

Abstract
Objective
We report a second family with autosomal dominant transportinopathy presenting with congenital or early-onset myopathy and slow progression, causing proximal and less pronounced distal muscle weakness.
Methods
Patients had clinical examinations, muscle MRI, EMG, and muscle biopsy studies. The MYOcap gene panel was used to identify the gene defect in the family. Muscle biopsies were used for histopathologic and protein expression studies, and TNPO3 constructs were used to study the effect of the mutations in transfected cells.
Results
We identified a novel heterozygous mutation, c.2757delC, in the last part of the transportin-3 (TNPO3) gene in the affected family members. The mutation causes an almost identical frameshift affecting the stop codon and elongating the C-term protein product of the TNPO3 transcript, as was previously reported in the first large Spanish-Italian LGMD1F kindred. TNPO3 protein was increased in the patient muscle and accumulated in the subsarcolemmal and perinuclear areas. At least one of the cargo proteins, the splicing factor SRRM2 was normally located in the nucleus. Transiently transfected mutant TNPO3 constructs failed to localize to cytoplasmic annulate lamellae pore complexes in cells.
Conclusions
We report the clinical, molecular genetic, and histopathologic features of the second transportinopathy family. The variability of the clinical phenotype together with histopathologic findings suggests that several molecular pathways may be involved in the disease pathomechanism, such as nucleocytoplasmic shuttling, protein aggregation, and defective protein turnover.
The limb-girdle muscular dystrophies form a heterogeneous group of genetically transmitted myopathies with predominantly proximal, progressive muscle weakness.1 To date, 8 forms of dominant limb-girdle muscular dystrophy with known genetic cause have been identified; however, a new nomenclature has recently been proposed,2 in which only 4 dominant forms fulfilled the required criteria: LGMD D1 DNAJB6 related, D2 TNPO3 related, D3 heterogeneous nuclear ribonucleoprotein D like related, and D4 calpain3 related.
A dominant mutation in the TNPO3 gene was found to cause LGMD1F in a large Spanish-Italian family with proximal limb and axial muscle weakness.3,4 The causative mutation, c.2771delA p.*924Cext*15 in exon 22, results in the extension of the reading frame by 15 additional amino acids. There was wide variability in the age at onset and disease severity,3 and also, nonpenetrance was observed.5 Muscle weakness and atrophy of the lower limbs were prominent. Additional features were dysphagia, arachnodactyly, joint contractures, scapular winging, and hyperlordosis in some of the patients.3,6 Muscle histopathology was characterized by myopathic changes, including nuclear pathology, myofibrillar protein accumulation in the cytoplasm, and rimmed vacuolar pathology corresponding to accumulated autophagosomal membranes at the ultrastuctural level.6,7
Transportin-3 (TNPO3) belongs to the importin beta superfamily. It facilitates the nuclear import of Ser/Arg-rich (SR) proteins.8 SR motifs are commonly found on RNA-binding proteins associated with splicing. TNPO3 has also been identified as essential for HIV infection, and loss of TNPO3 function is protective against HIV.9 The role of TNPO3 in skeletal muscle and how mutations affect its function and lead to muscle disease have not been described.
Patients
A Swedish family with 3 patients representing subsequent generations, the proband (II-1), his mother (I-3), and his son (III-1), was included in this study (figure 1). The patients were followed up since early childhood because of walking difficulties or hypotonia at birth. All underwent neurologic examinations, muscle biopsy, and muscle MRI studies. EMG findings and creatine kinase (CK) levels were available in the proband and his mother. Muscle biopsies were performed at different time points: for I-3, at ages 31 and 48 years (both from the tibialis anterior muscle, TA); for II-1, at ages 3, 24, and 35 years (all from the TA); and for III-1, at age 16 months (vastus lateralis).
Figure 1. Pedigree.
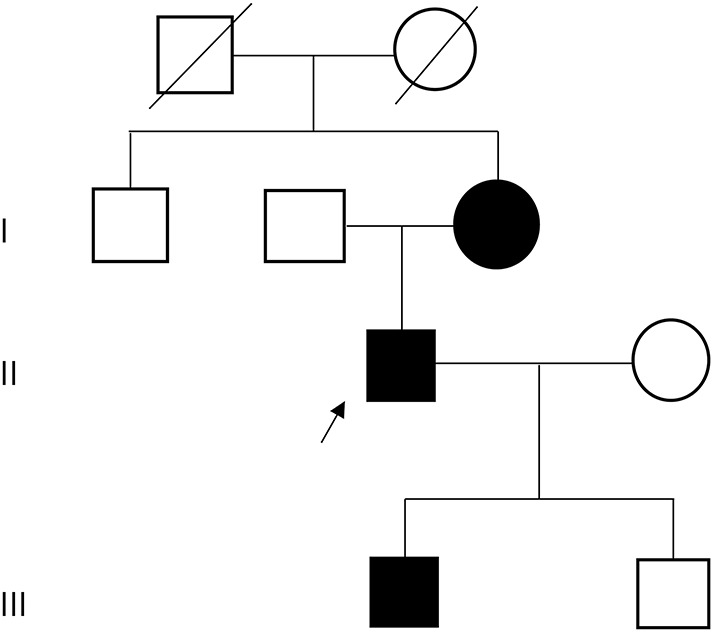
The affected family members were included in this study. The proband (II-1) is indicated with an arrow.
Standard protocol approvals, registrations, and patient consents
All participants provided appropriate consent, and the study was approved by the IRB of Tampere University Hospital.
Methods
Molecular genetics
Targeted massively parallel sequencing was performed for DNA samples of patients II-1 and III-1, as previously described,10 and sample I-3 was Sanger sequenced. The sequencing library was enriched using the probes of MYOcap v3 gene panel that is targeted to the exons of 265 genes known or predicted to cause muscular dystrophy or myopathy.
Histologic techniques
Snap-frozen muscle biopsies were processed into sections for histologic and immunohistochemical stainings. Conventional hematoxylin and eosin (H&E), Herovici, modified Gomori trichrome, and nicotinamide adenine dinucleotide tetrazolium reductase staining techniques were applied.11
For immunohistochemistry (IHC), the Ventana GX automated immunostainer was used to get 3,3′-diaminobenzidine immunolabeling, followed by hematoxylin and bluing counterstain (all by Roche Tissue Diagnostics/Ventana Medical Systems, Tucson, AZ).
Immunofluorescent (IF) stainings were performed manually. The frozen muscle sections were fixed in 4% paraformaldehyde for 15 minutes, permeabilized with 0.05% Triton X-100 in phosphate buffered saline (PBS) for 10 minutes, and blocked with 2% bovine serum albumin (BSA) in PBS for 30 minutes. Primary antibody incubation was performed overnight at 8°C. After PBS washes, Alexa-488 and Alexa-546 Fluor–conjugated secondary antibodies were used for detection at room temperature (RT) for 1 hour. Hoechst nuclear conterstain was performed before mounting in Fluoromount (Sigma-Aldrich) medium. The following primary antibodies were used: polyclonal anti-TNPO3 antibody (C-term) (Abcam ab109386), polyclonal anti-p62 (Millipore/Sigma P0067), monoclonal anti-TAR DNA-binding protein 43 (TDP-43) (Sigma-Aldrich WH0023435M1-1), polyclonal anti-ubiquitin (Dako Z0458), polyclonal anti-myotilin (ProteinTech 10731-1-AP), monoclonal anti-desmin (Abcam ab32362), monoclonal anti-alpha-B-crystallin (CRYAB) (Leica Biosystems NCL-ABCrys-512), monoclonal anti-tropomyosin (Abcam ab7786), polyclonal anti-CHCHD10 (Novus Biologicals NBP1-91169), polyclonal anti-RNA binding motif protein 4 (RBM4) (Atlas Antibodies HPA047849), and polyclonal anti-serine/arginine repetitive matrix 2 (SRRM2) (Abcam ab122719).
Western blotting
Frozen muscle biopsies were homogenized in Laemmli sample buffer and heated at 98°C for 5 minutes to prepare tissue lysates. Conventional sodium dodecyl sulphate polyacrylamide gel electrophoresis and Western blotting protocols were used, with 4%–20% precast TGX (Bio-Rad Laboratories, Hercules, CA) and Trans-Blot Turbo System (Bio-Rad) for protein transfer onto polyvinylidene difluoride membranes. For immunodetection, the membrane was incubated overnight at 8°C with anti-TNPO3 antibody (Abcam ab109386) at 1/500 dilution in tris-buffered saline with tween-20/5% skimmed milk powder. The next day, after horseradish peroxidase-conjugated secondary antibody incubation for 1 hour at RT, the bands were detected using enhanced chemiluminescence (SuperSignal West Femto Maximum Sensitivity Substrate, Thermo Fisher Scientific). After blotting, the gels were recovered and stained with Coomassie blue for myosin heavy chain, which was used as a loading control. ChemiDoc reader and ImageLab software (Bio-Rad) were used to obtain images and for calculating the relative quantities and molecular weights of the bands.
Plasmids
Wild-type (WT) hemagglutinin (HA)-TNPO3 was provided by Nathaniel Landau of New York University. The C-terminal extension constructs “+15” and “RG” were made by insertion mutagenesis of the HA-TNPO3 plasmid using the Q5 Site-Directed Mutagenesis Kit (NEB E0554S) and the following primers:

Immunofluorescence
HeLa cells were grown on glass coverslips in a 12-well plate. The next day, cells were transfected with 1.0 μg of plasmid using Lipofectamine 2000 and following the manufacturer's protocol. Twenty-four hours later, cells were fixed in ice-cold 4% paraformaldehyde in PBS for 15 minutes at RT. Cells were then washed with PBS 3 times for 5 minutes each and permeabilized with 0.2% Triton X-100 in PBS for 10 minutes at RT.
Blocking and detection of hemagglutinin only
Cells were incubated in blocking buffer made up of 1% BSA in PBS for 1 hour at RT. Coverslips were then transferred to a humidified chamber and incubated with Alexa Fluor 488-conjugated mouse anti-HA antibody diluted 1/200 in blocking buffer for either 2 hours at RT or overnight at 4°C. Coverslips were washed with PBS 3 times for 5 minutes each at RT. They were then carefully dipped in molecular-grade water for 10 seconds and mounted on microscope slides using Mowiol mounting medium.
Blocking and double-labeling of both marker and hemagglutinin
Cells were incubated in Blocking Buffer A (PBS + 1% BSA + 5% serum of the secondary antibody host) for 1 hour at RT. Coverslips were then transferred to a humidified chamber and incubated overnight with anti-HA, anti-T-cell-restricted intracellular antigen-1 (TIA1), RanGAP1 (Santa Cruz Biotechnology), or Mab414 (BioLegend) in Blocking Buffer A. The next day, coverslips were washed 3 times for 5 minutes each with PBS and then incubated with Alexa Fluor 555–conjugated secondary antibody diluted 1/500 in Blocking Buffer A for 1 hour at RT. Cells were washed 3 times for 5 minutes each with PBS and then incubated in Blocking Buffer B (PBS + 1% BSA + 5% serum of the primary antibody host) for 1 hour at RT. Coverslips were then incubated for 2 hours at RT with Alexa Fluor 488–conjugated mouse anti-HA antibody diluted 1/200 in Blocking Buffer B. Coverslips were washed with PBS 3 times for 5 minutes each at RT. They were then carefully dipped in molecular-grade water for 10 seconds and mounted on microscope slides using Mowiol mounting medium.
Arsenite assay
Twenty-four hours after transfection, media were removed from the coverslips and replaced with warm media containing 0.5 mM arsenite or an equivalent volume of PBS. Cells were incubated for 45 minutes in a humidified 37°C, 5% CO2 incubator. After incubation, media were aspirated, and cells were washed once with PBS before fixing and staining as described above.
Quantification of TNPO3 foci
Fixed and stained U2OS cells were viewed on low magnification, and transfected cells were scored as either TNPO3 foci positive or TNPO3 foci negative. A minimum of 50 cells were counted for each condition, and at least 3 independent experiments were conducted.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
The proband, a 44-year-old man (II-1), was first examined at age 13 months because of unsteady waddling gait and abnormal head control. At age 3 years, he had mild myopathic facies, narrow palate, and generalized muscle weakness and atrophy. There were also absent tendon reflexes and Gower sign present when rising from the floor. Slight extension contracture was observed in the right elbow, otherwise joint contractures or hyperlordosis were not present. Cognitive functions were normal. During childhood, he was unable to run or walk on toes and heels. The most pronounced weakness was present in ankle dorsiflexion and knee and hip flexion. EMG showed no abnormalities, muscle enzymes were slightly elevated, whereas other routine laboratory tests yielded normal results. His functional abilities were stable and weakness nonprogressive until early adulthood. After that, weakness slowly progressed to encompass most upper and lower limb muscles, although more markedly proximal muscles.
His mother (I-3) was able to walk at 16 months and had proximal and distal lower limb and neck flexor weakness and Gower sign. She also had proximal upper limb weakness. Hyperlaxity of most joints was noted. The CK level was normal, and EMG was suggestive of myopathy. During childhood, generalized muscle weakness was noted. Achilles tendon reflex was normal, others were absent. She could walk on toes but not on heels. Her disease was also stable until adulthood, after which it slowly progressed. She started to use a wheelchair at age 64 years.
The son (III-1) was hypotonic at birth and had slightly high palate, no contractures. There was no facial weakness or atrophy or hypertrophy of muscles. Tendon reflexes and routine laboratory tests were normal. He started to walk with waddling gait at 13 months. Gower sign was present, and slight girdle weakness and axillar hypotonia were observed. At age 7 years, he could not run and had difficulties walking on uneven ground. Trendelenburg was positive. He had no hyperlordosis or scoliosis. The Medical Research Council scale score was 3–4/5 in all proximal and distal limb muscles. His vital capacity was 75% at age 8 years.
Muscle imaging
Muscle MRI of the proband showed generalized and severe diffuse fatty degenerative changes in all pelvic and thigh muscles and slightly less severe but still diffuse changes in all the distal lower limb muscles (figure 2). The changes in the pelvic and thigh muscles were also severe in the mother, although less severe in the hamstrings and anterior compartment of the lower limbs. Early diffuse degenerative changes were seen in all muscles of the son of the proband.
Figure 2. Muscle imaging.

Severe generalized fatty degenerative changes in the proband (A). The hamstring muscles and anterior compartment of the lower limbs were relatively spared in the mother (B). Mild and diffuse early changes were seen in the son of the proband more pronounced in the sartorius, gracilis, adductor magnus, and peroneus longus muscles (C).
Genetics
The analysis of the MYOcap sequencing data revealed a heterozygous variant c.2757delC p.(R920Gfs*20) in TNPO3 (NM_012470) in II-1 and III-1, and the same mutation was later confirmed with Sanger sequencing in I-3. The detected variant was not listed in the Exome Variant Server, Exome Aggregation Consortium, or 1000 Genomes databases. Other variants suitable for autosomal dominant inheritance were not detected.
Histopathology
The muscle biopsies obtained from the proband (II-1) and his mother (I-3) at adult age showed general myopathic changes including variation in fiber size and shape, fiber splitting, and numerous internal nuclei (figure 3, A and B). Both hypertrophic and atrophic fibers were present in large numbers, and prominent type 1 fiber predominance was observed (>90% type 1 fibers, whereas in the TA muscle, 60%–80% type 1 fibers is considered normal).12 Very few occasional necrotic and regenerating fibers were present. The nuclear pathology was remarkable, including swollen nuclei with central pallor (figure 3A), and accumulation of perinuclear/subsarcolemmal basophilic material was a frequent finding. Cytoplasmic abnormalities were observed as well, including rimmed vacuoles (figure 3B), sarcoplasmic basophilic material (Figure 3, D and F), and even some cytoplasmic bodies (figure 3D). Occasional ragged red fibers were encountered (figure 3F), and the number of Cytochrome C oxidase-negative fibers was slightly increased (5%–10% fibers Cytochrome C oxidase-negative in I-3 at age 48 years). Sporadic targetoid, whorled, and moth-eaten fibers were also observed (I-3 at age 48 years).
Figure 3. Histopathology.

Histopathology of patient II-1 TA biopsy (A–E, G–K, M–N, P) shows myopathic changes: fiber size variation and numerous internal nuclei, some with central pallor in H&E (A, arrowheads) and rimmed vacuoles in Herovici (B, arrowheads). Subsarcolemmal TNPO3 accumulation is observed in TNPO3 IHC staining (C, arrowheads). A fiber with small cytoplasmic bodies is seen in Gomori trichrome staining (D, black arrowhead) and a fiber with myofibrillar pathology (D, white arrowhead). In a serial section, mitochondrial NADH staining reveals an uneven staining pattern in central parts of the muscle fibers (E). Gomori trichrome of patient I-3 TA biopsy (F) shows a ragged red fiber (black arrowhead) and myofibrillar pathology (white arrowhead). Large (arrowhead) and small myotilin accumulations are observed in IHC staining (G). In IF double staining, p62 (green) and TDP-43 (red) colocalize in inclusion bodies (H, arrowheads) in the rimmed vacuolar fiber. The mitochondrial CHCHD10 shows subsarcolemmal accumulation in several muscle fibers (I). IF double staining of desmin (green) and alpha-B-crystallin (red) shows both cytoplasmic and subsarcolemmal overexpression and colocalization (J, arrowheads), as does desmin (green) and tropomyosin (red) in K. Notably, desmin (green) and tropomyosin (red) show overexpression in patient III-1 muscle at only age 16 months (L). In confocal microscopy, RBM4 (green) is often excluded from the nuclei (M, arrowheads) in the patient biopsies, whereas SRRM2 (green) in N shows strictly nuclear localization in the patient, a pattern similar to control muscles. Confocal analysis of TNPO3 shows normal nuclear localization in patient III-1 (O), but often perinuclear accumulation of TNPO3 in adult patient biopsy (P, arrowheads), which shows as a nuclear rim. (A–L), scale bar = 100 μm; (M–P), scale bar = 50 μm. IF = immunofluorescent; IHC = immunohistochemistry; TA = tibialis anterior; TNPO3 = transportin-3.
In biopsies taken at young age (II-1 and III-1 at 3 years and 16 months, respectively), there was fiber size variation but no internal nuclei. Sarcoplasmic abnormalities were not observed in routine histochemical stainings (H&E and Gomori), but the myofibrillar/cytoskeletal pathology became visible when immunostained for myotilin, desmin, and tropomyosin, showing several fibers with accumulation (figure 3L). However, the rimmed vacuolar pathology, as well as perinuclear basophilic masses, appeared later in the disease course and was not present at childhood. Type 1 fiber predominance was present as early as at age 3 years (II-1).
Ultrastuctural examination of I-3 and II-1 showed subsarcolemmal deposits of amorphic material and degenerated mitochondria and sometimes membranous structures (data not shown). Areas of myofibrillar disorganization, including Z-streaming, were encountered, as well as sporadic fibers with minicore-like pathology. The nuclei were large and showed atypical morphology.
Immunohistochemical analysis showed that TNPO3 accumulated subsarcolemmally, surrounding the myonuclei, corresponding to the perinuclear basophilic material observed in H&E and Herovici (figure 3C). Other markers present in these subsarcolemmal masses included desmin, tropomyosin, CRYAB, and mitochondrial protein CHCHD10 (in addition, some lysosomal associated membrane protein 2-positive granules were observed in the perinuclear location.) The accumulated myofibrillar material in the cytoplasm stained for desmin, myotilin, CRYAB, ubiquitin, and p62/sequestosome 1 (SQSTM1) and, to a lesser extent, for tropomyosin and lysosomal associated membrane protein 2. Rimmed vacuolar fibers were positive for p62/SQSTM1, microtubule-associated proteins 1A/1B light chain 3, TDP-43, and ubiquitin (figure 3H).
No gross abnormality of nuclear markers matrin-3 and emerin was observed with the IF technique. Confocal analysis showed diffuse to granular nuclear localization of TNPO3 in the control muscle and patient III-1 (age 16 months) (figure 3O), whereas in II-1, variable nuclear and/or perinuclear localization was observed (figure 3P). A similar staining pattern was observed with RBM4 (figure 3M), one of the splicing factors TNPO3 binds and translocates to the nucleus. Another Ser/Arg-rich splicing factor, SRRM2, showed normal nuclear (speckle) localization in patient II-1, with no perinuclear accumulation (figure 3N).
Western blotting
In Western blotting, one band of approximately 96 kD was observed in both controls and patients (figure 4A); hence, we could not differentiate the mutant isoform from the WT by gel migration. However, when the protein bands were quantitated, we observed a marked (two- to threefold) increase in the TNPO3 protein levels in patients II-1 and III-1 compared with controls (figure 4B).
Figure 4. Western blotting.
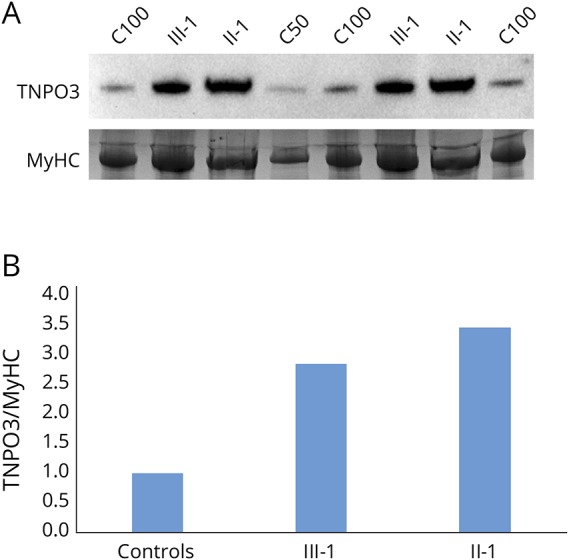
(A) Western blotting shows approximately 3-fold increase in TNPO3 protein expression in the patient samples analyzed (III-1 and II-1) compared with the pooled control sample (C100 and C50). (B) The bands were quantitated by calculating the relative quantities of TNPO3/MyHC in each sample and normalizing the control samples average to 1. MyHC = myosin heavy chain; TNPO3 = transportin-3.
Functional studies
Immunoblotting of Hela cells following transient transfection with plasmids expressing HA-tagged TNPO3-WT, TNPO3 carrying the previously reported 15 amino acid C-terminal extension (HA-TNPO3+15) or the currently reported frameshift mutation (HA-TNPO3-RG) (figure 5A) revealed similar expression levels and a slight increase in the molecular weight of HA-TNPO3+15 and HA-TNPO3-RG as expected (figure 5B). Immunofluorescence of exogenously expressed TNPO3 using an HA antibody revealed similar nuclear localization in addition to cytosolic puncta that were significantly enriched in HA-TNPO3-WT–expressing cells compared with TNPO3 mutants (figure 5, C and D).
Figure 5. Expression and localization of HA-tagged TNPO3 constructs.

(A) Schematic of HA-tagged TNPO3-WT, the previously reported LGMD1F mutation TNPO3+15 or the currently described frameshift mutation TNPO3-RG. (B) Immunoblot with anti-HA, TNPO3, or GAPDH of HeLa cell lysates transfected with HA-TNPO3-WT, HA-TNPO3+15, or HA-TNPO3-RG. (C) HA immunofluorescence of HeLa cells transiently transfected with HA-TNPO3-WT, HA-TNPO3+15, or HA-TNPO3-RG. HA-positive cytosolic puncta (arrows). (D) Quantitation of the percent of cells in C with cytosolic HA-TNPO3 puncta (*** p = < 0.0001). (E) Immunofluorescence using antibodies to HA (green) and TIA1 (red) of HeLa cells transfected with TNPO3-WT or TNPO3-RG pre- and post-stress granule induction with arsenite. HA-positive/TIA1-negative puncta (arrowheads). (F) Immunofluorescence using antibodies to HA (red) and RanGAP1 (green) or nucleoporin antibody Mab414 (green) of HeLa cells transfected with TNPO3-WT or TNPO3-RG. Cytosolic puncta (arrowheads). Scale bars (C, E, and F) = 20 μm. TNPO3 = transportin-3; WT = wild type.
To more clearly identify the cytosolic structure containing HA-TNPO3-WT, we performed dual immunofluorescence with an antibody to the stress granule (SG) marker TIA1 because it has recently been reported that nuclear import receptors associate with SGs. Although HA-TNPO3-WT and HA-TNPO3-RG do localize to a subset of TIA1-positive SGs, the larger cytosolic HA-TNPO3-WT puncta do not colocalize even when cells are treated with arsenite to induce SGs in HeLa cells (figure 5E).
We reasoned that cytosolic TNPO3 puncta may colocalize with nuclear pore complexes (PCs) that assemble in the cytoplasm at structures termed annulate lamellae pore complexes (ALPCs). In addition to nucleoporins, ALPCs contain much of the machinery necessary for nuclear transport including RanGAP1 and both nuclear import and export receptors. Co-immunofluorescence with antibodies to nuclear PCs (Mab414) or RanGAP1 and HA in HeLa cells transfected with HA-TNPO3-WT or HA-TNPO3-RG demonstrated strong colocalization at ALPCs in only TNPO3-WT–expressing cells.
Discussion
Only 1 family with TNPO3 mutation causing LGMD1F has been reported to date.3,4,13 In addition, a patient with sporadic LGMD harboring a completely different heterozygous missense mutation in the C-terminal part of TNPO3 has been described4,14 The follow-up study on the large family found that the mutation c.2771delA segregated in the total of 45 individuals of 115 studied family members with the estimated penetrance rate of 84.7%.5 Age at onset ranged from less than 1 year to 58 years. Based on the onset and the rate of progression, a juvenile form (onset before age 15 years, severe disability) and an adult form (onset in the third to fourth decades, slower progression) were presented.3,12 In the juvenile group, 6 patients had onset at age 1 year with delayed early motor skills. Although our patients had very early-onset proximal weakness or congenital hypotonia, the disease was stable during childhood and slow progression started in adulthood. The youngest of our patients had a mild decrease in respiratory functions; none had cardiomyopathy or hyperlordosis. Also, muscle MRI findings have been variable with main involvement of the vastus lateralis, rectus femoris, sartorius, and gracilis muscles and at later stages of the calf and peroneus longus muscles.3,6
We report the second transportinopathy family with a novel but almost identical frameshift mutation in the TNPO3 gene compared with the one identified in the first family.3,12 The congenital very early-onset generalized limb weakness and slow progression in our patients differed from the phenotype of most of the patients in the primary family, although some patients with congenital disease also were encountered. Two of our patients had already very severe fatty degeneration in most of their lower limb muscles making comparison difficult, whereas earliest involvement can be observed in the sartorius, gracilis, adductor magnus, and peroneus longus muscles (figure 2C). In contrast to the previous reports, we found that TNPO3 protein was quantitatively increased in the patient muscle and accumulated in the subsarcolemmal and perinuclear areas in the muscle fibers.
The detected mutation c.2757delC is predicted to cause a frameshift and transfer the stop codon (p.R920Gfs*20). The predicted outcome on protein level is almost identical to the original TNPO3 mutation c.2771delA p.*924Cext*15 (the amino acid sequence changed by the mutations is underlined):

Actually, there is just a 5 amino acid difference between the 2 protein products. Hence, it is expected that the pathomechanisms are largely shared as well. Indeed, we found that expression of the previously reported TNPO3 variant behaved similarly to our reported variant in cell culture. Specifically, TNPO3-WT localizes to the nuclear envelope and a cytosolic organelle termed annulate lamellae (ALs). ALs are cytosolic accumulations of nuclear PCs and the molecular machinery necessary for nuclear import/export. The function of ALs and ALPCs is unclear but may serve as repository necessary for replacement of nuclear pores and import/export components. The effect of TNPO3 mutations on nuclear pore function remains to be established. TNPO3 is a nuclear transport receptor, regulating nucleocytoplasmic shuttling of several S/R-rich splicing factors. Perinuclear accumulation and simultaneous nuclear depletion of TNPO3 and RBM4 observed in the patient muscle biopsy are in line with these mechanisms. Consequently, processing of specific mRNA species may be altered, leading to aberrant splicing and expression of muscle genes. However, the nuclear localization of SRRM2 indicates that these transcription factors have also other alternatives to TNPO3 for nuclear entry. Physical accumulation of TNPO3 in the perinuclear/subsarcolemmal regions, shown by IHC and Western blotting, together with other proteins and organelles, could disturb normal nuclear functions in several ways. Indeed, the patient biopsies show nuclear anomalies.
Myofibrillar pathology was present in the patient biopsies in this study, although less extensively than in conventional myofibrilliar myopathies such as myotilinopathy or zaspopathy,15 shown by accumulation of myotilin and desmin, as reported before in the Spanish family.6 As a novel finding, we found that the myofibrillar changes were present as early as at age 16 months, which is in line with the congenital onset of symptoms. Notably, we did not observe perinuclear TNPO3 accumulation or depletion from nuclei (or other abnormal localization) at that early stage, suggesting that this pathology develops later and may contribute to later events in the muscle pathology. Rimmed vacuolar pathology with p62/SQSTM1, TDP-43, microtubule-associated proteins 1A/1B light chain 3, and valocin-containing protein-positive material and accumulated autophagosomal membranes at the ultrastructural level indicate induction of the autophagosomal degradation pathway. Again, the number of rimmed vacuolated fibers was rather low. Defects in RNA metabolism are connected to defective autophagy and rimmed vacuolar pathology, as seen in multisystem proteinopathy pathologies.16
The muscle histopathologic findings and experimental data so far implicate that there may be several possible mechanisms involved in the transportinopathy molecular pathophysiology. To elucidate the pathogenic effects of the described TNPO3 mutations, studies clarifying how they affect the nuclear import/export dynamics will be required. In addition, the splicing of muscle genes, especially those regulated by the TNPO3 cargo proteins, needs to be analyzed. However, toxic proteins often shed little or no light on the functions of the normal cognate.
Glossary
- AL
annulate lamella
- ALPC
annulate lamellae pore complex
- BSA
Bovine serum albumin
- CK
creatine kinase
- DAB
diaminobenzinide
- HA
hemagglutinin
- IF
immunofluorescent
- IHC
immunohistochemistry
- PBS
Phosphate buffered saline
- PC
pore complex
- RBM4
RNA binding motif protein 4
- RT
room temperature
- SG
stress granule
- SR
serine/arginine rich
- SRRM2
SR repetitive matrix 2
- SQSTM1
sequestosome 1
- TA
tibialis anterior
- TDP-43
TAR DNA-binding protein 43
- TIA1
T-cell-restricted intracellular antigen-1
- TNPO
transportin-3
- WT
wild type
Appendix. Authors
Study funding
The study was funded by the Finnish Medical Foundation (J.P.), the Academy of Finland (B.U.), the Sigrid Jusélius Foundation (B.U.), and the Folkhälsan Research Foundation (B.U.). This study is not industry sponsored.
Disclosure
Disclosures available: Neurology.org/NG.
References
- 1.Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol 2014;33:1–12. [PMC free article] [PubMed] [Google Scholar]
- 2.Straub V, Murphy A, Udd B. LGMD workshop study group. 229th ENMC international workshop: limb girdle muscular dystrophies—nomenclature and reformed classification Naarden, the Netherlands, 17–19 March, 2017. Neuromuscul Disord 2018;28:702–710. [DOI] [PubMed] [Google Scholar]
- 3.Melià MJ, Kubota A, Ortolano S, et al. Limb-girdle muscular dystrophy 1F is caused by a microdeletion in the transportin 3 gene. Brain 2013;136:1508–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torella A, Fanin M, Mutarelli M, et al. Next-generation sequencing identifies transportin 3 as the causative gene for LGMD1F. PLoS One 2013;8:e63536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fanin M, Peterle E, Fritegotto C, et al. Incomplete penetrance in limb-girdle muscular dystrophy type 1F. Muscle Nerve 2015;52:305–306. [DOI] [PubMed] [Google Scholar]
- 6.Peterle E, Fanin M, Semplicini C, Padilla JJ, Nigro V, Angelini C. Clinical phenotype, muscle MRI and muscle pathology of LGMD1F. J Neurol 2013;260:2033–2041. [DOI] [PubMed] [Google Scholar]
- 7.Cenacchi G, Peterle E, Fanin M, Papa V, Salaroli R, Angelini C. Ultrastructural changes in LGMD1F. Neuropathology 2013;33:276–280. [DOI] [PubMed] [Google Scholar]
- 8.Maertens GN, Cook NJ, Wang W, et al. Structural basis for nuclear import of splicing factors by human Transportin 3. Proc Natl Acad Sci USA 2014;111:2728–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bin Hamid F, Kim J, Shin CG. Cellular and viral determinants of retroviral nuclear entry. Can J Microbiol 2016;62:1–15. [DOI] [PubMed] [Google Scholar]
- 10.Evilä A, Arumilli M, Udd B, Hackman P. Targeted next-generation sequencing assay for detection of mutations in primary myopathies. Neuromuscul Disord 2016;26:7–15. [DOI] [PubMed] [Google Scholar]
- 11.Dubowitz V, Sewry CA. Muscle Biopsy—A Practical Approach. 3 ed London: Saunders Elsevier; 2007. [Google Scholar]
- 12.Dahmane R, Djordjevic S, Simunic B, Valencic V. Spatial fiber type distribution in normal human muscle Histochemical and tensiomyographical evaluation. J Biomech 2005;38:2451–2459. [DOI] [PubMed] [Google Scholar]
- 13.Gamez J, Navarro C, Andreu AL, et al. Autosomal dominant limb-girdle muscular dystrophy: a large kindred with evidence for anticipation. Neurology 2001;56:450–454. [DOI] [PubMed] [Google Scholar]
- 14.Gibertini S, Ruggieri A, Saredi S, et al. Long term follow-up and further molecular and histopathological studies in the LGMD1F sporadic TNPO3-mutated patient. Acta Neuropathol Commun 2018;6:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selcen D. Myofibrillar myopathies. Neuromuscul Disord 2011;21:161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013;153:1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.