

Abstract
Branched chained amino acids (BCAA) are essential components of the human diet and important nutrient signals, which regain particular interest in recent years with the avenue of metabolomics studies suggesting their potential role as biomarkers. There is now compelling evidence for predictive role of BCAA in progression of diabetes, but causality relationship is still debated concerning insulin resistance and genetic versus non-genetic pathogenesis. Mendelian randomization studies in large cohorts of diabetes indicated pathogenic role of PPM1K (protein phosphatase Mg2+/Mn2+ dependent 1K) on Chr 4q22.1 gene, encoding for a phosphatase that activates BCKDH (branched chain keto acid dehydrogenase) complex. Recent studies indicated that insulin rapidly and dose-dependently regulates gene expression of the same complex, but the relationship with systemic insulin resistance and glucose levels is complex. Rare genetic syndromes due to Mendelian mutations in key genes in BCAA catabolism may be good models to understand potential role of gene of BCAA catabolism. However, in studying complex disorders geneticists are faced to complete new aspects of metabolic regulation complicating understanding genetics of obesity, diabetes or metabolic syndrome. A review of genetic syndromes of BCAA metabolism suggests that insulin resistance is not present, except rare cases of methylmalonic aciduria due to MUT (methylmalonyl-coA mutase) gene on Chr 6p12.3. Another aspect that complicates understanding is the new role of central nervous system (CNS) in insulin resistance. For a long time the hypothalamic hunger/satiety neuronal system was considered a key site of nutrient regulation. Genes may also affect the brain rewarding system (BRS) that would regulate food intake by modulating the motivation to obtain food and considering hedonic properties. Nutrigenomic and nutrigenetic investigations taking into account concurrently BCAA intake, metabolic regulation and gene variation have large perspectives to merge genetic and nutritional understanding in complex disorders.
Keywords: branched chain amino acids; BCAA; insulin resistance; gene, Mendelian syndromes; brain rewarding; nutrigenomics
INTRODUCTION
Branched chain amino acids (BCAA) including leucine (Leu), isoleucine (Ile) and valine (Val) are essential amino acids abundant in the human diet, which play a major role as nutrient signals. In athletic community, BCAA gained particular interest since they can stimulate protein synthesis in the muscle (1). In human pathology, increased levels of circulating BCAA were associated with obesity, insulin resistance and type 2 diabetes (T2D) (2). Felig et al. made the first observations in patients with obesity (3). Since then most of studies on BCAA were focused on signaling role in peripheral cells (muscle, liver and adipocyte) and at the level of Central Nervous System (CNS).
With the avenue of metabolomics investigations, BCAA regain particular interest as potential biomarkers for the development of T2D, although the relationship with various components of this disease became more complex than initially expected, particularly concerning insulin resistance (4). At the cellular level, a clearly established role of BCAA concerns signaling through mTOR (mechanistic target of rapamycin), which is the central regulator of nutrient signals (5). Investigators are faced to complex interactions and high diversity of sites of action such as liver, gastrointestinal tract and CNS. In the brain there are particular neurons that represent special regulatory areas (6, 7). Finally, new sources of BCAA appeared from the activity of bacteria in the microbiome, thus adding new routes of regulation in human pathology (8, 9).
At present, there is clear evidence for the role of BCAA in pathogenesis of obesity, diabetes and insulin resistance (2). To understand such diseases, severe genetic syndrome due to Mendelian (coding) mutations in key enzymes in BCAA metabolism represented good models. What remains less clear is the contribution of genetic variation of key genes in complex disorders (8). Geneticists should approach this new field of investigation of non-Mendelian heritability. In this short review we would like to dissect some novel aspects of the regulation of BCAA metabolism in the perspective of genetic investigation in complex disorders such as obesity, T2D or metabolic syndrome. This area of research entered in the focus of MEDIGENE European program (2012-2016) (https://cordis.europa.eu/result/rcn/195753_fr.html) intended to investigate the BCAA metabolism by controlling intake (diet), investigate regulation and genetic defects, thus engaging researchers in the new field of nutrigenomics and personalized nutrition. The reader is invited to consult several review articles in the field (2, 6, 10-13) and for genetic diseases the public OMIM database using gene or disease name (https://www.omim.org).
Metabolism of BCAA
In the human diet BCAA are abundant representing 15-25% of the total protein intake. Table 1 indicates normal requirements in BCAA in humans following recommendations of FAO and AFSSA in France (14, 15). BCAA are absorbed in the intestine and then rapidly transported through blood to peripheral tissue where they are metabolized, mainly in the muscle, liver, adipocyte and brain. BCAA share a common transport mechanism, in which several transporters are involved (16). The main neutral transport is assured by a series of exo-transporters, the most important being LAT1/4f2hc encoded by the SLC7A5 gene (solute carrier family 7 member 5) located on Chr 16q24.2. Transport system is also encoded by a second gene SLC3A2 (solute carrier family 3 member 2), which is located on Chr 11q12.3. Some other transporters were described more or less specific for BCAA such as SLC6A15, SLC7A6/ SLC3A2, SLC7A7/ SLC3A2 and SLC43A1 (17). At the cellular level, the fate of BCAA is either protein synthesis or shuttled to mitochondria for oxidation.
Table 1.
Daily requirements of BCAA in humans following international and French organizations
| Amino acid | Requirements (mg/kg/day) | |
| FAO/WHO/UNU (2007) | AFSSA | |
| Isoleucine | 20 | 18 |
| Leucine | 39 | 39 |
| Valine | 26 | 18 |
Inborn errors of BCAA metabolism
The first step in the degradation of BCAA is conducted by BCAT (branched chain amino-transferase) having vitamin B6 as cofactor and in the presence of 2-ketoglutarate that transfer the amino group thus producing glutamate (18-20). This first step represents one of potential sites of regulation of BCAA catabolism and pathology. BCAT enzymes are found in two isoforms, one cytosolic (BCATc) or BCAT1 found in the brain tissue and a second mitochondrial (BCATm) or BCAT2 operating in the muscle and liver. Through BCAT activity, leucine is transformed in 2-keto-isocaproate, valine is metabolized in 2-keto-isovalerate and isoleucine in 2-keto-3-methyl-glutarate, by reversible reactions (Fig. 1).
Figure 1.
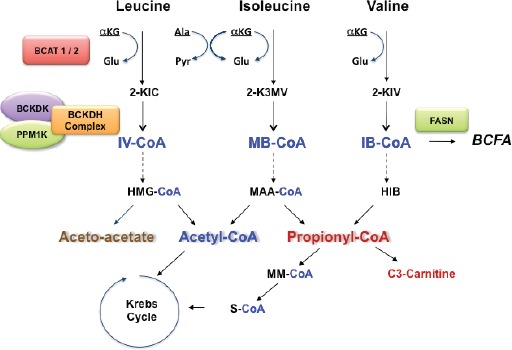
Major steps in the catabolism of BCAA. The first step in BCAA catabolism is determined by reversible transamination by the BCAT enzymes followed by irreversible oxidation by the BCKDH complex. Downstream metabolism is then independent for three amino acids, ketogenic for leucine and isoleucine (aceto acetate and acetyl Co-A) and glucogenic for valine with the formation of propionyl-CoA. Acetyl Co-A and succinyl-CoA enter then in the Krebs cycle for energy generation and gluconeogenesis or as precursors of lipogenesis and ketone bodies formation. Not indicated in the figure is AACS (acetoacetyl-CoA synthetase) located on Chr 12q24.31, which is a ketone body-utilizing ligase with a role in lipid synthesis through the non-oxidative pathway.
There are several known disease-causing mutations in BCAT2 gene located on Chr 19q13.33 (MIM 113530). The disorder is autosomal recessive and affected patients presented headache and memory impairment and display heterozygous BCAT2 gene mutations (e.g. Arg170Gln and Glu264Lys). The disease is characterized by increasing level of valine and leucine (21). Affected patients respond to treatment by vitamin B6, particularly by decreasing levels of BCAA. Patients with BCAT2 mutation also display neurologic abnormalities, which are explained by deficient BCATm in the brain. It should be mentioned that in the brain, BCATm is located in the endothelial cells and astrocytes while cytosolic BCATc is restricted to neurons. There are reports indicating increased expression of BCAT in Alzheimer disease or some other forms of dementia (10).
Figure 2.
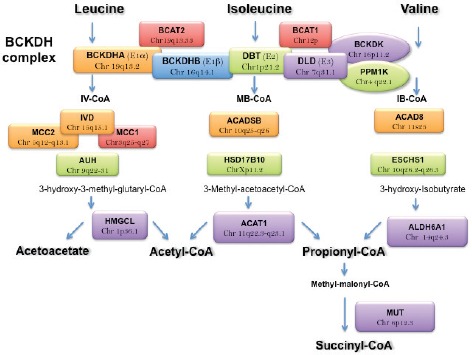
Major Mendelian diseases and location of related genes in the catabolism of BCAA. Mendelian diseases can be classified as function of genes involved in four steps: 1) gene involved in reversible transamination; 2) irreversible oxidation by the BCKDH complex including the BCKD kinase and PPM1K; 3) a series of rare diseases of intermediates in leucine, isoleucine and valine pathways; 4) genes involved in the distal phase of BCAA metabolism like the MUT gene.
The second important step in BCAA catabolism is the oxidative decarboxylation to acyl-CoA conducted by the BCKDH complex (22-24). This multi-subunits complex represents the rate limiting step enzyme. The complex is composed of E1, E2 and E3 subunits. The produced acyl-CoA are specific for each amino acid as following: iso-valeryl (IV)-CoA from leucine, 2-methy-butyryl (MB)-CoA from isoleucine and isobutyryl (IB)-CoA from valine (Fig. 1). The reaction is complex and starts with decarboxylation by E1 subunit, then transfer of acyl group by E2 with formation of acyl-CoA ester and finally the re-oxidation by E3 subunit with the formation of NADH. The reaction requires thiamine and Mg. In humans, the mitochondrial BCAT is cooperatively interacting with E1 subunit of BCKH complex (10). BCKDH complex is operating in the liver, kidney and adipose tissue but its regulation appears mainly in the liver. Hepatic expression of BCKDH is regulated by insulin leading to decrease of plasma levels of BCAA (25). In the liver, insulin modulates the expression of hepatic BCKDH enzyme and acts also by decreasing its phosphorylation. The same mechanism is operating in adipocyte, but to a lesser extent (25).
Genetic defects in the BCKDH complex can be understood in the context of various locations of genes responsible for E1, E2 and E3 subunits. Besides the BCKDH kinase, another gene, PPM1K (protein phosphatase Mg2+/Mn2+ dependent 1K) on Chr 4q22.1 encodes a phosphatase that activates the BCKD complex by dephosphorylating E1α subunit in mitochondria (26). In humans, genetic defects in BCKDH complex are known as Maple Syrup Urine Disease (MSUD). This autosomal recessive disorder ((MIM 248600) is due to multiple mutation in different genes (27). As indicated in Table 2, there are forms as function of the gene involved. MSUD is characterized by neurological and developmental delay, encephalopathy and feeding alterations with typical odor (maple syrup) of urine. Levels of BCAA are elevated with toxic effects on the muscle and brain. Among BCAAs, leucine is particularly toxic to brain affecting water homeostasis, producing swelling and depleting glutamate levels within the brain. Clinical symptoms remain variable as function of the genetic defect (28). The classical form occurs in the neonatal period, when the activity of the complex is less than 2%. Mild forms can occur later in life, in which there is almost 30% residual activity (28). There are around 200 homozygous or compound heterozygous mutations identified in MSUD (MIM 248600).
Table 2.
Diseases involving the second irreversible step in BCAA metabolism
| Disease |
Subunit involved (or regulators) |
Location |
MIM# Phenotype |
Gene name |
| MSUD, Type Ia | E1α | 19q13.2 | 248600 | BCKDHA |
| MSUD, Type Ib | E1β | 16q14.1 | 248600 | BCKDHB |
| MSUD, Type II | E2 | 1p21.2 | 248600 | DBT |
| MSUD, Type III | E3 | 7q31.1 | 246900 | DLD |
| MSUD, Mild variant | PPM1K | 4q22.1 | 615135 | PPM1K |
| Deficient BCKDK | Kinase | 16q11.2 | 614923 | BCKDK |
Another group of genetic diseases are due to mutations in the intermediate steps of the BCAA degradation, before the production of aceto-acetate, acyl-CoA and propionyl Co-A (Fig. 1). These diseases are specific for each amino acid, leucine, isoleucine and valine; therefore they will be presented for each pathway (10).
Leucine pathway. In the leucine pathway, there are several steps of degradation catalyzed by dehydrogenases: isovaleryl-CoA dehydrogenase (IVD) on Chr 15q15.1, 2 decarboxylases (MCC1 and MCC2) and a hydratase (AUH). Isovaleric acidemia is generally caused by homozygous mutations in the IVD gene (MIM 243500). The IVD is a flavoenzyme, which was purified from human liver. The deficiency in IVD is characterized by severe neonatal ketoacidosis, but there are also mild forms with intermittent episodes of ketoacidosis in the childhood. The inborn error is rare (1:62000-250 000 births) and may coexist with Angelman syndrome. The prognosis is severe often leading to death (mortality 33%). Clinically, patients were described with psychomotor abnormalities, a particular odor of “sweety feet” and aversion to dietary protein (29). Hepatomegaly was noted while on biological profile the main feature was hyperglycemia with an elevated urinary excretion of isovaleryl glycine. Symptoms are explained by the toxic effect of isovaleric acid on the brain. Accumulation of isovaleryl-CoA leads to the formation of isovaleryl carnitine, which is secondarily reduced to free carnitine in the plasma. False positive tests with increased C5-carnitine may be due to maternal therapy with antibiotics containing pivalic acid. In the presence of hyperglycemia, there is a risk to confound the disease with diabetic ketoacidosis. Several recurrent mutations were described in this disease.
Another disease is 3-methylcrotonylglycinuria Type I (MIM 210200) due to mutations in the MCC1 (3-methylcrotonyl-CoA carboxylase I) on Chr 3q27.1, which is different from 3-methylcrotonylglycinuria Type II due to MCC2 (3-methylcrotonyl-coA carboxylase II) gene on Chr 5q13.2 (30). MCC deficiency mutations were found in non-consanguineous Tunisian-Jewish family, and patients from Argentina and Bavaria (frequency 1:84,700), but the disease presents low expressivity and penetrance. The highest incidence was described in Faroe Islands (1:2400) where patients are caring a homozygous mutation. Affected individuals present increased blood and urine 3-hydroxy-isovaleryl carnitine (C5-carnitine) and episodes of metabolic acidosis accompanied by hypoglycaemia and hyperamonemia (10). Type II disease was described in Amish/Mennonite population of Lancaster County (Pennsylvania) but also in consanguineous Turkish, Swiss and Japanese families (MIM 210200).
In the next step of leucine pathway, the best known disease is 3-methylglutaconic aciduria or MGCA (MIM 250950) due to mutation in the AUH gene (Chr 9q22-31). The AUH encodes for 3-methylglutaconyl-CoA hydratase, which transforms 3-methyl-glutaconyl-CoA to 3-hydroxy-3-methyl-glutaryl-CoA (31). The primary MGCA Type 1 is an autosomal recessive disorder, occurring in infancy or childhood, characterized by abnormalities in speech, hyperchloremic acidosis with learning disabilities, seizures and cerebellar abnormalities. Leucine loading in the diet, which increases the 3-methyl-glutaconate in the urine, may be used to differentiate this form from other types of MGCA. The classical form is the methylglutaconic aciduria Type 1 due to mutations in AUH, but there are other syndromes described (32). The Barth syndrome, Type II MGCA is due to mutations in the tafazzin gene on Chr Xq28 and characterized by cardiac and skeletal myopathy (mitochondrial), short stature and recurrent infections. Tafazzin is a transacylase remodeling the immature cardiolipin to a form containing tetralinoleoyl compounds. Mutations were identified in English and Japanese populations. Methylglutaconic aciduria Type III also called Costeff syndrome is caused by mutations in the OPA3 (optic atrophy 3 with cataract) gene on Chr 19q13.32 with optic atrophy, spastic paraplegia and movement abnormalities (10, 32). Splice site mutations were found in Iraqi Jewish, French and Kurdish families. Methylglutaconic aciduria Type IV regroups several heterogeneous disorders with psychomotor retardation. Type V MGCA is caused by mutations in DNAJC19 (member 19, subfamily c of DNAJ/HSP40 homolog) gene on Char 3q26 with cardiomyopathy and conduction defects cerebellar ataxia, testicular dysgenesis and growth failure. The disorder was found in Canadian Dariusleut Hutterite and Finnish populations. Type VI disease is due to mutation in SERAC1 (serine active site-containing protein 1) gene on Chr 6q25 with deafness, encephalopathy and Leigh-like syndrome identified in consanguineous Turkish, Pakistani, Dutch and Iraqi families. Type VII is caused by mutations in CLBP (caseinolytic peptidase B) gene on Chr 11q13 characterized by cataracts, neurologic involvement and neutropenia. Type VIII disease is caused by mutations in HTRA2 (serine peptidase 2) gene on Chr 2p13, which encodes for a serine protease in the inter-membrane mitochondrial space. Mutations were found in 3 infant sibs from consanguineous parents of Druze origin (themselves from parents of Ashkenazi Jewish descents) as well as in a family of consanguineous Mexican parents. A previous report indicated the presence of mutations (or polymorphisms) in German patients with Parkinson disease, but another cohort did not contain mutations. Finally the Type IX disorder is due to mutation sin TIMM50 (yeast homologue of translocase of inner mitochondrial membrane 50) gene on Chr 19q13.2, which was described in consanguineous parents from Bedouin or Muslim families caring the Thr252Met and Arg217Trp substitutions (MIM 250950).
The transformation of 3-hydroxy-3-methyl-glutaryl-CoA in acetoacetate and acetyl-CoA is regulated by a lyase, called HMGCL (3-hydroxy-3-methylglutaryl-coA lyase) located on Chr 1p36.11 (MIM 246450). The HMGCL deficiency or hydroxymethylglutaric aciduria is a rare autosomal recessive disorder of metabolic acidosis without urinary excretion of ketone bodies and hypoglycemia (33). There is an increased urinary level of methylglutaric and hydroxyisovaleric acids. The disease was described in 1976 and may occur by uniparental isodisomy of Chr 1. Patients display irritability, are lethargic and evolve toward coma. Cases were described in Australia and Morocco and Arab-Bedouin families. Other patients were described in Europe from Turkish families and from Belgium, Germany, Netherlands and Switzerland. The highest incidence was found in Saudi Arabia (16%). In some populations, 7% of patients present the Reye syndrome. Dietary supplementation with carnitine may be beneficial but treatment imposes leucine restriction in the diet (34).
Isoleucine pathway. In the isoleucine pathway of degradation, the first step after the BCKDH is regulated by the branched-chain acyl CoA dehydrogenase (ACADSB) on Chr 10q26.13 leading to Tiglyl-CoA from 2-methyl-butyryl-CoA. Deficiency in this enzyme generates 2-methylbutyrylglycinuria due to homozygous or heterozygous mutations (MIM 610006). This inborn error occurs in infancy, very often asymptomatic or with delayed development and neurologic signs (35). Mutations were described in Pakistani, European and Eritrean or Somalia, Turkish, Lebanese Arab patients or from Hmong ethnic groups. The disorder is characterized by muscle atrophy and strabismus, lethargy, hypothermia, hypoglycaemia and mental retardation with elevated C5-carnitine levels (35).
The transformation of 2-Methyl-3-hydroxy-Butyryl-CoA to 3-Methyl-acetoacetyl-CoA is catalyzed by 17-beta-hydroxysteroid dehydrogenase (HSD17B10) located on Chr Xp11.22 (MIM 300256). The enzyme is multifunctional with a wide spectrum of substrates like steroids, isoleucine, and fatty acids with preference for short-chain methyl-branched acyl-CoA (36). Different mutations were found in HSD10 deficiency displaying mental retardation, choreoathetosis and retinal degeneration. Mutations were described in East Asian or Spanish descents or Caucasians. Typically, patients show lactic acidosis, hypoglycemia and hyperammonemia. The load in isoleucine results in an increased excretion of 2-methyl-3-hydroxybutyrate, tiglyl-glycine and increased plasma levels of isoleucine.
Alpha-methylacetoacetic aciduria is induced by deficiency in ACAT1 gene (acetyl-CoA acetyltransferase-1) located on Chr 11q22.3 and characterized by urinary excretion of 2-methyl-3-hydroxybutyric acid, 2-methylacetoacetic acid (or 2-butanone after spontaneous decarboxylation) and tiglylglycine (MIM 203750). Mutations were found in German, Dutch and Chilean families with recurrent episodes of ketoacidosis (37). ACAD8 (isobutyryl-coA dehydrogenase) catalyzes the transformation of isobutyryl-CoA in methyl-acrylyl-CoA (MIM 611283). The gene is located on Chr 11q25. The deficiency is rare. It was described in 1998 in patients with cardiomyopathy. L-carnitine supplementation in the diet can lead to catch-up growth in infants and improvement of the cardiac status (38).
Valine pathway. The pathology of valine pathway involves 4 enzymes in the degradation after the BCKDH step. The ECHS1 enzyme is a mitochondrial short-chain enoyl-coA hydratase (MIM 616277). Its gene is located on Chr 10q26.3 and the deficiency generates a delayed psychomotor development, neuro-degeneration, lactic acidosis and brain lesions (basal ganglia) (39). Mutations were described in patients of Greek ancestry or Japanese. HIBCH gene (formerly named HADH2) is located on Chr 2p32.2 encoding for 3-Hydroxy-isobutyryl-CoA hydrolase, which transforms to 3-hydroxy-isobutyrate. The autosomal deficiency is characterized by psychomotor development, neurodegeneration, lactic acidosis and brain lesions (MIM 250620). Mutations were detected in Pakistani and Tunisian populations. The ALDH6A1 gene (methylmalonate semialdehyde dehydrogenase) encodes for the enzyme transforming 3-hydroxy-isobutyrate to propionyl-CoA (MIM 614105). Patients may be asymptomatic or with developmental delay, dysmorphic features and delayed myelinisation. Mutations were found in Pakistani or European origin patients (40).
Diseases in distal steps of BCAA metabolism
In the distal steps of metabolism, the MUT gene (Chr 6p12.3) encodes for methylmalonyl-coA mutase, a mitochondrial enzyme catalyzing isomerization of methylmalonyl-CoA to succinyl-CoA (MIM 251000). The deficiency generates the methylmalonic aciduria (MMA). Patients usually present development retardation, chronic metabolic acidosis and hyperglycemia (41). Multiple mutations were described in French Canadians, French, Turkish and Thai patients. Seven mutations were seen exclusively in black patients. Mendelian mutations in Hispanics were associated with a particular haplotype downstream the gene (42). Symptoms depend on the residual activity of the enzyme. Initial studies suggested a toxic effect of methylmalonyl acid, but more recently, it was suggested that methylmalonyl-CoA operates as inhibitor of the pyruvate carboxylase, blocking the formation of oxaloacetate and phosphoenolpyruvate, thus impeding gluconeogenesis in the liver. This in turn would increase catabolism of lipids producing hypoglycemia and ketoacidosis. Of note, some previous cases of methylmalonyl aciduria have been described with severe insulin resistance, but molecular studies were not performed in these patients (43).
Due to diversity of genes involved in these Mendelian diseases, it is difficult to regroup clinical manifestations and therapeutic interventions for all BCAA. Diagnosis by geneticists is based on measurement of BCAA or their intermediates in the blood and urine by tandem mass spectrometry or detection on dried blood spots. At the therapeutic level, the clinician can modulate protein intake, the load of specific amino acids or to supplement diet with carnitine and glycine to detoxify plasma from intermediate metabolites (reviewed in Ref 11). The diet regimen should be in general high caloric while fasting should be avoided and if possible, complemented with continuous overnight feeding, thus keeping as good as possible the energy balance. Some amino acids (eg. tyrosine and tryptophan) or essential fatty acids (omega 3 polyunsaturated fatty acids) may improve by competition toxic effects of leucine in the brain. Carnitine supplementation is still under debate, but combination with glycine may be effective for detoxifying academia. Antioxidant therapy, Q10 and vitamin E have also shown as beneficial agents. Finally, in several cases of MSUD, liver transplantation was used with beneficial effects (11).
BCAA in complex disorders
Based on knowledge on above described genetic Mendelian diseases and considering the blood level of BCAA of their intermediates, geneticists are faced in complex disorders to complete new aspects of BCAA regulation, overpassing the simple variation in BCAA plasma level. In the last several years, BCAA deregulation in complex disorders became more complicated, albeit with interesting new aspects of metabolism and diagnosis possibilities (2, 25). The increased level of BCAA in obesity and diabetes was for a long time explained by possible defects in BCAA catabolism (2). The attention was drawn on potential defects in BCAT and BCKDH enzymes. A decrease in gene expression of BCAT2 and BCKDH E1α was detected in insulin resistant subjects (44). Moreover, it was demonstrated that adipose tissue was able to modulate circulating concentrations of BCAA in vivo (45). Similarly, it has been shown that human skeletal muscle BCAA degradation pathway was positively correlated with insulin sensitivity with a highest correlation for distal genes like MUT and ALDH6A1. A down-regulation of genes implicated in this pathway was observed in insulin resistant adipose tissue, (BCAT2, BCKDHB, MCCC1, MCCC2, AUH, HIBADH, ALDH6A1, ACAD8) (46). At a more complex level, defects in BCAA metabolism would contribute to impaired lipid metabolism (47).
In genetics, complex diseases were investigated either by searching alterations in BCAA levels associated with genes responsible for T2D or by more specific approaches such as Mendelian Randomization (MR). MR is a new method to evaluate causality in observational epidemiology indicating biomarkers causally implicated. Such MR analysis showed that alterations in BCAA metabolism in particular, in the rate-limiting-step catalyzed by BCKD complex have a causal role in the pathophysiology of T2D. By a Genome-Wide Association Study (GWAS), authors showed that among 10.5 millions genetic variants, several genomic regions statistically associated with BCAA levels, particularly variants upstream the gene PPM1K, encoding the phosphatase for activation of BCKD complex (48). PPM1K dephosphorylate E1α subunit and BCAA levels were associated with a higher risk of T2D (48). In PPM1K two SNP markers were identified (rs7678928, rs1440581) associated with isoleucine, valine and leucine levels. In the same study, other genes were found such as GCKR (glucokinase regulator) on Chr 2p23.3 by rs1260326, DDX19A (DEAD-box helicase 19A) on Chr 16q22.1 by rs75950518 and CBLN1 (cerebellin 1 precursor) on Chr 16q12.1 by the marker rs1420601 (48). These results were concordant with POUNDS trial, which found that under a high-fat diet for weight loss, subjects with the C allele of the variant rs1440581 upstream PPM1K gene have less benefits for weight and insulin resistance improvements (49). By a systems genetics approach, the gene PPM1K was also defined as a susceptibility gene for T2D (50). Mahendran et al. 2017 showed that BCAA plasma levels do not present a causative effect on insulin resistance (51). These explorations on BCAA were complemented with metabolomics studies based essentially on tandem liquid chromatography and mass spectrometry in plasma. Thus, studies on BCAA regained particular interest after the study of Newgard et al. in 2009 showing a relationship between BCAA plasma levels and insulin resistance (4).
The relationship between diet and BCAA metabolism remained contradictory in the literature. In human studies, high intake of BCAA was associated with an increased risk of incident T2D (52, 53) and insulin resistance. Furthermore, a positive correlation between dietary intake of BCAA and their plasma levels was shown (53). In parallel, consumption of a diet restricted in protein had positive effects on metabolic health and was able to reduce plasma levels of BCAA (54). In this last study, authors showed that young and healthy mice fed with diet restricted in BCAA presented improvements in body composition and glucose tolerance. More recently, another study in mice showed that reducing BCAA content in the context of a Western diet and diet-induced obesity was sufficient to improve metabolic health by reducing weight gain and ameliorating insulin resistance (55). Interestingly, weight improvement was explained by transient increase of FGF-21 (Fibroblast Growth Factor 21) blood levels, which in turn promoted energy expenditure (55).
Besides nutritional factors, alterations in BCAA metabolism may be associated with insulin resistance, metabolic dysfunction and high plasma level of BCAA or other related metabolites such as branched-chain ketoacids and acylcarnitines. In muscle, such alterations lead to a decrease in intermediates for TCA anaplerosis, resulting in incomplete oxidation of fatty acids and accumulation of acylcarnitines (56). This accumulation would induce mitochondrial stress and insulin resistance. The relationship with type 2 diabetes – as a causal effect on diabetes or insulin resistance - remained however controversial. A clear relation between BCAA and diabetes was confirmed in Diabetes Prevention Program (58) and more recently, it was shown that several drugs (e.g. empagliflozin) would be effective in the treatment of diabetes by increasing the short chain acylcarnitine (59). In obesity, diabetes and metabolic syndrome, several studies indicated that elevation of BCAA in the plasma may be used as biomarkers for development of hyperglycemia (57).
Brain involvement in BCAA regulation
While genetic investigation in complex disorders is permanently increasing, the geneticist is faced however with many other aspects related to brain regulation of BCAA. Leucine plays a major role in brain effects since (as in peripheral cells) leucine is a potent stimulator of mTOR. In peripheral tissues, leucine is an inhibitor of muscle protein breakdown stimulates insulin secretion of the β-cells and acts as allosteric activator of glutamate dehydrogenase. Besides these peripheral effects, leucine controls food intake by a specific mechanism. In the brain, leucine passes the blood brain barrier (BBB) and directly enters in neurons where, under the effect of BCATc, is transformed in Glu and α-keto-isocaproate (KIC) (6). Glutamate is then released at synapse interface and the not-used fraction of the neurotransmitter is recaptured by the astrocyte, which retransforms glutamate (glutamine synthetase GS) in Gln. In neuron the Gln is transformed back in Glu under the effect of glutaminase (GLA). The BBB is practically impermeable for the other two BCAA (6).
A recent report in mice indicated that insulin was able to decrease plasma level of BCAA by increasing BCKDH activity either by increasing its expression or by decreasing its phosphorylation, effects which were obtained in the liver tissue and independent from changes in the glycaemia (25). More interestingly, BCAA metabolism appeared to be regulated by neuroendocrine mechanisms involving mediobasal hypothalamus (6). Thus, infusion of 2-deoxyglucose (non-metabolisable analogue of glucose) in the 3rd ventricle induced glucopenia in the CNS triggering the counter regulation by autonomic nervous system with secretion of glucagon and corticosterone (6). These studies strongly suggest the involvement of brain in regulation of BCAA metabolism and recall the notion of brain insulin resistance (reviewed in Ref 61) and involvement of brain rewarding system (BRS). Indeed, in the CNS the hunger satiety circuit is regulated at the hypothalamic level by NPY/AGRP that stimulates eating (orexigenic) and by POMC/CART that inhibit (anorexigenic) 62). Both systems are under the effect of ghrelin and peptide YY (PYY) from the intestine, insulin from the pancreas and leptin from the adipocyte (63). The rewarding system is located at another level and formed of neuronal arrays that regulated hunger/satiety circuit. BRS is composed of Nucleus Accumbens (NA), Ventral Tegmental Area (VTA), amygdala and pre-frontal cortex, in which many neurotransmitters are involved (dopamine, serotonin, gamma-amino-butyric acid, glutamate and opioids). The best known is dopamine system. BRS regulates learning and memory, both related to hedonic food properties, increasing motivation to obtaining food reward (63). It is supposed that rewarding system bypass the hypothalamic regulation leading to alteration in emotions (eaters) in obesity, system that is stimulated by high dense energy food. In the hypothalamus, insulin induced orexigenic factors such as decreased neuropeptide Y (NPY), increased proopiomelanocortin (POMC) and agouti related neuropeptide (AgRP) and increased corticotropin releasing hormone (CRH) and thyrotropin releasing hormone (TRH), thus reducing the food intake (64). In the amygdala, the insulin system is also operating by reducing food intake through decrease NPY and increased oxytocin. The system may be disrupted in obesity under the influence of tumor necrosis factor (TNFα), interleukin 12B (IL-12B) and other factor from the periphery (65). What would be the role of BCAA in the BRS remains to be investigated but very likely the fibroblast growth factor 21 (FGF-21) plays a particular role. Taken together these new studies indicate that BCAA are sensed by the hypothalamus, which can induce modifications in the nutrient metabolism, mechanism that should be taken into account by geneticists in analyzing potential genetic defects in obesity, T2D or metabolic syndrome (66). Along this line, our laboratory was involved in genetic screening of genes involved in BCAA metabolisms and rewarding system through the FGF-21. As a comprehensive approach in the MEDIGENE program (67), we also developed a new database with the content in BCAA in the alimentary CIQUAL table, and integrated in MEDIPAD software, which can register electronically 24 h dietary surveys. The computer program is related to clinical, genetic and anthropologic databases and coordination activities (68) allowed the software to be used in all Mediterranean countries in different languages having the common server in France in nutrigenomic investigation.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Jäger R, Kerksick CM, Campbell BI, Cribb PJ, Wells SD, Skwiat TM, Purpura M, Ziegenfuss TN, Ferrando AA, Arent SM, Smith-Ryan AE, Stout JR, Arciero PJ, Ormsbee MJ, Taylor LW, Wilborn CD, Kalman DS, Kreider RB, Willoughby DS, Hoffman JR, Krzykowski JL, Antonio J. International Society of Sports Nutrition Position Stand: protein and exercise. J Int Soc Sports Nutr. 2017;14:20–25. doi: 10.1186/s12970-017-0177-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol. 2014;10(12):723–736. doi: 10.1038/nrendo.2014.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Felig P, Marliss E, Cahill GF., Jr Plasma amino acid levels and insulin secretion in obesity. N Engl J Med. 1969;281(15):811–816. doi: 10.1056/NEJM196910092811503. [DOI] [PubMed] [Google Scholar]
- 4.Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, Rochon J, Gallup D, Ilkayeva O, Wenner BR, Yancy WS, Jr, Eisenson H, Musante G, Surwit RS, Millington DS, Butler MD, Svetkey LP. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9(4):311–326. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014;24(7):400–406. doi: 10.1016/j.tcb.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sperringer JE1, Addington A2, Hutson SM2. Branched-Chain Amino Acids and Brain Metabolism. Neurochem Res. 2017;42(6):1697–1709. doi: 10.1007/s11064-017-2261-5. [DOI] [PubMed] [Google Scholar]
- 7.Yudkoff M. Brain metabolism of branched-chain amino acids. Glia. 1997;21(1):92–98. doi: 10.1002/(sici)1098-1136(199709)21:1<92::aid-glia10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 8.Bloomgarden Z. Diabetes and branched-chain amino acids: What is the link? J Diabetes. 2018;10(5):350–352. doi: 10.1111/1753-0407.12645. [DOI] [PubMed] [Google Scholar]
- 9.Saad MJ, Santos A, Prada PO. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology (Bethesda), 2016;31(4):283–293. doi: 10.1152/physiol.00041.2015. [DOI] [PubMed] [Google Scholar]
- 10.Adeva-Andany MM, López-Maside L, Donapetry-García C, Fernández-Fernández C, Sixto-Leal C. Enzymes involved in branched-chain amino acid metabolism in humans. Amino Acids. 2017;49:1005–1028. doi: 10.1007/s00726-017-2412-7. [DOI] [PubMed] [Google Scholar]
- 11.Manoli I, Venditti CP. Disorders of branched chain amino acid metabolism. Transl Sci Rare Dis. 2016;1(2):91–110. doi: 10.3233/TRD-160009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wasim M, Awan FR, Khan HN, Tawab A, Iqbal M, Ayesha H. Aminoacidopathies: Prevalence, Etiology, Screening, and Treatment Options. Biochem Genet. 2018;56(1-2):7–21. doi: 10.1007/s10528-017-9825-6. [DOI] [PubMed] [Google Scholar]
- 13.Blackburn PR, Gass JM, Vairo FPE, Farnham KM, Atwal HK, Macklin S, Klee EW, Atwal PS. Maple syrup urine disease: mechanisms and management. Appl Clin Genet. 2017;10:57–66. doi: 10.2147/TACG.S125962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.AFSSA Apport en protéines : consommation, qualité, besoins et recommandations. 2007:1–461. [Google Scholar]
- 15.FAO/WHO/UNU Expert Consultation Protein and amino acid requirements in human nutrition. World Health Organization technical report series. 2007;935:1–265. [PubMed] [Google Scholar]
- 16.Poncet N, Taylor PM. The role of amino acid transporters in nutrition. Curr Opinin Clin Nutr Metab Care. 2013;16(1):57–65. doi: 10.1097/MCO.0b013e32835a885c. [DOI] [PubMed] [Google Scholar]
- 17.Sahoo S, Aurich MK, Jonsson JJ, Thiele I. Membrane transporters in a human genome-scale metabolic knowledgebase and their implications for disease. Front Physiol. 2014;5:91. doi: 10.3389/fphys.2014.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutson SM, Sweatt AJ, LaNoue KF. Branched-Chain Amino Acid Metabolism : Implications for Establishing Safe Intakes. J Nutr. 2005;135(6 Suppl):1557S–1564S. doi: 10.1093/jn/135.6.1557S. [DOI] [PubMed] [Google Scholar]
- 19.Hutson SM, Wallin R, Hall TR. Identification of mitochondrial branched chain aminotransferase and its isoforms in rat tissues. J Biol Chem. 1992;267(22):15681–15686. [PubMed] [Google Scholar]
- 20.Bledsoe RK, Dawson PA, Hutson SM. Cloning of the rat and human mitochondrial branched chain aminotransferases (BCATm) Biochimica et Biophysica Acta. 1997;1339(1):9–13. doi: 10.1016/s0167-4838(97)00044-7. [DOI] [PubMed] [Google Scholar]
- 21.Wang XL1, Li CJ, Xing Y, Yang YH, Jia JP. Hypervalinemia and hyperleucine-isoleucinemia caused by mutations in the branched-chain-amino-acid aminotransferase gene. J Inherit Metab Dis. 2015;38(5):855–861. doi: 10.1007/s10545-015-9814-z. [DOI] [PubMed] [Google Scholar]
- 22.Chuang DT, Chuang JL, Wynn RM. Branched-Chain Amino Acids : Metabolism , Physiological Function, and Application Lessons from Genetic Disorders of Branched-Chain Amino. J Nutr. 2006;136(1 Suppl):243S–249S. doi: 10.1093/jn/136.1.243S. [DOI] [PubMed] [Google Scholar]
- 23.Chuang JL, Cox RP, Chuang DT. E2 transacylase-deficient (type II) maple syrup urine disease: Aberrant splicing of E2 mRNA caused by internal intronic deletions and association with thiamine-responsive phenotype. J Clin Invest. 1997;100(3):736–744. doi: 10.1172/JCI119586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chuang JL, Fisher CR, Cox RP, Chuang DT. Molecular basis of maple syrup urine disease: novel mutations at the E1 alpha locus that impair E1(alpha 2 beta 2) assembly or decrease steady-state E1 alpha mRNA levels of branched-chain alpha-keto acid dehydrogenase complex. Am J Hum Genet. 1994;55(2):297–304. [PMC free article] [PubMed] [Google Scholar]
- 25.Shin AC, Fasshauer M, Filatova N, Grundell LA, Zielinski E, Zhou JY, Scherer T, Lindtner C, White PJ, Lapworth AL, Ilkayeva O, Knippschild U, Wolf AM, Scheja L, Grove KL, Smith RD, Qian WJ, Lynch CJ, Newgard CB, Buettner C. Brain insulin lowers circulating BCAA levels by inducing hepatic BCAA catabolism. Cell Metab. 2014;20(5):898–909. doi: 10.1016/j.cmet.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Damuni Z, Reed LJ. Branched-chain alpha-keto acid dehydrogenase phosphatase and its inhibitor protein from bovine kidney. Methods Enzymol. 1988;166:321–329. doi: 10.1016/s0076-6879(88)66044-7. [DOI] [PubMed] [Google Scholar]
- 27.Morton DH, Strauss KA, Robinson DL, Puffenberger EG, Kelley RI. Diagnosis and treatment of maple syrup disease: a study of 36 patients. Pediatrics. 2002;109(6):999–1008. doi: 10.1542/peds.109.6.999. [DOI] [PubMed] [Google Scholar]
- 28.Blackburn PR, Gass JM, Vairo FPE, Farnham KM, Atwal HK, Macklin S, Klee EW, Atwal PS. Maple syrup urine disease: mechanisms and management. Appl Clin Genet. 2017;10:57–66. doi: 10.2147/TACG.S125962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vockley J, Ensenauer R. Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity. Am J Med Genet C Semin Med Genet. 2006;142C(2):95–103. doi: 10.1002/ajmg.c.30089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holzinger A, Roschinger W, Lagler F, Mayerhofer PU, Lichtner P, Kattenfeld T, Thuy LP, Nyhan WL, Koch HG, Muntau AC, Roscher AA. Cloning of the human MCCA and MCCB genes and mutations therein reveal the molecular cause of 3-methylcrotonyl-CoA: carboxylase deficiency. Hum Mol Genet. 2001;10(12):1299–1306. doi: 10.1093/hmg/10.12.1299. [DOI] [PubMed] [Google Scholar]
- 31.IJlst L, Loupatty FJ, Ruiter JP, Duran M, Lehnert W, Wanders RJ. 3-Methylglutaconic aciduria type I is caused by mutations in AUH. Am J Hum Genet. 2002;71(6):1463–1466. doi: 10.1086/344712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wortmann SB, Kluijtmans LA, Engelke UF, Wevers RA, Morava E. The 3-methylglutaconic acidurias: What’s new? J Inherit Metab Dis. 2012;35(1):13–22. doi: 10.1007/s10545-010-9210-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leung AA, Chan AK, Ezekowitz JA, Leung AK. A Case of Dilated Cardiomyopathy Associated with 3-Hydroxy-3-Methylglutaryl-Coenzyme A (HMG CoA) Lyase Deficiency. Case Rep Med. 2009:183125. doi: 10.1155/2009/183125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zafeiriou DI, Vargiami E, Mayapetek E, Augoustidou-Savvopoulou P, Mitchell GA. 3-Hydroxy-3-Methylglutaryl Coenzyme A Lyase Deficiency with Reversible White Matter Changes after Treatment. Pediatr Neurol. 2007;37(1):47–50. doi: 10.1016/j.pediatrneurol.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 35.Andresen BS, Christensen E, Corydon TJ, Bross P, Pilgaard B, Wanders RJ, Ruiter JP, Simonsen H, Winter V, Knudsen I, Schroeder LD, Gregersen N, Skovby F. Isolated 2-methylbutyrylglycinuria caused by short/branched-chain acyl-CoA dehydrogenase deficiency: identification of a new enzyme defect, resolution of its molecular basis, and evidence for distinct acyl-CoA dehydrogenases in isoleucine and valine metabolism. Am J Hum Genet. 2000;67(5):1095–1103. doi: 10.1086/303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ensenauer R, Niederhoff H, Ruiter JP, Wanders RJ, Schwab KO, Brandis M, Lehnert W. Clinical variability in 3-hydroxy-2-methylbutyryl-CoA dehydrogenase deficiency. Ann Neurol. 2002;51(5):656–659. doi: 10.1002/ana.10169. [DOI] [PubMed] [Google Scholar]
- 37.Tilbrook LK, Slater J, Agarwal A, Cyriac J. An unusual cause of interference in a salicylate assay caused by mitochondrial acetoacetyl-CoA thiolase deficiency. Ann Clin Biochem. 2008;45(Pt 5):524–526. doi: 10.1258/acb.2008.007202. [DOI] [PubMed] [Google Scholar]
- 38.Roe CR, Cederbaum SD, Roe DS, Mardach R, Galindo A, Sweetman L. Isolated isobutyryl-CoA dehydrogenase deficiency: An unrecognized defect in human valine metabolism. Mol Genet Metab. 1998;65(4):264–271. doi: 10.1006/mgme.1998.2758. [DOI] [PubMed] [Google Scholar]
- 39.Peters H, Buck N, Wanders R, Ruiter J, Waterham H, Koster J, Yaplito-Lee J, Ferdinandusse S, Pitt J. ECHS1 mutations in Leigh disease: a new inborn error of metabolism affecting valine metabolism. Brain. 2014;137(Pt 11):2903–2908. doi: 10.1093/brain/awu216. [DOI] [PubMed] [Google Scholar]
- 40.Sass JO, Walter M, Shield JP, Atherton AM, Garg U, Scott D, Woods CG, Smith LD. 3-Hydroxyisobutyrate aciduria and mutations in the ALDH6A1 gene coding for methylmalonate semialdehyde dehydrogenase. J Inherit Metab Dis. 2012;35(3):437–442. doi: 10.1007/s10545-011-9381-x. [DOI] [PubMed] [Google Scholar]
- 41.Oberholzer VG, Levin B, Burgess EA, Young WF. An inborn error of metabolism leading to chronic metabolic acidosis. Arch Dis Child. 1967;42(225):492–504. doi: 10.1136/adc.42.225.492. Methylmalonic aciduria. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Worgan LC, Niles K, Tirone JC, Hofmann A, Verner A, Sammak A, Kucic T, Lepage P, Rosenblatt DS. Spectrum of mutations in mut methylmalonic acidemia and identification of a common Hispanic mutation and haplotype. Hum Mutat. 2006;27(1):31–43. doi: 10.1002/humu.20258. [DOI] [PubMed] [Google Scholar]
- 43.Boeckx RL, Hicks JM. Methylmalonic acidemia with the unusual complication of severe hyperglycemia. Clin Chem. 1982;28:1801–1803. [PubMed] [Google Scholar]
- 44.Serralde-Zuniga AE, Guevara-Cruz M, Tovar AR, Herrera-Hernandez MF, Noriega LG, Granados O, Torres N. Omental adipose tissue gene expression, gene variants, branched-chain amino acids, and their relationship with metabolic syndrome and insulin resistance in humans. Genes Nutr. 2014;9(6):431. doi: 10.1007/s12263-014-0431-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herman MA, She P, Peroni OD, Lynch CJ, Kahn BB. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem. 2010;285(15):11348–56. doi: 10.1074/jbc.M109.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pietiläinen KH, Kaprio J, Borg P, Plasqui G, Yki-Järvinen H, Kujala UM, Rose RJ, Westerterp KR, Rissanen A. Physical inactivity and obesity: a vicious circle. Obesity (Silver Spring) 2008;16(2):409–414. doi: 10.1038/oby.2007.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lerin C, Goldfine AB, Boes T, Liu M, Kasif S, Dreyfuss JM, De Sousa-Coelho AL, Daher G, Manoli I, Sysol JR, Isganaitis E, Jessen N, Goodyear LJ, Beebe K, Gall W, Venditti CP, Patti ME. Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol Metab. 2016;5(10):926–936. doi: 10.1016/j.molmet.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lotta LA, Scott RA, Sharp SJ, Burgess S, Luan J, Tillin T, Schmidt AF, Imamura F, Stewart ID, Perry JR, Marney L, Koulman A, Karoly ED, Forouhi NG, Sjogren RJ, Naslund E, Zierath JR, Krook A, Savage DB, Griffin JL, Chaturvedi N, Hingorani AD, Khaw KT, Barroso I, McCarthy MI, O’Rahilly S, Wareham NJ, Langenberg C. Genetic Predisposition to an Impaired Metabolism of the Branched-Chain Amino Acids and Risk of Type 2 Diabetes: A Mendelian Randomisation Analysis. PLoS Med. 2016;13(11):e1002179. doi: 10.1371/journal.pmed.1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu M, Qi Q, Liang J, Bray GA, Hu FB, Sacks FM, Qi L. Genetic determinant for amino acid metabolites and changes in body weight and insulin resistance in response to weight-loss diets: the Preventing Overweight Using Novel Dietary Strategies (POUNDS LOST) trial. Circulation. 2013;127(12):1283–1289. doi: 10.1161/CIRCULATIONAHA.112.000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taneera J, Lang S, Sharma A, Fadista J, Zhou Y, Ahlqvist E, Jonsson A, Lyssenko V, Vikman P, Hansson O, Parikh H, Korsgren O, Soni A, Krus U, Zhang E, Jing XJ, Esguerra JL, Wollheim CB, Salehi A, Rosengren A, Renstrom E, Groop L. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 2012;16(1):122–134. doi: 10.1016/j.cmet.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 51.Mahendran Y, Jonsson A, Have CT, Allin KH, Witte DR, Jorgensen ME, Grarup N, Pedersen O, Kilpelainen TO, Hansen T. Genetic evidence of a causal effect of insulin resistance on branched-chain amino acid levels. Diabetologia. 2017;60(5):873–878. doi: 10.1007/s00125-017-4222-6. [DOI] [PubMed] [Google Scholar]
- 52.Zheng Y, Ceglarek U, Huang T, Li L, Rood J, Ryan DH, Bray GA, Sacks FM, Schwarzfuchs D, Thiery J, Shai I, Qi L. Weight-loss diets and 2-y changes in circulating amino acids in 2 randomized intervention trials. Am J Clin Nutr. 2016;103(2):505–511. doi: 10.3945/ajcn.115.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng Y, Li Y, Qi Q, Hruby A, Manson JE, Willett WC, Wolpin BM, Hu FB, Qi L. Cumulative consumption of branched-chain amino acids and incidence of type 2 diabetes. Int J Epidemiol. 2016;45(5):1482–1492. doi: 10.1093/ije/dyw143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fontana L, Cummings NE, Arriola Apelo SI, Neuman JC, Kasza I, Schmidt BA, Cava E, Spelta F, Tosti V, Syed FA, Baar EL, Veronese N, Cottrell SE, Fenske RJ, Bertozzi B, Brar HK, Pietka T, Bullock AD, Figenshau RS, Andriole GL, Merrins MJ, Alexander CM, Kimple ME, Lamming DW. Decreased Consumption of Branched-Chain Amino Acids Improves Metabolic Health. Cell Rep. 2016;16(2):520–530. doi: 10.1016/j.celrep.2016.05.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cummings NE, Lamming DW. Regulation of metabolic health and aging by nutrient-sensitive signaling pathways. Mol Cell Endocrinol. 2017;455:13–22. doi: 10.1016/j.mce.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Newgard CB. Metabolomics and Metabolic Diseases: Where Do We Stand? Cell Metab. 2017;25(1):43–56. doi: 10.1016/j.cmet.2016.09.018. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Batch BC, Hyland K, Svetkey LP. Branch chain amino acids: biomarkers of health and disease. Curr Opin Clin Nutr Metab Care. 2014;17(1):86–89. doi: 10.1097/MCO.0000000000000010. [DOI] [PubMed] [Google Scholar]
- 58.Walford GA, Ma Y, Clish C, Florez JC, Wang TJ, Gerszten RE, Diabetes Prevention Program Research Group Metabolite Profiles of Diabetes Incidence and Intervention Response in the Diabetes Prevention Program. Diabetes. 2016;65(5):1424–1433. doi: 10.2337/db15-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kappel BA, Lehrke M, Schütt K, Artati A, Adamski J, Lebherz C, Marx N. Effect of Empagliflozin on the Metabolic Signature of Patients With Type 2 Diabetes Mellitus and Cardiovascular Disease. Circulation. 2017;136(10):969–972. doi: 10.1161/CIRCULATIONAHA.117.029166. [DOI] [PubMed] [Google Scholar]
- 60.Kimball SR. Leucine-Induced Upregulation of Terminal Oligopyrimidine mRNA Translation in Skeletal Muscle: Just the Tip of the Iceberg? J Nutr. 2017;147(9):1603–1604. doi: 10.3945/jn.117.256289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heni M, Kullmann S, Preissl H, Fritsche A, Häring HU. Impaired insulin action in the human brain: causes and metabolic consequences. Nat Rev Endocrinol. 2015;11(12):701–711. doi: 10.1038/nrendo.2015.173. [DOI] [PubMed] [Google Scholar]
- 62.Koekkoek LL, Mul JD, la Fleur SE. Glucose-Sensing in the Reward System. Front Neurosci. 2017;11:716. doi: 10.3389/fnins.2017.00716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Panduro A, Rivera-Iñiguez I, Sepulveda-Villegas M, Roman S. Genes, emotions and gut microbiota: The next frontier for the gastroenterologist. World J Gastroenterol. 2017;23(17):3030–3042. doi: 10.3748/wjg.v23.i17.3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leigh SJ, Morris MJ. The role of reward circuitry and food addiction in the obesity epidemic: An update. Biol Psychol. 2018;131:31–42. doi: 10.1016/j.biopsycho.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 65.Areias MF, Prada PO. Mechanisms of insulin resistance in the amygdala: influences on food intake. Behav Brain Res. 2015;282:209–217. doi: 10.1016/j.bbr.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 66.Gannon NP, Schnuck JK, Vaughan RA. BCAA Metabolism and Insulin Sensitivity - Dysregulated by Metabolic Status? Mol Nutr Food Res. 2018;62(6):e1700756. doi: 10.1002/mnfr.201700756. [DOI] [PubMed] [Google Scholar]
- 67.Grigorescu F. New Genetic Approaches in Understanding Susceptibility for Metabolic Syndrome in Immigrant Populations Around Mediterranean Area. Acta Endocrinologica-Bucharest. 2012;8(1):87–98. [Google Scholar]
- 68.Grigorescu F. Medigene Program Rewarded with the European Stars Award 2017. Acta Endocrinologica-Bucharest. 2017;13(4):512–513. doi: 10.4183/aeb.2017.512. [DOI] [PMC free article] [PubMed] [Google Scholar]