

Abstract
Objective
We developed a novel, hybrid method combining both blue-native (BN-PAGE) and clear-native (CN-PAGE) polyacrylamide gel electrophoresis, termed BCN-PAGE, to perform in-gel activity stains on the mitochondrial electron transport chain (ETC) complexes in skin fibroblasts.
Methods
Four patients aged 46–65 years were seen in the Metabolic Clinic at Alberta Children's Hospital and investigated for mitochondrial disease and had BN-PAGE or CN-PAGE on skeletal muscle that showed incomplete assembly of complex V (CV) in each patient. Long-range PCR performed on muscle-extracted DNA identified 4 unique mitochondrial DNA (mtDNA) deletions spanning the ATP6 gene of CV. We developed a BCN-PAGE method in skin fibroblasts taken from the patients at the same time and compared the findings with those in skeletal muscle.
Results
In all 4 cases, BCN-PAGE in skin fibroblasts confirmed the abnormal CV activity found from muscle biopsy, suggesting that the mtDNA deletions involving ATP6 were most likely germline mutations that are associated with a clinical phenotype of mitochondrial disease.
Conclusions
The BCN-PAGE method in skin fibroblasts has a potential to be a less-invasive tool compared with muscle biopsy to screen patients for abnormalities in CV and other mitochondrial ETC complexes.
Mitochondrial diseases can have abnormal electron transport chain (ETC) dysfunction.1 Electrons are transferred through 5 protein complexes (I, II, III, IV, and V) that interact and form supercomplexes (respirasomes) in the inner mitochondrial membrane. Tissues with high energy requirements may be more vulnerable to disruption of ETC function caused by either nuclear DNA (nDNA) or mitochondrial DNA (mtDNA) mutations.2 Skeletal muscle biopsy is the preferred source to measure ETC protein integrity and function because of the higher mitochondrial density, but can be invasive.3
While a skin biopsy is less invasive,4,5 there have been some concerns whether existing procedures can represent function in skeletal muscle due to a lower mitochondrial density, activity and some metabolic defects are not expressed in skin fibroblasts.6 Most techniques involve measuring enzyme activity and using ETC protein immunoblotting to detect protein abundance7,8 and their utility in the clinical setting in patients with disease is not clear.9,10
Our aim was to determine whether low-level deletions found in a muscle samples also existed in cultured skin fibroblasts using either one or a combination of blue-native or clear-native polyacrylamide gel electrophoresis (BN-PAGE/CN-PAGE) of ETC proteins. We found that a hybrid method, blue-native and clear-native polyacrylamide gel electrophoresis (BCN-PAGE), was able to resolve the difficulty in abnormal detecting complex V (CV) patterns in patients with the disease.4,5
Methods
Standard protocol approvals, registrations, and patient consents
All experimental procedures of this study were performed in accordance with the regulations of the University of Calgary's Conjoint Health Research Ethics Board (REB13-0753) and the Declaration of Helsinki, and written informed patient consent was obtained.
Patients and tissues
The mitochondrial clinic at Alberta Children's Hospital uses a standard protocol for mitochondrial disease testing, which includes a muscle needle biopsy for light and electron microscopy11,12, mtDNA extraction for Kearns-Sayre syndrome Southern blot, targeted mutation analysis, long-range PCR for private deletions and point mutations, muscle for BN-PAGE or CN-PAGE, and skin biopsy for fibroblast culture. Four patients aged 46–65 years were seen in the Metabolic Clinic at Alberta Children's Hospital (Calgary, AB) and investigated for mitochondrial disease (table 1). Four controls aged 46–62 years were also selected from a bank of skin fibroblasts that had previously been investigated for and found not to have a diagnosis of an inborn error of metabolism or a mitochondrial disease through the Metabolic Clinic at Alberta Children's Hospital.
Table.
Patient characteristics at the time of diagnosis confirmation

Muscle and skin biopsies were performed as part of standard-of-care diagnostic procedures using a needle muscle biopsy.13 Approximately 150 mg total muscle sample was biopsied from the vastus lateralis before being snap frozen without preservatives and briefly stored in liquid nitrogen. A portion of muscle was sent for respiratory chain enzyme analysis and either BN-PAGE or CN-PAGE14,15 to either University of Colorado Denver Biochemical Genetics Laboratory (Aurora, CO) or Medical Neurogenetics Laboratories (Atlanta, GA) in accordance with the provincial health plan. The remaining muscle was sent for mtDNA sequencing and assessment (Sanger or next-generation sequencing (NGS) and Southern blot) at the University of Alberta—Molecular Diagnostic Laboratory (Edmonton, AB). BN-PAGE or CN-PAGE identified incomplete assembly of CV in each patient, characterized by the CV doublet (figure 1). The combined sequencing and Southern blot analyses performed on muscle tissue successfully identified 4 unique mtDNA deletions spanning the ATP6 gene of CV (table 1).16 Skin samples were collected using either a 4-mm circular punch biopsy or a 4 × 2-mm linear piece removed from the incision site of a muscle biopsy and transferred to the Biochemical Genetics Laboratory at Alberta Children's Hospital (Calgary, AB) for subsequent fibroblast culturing.
Figure 1. Clinical identification of incompletely assembled mitochondrial CV from skeletal muscle. A representative image identifying incomplete assembly of mitochondrial CV from skeletal muscle.
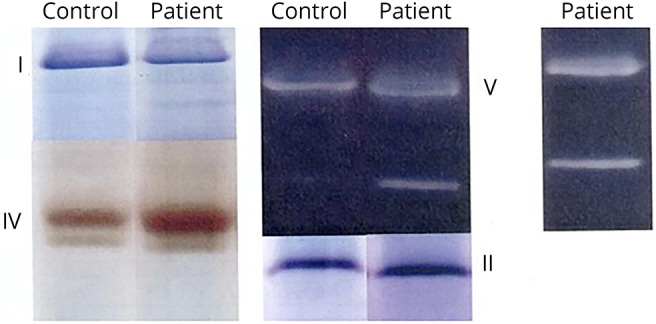
Clinical diagnostics of patient mitochondrial enzyme assemblies were performed using in-gel activity staining of BN-PAGE or CN-PAGE. Diagnostic assessments were conducted on patient skeletal muscle tissues with BN-PAGE14,15 performed at University of Colorado Denver Biochemical Genetics Laboratory (n = 3) (Aurora, CO) and CN-PAGE at MNG Laboratories (n = 1) (Atlanta, GA). Representative image pertains to the BN-PAGE cohort (patient 1). BN-PAGE or CN-PAGE = blue-native or clear-native polyacrylamide gel electrophoresis; CV = complex V; MNG = Medical Neurogenetics.
Skin fibroblast culture
Patient and control skin biopsy tissues were each passaged to P5 and expanded into T175 flasks as per the protocol used by the Biochemical Genetics Laboratory at Alberta Children's Hospital. Cellular media, composed of minimum essential medium with 2 mM glutamine (Life Technologies, Burlington, ON), 10% fetal bovine serum (Life Technologies), 1 mM sodium pyruvate (Life Technologies), 20 mM uridine (Life Technologies), and 100 U/mL penicillin-streptomycin, were removed and replaced after 3 successive days of cell incubation.17 Previously shown to reduce false-negative mtDNA results in skin fibroblasts, uridine and sodium pyruvate were added to the media to maintain survival of abnormal mitochondria.18 On reaching 80% confluence, skin fibroblasts were detached from their respective flasks using 3 mL 0.25% trypsin/1 mM EDTA (Life Technologies) and monitored under light microscopy to establish successful separation. Detached skin fibroblasts from 8 T175 flasks per patient/control cell line were aspirated with a 25-mL pipette and pooled in a 50-mL conical tube. Pooled cells were centrifuged at 200g for 5 minutes at room temperature (Beckman Spinchron R Centrifuge; Beckman Coulter, Ramsey, MN). The supernatant was discarded, and the pellet was carefully washed 3× with 2 mL sucrose buffer—stock solution (250 mM sucrose, 20 mM Tris, 0.1 mM EDTA, pH 7.4) (Sigma-Aldrich, Oakville, ON) before being resuspended in 5 mL of cold sucrose buffer (4°C). The cell suspension was then transferred into a glass and Teflon homogenizer and underwent 20 passes on ice. Homogenized cell suspension was then centrifuged at 3,000 rpm for 7 minutes at 4°C (Sorvall Legend Micro 21R Microcentrifuge; Thermo Fisher Scientific, Burlington, ON). The supernatant was then removed (pellet discarded) and centrifuged at 10,000 rpm for 10 minutes at 4°C (Sorvall Legend Micro 21R Microcentrifuge). The supernatant was then discarded, and the pellet was resuspended in 2 mL of cold (4°C) sucrose buffer before another centrifugation step at 10,000 rpm for 10 minutes at 4°C (Sorvall Legend Micro 21R Microcentrifuge). The supernatant was then discarded, and the pellet was resuspended in 250 μL of buffer C—stock solution (1.5 M aminocaproic acid, 50 mM Bis-Tris, pH 7.0) (Sigma-Aldrich). At this stage, a 10-μL aliquot was removed for protein quantification of skin fibroblast mitochondria, with bovine serum albumin as the standard, using the Bradford method (Bio-Rad, Hercules, CA). Solubilization of the mitochondrial membranes was then achieved by incubating 1.6 mg of the nonionic detergent n-Dodecyl β-D-maltoside (DDM—from a stock solution) per 1 mg mitochondrial protein for 20 minutes on ice. After solubilization, samples were centrifuged at 14,800 rpm for 30 minutes at 4°C (Sorvall Legend Micro 21R Microcentrifuge). The supernatant was discarded, and the pellet was resuspended in Brilliant Blue G (BBG) (Sigma-Aldrich)—300 µg BBG (from a stock solution) per 100 μg of mitochondrial protein. The mitochondrial protein product suspended in BBG was stored at −80°C until BCN-PAGE analysis.
Blue-native and clear-native polyacrylamide gel electrophoresis
BCN-PAGE was performed using an XCell SureLock Mini-Cell electrophoresis system at 4°C (Thermo Fisher Scientific). NativePAGE 4%–16% Bis-Tris gels (Thermo Fisher Scientific) were directly loaded with 40 µg mitochondrial protein per well using the stored solutions of mitochondrial protein product and BBG. Gels were run at 160 V for the first hour, and the electric field strength was then modified to 100 V for the remaining 4.5 hours, as this was found to optimize protein separation. Each electrophoresis experiment used a single batch of anode (50 mM Tricine, 15 mM Bis-Tris, pH 7.0) and cathode (50 mM Tricine, 15 mM Bis-Tris, 0.01% wt/vol DDM, pH 7.0) (Sigma-Aldrich) buffers without replacement.
In-gel enzyme activity staining
Following BCN-PAGE, adapted from previously published protocols, each individual oxidative phosphorylation (OXPHOS) protein complex was investigated for its respective enzymatic activity and protein complex assembly.19–21 Individual gels were run for each OXPHOS enzyme complex, with each gel containing all 4 control and all 4 patient samples. Starting with complex I, gels were completed in ascending order of associated OXPHOS enzyme complex number up to CV. Each activity stain was prepared fresh to an approximate volume of 20 mL (see below), and before scanning or imaging, each gel was rinsed thoroughly with water and mild agitation (3 × 10 minutes). For complex III, a protein abundance stain was used because the published activity stain is not deemed effective in skin fibroblasts.22
Complex I activity
The gel was preincubated in 2 mM Tris-HCl pH 7.0 (Sigma-Aldrich) for 15 minutes at room temperature with mild agitation. It was then transferred to a solution of 2 mM Tris-HCl pH 7.4 (Sigma-Aldrich) containing 0.1 mg/mL nicotinamide adenine dinucleotide (Sigma-Aldrich) and 0.25 mg/mL nitro blue tetrazolium (NBT) (Sigma-Aldrich) at room temperature with mild agitation. Banding began to develop within 2 hours, with optimal band visualization >24 hours.
Complex II activity
The gel was preincubated in 200 mM Tris-HCl pH 7.4 (Sigma-Aldrich) for 15 minutes at room temperature with mild agitation. The gel was then incubated in a solution of 200 mM Tris-HCl pH 7.4 (Sigma-Aldrich) containing 30 mM succinic acid (Sigma-Aldrich), 0.2 mM phenazine methosulfate (Sigma-Aldrich), 2 mM EDTA (Sigma-Aldrich), 2 mM potassium cyanide (Sigma-Aldrich), and 1.0 mg/mL NBT (Sigma-Aldrich) at room temperature with mild agitation. Banding developed within 12 hours.
Complex III protein abundance
The gel was preincubated in 5 mM Tris-HCl pH 7.4 (Sigma-Aldrich) for 15 minutes at room temperature with mild agitation. The gel was then incubated in approximately 20 mL of 1-Step tetramethybenzidine-Blotting Substrate Solution (Thermo Fisher Scientific) at room temperature with mild agitation. Banding developed within 6–12 hours, with optimal band visualization after 12 hours.
Complex IV activity
The gel was preincubated in 50 mM phosphate buffer pH 7.4 (Sigma-Aldrich) for 15 minutes at room temperature with mild agitation. It was then incubated in a solution of 50 mM phosphate buffer pH 7.4 (Sigma-Aldrich) containing 10 mg diaminobenzidine tetrahydrochloride (Sigma-Aldrich), 20 mg cytochrome c (Sigma-Aldrich), and 1.5 g sucrose (Sigma-Aldrich) at room temperature with mild agitation. Banding developed within 4–12 hours.
CV activity
The gel was rinsed thoroughly with water and mild agitation (3 × 10 minutes) before being preincubated in 50 mM Tris pH 8.6 (Sigma-Aldrich) for 1 hour at room temperature with mild agitation. During this time, a solution containing the following chemicals added in the following order was prepared: 35 mM Tris, 270 mM glycine, 14 mM MgSO4, and 8 mM ATP (Sigma-Aldrich). The solution was then adjusted to a pH of 7.8 before the addition of 0.2% Pb (NO3)2 (Sigma-Aldrich). Finally, the solution was adjusted to a pH of 8.6, and the gel was incubated at 37°C with mild agitation. Banding developed within 1–2 hours, with optimal band development after 18 hours.
Protein immunoblotting
Following BCN-PAGE, protein immunoblotting adapted from previously published work21 was performed using separate gels for patient and control samples. After being transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA), primary antibodies were used to probe the membranes overnight at 4°C on a shaking platform. Secondary antibodies were then incubated on the membrane for 1.5 hours at room temperature. Primary antibodies were as follows: Total OXPHOS (catalog no. ab110413; Abcam, Cambridge, MA) and β-actin (catalog no. ab8226; Abcam). Membranes were exposed to the chemiluminescent SuperSignal West Femto Maximum Sensitivity Substrate (Life Technologies) and imaged using the ChemiGenius imaging system (Syngene, Frederick, MD). Densitometry was performed using GeneTools (Syngene), with β-actin as a loading control.
Whole-exome sequencing
Two of the patients (1 and 4) also participated in our NGS study and had whole-exome sequencing performed. Exomes were sequenced using the 5500XL SOLiD System (Life Technologies), and exome enrichment was performed using Agilent's SureSelect XT Human All Exon V5 (Agilent Technologies). For secondary analysis, the sequencing data were uploaded to the Galaxy instance of University of Calgary (vpn.chgi.ucalgary.ca/), which used the Genome Analysis Tool Kit and sequence alignment map tools to generate a variant call file. Filtering and interpretative analysis of the resultant annotated variants were conducted in .xlsx format. The filtering strategy consisted of sequencing quality parameters (variant reads), frequency of the variant (≤MAF 0.01), zygosity, variant context, and computational evidence such as PolyPhen, scale-invariant feature transform, and genomic evolutionary rate profiling. The assembly used was GRCh37/hg19.
Statistical analyses
Statistical analysis was performed using GraphPad Prism for Windows, Version 7.02 (GraphPad Software Inc., La Jolla, CA). Differences between groups were determined by Student t tests where p < 0.05 was significant. Data are expressed as mean ± SEM.
Data availability
Anonymized data will be shared by request from any qualified investigators.
Results
In-gel enzyme activity staining
A representative image comparing BCN-PAGE resolved mitochondrial OXPHOS protein complexes between patient 1 and control 1 is reported in figure 2. The results of our modified BCN-PAGE assay demonstrate the ability to both successfully identify each of the 5 mitochondrial OXPHOS protein complexes and to recapitulate clinical diagnostic findings of incompletely assembled CV exclusively found in patient samples. Individualized details of each OXPHOS protein complex are described in the following sections.
Figure 2. Clinical identification of incompletely assembled mitochondrial CV from skin fibroblasts. A representative image identifying incomplete assembly of mitochondrial CV from skin fibroblasts.
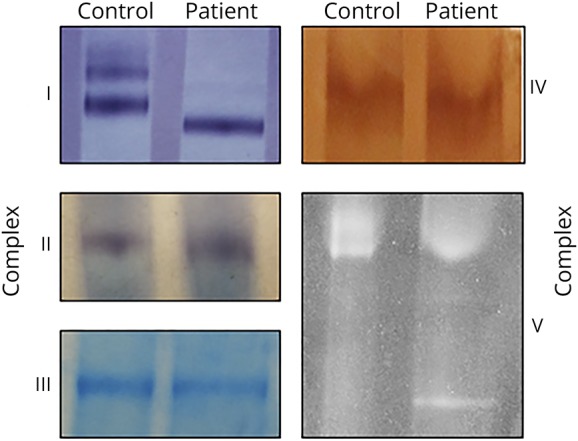
Assessment of patient mitochondrial enzyme assemblies was performed using in-gel activity staining of hybrid BCN-PAGE. Affirmation of previous diagnostic assessments was conducted on isolated mitochondria from skin fibroblasts of patients and controls (n = 4 per group). Representative image pertains to data from patient 1 and control 1 samples. BCN-PAGE = blue-native and clear-native polyacrylamide gel electrophoresis; CV = complex V.
Complex I activity
Clinical in-gel activity staining identified normal activity for complex I in each patient (n = 4) compared with an internal control sample. BCN-PAGE analysis identified a pronounced difference in protein complex assembly between patient and control samples (figure 2). A combination of lower and higher protein assembly existed for control samples, whereas only a lower assembly of complex I was present in the patients (n = 4, per group). The higher assembly is most likely supercomplexes, as documented previously.23
Complex II and complex IV activities
Clinical in-gel activity staining identified normal activity of both complex I and complex IV for each patient (n = 4, per complex) compared with an internal control sample. BCN-PAGE analysis corroborated these findings by discerning no visual differences between patient and control samples (n = 4, per group) (figure 2).
Complex III protein abundance
Clinical in-gel activity staining of complex III is commonly not reported because of reproducibility in fibroblasts. However, BCN-PAGE analysis can resolve a protein abundance of complex III using the 1-Step tetramethybenzidine-Blotting Substrate Solution (figure 2). No visual differences between protein abundance or assembly were identified between patient and control samples (n = 4, per group).
CV activity
Clinical in-gel activity staining identified normal activity of CV, although each patient sample also identified a stronger-than-normal single band of incompletely assembled CV (n = 4) compared with an internal control. BCN-PAGE analysis confirmed these findings by identifying a similarly misassembled CV doublet in patient samples, whereas each control sample presented as a single band (n = 4, per group) (figure 2).
Protein immunoblotting
Protein levels of individual mitochondrial OXPHOS protein complexes were assessed to provide a quantitative measure of protein abundance. Examination of the blots revealed no significant differences in protein abundance for mitochondrial OXPHOS complexes II, III, IV, or V (n = 4, per group) (figure 3). Interestingly, even in skin fibroblasts, we found abundant complex I in control samples compared with patients, potentially affirming visual contrast between patient and control samples from BCN-PAGE experiments.
Figure 3. Examination of OXPHOS proteins isolated from patient skin fibroblasts with incompletely assembled mitochondrial CV.
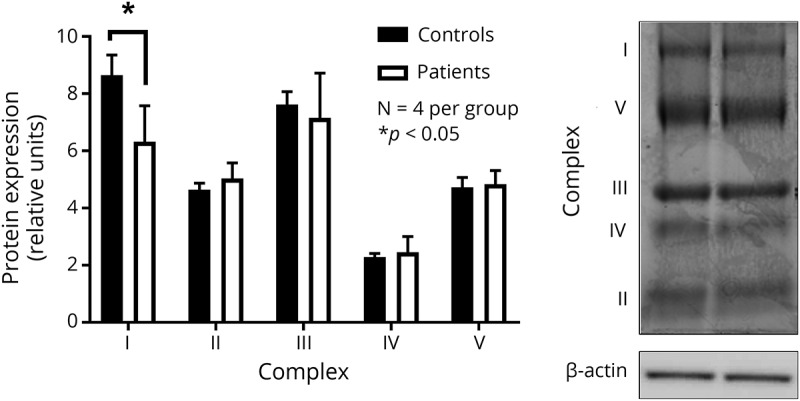
Assessment of mitochondrial OXPHOS proteins in mitochondria isolated from patients with mitochondrial CV deletions and controls, using skin fibroblasts. Results are accompanied by representative immunoblot images with n = 4 per group. Data are presented as mean ± SEM, with * indicating a significant difference between patient and control samples at p < 0.05. CV = complex V; OXPHOS = oxidative phosphorylation.
Whole-exome sequencing
Exome sequencing in patients 1 and 4 did not reveal any nuclear gene candidates to explain the phenotype or mitochondrial disease. However, long-range PCR of the region commonly deleted in the patients with Kearns-Sayre syndrome revealed a low level of mitochondrial genome deletions. Sanger sequencing of the deletion breakpoint revealed that all deletions involved generating a novel ATP6 protein that had the 3′ terminal portion of the gene fused to sequence in the hypervariable region. This fusion, if stable, would be predicted to cause the ATP6 protein to have a longer carboxy-terminal end.
Discussion
We present 4 cases of patients with a low level of mtDNA deletions in skeletal muscle and abnormal assembly of CV proteins. CV abnormalities can be associated with many pathogenic variants affecting both nDNA and mtDNA.24,25 In 3 of these patients, the level of heteroplasmy in skeletal muscle was below 10%, which raised the concern that the mtDNA deletions may be due to aging of the skeletal muscle and not due to a germline mitochondrial disease. We then modified established gel electrophoresis techniques to create a hybrid BCN-PAGE method that successfully detected an abnormal CV in the skin fibroblasts from all 4 patients. These results were consistent with the mtDNA mutation, which showed the presence of the same deletion in both muscle and skin and thereby reducing the likelihood of somatically acquired deletions in the skeletal muscle. Existing as a subset of inherited metabolic disorders, many mitochondrial diseases are characterized by a deficiency in OXPHOS function—which can result from either nDNA or mtDNA mutations.26 Diagnosis of individual mitochondrial diseases is complicated by the variability of clinical phenotypes and tissue-specific heteroplasmy of the mitochondrial genome, and thus, patients commonly require a multifaceted diagnostic approach examining tissues from multiple organ systems.27 Further complicating mitochondrial genome analysis, skeletal muscle can accumulate spontaneous mtDNA mutations, which persist because of the postmitotic nature of the tissue.28,29 For this reason, we developed a BCN-PAGE assay using mitogenic skin fibroblasts from patients who had potentially pathogenic mtDNA mutations in skeletal muscle.
Commonly performed during the clinical investigation of mitochondrial disease; BN-PAGE and CN-PAGE use traditional nondenaturing (native) electrophoresis to separate proteins based on their electrophoretic motility.30,31 The primary difference between the aforementioned techniques is the presence of anionic dyes or detergents to enhance protein migration by increasing the negative charge of the migrating proteins.32 This is commonly achieved through the addition of BBG, an anionic dye that is added to both cathode and sample buffers during the beginning of running a BN-PAGE gel, before being removed for the remainder of the assay. However, the addition of BBG has been shown to interfere with in-gel activity staining and downstream protein immunoblotting.32 On the other hand, CN-PAGE lacks BBG, and therefore, only acidic proteins migrate toward the anode.31 A modified CN-PAGE assay (high-resolution CN-PAGE) has also been developed where both anionic and nonionic detergents are added to the cathode buffer. This creates micelles, which alter the charge of the native proteins and facilitate migration toward the anode.33 However, the use of anionic detergents may compromise the assembly of mitochondrial OXPHOS protein complexes.
The development of a BCN-PAGE assay aimed to combine the beneficial aspects of the various BN- and CN-PAGE conditions while preserving both protein activity and protein complex assembly. Specifically, mitochondrial protein assembly was maintained during extraction using the mild, nonionic detergent (DDM) during membrane solubilization. Next, the cathode buffer of the BCN-PAGE assay was prepared without the addition of the anionic dye BBG, typically used in BN-PAGE. Instead, BBG was replaced with the nonionic detergent (DDM) to prevent BBG interference with in-gel activity staining and protein immunoblotting.32 The modified BCN-PAGE cathode buffer therefore acts to retain OXPHOS protein activity and protein complex assembly. The BCN-PAGE sample buffer also contains an optimized concentration of BBG. Acting as a charge-shift molecule, the anionic BBG dye permits protein complex migration toward the anode.31 In contrast to CN-PAGE protocols, BCN-PAGE does not use anionic detergents in the cathode or sample buffers, thus allowing native protein migration and complex formation.34 Finally, the duration of electrophoresis and electric field strength in BCN-PAGE were selected for optimal protein separation. Collectively, the combination of BN- and CN-PAGE techniques provides a more comprehensive examination of the mitochondrial OXPHOS protein activity and protein complex assembly.
The present study used a BCN-PAGE assay using skin fibroblasts as a supplement to corroborate clinical findings from skeletal muscle biopsies. Following a common mitochondrial disease workup, each patient had a skeletal muscle biopsy performed for biochemical analysis (figure 1), histology (data not shown), and molecular analysis (table 1), and a skin biopsy, which was banked for future use. Of interest, the hallmark doublet of incompletely assembled CV, as identified in clinical BN- or CN-PAGE results, was visualized in patient skin fibroblasts using BCN-PAGE (figure 2). These findings suggest that the mtDNA mutations of CV identified in muscle are likely of germline origin as opposed to being acquired somatically or as a result of clonal expansion.35 Furthermore, the systematic identification of complex I supercomplexes may be another advantage of our BCN-PAGE technique. These visualized differences in complex I protein assembly identified using BCN-PAGE (figure 2) were also elevated following protein immunoblotting (figure 3). This is consistent with the visual contrast between patient and control samples from BCN-PAGE experiments. BCN-PAGE using skin fibroblasts provides support for a germline mitochondrial genome mutation, and visualization of BCN-PAGE protein bands, after in-gel activity staining, may provide valuable information regarding mitochondrial protein abundance.
CV comprises an F0 and an F1 region, which can be further divided into 16 subunits, 2 of which are encoded by mtDNA. As part of the F0 region embedded in the inner mitochondrial membrane, subunits A and A6L are encoded by mtDNA genes ATP6 and ATP8, respectively. The functioning ATP6 protein forms a proton pore connecting the noncatalytic F0 region to the catalytically active F136. Apart from facilitating the movement of protons between each region, ATP6 protein also physically connects the F0 and F1 regions via the peripheral stalk. The existing hypothesis is that damage to subunit A may lead to impaired intramitochondrial protein translation, instability of CV, and finally dissociation of the F0 and an F1 regions—resulting in the visualization of CV as a doublet.36 However, identification of these subcomplexes in cultured skin fibroblasts from patients has been variable, with heteroplasmy levels >95%.36 Considering that 3 of our patients were identified to have low heteroplasmy (<10%), with the fourth having a heteroplasmy of 25%, we hypothesize that expression of the ATP6 protein may act as a dominant negative mutation.37 In other words, the mutant ATP6 protein binds with surrounding CV proteins, resulting in the effective sequestration of properly functioning CV.
Frequently used to complement existing diagnostic procedures for various inherited metabolic disorders, the skin punch biopsy is a simple procedure and can be used to study biochemical defects in patient skin fibroblasts.38 The gold standard for assessment of mitochondrial enzyme activity for suspected mitochondrial disease is, however, the skeletal muscle biopsy.39 Specifically, skeletal muscle is regarded as a superior tissue because it contains a high concentration of mitochondria,40 thereby lending to increased test sensitivity,3 and its function is commonly affected by mitochondrial disease.41 However, false-positive results may result from improper storage of tissue42 or spontaneous accumulation of mtDNA mutations,3 whereas false-negative results can be caused by sample selection bias.43 Although they exhibit a low bioenergetic capacity, skin fibroblasts can have varying levels of mtDNA heteroplasmy in relation to the age of the patient. Recent research has demonstrated that both DNA mutations and mtDNA heteroplasmy levels in skin fibroblasts remained unaltered following prolonged culture (passages 2–15), indicating that mtDNA mutations in skin fibroblasts are likely inherited.44 Contrarily, some research has identified limited utility for mtDNA depletion detection using skin fibroblasts.45 This demonstrates the importance of a comprehensive mitochondrial diagnostic approach in which multiple techniques should be applied. Combining this approach with other noninvasive approaches46 may allow a less invasive approach in an increasing number of cases, especially in diseases that may escape detection using classic methods of leukocyte DNA whole-exome or whole-genome sequencing.
Acknowledgment
The authors thank Elizabeth Newell for editing an earlier draft of this manuscript.
Glossary
- BBG
Brilliant Blue G
- ETC
electron transport chain
- BN-PAGE
blue-native polyacrylamide gel electrophoresis
- BCN-PAGE
blue- and clear-native polyacrylamide gel electrophoresis
- CN-PAGE
clear-native polyacrylamide gel electrophoresis
- CV
complex VC
- DDM
n-Dodecyl β-D-maltoside
- MNG
Medical Neurogenetics
- mtDNA
mitochondrial DNA
- NBT
nitro blue tetrazolium
- nDNA
nuclear DNA
- NGS
next-generation sequencing
- OXPHOS
oxidative phosphorylation
Author Contributions
C. Newell, A. Khan, D. Sinasac, S. Hume, J. Shearer, and I. Sosova designed and developed the research. C. Newell, A. Khan, M.W. Friederich, and I. Sosova conducted experiments and collected and analyzed data. C. Newell, A. Khan, J. Shoffner, J.L. Van Hove, and J. Shearer wrote the manuscript. All authors read and approved the final manuscript.
Study funding
This study was supported by Alberta Children's Hospital Research Foundation (ACHRF) and MitoCanada (A. Khan). This research was supported by PhD funding to C. Newell from MitoCanada and Alberta Innovates—Health Solutions MD/PhD Studentship. IS was a recipient of Alberta Children's Hospital Research Institute (ACHRI) Clinical Research Fellowship.
Disclosure
Disclosure available: Neurology.org/NG.
References
- 1.Parikh S, Goldstein A, Koenig MK, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med 2015;17:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorman GS, Chinnery PF, DiMauro S, et al. Mitochondrial diseases. Nat Rev Dis Primers 2016;2:16080. [DOI] [PubMed] [Google Scholar]
- 3.Haas RH, Parikh S, Falk MJ, et al. The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab 2008;94:16–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Analysis of mitochondrial subunit assembly into respiratory chain complexes using Blue Native polyacrylamide gel electrophoresis. Anal Biochem 2007;364:128–137. [DOI] [PubMed] [Google Scholar]
- 5.Wittig I, Carrozzo R, Santorelli FM, Schägger H. Supercomplexes and subcomplexes of mitochondrial oxidative phosphorylation. Biochim Biophys Acta 2006;1757:1066–1072. [DOI] [PubMed] [Google Scholar]
- 6.Emma F, Montini G, Parikh SM, Salviati L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol 2016;12:267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams SL, Scholte HR, Gray RG, Leonard JV, Schapira AH, Taanman JW. Immunological phenotyping of fibroblast cultures from patients with a mitochondrial respiratory chain deficit. Lab Invest 2001;81:1069–1077 . [DOI] [PubMed] [Google Scholar]
- 8.Kramer KA, Oglesbee D, Hartman SJ, et al. Automated spectrophotometric analysis of mitochondrial respiratory chain complex enzyme activities in cultured skin fibroblasts. Clin Chem 2005;51:2110–2116. [DOI] [PubMed] [Google Scholar]
- 9.Fiala GJ, Schamel WWA, Blumenthal B. Blue native polyacrylamide gel electrophoresis (BN-PAGE) for analysis of multiprotein complexes from cellular lysates. J Vis Exp 2011:24 Epub 2011 Feb 24. Available at: jove.com/index/Details.stp?ID=2164. Accessed March 1, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.da Cunha ES, Domingues CC, de Paula E. Modified native electrophoresis protocol for the solubilization and separation of mitochondrial protein complexes. Anal Biochem 2011;418:158–160. [DOI] [PubMed] [Google Scholar]
- 11.Bourgeois JM, Tarnopolsky MA. Pathology of skeletal muscle in mitochondrial disorders. Mitochondrion 2004;4:441–452. [DOI] [PubMed] [Google Scholar]
- 12.Sarnat HB, Marín-García J. Pathology of mitochondrial encephalomyopathies. Can J Neurol Sci 2005;32:152–166. [DOI] [PubMed] [Google Scholar]
- 13.Tarnopolsky MA, Pearce E, Smith K, Lach B. Suction-modified Bergström muscle biopsy technique: experience with 13,500 procedures. Muscle Nerve 2011;43:716–725. [DOI] [PubMed] [Google Scholar]
- 14.Coughlin CR, Scharer GH, Friederich MW, et al. Mutations in the mitochondrial cysteinyl-tRNA synthase gene, CARS2, lead to a severe epileptic encephalopathy and complex movement disorder. J Med Genet 2015;52:532–540. [DOI] [PubMed] [Google Scholar]
- 15.Chatfield KC, Coughlin CR, Friederich MW, et al. Mitochondrial energy failure in HSD10 disease is due to defective mtDNA transcript processing. Mitochondrion 2015;21:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 1975;98:503–517. [DOI] [PubMed] [Google Scholar]
- 17.Vangipuram M, Ting D, Kim S, Diaz R, Schüle B. Skin punch biopsy explant culture for derivation of primary human fibroblasts. J Vis Exp e3779. MyJoVE Corporation; Epub 2013 Jul 7. [DOI] [PMC free article] [PubMed]
- 18.Chretien D, Rustin P. Mitochondrial oxidative phosphorylation: pitfalls and tips in measuring and interpreting enzyme activities. J Inherit Metab Dis 2003;26:189–198. [DOI] [PubMed] [Google Scholar]
- 19.Smet J, De Paepe B, Seneca S, et al. Complex III staining in blue native polyacrylamide gels. J Inherit Metab Dis 2011;34:741–747. [DOI] [PubMed] [Google Scholar]
- 20.Van Coster R, Smet J, George E, et al. Blue native polyacrylamide gel electrophoresis: a powerful tool in diagnosis of oxidative phosphorylation defects. Pediatr Res 2001;50:658–665. [DOI] [PubMed] [Google Scholar]
- 21.Díaz F, Barrientos A, Fontanesi F. Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using blue native gel electrophoresis. Curr Protoc Hum Genet 2009;Chapter 19:Unit 19.4. Available at: ncbi.nlm.nih.gov/pubmed/19806591. Accessed March 23, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smet J, De Paepe B, Seneca S, et al. Complex III staining in blue native polyacrylamide gels. J Inherit Metab Dis 2011;34:741–747. [DOI] [PubMed] [Google Scholar]
- 23.Friederich MW, Erdogan AJ, Coughlin CR, et al. Mutations in the accessory subunit NDUFB10 result in isolated complex I deficiency and illustrate the critical role of intermembrane space import for complex I holoenzyme assembly. Hum Mol Genet 2016;26:ddw431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roels F, Verloo P, Eyskens F, et al. Mitochondrial mosaics in the liver of 3 infants with mtDNA defects. BMC Clin Pathol 2009;9:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tucci A, Liu Y-T, Preza E, et al. Novel C12orf65 mutations in patients with axonal neuropathy and optic atrophy. J Neurol Neurosurg Psychiatry 2014;85:486–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimmock DP, Lawlor MW. Presentation and diagnostic evaluation of mitochondrial disease. Pediatr Clin North Am 2017;64:161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang C, Ahmad K, Sue CM. The broadening spectrum of mitochondrial disease: shifts in the diagnostic paradigm. Biochim Biophys Acta 2014;1840:1360–1367. [DOI] [PubMed] [Google Scholar]
- 28.Wachsmuth M, Hübner A, Li M, Madea B, Stoneking M. Age-related and heteroplasmy-related variation in human mtDNA copy number. PLoS Genet 2016;12:e1005939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kazachkova N, Ramos A, Santos C, Lima M. Mitochondrial DNA damage patterns and aging: revising the evidences for humans and mice. Aging Dis 2013;4:337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schägger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem 1991;199:223–231. [DOI] [PubMed] [Google Scholar]
- 31.Schägger H, Cramer WA, von Jagow G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem 1994;217:220–230. [DOI] [PubMed] [Google Scholar]
- 32.Shukolyukov SA. Native electrophoresis in cell proteomic: BN-PAGE and CN-PAGE. Cell Tissue biol 2011;5:311–318. [Google Scholar]
- 33.Wittig I, Karas M, Schagger H. High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol Cell Proteomics 2007;6:1215–1225. [DOI] [PubMed] [Google Scholar]
- 34.Wittig I, Braun H-P, Schägger H. Blue native PAGE. Nat Protoc 2006;1:418–428. [DOI] [PubMed] [Google Scholar]
- 35.Tuppen HAL, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim Biophys Acta 2010;1797:113–128. [DOI] [PubMed] [Google Scholar]
- 36.Smet J, Seneca S, De Paepe B, et al. Subcomplexes of mitochondrial complex V reveal mutations in mitochondrial DNA. Electrophoresis 2009;30:3565–3572. [DOI] [PubMed] [Google Scholar]
- 37.Baquedano MS, Guercio G, Marino R, et al. Unique dominant negative mutation in the N-terminal mitochondrial targeting sequence of StAR, causing a variant form of congenital lipoid adrenal hyperplasia. J Clin Endocrinol Metab 2013;98:E153–E161. [DOI] [PubMed] [Google Scholar]
- 38.Ceuterick C, Martin J-J. Diagnostic role of skin or conjunctival biopsies in neurological disorders: an update. J Neurol Sci 1984;65:179–191. [DOI] [PubMed] [Google Scholar]
- 39.Desjardins CA, Gundersen-Rindal DE, Hostetler JB, et al. Comparative genomics of mutualistic viruses of Glyptapanteles parasitic wasps. Genome Biol 2008;9:R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stephenson EJ, Hawley JA. Mitochondrial function in metabolic health: a genetic and environmental tug of war. Biochim Biophys Acta 2014;1840:1285–1294. [DOI] [PubMed] [Google Scholar]
- 41.Taivassalo T, Haller RG. Exercise and training in mitochondrial myopathies. Med Sci Sports Exerc 2005;37:2094–2101. [DOI] [PubMed] [Google Scholar]
- 42.Shoffner JM. Mitochondrial myopathy diagnosis. Neurol Clin 2000;18:105–123. [DOI] [PubMed] [Google Scholar]
- 43.Koenig MK. Presentation and diagnosis of mitochondrial disorders in children. Pediatr Neurol 2008;38:305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang E, Wang X, Tippner-Hedges R, et al. Age-related accumulation of somatic mitochondrial DNA mutations in adult-derived human iPSCs. Cell Stem Cell 2016;18:625–636. [DOI] [PubMed] [Google Scholar]
- 45.Dimmock D, Tang LY, Schmitt ES, Wong LJ. Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin Chem 2010;56:1119–1127. [DOI] [PubMed] [Google Scholar]
- 46.Newell C, Hume S, Greenway SC, Podemski L, Shearer J, Khan A. Plasma-derived cell-free mitochondrial DNA: a novel non-invasive methodology to identify mitochondrial DNA haplogroups in humans. Mol Genet Metab 2018. Epub 2018 Oct 16. Available at: sciencedirect.com/science/article/pii/S1096719218303950. Accessed October 23, 2018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request from any qualified investigators.