

Abstract
Nitric oxide (NO) synthases (NOSs) catalyze the formation of NO from l-arginine. We have shown previously that the NOS enzyme catalytic cycle involves a large number of reactions but can be characterized by a global model with three main rate-limiting steps. These are the rate of heme reduction by the flavin domain (kr), of dissociation of NO from the ferric heme-NO complex (kd), and of oxidation of the ferrous heme-NO complex (kox). The reaction of oxygen with the ferrous heme-NO species is part of a futile cycle that does not directly contribute to NO synthesis but allows a population of inactive enzyme molecules to return to the catalytic cycle, and thus, enables a steady-state NO synthesis rate. Previously, we have reported that this reaction does involve the reaction of oxygen with the NO-bound ferrous heme complex, but the mechanistic details of the reaction, that could proceed via either an inner-sphere or an outer-sphere mechanism, remained unclear. Here, we present additional experiments with neuronal NOS (nNOS) and inducible NOS (iNOS) variants (nNOS W409F and iNOS K82A and V346I) and computational methods to study how changes in heme access and electronics affect the reaction. Our results support an inner-sphere mechanism and indicate that the particular heme-thiolate environment of the NOS enzymes can stabilize an N-bound FeIII-N(O)OO− intermediate species and thereby catalyze this reaction, which otherwise is not observed or favorable in proteins like globins that contain a histidine-coordinated heme.
Keywords: nitric oxide, nitric-oxide synthase, heme, enzyme mechanism, computational biology, thermodynamics, enzyme kinetics, dioxygenation, heme protein, heme-thiolate, reaction mechanism, redox reaction
Introduction
NO synthases (NOSs;4 EC 1.14.13.39) are homodimeric enzymes that catalyze formation of the biological messenger molecule nitric oxide (NO) from l-arginine (l-Arg) (1–3). Three main isoforms of NOS have been described in mammals: neuronal (nNOS), endothelial (eNOS), and inducible (iNOS). All isoforms share significant sequence homology and consist of two main modules: an N-terminal oxygenase domain (NOSoxy) that binds heme, tetrahydrobiopterin (H4B), and the substrate l-Arg and a C-terminal reductase domain that binds FMN, FAD, and NADPH. A calmodulin-binding region is located between NOSoxy and the reductase domain. NOS reductase domains are closely related to dual-flavin NADPH oxidoreductase enzymes like cytochrome 450 reductase, whereas proteins similar to the NOSoxy have been found in bacteria, plants, fungi, and basal metazoans (4, 5).
NO synthesis is a complex multistep process (6). The kinetics of NO synthesis are complicated by the fact that not all of the NO synthesized by NOS is effectively released out of the enzyme, but instead can remain bound to the heme and then be oxidized to nitrate. To describe this process, we have proposed a global mechanism that includes the NO biosynthetic reactions, the NO release step, and a futile cycling of NOS through its ferrous heme-NO complex without net NO release (Fig. 1A) (6–8). It is important to note that the futile cycle becomes an inherent property of the NOS catalytic cycle; as the electron transfer from the reductase domain to the heme is involved in the initial cycle step (kr) but also in the inactivation of the enzyme via the reduction of the ferric-NO complex (kr‴), it follows that an optimal value for kr that maximizes NO output exists for each NOS isoform (6). The model can explain a number of paradoxical catalytic behaviors observed in different NOS isoforms and mutants (6, 7, 9).
Figure 1.

Global model of NOS catalysis and relevance of kox rates. A, global model of NOS catalysis. The rates of heme reduction (kr, kr′, kr″, and kr‴), FeIIINO dissociation (kd), and FeIINO oxidation (kox) are the key rate-limiting steps, as oxygen binding and the catalytic steps (kcat1 and kcat2) are comparatively faster. B, effect of kox rates on instantaneous NO synthesis rates. The rate of NO synthesis versus time was simulated according to the global model using rates for nNOS at 25 °C (63) and varying kox values (0.01–1.0 s−1; values for each trace are indicated). The kox rate does not change the initial rates of NO synthesis but becomes more important in setting steady-state NO synthesis rates. C, effect of kox rates on steady-state NO synthesis rates (steady NO synthesis rate calculated at 10 s from simulations conducted as those in B).
The oxidation of the ferrous heme-NO complex of NOS (designated by kox in Fig. 1A) is actually an NO dioxygenation reaction that destroys the NO made by the NOS enzyme. It has received somewhat less attention than the other kinetic parameters of NOS catalysis, particularly the rate of heme reduction by the reductase domain (kr in Fig. 1A), or the rate of NO dissociation from the ferric heme-NO product complex (kd in Fig. 1A) (6–8). Nevertheless, the kox rate setting is important for NOS enzymes, because it helps to determine how much NOS enzyme accumulates as the ferrous heme-NO complex during steady state NO synthesis (Fig. 1, B and C), which in turn markedly impacts the apparent KmO2 and magnitude of the steady-state NO synthesis activity of the given NOS enzyme (Fig. 1, B and C) (6–8). Indeed, computer simulations of the global model in Fig. 1A have indicated how different kox settings can impact the steady-state rates of NO synthesis and apparent KmO2 of a given NOS. Such simulations have been helpful for explaining the catalytic phenotypes of various NOS mutants with altered kox settings (10), as well as understanding the three WT NOS isozymes, which possess different kox settings (6, 7). For example, the relatively fast oxidation rate of the ferrous heme-NO complex of iNOS (i.e. it has the highest kox setting) helps to explain why it displays an NO synthesis activity that is equivalent to or exceeding that of nNOS, even though the heme reduction rate (kr) in iNOS, which determines its inherent rate of NO biosynthesis, is 4–6 times slower than in nNOS (7).
Previous measures of kox were typically made at single O2 concentrations (i.e. using air- or O2-saturated buffers) (7, 8). We investigated the NO dioxygenase (NOD) reaction of nNOS and iNOS as a function of O2 concentration, using the bacterially expressed oxygenase domains (nNOSoxy and iNOSoxy) (11), which are useful surrogates for the full-length NOS enzymes in such studies (7, 8). We restricted our analysis to the catalytically relevant H4B– plus l-Arg–bound NOSoxy forms and found the following. (i) The nNOSoxy and iNOSoxy have a 100-fold difference in their respective kox settings (k1 = 230 m−1 s−1 for nNOSoxy and k1 = 26,500 m−1 s−1 for iNOSoxy, where kobs = k1[O2] + k−1). (ii) Their kinetic behaviors suggest an NOD reaction mechanism whereby the ferrous heme-NO complex of both proteins reacts directly with O2 to yield the ferric enzyme plus nitrate. This is novel and contrasts with the NOD reactions of globin ferrous heme-NO complexes (i.e. hemoglobin, myoglobin, bacterial NO dioxygenases), which are not directly reactive toward oxygen and instead have their NOD reactions rate-limited by an initial NO dissociation from the ferrous heme, which allows subsequent O2 binding to form a ferrous heme-O2 complex that reacts with NO in the distal pocket (12, 13). (iii) Because we observed no buildup of reaction intermediates in the NOSoxy reactions even at the highest possible O2 concentration, it appears that O2 access and/or its reactivity with the ferrous heme-NO complex is rate-limiting for the NOD reactions of the two H4B– and l-Arg–bound NOSoxy proteins.
Here, we expanded our study by investigating how substrate and cofactor occupancy, protein structural changes, and heme electronic features impact kox in iNOSoxy and nNOSoxy. In particular, we focus on factors that are known to modify the stability of the FeII-O2 and/or FeII-NO complexes of NOS, such as heme thiolate environment (10, 14, 15), NOS dimerization (16, 17), high-spin/low-spin heme transitions (18, 19), and H4B cofactor binding (20–22). Additionally, we examine the kox reaction using computational methods to predict the thermodynamics and the feasibility of the direct reaction of the ferrous NO adduct of NOS with O2, using a NOS active site model. These calculations were then repeated using a globin active site model to elucidate the observed differences in NOD reactivity between NOS and globins. The results increase our understanding of how kox is controlled in NOS enzymes and provide further insight into their NOD reaction mechanism.
Results
Effects of bound H4B and l-Arg
Reactions were run at 10 °C under different conditions of substrate or cofactor occupancy and across a range of O2 concentrations, using iNOSoxy and nNOSoxy proteins that we purified in the absence of l-Arg or H4B. Adding l-Arg or H4B alone or together to iNOSoxy stabilized its ferric high-spin form (23) and enabled the generation of a stable six-coordinate FeII-NO complex. In contrast, the l-Arg– and H4B–free iNOSoxy and nNOSoxy proteins did not form stable six-coordinate FeII-NO complexes and converted rapidly to five-coordinate FeII-NO complexes that reacted very slowly with O2, as reported previously (11). These were not studied further.
The oxidation reactions of the l-Arg– and H4B-bound iNOSoxy FeII-NO complexes were monophasic, with no buildup of detectable spectral intermediates, and formed ferric high-spin iNOSoxy as the product (Fig. 2A), identical to what we observed previously (11). Reaction of the iNOSoxy FeII-NO complex that only contained H4B proceeded at similar rates across the range of O2 concentrations (Fig. 3A) and gave essentially an identical kox rate constant as the H4B– and l-Arg–bound protein (Table 1). The reactions of the iNOSoxy FeII-NO complex that only contained l-Arg were again very similar regarding the spectral evolution (data not shown) but in this circumstance occurred at substantially increased rates relative to the two other conditions (Fig. 3A), giving a 2.7 times higher kox rate constant (Table 1). The rate versus oxygen concentration relationship was linear in all three conditions of substrate and cofactor occupancy (Fig. 3A). Our results suggest that bound H4B retards kox in iNOSoxy, whereas bound l-Arg does not, and that O2 access or reactivity remains rate-limiting under all three conditions.
Figure 2.

Reaction of ferrous heme-NO complexes with oxygen. A, WT iNOSoxy; B, WT nNOSoxy; C, V346I iNOSoxy; D, K82A iNOSoxy; E, W409F nNOSoxy (pH 7.6); F, W409F nNOSoxy (pH 9.5). All reactions were between the preformed ferrous-NO complex (around 5 μm) and 170 μm oxygen (final concentrations). The samples included H4B and l-Arg except K82A iNOSoxy, which corresponds to l-Arg alone. The initial spectrum is shown as a thick black line, and final spectra are indicated by thin black lines. Selected spectra during the reaction are shown in gray. The arrows indicate the direction of the absorbance change. Insets, absorbance changes for the wavelength maxima of the FeIINO species (around 436 nm; open symbols) and the FeIII species (around 396 nm; closed symbols).
Figure 3.
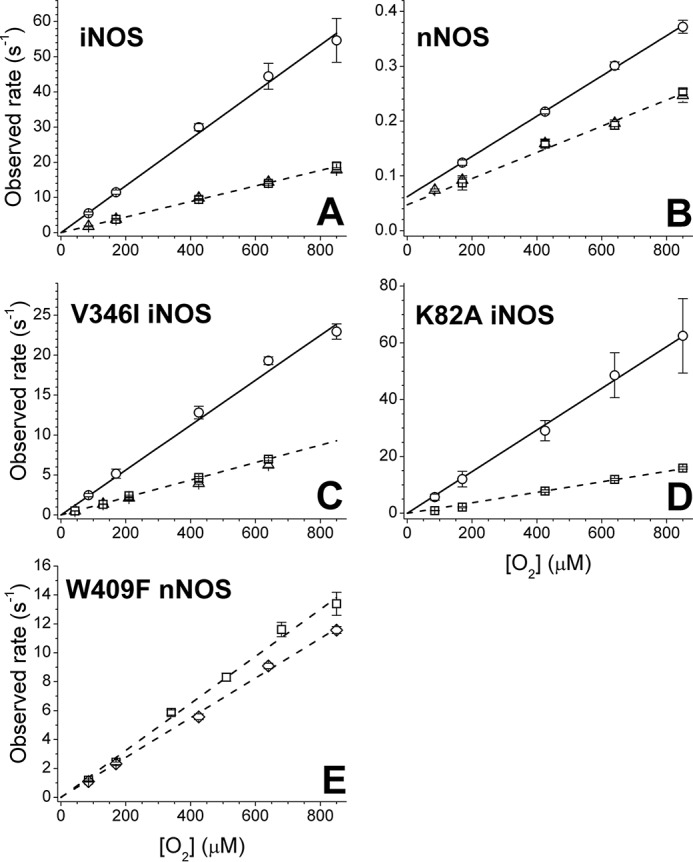
Dependence of the observed ferrous heme-NO oxidation rates versus oxygen concentration in the presence or absence of l-Arg and H4B. A, WT iNOSoxy; B, WT nNOSoxy; C, V346I iNOS; D, K82A iNOS; E, W409F nNOS. Points indicate the averaged observed rates calculated at single wavelengths for FeII-NO decay (around 436 nm) and FeIII buildup (around 396 nm). The substrates are as follows: −H4B, +l-Arg (circles); +H4B, +l-Arg (squares); and +H4B, −l-Arg (triangles). For nNOSoxy W409F, the rates in the presence of both H4B and l-Arg at pH 7.6 (squares) and pH 9.5 (diamonds) are shown. Lines indicate linear fits for the rates in the presence of only l-Arg (solid lines) or in the presence of both H4B and l-Arg (dashed lines). The fits for +H4B-only experiments (triangles) are almost identical to the experiments in the presence of both H4B and l-Arg and are not indicated.
Table 1.
Rates for the six-coordinate FeII-heme-NO oxidation for several NOSoxy forms
5C, five-coordinate FeII-NO complex.
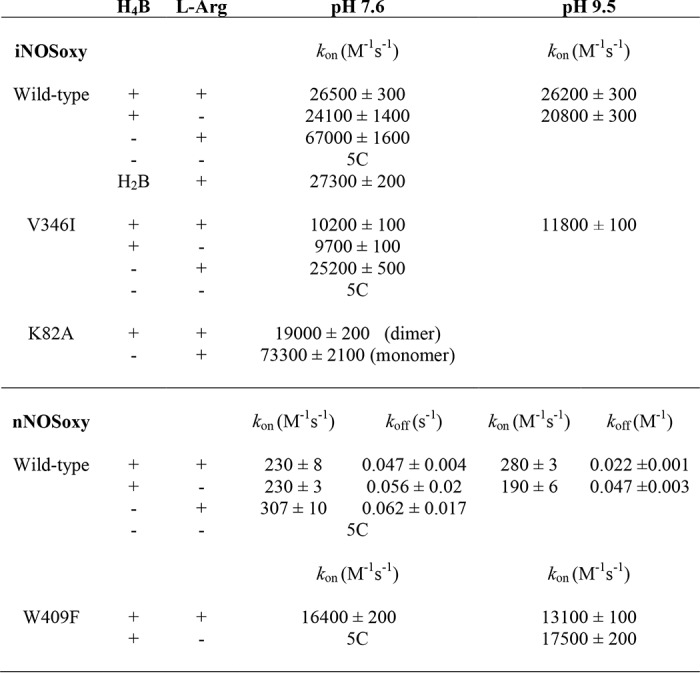
We performed additional experiments with an iNOSoxy containing dihydrobiopterin (H2B) instead of the native cofactor (H4B). H2B can occupy the same binding site in NOS enzymes, but it is redox-inactive during the NO synthesis process (22, 24). The oxidation reaction of the iNOSoxy FeII-NO complex in the presence of H2B and l-Arg yielded similar kinetic results to those obtained for H4B (Table 1). Thus, the biopterin reduction state does not influence its capacity to diminish the kox setting in iNOSoxy.
We similarly studied the kox reactions of the nNOSoxy FeII-NO complex under the three conditions as noted above. Overall, the same pattern was observed (Figs. 2B and 3B), although the magnitudes of kox obtained across the range of O2 concentrations were much less than for iNOSoxy reactions, as we previously observed (11). The nNOSoxy FeII-NO complex containing H4B alone, or along with l-Arg, had similar kox rates, and in the absence of H4B, the l-Arg–bound complex again showed increased kox rates (Fig. 3B). However, the increase seen for the H4B-free nNOSoxy was very modest compared with that seen for iNOSoxy and resulted in only a 30% increase in the kox rate constant (Table 1). Nevertheless, the data suggest that the kox reactions of nNOSoxy and iNOSoxy are similarly influenced by l-Arg– and/or H4B–binding site occupancy.
Effect of restricting access to the distal heme pocket
In iNOS, the magnitude of several kinetic parameters is sensitive to relatively small changes in the protein structure near the distal heme pocket (25). Replacing a conserved Val at the entrance to the distal heme pocket with the bulkier Ile altered the observed rates of O2 and NO binding to or dissociation from the iNOS heme, which could be explained by changes in ligand access to or egress from the distal heme pocket (25). These effects on heme-NO binding kinetics were also observed for a bacterial NOSoxy protein when similar substitutions were made (26). We therefore used the V346I mutation to study the effect of distal heme pocket changes on kox rates in iNOSoxy. The protein formed a stable six-coordinate FeII-NO complex in the presence of either H4B or l-Arg, and as we observed for the WT iNOSoxy, a five-coordinate FeII-NO complex appeared in the absence of H4B and l-Arg. The spectral evolution observed during reaction of a V346I iNOSoxy FeII-NO complex in the presence of l-Arg and H4B was very similar to that seen for WT iNOSoxy (Fig. 2C). Nevertheless, the kox reaction rates across the range of O2 concentrations were about 40% that of WT iNOS (Fig. 3C). The observed rates remained linearly dependent on O2 concentration, and the koff term was too small to be accurately determined. The calculated value for the kon rate constant was ∼10,200 m−1 s−1, which is 38% of the magnitude observed for WT under identical conditions (Table 1). The effects of individual H4B or l-Arg site occupancy were remarkably similar in the mutant compared with WT iNOSoxy (Fig. 3, A and C), with little difference in the kox rates observed when H4B was bound with or without l-Arg and a significant (2.5-fold) rate increase when only l-Arg was bound (Table 1). Our finding that the kox reaction is sensitive to the protein structure determining the opening to the distal heme pocket is consistent with O2 access being a rate-limiting feature of the kox reaction.
Effect of NOSoxy dimeric structure
NOSoxy subunit dimerization is required in the enzyme's post-translational assembly and is associated with specific protein structural changes that decrease the solvent exposure of the bound heme and help to create the l-Arg– and H4B–binding sites (20, 21). We therefore studied the kox reaction of the K82A iNOSoxy mutant, which is monomeric as purified but dimerizes in the presence of H4B (17). After incubating K82A iNOSoxy at high H4B and l-Arg concentrations (0.5 mm H4B, 20 mm l-Arg) to fully induce dimerization, the enzyme formed a stable six-coordinate FeII-NO complex, and the kinetics of FeII-NO oxidation reaction were comparable with WT iNOS, giving a linear dependence of kox versus O2 concentration (Fig. 3D) and a kon of ∼19,000 m−1 s−1 (Table 1). To study the reaction of the monomeric enzyme, we incubated K82A iNOSoxy with l-Arg alone, which is unable to promote dimerization in the mutant (17). In this case, the enzyme formed a FeII-NO complex that was a blend of five- and six-coordinate, with Soret bands at 393 nm and at 438 nm for the five- and six-coordinate FeII-NO complex, respectively (Fig. 2D). This mix of complexes was found to be stable over the experimental time frame. The reaction of the monomeric form of the K82A iNOSoxy FeII-NO complex with O2 proceeded with two observable phases (Fig. 2D): a fast phase associated with the loss of 438-nm absorbance and a concomitant increase at 398 nm and a second, much slower phase in which the Soret band shifted toward higher wavelengths. We assigned these changes to two reactions. The fast oxidation is that of the six-coordinate FeII-NO complex, with an absorbance decrease near 438 nm and buildup of high spin FeIII (396 nm), and a subsequent slow oxidation represents the conversion of the five-coordinate FeII-NO complex (386 nm) to low-spin FeIII (419 nm). Absorbance changes at 440 and 398 nm (Fig. 2D, inset) were fitted to a bi-exponential equation, and the fast rates were plotted versus O2 concentration (Fig. 3D). The observed rates for the fast phase (representing oxidation of the six-coordinate complex) were similar at either wavelength, giving kon of ∼73,300 m−1 s−1. This was significantly faster than for the H4B-bound dimeric form of K82A iNOSoxy and represented a 4-fold increase (Table 1). This kox rate constant was similar to what we observed for the WT iNOSoxy enzyme without H4B (see Table 1). Thus, the FeII-NO complexes in the iNOSoxy monomer and in the H4B-free iNOSoxy dimer were kinetically similar regarding their reactivity toward O2, despite any possible differences between their heme environments or ligand access. These results suggest a more open heme pocket in the absence of H4B.
Effect of modifying the heme midpoint reduction potential
NOS isoforms contain a conserved tryptophan (Trp-409 in nNOS) that is located on the proximal side of the heme that engages in a π-stacking interaction with the heme porphyrin and forms a hydrogen bond with the axial cysteine thiolate heme ligand (15). Studies using Trp-409 mutants suggested that the loss of the Trp-Cys hydrogen bond should increase the negative charge density on the cysteine thiolate and thus lower the heme midpoint reduction potential (10, 15). In fact, a number of studies on the homologous Trp residue of iNOS (27), eNOS (28), and bacterial NOS proteins (28–31) indicate a correlation between the strength of the hydrogen bonding to the thiolate, the heme midpoint potential, and the stability/reactivity of the FeII-NO/FeII-O2 complexes. To test this hypothesis, we utilized a spectroelectrochemical method (27) to measure and compare the heme midpoint potentials of WT and W409F nNOSoxy in the presence of both l-Arg and H4B. The fittings of the experimental values to the Nernst equation are shown in Fig. 4. The calculated Em values were −275 ± 4 mV for nNOSoxy WT, and −335 ± 4 mV for W409F nNOSoxy. The midpoint value we obtained for WT is close to previously reported values (23, 32, 33). Thus, the W409F substitution decreased the ferric/ferrous heme midpoint potential by −60 mV. This confirmed the predicted effect of the Trp-Cys hydrogen bond and made the W409F nNOSoxy mutant useful for our investigation.
Figure 4.
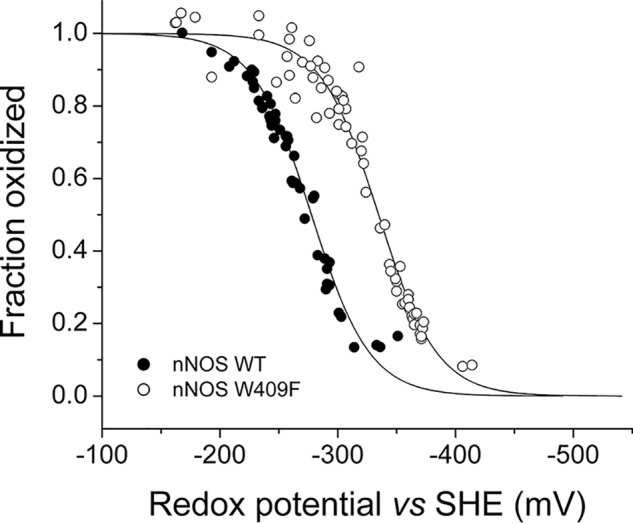
Potentiometric redox titration of WT and W409F nNOS oxygenase domains. The data are presented as the measured potential (E) versus the fraction oxidized. The solid lines represent the fit of the theoretical one-electron Nernst equation.
The W409F nNOSoxy mutant is known to form a six-coordinate FeII-NO complex that shifts rapidly to a mixture of a weaker six-coordinate complex and some five-coordinate complex (10, 18), similar to the behavior of K82A iNOSoxy described above. We measured kox rates as a function of O2 concentration for the FeII-NO complex of the W409F nNOSoxy mutant in the presence of l-Arg and H4B (Fig. 2E), focusing on the reciprocal absorbance changes associated with oxidation of the six-coordinate complex at 438 and 396 nm. The reaction was monophasic and proceeded with the typical spectroscopic transitions, but the observed rates were much higher than for the WT nNOSoxy enzyme (Fig. 3E). The kox rates increased linearly with oxygen concentration across the entire range and gave a value for kon of ∼16,400 m−1 s−1 (Table 1). This represents a 70-fold increase in kox value relative to WT (Table 1) and thus demonstrates that decreasing the ferric/ferrous heme midpoint potential in nNOSoxy has a large impact on the kox.
Effect of pH
Increasing the pH can impact two aspects of the kox reaction. First, it can strengthen the heme thiolate bond in W409F nNOSoxy and so stabilize the six-coordinate FeII-NO complex (18). Second, it may conceivably increase the stability of a putative heme-bound peroxynitrite intermediate that could form as an immediate product of the kox reaction (11). We therefore studied the kox reaction at pH 9.5 for several proteins: iNOSoxy, V346I iNOSoxy, nNOSoxy, and W409F nNOSoxy.
Increased pH did not alter the spectroscopic properties of the protein FeII-NO complexes except in W409F nNOSoxy, where it completely stabilized the six-coordinate complex having a Soret absorbance at 438 nm (Fig. 2F), as previously reported for the nNOSoxy W409Y mutant (18). Running the reactions at pH 9.5 did not alter the O2 dependence of the kox reaction for W409F nNOSoxy or for any of the other proteins, as their kox reactions remained monophasic with no detectable buildup of a reaction intermediate, and their observed kox rates still increased linearly with oxygen concentration across the entire range (data not shown). We also observed only small changes (within 20%) in the kox rate constants for the reactions run at pH 9.5 versus 7.4 (Table 1). Thus, increasing the pH had minimal effects on the O2 reactivity of the FeII-NO complexes and did not alter the reaction trajectory regarding stabilization of possible heme-bound intermediates.
Computational investigation of the kox reaction
To further understand the reaction of NOS FeII-NO complexes with O2, we have performed density functional theory (DFT) calculations on a computational model of the NOS active site. The model used for these calculations was constructed by taking the coordinates from the X-ray crystal structure reported of the FeII-NO complex of nNOS (34) and keeping only the FeII-NO protoporphyrin IX complex, the axial bound cysteinate (thiolate) ligand (Cys-415), the conserved axial tryptophan (Trp-409), and a glycine residue (Gly-417), which also forms a hydrogen bond with the axial thiolate ligand. Further details of the NOS active site model used for all calculations are provided under “Experimental procedures.” The geometry of this NOS FeII-NO truncated active site model was fully optimized using BP86/TZVP with water as the polarized solvent continuum (via a self-consistent reaction field approach). Fig. S1 shows the structure obtained from these calculations. A triplet O2 (3O2) molecule was then introduced into the system, with the unpaired elections of the 3O2 antiparallel to the unpaired electron of the S = ½ FeII-NO unit, to allow for potential bond formation between these two radical species. Initially, the O2 molecule was constrained at a distance of 3.26 Å from the nitrogen atom of the FeII-NO unit (see Fig. 5 for the optimized structure). Geometric parameters of this optimized structure are compared with the nNOS FeII-NO crystal structure in Table S1. This comparison shows that our simplified active site model replicates the bond distances and angles observed in the NOS crystal structure reasonably well, without including any distal secondary sphere amino acids. Using this optimized structure as the starting point (ON-OO distance = 3.26 Å), a potential energy surface (PES) scan was performed where the 3O2 was moved closer to the nitrogen atom of the FeII-NO unit until an ON-OO distance of 1.42 Å was reached (Fig. S2). From these calculations, initial movement of the 3O2 toward the FeII-NO unit is predicted to result in formation of a favorable van der Waals complex (ON-OO distance = 2.11 Å), which is −4.2 kcal/mol lower in energy than the starting structure. Movement of the O2 closer to the NO unit results in formation of a new O–N bond with a calculated activation barrier of 5.7 kcal/mol for formation of a ferric peroxynitrite complex (FeIII-N(O)OO−; the boldface letter indicates the atom bound to the Fe center) with a very shallow energy minimum for a structure with a ON-OO bond distance of 1.50 Å. Attempts to optimize this FeIII-N(O)OO− complex with an unconstrained ON-OO bond resulted in reformation of the FeII-NO/O2 van der Waals complex. From these calculations, the shallow energy minimum for the FeIII-N(O)OO− complex suggests that it is not a stable intermediate, and additional stabilization of the ON-OO bond is needed for this reaction pathway to become feasible. For comparative purposes, a similar PES scan was then repeated for a FeII-NO globin mimic (i.e. a ferrous heme nitrosyl with an axial imidazole ligand) to determine whether the DFT calculations could help explain the experimentally observed differences in reactivity between NOS and globin proteins. Further details and results of these calculations are provided in the supporting information. Comparing the heme-thiolate and heme-imidazole PES scans in Fig. S2, the energy differences between the FeII-NO/O2 complexes and the peroxynitrite intermediates (ON-OO distance = 1.42 Å) are +6.1 and +12.4 kcal/mol, respectively. In the globin model, no energy minimum for the peroxynitrite intermediate is observed.
Figure 5.
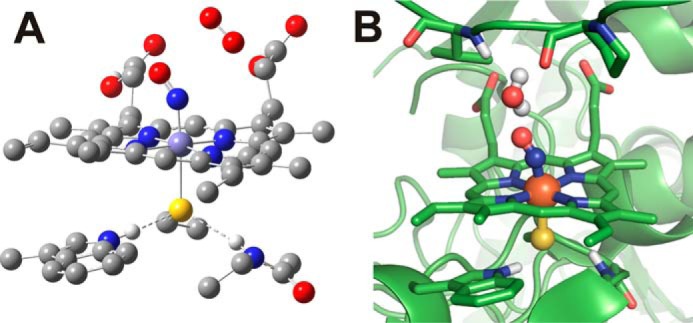
The NOS active site. A, the DFT-optimized (BP86/TZVP) structure of the FeII-NO nNOS active site model is shown with 3O2 at an ON-OO distance of 3.26 Å. The hydrogen bonds to the thiolate ligand are shown as dashed lines. B, crystal structure representation of the nNOS FeII-NO complex, showing the water molecule present in the active site (PDB code 2G6K). The water molecule is also involved in hydrogen bonding with Val-567 and Pro-565 (on top) in the active site. All protons have been omitted for clarity, except those involved in relevant hydrogen bonds.
For the NOS kox reaction to proceed through a peroxynitrite intermediate, further stabilization of this species is needed (to prevent the back reaction to O2 and the FeII-NO complex). One possibility in this regard would be protonation of the newly formed peroxynitrite ligand. Free peroxynitrite has a pKa of 6.8 (35), meaning it could be converted to peroxynitrous acid under biological conditions. Protonation of our FeIII-N(O)OO− complex in the NOS model in silico and optimization of the structure resulted in a stable ferric-peroxynitrous acid complex (FeIII-N(O)OOH) with a ON-OO bond length of 1.483 Å, without the need to constrain the ON-OO bond for the optimization. However, the fact that experimentally, only a small pH dependence of the NOD reaction in NOS is observed (Table 1) argues against this possibility.
Alternatively, we hypothesized that instead of protonation of the peroxynitrite ligand, a simple hydrogen bond to this species could be sufficient for stabilization, allowing the NOD reaction to proceed. To test this possibility, a water molecule was placed in close proximity to the peroxynitrite complex, and the structure was then re-optimized without constraining the peroxynitrite ON-OO bond. This resulted in a stable structure with only a slightly elongated ON-OO bond of 1.508 Å (see Fig. 6, inset). In this structure, the H2O molecule is predicted to make a reasonably strong hydrogen bond to the anionic ONOO− ligand with an OH-OONO distance of 1.69 Å and O-H-O(ONO) angle of 176.7°. This structure is calculated to be 8.1 kcal/mol lower in energy relative to the complex without the H2O hydrogen bond (ON-OO = 1.50 Å). A PES scan was then performed with the inclusion of the H2O molecule as shown in Fig. 6 (black points). Results from this PES scan predict that the ferric peroxynitrite intermediate is now only 1.3 kcal/mol higher in energy relative to the FeII-NO/O2 van der Waals complex (observed here at ON-OO = 2.108 Å), and that the activation energy barrier for formation of the peroxynitirite intermediate is reduced to 2.4 kcal/mol relative to the van der Waals complex. Together, these results indicate that formation and stabilization of a heme-thiolate FeIII-ONOO− intermediate would be quite feasible in NOS when assisted by a hydrogen bond from a water molecule. This is certainly a reasonable possibility, considering that water molecules have been identified in the NOS active site by X-ray crystallography, as shown in Fig. 5B (34). To compare the NOS model to globins, we built a model including the distal histidine conserved in globins (see details in the supporting information and Fig. S3). A PES scan of the globin model (Fig. 6, red points) indicates that the distal imidazole does not stabilize the N-bound peroxynitrite complex, in agreement with the experimental observation that reaction of 3O2 with the NO adduct of a ferrous globin requires initial dissociation of NO (and binding of O2 to the heme) for the reaction to proceed.
Figure 6.
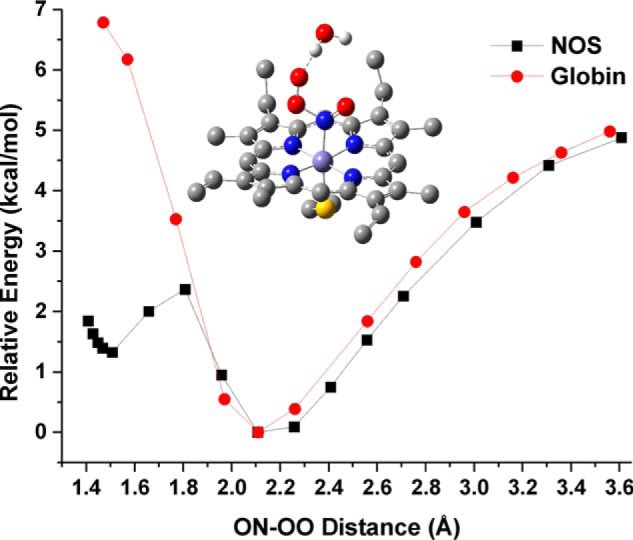
Comparison of the PES scan results for the NOS and globin active site models. Shown are the relative energies obtained from the PES scans for the reaction of the corresponding FeII-NO complexes with 3O2. Black line, the NOS active site model with the active site H2O hydrogen bond donor. Red line, the globin active site model containing the distal His (here 5-ethylimidazole) residue. Each point represents a fully optimized structure. The minimum energy calculated for the FeII-NO/O2 van der Waals complex of each model was set to 0 kcal/mol to allow for a direct comparison of the relative energies for formation of a ferric peroxynitrite intermediate. Middle, fully optimized structure of the FeIII-N(O)OO−/H2O heme-thiolate complex (ON-OO distance = 1.508 Å). The two axial hydrogen bond donors to the thiolate, the heme carboxylic acid side chains, and all protons (except those of the H2O molecule) have been removed for clarity.
Nature of the peroxynitrite intermediate
In the NOD reaction of globin proteins, where NO reacts with oxyglobin to generate nitrate, the reaction likely proceeds through formation of a terminal O-bound peroxynitrite intermediate (FeIII-OONO−). Other computational studies have investigated the formation and conversion of this intermediate to nitrate and have concluded that a sequential isomerization mechanism is the most thermodynamically feasible pathway (over a concerted mechanism) (36, 37). In this mechanism, the ferric globin peroxynitrite complex (FeIII-OONO−) undergoes O-O(NO) bond homolysis, resulting in formation of an FeIV=O complex (Compound II in the deprotonated state) and the nitrogen dioxide radical (•NO2), which then quickly recombine to generate a ferric-nitrate complex (FeIII-NO3−). For a similar sequential isomerization mechanism to occur for the proposed NOS N-bound peroxynitrite intermediate, a switch in the peroxynitrite coordination mode would first need to occur. To help determine the feasibility of this possibility, the geometries of the three other binding modes of peroxynitrite to our heme-thiolate NOS model were optimized to allow for a comparison of their relative energies (see Table 2). Full optimization of the three additional peroxynitrite coordination modes was possible without constraining the ON-OO bond. Geometry optimization of the structures in the presence of the H2O hydrogen bond donor shows that the terminal O-bound complex (FeIII-OONO−) is the most favorable binding mode, which is 5.8 kcal/mol lower in energy than the stable N-bound structure. The (ON)O(O)-bound peroxynitrite complex is 4.8 kcal/mol higher in energy than the N-bound species (and 10.6 kcal/mol higher in energy than the terminal OONO-bound complex), and the O(NOO)-bound peroxynitrite complex is 5.2 kcal/mol higher in energy than the N-bound species (+11.0 kcal/mol compared to the OONO-bound complex). Note that the relative energies of the different peroxynitrite complexes in the absence of a hydrogen bond are also listed in Table 2.
Table 2.
Comparison of the calculated (BP86/TZVP) bond distances and relative energies of the nitrate complexes and of the possible peroxynitrite coordination modes, bound to the ferric heme-thiolate NOS model
Bottom rows, with the H2O hydrogen bond donor; top rows, without the H2O hydrogen bond donor. The coordinating atom of the peroxynitrite ligand is indicated in boldface type.
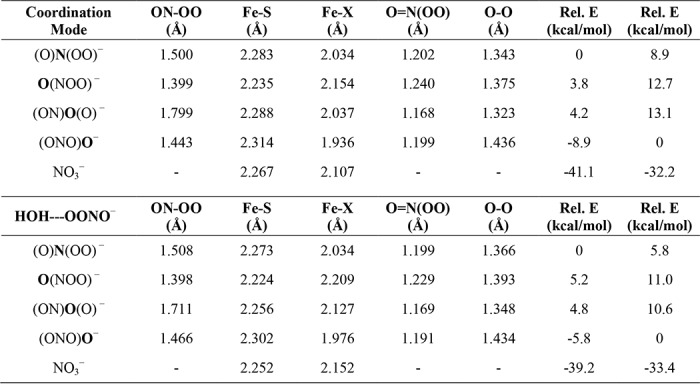
This analysis suggests a mechanism whereby the initially generated N-bound peroxynitrite molecule would dissociate from the ferric heme and rebind through its terminal oxygen to form the most energetically favorable species (Fig. 7). Indeed, optimization of the N-bound FeIII-N(O)OO− complex in the high-spin state resulted in dissociation of the ONOO−/H2O ligand. As dissociation of the N-bound peroxynitrite ligand would result in a conversion of the heme Fe from low-spin to high-spin, it is plausible that this transition would allow adequate time for rotation of the unbound peroxynitrite in the NOS active site, which would then allow rebinding of peroxynitrite in its most favorable coordination mode (i.e. terminal O-bound FeIII-OONO−). The subsequent conversion of bound peroxynitrite to nitrate could occur by the same mechanism as proposed for the NOD reaction in globins (36, 37). The end product of this reaction, a low-spin ferric nitrate complex (FeIII-NO3−), was calculated to be 33.4 kcal/mol lower in energy than the terminal O-bound FeIII-OONO− complex. The relative energetics of the steps and species proposed in this mechanism are shown in Fig. 8.
Figure 7.

Proposed reaction pathways for the kox reaction based on the DFT results. Structures in the proposed reaction pathway are shown in black; structures in the alterative pathway are shown in red. Selected species are named as follows. S.P., starting point (FeII-NO + 3O2, N-O distance = 3.6 Å); N-PN, N-bound peroxynitrite complex (FeIII-N(O)OO−); O1-PN, cis FeIII-(ON)O(O)− complex; cis O2-PN, the terminal O-bound cis FeIII-OONO− complex; trans O2-PN, trans O-bound FeIII-OONO− complex; NO3−, ferric O-bound nitrate complex (FeIII-NO3−).
Figure 8.
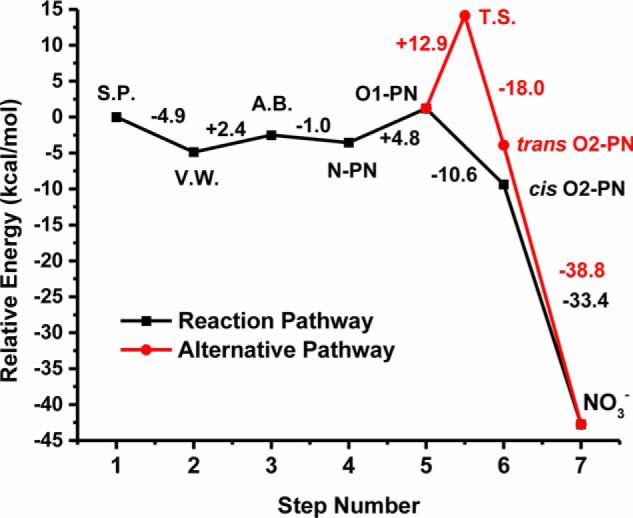
Summary of DFT reaction pathways. Comparison of the relative energies of all major reaction steps determined from the DFT calculations for the reaction of the NOS active site model FeII-NO complex with 3O2, leading to peroxynitrite (PN). All points represent fully optimized structures with the inclusion of the hydrogen-bonding H2O molecule. The black points represent the formation of the N-bound peroxynitrite complex (FeIII-N(O)OO−; N-PN) and the switch in PN coordination mode to the cis FeIII-(ON)O(O)− complex (O1-PN) and then to the terminal O-bound cis FeIII-OONO− complex (cis O2-PN; TS not determined). The red points represent the alternative reaction pathway in which the cis FeIII-(ON)O(O)− complex (O1-PN) is converted to the trans O-bound FeIII-OONO− complex (trans O2-PN), and then to the ferric O-bound nitrate complex (FeIII-NO3−; NO3−). The relative changes in energy (kcal/mol) from each step to the next (from left to right) are given between the respective reaction points and are listed in Table S2. S.P., starting point (FeII-NO + 3O2, N-O distance = 3.6 Å); V.W., the FeII-NO/O2 van der Waals complex (ON-OO = 2.108 Å); A.B., the calculated activation barrier (ON-OO = 1.81 Å) for formation of the N-bound cis FeIII-N(O)OO− complex; T.S., the calculated transition state structure for the alternative pathway.
Alternatively, the calculated structure of the (ON)O(O)− O-bound peroxynitrite complex has a significantly elongated OO–NO bond of 1.711 Å in the presence of the hydrogen bond. This suggests that if the generated N-bound peroxynitrite would first rotate into this alternative FeIII-(O)ONO− binding mode, then cleavage of the ON-OO bound could occur, leading to migration of NO to the terminal oxygen, resulting in the trans O-bound peroxynitrite complex (FeIII-OONO−). The trans FeIII-OONO− complex is calculated to be −5.1 kcal/mol lower in energy relative to the FeIII-(O)O(NO)− starting complex. Following the mechanism of the NOD reaction for globins, this would then lead to O-O(NO) bond homolysis and formation of an FeIV=O complex and •NO2. In this case, the nitrogen atom of the released •NO2 would be in an ideal position to undergo radical rebound with the FeIV=O intermediate, to give nitrate (Fig. 7). Details and results of the calculations performed to investigate this alternative pathway for formation of a trans FeIII-OONO− complex are provided in the supporting information. The breaking of the FeIII-(O)ONO− O–N bond and conversion to the trans FeIII-OONO− complex is calculated to have an activation barrier of 12.9 kcal/mol and is therefore energetically feasible. Fig. 8 shows the energetics of this alternative pathway compared with the mechanism that was discussed previously.
Discussion
NOS catalysis involves two different pathways that either lead to the production of NO via reaction of the enzyme heme-oxy species and release of NO from the FeIII-NO species (productive cycle) or lead to the production of other nitrogen oxides via formation of an enzyme FeII-NO complex (futile cycle, Fig. 1A) (6). The rate of oxidation of the FeII-NO complex (kox) was shown to be a key factor in determining the distribution and cycling of enzyme species during catalysis (6, 7). Our results indicate that the reactivity of the FeII-NO complex can be finely tuned by the NOS heme environment.
Oxygen accessibility and kox
Oxygen binding to the NOS ferrous heme is extremely fast (higher than 1 × 105 m−1 s−1 (38, 39)). Nevertheless, the kox rates obtained in this work are 1–4 orders of magnitude smaller. This difference might be due to an electrostatic, repulsive effect of the heme-bound NO, but it could also indicate that oxygen entry in the heme pocket is not the rate-limiting step. Thus, for WT nNOS, oxygen access is not a rate-limiting factor as the nNOS W409F mutant is able to catalyze a much faster reaction without altering the heme distal pocket. This suggests that in WT nNOS, the [FeII-NO···O2] complex is not very reactive, and oxygen can diffuse out of the heme pocket without reacting. On the other hand, for the iNOS enzyme, there is a marked dependence of the rates on O2 concentration, which suggests that oxygen entry and formation of the [FeII-NO···O2] complex is almost rate-limiting. The V346I mutant shows that a small reduction in the size of the heme pocket can cause a 2-fold decrease in the rates compared with WT iNOS. It must be noted that this effect may be due to changes not only in oxygen accessibility but also in the stability of the [FeII-NO···O2] complex.
Further evidence for the influence of dioxygen accessibility on iNOS kox rates is the increase observed in the absence of H4B. Although H4B has a significant effect on the electronic properties of the heme, it can also modify heme accessibility. These H4B effects on heme ligand access are likely related to structural changes that occur upon H4B binding within the NOSoxy domain, which also help create the dimer interface and thus promote dimerization of the eukaryotic NOS enzymes. Such structural changes caused by H4B are indicated by a comparison of the H4B-bound NOSoxy dimer structure with the structures of the iNOSoxy monomer (40) and the H4B-free, dimeric form of Bacillus subtilis NOS (16). In our present study, it is apparent that the different kox rates that we observed for the K82A iNOSoxy mutant in its monomeric versus H4B-bound dimeric form can be attributed to the absence or presence of H4B. Although we were unable to study the oligomerization effect separately from the presence of H4B, the bulk of evidence suggests that the oligomerization state of the NOS protein plays a smaller role in determining its kox than do structural changes within the dimeric NOS that are caused by H4B binding itself.
The proximal cysteine environment and kox rates
A number of reports have proven the relevance of the Trp-409 residue in nNOS (10, 14, 15, 18). Our results show that replacement of Trp-409 with phenylalanine causes a decrease in the redox potential and significant changes in the oxidation of the FeII-NO complex. The nNOS W409F mutant behaves similarly to WT iNOS, with a high kox rate and a marked oxygen dependence of the rates. The higher reactivity of the mutant can be attributed to the more reducing heme iron in the mutant, which accelerates the formation of the peroxynitrite intermediate. Increasing the pH stabilizes the six-coordinate FeII-NO complex but still allows high kox rates. A likely explanation of this effect is that the more alkaline pH can prevent the protonation of the cysteine, which is linked in computational studies to the cleavage of the Fe–S bond (41) as proposed previously (18). It remains unclear why the stabilization was previously found only for the W409Y mutant, but the proposed formation of a hydrogen bond between W409Y and the cysteine may not be the main factor, as (i) we were able to detect the same effect in the W409F mutant, and (ii) the crystal structure indicates that unless an important rearrangement occurs, the -OH group of the tyrosine will not be in an adequate geometry for a hydrogen bond with the cysteine, although the OH group will be in close vicinity of the sulfur and can have an electrostatic effect. Interestingly, the effects are in agreement with studies of the homologous tryptophan in bacterial NOS proteins (28–31) (Table S2). In both B. subtilis NOS and Staphylococcus aureus NOS, the heme redox potential decreases in the sequence His > Trp(WT) > Phe (29, 31). The histidine mutants show features consistent with stronger FeII-NO/FeII-O2 complexes (stronger σ bonding between the Fe and the diatomic ligand) and slower kox rates. Alternatively, the elimination of the hydrogen bond to the cysteine thiolate in the Phe mutant leads to weaker σ-bonding and increased π-backbonding in the interaction with distal ligands (29). Thus, the Phe mutants show increased reactivity of their FeII-NO and FeII-O2 complexes. In summary, from the nNOSoxy results, it appears that an increased basicity of the proximal ligand can induce a greater push effect and a higher FeII-NO reactivity, consistent with other observations in different NOS proteins (27–31). We observe a loss of the thiolate bond upon NO binding associated with mutants with very low heme midpoint potentials. Consequently, FeII-NO oxidation can be modulated, but only within certain constraints of heme-thiolate bond viability.
Mechanistic implications
We have reported previously that NOS enzymes are able to catalyze the fast oxidation of FeII-NO complexes, whereas most heme enzymes known to catalyze a similar process either catalyze the reaction at a much slower rate (neuroglobin, hemopexin) or proceed via a mechanism involving the reaction of FeII-O2 with NO (hemoglobin, myoglobin, flavohemoglobin) (11).
The reaction of FeII-NO with O2 may include formation of a transient FeIII-N(O)OO− intermediate (Fig. 7) (12, 42, 43). This species has been proposed for the symmetrical reaction of oxyhemoglobin with nitric oxide at alkaline pH, but not under neutral or acidic conditions (43). This has been related to the pH-dependent stability of cis-peroxynitrite. Depending on the geometry, the dissociation of this intermediate can hypothetically produce either cis- or trans-peroxynitrite. Trans-peroxynitrite would rearrange to nitrate fast, whereas cis-peroxynitrite is more stable. The stability of cis-peroxynitrite is highly dependent on pH, as protonation to peroxynitrous acid (ONOOH) lowers the barrier for isomerization (44).
Overall, our results indicate that the effects of increased pH, if any, are negligible. Thus, according to the proposed reaction scheme (Fig. 7; see also Figs. S4–S6 and Table S3), our data suggest that any step involving peroxynitrite formation is not rate-limiting.
Our previous mechanistic investigation of nNOS- and iNOS-heme-nitrosyl oxidation points rather to a fast and direct reaction with dioxygen (11). A proposed alternative mechanism suggested the possible involvement of an outer-sphere electron transfer step between incoming molecular oxygen and the FeII-NO heme complex of iNOSoxy or nNOSoxy (11). A comparison of outer-sphere electron transfer rates with observed kox rates informs on this argument. In this regard, it is possible to estimate the expected rate of a limiting case of outer-sphere electron transfer (kouter-sphere) between molecular oxygen and the heme-nitrosyl complex in nNOS using the simplified Marcus cross-relationship: kouter-sphere = (k11·k22·Keq·f12)½ (45). In this case, k11 is the self-exchange rate for the O2/O2·− couple (∼450 m−1 s−1) (46); k22 is the self-exchange for the nNOS-heme-ferrous/ferric nitrosyl couple. Keq (∼2.6 × 10−6 at 25 °C) is the thermodynamic equilibrium constant of redox exchange between the O2/O2·− couple (E0′ = −0.330 V) and the nNOS ferrous/ferric heme-nitrosyl couple (E0′ ≈ 0.0 V) (32). The factor f12 is a function of the two self-exchange rates k11 and k22 and is given by the equation, ln(f12) = (ln(Keq))2/4(ln(k11·k22/Z) (where Z is the collision frequency ∼1011). Although the self-exchange rate of the nNOS ferrous/ferric heme-nitrosyl is not known, it is possible to use estimates based on measured rates in other heme proteins. Measured self-exchange rates for various cytochromes show a range extending over 5 orders of magnitude (from 102 to ∼107 m−1 s−1) (47). Much lower rates have also been reported for other systems (e.g. cytochrome c peroxidase k22 = 1.9 × 10−9 m−1 s−1) (48). Although there are no data on self-exchange rates in cytochromes P450, these are expected to be small. In general, metalloproteins in which redox changes are associated with larger reorganization energies exhibit slow electron self-exchange rates. Resonance Raman studies point to changes in heme deformation when going from heme-FeIII-NO to heme-FeII-NO in NOSs (19, 49) as well as a significant change in the Fe-N-O angle (50). It is thus expected that the self-exchange rate for the nNOS-Fe-heme-nitrosyl would not exceed the moderately fast hemeprotein cases with less reorganization (e.g. k22 ≈ 3 × 103 m−1 s−1). Based on this estimate and the values for the other parameters mentioned above, one can estimate an expected outer-sphere electron transfer rate of k12(calc) ≈ 3 × 10−4 s−1 using an [O2] = 270 μm. This rate is 400 times slower than the observed rate for ferrous heme nitrosyl oxidation (kox = 0.115 s−1 at 25 °C) (the ratio is even higher than 400 times at higher temperatures). Fig. S7 shows that this relationship holds even if we assume self-exchange rates for the nNOS-heme ferrous/ferric-nitrosyl couple as high as 106 m−1 s−1 or even 108 m−1 s−1. An outer-sphere electron transfer step limiting kox is therefore unlikely. Instead, the analysis suggests that, either a direct attack of O2 on the bound heme-nitrosyl or, alternatively, an inner-sphere electron transfer complex, leading to a structured transition state, is more likely to be the mechanism that governs kox in nNOS.
Discussion of DFT results
Taken together, the results from our DFT calculations are in agreement with the conclusion that FeII-NO oxidation in NOS (but not globins) occurs via direct attack of 3O2 on the FeII-NO complex. Our DFT results show that the reaction of the NOS heme-thiolate FeII-NO complex with 3O2 generates a stable, N-bound FeIII-N(O)OO− intermediate. In contrast, a direct reaction of globin FeII-NO complexes with 3O2 is not supported by our DFT analysis. This is in agreement with the similar rates observed for globin FeII-NO oxidation and NO dissociation and further supports the idea that globin FeII-NO oxidation occurs through a rate-limiting NO/O2 ligand exchange process, followed by the typical NOD reaction between the oxyglobin complex and NO. In NOS, such a ligand exchange is not necessary.
Our results show that stabilization of the N-bound FeIII-N(O)OO− intermediate can actually be accomplished by a simple hydrogen bond, which is in good agreement with our kinetic data. Based on this result, we re-examined the crystal structure of the nNOS FeII-NO complex, which in fact shows a H2O molecule directly above the FeII-NO unit in the active site (Fig. 5B) (34). In this structure, the H2O appears to be in hydrogen-bonding distance of the oxygen atom of NO and one of the nitrogen atoms of l-Arg, which was co-crystallized in the active site. H2O molecules are also observed directly above the heme in the crystal structure of ferric iNOSox (PDB code 1NOD) (51), again indicating that H2O molecules are commonly found in the active site of NOS, close to the heme, and in the needed position to interact with a peroxynitrite ligand. Based on these observations, it seems highly likely that a H2O molecule would be available to form the proposed hydrogen bond with the newly formed ONOO− ligand under standard catalytic conditions. We thus conclude that this is the most likely scenario for the stabilization of an N-bound FeIII-N(O)OO− intermediate, which is unique to NOS enzymes.
Finally, we investigated two pathways in which the peroxynitrite coordination mode could change from the initial, N-coordinated form to the energetically most favored, terminal oxygen atom–bound FeIII-OONO− form. We propose that this would then allow NOS-assisted sequential isomerization to nitrate through a similar mechanism as the NOD reaction proposed for globin proteins (Figs. 7 and 8; see also Figs. S4–S6 and Table S3). Our DFT calculations predict that formation of a heme-thiolate FeIII-NO3− complex would be 33.4 kcal/mol lower in energy than the terminal cis O-bound FeIII-OONO− complex and 38.8 kcal/mol lower in energy than the trans FeIII-OONO− complex, which is in agreement with the fast second order rate constants determined for the iNOS kox reaction, which indicates that there is a large driving force for nitrate formation, once peroxynitrite has been generated. These results are also in good agreement with gas-phase calculations at the UB3LYP/6-311G(d,p) level of theory, reported for the NOD reaction in globins, where the heme-imidazole FeIII-NO3− complex was predicted to be 28.2 kcal/mol lower in energy than the cis FeIII-OONO− peroxynitrite complex (37).
Experimental procedures
Reagents
H4B and H2B were purchased from Schircks Laboratories (Jona, Switzerland). Oxygen and nitrogen gas (Medipure grade) were from Praxair (Danbury, CT), and NO gas was from Linde LLC (Murray Hill, NJ). Other reagents were purchased from sources reported previously (11, 15).
Protein expression and purification
The oxygenase domains of rat nNOS and mouse iNOS were overexpressed in Escherichia coli BL21 (DE3) cells carrying the corresponding gene in the pCWori vector (38, 52). WT and mutant proteins were purified as described previously (15, 17, 25, 38, 52). Protein concentration was determined from the absorbance at 445 nm of the ferrous heme-CO complex, using an extinction coefficient of 74 mm−1 cm−1 (53).
Ferrous heme-NO oxidation
The reaction was studied as described previously (11). Briefly, ferrous heme-NO complexes were prepared by titrating anaerobic solutions of ferric protein with dithionite and then with NO from a NO-saturated solution. The titrations were monitored using either a Cary100 or a Shimadzu UV-2401 PC spectrophotometer. The resulting protein FeII-NO complexes were rapidly mixed with O2-containing buffer with the same composition as the protein buffer (including H4B and/or l-Arg if present in the protein buffer). Reactions were carried out in a Hi-Tech SF-61 stopped-flow instrument (Hi-Tech Scientific, Salisbury, UK) equipped with a diode array detector. Experiments were performed at 10 °C. Sample solutions contained 10 μm NOS in 40 mm EPPS buffer, pH 7.6 (or 40 mm CHES buffer, pH 9.5), 10% glycerol, 150 mm NaCl, and 0.5 mm EDTA. Depending on the experiment, different combinations of H4B or l-Arg were added. The concentrations used were 20 μm H4B and/or 2.5 mm l-Arg unless stated otherwise. To study the oxygen dependence of the rates, different concentrations of oxygen were produced by mixing oxygen- and nitrogen-saturated buffers.
Kinetic analysis
Reaction rates were calculated from the absorbance changes at the wavelength of the Soret peak of FeIII-heme (∼396 nm, ferric heme recovery rate) and heme FeII-NO (∼436 nm, ferrous heme-NO decay). Data were fitted to a single-exponential equation. The oxygen dependence of the observed rates versus O2 concentration was in all cases best fit to a linear equation in the form, kobs = k1 [O2] + k−1, where k1 and k−1 are the apparent rates of O2 association/dissociation to/from the FeII-NO complex. For iNOSoxy proteins and nNOS W409F mutant, the value of k−1 is much smaller than k1 and cannot be determined accurately. For these proteins, a linear fit through zero in the form kobs = k1 [O2] was preferred.
Calculation of potential shift induced by NO binding
The dissociation constants for a ligand are related to the shift in redox potential, because the Nernst equation can be rewritten as follows (54),
| (Eq. 1) |
where E°c is the redox potential for the heme-NO complex, E° is the potential of the heme in the absence of NO, and KdIII and KdII are the dissociation constants for NO from the FeIII and FeII states, respectively.
Redox potentiometry
The redox potentials for the NOS heme FeIII/FeII couples were determined as previously described in detail (27, 55, 56). The experiments were carried out in a glove box (Belle Technology, Dorset, UK) under nitrogen atmosphere with oxygen levels below 5 ppm. Absorption spectra were recorded in a Cary 50 spectrophotometer using a dip probe detector, and the potentials were monitored using an Accumet AB15 potentiometer (Fisher Scientific) coupled to an Ag/AgCl electrode saturated with 4 m KCl. Protein samples were ∼10 μm in potassium phosphate buffer, 100 mm, pH 7.0, with 125 mm NaCl, 25 μm H4B, and 2.5 mm l-Arg. 1–5 μm anthraquinone 2-sulfonate (Em = −225 mV), phenosafranine (Em = −252 mV), and benzyl viologen (Em = −358 mV) were used as redox mediators. Measurements were done at 15 ± 1 °C. The fraction of protein oxidized was plotted versus the redox potential and fitted to the Nernst equation using Origin version 7.5 software (OriginLab, Northampton, MA). Reported values for the redox potential measurements are mean values ± S.D. of three determinations. Actual errors may be larger, as other factors such as electrode accuracy or equilibration errors are not factored in the calculations.
Computational methods
All DFT calculations were performed with the Linux version of the program Gaussian 09 (57). All geometry optimizations of the DFT models were performed at the UBP86/TZVP level, using the unrestricted formalism of Becke's 1988 exchange functional (58) and the gradient corrections of Perdew, along with his 1981 local correlation functional P86 (BP86) (59). TZVP is a triple ζ polarized basis set by Ahlrichs and co-workers (60, 61). All calculations were performed using the polarizable continuum model as implemented in Gaussian 09 with water selected as the solvent.
The NOS active site model used for all DFT calculations was generated by starting with the coordinates from the X-ray crystal structure of the FeII-NO complex of nNOS (PDB code 2G6K) (34) and keeping only the coordinates of the Fe-NO protoporphyrin IX complex, Cys-415, Trp-409, and Gly-417. The Cys-415 and Trp-409 residues where then truncated to their side chains, an ethanethiolate and a 3-methylindole, respectively. Gly-417 was converted to an N-ethylacetamide to better mimic its structure in the NOS active site (i.e. as an amide group, with the peptide bond present to its neighboring amino acid Val-416). As indicated in Fig. S1, the coordinates of the terminal carbon atoms of the ethanethiolate, the N-ethylacetamide, the protoporphyrin IX propionic acid groups, and the methyl group of 3-methylindole were frozen to conserve their native positions found in the NOS active site. All other atoms were left unconstrained, and the geometry of the structure was fully optimized, with a total charge = −1 and a total multiplicity = 2. After this initial geometry optimization, the coordinates of the protoporphyrin IX propionic acid oxygens and of the protons of the frozen methyl groups were frozen as well for all following calculations. This was necessary to prevent random rotations of the methyl and carboxylic acid groups that were observed in early calculations, which increase computational time without having any relevance for the NOD reaction studied here. Triplet oxygen (3O2) was then introduced into the optimized geometry of the NOS FeII-NO complex, with the triplet spin of the O2 molecule antiparallel to the doublet spin of the FeII-NO complex to allow for bond formation between these two radicals. To achieve this, the wave function of the overall complex was constructed with a total multiplicity = 2 and a total charge = −1, and four fragments were generated for the different components of the NOS model. An “initial guess” was generated and then used to start geometry optimizations or PES scan calculations. The charge and multiplicity of the four fragments were assigned as follows: (i) FeII-NO and 3O2 (charge = 2, multiplicity = 2); (ii) the ethanethiolate ligand (−1, 1); (iii) protoporphyrin IX (−2, 1); (iv) the 3-methylindole and N-ethylacetamide (0, 1). For successful generation of the correct initial wave function, with the S = ½ heme FeII-NO complex and 3O2 with antiparallel spins, an ON-OO distance of at least 3.26 Å was found to be necessary. This distance was thus used as the starting point in the following PES scan calculations.
The globin active site model used for the DFT calculations was generated from the crystal structure of a WT Mb FeII-NO complex (PDB code 1NPF) (62). Here, we used the coordinates of the FeII-NO protoporphyrin IX complex, the axial His-93 and the distal His-64 residues. Both His-93 and His-64 were truncated to the relevant side chains and modeled as 5-ethylimidazole groups (see Fig. S3). The coordinates of the terminal carbon atoms of the 5-ethylimidazole groups and of the protoporphyrin IX propionic acids were frozen, and the rest of the coordinates were fully optimized. In the next step, the carboxylic acid groups were converted to methyl groups to prevent undesired interactions of the carboxylic acids with the 5-ethylimidazole groups based on their native positioning, which does not naturally occur in Mb due to the formation of other interactions (i.e. salt bridges) between the carboxylate propionate groups and neighboring amino acids not included in our DFT model. The model was then re-optimized, and this model was then used for all following calculations. This is the same overall approach as that for the NOS model. A procedure analogous to that for the NOS model was also used to construct the intended wave function of the FeII-NO globin model with added 3O2 (see above). Here, a total multiplicity = 2 and a total charge = 0 were used for the calculation. For the successful generation of the wave function with a doublet FeII-NO complex and a triplet O2 with antiparallel electron spins, it was necessary to first use an ON-OO distance of ∼3 Å. From this starting structure, the PES scan was then conducted.
Author contributions
J. T., J. S., N. L., and D. J. S. conceptualization; J. T., A. P. H., J. S., N. L., and D. J. S. formal analysis; J. T., N. L., and D. J. S. funding acquisition; J. T., A. P. H., J. S., N. L., and D. J. S. investigation; J. T., A. P. H., J. S., N. L., and D. J. S. methodology; J. T., A. P. H., J. S., N. L., and D. J. S. writing-original draft; J. T., A. P. H., J. S., N. L., and D. J. S. writing-review and editing; N. L. and D. J. S. resources; N. L. and D. J. S. supervision; D. J. S. project administration.
Supplementary Material
This work was supported by National Institutes of Health Grant GM51491 (to D. J. S.) and by National Science Foundation Grant CHE-1464696 (to N. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S3 and Figs. S1–S7.
- NOS
- nitric-oxide synthase
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- EPPS
- 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid
- FeII-NO
- ferrous heme-NO complex
- FeIII-NO
- ferric heme-NO complex
- H4B
- (6R)-5,6,7,8-tetrahydro-l-biopterin
- H2B
- 7,8-dihydro-l-biopterin
- kr
- reduction rate of the heme by the reductase (flavoprotein) domain of NOS
- kd
- dissociation rate of NO from the ferric heme-NO complex of NOS
- kox
- oxidation rate of the ferrous heme-NO complex of NOS
- kon
- association rate for the binding of O2 to the ferrous heme-NO complex
- koff
- dissociation rate for the ferrous heme-NO-O2 complex
- nNOS
- neuronal nitric-oxide synthase
- NOSoxy
- oxygenase domain of nitric-oxide synthase
- nNOSoxy
- oxygenase domain of the neuronal nitric-oxide synthase
- iNOS
- inducible nitric-oxide synthase
- iNOSoxy
- oxygenase domain of the inducible nitric-oxide synthase
- eNOS
- endothelial nitric-oxide synthase
- NOD
- nitric oxide dioxygenation
- DFT
- density functional theory
- PES
- potential energy surface
- PDB
- Protein Data Bank.
References
- 1. Alderton W. K., Cooper C. E., and Knowles R. G. (2001) Nitric oxide synthases: structure, function and inhibition. Biochem. J. 357, 593–615 10.1042/0264-6021:3570593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gorren A. C., and Mayer B. (2007) Nitric-oxide synthase: a cytochrome P450 family foster child. Biochim. Biophys. Acta 1770, 432–445 10.1016/j.bbagen.2006.08.019 [DOI] [PubMed] [Google Scholar]
- 3. Stuehr D. J. (1999) Mammalian nitric oxide synthases. Biochim. Biophys. Acta 1411, 217–230 10.1016/S0005-2728(99)00016-X [DOI] [PubMed] [Google Scholar]
- 4. Crane B. R. (2008) The enzymology of nitric oxide in bacterial pathogenesis and resistance. Biochem. Soc. Trans. 36, 1149–1154 10.1042/BST0361149 [DOI] [PubMed] [Google Scholar]
- 5. Santolini J. (2019) What does “NO-Synthase” stand for? Front. Biosci. (Landmark Ed.) 24, 133–171 10.2741/4711 [DOI] [PubMed] [Google Scholar]
- 6. Stuehr D. J., Santolini J., Wang Z. Q., Wei C. C., and Adak S. (2004) Update on mechanism and catalytic regulation in the NO synthases. J. Biol. Chem. 279, 36167–36170 10.1074/jbc.R400017200 [DOI] [PubMed] [Google Scholar]
- 7. Santolini J., Meade A. L., and Stuehr D. J. (2001) Differences in three kinetic parameters underpin the unique catalytic profiles of nitric-oxide synthases I, II, and III. J. Biol. Chem. 276, 48887–48898 10.1074/jbc.M108666200 [DOI] [PubMed] [Google Scholar]
- 8. Santolini J., Adak S., Curran C. M., and Stuehr D. J. (2001) A kinetic simulation model that describes catalysis and regulation in nitric-oxide synthase. J. Biol. Chem. 276, 1233–1243 10.1074/jbc.M006858200 [DOI] [PubMed] [Google Scholar]
- 9. Salerno J. C., and Ghosh D. K. (2009) Space, time and nitric oxide–neuronal nitric oxide synthase generates signal pulses. FEBS J. 276, 6677–6688 10.1111/j.1742-4658.2009.07382.x [DOI] [PubMed] [Google Scholar]
- 10. Adak S., Wang Q., and Stuehr D. J. (2000) Molecular basis for hyperactivity in tryptophan 409 mutants of neuronal NO synthase. J. Biol. Chem. 275, 17434–17439 10.1074/jbc.M000846200 [DOI] [PubMed] [Google Scholar]
- 11. Tejero J., Santolini J., and Stuehr D. J. (2009) Fast ferrous heme-NO oxidation in nitric oxide synthases. FEBS J. 276, 4505–4514 10.1111/j.1742-4658.2009.07157.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Møller J. K., and Skibsted L. H. (2004) Mechanism of nitrosylmyoglobin autoxidation: temperature and oxygen pressure effects on the two consecutive reactions. Chemistry 10, 2291–2300 10.1002/chem.200305368 [DOI] [PubMed] [Google Scholar]
- 13. Herold S., and Röck G. (2005) Mechanistic studies of the oxygen-mediated oxidation of nitrosylhemoglobin. Biochemistry 44, 6223–6231 10.1021/bi0475929 [DOI] [PubMed] [Google Scholar]
- 14. Adak S., and Stuehr D. J. (2001) A proximal tryptophan in NO synthase controls activity by a novel mechanism. J. Inorg. Biochem. 83, 301–308 10.1016/S0162-0134(00)00176-8 [DOI] [PubMed] [Google Scholar]
- 15. Adak S., Crooks C., Wang Q., Crane B. R., Tainer J. A., Getzoff E. D., and Stuehr D. J. (1999) Tryptophan 409 controls the activity of neuronal nitric-oxide synthase by regulating nitric oxide feedback inhibition. J. Biol. Chem. 274, 26907–26911 10.1074/jbc.274.38.26907 [DOI] [PubMed] [Google Scholar]
- 16. Pant K., and Crane B. R. (2005) Structure of a loose dimer: an intermediate in nitric oxide synthase assembly. J. Mol. Biol. 352, 932–940 10.1016/j.jmb.2005.07.070 [DOI] [PubMed] [Google Scholar]
- 17. Ghosh D. K., Crane B. R., Ghosh S., Wolan D., Gachhui R., Crooks C., Presta A., Tainer J. A., Getzoff E. D., and Stuehr D. J. (1999) Inducible nitric oxide synthase: role of the N-terminal β-hairpin hook and pterin-binding segment in dimerization and tetrahydrobiopterin interaction. EMBO J. 18, 6260–6270 10.1093/emboj/18.22.6260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Couture M., Adak S., Stuehr D. J., and Rousseau D. L. (2001) Regulation of the properties of the heme-NO complexes in nitric-oxide synthase by hydrogen bonding to the proximal cysteine. J. Biol. Chem. 276, 38280–38288 10.1074/jbc.M105341200 [DOI] [PubMed] [Google Scholar]
- 19. Rousseau D. L., Li D., Couture M., and Yeh S.-R. (2005) Ligand–protein interactions in nitric oxide synthase. J. Inorg. Biochem. 99, 306–323 10.1016/j.jinorgbio.2004.11.007 [DOI] [PubMed] [Google Scholar]
- 20. Baek K. J., Thiel B. A., Lucas S., and Stuehr D. J. (1993) Macrophage nitric oxide synthase subunits: purification, characterization, and role of prosthetic groups and substrate in regulating their association into a dimeric enzyme. J. Biol. Chem. 268, 21120–21129 [PubMed] [Google Scholar]
- 21. Klatt P., Schmidt K., Lehner D., Glatter O., Bächinger H. P., and Mayer B. (1995) Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrahydrobiopterin and l-arginine in the formation of an SDS-resistant dimer. EMBO J. 14, 3687–3695 10.1002/j.1460-2075.1995.tb00038.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Presta A., Siddhanta U., Wu C., Sennequier N., Huang L., Abu-Soud H. M., Erzurum S., and Stuehr D. J. (1998) Comparative functioning of dihydro- and tetrahydropterins in supporting electron transfer, catalysis, and subunit dimerization in inducible nitric oxide synthase. Biochemistry 37, 298–310 10.1021/bi971944c [DOI] [PubMed] [Google Scholar]
- 23. Presta A., Weber-Main A. M., Stankovich M. T., and Stuehr D. J. (1998) Comparative effects of substrates and pterin cofactor on the heme midpoint potential in inducible and neuronal nitric oxide synthases. J. Am. Chem. Soc. 120, 9460–9465 10.1021/ja9740374 [DOI] [Google Scholar]
- 24. Bec N., Gorren A. C., Voelker C., Mayer B., and Lange R. (1998) Reaction of neuronal nitric-oxide synthase with oxygen at low temperature: evidence for reductive activation of the oxy-ferrous complex by tetrahydrobiopterin. J. Biol. Chem. 273, 13502–13508 10.1074/jbc.273.22.13502 [DOI] [PubMed] [Google Scholar]
- 25. Wang Z. Q., Wei C. C., Sharma M., Pant K., Crane B. R., and Stuehr D. J. (2004) A conserved Val to Ile switch near the heme pocket of animal and bacterial nitric-oxide synthases helps determine their distinct catalytic profiles. J. Biol. Chem. 279, 19018–19025 10.1074/jbc.M311663200 [DOI] [PubMed] [Google Scholar]
- 26. Whited C. A., Warren J. J., Lavoie K. D., Weinert E. E., Agapie T., Winkler J. R., and Gray H. B. (2012) Gating NO release from nitric oxide synthase. J. Am. Chem. Soc. 134, 27–30 10.1021/ja2069533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tejero J., Biswas A., Wang Z. Q., Page R. C., Haque M. M., Hemann C., Zweier J. L., Misra S., and Stuehr D. J. (2008) Stabilization and characterization of a heme-oxy reaction intermediate in inducible nitric-oxide synthase. J. Biol. Chem. 283, 33498–33507 10.1074/jbc.M806122200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lang J., Driscoll D., Gélinas S., Rafferty S. P., and Couture M. (2009) Trp180 of endothelial NOS and Trp56 of bacterial saNOS modulate σ bonding of the axial cysteine to the heme. J. Inorg. Biochem. 103, 1102–1112 10.1016/j.jinorgbio.2009.05.011 [DOI] [PubMed] [Google Scholar]
- 29. Lang J., Santolini J., and Couture M. (2011) The conserved Trp-Cys hydrogen bond dampens the “push effect” of the heme cysteinate proximal ligand during the first catalytic cycle of nitric oxide synthase. Biochemistry 50, 10069–10081 10.1021/bi200965e [DOI] [PubMed] [Google Scholar]
- 30. Brunel A., Wilson A., Henry L., Dorlet P., and Santolini J. (2011) The proximal hydrogen bond network modulates Bacillus subtilis nitric-oxide synthase electronic and structural properties. J. Biol. Chem. 286, 11997–12005 10.1074/jbc.M110.195446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hannibal L., Somasundaram R., Tejero J., Wilson A., and Stuehr D. J. (2011) Influence of heme-thiolate in shaping the catalytic properties of a bacterial nitric-oxide synthase. J. Biol. Chem. 286, 39224–39235 10.1074/jbc.M111.286351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ost T. W., and Daff S. (2005) Thermodynamic and kinetic analysis of the nitrosyl, carbonyl, and dioxy heme complexes of neuronal nitric-oxide synthase. The roles of substrate and tetrahydrobiopterin in oxygen activation. J. Biol. Chem. 280, 965–973 10.1074/jbc.M411191200 [DOI] [PubMed] [Google Scholar]
- 33. Gao Y. T., Smith S. M., Weinberg J. B., Montgomery H. J., Newman E., Guillemette J. G., Ghosh D. K., Roman L. J., Martasek P., and Salerno J. C. (2004) Thermodynamics of oxidation-reduction reactions in mammalian nitric-oxide synthase isoforms. J. Biol. Chem. 279, 18759–18766 10.1074/jbc.M308936200 [DOI] [PubMed] [Google Scholar]
- 34. Li H., Igarashi J., Jamal J., Yang W., and Poulos T. L. (2006) Structural studies of constitutive nitric oxide synthases with diatomic ligands bound. J. Biol. Inorg. Chem. 11, 753–768 10.1007/s00775-006-0123-8 [DOI] [PubMed] [Google Scholar]
- 35. Pryor W. A., and Squadrito G. L. (1995) The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am. J. Physiol. 268, L699–L722 10.1152/ajplung.1995.268.5.L699 [DOI] [PubMed] [Google Scholar]
- 36. Blomberg L. M., Blomberg M. R., and Siegbahn P. E. (2004) A theoretical study of myoglobin working as a nitric oxide scavenger. J. Biol. Inorg. Chem. 9, 923–935 10.1007/s00775-004-0585-5 [DOI] [PubMed] [Google Scholar]
- 37. Carabet L. A., Guertin M., Lagüe P., Lamoureux G. (2017) Mechanism of the nitric oxide dioxygenase reaction of Mycobacterium tuberculosis hemoglobin N. J. Phys. Chem. B 121, 8706–8718 10.1021/acs.jpcb.7b06494 [DOI] [PubMed] [Google Scholar]
- 38. Abu-Soud H. M., Gachhui R., Raushel F. M., and Stuehr D. J. (1997) The ferrous-dioxy complex of neuronal nitric oxide synthase: divergent effects of l-arginine and tetrahydrobiopterin on its stability. J. Biol. Chem. 272, 17349–17353 10.1074/jbc.272.28.17349 [DOI] [PubMed] [Google Scholar]
- 39. Marchal S., Gorren A. C., Sørlie M., Andersson K. K., Mayer B., and Lange R. (2004) Evidence of two distinct oxygen complexes of reduced endothelial nitric oxide synthase. J. Biol. Chem. 279, 19824–19831 10.1074/jbc.M313587200 [DOI] [PubMed] [Google Scholar]
- 40. Crane B. R., Arvai A. S., Gachhui R., Wu C., Ghosh D. K., Getzoff E. D., Stuehr D. J., and Tainer J. A. (1997) The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science 278, 425–431 10.1126/science.278.5337.425 [DOI] [PubMed] [Google Scholar]
- 41. Fernández M. L., Martí M. A., Crespo A., and Estrin D. A. (2005) Proximal effects in the modulation of nitric oxide synthase reactivity: a QM-MM study. J. Biol. Inorg. Chem. 10, 595–604 10.1007/s00775-005-0004-6 [DOI] [PubMed] [Google Scholar]
- 42. Arnold E. V., and Bohle D. S. (1996) Isolation and oxygenation reactions of nitrosylmyoglobins. Methods Enzymol. 269, 41–55 10.1016/S0076-6879(96)69008-9 [DOI] [PubMed] [Google Scholar]
- 43. Herold S. (1999) Mechanistic studies of the oxidation of pyridoxalated hemoglobin polyoxyethylene conjugate by nitrogen monoxide. Arch. Biochem. Biophys. 372, 393–398 10.1006/abbi.1999.1534 [DOI] [PubMed] [Google Scholar]
- 44. Tsai J.-H. M., Harrison J. G., Martin J. C., Hamilton T. P., van der Woerd M., Jablonsky M. J., and Beckman J. S. (1994) Role of conformation of peroxynitrite anion (ONOO−) with its stability and toxicity. J. Am. Chem. Soc. 116, 4115–4116 10.1021/ja00088a072 [DOI] [Google Scholar]
- 45. Marcus R. A., and Sutin N. (1985) Electron transfers in chemistry and biology. Biochim. Biophys. Acta Rev. Bioenerg. 811, 265–322 10.1016/0304-4173(85)90014-X [DOI] [Google Scholar]
- 46. Lind J., Shen X., Merenyi G., and Jonsson B. (1989) Determination of the rate constant of self-exchange of the oxygen O2/O2-couple in water by 18O/16O isotope marking. J. Am. Chem. Soc. 111, 7654–7655 10.1021/ja00201a078 [DOI] [Google Scholar]
- 47. Simonneaux G., and Bondon A. (2005) Mechanism of electron transfer in heme proteins and models: the NMR approach. Chem. Rev. 105, 2627–2646 10.1021/cr030731s [DOI] [PubMed] [Google Scholar]
- 48. Cheung E., and English A. M. (1995) Reductions by ferrocytochrome c peroxidase: 5. Kinetics of ferricyanide reduction. Can. J. Chem. 73, 1181–1186 10.1139/v95-145 [DOI] [Google Scholar]
- 49. Li D., Stuehr D. J., Yeh S. R., and Rousseau D. L. (2004) Heme distortion modulated by ligand-protein interactions in inducible nitric-oxide synthase. J. Biol. Chem. 279, 26489–26499 10.1074/jbc.M400968200 [DOI] [PubMed] [Google Scholar]
- 50. Pant K., and Crane B. R. (2006) Nitrosyl-heme structures of Bacillus subtilis nitric oxide synthase have implications for understanding substrate oxidation. Biochemistry 45, 2537–2544 10.1021/bi0518848 [DOI] [PubMed] [Google Scholar]
- 51. Crane B. R., Arvai A. S., Ghosh D. K., Wu C., Getzoff E. D., Stuehr D. J., and Tainer J. A. (1998) Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science 279, 2121–2126 10.1126/science.279.5359.2121 [DOI] [PubMed] [Google Scholar]
- 52. Ghosh D. K., Wu C., Pitters E., Moloney M., Werner E. R., Mayer B., and Stuehr D. J. (1997) Characterization of the inducible nitric oxide synthase oxygenase domain identifies a 49 amino acid segment required for subunit dimerization and tetrahydrobiopterin interaction. Biochemistry 36, 10609–10619 10.1021/bi9702290 [DOI] [PubMed] [Google Scholar]
- 53. Stuehr D. J., and Ikeda-Saito M. (1992) Spectral characterization of brain and macrophage nitric oxide synthases: cytochrome P-450-like hemeproteins that contain a flavin semiquinone radical. J. Biol. Chem. 267, 20547–20550 [PubMed] [Google Scholar]
- 54. Andersen J. F., Ding X. D., Balfour C., Shokhireva T. K., Champagne D. E., Walker F. A., and Montfort W. R. (2000) Kinetics and equilibria in ligand binding by nitrophorins 1–4: evidence for stabilization of a nitric oxide-ferriheme complex through a ligand-induced conformational trap. Biochemistry 39, 10118–10131 10.1021/bi000766b [DOI] [PubMed] [Google Scholar]
- 55. Konas D. W., Takaya N., Sharma M., and Stuehr D. J. (2006) Role of Asp1393 in catalysis, flavin reduction, NADP(H) binding, FAD thermodynamics, and regulation of the nNOS flavoprotein. Biochemistry 45, 12596–12609 10.1021/bi061011t [DOI] [PubMed] [Google Scholar]
- 56. Ilagan R. P., Tiso M., Konas D. W., Hemann C., Durra D., Hille R., and Stuehr D. J. (2008) Differences in a conformational equilibrium distinguish catalysis by the endothelial and neuronal nitric-oxide synthase flavoproteins. J. Biol. Chem. 283, 19603–19615 10.1074/jbc.M802914200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Frisch M., Trucks G., Schlegel H., Scuseria G., Robb M., Cheeseman J., Scalmani G., Barone V., Mennucci B., Petersson G., Nakatsuji H., Caricato M., Li X., Hratchian H., Izmaylov A., et al. (2009) Gaussian 09, revision a.02, Gaussian Inc., Wallingford, CT [Google Scholar]
- 58. Becke A. D. (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 38, 3098–3100 10.1103/PhysRevA.38.3098 [DOI] [PubMed] [Google Scholar]
- 59. Perdew J. P. (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter 33, 8822–8824 10.1103/PhysRevB.33.8822 [DOI] [PubMed] [Google Scholar]
- 60. Schäfer A., Huber C., and Ahlrichs R. (1994) Fully optimized contracted Gaussian basis sets of triple ζ valence quality for atoms Li to Kr. J. Chem. Phys. 100, 5829–5835 10.1063/1.467146 [DOI] [Google Scholar]
- 61. Schäfer A., Horn H., and Ahlrichs R. (1992) Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577 10.1063/1.463096 [DOI] [Google Scholar]
- 62. Copeland D. M., West A. H., and Richter-Addo G. B. (2003) Crystal structures of ferrous horse heart myoglobin complexed with nitric oxide and nitrosoethane. Proteins 53, 182–192 10.1002/prot.10495 [DOI] [PubMed] [Google Scholar]
- 63. Haque M. M., Tejero J., Bayachou M., Wang Z. Q., Fadlalla M., and Stuehr D. J. (2013) Thermodynamic characterization of five key kinetic parameters that define neuronal nitric oxide synthase catalysis. FEBS J. 280, 4439–4453 10.1111/febs.12404 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.