hydrolase (GH) families, but shows significant similarity to certain eukaryotic proteins with unknown functions. The recombinant TfSGL (TfSGLr) specifically hydrolyzed linear and cyclic β-1,2-glucans to sophorose (Glc-β–1,2-Glc) as a main product. TfSGLr hydrolyzed reducing-end–modified β-1,2-gluco-oligosaccharides to release a sophoroside with the modified moiety. These results indicate that TfSGL is an endo-type enzyme that preferably releases sophorose from the reducing end of substrates. Stereochemical analysis demonstrated that TfSGL is an inverting enzyme. The overall structure of TfSGLr includes an (α/α)6 toroid fold. The substrate-binding mode was revealed by the structure of a Michaelis complex of an inactive TfSGLr mutant with a β-1,2-glucoheptasaccharide. Mutational analysis and action pattern analysis of β-1,2-gluco-oligosaccharide derivatives revealed an unprecedented catalytic mechanism for substrate hydrolysis. Glu-262 (general acid) indirectly protonates the anomeric oxygen at subsite −1 via the 3-hydroxy group of the Glc moiety at subsite +2, and Asp-446 (general base) activates the nucleophilic water via another water. TfSGLr is apparently different from a GH144 SGL in the reaction and substrate recognition mechanism based on structural comparison. Overall, we propose that TfSGL and closely-related enzymes can be classified into a new family, GH162.
Keywords: fungi; oligosaccharide; enzyme catalysis; enzyme structure; glycoside hydrolase; β-1,2-glucan; β-1,2-gluco-oligosaccharide; endo-β-1,2-glucanase; novel glycoside hydrolase family; Talaromyces funiculosus
Introduction
There are a great variety of carbohydrates in nature, which play various important physiological roles. In correspondence with such variety, the biochemical functions and structures of enzymes related to carbohydrates are very diverse. These enzymes are basically classified based on amino acid sequence similarity in the Carbohydrate-Active enZYmes Database (CAZy)2 (http://www.cazy.org/)3 (1). The glycoside hydrolase (GH) family, the largest group in CAZy, is expanding, e.g. seven novel GH families (GH137–143) were recently found in the Bacteroides thetaiotaomicron rhamnogalacturonan-II degradome (2). However, exploration of GH enzymes still has not caught up with the wide structural variety of carbohydrates.
β-1,2-Glucan is composed of a β-1,2-linked glucosyl backbone and is mainly found in bacterial cyclic polysaccharides produced by Gram-negative bacteria such as Brucella and Rhizobium (3–7). Cyclic β-1,2-glucans play roles as symbiotic or virulence factors inhibiting the immune responses of plants and in induction of splenomegaly in the mice (8–12). Roles of modulation of intracellular osmotic pressure and intracellular iron homeostasis have also been reported (13–15). Recently, cyclic β-1,2-glucan was also found in Eukaryotes such as Chlorella pyrenoidosa, an edible green microalga beneficial for immunostimulatory activity (16). There is some variety in the degree of polymerization (DP) of cyclic β-1,2-glucans. C. pyrenoidosa, Brucella abortus, and Agrobacterium tumefaciens (classified into Rhizobium radiobacter to date) produce cyclic β-1,2-glucans with DP of ∼20, whereas Rhizobium meliloti produces cyclic β-1,2-glucan with DP of around 40 (3–5, 16). In B. abortus, the DP of cyclic β-1,2-glucans is controlled by the C-terminal domain acting on β-1,2-gluco-oligosaccharides (sophorooligosaccharides, Sopns, where n denotes DP) in cyclic β-1,2-glucan synthase (17, 18).
Despite the important physiological functions of β-1,2-glucans in nature, β-1,2-glucan-associated enzymes and proteins have been explored much less than other GH enzymes that degrade polysaccharides such as β-1,3-glucan and cellulose (β-1,4-glucan), due to the difficulty in practical preparation of cyclic β-1,2-glucans. This may be attributed to the fact that cyclic β-1,2-glucan synthase is a transmembrane enzyme and uses UDP-glucose, which is a more expensive starting material than glucosides such as sucrose and starch, as a substrate (17).
Recently, GH94 1,2-β-d-oligoglucan phosphorylase (SOGP, EC 2.4.1.333) was found in Listeria innocua (19). SOGP from L. innocua (LiSOGP) reversibly phosphorolyzes Sopns with a DP of 3 or more. A large-scale preparation method for linear β-1,2-glucan (unless otherwise noted, β-1,2-glucans represent a linear form) involving LiSOGP has been established, which overcomes the limited availability of β-1,2-glucans (20). Furthermore, investigation of biochemical functions of GH3 β-glucosidase (BGL) and Sopns-binding protein in the LiSOGP gene cluster (LiBGL and LiSO-BP, respectively) revealed that the gene cluster is involved in metabolism of Sopns (21, 22). A β-glucosidase from B. thetaiotaomicron, a LiBGL homolog, has been identified as a Sopns-degrading β-glucosidase (23). In addition, the structure–function relationships of these enzymes and protein have also been unveiled (21–24).
Compared with the exo-type enzymes acting on β-1,2-glucan and Sopns, endo-β-1,2-glucanases (SGLs) have not been well studied. In 2017, Cpin6279 protein from Chitinophaga pinensis (CpSGL), a soil Gram-negative bacterium, was first identified as an SGL with a novel amino acid sequence, demonstrating a novel family, GH144 (25). Then, a sophorosyl hydrolase (nonreducing end), which releases sophorose from the nonreducing end of Sopns (PdSGL), was found as an exo-type enzyme in Parabacteroides distasonis, an intestinal bacterium, among the GH144 homologs (26).
GH144 homologs are distributed mainly in Gram-negative bacteria. In Eukaryotes, SGL activities have been reported in some fungi such as Aspergillus, Penicillium species, and Acremonium sp. 15 (27, 28). However, the distribution of eukaryotic SGLs and their relationship with bacterial SGLs are unclear. In this paper, we identified a Talaromyces funiculosus (formerly Penicillium funiculosum) SGL (TfSGL) with a novel sequence, characterized the recombinant TfSGL (TfSGLr), and further determined the 3D structure of TfSGLr.
Results and discussion
Purification of an SGL from fungi
Among fungi known to inducibly secrete SGLs by cyclic β-1,2-glucans, we examined the SGL activities in culture filtrates of T. funiculosus grown in the presence of linear β-1,2-glucan as a sole carbon source. Because the culture filtrate of T. funiculosus degraded the linear β-1,2-glucan, we purified the secreted TfSGL by three chromatographies. Although the SGL fractions contained BGL activity during the purification steps, SGL activity was completely separated from BGL activity by size-exclusion chromatography (SEC), the final purification step (Fig. 1A and Table 1), suggesting that the purified enzyme is an actual SGL. In Fig. 1A, DPs of the reaction products were somewhat obscure, which might be attributed to a slight smiling effect by NaCl in the enzyme solutions. The fractions containing the target enzyme gave a single band that migrated at ∼60 kDa on SDS-PAGE, indicating that TfSGL had been successfully purified to homogeneity. On SEC analysis, TfSGL eluted at 59 kDa (Fig. S1), suggesting that TfSGL is a monomeric enzyme. The purified TfSGL is highly specific for β-1,2-glucan among the polysaccharides examined (data not shown).
Figure 1.

Purification of TfSGL from a culture filtrate. A, top, SDS-PAGE of eluted fractions in the final purification step (SEC). Middle, TLC analysis of SGL activity in the fractions. BGL activity was inhibited by GDL. Lane M represents a mixture of 0.2% sugars (Glc and Sop2–4), and the numbers to the left of the TLC plates represent DP of Sopns. Bottom, BGL activity in the fractions. pNP-Glc was used as a substrate. Upward and downward arrows indicate correspondence of lanes among the three parts. B, protein (left) and sugar-chain (right) staining of TfSGL. Lanes 1–5 represent protein standard markers, glycoprotein standard markers, glycopeptidase F, and TfSGL without and with treatment of glycopeptidase F, respectively.
Table 1.
Purification of TfSGL
| Purification step | Total protein (mg) | β-1,2-Glucan–degrading activity |
BGL activity |
||||
|---|---|---|---|---|---|---|---|
| Total activity (U) | Specific activity (U/mg) | Yield (%) | Purification (fold) | Total activity (U) | Specific activity (U/mg) | ||
| Culture filtrate | 4.0 | 1.3 | 0.32 | 100 | 1.0 | 0.60 | 0.15 |
| HiTrapTM butyl HP (hydrophobic) | 0.22 | 0.29 | 1.4 | 23 | 4.2 | 0.045 | 0.21 |
| RESOURCETMQ (anion-exchange) | (0.086)a | 0.14 | (1.7)a | 11 | (5.2)a | 0.0035 | 0.041 |
| SuperdexTM 200GL (size-exclusion) | (0.13)a | 0.14 | (1.1)a | 11 | (3.5)a | <0.0002 | <0.002 |
a The values related to protein concentrations are shown in parentheses, as the low protein concentrations made the quantification inaccurate.
Glycosylated protein analysis of the purified enzyme
To determine whether TfSGL is glycosylated, periodic acid-Schiff stain analysis of the purified enzyme was performed. The enzyme was detected on both protein and sugar-chain staining, indicating that TfSGL is a glycoprotein (Fig. 1B). After the purified enzyme had been incubated with glycopeptidase F, which specifically cleaves N-glycans in glycoproteins, TfSGL was detected only on protein staining at ∼50 kDa. Therefore, TfSGL is a glycoprotein possessing only N-glycan of ∼10 kDa.
Analysis of action patterns on β-1,2-glucans and Sopns
The action pattern of TfSGL on linear β-1,2-glucan was determined by TLC analysis. TfSGL hydrolyzed β-1,2-glucan to Sop2 as a main product, implying that TfSGL preferably releases Sop2 from either end of β-1,2-glucan (Fig. 2A). In addition, Sop4 was accumulated as a minor product. Sopns with a DP of 5 or more were not visible in the TLC plate at a late stage of the reaction, although Sop5 was temporarily accumulated at the middle stage of the reaction probably due to slow hydrolysis of Sop5 by TfSGL (Fig. 2A). TfSGL hydrolyzed Sop5 to Sop2 and Sop3 but did not degrade Sop3 or Sop4 (Fig. 2B). These results indicate that TfSGL hydrolyzes Sopns with DP of 5 or more to Sop2 as a main product.
Figure 2.

TLC analysis of action patterns on β-1,2-glucan (A) and Sop3–5 (B) with TfSGL. Lane M represents a mixture of 0.2% Sop2–5. The numbers beside the TLC plates represent DP of Sopns. The purified TfSGL (1.9 and 3.8 μg/ml for hydrolytic reactions of β-1,2-glucan and Sopns, respectively) was incubated in 100 mm acetate-Na buffer (pH 5.0) containing 0.2% β-1,2-glucan or 5 mm Sopns with DP of 3–5 at 20 °C. Arrows represent β-1,2-glucan used for reactions. The origins of the TLC plates are shown as horizontal lines denoted by asterisks.
Sequence analysis
Because identification of the internal amino acid sequence of the purified enzyme was unsuccessful by LC-tandem MS analysis, N-terminal and internal peptide sequencing was performed. The obtained N-terminal and two internal peptide sequences are shown in Fig. 3. Then, we cloned a region containing the whole TfSGL gene of genomic DNA (gDNA) and complementary DNA (cDNA) (assigned accession number by DNA Data Bank of Japan, LC430902). The predicted transcription start site and the predicted polyadenylation signal sequence were found ∼100 bp upstream of the initiation codon and ∼300 bp downstream of the termination codon, respectively. The N-terminal signal peptide in TfSGL was predicted to have 18 residues, whereas the actual N terminus of TfSGL is the 22nd residue. Several plausible N-glycosylation sites predicted in TfSGL are asparagine residues (residues 41, 60, 120, 143, 194, 197, 237, 361, and 384), with this being consistent with the results of glycosylated protein analysis of TfSGL. Prediction of GPI-modification sites in TfSGL was performed because some TfSGL homologs are annotated as GPI-anchored proteins. However, no predicted GPI-modification site was found in TfSGL or these homologs. This is consistent with the fact that internal peptide 2 was quite near the C terminus.
Figure 3.

Scheme of cloning of a gDNA region encoding the whole TfSGL gene. The coding DNA sequence (CDS) regions shown in the square boxes represent exons of the TfSGL gene. The regions of the N-terminal peptide and internal peptides 1 and 2 are shown as black, gray, and dotted patterns, respectively. The regions around the peptides are shown as an enlarged view. The obtained N-terminal and two internal peptide sequences are shown below. The first (G) and 7th (X) residue in the internal peptide 2 is a presumed and an undetermined residue, respectively. Only the 6th residue in the N-terminal peptide and the 7th residue in the internal peptide 2 were replaced with cysteine residues in the deduced amino acid sequence of TfSGL. The differences in the sequences are attributed to the fact that cysteine cannot generally be detected unless it is pyridylethylated. The degenerate and specific PCR primers used for cloning are represented with thin and bold arrows, respectively. Primer pairs used for degenerate PCR and specific PCR are boxed with dotted and solid lines, respectively. The numbers above the coding DNA sequence boxes and beside the primers represent the nucleotide numbers from the start codon on gDNA.
Phylogenetic analysis
We constructed a phylogenetic tree, as shown in Fig. 4. The phylogenetic tree comprised only proteins annotated as hypothetical, unnamed, or GPI-anchored proteins. Although several homologs are annotated as glycosyl hydrolase family proteins, the annotations depend on the GH1 domain fused with regions homologous to TfSGL, and no annotation is given to the homologous regions. The biochemical functions of these TfSGL homologs are unknown, and they exhibit no sequence similarity to any known GH families. In addition, even structural prediction with InterPro (https://www.ebi.ac.uk/interpro/)3 did not allow classification of TfSGL into any of the families. Almost all of the homologs are from Eukaryotes with some exceptions, such as a homolog from Elusimicrobia bacterium, which was found from low O2 conditions as an environmental uncultured species (Fig. 4). No TfSGL homolog was found in Talaromyces marneffei or Talaromyces stipitatus, strains used for a Mascot search in the LC-tandem MS analysis.
Figure 4.

Phylogenetic tree for TfSGL homologs. The phylogenetic tree for TfSGL homologs was prepared using TfSGL homologs exhibiting at least 35% sequence identity and 50% coverage. Proteins are represented as GenBankTM or NCBI reference sequence accession numbers with the corresponding organism names. Ascomycota, highlighted in gray; Basidiomycota, dotted pattern; Elusimicrobia, pattern of horizontal lines; Dinoflagellata, pattern of diagonal lines with the left side up; Heterolobosea, pattern of vertical and horizontal lines; Ciliophora, pattern of vertical lines; Euglenozoa, pattern of diagonal lines with the right side up; Choanozoa, pattern of squares; Mycetozoa, pattern of diamonds. Letters prior to the organism names represent annotations; A, GPI-anchored protein; B, unnamed, hypothetical, or predicted protein. Black diamonds represent TfSGL homologs fused with the GH1 domain. The GH1 domains were excluded for preparation of the phylogenetic tree. The scale bar represents the number of substitutions per amino acid site. Asterisks represent homologs whose sequences were used for sequence alignment with TfSGL in Fig. 10.
General properties of TfSGLr
TfSGLr was successfully produced by Pichia pastoris. The purified enzyme migrated as a single band with a similar molecular mass to that of the native TfSGL on SDS-PAGE (Fig. S2A). Because TfSGLr showed hydrolytic activity toward β-1,2-glucan like the native TfSGL, β-1,2-glucan was used as a substrate to investigate the pH and temperature profiles. TfSGLr was stable at pH 4.0–7.0 and up to 30 °C. TfSGLr exhibits the highest activity at 60 °C and at pH 4.0–4.5 (Fig. S2, B and C).
Substrate specificity of TfSGLr
To determine the substrate specificity of the TfSGLr, hydrolytic activity toward various polysaccharides was examined. The enzyme acted on linear β-1,2-glucan specifically (specific activity was 17 units/mg as Sop2-releasing activity). Then, the chain-length specificity of the enzyme for Sopns was examined. The enzyme hydrolyzed Sop5 to Sop2 and Sop3, and Sop6 to Sop2 and Sop4 as main products, with Sop3 as a minor product, and Sop7 to equal amounts of Sop2–5. The enzyme did not hydrolyze Sop3 or Sop4 (Fig. 5A). These catalytic properties are essentially the same as those of the native TfSGL. The hydrolytic velocity toward Sop5 was ∼5 times lower than that toward Sop6. These results suggest that TfSGLr hydrolyzes β-1,2-glucan with a DP of 5 or more but requires DP of at least 6 for efficient hydrolysis.
Figure 5.

TLC analysis of hydrolysates from Sopns (A), β-1,2-glucans (B), and Sopn analogs (C) obtained with TfSGLr. Lanes M1–5 represent sugar markers containing 0.2% each gluco-oligosaccharide; M1, Glc and Sop2–7; M2, Glc and Sop2, 3, 5–7, 9, 11; M3, Glc and Sop4, 8, 10, 11; M4, rSop2, 4, 6, 8, 10; and M5, rSop3, 5, 7, 9, 11. The substrates used for the reactions are shown above the TLC plates. Each substrate (0.2%) was hydrolyzed with TfSGLr (19.3 μg/ml for hydrolysis of Sop5 and rSop6 and 3.2 μg/ml for the other substrates). The origins of the TLC plates are shown as horizontal lines denoted by asterisks. The numbers beside the TLC plates represent DP of Sopns. B, arrows indicate β-1,2-glucans used for reactions. C, numbers with the letter r beside the TLC plates represent rSopns. The pattern diagrams of rSopns are shown below the TLC plates. The open and closed circles in the pattern diagrams represent Glc and the reduced Glc moieties, respectively. Cleavage sites are represented as arrowheads.
Kinetic parameters for β-1,2-glucan and Sop5
The kinetic parameters of TfSGLr were determined using β-1,2-glucan and Sop5 as substrates (Fig. 6). These results show that Sop5 is a much less preferable substrate than β-1,2-glucan. These parameters for β-1,2-glucan and Sop5 are comparable with those for CpSGL (25). In addition, the activity of general endo-type GH enzymes toward their substrates is also similar to that of TfSGLr toward β-1,2-glucan (29, 30).
Figure 6.

Kinetic parameters of TfSGLr for Sop5 (left) and β-1,2-glucan (right). The data plotted as open and closed circles are regressed with the substrate inhibition equation (left) and Michaelis-Menten equation (right), respectively. The regressed lines are shown as solid lines. A molar concentration of β-1,2-glucan is calculated using average DP of the β-1,2-glucan. The values when the concentrations of β-1,2-glucan are represented by mg/ml are attached in parentheses.
Action patterns of TfSGLr
TfSGLr hydrolyzed both cyclic and linear β-1,2-glucans at similar velocity, indicating that TfSGLr is an endo-type enzyme (Fig. 5B). A main product released from linear β-1,2-glucan was Sop2. Sop3–5 were also produced as minor products after a 1-h reaction. To determine which ends of the linear β-1,2-glucan are hydrolyzed by TfSGLr, the action patterns on Sopns modified at the reducing-end by NaBH4 (rSopns) were investigated. Although Sop2 was not observed at the early stage of the reaction, rSop3 was found to be released from rSop6–8 despite the modification of the reducing end (Fig. 5C). Considering that rSop2 was not produced, rSop3 is likely released as a disaccharide by TfSGLr, probably because the modified moiety adopting an open chain form is not recognized by TfSGLr. In addition, there was no difference in hydrolytic velocity for the substrates regardless of the modification at the reducing end of the substrates (Fig. 5, A and C), suggesting that modification of the substrates does not affect reaction velocity. These results suggest that TfSGL is an endo-type enzyme but quite preferably releases Sop2 from the reducing end of β-1,2-glucan. This is a different property from that of PdSGL, which releases Sop2 from the nonreducing end of linear β-1,2-glucan (26).
Stereochemistry of hydrolysis catalyzed by TfSGLr
To determine the reaction mechanism of TfSGLr, the anomeric configurations of hydrolysates released from β-1,2-glucan were examined by 1H NMR. The β-anomer signal derived from the anomeric axial proton at the reducing end in liberated Sopns (H1βR, around δ 4.7) overlapped with chemical shifts derived from water and anomeric axial protons in internal glucose (Glc) moieties. Thus, the signals derived from the anomeric proton of the α-anomer of the released Sopns (H1αR, around δ 5.4) and the C2 proton at the nonreducing end of all the reaction products (H2NR, around δ 3.3) were used for stereochemical analysis as described by Abe et al. (25). Both peak areas increased immediately after addition of the enzyme (Fig. 7, A and B). Then, the former peak area gradually decreased during the reaction due to the nonenzymatic mutarotation of the products, whereas the latter gradually increased probably due to slow hydrolysis of Sop5, which was accumulated as shown in Fig. 5B. This result suggests that TfSGL is an inverting enzyme. We also examined the change of the degree of optical rotation during the hydrolysis of β-1,2-glucan by TfSGLr. An increase in the degree of optical rotation at the early stage of the reaction and a dramatic decrease in it in addition to aqueous ammonia was observed (Fig. 7C), with this being the same pattern as that of CpSGL, an inverting enzyme (25). This result also supports an inverting mechanism of TfSGL.
Figure 7.

Stereochemical analysis of TfSGLr. A, time-course analysis of a 1H NMR spectrum of the reaction mixture during hydrolysis of β-1,2-glucan with the average DP of 25 by TfSGLr. Encircled protons in the reaction products represent the positions of protons used for determination of stereochemical analysis. Signal-H1αR and signal-H2NR correspond to those of α-anomeric products and the total of α- and β-anomeric products, respectively. B, time-course of the ratio of the integral values of chemical shifts derived from signal-H1αR and signal-H2NR to an internal standard. The ratios corresponding to these chemical shifts are shown in a solid line and closed circles, and a dashed line and open circles, respectively. C, time-course of degree of optical rotation during hydrolysis of β-1,2-glucan with the average DP of 25 by TfSGLr. The arrow shows the time when aqueous ammonia was added.
Overall structure of TfSGLr
The apo structure of TfSGLr was determined at 2.0 Å resolution using the iodide single-wavelength anomalous diffraction-phasing method (Table S1 and Fig. 8). There are two molecules in an asymmetric unit, and their root mean square deviation (RMSD) value was determined to be 0.1 Å. The structure consists of mainly antiparallel α-helices forming inner and outer rings, indicating that TfSGLr folds into a single (α/α)6-barrel domain (Fig. 8). A structural homology search using the Dali server (31) showed that the overall structure of TfSGLr is similar to those of GH144 family members, including CpSGL (Protein Data Bank (PDB) code 5GZH) (Table S2). However, TfSGLr shows remarkably low amino acid sequence identities with GH144 proteins according to structure-based alignment with the PDBeFold server (less than 11%) (32, 33). TfSGLr also exhibits structural similarities to other GH family enzymes, a GH126 α-amylase from Clostridium perfringens (PDB code 3REN) (34), and a GH15 glucoamylase from Saccharomycopsis fibuligera (PDB code 1AYX). The similarities are lower than those of the GH144 enzymes according to Z-scores (RMSD values) (Table S2). TfSGLr has a cleft crossing the surface of the structure, and there is a large pocket at the center of the cleft (Fig. 8C). The pocket is located at the center of the (α/α)6-barrel as in most (α/α)6-barrel GH enzymes.
Figure 8.
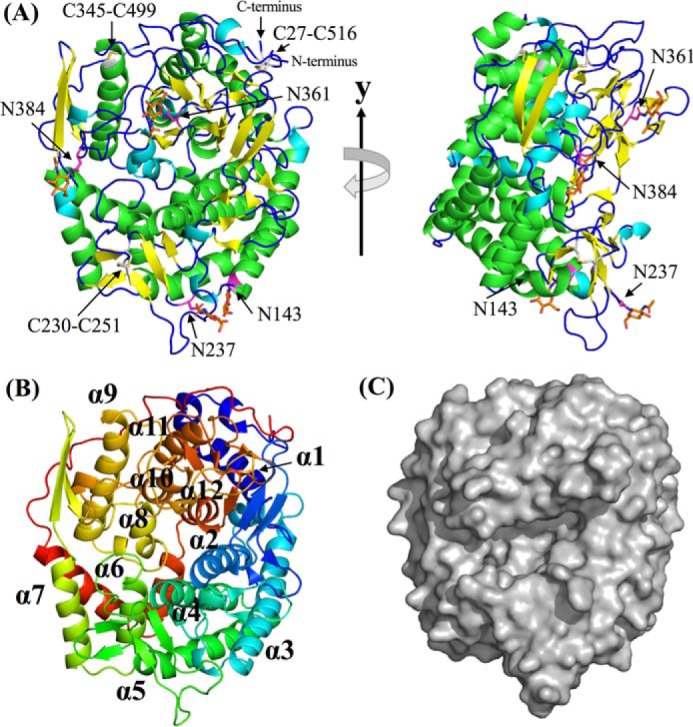
Overall structure of TfSGLr. A, front view of the overall apo TfSGLr structure (left) and the structure rotated by 90° around the y axis (right). The α-helices, 310-helices, β-strands, and loop are shown in green, cyan, yellow, and blue, respectively. Four asparagine residues (Asn-143, Asn-237, Asn-361, and Asn-384) bonded with GlcNAc, and the GlcNAc moieties and residues constituting three disulfide bonds (Cys-27–Cys-516, Cys-230–Cys-251, and Cys-345–Cys-499) are represented by magenta, orange, and white sticks, respectively. B, order of helices constituting an (α/α)6-barrel in TfSGLr. The barrel is represented by a rainbow cartoon. The helices in TfSGLr are numbered in order from the N terminus. C, surface of the overall structure of apo TfSGLr.
Complex structure with Sop2 and Glc
In the obtained complex structure with Sop2 and Glc by soaking a TfSGLr crystal in Sop2, the electron density of a Sop2 molecule was clearly observed at the center of the cleft (Fig. 9A). Considering that TfSGLr produces mainly Sop2 from the reducing end of linear β-1,2-glucan, the Sop2 molecule seems to be located at subsites +1 to +2. This is supported by the TfSGLr structure in complex with Sop7, as described below.
Figure 9.

Complexes with Sop2 and Glc in the WT and Sop7 in the E262Q mutant. A and B, substrate-binding modes of TfSGLr–Sop2 and Glc complex (A), and the E262Q–Sop7 complex (B). The numbers beside the substrate represent the positions of subsites. The Fo − Fc omit maps for Sop2, Glc, and Sop7 are shown at the 4σ contour level and represented by a blue mesh. The blue dotted lines represent the hydrogen bonds between the ligands and the enzymes. A, observed Sop2 and Glc in chain B are represented by white sticks. Residues involved in interaction with Sop2 and Glc in the complex, and the corresponding residues in the superimposed apo structure of TfSGLr are represented by green and light blue sticks, respectively. A blobby electron density beyond the anomeric hydroxy group of the Glc molecule at subsite −3 was omitted from this figure. B, observed Sop7 moiety of β-1,2-glucan with the average DP of 25 in the complex of the E262Q mutant is represented by a yellow-green stick. Residues involved in interaction with Sop7 in the E262Q mutant are represented by brown sticks. C, enlarged view of the skewed Glc moiety in the Sop7 molecule. A water near the anomeric carbon of the Glc moiety is represented by a red sphere. D, surface representation of the catalytic center with Sop7 in the E262Q mutant. The catalytic pocket is enclosed by a red dotted line. Ligands in the Sop2 and Glc complex are represented by a white stick when the Sop2 and Glc complex and the Sop7 complex are aligned.
The Glc moiety at the nonreducing end of the Sop2 molecule forms hydrogen bonds with Trp-155 and Asp-177 (Fig. 9A). The hydrophobic side chain of Leu-176 interacts with the pyranose ring of the Glc moiety. In addition, the aromatic ring of Trp-169 undergoes a hydrophobic interaction with the C6 atom of the Glc moiety. The Glc moiety at the reducing end of the Sop2 molecule forms six hydrogen bonds with Asp-259, Glu-262, and His-316. The side chain of His-316 adopts alternative conformations in the apo TfSGLr, unlike in the case of the Sop2 complex (Fig. 9A). Two aromatic residues (Tyr-311 and Trp-312) undergo hydrophobic stacking interactions with C6 and the pyranose ring in the Glc moiety, respectively.
A relatively weak electron density that can be fitted with a Glc molecule was also observed in the pocket. The Glc molecule forms hydrogen bonds with Asp-72, Lys-94, Asn-360, and Asp-446 (Fig. 9A). The aromatic rings of Trp-155 and Tyr-373 undergo hydrophobic stacking interactions with the pyranose ring of the Glc molecule.
Determination of subsite positions in the complex structure with Sop7
To elucidate a more detailed substrate recognition mechanism by obtaining the structure of the Michaelis complex of TfSGLr, we searched for an inactive TfSGLr mutant. Because mutation of Glu-262 in the substrate-binding pocket to glutamine abolished the activity of TfSGL toward β-1,2-glucan, we soaked a crystal of the E262Q mutant in β-1,2-glucan. In each catalytic pocket of two molecules (chain A or B) in the asymmetric unit of the complex, the electron density of a Sop5 or Sop7 molecule was observed, respectively. Therefore, chain B was used for description of the complex. Among the Glc moieties of the Sop7 molecule, the electron density of a Sop6 moiety was clearly observed (Fig. 9B), whereas that of the remaining Glc moiety was ambiguous.
It should be noted that the fourth Glc moiety from the nonreducing end of the Sop7 molecule forms a skew-boat conformation (1S3) (Fig. 9C) according to Cremer-Pople parameters (ϕ (°), θ (°), and Q (Å) of the Glc moiety are 204.341, 81.202, and 0.713, respectively) (35). In contrast, the other Glc moieties form a 4C1 conformation. In GH5 retaining endo-β-1,4-glucanase from Bacillus agaradhaerens and GH63 inverting α-glycosidase from Escherichia coli, the Glc moieties of their substrates at subsite −1 adopt a 1S3 conformation (36, 37). This twisted conformation enables a nucleophile to be located where nucleophilic attack to an anomeric carbon is possible. In TfSGLr, a water molecule is located (3.1 Å) near the anomeric carbon of the Glc moiety with a 1S3 conformation. The angle formed by this water, the anomeric carbon, and an oxygen atom of the glycosidic bond between subsites −1 and +1 is 161.1°, which is suitable for nucleophilic attack (in-line-attack) on the anomeric carbon. These observations strongly suggest that the position of the fourth Glc moiety is subsite −1 and that TfSGL accommodates the Sop7 molecule at subsites −4 to +3, a Michaelis complex being formed (Fig. 9B).
Substrate-binding mode in the Sop7 complex
The Glc moieties of Sop7 at subsites +1 and +2 well overlapped those of the Sop2 molecule in the TfSGLr–Sop2 complex, and this is consistent with the presumed subsite positions of the Sop2 molecule in the TfSGLr–Sop2 complex described above (Fig. 9D). The Glc molecule in the TfSGLr–Sop2 complex is also well-superimposed with the Glc moiety at subsite −3 in the TfSGLr–Sop7 complex. Thus, the Glc moieties of Sop7 at subsites −3, +1, and +2 are firmly recognized in almost the same way as in the Sop2 complex (Fig. 9, A and B). The Glc moieties at subsites −3 to +2 are apparently accommodated in the large substrate pocket, which looks like a “gravy boat” (Fig. 9D).
Unlike subsites −3, +1, and +2, subsites −2 and −1 seem to be unfavorable factors for substrate binding. The Glc moiety at subsite −2 forms only one hydrogen bond with Ser-375. The twisted glycosidic bond between subsites −1 and +1 (see below), and the twisted conformation of the Glc moiety at subsite −1 also make the binding unstable. These observations imply the importance of subsite −3 for substrate binding on the minus subsite side. Taken together with the requirement of subsite +2 for catalysis, as described later, structural observations suggest that at least a DP of 5 is needed for Sopn to act as a substrate. This is consistent with the finding that Sop5 is a minimum substrate of TfSGLr (Fig. 5A).
The 2- and 1-hydroxy groups of the Glc moieties at subsites −4 and +3, respectively, face the solvent (Fig. 9D), indicating that TfSGL can accommodate a substrate extending beyond both subsites −4 and +3. This observation is consistent with the endolytic property of TfSGL.
At subsite −4, only a hydrogen bond with Trp-169 and a hydrophobic stacking interaction with the aromatic ring of Trp-155 were found, whereas no interaction was observed at subsite +3. This difference is consistent with the observed action patterns that Sop6 was preferentially hydrolyzed to Sop4 and Sop2. Judging from these observations, the binding mode of Sop7 is consistent with the characteristics of TfSGL, suggesting that binding of Sop7 is productive.
Most of the substrate recognition residues are conserved among TfSGL homologs except that two residues in TfSGLr (Tyr-311 and Ser-375) exhibit a little variety (Fig. 10). This implies that the function and structure of TfSGL are conserved among TfSGL homologs.
Figure 10.

Multiple sequence alignment of TfSGL and its homologs. Multiple alignment using TfSGL homologs with at least 37% sequence identity was carried out. The homologs are represented as GenBankTM or NCBI reference sequence accession numbers. The symbols below the sequences are represented as follows: open circles, candidates for catalytic residues of TfSGLr; closed circles, residues involved in substrate recognition; closed triangles, N-glycosylated asparagine residues; and closed stars, the general acid (Glu-262) and the general base (Asp-446) of TfSGL. The same numbers below sequences represent disulfide bond pairs. Residue numbers above sequences are based on the amino acid sequence of the native TfSGL. The secondary structures of TfSGLr are shown above the sequence. The order of helices constituting (α/α)6 barrels in TfSGLr is shown in parentheses.
Comparison of conformations of ligands between β-1,2-glucan–associated proteins
The conformations of Sop3–5 have been reported as ligands in the complex structures of LiSO-BP (22). In the complexes, the Sop3–5 molecules adopt stable conformations, and the Glc moieties in the ligands line up in a zigzag manner. Compared with these ligands, a feature of the Sop7 molecule in TfSGLr is a twisted (1S3) conformation at subsite −1, as described above. It is observed that the anomeric hydroxy group in the Glc moiety is located at a lifted position when pair-fitting of the pyranose rings of substrates at subsites −3 to −1 in TfSGLr and at units A to C (the Sop3 moiety from the nonreducing end of Sop5) in LiSO-BP is performed (Fig. S3). The Glc moiety of subsite +1 in the TfSGLr–Sop7 complex is rotated by ∼100° against that in the LiBGL–Sop2 complex, suggesting that the relative positions of subsite +1 against subsite −1 are quite different between the two enzymes (Fig. S3). The difference in the position of subsite +1 makes subsites −2 to +1 crowded in the TfSGLr–Sop7 complex (Fig. 9B). A “gravy boat”-like large pocket might be required to accommodate substrates with the twisted conformation (Fig. 9D). Unlike TfSGLr, Sopns are sandwiched by the closure motion of two domains in LiSO-BP, and LiBGL has a narrow “coin slot”-like structure at subsite +1 (a pyranose ring is sandwiched by two aromatic residues) (21, 22). In the case of SOGP from Lachnoclostridium phytofermentans, a Glc moiety at subsite +1 is flipped at the position corresponding to subsite +1 in LiBGL (21, 24). The structure of TfSGLr exhibits a novel substrate-binding mode among β-1,2-glucan–associated enzymes and proteins.
Mutational analysis of candidates for a general base
In general, an inverting GH enzyme hydrolyzes a glycosidic bond in its substrate through a single-displacement mechanism using two acidic residues (Fig. 11A). A general base activates a water for nucleophilic attack on the anomeric carbon, whereas a general acid protonates the glycosidic bond oxygen directly (38). However, no acidic residue directly interacts with the nucleophilic water molecule or is located within the proton transfer distance range from the glycosidic bond oxygen atom at the cleavage site in the case of TfSGLr (Fig. 11B). Therefore, TfSGLr unlikely utilizes a canonical reaction mechanism.
Figure 11.

Candidate residues for catalysis by TfSGLr. A, canonical reaction scheme for inverting glycoside hydrolases. B, candidate residues for catalysis. The Glc moieties at subsites −1 to +2 in the Sop7 complex of the E262Q mutant are represented by yellow-green sticks. The positions of subsites are labeled with numbers. The blue and gray dotted lines represent hydrogen bonds and longer hydrogen bonds (over 3.5 Å) related to a water network in TfSGLr, respectively. The distances of the hydrogen bonds are shown beside the lines. A red dotted line represents the position of nucleophilic attack. Residues corresponding to each mutant are represented as sticks and are color-coded based on the relative hydrolytic activity toward β-1,2-glucan (<0.1%, magenta; 0.1–10%, yellow; and >10%, green). The 3-hydroxy groups of the Glc moieties at subsites +1 and +2 and the oxygen atom at the cleavage site are highlighted in blue and red circles, respectively. C, TLC analysis of the activities of TfSGLr mutants toward β-1,2-glucan. Lane M represents markers each containing 0.2% Sop2–5 and β-1,2-glucan. The mutants used for the reactions are shown above the TLC plates. Each TfSGLr mutant (0.2 mg/ml) was incubated in 100 mm acetate-Na buffer (pH 4.0) containing 0.2% β-1,2-glucan at 30 °C. The origins of the TLC plates are shown as horizontal lines denoted by asterisks. The positions of β-1,2-glucan are shown by arrows.
As to the reaction pathway for a general base, only Tyr-373 interacts with the nucleophilic water among the amino acid residues (Fig. 11B). Furthermore, there is no acidic residue hydrogen-bonded with Tyr-373. Therefore, we added the following residues that interact indirectly with the nucleophilic water via multiple waters as candidate residues for catalysis: His-316, Ser-358, Tyr-396, Glu-430, Thr-444, and Asp-446. Glu-180 was also added as a candidate because it is an acidic residue located near subsite −1 (Fig. 11B). These candidate residues, except Thr-444, are highly conserved among TfSGL homologs (Fig. 10). Therefore, mutational analysis of all these residues was carried out for identification of the general base, although the T444A mutant was not expressed as a soluble protein. The D446N mutant showed no activity toward β-1,2-glucan on colorimetric assay or TLC analysis (Fig. 11C and Table 2). The relative activity of the E430A mutant was less than 1%, which could also be regarded as the activity level of a catalytic mutant. Even when the 3-methyl-2-benzothiazolinone hydrazone (MBTH) method (39), the most sensitive method for quantification of Sopns, was used for assay, the relative activity of the E430A mutant did not show a change (Table 2). However, it is difficult to regard Glu-430 as a general base, because there needs to be three additional waters to reach the nucleophilic water, and one of the hydrogen bond lengths between these waters (3.6 Å) is longer than a general one. Although there is also a proton network from Glu-430 to the nucleophilic water via Tyr-373, proton transfer unlikely takes place via this route. This is because the Y373F mutant retained too high hydrolytic activity toward β-1,2-glucan as a residue involved in the reaction pathway (Table 2). In addition, the hydrolytic activity of the E430A mutant detected by TLC analysis was apparently higher than those of D177N and E262Q mutants (the candidates for a general acid described below) and the D446N mutant (Fig. 11C). The other candidates (His-316, Ser-358, and Tyr-396) are also unlikely general bases. This is because the mutants (H316Q, S358A, and Y396F) retained sufficient hydrolytic activity toward β-1,2-glucan, although the H316A mutant showed remarkably decreased activity. These results suggest that Asp-446 probably acts as a general base via two water molecules.
Table 2.
Specific activities of the wildtype and mutant TfSGLr for β-1,2-glucan
| Mutant | Specific activitya (U/mg) | Relative activityb (%) |
|---|---|---|
| Wildtype | 17 (18) | 100 (100) |
| Candidates for general acid | ||
| D177N | NDc | ND |
| E262Q | ND | ND |
| Candidates for general base | ||
| Acidic residues | ||
| E180A | 1.2 (4.0) | 7.3 (22) |
| E430A | 0.11 (0.11) | 0.63 (0.61) |
| D446N | ND | ND |
| Nonacidic residues | ||
| H316A | 0.02 (0.05) | 0.12 (0.27) |
| H316Q | 1.5 (3.1) | 9.0 (17) |
| S358A | 12 | 70 |
| Y373F | 1.2 (2.3) | 7.1 (13) |
| Y396F | 9.8 | 58 |
a Specific activity of TfSGLr was calculated by measuring the amount of Sop2 produced from β-1,2-glucan using the GOPOD method. Specific activity and relative activity calculated by measuring the amount of produced Sopns using the MBTH method are shown in parentheses.
b Specific activities of wildtype TfSGLr calculated by the GOPOD and MBTH methods were defined as 100% relative activities.
c ND represents the data of less than 0.02 units/mg (0.01% relative activity).
Because Tyr-373 interacting with the nucleophilic water is not located between Asp-446 and the nucleophilic water, Tyr-373 probably stabilizes the position of the nucleophilic water. The drastically decreased activities of the H316A and E430A mutants might imply the importance of the water network for catalytic efficiency.
Mutational analysis of candidates for a general acid
As to the reaction pathway for a general acid, only 3-hydroxy groups in the Glc moieties at subsites +1 and +2 are found within the proton transfer distance range from the oxygen atom in the cleavage site. The 3-hydroxy groups at subsites +1 and +2 form hydrogen bonds with Asp-177 and Glu-262, respectively. Although Asp-177 and Glu-262 are located too far from the glycosidic bond's oxygen atom for direct proton transfer (distances of 4.8 and 4.4 Å, respectively), these residues are highly conserved among TfSGL homologs (Fig. 10). Therefore, it is predicted that Asp-177 and/or Glu-262 act as a general acid via the hydroxy groups of the Glc moieties. TLC analysis showed that no hydrolytic products derived from linear β-1,2-glucan by the E262Q mutant were detected, whereas Sop2 was slowly produced by the D177N mutant (Fig. 11C). However, according to the colorimetric assay, the activities of both the D177N and E262Q mutants toward β-1,2-glucan were undetectable (Table 2). This finding suggests that both Asp-177 and Glu-262 still could be catalytic residues. Therefore, the action patterns of TfSGLr on Sopns with the appropriate 3-hydroxy groups deoxygenated were analyzed as described below.
Action pattern analysis of 3-deoxy Sopn-derivatives
To determine the actual general acid of TfSGL, the action patterns for Sop5–6 deoxygenated at their 3-hydroxy groups at the first or second Glc moiety from the reducing end (3dSop5–6 and 3′dSop5, respectively) were investigated (Fig. 12). 3′dSop2 was released from 3′dSop5 by TfSGLr, whereas 3dSop5 was hardly hydrolyzed even in the presence of an excessive amount of TfSGLr (Fig. 12, A and B). We also examined the action pattern for 3dSop6, a substrate with a preferable chain length. As a result, 3dSop3 and Sop3 were released from 3dSop6 without production of 3dSop2 and Sop4 (Fig. 12C). This finding indicates that deoxygenation of the 3-hydroxy group of the Glc moiety at subsite +2 completely inhibits the cleavage of the substrate despite that the glycosidic bond between the second and third Glc moieties from the reducing end is the preferable cleavage site in Sop6 (Figs. 5 and 12D). These results exclusively suggest that Glu-262 acts as a general acid via the 3-hydroxy group of the Glc moiety at subsite +2.
Figure 12.

HPLC analysis of fluorescently labeled hydrolysates are from 3dSop5 (A), 3′dSop5 (B), and 3dSop6 (C) obtained with TfSGLr. A–C, panels at top show the retention times of peaks derived from the fluorescently labeled Glc and Sop2–6. The numbers above the peaks represent the DPs of the sugars. The second panels from top show the retention times of peaks derived from the fluorescently labeled 3dSop2, 3′dSop2, and 3dSop3, respectively. The third and fourth panels from top show the retention times of peaks derived from the fluorescently labeled reaction mixtures when TfSGLr was incubated with each derivative at 30 °C for 0, 180, or 30 min, respectively. The pattern diagrams of oligosaccharides corresponding to the peaks are shown above the peaks. The open and closed circles in the pattern diagrams represent the Glc and 3-deoxy-Glc moieties, respectively. The asterisks denote peaks derived from the fluorescent labeling reagent. D, summary of the results of action pattern analysis. N.D. represents that no product cleaved at the cleavage site was detected.
Molecular dynamics simulation of TfSGLr
To exclude the possibility that the crystal structures of TfSGL are affected by crystal packing, molecular dynamics (MD) simulations of the apo structures (see “Experimental procedures”) and the Sop7 complex of TfSGLr were performed. Neither the TfSGLr structure nor the Sop7-complex structure significantly changed during the simulations with the average RMSD values of 0.8–1.3 Å (Fig. S4). The Cα root mean square fluctuation (RMSF) values of residues around the catalytic pocket of TfSGLr were lower than 1.0 Å (Table S3 and Fig. S4). Principal component analysis (PCA) and cluster analysis demonstrated that a representative MD structure of the complex and its crystal structure are almost the same with the Cα RMSD of 0.6 Å and that a representative MD structure of the apo state showed a slight opening movement of the catalytic pocket relative to the apo crystal structure (Fig. S5). These results suggest that crystal packing does not affect the structure of TfSGL in complex with Sop7.
Noncanonical reaction mechanism of TfSGL
The proposed catalytic reaction of TfSGL proceeds as follows. Glu-262 (general acid) indirectly protonates the anomeric oxygen at subsite −1 via the 3-hydroxy group of the Glc moiety at subsite +2. Asp-446 (general base) indirectly activates the water for nucleophilic attack via another water (Fig. 13). A proton relay mechanism in TfSGL is called a “Grotthuss”-style mechanism (40) and is a noncanonical one in GH enzymes.
Figure 13.

Reaction mechanism of TfSGL. The Glc moieties at subsites −1 and +2 are highlighted in gray. Curved arrows represent the pathway for proton transfer, and the dotted lines represent hydrogen bonds involved in catalysis. The parts in the substrate that are not directly involved in the reaction mechanism are omitted.
The pKa prediction of Glu-262 and Asp-446 was performed by the PROPKA3.0 (41) server using the protein moiety in the Sop2-complex structure. The pKa values of Glu-262 (8.22 and 8.21 in chain A and B, respectively) were clearly higher than those of Asp-446 (3.37 and 3.35, respectively), which is consistent with the proposed TfSGL reaction mechanism. The difference in the pKa values may be mainly attributed to a decrease in pKa value of Asp-446 by a positive charge of Lys-94 close to Asp-446 and an increase in pKa value of Glu-262 by negative charges of Asp-259 and Glu-317 close to Glu-262.
In GH130 4-O-β-d-mannosyl-d-glucose phosphorylase from Bacteroides fragilis, the 3-hydroxy group in the substrate at subsite −1 is found within the range of hydrogen bond interaction with the acidic residue and the glycosidic bond oxygen (42). Based on the structural features and mutational analysis of the GH130 enzyme, it is postulated that the 3-hydroxy group plays a role as a mediator of protonation. This mechanism is the same as that of TfSGL in that a proton is relayed via a substrate hydroxy group. However, the two enzymes use different hydroxy groups.
Several retaining and inverting enzymes follow a Grotthuss mechanism, in which a general base remotely activates the nucleophilic water via another water molecule (GH6, GH101, and GH136) (43–45). For example, cellobiohydrolase from Trichoderma reesei (Cel6A) is an inverting GH and is considered to follow a Grotthuss mechanism. TfSGLr is similar to Cel6A in that another water molecule mediates proton transfer. However, the positions of general bases in the two enzymes are quite different.
There are other GHs that use a noncanonical mechanism. The substrate-assisted mechanism (GH18, GH20, GH56, GH84, GH85, GH92, and GH103) is one of the representative examples (46–52). Catalytic mechanisms with catalytic acidic residues replaced by nonacidic residues have been reported for some GH enzymes (GH3, GH33, GH34, GH45, GH55, GH83, GH95, GH117, GH127, GH143, and GH145) (2, 53–61). However, GH families whose catalytic mechanisms have been clearly identified possess either canonical catalytic residue. Although a C6 hydroxy proton-mediated pathway in the deglycosylation step is proposed for GH103 enzymes, there seems to be ambiguity due to the lack of a Michaelis complex structure. The reaction mechanism of TfSGL is quite unique in that both reaction pathways involving a general acid and a general base are noncanonical.
Comparison of the TfSGLr and CpSGL structures
Because TfSGLr and CpSGL exhibit the same substrate specificity and show structural similarity despite their exclusively different amino acid sequences, we compared the overall structures of the two enzymes. The positions of helices constituting (α/α)6 barrels are similar between them, suggesting a relationship in structural evolution (Fig. 14A) (15). Although the ligands in both enzymes deviated in the superimposition of the overall structures, these ligands are well-superimposed by fitting the corresponding Glc moieties between the two enzymes. The substrate-binding pockets of TfSGLr and CpSGL are similar in that the Sop5 moiety in TfSGLr fits well in the substrate pocket of CpSGL. There is a difference in the shape of the pocket between the two enzymes (Fig. 14B). The α-face side of the Glc moiety at subsite +2 in TfSGLr is narrowed due to hydrophobic interaction with Tyr-311 and Trp-312, although this space in CpSGL is wide open and Phe-204 sticks out a little to the β-face side of the same Glc moiety in CpSGL. Such a difference might be related to the preferential Sop2 release from β-1,2-glucans for TfSGL, unlike CpSGL that hydrolyzes β-1,2-glucans in a random manner to release Sop2–5.
Figure 14.
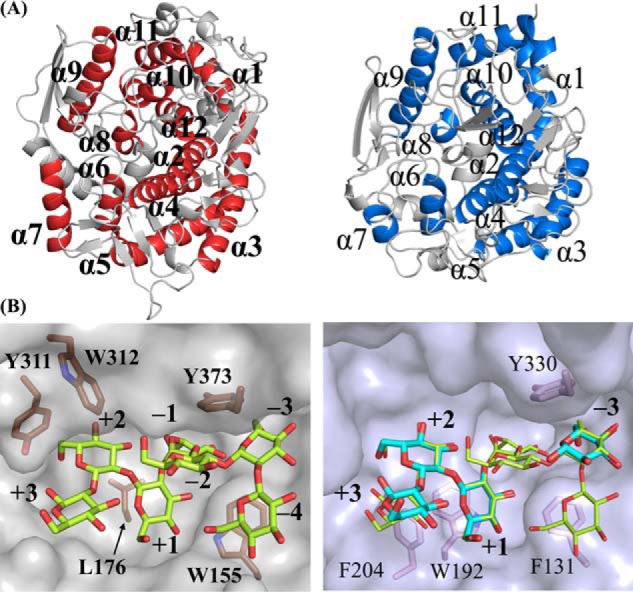
Comparison of structures between TfSGLr and CpSGL complexes. The positions of subsites are represented as numbers. The ligands bound with TfSGLr and CpSGL (PDB code 5GZK) are shown as yellow-green and cyan sticks, respectively. Residues involved in hydrophobic recognition of substrates and/or formation of the catalytic pockets in TfSGLr and CpSGL are shown as brown and pink sticks, and the residues are labeled with numbers in bold and regular type, respectively. A, comparison of overall structures between TfSGLr (left) and CpSGL (right). The two structures are superimposed based on their overall structures. The helices constituting (α/α)6 barrels in TfSGLr and CpSGL are colored red and blue, respectively. The helices in TfSGLr and CpSGL are numbered in order from the N termini and are represented by bold and regular letters, respectively. B, comparison of the catalytic pockets between the TfSGLr and CpSGL complexes. Because the ligands in CpSGL are deviated 2–5 Å from the corresponding moieties in TfSGLr and the Glc moieties of Sop7 in TfSGLr at subsites −3, +1, and +2 are strongly bound, the two enzymes were aligned by superimposing the Glc moieties at subsites −3, +1, and +2 in both enzymes. Both corresponding ligand pairs are well-superimposed on 17.8° rotation of CpSGL. TfSGLr (left) and CpSGL (right) are shown as semi-transparent gray and light blue surface models. The Sop7 molecule is superimposed in the right panel.
We compared the reaction mechanisms of TfSGL and CpSGL, although the catalytic mechanism of CpSGL has not been determined. The catalytic residues in TfSGLr and candidate residues in CpSGL are shown in Figs. 15 and 16A. The carboxyl groups of a general acid in TfSGLr (Glu-262) and Glu-211 in CpSGL are well-superimposed, although the positions of their main chains are different. Both residues are highly conserved in their respective homologs. In addition, these two acidic residues were found to be conserved on structure-based pairwise alignment of TfSGLr and CpSGL (Fig. 15). It has been reported that the activity of the E211Q mutant toward β-1,2-glucan is drastically decreased as compared with the WT CpSGL (the relative activity is 0.15%) (25). These facts imply that CpSGL homologs may have the same general acid as TfSGL. On the contrary, a catalytic base in TfSGL (Asp-446) is substituted with Ile-399 in CpSGL. Ile-399 is highly conserved as a hydrophobic residue (Ile, Leu, and Val) in GH144 homologs. Furthermore, candidate residues for catalysis in CpSGL (Asp-135, Asp-139, and Glu-142) are located at the positions where proton transfer only via water molecules from a nucleophilic water is impossible according to the superimposed Sop5 moiety in TfSGLr. These observations suggest that the general bases are obviously different between TfSGL and CpSGL despite the undetermined reaction mechanism of CpSGL.
Figure 15.

Structure-based alignment of TfSGLr and CpSGL. The alignment based on the structures of TfSGLr and CpSGL was performed using the apo structures of TfSGLr and CpSGL (PDB code 5GZH). The secondary structures of TfSGLr and CpSGL are shown above and below the sequences, respectively. The orders of helices constituting (α/α)6-barrels in both enzymes are shown in parentheses. The amino acid sequences of both enzymes are numbered based on those of the respective native enzymes. The black and white stars represent candidate residues in TfSGLr and CpSGL for catalysis, respectively.
Figure 16.

Comparison of acidic residues (A) and substrate recognition residues at the subsite minus (B) and plus (C) sides between TfSGLr and CpSGL. The substrate cleavage site and 3-hydroxy group of the Glc moiety at subsite +2 are highlighted in semi-transparent red and blue circles, respectively. The positions of subsites in TfSGLr and CpSGL are represented as numbers. The ligands bound with TfSGLr and CpSGL are shown as yellow-green and cyan sticks, respectively. Residues in TfSGLr and CpSGL are shown as brown and pink sticks, and their residue numbers are denoted by bold and regular numerals, respectively. A, The blue and gray dotted lines represent the catalytic pathway and the candidate pathway ruled out on action pattern analysis of TfSGL, respectively. B and C, hydrogen bonds between residues and ligands in TfSGLr and CpSGL are shown as blue and red dotted lines, respectively. Residues for the hydrophobic interactions with the substrates (Leu-176, Tyr-311, Trp-312, and Tyr-373 in TfSGLr; and Trp-192, Phe-204, and Tyr-330 in CpSGL) are omitted in B and C, and Trp-155 in TfSGLr and Phe-131 in CpSGL are omitted only in B, because these residues can be compared in Fig. 14B.
The overall positions of substrate recognition residues in TfSGL and CpSGL are quite different (Fig. 16, B and C), although both enzymes have comparable hydrogen bonds at subsites −3, +1, and +2. At subsite −1, where no ligands are observed in CpSGL, there are three potential hydrogen bonds with His-119, Glu-142, and Glu-211, whereas TfSGLr forms five hydrogen bonds. In addition, there is no potential hydrogen bond at subsite −2 in CpSGL. Meanwhile, there are only two similar parts as follows: the Glc moieties are stacked on the aromatic residues at subsite −3 (Fig. 14B), and the carboxylate groups of Glu-262 in TfSGL and Glu-211 in CpSGL are located at almost the same position.
Overall, TfSGLr has the same substrate specificity, and a similar overall structure and shape of the catalytic pocket as CpSGL. However, TfSGLr is clearly different from CpSGL in primary sequence, the positions of the base catalysts, and most substrate recognition residues. Therefore, TfSGL and its homologs should be classified into a novel family, GH162.
Speculated physiological roles of TfSGL homologs
Almost all of the TfSGL homologs are distributed in Eukaryotes, especially in the Ascomycota, Basidiomycota, and Mycetozoa. Among the Ascomycota possessing TfSGL homologs, there are many species related to the rhizosphere such as Talaromyces verruculosus (62). Fusarium oxysporum is a plant-pathogenic fungus, and Oidiodendron maius is known as an endophyte on azaleas. Metarhizium species and Beauveria bassiana are insect pathogens but reside in the rhizosphere, where they supply nitrogen to plants from insects. Considering that cyclic β-1,2-glucans are produced in the rhizosphere by plant symbionts such as Rhizobium, TfSGL homologs in such species might be involved in residence in the rhizosphere by metabolizing cyclic β-1,2-glucans.
Besides them, many pathogenic, symbiotic, and predatory species are known. In the Ascomycota, Cordyceps confragosa, Torrubiella hemipterigena, Aschersonia aleyrodis, and Tolypocladium ophioglossoides are fungi parasitic on specific insects and mushrooms. Pochonia chlamydosporia and Purpureocillium lilacinum are nematophagous fungi. Mycetozoa species prey on bacteria. In addition, there are also Ciliophora species such as a predator of bacteria (Paramecium tetraurelia) and a symbiotic Eukaryote found with a coral (Symbiodinium microadriaticum). It has been reported that some Paramecium spp. such as Paramecium bursaria form symbiotic relationships with symbiotic Chlorella (63). Considering the fact that cyclic β-1,2-glucans are reported to reduce the immune responses of hosts (9), TfSGL homologs might be related to interactions with other organisms.
Talaromyces cellulolyticus, a species closely-related to T. funiculosus, possesses a TfSGL homolog (GAM34680.1; the amino acid sequence identity with TfSGL is 99%). There are BGL (GH1 and GAM34681.1) and sugar transporter (GAM34682.1) genes in the vicinity of the gene encoding the TfSGL homolog. The BGL and transporter genes are also highly conserved in the vicinity of many TfSGL gene homologs. In addition, BGLs from T. reesei, an Ascomycota fungus, Trire2_120749 and Trire2_22197 (the amino acid sequence identities with the BGL from T. cellulolyticus are 49 and 69%, respectively) are highly up-regulated in the presence of Sop2 (64). Therefore, TfSGL and its homologs may play a role in the metabolism of β-1,2-glucans with the cooperative action of the BGLs and transporters, although the degrading and binding activities of these putative BGLs and transporters have not been examined.
This study makes a significant contribution to expansion of GH families and the variety of reaction mechanisms for GH enzymes. Furthermore, this study will help us to clarify the molecular evolution of SGLs from Prokaryotes and Eukaryotes. The unique distribution of the new GH162 family will lead to exploration of the physiological roles of GH162 enzymes.
Experimental procedures
Materials
T. funiculosus (NBRC100958) and P. pastoris (KM71H) were purchased from the National Institute of Technology and Evaluation (NITE, Tokyo, Japan) and Thermo Fisher Scientific, respectively. Linear β-1,2-glucans with the average DP of 25 and 77 (unless otherwise noted, the average DP of β-1,2-glucans is 77) and Sopns with DP of 2–11 were prepared using LiSOGP and CpSGL, as described previously (20, 25). Cyclic β-1,2-glucan with DP of 17–24 was kindly donated by Dr. M. Hisamatsu of Mie University (65). rSopns with DP of 6–11 were prepared by modifying Sopns at the reducing end with NaBH4 treatment as described by Shimizu et al. (26). Approximately 10% Sopn solutions were added to 1 m NaBH4 of 20 μl, followed by incubation at room temperature for 5 min or more. After 3 m acetate (15 μl) had been added to them, the rSopns were precipitated with isopropyl alcohol (1 ml). Each pellet was dissolved in a small amount of water. Then, the samples were precipitated again with isopropyl alcohol and dried up. Laminarin and carboxymethyl (CM)-cellulose were purchased from Sigma. CM-pachyman, CM-curdlan, lichenan, β-glucan from barley, tamarind xyloglucan, glucomannan, arabinogalactan, arabinan, and polygalacturonic acid were purchased from Megazyme (Wicklow, Ireland). Pustulan was purchased from Calbiochem.
Purification of SGL from T. funiculosus
T. funiculosus was grown in 2 liters of medium (comprising 0.5% β-1,2-glucan with the average DP of 25), 2 g/liter KH2PO4, 1.4 g/liter (NH4)2SO4, 0.3 g/liter Mg2SO4·7H2O, 0.4 g/liter CaCl2·2H2O, 0.3 g/liter urea, 5 mg/liter FeSO4·7H2O, 18 mg/liter MnCl2·4H2O, 20 mg/liter CoCl2·6H2O, 17 mg/liter ZnCl2, and 100 μg/ml ampicillin; adjusted to pH 6.0) (28) at 30 °C in a shaking incubator (150 rpm) for 3 days. This culture was filtrated using a gauze cloth and a glass filter and concentrated using a Vivaflow 200 (10,000 molecular weight cutoff) (Sartorius, Gottingen, Germany). An ammonium sulfate solution containing 50 mm acetate-Na buffer (pH 5.5) was added to obtain 40% saturated ammonium sulfate concentration. The supernatant filtrated using a Ministart® syringe filter (Sartorius) was loaded onto a HiTrapTM butyl HP column (5 ml; GE Healthcare, Buckinghamshire, UK) equilibrated with 50 mm acetate-Na buffer (pH 5.5) and 40% saturated ammonium sulfate (buffer A). After the unbound proteins had been washed with buffer A, a target protein was eluted using a linear gradient of ammonium sulfate (40–0% saturated concentration). All fractions containing SGL activity were collected, buffered with 50 mm MOPS-NaOH buffer (pH 7.0), and then concentrated using Amicon Ultra 30,000 molecular weight cutoff (Merck Millipore, Darmstadt, Germany). Next, the sample was loaded onto a RESOURCETMQ column (1 ml; GE Healthcare) equilibrated with 50 mm MOPS-NaOH buffer (pH 7.0). After unbound proteins had been washed out with the same buffer, the target enzyme was eluted with a linear gradient of 0–1 m NaCl in 50 mm MOPS-NaOH buffer (pH 7.0). Finally, the enzyme solution concentrated with Amicon Ultra 30,000 molecular weight cutoff to 2 mg/ml (500 μl) was loaded onto a SuperdexTM 200GL column (24 ml; GE Healthcare) equilibrated with 50 mm acetate-Na buffer (pH 5.5) containing 150 mm NaCl, and then the target enzyme was eluted with the same buffer. All purification steps were carried out using an AKTA prime plus chromatography system (GE Healthcare).
Assay of SGL and BGL activity in the purification steps
To detect SGL activity in fractionated samples during the purification steps, the reaction was performed in 100 mm acetate-Na buffer (pH 5.5) containing 0.5% β-1,2-glucan and 50 mm d(+)-glucono-1,5-lactone (GDL, Wako, Osaka, Japan) (pH 5.5) as an inhibitor of BGL and 35% (v/v) each enzyme solution at 30 °C for an hour and was stopped at 100 °C for 5 min. The Sopns released from β-1,2-glucan were detected by TLC. In the case of the BGL assay, the reactions were performed using 5 mm p-nitrophenyl-β-d-glucopyranoside (pNP-Glc, Wako) without GDL. After 9 volumes of 0.5 m Na2CO3 had been added to a reaction mixture, the mixture was added to a 96-well microtiter plate (Sigma), and then the absorbance at 405 nm of the sample was measured using a Spectramax 190 (Molecular Devices, CA) (unless otherwise noted, the absorbance of samples was measured with this instrument). One unit was defined as the amount of the enzyme required to release 1 μmol of pNP from pNP-Glc in a minute. The amount of pNP was calculated using the molar extinction coefficient of pNP (= 18,200 m−1 cm−1).
The p-hydroxybenzoic acid hydrazide (PAHBAH) method was used for evaluation of β-1,2-glucan–degrading activity (66). It should be noted that the evaluated activity includes BGL activity, because this method measures the reducing power of the products. Reactions were performed with 5% (v/v) of the enzyme solution in 100 mm acetate-Na buffer (pH 5.5) containing 0.5% β-1,2-glucan at 30 °C for an hour, and then 4 volumes of a PAHBAH solution (comprising 10 mg/ml PAHBAH, 0.1 m HCl, and 0.4 m NaOH) were added to the reaction mixtures. After heat treatment at 100 °C for 5 min, the absorbance at 405 nm was measured. Glc was used as a standard for reducing sugar, and 1 unit was defined as the amount of the enzyme required to release 1 μmol of the Glc equivalent reducing power of Sopns in a minute.
SDS-PAGE and glycosylated protein analyses
The fractionated samples obtained during the purification were separated on 10% SDS-polyacrylamide gels (67). The gels were stained with 2D-silver stain reagent (Cosmo Bio, Tokyo, Japan). In the case of the recombinant enzymes, Coomassie Brilliant Blue R-250 was used for staining. Precision Plus ProteinTM unstained standards (Bio-Rad) were used as molecular weight markers. Glycosylated protein analysis of the purified native TfSGL was performed as described below. N-Linked glycans in the native TfSGL were completely removed using glycopeptidase F (Takara Bio, Shiga, Japan) under denaturation conditions. After the samples had been loaded for SDS-PAGE, glycans and proteins in the gels were stained with a Pro-Q Emerald 300 gel stain kit (Thermo Fisher Scientific) and SYPRO® Ruby protein gel stain (Thermo Fisher Scientific), respectively, according to the manufacturer's instructions. Avidin (Wako) and glucose oxidase (Oriental Yeast, Tokyo, Japan) were used as glycoprotein markers.
TLC analysis
The native TfSGL and TfSGLr were incubated in 100 mm acetate-Na buffer (pH 4.0) containing 0.2% each substrate (Sopns with DP of 3–7, rSopns with DP of 6–8, cyclic β-1,2-glucan, and linear β-1,2-glucan) at 30 °C. After heat treatment at 100 °C for 5 min, the reaction mixtures (0.5 μl) were spotted onto TLC Silica Gel 60 F254 (Merck Millipore) plates. The plates were developed with 75% acetonitrile. As for hydrolysates of Sopns and rSopns, the plates were developed twice with a solution (acetonitrile/acetic acid/isopropyl alcohol/deionized water = 17:4:4:3). Then, the plates were soaked in a 5% (w/v) sulfuric acid/ethanol solution and heated in an oven until the spots were visualized clearly.
Size-exclusion chromatography
SEC analysis was performed as described above. Ovalbumin (44 kDa), conalbumin (75 kDa), aldolase (158 kDa), ferritin (440 kDa), and thyroglobulin (669 kDa) (GE Healthcare) were used as molecular weight markers. Blue dextran 2000 (2,000 kDa) was used to determine the void volume of the column. The molecular weight of TfSGL was calculated using Equation 1,
| (Eq. 1) |
where Kav is the gel-phase distribution coefficient; Ve is the volume required to elute each protein; Vo is the volume required to elute blue dextran 2000; and Vt is the bed volume of the column.
Identification of an SGL gene from T. funiculosus
To identify the amino acid sequence encoding an SGL gene, analysis of N-terminal and internal amino acid sequences of the purified enzyme was entrusted to Genostaff (Tokyo, Japan) and APRO Life Science Institute (Tokushima, Japan), respectively. Degenerate primers were designed based on the results of these sequence analysis (Table S4).
gDNA and total RNA were isolated from T. funiculosus cells grown in 100 ml of medium containing a 0.5% linear β-1,2-glucan with the average DP of 25 as a sole carbon source at 30 °C for 3 days (28). The collected cells were suspended in deionized water, frozen with liquid nitrogen, and then ground into powder. For gDNA preparation, a powdered sample was resuspended in the extraction buffer comprising 100 mm NaCl, 1% SDS, 2% Triton X-100, 2 units of RNase A, and 10 mm Tris-EDTA buffer (10 mm Tris-HCl and 1 mm EDTA) (pH 8.0). The solution was mixed with phenol/chloroform, and then ethanol was added to the water phase to precipitate the gDNA. After the precipitate had been dissolved in deionized water, 2 units of RNase A was added to the solution. This solution was incubated at 37 °C for 1.5 h and then mixed with phenol/chloroform. The gDNA was precipitated by the addition of isopropyl alcohol to the water phase. The pellet was washed with 70% ethanol and then dissolved in the Tris-EDTA buffer after drying up. The total RNA was extracted from the disrupted cells using ISOGEN (Wako), and the cDNA was synthesized from the total RNA using ReverTra Ace quantitative PCR RT Master Mix with a gDNA remover (Toyobo, Osaka, Japan), according to the manufacturer's instructions.
PCR was carried out using TaKaRa Ex TaqHS (Takara Bio). Basically, the reactions were performed under the following conditions: one cycle of denaturation at 98 °C for 2 min, 35 cycles of 98 °C for 10 s, 50 °C for 30 s, and 72 °C for 0.5–1.5 min, and then one cycle of 72 °C for 3.5 min. The scheme for cloning of the whole TfSGL gene was shown in Fig. 3. To amplify the gDNA region between internal peptides 1 and 2, primary PCR was performed using an F1-R1 primer pair (annealing temperature of 45 °C) and gDNA as a template, and the amplified product was purified using a High Pure PCR product purification kit (Roche Applied Science, Basel, Switzerland). Then, nested PCR was performed using an F2–R2 primer pair and the purified primary PCR product as a template. This PCR product was purified and sequenced. R3 and R4 primers were designed as specific primers based on the sequence. A degenerated primer F3 was designed based on the N-terminal peptide sequence. Primary and nested PCR were performed as described above using an F3–R3 primer pair and an F3–R4 primer pair (extension time, 2 min) in that order, and then the amplified product was purified and sequenced. Finally, F4 and R5 primers were designed based on the analyzed nucleotide sequence to amplify the 5′- and 3′-regions of the gene encoding TfSGL. After gDNA had been digested with EcoRI or HindIII (Takara Bio), self-ligation of the purified digests was performed using Ligation High, Version 2 (Toyobo). Inverse PCR was performed using an F4–R5 primer pair (annealing temperature of 60 °C, extension time of 3 min and 30 cycles) and the self-ligated sample as a template. The PCR product was purified and sequenced. Then, partial regions in the TfSGL gene were amplified using specific primer pairs (F5–R5, F6–R6, and F7–R7) and cDNA as a template to cover the whole ORF, and then the PCR product was sequenced to obtain the whole TfSGL gene sequence. The ORF, its transcription start site, and the polyadenylation signal sequence in the sequenced gDNA region were predicted with Softberry (http://www.softberry.com/)3 (68).
Sequence analysis
The N-glycosylation site, GPI modification site, and N-terminal signal peptide in TfSGL were predicted with the NetNGlyc 1.0 Server (http://www.cbs.dtu.dk/services/NetNGlyc/),3 big-PI Fungal Predictor GPI Modification Site Prediction in Fungi (http://mendel.imp.ac.at/gpi/fungi_server.html)3 (69), and the SignalP 4.1 Server (http://www.cbs.dtu.dk/services/SignalP/)3 (70), respectively. TfSGL homologs, which were used for phylogenetic analysis, were collected using the BLAST program (NCBI, http://www.ncbi.nlm.nih.gov/), and the multiple alignment was carried out using the ClustalW program (http://clustalw.ddbj.nig.ac.jp/).3 Phylogenetic analysis of TfSGL homologs was carried out using the neighbor joining method (MEGA7 program) (http://www.megasoftware.net/)3 (71). GPI modification sites in TfSGL homologs annotated as GPI-anchor proteins were predicted with the same server as described above.
Cloning, expression, and purification of TfSGLr using P. pastoris
The TfSGL gene without the region encoding the residues at the N terminus (1–21 amino acids) was amplified by PCR using KOD-Plus (Toyobo) as a DNA polymerase according to the manufacturer's instructions. First, PCR was performed using an F8–R8 primer pair and the cDNA pool of T. funiculosus as a template. Second, PCR was performed to add a His6 tag to the N terminus of the TfSGL gene using an F9–R9 primer pair. The amplified product digested with EcoRI and NotI (Takara Bio) was ligated into the pPICZαB vector (Thermo Fisher Scientific) digested with the same restriction enzymes to fuse the secretion signal peptide for P. pastoris at the N terminus of the His6 tag. The plasmid produced in E. coli XL1-blue was linearized with PmeI (New England Biolabs). The plasmid sample was purified with a phenol/chloroform/isoamyl alcohol solution (Sigma) and by ethanol precipitation. The purified plasmid was transformed into P. pastoris in the presence of 0.5 mg/ml Zeocin. TfSGLr was expressed according to the manufacturer's instructions for an EasySelectTM Pichia expression kit (Thermo Fisher Scientific). The transformant was harvested using 2 liters of buffered minimal glycerol medium at 30 °C for about 18 h (until A600 = 2–6), and then the cells were suspended in 200 ml of buffered minimal methanol medium. Methanol (0.5%) was added to the suspension to induce TfSGLr, and enzyme induction was performed at 30 °C for 4–6 days. MOPS-NaOH buffer containing NaCl was added to the culture filtrate until the concentrations reached 500 mm NaCl and 50 mm MOPS-NaOH buffer (pH 7.0), and then the mixture was filtrated with a 0.45-μm filter (Sartorius). The sample was loaded onto a HisTrap FF crude column (5 ml; GE Healthcare) equilibrated with 50 mm MOPS-NaOH buffer (pH 7.0) containing 500 mm NaCl (buffer B). After unbound proteins had been washed out using the same buffer containing 10 mm imidazole, TfSGLr was eluted using a linear gradient of imidazole concentration (10–300 mm) in buffer B. The enzyme solution was buffered with 5 mm acetate-Na buffer (pH 4.0) using Amicon Ultra 30,000 molecular weight cutoff. The absorbance at 280 nm of the sample was measured using a spectrophotometer V-560 (Jasco, Tokyo, Japan), and the concentration of the enzyme was calculated using the theoretical molecular mass of TfSGLr (56,554 Da) and a molar extinction coefficient of 123,020 m−1·cm−1 (72).
General properties
To determine the optimum pH, TfSGLr (3.2 μg/ml) was incubated in 100 mm various buffers containing 0.2% β-1,2-glucan at 30 °C for 15 min and then heated at 100 °C for 5 min to stop the reaction. The reducing power of Sopns released from the β-1,2-glucan was measured by the PAHBAH method. The optimum temperature for TfSGLr was determined after the enzyme had been reacted in 100 mm acetate-Na buffer (pH 4.0) at each temperature (0–70 °C). To examine the pH stability for TfSGLr, the purified enzyme (6.4 μg/ml) was incubated in 20 mm various buffers at 30 °C for an hour, and then the reaction was carried out in 100 mm acetate-Na buffer (pH 4.0) containing 0.2% β-1,2-glucan at 30 °C for 15 min. To determine the temperature stability, TfSGLr (6.4 μg/ml) was incubated in 100 mm acetate-Na buffer (pH 4.0) at each temperature (0–70 °C) for an hour, and then the reaction was carried out under the same conditions as for pH stability.
Substrate specificity
TfSGLr (3.2 μg/ml) was incubated in 100 mm acetate-Na buffer (pH 4.0) containing each substrate (0.2% β-glucan, 0.1% glucomannan, 0.2% tamarind xyloglucan, 0.2% arabinan, 0.1% polygalacturonic acid, 0.0125% lichenan, 0.0125% laminarin, 0.05% pustulan, 0.1% CMC, 0.05% CM-curdlan, 0.025% CM-pachyman, 0.006% arabinogalactan, or 0.2% β-1,2-glucan) at 30 °C for 15 min, and then the amount of oligosaccharides released from each polysaccharide was measured by the PAHBAH method. Glc was used as a standard. The GOPOD method was used for determination of activity releasing Sop2 from the β-1,2-glucan, as described below. The reaction mixtures were incubated with BGL (1 mg/ml) from almonds (Oriental Yeast) at 50 °C for 2 h in 100 mm acetate-Na buffer (pH 5.0) to hydrolyze Sop2 specifically. Finally, the GOPOD solution (9 volumes of the mixture) was added to the mixture, followed by incubation at 45 °C for 20 min. The amounts of Glc in the samples were calculated by measuring the absorbance at 510 nm of the samples. Glc was used as a standard. One unit was defined as the amount of the enzyme required to release 1 μmol of Sop2 in a minute.
Kinetic analysis
To determine the kinetic parameters of TfSGLr for β-1,2-glucan and Sop5, Sop2 released from these substrates was quantified. After TfSGLr (1.5 and 7.5 μg/ml for hydrolysis of β-1,2-glucan and Sop5, respectively) had been incubated at 30 °C for 10 min in 20 mm acetate-Na buffer (pH 4.0) containing various concentrations of Sop5 (0.12–2.5 mm) or β-1,2-glucan (5.0 × 10−3 −0.13 mm (= 6.3 × 10−2 −1.6 (mg/ml)), the reactions were stopped by heat treatment at 100 °C for 5 min. Sop2-releasing activity was determined using the GOPOD method, as described in the previous section. The experimental data obtained on hydrolysis of β-1,2-glucan and Sop5 were fitted to the Michaelis-Menten Equation 2,
| (Eq. 2) |
where v is initial velocity; [E] is enzyme concentration; kcat is turnover number; [S] is β-1,2-glucan concentration; and Km is Michaelis constant, and the substrate inhibition Equation 3,
| (Eq. 3) |
where v is initial velocity; [E] is enzyme concentration; kcat is turnover number; [S] is Sop5 concentration; Km is Michaelis constant; and Ki is substrate inhibition constant. The KaleidaGraph program Version 3.51 was used for their regression analysis.
Stereochemical analysis of the reaction products by 1H NMR
To determine which reaction mechanism TfSGL follows, an anomer inverting or retaining mechanism, 1H NMR analysis was performed on the reaction products released from β-1,2-glucan by TfSGLr. TfSGLr (0.12 mg/ml) was incubated at room temperature in 5 mm acetate-Na buffer (pH 4.0) containing 1.5% β-1,2-glucan with the average DP of 25 and 90% D2O. 1H NMR spectra of the sample were recorded using a Bruker Advance 600 spectrometer (Bruker BioSpin, Rheinstetten, Germany). The assignment of the signals of Sop2–4 and linear β-1,2-glucan was based on the previous studies (19, 25). Acetate in the buffer was used as an internal standard for calculation of the relative peak area. To use acetone as an external standard for calibration of chemical shifts, as performed in the previous study (25), a droplet of acetone was added to the reaction mixture after the reaction had finished. Because chemical shifts derived from the C1 proton at the reducing end of β-anomer products are completely masked by the chemical shift derived from H2O, the chemical shift of the C2 proton at the nonreducing end was used as the total signal of α- and β-anomers of the reaction products. The C2 protons were observed at almost the same chemical shifts regardless of the anomer, as described by Abe et al. (25).
Polarimetric analysis of the reaction products
To confirm the reaction mechanism of TfSGL, the time course of the degree of optical rotation in the reaction mixture was monitored. TfSGLr (1 mg/ml) was incubated at room temperature in 20 mm acetate-Na buffer (pH 4.0) containing 2% β-1,2-glucan with the average DP of 25, and the degree of the optical rotation of the reaction mixture was measured using a Jasco p1010 polarimeter (Jasco). Several droplets of 35% aqueous ammonia were added at 235 s after the reaction start to enhance mutarotation of the anomers.
Analysis of the reaction products from 3-deoxy Sopn-derivatives with TfSGLr
3dSop5–6 and 3′dSop5 were synthesized using LiSOGP from 3 and 3′-deoxy-Sop2, which are Sop2 derivatives having 3-deoxyglucose moieties at the reducing and nonreducing ends, respectively. These Sop2 derivatives were prepared as described previously (73). The reaction for synthesis of 3dSop5–6 or 3′dSop5 was performed in 50 mm MOPS-NaOH buffer (pH 7.5) containing 10 or 25 mm α-d-glucose-1-phosphate, 5 mm of each Sop2 derivative, and 2 mg/ml LiSOGP at 30 °C for an hour or 8 days, respectively. The reactions were stopped at 80 °C for 5 min, and the supernatants were collected after centrifugation. The supernatants were added to AmberliteTM MB-4 (Organo, Tokyo, Japan) to remove unreacted α-d-glucose-1-phosphate. These samples were concentrated to 10 μl with a centrifugal evaporator EC-57CS (Sakuma, Tokyo, Japan). Then, 3dSop2–6 and 3′dSop2–5 were separated with a Shimadzu LC-9A high-performance liquid chromatograph equipped with an SCL-6B system controller and SIL-6B autoinjector (Shimadzu, Kyoto, Japan) using an Asahipak NH2P-50 4E column (Shodex, Tokyo, Japan) with distilled water and acetonitrile (= 35:65 (v/v)) as the eluent at a flow rate of 1 ml/min and with detection with a Refractive Index Detector (RI-8010) (Tosoh Bioscience, Tokyo, Japan). The enzymatic reactions of TfSGLr for 3dSop5 and 3′dSop5 were performed in 50 mm acetate-Na buffer (pH 4.0) containing 2 mg/ml TfSGLr and each derivative at 30 °C for 180 min. When using 3dSop6 as a derivative, the concentration of TfSGLr and the reaction time were changed to 5 μg/ml and 30 min, respectively. Although the amounts of 3dSop5–6 and 3′dSop5 were too small to be determined, their concentrations in the reaction mixtures are estimated to have been 0.5, 2.5, and 4.2 μm, respectively, based on comparison with the peak area of standard Sop5–6. These samples were concentrated with a centrifugal evaporator, and the reducing ends of sugars were fluorescently labeled with 2-aminobenzamide. First, 2-aminobenzamide (136 mg) was added to sodium cyanoborohydride (35 mg), and then they were dissolved in methanol (350 μl) and acetic acid (41 μl) to prepare the fluorescent reagent. The samples (10 μl) were mixed with 40 μl of the fluorescent reagent, followed by incubation at 80 °C for an hour. The mixtures were extracted with chloroform (200 μl) and distilled water (200 μl) after cooling to room temperature. The standard solutions (containing 1 mm Glc and Sop2–6, 3dSop2, 3dSop3, 3dSop5, 3dSop6, 3′Sop2, or 3′dSop5) were labeled in the same way. Finally, the supernatants were collected by centrifugation. The solutions (3dSop5–6 and 3′dSop5) were dried up and then the samples were dissolved in distilled water (20 μl). The fluorescently-labeled samples of 10 μl were loaded onto a TSKgel Amide-80 column (Tosoh Bioscience) and separated and analyzed with a Waters W600 HPLC solvent delivery system (Waters) with detection with a 2475 fluorescence detector (Waters) at 422 nm.
Crystallography
To remove N-glycans from the purified TfSGLr, the enzyme (5 mg) was treated with 50 units of endoglycosidase H (New England Biolabs) in 50 mm citric acid buffer (pH 5.5) at 20 °C overnight. The sample was purified using a HisTrap FF crude column as described above. The enzyme solution was buffered with 5 mm acetate-Na buffer (pH 4.0) using Amicon Ultra 30,000 molecular weight cutoff and then concentrated to 20 mg/ml. The initial screening for TfSGLr crystal was performed using Wizard classic 1 and 2 block (Rigaku, Tokyo, Japan). The initial crystal condition for TfSGLr was a mixture of TfSGLr (10 mg/ml, 1 μl) and a solution (1 μl, comprising 0.1 m sodium phosphate dibasic/citrate buffer (pH 4.2), 0.2 m NaCl, and 20% (w/v) PEG 8000). Iodinated derivatives of TfSGLr crystals were prepared by vaporizing iodine labeling (74) to determine the initial phase. The TfSGLr crystals for the labeling were prepared under a modified condition as follows. The enzyme solution (20 mg/ml, 1 μl) was mixed with 3 volumes of a reservoir solution comprising 0.1 m phosphate-citrate buffer (pH 4.8), 0.1 m NaCl, and 20% (w/v) PEG 3350, followed by incubation at 25 °C for 5 days. A drop of iodine/potassium iodide solution comprising 135 mg/ml iodine and 250 mg/ml potassium iodide (1.5 μl) was placed beside a drop of TfSGLr. The drop was incubated at 25 °C for 3 days to label the TfSGLr with iodine (74). The apo crystal for data collection was obtained at 25 °C for 3 days by mixing TfSGLr (20 mg/ml, 0.8 μl) and a reservoir solution (3 μl, containing 0.1 m citrate-Na buffer (pH 4.1), 0.1 m NaCl, and 20% (w/v) PEG 2000).
The crystal for the Sop2 complex was obtained at 25 °C for 3 days by mixing TfSGLr (20 mg/ml, 1 μl) and a reservoir solution (3 μl, comprising 0.1 m phosphate-citrate buffer (pH 4.6), 0.1 m NaCl, and 20% (w/v) PEG 3350). The crystal for the Sop7 complex was obtained at 25 °C for 3 days by mixing the E262Q mutant (10 mg/ml, 1.6 μl) and a reservoir solution (3 μl, comprising 0.1 m citrate-Na buffer (pH 4.0), 0.1 m NaCl, and 25% (w/v) PEG 2000). These crystals were soaked in the reservoir solution supplemented with 25% (w/v) PEG 400 for cryoprotection and then soaked in a solution containing 50 mm Sop2 or 5% (w/v) β-1,2-glucan with the average DP of 25, respectively. Each crystal was soaked in the reservoir solution supplemented with 25% (w/v) PEG 400 as a cryoprotectant and kept at 100 K in a nitrogen-gas stream during data collection. All X-ray diffraction data were collected on a beamline (BL-5A) at Photon Factory (Tsukuba, Japan).
The initial phase information was obtained by the iodide single-wavelength anomalous diffraction phasing method using the diffraction data for the iodinated TfSGLr crystal collected at 1.5 Å. The program used was crank2 program (shelxc, shelxd, refmac, solomon, multicomb, buccaneer, parrot) in ccp4 (http://www.ccp4.ac.uk/).3 The diffraction data for the apo TfSGLr crystal and the ligand-bound TfSGLr crystals were collected at 1.0 Å and processed with X-ray Detector Software (http://xds.mpimf-heidelberg.mpg.de/)3 (75) and the aimless program (http://www.ccp4.ac.uk/).3 First, the apo TfSGLr structure was determined using the initial phase of TfSGLr; second, the other structures of TfSGLr were revealed by molecular replacement using the apo TfSGLr as a model structure. The molecular replacement, auto model building, and refinement were performed using the molrep program, buccaneer program, refmac5 program and coot program, respectively (http://www.ccp4.ac.uk/)3 (76–79). Structural comparison of the two molecules in an asymmetric unit was performed with the Dali pairwise comparison server (http://ekhidna.biocenter.helsinki.fi/dali_lite/start)3 (80). A structural homology search was performed with the Dali server (http://ekhidna.biocenter.helsinki.fi/dali_server/)3 (31). The secondary structure was assigned with the DSSP program (http://swift.cmbi.ru.nl/gv/dssp/)3 (81), and the multiple alignment and the structure-based alignment, including that of the secondary structures, were visualized using the ESPript 3.0 server (http://espript.ibcp.fr/ESPript/ESPript/)3 (82). All structures in the figures were designed with the PyMOL program (https://pymol.org/2/).3 The overall structures of TfSGLr and CpSGL were superimposed with the PDBeFold server (http://www.ebi.ac.uk/msd-srv/ssm/)3 (32, 33).
MD simulation
Initial structures for MD simulation were taken from the crystal structures of TfSGL in the apo form (6IMU, chain A) and in complex with Sop7 (6IMW, chain B). Because the apo crystal structure has two alternative conformations of His-316 (Fig. 9A), one adopts a different orientation from the Sop7 complex and the other adopts the same orientation with the Sop7 complex, the two initial structures (termed apo1 and apo2, respectively) were prepared. The N- and C-terminal residues were capped with acetyl and N-methyl groups, respectively. N-Acetylglucosamines (GlcNAc) derived from N-linked glycan were removed from the structures. Disulfide bonds were introduced between Cys-27 and Cys-516, between Cys-230 and Cys-251, and between Cys-345 and Cys-499. The mutation of catalytic acid residue (E262Q) in the Sop7 complex was manually reverted to that of the WT. The protonation states of amino acid residues were determined basically based on PROPKA 3.1 (41) (assuming pH 4.0), except for a few residues of which the protonation states were determined based on visual inspection of the hydrogen bond network. In the apo2 and the Sop7 complex, His-57 and His-316 were protonated only on the Nϵ2 atoms, and the other histidine residues were protonated on both the Nδ1 and Nϵ2 atoms. In apo1, His-57 was protonated only on the Nϵ2 atoms, and the other histidine residues were protonated on both the Nδ1 and Nϵ2 atoms. Glu-24, Glu-36, Glu-82, Glu-144, Glu-180, Glu-193, Glu-226, Glu-246, Glu-262, Glu-286, Glu-323, Glu-389, Glu-430, Glu-478, Glu-481, and Glu-503 of the apo and Sop7 complex structures were protonated, whereas other glutamate residues were deprotonated. In both the apo structures, Asp-48, Asp-111, Asp-168, Asp-177, Asp-234, Asp-259, Asp-278, Asp-369, and Asp-405 were protonated, and the other aspartate residues were deprotonated. In the Sop7 complex, Asp-48, Asp-111, Asp-177, Asp-234, Asp-259, Asp-328, and Asp-405 were protonated, and the other aspartate residues were deprotonated. All the arginine and lysine residues were protonated. The systems for MD simulation were constructed by immersing the initial models in cubic water boxes where the distance between protein atoms and the closest boundary was at least 10 Å. Chloride ions were added to the systems for neutralization. The LEaP module of AmberTools 18 (83) was used to construct the systems. Amber ff14SB (84) and GLYCAM06j (85) force-field parameters and the TIP3P model (86) were used for the protein, carbohydrate, and water, respectively.
MD simulations were performed using GROMACS 2018 (87). After energy minimization and equilibration, a production MD run was carried out for 100 ns. During the MD simulation, the temperature was controlled at 303 K using the velocity rescaling thermostat (88), and the pressure was controlled at 1.0 × 105 pascal using the weak coupling method (89). Bond lengths involving the hydrogen atoms were constrained using the LINCS algorithm (90) to allow the use of a 2-fs time step. Electrostatic interaction was calculated using the particle mesh Ewald method (91). MD trajectories were recorded every 10 ps.
Cα RMSD and RMSF values were calculated using the gmx_rmsd and gmx_rmsf modules of GROMACS, respectively. PCA was carried out for a combined ensemble composed of ensembles from the simulations of the apo2 and the Sop7 complex using the method described previously (92). Conformational distributions of the apo2 and the Sop7 complex were calculated by separately projecting the trajectories onto the principal axes. The gmx_rmsf, gmx_covar, and gmx_anaeig modules were used for PCA. Representative structures from the MD ensembles were determined based on cluster analysis using the gmx_cluster module with the Cα RMSD cutoff of 0.8 Å.
Mutational analysis
The plasmids of TfSGLr mutants were constructed using a PrimeSTAR mutagenesis basal kit (Takara Bio) according to the manufacturer's instructions. PCRs were performed using appropriate primer pairs (Table S4) and the TfSGLr plasmid as a template. The transformation to P. pastoris and the expression and purification of TfSGLr mutants were performed in the same way as WT TfSGLr. The enzymatic reactions were performed basically in the same way as for determination of substrate specificity. The concentrations of mutants (0.001–2.6 mg/ml) and reaction time (0–2.5 h) were changed depending on the mutants. Color development was performed using the GOPOD method as described under “Kinetic analysis.” For several mutants, the MBTH method (colorimetric determination method for Sopns) (39) was also used for determination of specific activity, as described below.
MBTH method
The WT TfSGLr and TfSGLr mutants (0.001–2.6 mg/ml) were incubated in 20 mm acetate-Na buffer (pH 4.0) containing 0.2% β-1,2-glucan at 30 °C for 0–2.5 h. After appropriate intervals, 15-μl aliquots of the reaction mixtures were taken and heated at 100 °C for 5 min. The samples were mixed with 15 μl of 0.5 n NaOH and then an MBTH solution (15 μl, comprising 3 mg/ml MBTH and 1 mg/ml 1,4-DTT). The mixtures were incubated at 80 °C for 15 min. A solution (30 μl, comprising 0.5% FeNH4(SO4)2, 0.5% H3NSO3 and 0.25 N HCl) was added to the mixtures, and then 75 μl of distilled water was also added after cooling to room temperature. The absorbance at 620 nm was measured. Sop2 (0.5–2.5 mm) was used as a standard for reducing sugar, and 1 unit was defined as the amount of the enzyme required to release 1 μmol of the Sop2 equivalent reducing power in a minute.
Author contributions
N. T., M. N., K. A., T. T., A. M., S. Komba, H. N., and H. T. data curation; N. T. and M. N. formal analysis; N. T., M. N., and H. T. validation; N. T., M. N., M. N.-N., H. M., S. Kamisuki, H. A., Y. T., N. S., K. A., T. T., T. Y., and S. Komba investigation; N. T., M. N., M. N.-N., H. M., S. Kamisuki, H. A., Y. T., N. S., K. A., T. T., F. S., T. K., and S. Komba methodology; N. T. and M. N. writing-original draft; N. T., M. N., M. N.-N., H. M., S. Kamisuki, H. A., Y. T., N. S., K. A., T. T., A. M., T. Y., F. S., T. K., S. Komba, H. N., and H. T. writing-review and editing; M. N. and H. T. conceptualization; M. N., H. A., Y. T., N. S., K. A., S. Komba, H. N., and H. T. resources; M. N., A. M., and H. T. supervision; M. N., H. N., and H. T. project administration.
Supplementary Material
Acknowledgments
We are grateful to Dr. M. Kitaoka for the substrate preparation and Dr. M. Hisamatsu and Dr. N. Isono for providing the cyclic β-1,2-glucan. We also thank Dr. S. Fushinobu, Dr. T. Arakawa, Dr. N. Watanabe, and Dr. T. Hikage for the useful advice and assistance regarding structural analysis. We appreciate the help of all the staff at the Photon Factory for the X-ray data collection (Proposal No. 2016G619).
This work was supported in part by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant JP18am0101107. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5 and Tables S1–S4.
The atomic coordinates and structure factors (codes 6IMU, 6IMV, and 6IMW) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The nucleotide sequences of gDNA and cDNA for TfSGL gene have been deposited into the DNA Data Bank of Japan database under accession number LC430902.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- CAZy
- Carbohydrate-Active enZYmes Database
- GH
- glycoside hydrolase
- DP
- degree of polymerization
- Sopn
- sophoro-oligosaccharide (β-1,2-gluco-oligosaccharide, n denotes DP)
- SOGP
- 1,2-β-d-oligoglucan phosphorylase
- LiSOGP
- SOGP from L. innocua
- BGL
- β-glucosidase
- LiBGL
- BGL from L. innocua
- LiSO-BP
- Sopns-binding protein from L. innocua
- SGL
- endo-β-1,2-glucanase
- CpSGL
- SGL from C. pinensis
- PdSGL
- sophorosyl hydrolase from P. distasonis
- TfSGL
- SGL from T. funiculosus
- TfSGLr
- recombinant TfSGL
- SEC
- size-exclusion chromatography
- rSopn
- reducing end–modified Sopn (n denotes DP)
- Glc
- glucose
- RMSD
- root mean square deviation
- PDB
- Protein Data Bank
- MBTH
- 3-methyl-2-benzothiazolinone hydrazine
- 3dSopn
- Sopn deoxygenated at the 3-hydroxy group of the Glc moiety at the reducing end (n denotes DP)
- 3′dSopn
- Sopn deoxygenated at the 3-hydroxy group at the second Glc moiety from the reducing end (n denotes DP)
- MD
- molecular dynamics
- Cel6A
- cellobiohydrolase from T reesei
- CM
- carboxymethyl
- GDL
- d(+)-glucono-1,5-lactone
- pNP-Glc
- p-nitrophenyl-β-d-glucopyranoside
- PAHBAH
- p-hydroxybenzoic acid hydrazide
- RMSF
- root mean square fluctuation
- PCA
- principal component analysis
- GPI
- glycosylphosphatidylinositol.
References
- 1. Henrissat B. (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 280, 309–316 10.1042/bj2800309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ndeh D., Rogowski A., Cartmell A., Luis A. S., Baslé A., Gray J., Venditto I., Briggs J., Zhang X., Labourel A., Terrapon N., Buffetto F., Nepogodiev S., Xiao Y., Field R. A., et al. (2017) Complex pectin metabolism by gut bacteria reveals novel catalytic functions. Nature 544, 65–70 10.1038/nature21725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dell A., York W. S., McNeil M., Darvill A. G., and Albersheim P. (1983) The cyclic structure of β-d-(1→2)-linked d-glucans secreted by Rhizobia and Agrobacteria. Carbohydr. Res. 117, 185–200 10.1016/0008-6215(83)88086-0 [DOI] [Google Scholar]
- 4. Koizumi K., Okada Y., Utamura T., Hisamatsu M., and Amemura A. (1984) Further studies on the separation of cyclic (1→2)-β-d-glucans (cyclosophoraoses) produced by Rhizobium meliloti IFO 13336, and determination of their degrees of polymerization by high-performance liquid chromatography. J. Chromatogr. 299, 215–224 10.1016/S0021-9673(01)97833-1 [DOI] [Google Scholar]
- 5. Bundle D. R., Cherwonogrodzky J. W., and Perry M. B. (1988) Characterization of Brucella polysaccharide B. Infect. Immun. 56, 1101–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Breedveld M. W., and Miller K. J. (1995) Synthesis of glycerophosphorylated cyclic (1,2)-β-glucans in Rhizobium meliloti strain 1021 after osmotic shock. Microbiology 141, 583–588 10.1099/13500872-141-3-583 [DOI] [PubMed] [Google Scholar]
- 7. Hisamatsu M. (1992) Cyclic (1→2)-β-d-glucans (cyclosophorans) produced by Agrobacterium and Rhizobium species. Carbohydr. Res. 231, 137–146 10.1016/0008-6215(92)84014-J [DOI] [PubMed] [Google Scholar]
- 8. Dylan T., Ielpi L., Stanfield S., Kashyap L., Douglas C., Yanofsky M., Nester E., Helinski D. R., and Ditta G. (1986) Rhizobium meliloti genes required for nodule development are related to chromosomal virulence genes in Agrobacterium tumefaciens. Proc. Natl. Acad. Sci. U.S.A. 83, 4403–4407 10.1073/pnas.83.12.4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rigano L. A., Payette C., Brouillard G., Marano M. R., Abramowicz L., Torres P. S., Yun M., Castagnaro A. P., Oirdi M. E., Dufour V., Malamud F., Dow J. M., Bouarab K., and Vojnov A. A. (2007) Bacterial cyclic β-(1,2)-glucan acts in systemic suppression of plant immune responses. Plant Cell 19, 2077–2089 10.1105/tpc.106.047944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dylan T., Nagpal P., Helinski D. R., and Ditta G. S. (1990) Symbiotic pseudorevertants of Rhizobium meliloti ndv mutants. J. Bacteriol. 172, 1409–1417 10.1128/jb.172.3.1409-1417.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roset M. S., Ibañez A. E., de Souza Filho J. A., Spera J. M., Minatel L., Oliveira S. C., Giambartolomei G. H., Cassataro J., and Briones G. (2014) Brucella cyclic β-1,2-glucan plays a critical role in the induction of splenomegaly in mice. PLoS ONE 9, e101279 10.1371/journal.pone.0101279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Altabe S., Iñón de Iannino N., de Mendoza D., and Ugalde R. A. (1990) Expression of the Agrobacterium tumefaciens chvB virulence region in Azospirillum spp. J. Bacteriol. 172, 2563–2567 10.1128/jb.172.5.2563-2567.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dylan T., Helinski D. R., and Ditta G. S. (1990) Hypoosmotic adaptation in Rhizobium meliloti requires β-(1→2)-glucan. J. Bacteriol. 172, 1400–1408 10.1128/jb.172.3.1400-1408.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miller K. J., Kennedy E. P., and Reinhold V. N. (1986) Osmotic adaptation by Gram-negative bacteria: possible role for periplasmic oligosaccharides. Science 231, 48–51 10.1126/science.3941890 [DOI] [PubMed] [Google Scholar]
- 15. Javvadi S., Pandey S. S., Mishra A., Pradhan B. B., and Chatterjee S. (2018) Bacterial cyclic β-(1,2)-glucans sequester iron to protect against iron-induced toxicity. EMBO Rep. 19, 172–186 10.15252/embr.201744650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reyes Suárez E., Bugden S. M., Kai F. B., Kralovec J. A., Noseda M. D., Barrow C. J., and Grindley T. B. (2008) First isolation and structural determination of cyclic β-(1→2)-glucans from an alga, Chlorella pyrenoidosa. Carbohydr. Res. 343, 2623–2633 10.1016/j.carres.2008.07.009 [DOI] [PubMed] [Google Scholar]
- 17. Guidolin L. S., Ciocchini A. E., Iñón de Iannino N., and Ugalde R. A. (2009) Functional mapping of Brucella abortus cyclic β-1,2-glucan synthase: identification of the protein domain required for cyclization. J. Bacteriol. 191, 1230–1238 10.1128/JB.01108-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ciocchini A. E., Guidolin L. S., Casabuono A. C., Couto A. S., de Iannino N. I., and Ugalde R. A. (2007) A glycosyltransferase with a length-controlling activity as a mechanism to regulate the size of polysaccharides. Proc. Natl. Acad. Sci. U.S.A. 104, 16492–16497 10.1073/pnas.0708025104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakajima M., Toyoizumi H., Abe K., Nakai H., Taguchi H., and Kitaoka M. (2014) 1,2-β-Oligoglucan phosphorylase from Listeria innocua. PLoS ONE 9, e92353 10.1371/journal.pone.0092353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abe K., Nakajima M., Kitaoka M., Toyoizumi H., Takahashi Y., Sugimoto N., Nakai H., and Taguchi H. (2015) Large-scale preparation of 1,2-β-glucan using 1,2-β-oligoglucan phosphorylase. J. Appl. Glycosci. 62, 47–52 10.5458/jag.jag.JAG-2014_011 [DOI] [Google Scholar]
- 21. Nakajima M., Yoshida R., Miyanaga A., Abe K., Takahashi Y., Sugimoto N., Toyoizumi H., Nakai H., Kitaoka M., and Taguchi H. (2016) Functional and structural analysis of a β-glucosidase involved in β-1,2-glucan metabolism in Listeria innocua. PLoS ONE 11, e0148870 10.1371/journal.pone.0148870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abe K., Sunagawa N., Terada T., Takahashi Y., Arakawa T., Igarashi K., Samejima M., Nakai H., Taguchi H., Nakajima M., and Fushinobu S. (2018) Structural and thermodynamic insights into β-1,2-gluco-oligosaccharide capture by a solute-binding protein in Listeria innocua. J. Biol. Chem. 293, 8812–8828 10.1074/jbc.RA117.001536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ishiguro R., Tanaka N., Abe K., Nakajima M., Maeda T., Miyanaga A., Takahashi Y., Sugimoto N., Nakai H., and Taguchi H. (2017) Function and structure relationships of a β-1,2-gluco-oligosaccharide-degrading β-glucosidase. FEBS Lett. 591, 3926–3936 10.1002/1873-3468.12911 [DOI] [PubMed] [Google Scholar]
- 24. Nakajima M., Tanaka N., Furukawa N., Nihira T., Kodutsumi Y., Takahashi Y., Sugimoto N., Miyanaga A., Fushinobu S., Taguchi H., and Nakai H. (2017) Mechanistic insight into the substrate specificity of 1,2-β-oligoglucan phosphorylase from Lachnoclostridium phytofermentans. Sci. Rep. 7, 42671 10.1038/srep42671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abe K., Nakajima M., Yamashita T., Matsunaga H., Kamisuki S., Nihira T., Takahashi Y., Sugimoto N., Miyanaga A., Nakai H., Arakawa T., Fushinobu S., and Taguchi H. (2017) Biochemical and structural analyses of a bacterial endo-β-1,2-glucanase reveal a new glycoside hydrolase family. J. Biol. Chem. 292, 7487–7506 10.1074/jbc.M116.762724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shimizu H., Nakajima M., Miyanaga A., Takahashi Y., Tanaka N., Kobayashi K., Sugimoto N., Nakai H., and Taguchi H. (2018) Characterization and structural analysis of a novel exo-type enzyme acting on β-1,2-gluco-oligosaccharides from Parabacteroides distasonis. Biochemistry 57, 3849–3860 10.1021/acs.biochem.8b00385 [DOI] [PubMed] [Google Scholar]
- 27. Kitahata S., and Edagawa S. (1987) Cyclic (1→2)-β-d-glucan-hydrolyzing enzymes from Acremonium sp. 15 purification and some properties of endo-(1→2)-α-d-glucanase and β-d-glucosidase. Agric. Biol. Chem. 51, 2701–2708 10.1080/00021369.1987.10868457 [DOI] [Google Scholar]
- 28. Reese E. T., Parrish F. W., and Mandels M. (1961) β-d-1,2-Glucanases in fungi. Can. J. Microbiol. 7, 309–317 10.1139/m61-038 [DOI] [PubMed] [Google Scholar]
- 29. Tuohy M. G., Puls J., Claeyssens M., Vrsanská M., and Coughlan M. P. (1993) The xylan-degrading enzyme system of Talaromyces emersonii: novel enzymes with activity against aryl β-d-xylosides and unsubstituted xylans. Biochem. J. 290, 515–523 10.1042/bj2900515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Furtado G. P., Ribeiro L. F., Lourenzoni M. R., and Ward R. J. (2013) A designed bifunctional laccase/β-1,3–1,4-glucanase enzyme shows synergistic sugar release from milled sugarcane bagasse. Protein Eng. Des. Sel. 26, 15–23 10.1093/protein/gzs057 [DOI] [PubMed] [Google Scholar]
- 31. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 10.1093/nar/gkq366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 10.1107/S0907444904026460 [DOI] [PubMed] [Google Scholar]
- 33. Krissinel E., and Henrick K. (2004) Common subgraph isomorphism detection by backtracking search. Software Pract. Exper. 34, 591–607 10.1002/spe.588 [DOI] [Google Scholar]
- 34. Ficko-Blean E., Stuart C. P., and Boraston A. B. (2011) Structural analysis of CPF_2247, a novel α-amylase from Clostridium perfringens. Proteins 79, 2771–2777 10.1002/prot.23116 [DOI] [PubMed] [Google Scholar]
- 35. Cremer D., and Pople J. A. (1975) A general definition of ring puckering coordinates. J. Am. Chem. Soc. 97, 1354–1358 10.1021/ja00839a011 [DOI] [Google Scholar]
- 36. Davies G. J., Mackenzie L., Varrot A., Dauter M., Brzozowski A. M., Schülein M., and Withers S. G. (1998) Snapshots along an enzymatic reaction coordinate: analysis of a retaining β-glycoside hydrolase. Biochemistry 37, 11707–11713 10.1021/bi981315i [DOI] [PubMed] [Google Scholar]
- 37. Miyazaki T., Nishikawa A., and Tonozuka T. (2016) Crystal structure of the enzyme-product complex reveals sugar ring distortion during catalysis by family 63 inverting α-glycosidase. J. Struct. Biol. 196, 479–486 10.1016/j.jsb.2016.09.015 [DOI] [PubMed] [Google Scholar]
- 38. McCarter J. D., and Withers S. G. (1994) Mechanisms of enzymatic glycoside hydrolysis. Curr. Opin. Struct. Biol. 4, 885–892 10.1016/0959-440X(94)90271-2 [DOI] [PubMed] [Google Scholar]
- 39. Kobayashi K., Aramasa H., Nakai H., Nakajima M., and Taguchi H. (2018) Colorimetric determination of β-1,2-gluco-oligosaccharides for an enzymatic assay using 3-methyl-2-benzothiazolinonehydrazone. Anal. Biochem. 560, 1–6 10.1016/j.ab.2018.08.021 [DOI] [PubMed] [Google Scholar]
- 40. de Grotthuss C. J. (2006) Memoir on the decomposition of water and of the bodies that it holds in solution by means of galvanic electricity. Biochim. Biophys. Acta 1757, 871–875 10.1016/j.bbabio.2006.07.004 [DOI] [PubMed] [Google Scholar]
- 41. Olsson M. H., Søndergaard C. R., Rostkowski M., and Jensen J. H. (2011) PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 10.1021/ct100578z [DOI] [PubMed] [Google Scholar]
- 42. Nakae S., Ito S., Higa M., Senoura T., Wasaki J., Hijikata A., Shionyu M., Ito S., and Shirai T. (2013) Structure of novel enzyme in mannan biodegradation process 4-O-β-d-mannosyl-d-glucose phosphorylase MGP. J. Mol. Biol. 425, 4468–4478 10.1016/j.jmb.2013.08.002 [DOI] [PubMed] [Google Scholar]
- 43. Koivula A., Ruohonen L., Wohlfahrt G., Reinikainen T., Teeri T. T., Piens K., Claeyssens M., Weber M., Vasella A., Becker D., Sinnott M. L., Zou J. Y., Kleywegt G. J., Szardenings M., Ståhlberg J., and Jones T. A. (2002) The active site of cellobiohydrolase Cel6A from Trichoderma reesei: the roles of aspartic acids D221 and D175. J. Am. Chem. Soc. 124, 10015–10024 10.1021/ja012659q [DOI] [PubMed] [Google Scholar]
- 44. Gregg K. J., Suits M. D., Deng L., Vocadlo D. J., and Boraston A. B. (2015) Structural analysis of a family 101 glycoside hydrolase in complex with carbohydrates reveals insights into its mechanism. J. Biol. Chem. 290, 25657–25669 10.1074/jbc.M115.680470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamada C., Gotoh A., Sakanaka M., Hattie M., Stubbs K. A., Katayama-Ikegami A., Hirose J., Kurihara S., Arakawa T., Kitaoka M., Okuda S., Katayama T., and Fushinobu S. (2017) Molecular insight into evolution of symbiosis between breast-fed infants and a member of the human gut microbiome Bifidobacterium longum. Cell Chem. Biol. 24, 515–524.e5 10.1016/j.chembiol.2017.03.012 [DOI] [PubMed] [Google Scholar]
- 46. Synstad B., Gåseidnes S., Van Aalten D. M., Vriend G., Nielsen J. E., and Eijsink V. G. (2004) Mutational and computational analysis of the role of conserved residues in the active site of a family 18 chitinase. Eur. J. Biochem, 271, 253–262 10.1046/j.1432-1033.2003.03923.x [DOI] [PubMed] [Google Scholar]
- 47. Vocadlo D. J., and Withers S. G. (2005) Detailed comparative analysis of the catalytic mechanisms of β-N-acetylglucosaminidases from families 3 and 20 of glycoside hydrolases. Biochemistry 44, 12809–12818 10.1021/bi051121k [DOI] [PubMed] [Google Scholar]
- 48. Marković-Housley Z., Miglierini G., Soldatova L., Rizkallah P. J., Müller U., and Schirmer T. (2000) Crystal structure of hyaluronidase, a major allergen of bee venom. Structure 8, 1025–1035 10.1016/S0969-2126(00)00511-6 [DOI] [PubMed] [Google Scholar]
- 49. Dennis R. J., Taylor E. J., Macauley M. S., Stubbs K. A., Turkenburg J. P., Hart S. J., Black G. N., Vocadlo D. J., and Davies G. J. (2006) Structure and mechanism of a bacterial β-glucosaminidase having O-GlcNAcase activity. Nat. Struct. Biol. 13, 365–371 10.1038/nsmb1079 [DOI] [PubMed] [Google Scholar]
- 50. Abbott D. W., Macauley M. S., Vocadlo D. J., and Boraston A. B. (2009) Streptococcus pneumoniae endohexosaminidase D, structural and mechanistic insight into substrate-assisted catalysis in family 85 glycoside hydrolases. J. Biol. Chem. 284, 11676–11689 10.1074/jbc.M809663200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tiels P., Baranova E., Piens K., De Visscher C., Pynaert G., Nerinckx W., Stout J., Fudalej F., Hulpiau P., Tännler S., Geysens S., Van Hecke A., Valevska A., Vervecken W., Remaut H., and Callewaert N. (2012) A bacterial glycosidase enables mannose-6-phosphate modification and improved cellular uptake of yeast-produced recombinant human lysosomal enzymes. Nat. Biotechnol. 30, 1225–1231 10.1038/nbt.2427 [DOI] [PubMed] [Google Scholar]
- 52. Reid C. W., Legaree B. A., and Clarke A. J. (2007) Role of Ser216 in the mechanism of action of membrane-bound lytic transglycosylase B: further evidence for substrate-assisted catalysis. FEBS Lett. 581, 4988–4992 10.1016/j.febslet.2007.09.037 [DOI] [PubMed] [Google Scholar]
- 53. Macdonald S. S., Blaukopf M., and Withers S. G. (2015) N-Acetylglucosaminidases from CAZy family GH3 are really glycoside phosphorylases, thereby explaining their use of histidine as an acid/base catalyst in place of glutamic acid. J. Biol. Chem. 290, 4887–4895 10.1074/jbc.M114.621110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Watts A. G., Oppezzo P., Withers S. G., Alzari P. M., and Buschiazzo A. (2006) Structural and kinetic analysis of two covalent sialosyl-enzyme intermediates on Trypanosoma rangeli sialidase. J. Biol. Chem. 281, 4149–4155 10.1074/jbc.M510677200 [DOI] [PubMed] [Google Scholar]
- 55. Vocadlo D. J., and Davies G. J. (2008) Mechanistic insights into glycosidase chemistry. Curr. Opin. Chem. Biol. 12, 539–555 10.1016/j.cbpa.2008.05.010 [DOI] [PubMed] [Google Scholar]
- 56. Nakamura A., Ishida T., Kusaka K., Yamada T., Fushinobu S., Tanaka I., Kaneko S., Ohta K., Tanaka H., Inaka K., Higuchi Y., Niimura N., Samejima M., and Igarashi K. (2015) “Newton's cradle” proton relay with amide-imidic acid tautomerization in inverting cellulase visualized by neutron crystallography. Sci. Adv. 1, e1500263 10.1126/sciadv.1500263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bianchetti C. M., Takasuka T. E., Deutsch S., Udell H. S., Yik E. J., Bergeman L. F., and Fox B. G. (2015) Active site and laminarin binding in glycoside hydrolase family 55. J. Biol. Chem. 290, 11819–11832 10.1074/jbc.M114.623579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nagae M., Tsuchiya A., Katayama T., Yamamoto K., Wakatsuki S., and Kato R. (2007) Structural basis of the catalytic reaction mechanism of novel 1,2-α-l-fucosidase from Bifidobacterium bifidum. J. Biol. Chem. 282, 18497–18509 10.1074/jbc.M702246200 [DOI] [PubMed] [Google Scholar]
- 59. Hehemann J. H., Smyth L., Yadav A., Vocadlo D. J., and Boraston A. B. (2012) Analysis of keystone enzyme in agar hydrolysis provides insight into the degradation of a polysaccharide from red seaweeds. J. Biol. Chem. 287, 13985–13995 10.1074/jbc.M112.345645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ito T., Saikawa K., Kim S., Fujita K., Ishiwata A., Kaeothip S., Arakawa T., Wakagi T., Beckham G. T., Ito Y., and Fushinobu S. (2014) Crystal structure of glycoside hydrolase family 127 β-l-arabinofuranosidase from Bifidobacterium longum. Biochem. Biophys. Res. Commun. 447, 32–37 10.1016/j.bbrc.2014.03.096 [DOI] [PubMed] [Google Scholar]
- 61. Munoz-Munoz J., Cartmell A., Terrapon N., Henrissat B., and Gilbert H. J. (2017) Unusual active site location and catalytic apparatus in a glycoside hydrolase family. Proc. Natl. Acad. Sci. U.S.A. 114, 4936–4941 10.1073/pnas.1701130114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Miao F., Yang R., Chen D. D., Wang Y., Qin B. F., Yang X. J., and Zhou L. (2012) Isolation, identification and antimicrobial activities of two secondary metabolites of Talaromyces verruculosus. Molecules 17, 14091–14098 10.3390/molecules171214091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Karakashian S. J. (1975) Symbiosis in Paramecium bursaria. Symp. Soc. Exp. Biol. 29, 145–173 [PubMed] [Google Scholar]
- 64. Dos Santos Castro L., Pedersoli W. R., Antoniêto A. C. C., Steindorff A. S., Silva-Rocha R., Martinez-Rossi N. M., Rossi A., Brown N. A., Goldman G. H., Faça V. M., Persinoti G. F., and Silva R. N. (2014) Comparative metabolism of cellulose, sophorose and glucose in Trichoderma reesei using high-throughput genomic and proteomic analyses. Biotechnol. Biofuels 7, 41 10.1186/1754-6834-7-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hisamatsu M., Amemura A., Harada T., Koizumi K., Utamura T., and Okada Y. (1984) Cyclic (1→2)-β-d-glucan produced by Agrobacterium and Rhizobium: the structure and the distribution of molecular weight. J. Jpn. Soc. Starch Sci. 31, 117–123 10.5458/jag1972.31.117 [DOI] [Google Scholar]
- 66. Lever M. (1972) A new reaction for calorimetric determination of carbohydrates. Anal. Biochem. 47, 273–279 10.1016/0003-2697(72)90301-6 [DOI] [PubMed] [Google Scholar]
- 67. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 68. Solovyev V., Kosarev P., Seledsov I., and Vorobyev D. (2006) Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 7, Suppl. 1, S10.1–12 10.1186/gb-2006-7-s1-s10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Eisenhaber B., Schneider G., Wildpaner M., and Eisenhaber F. (2004) A sensitive predictor for potential GPI lipid modification sites in fungal protein sequences and its application to genome-wide studies for Aspergillus nidulans, Candida albicans, Neurospora crassa, Saccharomyces cerevisiae, and Schizosaccharomyces pombe. J. Mol. Biol. 337, 243–253 10.1016/j.jmb.2004.01.025 [DOI] [PubMed] [Google Scholar]
- 70. Petersen T. N., Brunak S., von Heijne G., and Nielsen H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 71. Kumar S., Stecher G., and Tamura K. (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pace C. N., Vajdos F., Fee L., Grimsley G., and Gray T. (1995) How to measure and predict the molar absorption-coefficient of a protein. Protein Sci. 4, 2411–2423 10.1002/pro.5560041120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tanaka N., Nakajima M., Aramasa H., Nakai H., Taguchi H., Tsuzuki W., and Komba S. (2018) Synthesis of three deoxy-sophorose derivatives for evaluating the requirement of hydroxy groups at position 3 and/or 3′ of sophorose by 1,2-β-oligoglucan phosphorylases. Carbohydr. Res. 468, 13–22 10.1016/j.carres.2018.08.005 [DOI] [PubMed] [Google Scholar]
- 74. Miyatake H., Hasegawa T., and Yamano A. (2006) New methods to prepare iodinated derivatives by vaporizing iodine labelling (VIL) and hydrogen peroxide VIL (HYPER-VIL). Acta Crystallogr. D Biol. Crystallogr. 62, 280–289 10.1107/S0907444905041909 [DOI] [PubMed] [Google Scholar]
- 75. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 77. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 10.1107/S0907444906022116 [DOI] [PubMed] [Google Scholar]
- 78. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 79. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 80. Hasegawa H., and Holm L. (2009) Advances and pitfalls of protein structural alignment. Curr. Opin. Struct. Biol. 19, 341–348 10.1016/j.sbi.2009.04.003 [DOI] [PubMed] [Google Scholar]
- 81. Kabsch W., and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
- 82. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Case D. A., Ben-Shalom I. Y., Brozell S. R., Cerutti D. S., Cheatham T. E. 3rd., Cruzeiro V. W. Darden T. A., Duke R. E., Ghoreishi D., Gilson M. K., Gohlke H., Goetz A. W., Greene D., Harris R., Homeyer N., et al. (2018) AMBER 2018. University of California, San Francisco, CA [Google Scholar]
- 84. Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., and Simmerling C. (2015) ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kirschner K. N., Yongye A. B., Tschampel S. M., González-Outeiriño J., Daniels C. R., Foley B. L., and Woods R. J. (2008) GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 29, 622–655 10.1002/jcc.20820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 87. Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., and Lindahl E. (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- 88. Bussi G., Donadio D., and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 10.1063/1.2408420 [DOI] [PubMed] [Google Scholar]
- 89. Berendsen H. J. C., Postma J. P., van Gunsteren W. F., DiNola A., and Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 10.1063/1.448118 [DOI] [Google Scholar]
- 90. Hess B. (2008) P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4, 116–122 10.1021/ct700200b [DOI] [PubMed] [Google Scholar]
- 91. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 10.1063/1.470117 [DOI] [Google Scholar]
- 92. Terada T., and Kidera A. (2012) Comparative molecular dynamics simulation study of crystal environment effect on protein structure. J. Phys. Chem. B. 116, 6810–6818 10.1021/jp2125558 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.