

Abstract
Native mass spectrometry continues to develop as a significant complement to traditional structural biology techniques. Within native MS, surface-induced dissociation (SID) has been shown to be a powerful activation method for the study of non-covalent complexes of biological significance. High-resolution mass spectrometers have become increasingly adapted to the analysis of high-mass ions and have demonstrated their importance in understanding how small mass changes can affect the overall structure of large biomolecular complexes. Herein we demonstrate the first adaptation of surface-induced dissociation in a modified high-mass range, high-resolution Orbitrap mass spectrometer. The SID device was designed to be installed in the Q-Exactive series of Orbitrap mass spectrometers with minimal disruption of standard functions. The performance of the SID-Orbitrap instrument has been demonstrated with several protein complex and ligand-bound protein complex systems ranging from 53 to 336 kDa. We also address the effect of ion source temperature on native protein-ligand complex ions as assessed by SID. Results are consistent with previous findings on quadrupole time-of-flight instruments and suggest that SID coupled to high-resolution MS is well-suited to provide information on the interface interactions within protein complexes and ligand-bound protein complexes.
Graphical Abstract

Introduction
Elucidating information about the quaternary structure of biological macromolecular complexes is central to our understanding of biological functions.1 While x-ray crystallography, NMR and cryo-electron microscopy are considered the dominant analytical techniques in structural biology, native mass spectrometry has proven itself as an important complementary technique.2–4 Although the three former techniques can often obtain atomic resolution structures, native MS carries advantages in sensitivity, speed, tolerance of sample and/or structural heterogeneity, and tolerance of a wide range of molecular sizes. In the field of native MS, time-of-flight (TOF) MS emerged early on as the method of choice for mass analysis.5,6 This is due in part to the inherently broad mass range of the TOF mass analyzer which makes it an obvious choice for the study of large biomacromolecules. Although the TOF analyzer served native MS extremely well for over a decade, higher resolution instruments equipped with Fourier transform ion cyclotron resonance (FT-ICR) or Orbitrap™ mass analyzers have become increasingly amenable to the analysis of large biomolecules.7–9 Advancements in native MS with Orbitrap platforms have demonstrated the potential of studying systems ranging to the megadalton scale at high-resolution.10,11
While important information about a multi-subunit complex such as molecular weight, binding kinetics, etc. can be determined from an MS experiment, the native MS toolbox is further extended through tandem MS (MS/MS) experiments. The power of native MS/MS stems from the ability to introduce non-covalently linked macromolecular complexes into the gas phase, select a specific ion (oligomeric state, ligand-bound state, etc.) and subsequently dissociate that species into subcomplexes prior to mass analysis. This workflow allows the determination of subunit identity, composition, stoichiometry, connectivity, topology, as well as ligand identity, stoichiometry, and binding location within a biomacromolecular complex.12–14 A wide range of activation techniques are utilized to probe the structure of protein complexes, each with its own advantages. Collision-induced dissociation (CID) is the most common activation method, as it comes standard on almost all commercially available tandem mass spectrometers. CID involves accelerating ions through a collision cell filled with inert gas such as nitrogen, with enough energy to induce fragmentation of the ion.15 Subjecting protein complexes to this multi-step, incremental energy deposition generally results in the unfolding/restructuring of the complex and ejection of a highly charged monomer, leaving behind the complementary (n-1)mer with a disproportionately low charge with respect to mass.16–18 Such experiments can be leveraged to gain information on stoichiometry and gas phase stability of a given complex.15,19 Electron transfer and electron capture dissociation (ETD, ECD) techniques have also been used, often resulting in backbone cleavages within a protein complex which can be useful for surface mapping and proteoform sequencing,7,20–22 and less commonly leading to direct dissociation of the non-covalent complex.23 Ultraviolet photodissociation (UVPD) has similarly been shown to provide sequence information of protein subunits within a complex and has also been demonstrated to break noncovalent interactions between subunits to yield subcomplexes.24–26
Surface-induced dissociation (SID) has emerged as a powerful collision-based activation method for probing protein complex structures in the gas phase, yielding fragmentation that provides information complementary to that gained from the activation methods mentioned previously.27 Rather than subjecting ions to multiple low-energy collisions with background gas, SID experiments direct ions into an inert, rigid surface to impart energy into the analyte in a single, high-energy step. SID generally results in a more symmetric partitioning of charge in the produced subcomplexes relative to CID28 and UVPD.26 As observed using ion mobility (IM)-MS instruments, subcomplexes generated via SID are generally compact after dissociation, with collision cross sections reflective of the biomolecular (sub)structures.29 Additionally, SID of ligand-bound protein complexes often results in subcomplexes with ligands retained in the binding pocket, which is often not the case for CID.30,31 These features make SID a useful tool to probe biomolecular complexes without solved structures.32–34
Until recently, work involving SID of protein complexes has largely been carried out on TOF platforms.35,36 With growing utilization of native MS for structural characterization of protein complexes, it has become apparent that greater mass resolution is often desirable and allows for a deeper insight into a given biological system. For example, measurement of mass shifts resulting from post-translational modifications and/or the binding of small ligands or cofactors becomes possible at the protein complex level at sufficient resolution.37,38 Each type of mass analyzer (TOF, FT-ICR, Orbitrap) has advantages and disadvantages, and the choice of instrument will often depend on the question being addressed. Our group has recently demonstrated SID on an FT-ICR platform, achieving unparalleled mass resolution of SID-generated subcomplexes.39 However, we believe that activation methods such as SID should be vendor and platform neutral and that SID on Orbitrap platforms optimized for increased m/z range will also be beneficial to mass spectrometry users.
Herein, we present a combination of surface-induced dissociation and high-resolution mass analysis as applied to the study of protein complexes. An Exactive™ Plus Extended Mass Range (EMR) Orbitrap instrument (Thermo Fisher Scientific, Bremen, Germany), modified with a high-mass quadrupole filter, was fitted with an SID device inspired by previous designs described in works by Wysocki et al.35,36 As with previous designs, the new device enables the instrument to perform SID experiments without interrupting the capability or performance of MS-only and CID experiments. The SID-Orbitrap platform was tested using several protein complexes with known quaternary structure. The resolving power of the Exactive Plus EMR platform was displayed through scrutiny of fragmentation products, revealing disparate oligomeric states of subunits that overlap one another in nominal m/z space and bound ligands retained through the SID process. Clear assignment of this nature was not possible in previous TOF studies without the assistance of IM.30 Additional work investigating the localization of ligand binding by SID using the instrument described here was recently published and highlights additional applications and capabilities of the SID-Orbitrap instrument.31 The work presented here expands upon the utility of FT-based mass analysis in the field of native MS.
Experimental Section
SID Device Design
An SID device 4 cm in length was designed to replace the short multipole between the selection quadrupole and the C-trap in the modified EMR platform (Figure 1a–c). Importantly, this design will allow the device to also be used on the Q Exactive line of mass spectrometers, including the Q-Exactive UHMR instrument. The electrode assembly was designed in Autodesk Inventor and its performance was optimized using SIMION 8.1 software. Initial simulation parameters were chosen to reflect reasonable kinetic energy distributions and spatial distributions of precursor ions to better represent the ion beam within the mass spectrometer. Precursor and product ions were simulated using charge and mass values previously observed from SID experiments on various instrument platforms. An initial kinetic energy of 30–100 eV for precursor ions entering the SID device was used to simulate surface collisions over a range of lab frame energies. Because dissociation of precursor ions may occur after collision with the surface and before or after exiting the SID device, transmission of both precursor and product ions was considered following surface collisions. Upon collision with the surface, ions representative of product and precursor mass and charge values were formed, with kinetic energies ranging between 5–20% of the initial precursor kinetic energy. Collisions with background gas were not considered.
Figure 1.

(a) Technical drawing of the SID device with mounting brackets. (b) A simplified cross-section depiction of the SID device showing a right-to-left ion trajectory in SID mode. (c) A diagram of the modified EMR platform with the prototype SID device installed between the quadrupole and C-trap.
Fabrication of the device was carried out in-house by the OSU Department of Chemistry and Biochemistry machine shop, using aluminum for all electrodes and polyether ether ketone (PEEK) polymer to mount and properly space the electrodes. The original ion path remains unobstructed to allow for minimal impact on MS only (i.e. no activation) experiments as well as CID (HCD) experiments. When SID is desired, the front-end electrodes of the SID device are tuned to direct ions to the surface at the top of the device. A detailed description of the surface used for collisions can be found elsewhere.35 Briefly, a glass surface was coated with a 10 Å titanium layer, a 1000 Å gold layer, and subsequently modified with a fluorocarbon self-assembled monolayer. Detailed drawings, and voltages applied to the SID device can be found in Figure S1.
Mass Spectrometry
All experiments were carried out on an Exactive Plus EMR instrument that had been previously been modified with a high-mass quadrupole filter that replaced the original transfer multipole, similar to the modifications presented by Dyachenko et al.40 All samples were ionized via static nano-electrospray ionization using in-house pulled borosilicate capillaries and supplied with voltage via direct contact with a platinum wire. MS-only and CID experiments remained widely unaffected by the presence of the SID device in the instrument. In transmission mode, voltages are applied to the entrance and exit lens stacks on the SID device to assist with ion beam focusing between the quadrupole and the C-trap. A small loss of transmission is observed due to the lack of the rf-confining multipole; however, it should be noted that the SID device remains installed on our instrument without negatively affecting MS-only or CID experiments. Voltages applied to ion transfer optics throughout the mass spectrometer, and HCD cell pressure, were adjusted to obtain optimal ion transmission while minimizing unintentional ion activation. Unless otherwise noted, CID was performed in the HCD cell, and all spectra were collected at 17,500 resolution as defined at 200 m/z. Quantitation of ligand-bound species and average charge state calculations were conducted using Protein Metrics Intact Mass deconvolution software41 and peak area integration respectively.
SID is accomplished by applying repulsive and attractive DC voltages to the front bottom and front top lenses respectively to guide the ion beam to the surface for collisions. Subsequently, the ion beam is extracted off the surface and guided into the C-trap region by the rear lenses within the SID device. Voltages applied to the SID device are provided from a ten channel DC power supply (Ardara Technologies, Ardara, PA). Detailed SID device tune settings for MS and MS/MS experiments can be found in Table S1. The fragment ions produced by SID can be trapped in the C-trap or allowed into the HCD cell for collisional cooling before returning to the C-trap region to be injected into the Orbitrap mass analyzer. The SID acceleration voltage is defined as the difference between the first SID entrance lens and the surface. The SID voltage range is afforded by applying a negative voltage to the C-trap DC offset during ion injection and accordingly increasing the available ∆V between SID entrance lenses and the surface (Figure S2a–b). To achieve higher energy SID capabilities, a modification to the ion optics board on the EMR instrument was required. Specifically, the voltage for the C-trap DC offset during ion injection steps had to be supplied by an external power supply to allow a larger range of voltages that can be applied to the C-trap offset during ion injection, and therefore increase the acceleration voltage range of SID up to approximately 235 V. To increase the ease of use of the instrument, we designed a circuit that allows the user to switch between the on-board power supply for MS and HCD experiments, and the external power supply for SID experiments (Figure S2c). It should be added that these modifications are relatively unobtrusive to the instrument and can be easily removed to produce a standard configuration, although removal of the SID device is not necessary for typical MS and CID experiments.
Materials
Protein complexes that have previously been characterized by SID in Q-TOF platforms,30,42 were chosen to demonstrate the SID capabilities of the Orbitrap instrument modified in this work. Streptavidin (SA) was purchased from Thermo Scientific™ Pierce™ (Rockford, IL, USA) and glutamate dehydrogenase (GDH) from bovine liver was purchased from Millipore Sigma (St. Louis, MO, USA). The protein samples were buffer exchanged into 100 mM ammonium acetate using Micro Bio-Spin P6 spin columns with a 6 kDa cutoff (Bio-Rad, Hercules, CA, USA), and subsequently adjusted with triethyl ammonium acetate (TEAA, Millipore Sigma) to a final buffer concentration of 80 mM ammonium acetate/20 mM TEAA. The addition of TEAA is used as a charge reducing reagent as it has been shown that reduced-charge precursor ions generally produce subcomplexes by SID that are more reflective of the complexes’ native structure.43 Protein concentrations were measured by A280 using a Nanodrop™ 2000C spectrophotometer (Thermo Fisher Scientific, Wilmington DE, USA). The samples were diluted to a protein complex concentration of 2 μM (SA tetramer) and 5 μM (GDH hexamer) for mass spectrometry experiments. Streptavidin-biotin binding experiments were performed under identical conditions, with biotin (Millipore Sigma) added to make a 1.5:1 biotin to streptavidin monomer molar ratio.
Results and Discussion
Streptavidin
Streptavidin serves as a model system for SID experiments. It is a 53 kDa D2 symmetric homotetramer, consisting of subunits arranged as a dimer of dimers (Figure 2a). Upon activation by CID, streptavidin tetramer dissociates into highly charged monomers and relatively low charged trimers (Figure S3).44 This behavior where one subunit unfolds and takes with it a disproportionally large amount of charge is typical of CID due to its multistep energy deposition process.16–18 Importantly, this dissociation result from CID does not support the known dimer of dimers subunit arrangement. In contrast, previous work has shown that SID of streptavidin tetramer results in the production of dimers at low energy, and monomers at high energy, consistent with the interface areas and complex geometry determined by crystal structure.30
Figure 2.

(a) Interface areas of streptavidin tetramer as determined by PISA analysis (1SWB).58 (b) Mass spectrum of streptavidin tetramer under charge reducing conditions. (c) SID spectrum (45 V, 495 eV) of the 11+ tetramer. (d) A zoomed-in segment of (c) at 140,000 resolution setting, revealing the isotope distributions of 3+ monomer and 6+ homodimer. (e) A zoomed in segment of (c) at 17,500 resolution setting, showing non-cleaved N-terminal methionine(s) and sodium adducts on 3+ monomer and 6+ dimer.
Charge states 11+ through 9+ are dominant for streptavidin tetramer that is ionized from ammonium acetate: TEAA solution. The 11+ charge state was selected using the quadrupole mass filter, and fragmented by SID (45 V, 495 eV) forming primarily dimers of charge state 5+ and 6+ (Figure 2b, c). Some ambiguity of product assignment in past SID experiments with TOF platforms has been resolved with the use of ion mobility to separate oligomeric states that overlap in m/z space (e.g. 3+ monomer and 6+ homodimer). In this instance, the high-resolution capabilities of the Orbitrap analyzer allowed for the direct observation of two SID products that overlap in nominal m/z space. Figure 2d shows the same 45 V (495 eV) SID spectrum recorded at high-resolution (140,000 resolution setting, 512 ms transient) displaying two isotope distributions over the same nominal m/z range, indicating the presence of 3+ monomers and 6+ homodimers. It should be noted that for higher resolution measurements, the intensities of higher order oligomers are artificially decreased relative to monomers due to loss of coherence of the ion packet within the Orbitrap analyzer from increased collisions with background gas for the species with larger cross sections and higher charge.45 In fact, this phenomenon is so apparent that Sanders et al. have leveraged the transient decay rate of protein ions in the Orbitrap to make collision cross section measurements.46 For this reason, we use low resolution (8,750–35,000 as defined at 200 m/z) for acquisition of protein complexes, and only use higher resolution for specific questions (i.e. small ligand binding, PTMs, etc.) being careful not to draw conclusions from the intensities of species at high-resolution.
The bias favoring lower oligomeric states at high resolution is easily observed by comparing isotope distributions at high resolution in Figure 2d to a zoomed-in perspective of the 3+ monomer and 6+ dimer from a lower resolution measurement in Figure 2e. While isotope distributions are no longer available at lower resolution, the presence of multiple oligomeric states can still be detected by examining the abundance of unremoved N-terminal methionine (Met) and sodium adducts on each signal. Streptavidin subunits can have incomplete Met removal on the N-terminus during protein expression, resulting in an observable distribution of peaks spaced by 131 Da.47 A given oligomeric state of streptavidin will retain zero to n non-cleaved Met residues (with n indicating the oligomeric state). Although +3 monomer and +6 dimer overlap in m/z space, dimer with one N-terminal methionine can easily be differentiated from monomer with one N-terminal methionine. Furthermore, the presence of sodium adducts on each species can also be used to estimate the amount of dimer and monomer present in the SID spectrum (Figure 2e). Using this approach to assign oligomeric states of SID products, we observed that the ratio of streptavidin dimer to monomer is greater at lower resolution than at higher resolution. This bias as a function of transient time is even more obvious when comparing the SID products of 11+ streptavidin over a range of SID energies at both low and high-resolution (Figure S4).
Ion source temperature effects
Great emphasis in native MS lies in the retention of the physiological structure/structural features of proteins and protein complexes during analysis by using gentle instrument conditions for ionization, ion transfer and detection. Unfortunately, as a result of such gentle instrument conditions, native ions have an increased tendency to carry adducts from non-volatile salts and buffers. Excessive adducting often reduces the “apparent resolution” of the instrument far below what is attainable under denaturing conditions.48 The source region on the Exactive line of instruments is often held at relatively high temperature (200–250 °C) to improve sensitivity and declustering of the ions as they enter the mass spectrometer. Although this drastically improves the apparent resolution and sensitivity of the instrument, it is often a concern that high source temperatures may result in restructuring of ions,49 similar to what has been observed at low collision-induced dissociation energies.50 Here we use SID in an attempt to determine if high ion source temperatures affect the structure of protein complex ions.
In previous studies, SID has been shown to differentiate between native-like ions and ions that have undergone gross structural changes (i.e. collapse, expansion) via pre-activation of the precursor ions, prior to dissociation.51 Notable differences in SID fragmentation were observed when precursor ions were activated prior to SID when compared to fragmentation without pre-activation. This same sensitivity to pre-activation is observed with the new SID device within the Q-Exactive EMR platform, as expected – SID measures the system as it is presented, providing dissociation patterns that vary with extent of pre-activation.
We chose to use the interaction of streptavidin tetramer with biotin to determine if ion source temperature affects the structure of the streptavidin tetramer. The streptavidin-biotin interaction is primarily mediated through hydrogen bonding and van der Waals interactions, although interactions with aromatic interactions also play a role.52 Previous studies have suggested that the solution-specific interactions between streptavidin and biotin are largely preserved upon introduction into the gas phase.53 When streptavidin tetramer bound with four biotin ligands was analyzed over a range of ion source temperatures, it was observed that tetramer with four bound biotin was the dominant species even at source temperatures up to 300 °C (Figure 3a, S5). In dissociation experiments, 11+ streptavidin tetramer was selected and subjected to 30 V (330 eV) of SID. As with previous reports, dimer products were observed both with and without biotin retained (Figure S6).30,39 The Orbitrap EMR platform provides baseline resolution of the apo and holo species in m/z space, allowing for easy relative quantitation of ligand retention. The same SID experiment was performed with ion source temperatures ranging from 120 to 300 °C, resulting in a greater loss of biotin from SID products as the ion source temperature was increased (Figure 3b, Figure S6). In contrast, even at the lowest ion source temperatures, when the 11+ streptavidin-biotin tetramer ion is subjected to CID, the biotin is completely lost before subunit dissociation occurs and exclusively apo-monomer and apo-trimer are observed at higher CID energies. (Figure S7). Interestingly, the symmetry of charge partitioning of SID products does not significantly change as a function of ion source temperature, in contrast to an increase in asymmetric charge partitioning that might have been expected if significant structural changes had occurred. (Figure S8). To determine if the loss of the biotin ligand could be an effect of precursor ion energy rather than a gross structural change, we collected SID spectra of 11+ streptavidin-biotin tetramer at additional SID energies with an ion source temperature of 120 °C (Figure 3c, S9). These results made it obvious that biotin retention is drastically affected by the energy of the ion-surface collision process, with almost complete biotin loss observed at SID energies of 550 eV (50 V). As no significant increase in asymmetric charge partitioning of the SID produced dimers is observed at high ion source temperatures, it is possible that the loss of biotin at high source temperatures is due to increased vibrational energy of the precursor ion prior to SID, and not necessarily a significant structural change during pre-activation. This finding is consistent with gross structure retention at elevated source temperature as presented in a recent publication that describes the use of the SID-Orbitrap EMR instrument to monitor ligand retention in protein complexes.31 Although we do not observe evidence of any gross structural changes during the pre-activation step, further investigations will need to be completed to fully understand the complex-dependent effect of ion source temperature on the structure of native protein complexes in the Exactive line of mass spectrometers. Further technological advances such as the implementation of ion mobility on the Exactive mass spectrometers may be critical in this understanding.54
Figure 3.

Plots comparing the relative degree of biotin retention on (a) streptavidin tetramers as a function of ion source temperature, (b) dimers produced by SID (30 V, 330 eV) of the streptavidin tetramers shown in part (a) as a function of ion source temperature, and (c) dimers produced from SID of streptavidin-biotin tetramer as a function of SID acceleration voltage at a constant ion source temperature of 120 °C. In all cases, error bars are representative of the standard deviation of triplicate measurements.
L-Glutamate Dehydrogenase
Bovine glutamate dehydrogenase (GDH) is a 333.6 kDa homohexameric enzyme arranged as a stacked dimer of trimers with a coiled antenna protruding from and stabilizing each trimer.55,56 Previous studies have shown that GDH is difficult to dissociate in the gas phase by CID, often requiring high CID energies, and high precursor charge states, resulting in the ejection of an unfolded monomer that is not representative of the initial subunit arrangement.42,57 In contrast, it has previously been shown that SID of GDH hexamer on a Q-IM-TOF platform primarily resulted in the dissociation of GDH hexamer into trimers with secondary dissociation into monomers.42 Furthermore, Ma et al. showed that under charge reducing conditions (addition of TEAA), the trimers and monomers produced from SID of GDH hexamer remain compact and have collision cross sections similar to those expected for native-like GDH subcomplexes clipped from the crystal structure of hexamer. These results show that unlike CID, SID can be used to elucidate the topology of GDH hexamer.
Nanoelectrospray of GDH hexamer in ammonium acetate:TEAA solution results in a charge state distribution with the 27+ charge state as the dominant peak (Figure S10a). Under these charge-reducing conditions, the maximum CID acceleration voltage of 200 V on the EMR platform does not result in dissociation of the 28+ hexamer, but instead only leads to minor charge stripping (Figure S10b). In contrast, subjecting the 28+ GDH hexamer to 175 V (4,900 eV) SID yields abundant trimer and a bimodal charge state distribution of monomer subcomplexes, as well as charge stripped hexamer (Figure 4a) The presence of symmetrically charged trimers supports the hypothesis by Ma et al. that GDH fragments by SID to form trimers without significant structural rearrangement.42 Higher energy SID (235 V, 6,580 eV) results in an increase in the low-charged monomer distribution, (Figure 4b). It is unsurprising that the monomer ions do not exhibit the symmetric charge partitioning typical of SID products, as the intertwined sections of each monomer need to unravel prior to dissociation. We hypothesize that the higher charged monomers likely originate from direct dissociation of hexamer to monomer and pentamer, where the complementary pentamers are not detected due to decreased transmission at high m/z in the EMR instrument. It is possible that the lower charged monomers are a result of secondary dissociation from trimers, or that they are a product of a different gas-phase conformation of the hexamer ions. These results suggest that the major products of SID reflect the topology of GDH hexamer, as the interactions between the trimer-trimer interface are broken at moderate energies, and the interactions between individual subunits within the trimer that are strengthened by the coiled helical tails present within each trimer are broken at higher energies. The discrimination of high m/z products on the EMR instrument is expected to be less of a problem on the recently-released Q Exactive UHMR instrument and this will be tested and reported once we have optimized SID in that instrument. In addition, the high-resolution capabilities of the Orbitrap instrument allow easy assignment of GDH subcomplexes without the use of additional methods such as ion mobility.
Figure 4.
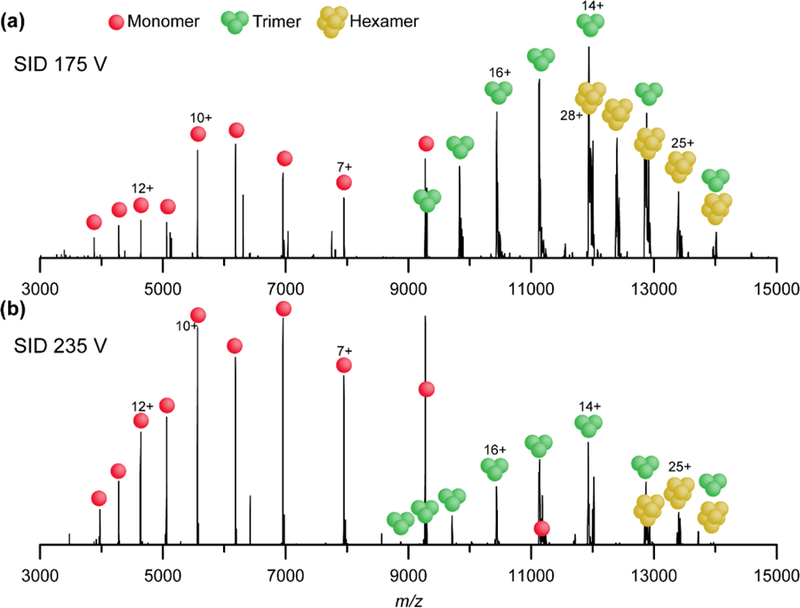
SID spectrum of +28 GDH hexamer at (a) 175 V and (b) 235 V SID acceleration voltage, resulting in trimers and monomers that are indicative of the overall dimer of trimers arrangement of GDH hexamer.
Conclusions
SID has been implemented in place of the transfer multipole in an Exactive Plus EMR Orbitrap instrument that has also been modified with a quadrupole mass filter. The SID device can be operated in place of the transfer multipole for ion transmission or can be used for surface collisions. Importantly, the SID device does not significantly affect ion transmission during CID or MS-only experiments, allowing the device to remain installed in the instrument for non-SID experiments. Well-studied multimeric protein and protein-ligand model complexes with different overall structures were successfully fragmented by SID and compare favorably with results previously acquired on time-of-flight platforms. The high mass resolving power inherent to the Orbitrap platform makes it possible to resolve oligomeric states with different mass and charge states that overlap in nominal m/z space by directly resolving the isotope envelope of each species. Furthermore, the extent of ligand binding to protein that was previously difficult to determine in SID experiments performed on TOF platforms is now clearly identifiable. The SID-Orbitrap platform has the potential to probe the effects of small ligands or protein modifications on the overall structure and stability of biomacromolecular complexes and is a powerful combination for the study of biomolecules of unknown structures.
Supplementary Material
Acknowledgements
Funding: The authors gratefully acknowledge financial support from the National Science Foundation (NSF 1455654 to V.H.W.) and from the National Institutes of Health (NIH P41GM128577 to V.H.W.).
The authors thank Mr. Larry Antal (OSU Department of Chemistry and Biochemistry Machine Shop), Mr. Eric Jackson (OSU Department of Chemistry and Biochemistry Instrument Support Group) for assistance with SID device manufacture and switching circuit design; Maria Reinhardt-Szyba, Dmitry Boll, Alexander Kholomeev, Jan-Peter Hauschild, Eugen Damoc (Thermo Fisher Scientific) for assistance and helpful discussions regarding instrument modifications; Randy Pedder (Ardara Technologies) for helpful electronics discussions and support; Royston Quintyn and Lindsay Morrison for initial SID experiments on the Orbitrap platform using a different design; Marshall Bern (Protein Metrics Inc.) for assistance with spectral deconvolution and relative quantitation; and Jacob McCabe (Texas A&M University) and Benjamin Jones (The Ohio State University) for assistance with batch processing of average charge state calculations.
Footnotes
Associated Content
Supporting Information: Detailed SID device drawings and voltages. Diagram of switching circuit for external power supply. MS-only and CID spectra of streptavidin and GDH. Spectra of streptavidin acquired over a range of SID energies. Spectra of biotin bound to streptavidin tetramer before SID and dimers after SID. Comparison of biotin binding to streptavidin dimer as a function of ion source temperature and SID energy. CID of streptavidin-biotin tetramer. Average charge state values for streptavidin-biotin dimers.
Conflict of Interest Disclosure
J.D.G., M.E.B, A.A.M, and S.R.H. are employees of Thermo Fisher Scientific Inc.
References
- (1).Spirin V; Mirny LA Protein Complexes and Functional Modules in Molecular Networks. Proc. Natl. Acad. Sci 2003, 100 (21), 12123–12128. 10.1073/pnas.2032324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Heck AJR Native Mass Spectrometry: A Bridge between Interactomics and Structural Biology. Nat. Methods 2008, 5 (11), 927–933. 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- (3).Liko I; Allison TM; Hopper JT; Robinson CV Mass Spectrometry Guided Structural Biology. Curr. Opin. Struct. Biol 2016, 40, 136–144. 10.1016/j.sbi.2016.09.008. [DOI] [PubMed] [Google Scholar]
- (4).Lössl P; van de Waterbeemd M; Heck AJ The Diverse and Expanding Role of Mass Spectrometry in Structural and Molecular Biology. EMBO J 2016, e201694818 10.15252/embj.201694818. [DOI] [PMC free article] [PubMed]
- (5).Sobott F; Hernández H; McCammon MG; Tito MA; Robinson CV A Tandem Mass Spectrometer for Improved Transmission and Analysis of Large Macromolecular Assemblies. Anal. Chem 2002, 74 (6), 1402–1407. 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- (6).van den Heuvel RHH; van Duijn E; Mazon H; Synowsky SA; Lorenzen K; Versluis C; Brouns SJJ; Langridge D; van der Oost J; Hoyes J; et al. Improving the Performance of a Quadrupole Time-of-Flight Instrument for Macromolecular Mass Spectrometry. Anal. Chem 2006, 78 (21), 7473–7483. 10.1021/ac061039a. [DOI] [PubMed] [Google Scholar]
- (7).Li H; Wolff JJ; Van Orden SL; Loo JA Native Top-Down Electrospray Ionization-Mass Spectrometry of 158 KDa Protein Complex by High-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem 2014, 86 (1), 317–320. 10.1021/ac4033214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rose RJ; Damoc E; Denisov E; Makarov A; Heck AJ R. High-Sensitivity Orbitrap Mass Analysis of Intact Macromolecular Assemblies. Nat. Methods 2012, 9 (11), 1084–1086. 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- (9).Fort KL; Waterbeemd M. van de; Boll D; Reinhardt-Szyba M; Belov ME; Sasaki E; Zschoche R; Hilvert D; Makarov AA; Heck AJR Expanding the Structural Analysis Capabilities on an Orbitrap-Based Mass Spectrometer for Large Macromolecular Complexes. Analyst 2017. 10.1039/C7AN01629H. [DOI] [PubMed]
- (10).Snijder J; van de Waterbeemd M; Damoc E; Denisov E; Grinfeld D; Bennett A; Agbandje-McKenna M; Makarov A; Heck AJR Defining the Stoichiometry and Cargo Load of Viral and Bacterial Nanoparticles by Orbitrap Mass Spectrometry. J. Am. Chem. Soc 2014, 136 (20), 7295–7299. 10.1021/ja502616y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).van de Waterbeemd M; Fort KL; Boll D; Reinhardt-Szyba M; Routh A; Makarov A; Heck AJ R. High-Fidelity Mass Analysis Unveils Heterogeneity in Intact Ribosomal Particles. Nat. Methods 2017, 14 (3), 283–286. 10.1038/nmeth.4147. [DOI] [PubMed] [Google Scholar]
- (12).Sharon M How Far Can We Go with Structural Mass Spectrometry of Protein Complexes? J. Am. Soc. Mass Spectrom 2010, 21 (4), 487–500. 10.1016/j.jasms.2009.12.017. [DOI] [PubMed] [Google Scholar]
- (13).Skinner OS; Haverland NA; Fornelli L; Melani RD; Vale LHFD; Seckler HS; Doubleday PF; Schachner LF; Srzentić K; Kelleher NL; et al. Top-down Characterization of Endogenous Protein Complexes with Native Proteomics. Nat. Chem. Biol 2017, nchembio.2515 10.1038/nchembio.2515. [DOI] [PMC free article] [PubMed]
- (14).Skinner OS; Havugimana PC; Haverland NA; Fornelli L; Early BP; Greer JB; Fellers RT; Durbin KR; Do Vale LHF; Melani RD; et al. An Informatic Framework for Decoding Protein Complexes by Top-down Mass Spectrometry. Nat. Methods 2016, 13 (3), 237–240. 10.1038/nmeth.3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Benesch JLP; Ruotolo BT; Simmons DA; Robinson CV Protein Complexes in the Gas Phase: Technology for Structural Genomics and Proteomics. Chem. Rev 2007, 107 (8), 3544–3567. 10.1021/cr068289b. [DOI] [PubMed] [Google Scholar]
- (16).Felitsyn N; Kitova EN; Klassen JS Thermal Decomposition of a Gaseous Multiprotein Complex Studied by Blackbody Infrared Radiative Dissociation. Investigating the Origin of the Asymmetric Dissociation Behavior. Anal. Chem 2001, 73 (19), 4647–4661. 10.1021/ac0103975. [DOI] [PubMed] [Google Scholar]
- (17).Jurchen JC; Williams ER Origin of Asymmetric Charge Partitioning in the Dissociation of Gas-Phase Protein Homodimers. J. Am. Chem. Soc 2003, 125 (9), 2817–2826. 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Popa V; Trecroce DA; McAllister RG; Konermann L Collision-Induced Dissociation of Electrosprayed Protein Complexes: An All-Atom Molecular Dynamics Model with Mobile Protons. J. Phys. Chem. B 2016, 120 (23), 5114–5124. 10.1021/acs.jpcb.6b03035. [DOI] [PubMed] [Google Scholar]
- (19).Dixit SM; Polasky DA; Ruotolo BT Collision Induced Unfolding of Isolated Proteins in the Gas Phase: Past, Present, and Future. Curr. Opin. Chem. Biol 2018, 42, 93–100. 10.1016/j.cbpa.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lermyte F; Sobott F Electron Transfer Dissociation Provides Higher-Order Structural Information of Native and Partially Unfolded Protein Complexes. PROTEOMICS 2015, 15 (16), 2813–2822. 10.1002/pmic.201400516. [DOI] [PubMed] [Google Scholar]
- (21).Zhang H; Cui W; Wen J; Blankenship RE; Gross ML Native Electrospray and Electron-Capture Dissociation FTICR Mass Spectrometry for Top-Down Studies of Protein Assemblies. Anal. Chem 2011, 83 (14), 5598–5606. 10.1021/ac200695d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Skinner OS; McAnally MO; Van Duyne RP; Schatz GC; Breuker K; Compton PD; Kelleher NL Native Electron Capture Dissociation Maps to Iron-Binding Channels in Horse Spleen Ferritin. Anal. Chem 2017, 89 (20), 10711–10716. 10.1021/acs.analchem.7b01581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Geels RBJ; van der Vies SM; Heck AJR; Heeren RMA Electron Capture Dissociation as Structural Probe for Noncovalent Gas-Phase Protein Assemblies. Anal. Chem 2006, 78 (20), 7191–7196. 10.1021/ac060960p. [DOI] [PubMed] [Google Scholar]
- (24).O’Brien JP; Li W; Zhang Y; Brodbelt JS Characterization of Native Protein Complexes Using Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc 2014, 136 (37), 12920–12928. 10.1021/ja505217w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Morrison LJ; Brodbelt JS 193 Nm Ultraviolet Photodissociation Mass Spectrometry of Tetrameric Protein Complexes Provides Insight into Quaternary and Secondary Protein Topology. J. Am. Chem. Soc 2016. 10.1021/jacs.6b03905. [DOI] [PMC free article] [PubMed]
- (26).Tamara S; Dyachenko A; Fort KL; Makarov AA; Scheltema RA; Heck AJR Symmetry of Charge Partitioning in Collisional and UV Photon-Induced Dissociation of Protein Assemblies. J. Am. Chem. Soc 2016. 10.1021/jacs.6b05147. [DOI] [PMC free article] [PubMed]
- (27).Zhou M; Wysocki VH Surface Induced Dissociation: Dissecting Noncovalent Protein Complexes in the Gas Phase. Acc. Chem. Res 2014, 47 (4), 1010–1018. 10.1021/ar400223t. [DOI] [PubMed] [Google Scholar]
- (28).Beardsley RL; Jones CM; Galhena AS; Wysocki VH Noncovalent Protein Tetramers and Pentamers with “ n “ Charges Yield Monomers with n /4 and n /5 Charges. Anal. Chem 2009, 81 (4), 1347–1356. 10.1021/ac801883k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhou M; Dagan S; Wysocki VH Protein Subunits Released by Surface Collisions of Noncovalent Complexes: Nativelike Compact Structures Revealed by Ion Mobility Mass Spectrometry. Angew. Chem. Int. Ed 2012, 51 (18), 4336–4339. 10.1002/anie.201108700. [DOI] [PubMed] [Google Scholar]
- (30).Quintyn RS; Yan J; Wysocki VH Surface-Induced Dissociation of Homotetramers with D2 Symmetry Yields Their Assembly Pathways and Characterizes the Effect of Ligand Binding. Chem. Biol 2015, 22 (5), 583–592. 10.1016/j.chembiol.2015.03.019. [DOI] [PubMed] [Google Scholar]
- (31).Busch F; VanAernum ZL; Ju Y; Yan J; Gilbert JD; Quintyn RS; Bern M; Wysocki VH Localization of Protein Complex Bound Ligands by Surface-Induced Dissociation High-Resolution Mass Spectrometry. Anal. Chem 2018, 90 (21), 12796–12801. 10.1021/acs.analchem.8b03263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Song Y; Nelp MT; Bandarian V; Wysocki VH Refining the Structural Model of a Heterohexameric Protein Complex: Surface Induced Dissociation and Ion Mobility Provide Key Connectivity and Topology Information. ACS Cent. Sci 2015, 1 (9), 477–487. 10.1021/acscentsci.5b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Romano CA; Zhou M; Song Y; Wysocki VH; Dohnalkova AC; Kovarik L; Paša-Tolić L; Tebo BM Biogenic Manganese Oxide Nanoparticle Formation by a Multimeric Multicopper Oxidase Mnx. Nat. Commun 2017, 8 (1), 746 10.1038/s41467-017-00896-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zhou M; Yan J; Romano CA; Tebo BM; Wysocki VH; Paša-Tolić L Surface Induced Dissociation Coupled with High Resolution Mass Spectrometry Unveils Heterogeneity of a 211 KDa Multicopper Oxidase Protein Complex. J. Am. Soc. Mass Spectrom 2018, 1–11. 10.1007/s13361-017-1882-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Galhena AS; Dagan S; Jones CM; Beardsley RL; Wysocki VH Surface-Induced Dissociation of Peptides and Protein Complexes in a Quadrupole/Time-of-Flight Mass Spectrometer. Anal. Chem 2008, 80 (5), 1425–1436. 10.1021/ac701782q. [DOI] [PubMed] [Google Scholar]
- (36).Zhou M; Wysocki VH Surface-Induced Dissociation of Ion Mobility-Separated Noncovalent Complexes in a Quadrupole/Time-of-Flight Mass Spectrometer. Anal. Chem 2012, 84 (14), 6016–6023. 10.1021/ac300810u. [DOI] [PubMed] [Google Scholar]
- (37).Gault J; Donlan JAC; Liko I; Hopper JTS; Gupta K; Housden NG; Struwe WB; Marty MT; Mize T; Bechara C; et al. High-Resolution Mass Spectrometry of Small Molecules Bound to Membrane Proteins. Nat. Methods 2016, 13 (4), 333–336. 10.1038/nmeth.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Liko I; Degiacomi MT; Lee S; Newport TD; Gault J; Reading E; Hopper JTS; Housden NG; White P; Colledge M; et al. Lipid Binding Attenuates Channel Closure of the Outer Membrane Protein OmpF. Proc. Natl. Acad. Sci 2018, 201721152 10.1073/pnas.1721152115. [DOI] [PMC free article] [PubMed]
- (39).Yan J; Zhou M; Gilbert JD; Wolff JJ; Somogyi Á; Pedder RE; Quintyn RS; Morrison LJ; Easterling ML; Paša-Tolić L; et al. Surface-Induced Dissociation of Protein Complexes in a Hybrid Fourier Transform Ion Cyclotron Resonance Mass Spectrometer. Anal. Chem 2017, 89 (1), 895–901. 10.1021/acs.analchem.6b03986. [DOI] [PubMed] [Google Scholar]
- (40).Dyachenko A; Wang G; Belov M; Makarov A; de Jong RN; van den Bremer ETJ; Parren PWHI; Heck AJR Tandem Native Mass-Spectrometry on Antibody–Drug Conjugates and Submillion Da Antibody–Antigen Protein Assemblies on an Orbitrap EMR Equipped with a High-Mass Quadrupole Mass Selector. Anal. Chem 2015, 87 (12), 6095–6102. 10.1021/acs.analchem.5b00788. [DOI] [PubMed] [Google Scholar]
- (41).Bern M; Caval T; Kil YJ; Tang W; Becker C; Carlson E; Kletter D; Sen KI; Galy N; Hagemans D; et al. Parsimonious Charge Deconvolution for Native Mass Spectrometry. J. Proteome Res 2018, 17 (3), 1216–1226. 10.1021/acs.jproteome.7b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ma X; Zhou M; Wysocki VH Surface Induced Dissociation Yields Quaternary Substructure of Refractory Noncovalent Phosphorylase B and Glutamate Dehydrogenase Complexes. J. Am. Soc. Mass Spectrom 2014, 25 (3), 368–379. 10.1007/s13361-013-0790-y. [DOI] [PubMed] [Google Scholar]
- (43).Zhou M; Dagan S; Wysocki VH Impact of Charge State on Gas-Phase Behaviors of Noncovalent Protein Complexes in Collision Induced Dissociation and Surface Induced Dissociation. The Analyst 2013, 138 (5), 1353 10.1039/c2an36525a. [DOI] [PubMed] [Google Scholar]
- (44).Schwartz BL; Bruce JE; Anderson GA; Hofstadler SA; Rockwood AL; Smith RD; Chilkoti A; Stayton PS Dissociation of Tetrameric Ions of Noncovalent Streptavidin Complexes Formed by Electrospray Ionization. J. Am. Soc. Mass Spectrom 1995, 6 (6), 459–465. 10.1016/1044-0305(95)00191-F. [DOI] [PubMed] [Google Scholar]
- (45).Makarov A; Denisov E Dynamics of Ions of Intact Proteins in the Orbitrap Mass Analyzer. J. Am. Soc. Mass Spectrom 2009, 20 (8), 1486–1495. 10.1016/j.jasms.2009.03.024. [DOI] [PubMed] [Google Scholar]
- (46).Sanders JD; Grinfeld D; Aizikov K; Makarov A; Holden DD; Brodbelt JS Determination of Collision Cross-Sections of Protein Ions in an Orbitrap Mass Analyzer. Anal. Chem 2018, 90 (9), 5896–5902. 10.1021/acs.analchem.8b00724. [DOI] [PubMed] [Google Scholar]
- (47).Wingfield PT N-Terminal Methionine Processing. Curr. Protoc. Protein Sci 2017, 88 (1), 6.14.1–6.14.3. 10.1002/cpps.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Lössl P; Snijder J; Heck AJR Boundaries of Mass Resolution in Native Mass Spectrometry. J. Am. Soc. Mass Spectrom 2014, 25 (6), 906–917. 10.1007/s13361-014-0874-3. [DOI] [PubMed] [Google Scholar]
- (49).Pacholarz KJ; Barran PE Distinguishing Loss of Structure from Subunit Dissociation for Protein Complexes with Variable Temperature Ion Mobility Mass Spectrometry. Anal. Chem 2015, 87 (12), 6271–6279. 10.1021/acs.analchem.5b01063. [DOI] [PubMed] [Google Scholar]
- (50).Benesch JLP Collisional Activation of Protein Complexes: Picking up the Pieces. J. Am. Soc. Mass Spectrom 2009, 20 (3), 341–348. 10.1016/j.jasms.2008.11.014. [DOI] [PubMed] [Google Scholar]
- (51).Quintyn RS; Zhou M; Yan J; Wysocki VH Surface-Induced Dissociation Mass Spectra as a Tool for Distinguishing Different Structural Forms of Gas-Phase Multimeric Protein Complexes. Anal. Chem 2015, 87 (23), 11879–11886. 10.1021/acs.analchem.5b03441. [DOI] [PubMed] [Google Scholar]
- (52).Stenkamp RE; Trong IL; Klumb L; Stayton PS; Freitag S Structural Studies of the Streptavidin Binding Loop. Protein Sci 1997, 6 (6), 1157–1166. 10.1002/pro.5560060604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Deng L; Broom A; Kitova EN; Richards MR; Zheng RB; Shoemaker GK; Meiering EM; Klassen JS Kinetic Stability of the Streptavidin–Biotin Interaction Enhanced in the Gas Phase. J. Am. Chem. Soc 2012, 134 (40), 16586–16596. 10.1021/ja305213z. [DOI] [PubMed] [Google Scholar]
- (54).Poltash ML; McCabe JW; Shirzadeh M; Laganowsky A; Clowers BH; Russell DH Fourier Transform-Ion Mobility-Orbitrap Mass Spectrometer: A Next-Generation Instrument for Native Mass Spectrometry. Anal. Chem 2018. 10.1021/acs.analchem.8b02463. [DOI] [PMC free article] [PubMed]
- (55).Peterson PE; Smith TJ The Structure of Bovine Glutamate Dehydrogenase Provides Insights into the Mechanism of Allostery. Structure 1999, 7 (7), 769–782. 10.1016/S0969-2126(99)80101-4. [DOI] [PubMed] [Google Scholar]
- (56).Smith TJ; Peterson PE; Schmidt T; Fang J; Stanley CA Structures of Bovine Glutamate Dehydrogenase Complexes Elucidate the Mechanism of Purine Regulation11Edited by I. A. Wilson. J. Mol. Biol 2001, 307 (2), 707–720. 10.1006/jmbi.2001.4499. [DOI] [PubMed] [Google Scholar]
- (57).Han L; Ruotolo BT Ion Mobility-Mass Spectrometry Differentiates Protein Quaternary Structures Formed in Solution and in Electrospray Droplets. Anal. Chem 2015, 87 (13), 6808–6813. 10.1021/acs.analchem.5b01010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Krissinel E; Henrick K Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol 2007, 372 (3), 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.