

Abstract
The maintenance of bone mass is critically dependent on the balance between bone formation by osteoblasts and bone resorption by osteoclasts, processes in which osteocytes play also an important role. The activities of these bone cells are regulated by a variety of endocrine and paracrine factors of which sex steroids, parathyroid hormone, 1.25(OH)2-vitamin D3, glucocorticoids, retinoids and thyroid hormones are the most well known systemic factors. To the long list of locally acting factors belong cytokines and growth factors. This list was extended some 15 years ago by the discovery of the very important role of the WNT signalling system for the maintenance of bone mass. The first evidence of its role was the findings that mutations in the LRP5 gene, encoding a co-receptor in WNT-signaling, could result in either gain or loss of bone mass, i.e. either high bone mass or osteoporosis. This was a most unexpected observation since no indications existed prior to this discovery that the WNT signalling system had a role in bone remodeling. Since then, many observations have been made demonstrating the important role of different WNTs in regulating bone formation and resorption. Interestingly, some of these findings have demonstrated that trabecular and cortical bone are regulated by different mechanisms. It is the aim of the present overview to give the readers an insight into the WNT signalling system and its role in bone remodeling.
Keywords: WNTs, bone, sclerostin, bone resorption, osteoclasts
INTRODUCTION
The WNT signalling system was initially discovered independently by scientists in the embryology and tumor biology fields. During the 1970´s the Nobel laureates Nüsslein-Volhard and Wieschaus described several genes in Drosophila melanogaster which were regulating the segments and polarity of the fruit fly. A mutation in one of these genes resulted in flies without wings and this gene was called Wingless (1). Since the 1960´s, the tumor biologists had been trying to understand how certain retroviruses could induce mouse mammary tumor cells to grow and during the 1980´s the Nobel laureate Varmus and his co-worker Nusse discovered that these viruses induced expression of a gene in the mammary cells which they thought was causing the tumorigenic growth and called it Int1 (first common integration site)(2). Cloning of the two genes revealed that Int1 was identical to Wingless and that the same gene was involved in embryonic development and cancer (3). In 1991 it was agreed that the Int1/Wingless family should be denoted Wnt (Wingless-related integration site)(4). At that time, it was not understood how the proteins produced under the control of the Wnt genes regulated embryological development in the fruit fly or mammary tumor growth. Later on, it was found that the WNT proteins bind to receptors in the cell membrane made up by co-receptors called low density lipoprotein receptor-related proteins (LRPs) and signalling receptor components called Frizzleds. During that time, there were no indications that bone cells could be regulated by the WNT proteins.
In the later part of the 1990´s, three groups found that a gene in chromosome 11q12-13 was associated with bone mass. In 1996, Warman et al. reported that patients with osteoporosis pseudoglioma syndrome (OPPG) were associated with mutations in this chromosome (5). The following year, Johnson et al. reported that the unusually high bone mass in the entire skeleton in certain families was assigned to the same chromosome(6). The year after, Heaney et al. showed that autosomal recessive osteopetrosis with high bone mass, progressive deafness and blindness was linked to microsattelite markers on chromosome 11q12-13 (7). These studies did not reveal which gene in this chromosome was regulating bone mass.
In 2001, Gong et al. reported that OPPG was caused by six homozygous frame shift or non-sense mutations in the LRP5 gene located in chromosome 11q12-13 (8). Next year, Little et al. described that all individuals in a family with the high bone mass syndrome (total body Z score of 4.91) had a mutation in exon 3 of the LRP5 gene which resulted in a substitution of the amino acid glycine to valine in the extracellular part of LRP5 (9). The same year, Boyden et al. could show that the same mutation was the cause of the high bone mass syndrome in another family (10). Since serum markers of bone resorption were unchanged, whereas markers of bone formation were increased, it was likely that the enhanced bone mass was due to increased osteoblast activity. None of these three studies, however, could determine if the mutation directly affected the osteoblasts or if the mutation affected other cells in the body indirectly regulating osteoblasts. All three groups showed that LRP5 was expressed by osteoblasts. It was also reported that WNT proteins could stimulate osteoblast activities and that the high bone mass mutation was associated with decreased binding of an inhibitor to the extracellular part of LRP5. These findings suggested that a gain-of-function mutation in the LRP5 gene caused the high bone mass syndrome and that a variety of loss-of-function mutations in the same gene caused the OPPG syndrome. Thus, the knowledge on the importance of the WNT signalling system in bone biology started with the discovery of the role of the co-receptor LPR5. In subsequent studies, the role of Frizzled receptors, different WNT proteins as well as different WNT signalling inhibitors for osteoblast and osteoclast activities has been extensively studied.
It is the aim of the present overview to give the readers a simplified overview of the WNT signalling system and in some more detail to describe how this system can regulate bone mass. For detailed reviews on these topics, the interested readers are referred to more extensive reviews (11-14).
WNT signalling pathways
The basic principles involved in WNT signalling are rather similar to those for many hormones, cytokines and growth factors acting through cell surface receptors. Thus, WNTs, present as proteins in the extracellular milieu, act as ligands binding to the co-receptors LRPs and to signalling receptors Frizzleds, present in cell membranes. LRPs are so called single-pass proteins, whereas Frizzleds belong to the family of seven-transmembrane signalling receptors. The complexity of this system is due to the presence of several LRPs (LRP4-6), 19 different WNTs, 10 different Frizzleds and several inhibitors.
When the heterotrimeric complex of WNT-LRP-Frizzled is activated by the binding of WNTs, Frizzled signalling in the cell cytosol inactivates the so-called “destruction complex” and, thereby, a protein called ß-catenin becomes activated (Fig. 1). This protein translocates from the cytosol to the nucleus where it will remove the inhibitor Groucho from the transcription factor complex TCF/LEF. Subsequently, TCF/LEF binds to the promoter regions of different genes and, thereby, regulates gene transcription, in the case of osteoblasts stimulating genes which are involved in bone formation. This WNT signalling pathway is called canonical WNT signalling pathway or the ß-catenin pathway.
Figure 1.
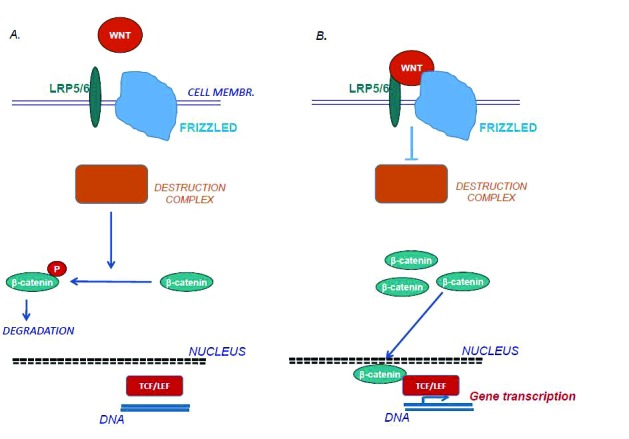
A. When the co-receptor LRP and the signaling receptor Frizzled are not activated by WNTs, the heterotetrameric destruction complex made up of the four components Axin, adenomatous polyposis coli, casein kinase and the constitutively active enzyme GSK3, phosphorylates β-catenin in the cytosol. This makes β-catenin directed to proteasomal degradation. B. When WNTs bind to LRP and Frizzled, the destruction complex becomes inactivated and non-phosphorylated β-catenin accumulates in the cytosol and translocates to the nuclei. In the nuclei, β-catenin activates the transcription factor complex TCF/LEF which binds to promoter regions of many genes including genes in osteoblasts which are involved in new bone formation.
The complexity of the WNT signalling pathway is not only made up by the existence of different WNTs, LRPs, Frizzleds and inhibitors, but also by the fact that WNT, in addition to the canonical pathway, can regulate cell activities also by other signalling mechanisms, so called non-canonical WNT signalling. In the planar cell polar pathway, WNTs form a complex with either one of the co-receptors ROR2 or RYK and Frizzled, which leads to activation of the small G-proteins Rho or Rac1. In the WNT/calcium pathway, WNTs activate phospholipase C which will cause changes in the intracellular levels of calcium and activation of MAPK (mitogen-activated protein kinases).
Although the role of the WNT signalling pathways in bone remodeling has attracted substantial interest during the past decade, we do not know which of the receptor components are most crucial for the regulation of osteoblast and osteoclast activities, nor do we know which cells produce the WNTs that regulate bone cells and what is regulating their production. We know, however, how certain WNTs can affect osteoblasts and osteoclasts. We also know how some of the inhibitors are important regulators of bone mass through their effects on WNT signalling. In the following sections, examples of how certain WNTs can affect bone cells will be given. In addition, certain inhibitors and their roles will be described.
Inhibitors of WNT signalling
Like in most biological systems, WNT signalling is controlled both by stimulators and inhibitors. In the case of canonical WNT signalling, the inhibitors can act either by sequestering soluble WNTs or by binding to the extracellular part of LRP5 and, thereby, interfering with the binding of WNTs (Fig. 2). To the first category, which also can act as inhibitors of non-canonical pathways, belongs secreted Frizzled-related proteins 1-5 (sFRP1-5) and WNT inhibitory factor 1 (WIF1). The other category includes sclerostin and Dickkopf 1-4 (DKK1-4).
Figure 2.
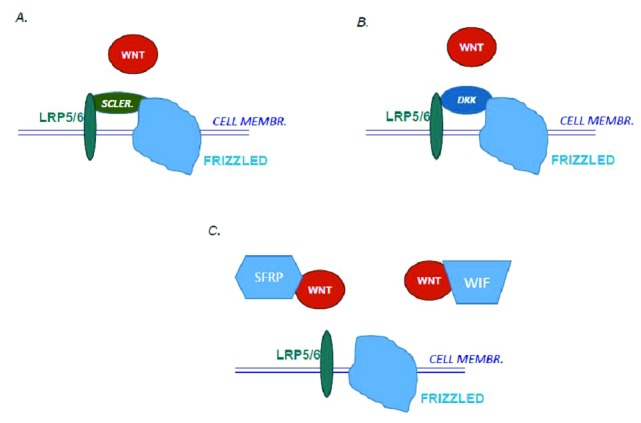
Different inhibitors can interact with WNT signaling. A. Sclerostin (Scler.) acts by binding to the extracellular part of the co-receptor LRP and thereby inhibits the binding of WNTs. Loss-of-function mutations in the SOST gene encoding sclerostin enhances binding of WNT and thereby increases WNT signalling and bone mass in patients with van Buchem´s disease and sclerosteosis. B. Dickkopf 1-4 (DKK) also binds to the extracellular part of the co-receptor LRP and inhibits binding of WNTs. C. Secreted Frizzled-related proteins (SFRP1-5) and WNT inhibitory protein (WIF) act as decoy receptors to bind soluble WNTs. Mutations in the SFRP4 gene are the cause of low bone mass in patients with Pyle´s disease.
Sclerostin is encoded by the SOSTgene, a gene which was found to be mutated in diseases with high bone mass such as van Buchem´s disease, sclerosteosis and autosomal dominant craniodiaphyseal dysplasia (15-17). Initially, it was not known which protein this gene was encoding but it was suspected that it could encode an inhibitor of bone morphogenetic proteins (BMPs). More recently, it was found that SOST encodes the protein sclerostin which acts as an inhibitor of WNT canonical signalling. As a consequence of the mutations, either less amount of sclerostin is produced or the mutated protein binds less well to the extracellular part of LRPs. Importantly, although WNT signalling seems to regulate a large variety of different cell types in the body, sclerostin seems to be expressed mainly by osteocytes (see further below for the therapeutic implications)(18). The common view is that sclerostin is secreted from osteocytes in their lacuna to osteoblasts on the bone surfaces. Decreased sclerostin expression or function lead to increased binding of WNTs in osteoblasts, enhanced activation of canonical WNT signalling and, therefore, more active bone forming osteoblasts. Patients with these mutations exhibit dense bone in all parts of the skeleton but the most striking phenotype is large jaw bones and very thick skull bones. Decreased sclerostin expression has been associated with both loading induced new bone formation (19) and the anabolic activity of intermittent parathyroid hormone (PTH) injections (20).
Experimental studies have shown that DKK1 can also act as an inhibitor of WNT canonical signalling in vitro and in vivo (21). In humans, increased DKK1 has been associated with decreased bone formation in the osteolytic lesions observed in patients with multiple myeloma (22).
Pyle´s disease, which is a skeletal disease with high incidence of fractures is characterized by a skeleton with thin cortices and dense trabecular bone. Recently it has been shown that this disease is caused by recessive mutations in the SFRP4 gene (23). Similar to the skeleton in humans with Pyle´s disease, deletion of the Sfrp4 gene in mice resulted in thin cortical bone, enhanced amount of trabecular bone in the metaphyses and weaker bones. Mechanistic evidence suggested that the reduced cortex is caused by a combination of enhanced bone resorption and decreased bone formation. Thus, SFRP4 was shown to act as an inhibitor of non-canonical signalling potentiating the osteoclastogenic effect of RANKL and as an inhibitor of non-canonical signalling affecting a BMP pathway associated with decreased cortical bone mass.
The role of WNT signalling in bone formation
The clinical studies revealing the important role of LRP5 for bone mass could not prove that the LRP5/WNT/Frizzled signalling was active directly in osteoblasts to regulate bone mass. The evidence showing that this signalling system can affect bone mass comes from mouse genetic studies in which LRPs and WNTs have been either genetically deleted or overexpressed globally in all cells or specifically in osteoblasts.
LRPs
Global deletion of the Lrp5 gene in mice results in decreased bone mass due to decreased number of active osteoblasts. Bone mass decreased to such an extent that some mice were limping due to spontaneous fractures in tibiae (24). Overexpression of the gene encoding human wild type LRP5 in osteoblasts results in increased bone mass in mice (25). This was much more pronounced when LRP5 with the G171V gain-of-function mutation, found to be associated with high bone mass in humans, is expressed in mice. Both trabecular and cortical bone mass was increased and this was due to more active osteoblasts with no effect on osteoclasts (25), in line with the analyses of bone formation and bone resorption markers in patients with LRP5 mutations.Further evidence for a direct role of LRP5/WNT/Frizzled signalling in osteoblasts comes from the elegant studies by Cui et al. (26). In mice with high bone mass mutations in Dmp1-Cre expressing cells (i.e. in late osteoblasts/osteocytes), bone mass is increased due to increased bone formation. In contrast, deletion of the mouse Lrp5 gene driven by Dmp1 results in decreased bone mass. High bone mass mutations in humans driven by Prxx1 result in increased bone mass in the femur, but not in vertebrae, because Prxx1 is expressed only by osteoblasts in appendicular skeleton but not in osteoblasts in the axial skeleton.
The mutations causing high bone mass are present in the extracellular part of LRP5 and decrease the sensitivity to inhibitors such as DKK1 (27) and sclerostin(28), resulting in increased binding of WNT and thereby enhanced LRP5/WNT/Frizzled signalling.
LRP6 is also expressed in osteoblasts and has overlapping but not identical functions with LRP5. In contrast to LRP5, mice with deletion of both alleles of Lrp6 die. However, mice with deletion of one of alleles of Lrp6 survive and have decreased bone mass (29). Mice with a spontaneous Lrp6 loss-of-function mutation have decreased bone mass, interestingly not due to decreased bone formation but to enhanced bone resorption (30).
Also LRP4 has been found to be expressed in osteoblasts and Lrp4 mutations in two patients with high bone mass could be linked to decreased sensitivity to sclerostin inhibition (31).
WNTs and canonical signalling
The WNT proteins and the different WNT-signalling pathways have important roles in the development, including the skeleton, as well as being important for bone mass in adults. The amount of bone tissue is dependent on the balance between bone formation and bone resorption and the understanding of how WNTs regulate bone mass requires the knowledge how different WNTs and different signal pathways regulate differentiation and function of osteoblasts and osteoclasts and how these cell types interact(13,14). WNTs regulate osteoblast differentiation from mesenchymal stem cells at different levels. Thus, WNTs have been shown to positively regulate bone formation by inducing osteoblastic differentiation of mesenchymal stem cells, increase osteoblast proliferation and inhibit osteoblast apoptosis, while negatively regulating adipocyte and chondrocyte differentiation. WNTs also affect osteoclast differentiation directly and indirectly (see further below). There are many difficulties to resolve before the WNT-signalling system can be therapeutically used. Despite these problems, antibody neutralization of the canonical WNT antagonist sclerostin seems to be one possibility (see further below).
Although it is clear from clinical and experimental studies on LRP5 that canonical WNT signalling has a crucial role for bone mass by regulating bone formation through effects in osteoblasts, it is not clear which WNTs are the most important ligands activating this pathway. As a consequence, it is not known which are the important WNT producing cells involved in bone formation. Nor is it known which are the important Frizzled receptors in osteoblasts causing bone formation. There are no reasons to believe that WNT are systemic endocrine acting factors and it is most likely that cells in the bone compartment produce the bone stimulatory WNTs. The knowledge on WNTs stimulating bone formation is mainly derived from experimental studies in mice although some human genetic data are also available (see further below).
The findings in mice lacking the Wls gene in osteoblasts nicely illustrate that WNTs produced and released by osteoblasts are important for bone mass. Wntless (Wls) is an intracellular chaperone protein which is important specifically for secretion of WNT proteins to the extracellular milieu (32). As could be expected, given the important roles of WNTs for embryonic development, mice lacking the Wls gene die early during embryonic development. However, deletion of Wls specifically in osteoblasts results in mice that survive (33). These mice have large reductions in both trabecular and cortical bone mass, in appendicular as well as axial skeleton, including skull bones. Bone mass is reduced to such an extent that mice exhibit spontaneous fractures. These observations demonstrate that WNTs produced by osteoblasts are important for skeletal development and indicate that osteoblastic WNTs act by auto- or paracrine mechanisms to control bone mass. The reduction of bone seems to be dependent on a combination of decreased bone formation and increased bone resorption since a decreased number of bone forming osteoblasts and an increased number of osteoclasts were observed. Observations in vivo and in vitro indicate that the decreased bone formation is due to decreased canonical WNT signalling in osteoblasts, in agreement with the findings showing the importance of LRP5 for bone mass.
WNT1, WNT 3a, WNT6 and WNT10b are three stimulators of canonical WNT signalling in many cell types including osteoblasts. WNT1, which is expressed in osteoblasts, has been shown to bind to LRP5 and to activate canonical WNT signalling (34). WNT3a, which is not expressed in osteoblasts, can act on these cells to increase their differentiation in vitro (35, 36). Interestingly, when Wnt3a is over-expressed in multiple myeloma cells and these cells are injected in mice, the osteoclast rich skeletal lesions are not developed (37). It is not known, however, if WNT3a acts on osteoblasts or osteoclasts to inhibit these lesions. Although WNT3a activates canonical WNT signalling in osteoblasts (33) and other cells, this ligand has been shown to activate also non-canonical WNT signalling in osteoblasts (38). WNT6 has been shown to increase osteoblastic differentiation while inhibiting adipocyte differentiation in vitro depending on ß-catenin, indicating that the effects are due to canonical WNT signalling (39). Wnt10b, which is expressed at increasing levels in differentiating osteoblasts (33), is the best characterized stimulator of canonical WNT-induced bone formation. WNT10b induces osteoblast differentiation in vitro (39, 40). Overexpression of the Wnt10b gene in bone marrow results in enhanced trabecular and cortical bone mass in long bones, hip and vertebrae resulting in bone with enhanced strength (40). These mice are resistant to ovariectomy induced osteoporosis. Increased Wnt10b expression specifically in osteoblasts also results in increased bone mass, which is due mainly to enhanced osteoblastogenesis (41). Bone mass is enhanced to such a degree that tooth eruption is impaired. The role of WNT10b for bone mass is further demonstrated by the observation that global deletion of the Wnt10b gene results in a decreased amount of trabecular bone (40,42). Together, these observations demonstrate the important role of canonical WNT signalling for bone mass in mice.
WNTs and non-canonical signalling
As pointed out above, WNTs can also signal through non-canonical signalling and there are suggestions from mice experiments that this pathway is also important for bone mass. Bone mass has been shown to be affected by three WNTs signalling through non-canonical pathway to regulate osteoblastic bone formation. Thus, enhanced expression of Wnt4 increases osteoblast differentiation in vitro (43). This has also been shown in vivo in mice overexpressing Wnt4 resulting in increased trabecular bone mass (44). This response was found to be due not only to enhanced bone formation, but also to decreased bone resorption. These mice were less susceptible to bone loss caused by estrogen deficiency or overexpression of TNF-α. WNT5a, which is expressed by osteoblasts (33), is important for embryonic development and mice lacking both alleles for Wnt5a (Wnt5a-/-) die perinatally. However, mice lacking only one allele (Wnt5a+/-) survive and have decreased trabecular bone mass since bone formation is decreased and bone resorption enhanced (45). WNT7b is not expressed by osteoblasts but can induce their differentiation from mesenchymal stem cells in vitro (38,46) and enhance bone formation and trabecular bone mass when overexpressed in vivo specifically in osteoblasts (47). These mice form so much bone that the bone marrow space is reduced and mice develop splenomegaly similar to osteopetrotic mice. In agreement with these observations, deletion of the Wnt7b gene results in decreased bone formation (38). These observations further illustrate the complexity of WNT signalling affecting bone formation.
The role of WNT signalling in bone resorption
The possibility to use pharmaceuticals increasing bone mass through WNT signalling not only requires knowledge on how such compounds affect canonical - and non-canonical - induced regulation of bone formation by osteoblasts, but also knowledge how the compounds affect osteoclast formation and activity, directly through effects on osteoclast progenitors or mature osteoclasts, but also indirectly through osteoblasts.
Differentiation of multinucleated, mature, bone resorbing osteoclasts from mononucleated osteoclast progenitors is critically dependent on activation of the receptor RANK (receptor activator of NF-κB) on the osteoclast progenitor cells by the ligand RANKL. RANKL can be produced locally in bone by osteoblasts/osteocytes, in which the expression of this cytokine is activated by stimulators of bone resorption such as parathyroid hormone or 1.25(OH)2-vitamin D3. The osteoclastogenic effect of RANKL is balanced by the decoy receptor osteoprotegerin (OPG), which binds to RANKL and inhibits the interaction with RANK (48, 49). OPG is produced by a variety of cell types including osteoblasts and its expression is negatively regulated by hormones stimulating bone resorption. The expression of OPG is also enhanced by canonical WNT signalling (50-52) and, therefore, the osteoanabolic effect of this signalling pathway is often due not only to increased bone formation but also to decreased bone resorption. We have demonstrated that also WNT16 can increase expression of OPG in osteoblasts (53). Since WNT16 can activate both canonical and non-canonical WNT signalling in osteoblasts it is unclear which pathway mediates the effect of WNT16.
There is also evidence that WNTs can act directly on osteoclast progenitor cells. Mice lacking one allele of Wnt5a have a decreased bone mass due to the decreased number of bone forming osteoblasts as discussed above (45). These mice also have a decreased number of osteoclasts, which does not explain the skeletal phenotype but suggests that WNT5a may regulate osteoclastogenesis. In line with this view, WNT5a potentiates RANKL-induced osteoclast formation of bone marrow macrophages. The authors demonstrate that WNT5a signalling in osteoclast progenitor cells is mediated by binding to the co-receptor ROR2 and subsequent non-canonical signalling, indicating that ROR2 is part of a positive regulation of osteoclastogenesis. Further evidence was gained by the finding that deletion of one allele of ROR2 results in an increased bone mass due to the decreased number of osteoclasts. In addition, it was found that ROR2 is not only involved in physiological regulation of osteoclast formation, but also in inflammation induced osteoclastogenesis. Thus, administration of soluble ROR2 protein, which acts as decoy receptor for WNT5a, inhibits excessive osteoclast formation in collagen-induced arthritis in mice.
Clinical studies have shown that a functional signal nucleotide polymorphism in the WNT16 gene is associated with cortical bone mass and increased susceptibility to fractures in humans (see further below). We have demonstrated that WNT16, expressed by osteoblasts, acts paracrinally on osteoclast progenitors in cortical bone and negatively regulate osteoclast formation causing an increase of cortical but not trabecular bone (53). Thus, mice in which the Wnt16 gene was deleted globally have a very thin cortex, but normal trabecular bone in long bones and vertebrae. The cortex was so thin that these mice developed spontaneous fractures, similar to mice in which the Lrp5 and Wls gene were deleted. Since WNT16 is expressed by osteoblasts, we next asked ourselves if it is WNT16 in these cells that is important for cortical bone mass. When Wnt16 was knocked out specifically in osteoblasts the mice also exhibited thin cortical bone with no change in trabecular bone, but when Wnt16 was deleted in osteocytes the mice were normal. The decreased cortical thickness was found to be associated with the increased number of cortical osteoclasts, whereas the number of osteoblasts and bone formation was normal. We next could show that the increase of osteoclasts was due to that WNT16 protein inhibits osteoclast formation by interfering with the signal mechanism by which the RANK receptor differentiates osteoclast progenitor cells from mature osteoclasts. This observation was made using both mouse and human osteoclast progenitor cells. WNT16-induced inhibition of osteoclastogenesis was mediated by non-canonical signalling. Thus, WNT16 can inhibit osteoclast formation both indirectly by increasing OPG in osteoblasts (see above) and by directly interfering with osteoclast differentiation. We also could show that inflammation induced bone resorption could be blocked by local treatment with WNT16 protein. Since the commonly used osteoporosis drugs mainly inhibit fractures in vertebrae, rich in trabecular bone, but are less effective to inhibit fractures in non-vertebral bones, rich in cortical bone, there is a medical need to develop drugs that more effectively increase cortical bone mass. It is possible that increasing WNT16 may be a mechanism to enhance cortical thickness reducing the risk for non-vertebral fractures. The importance of developing such drugs is demonstrated by the fact that fractures in elderly individuals often occur at sites suffering from cortical-bone fragility. The fact that WNT16 is important for bone mass in adult mice strengthens the view that increased WNT16 could be a potential mechanism for treatment of osteoporosis in humans. Thus, we have shown that induced deletion of Wnt16 in adult mice also results in decreased cortical bone mass (54).
Since we could observe that estrogen treatment increased Wnt16 expression in cortical bone we next addressed the issue if WNT16 could be involved in the mechanisms by which estrogen preserves bone mass. However, using mice in which the Wnt16 gene was over-expressed specifically in osteoblasts we observed that ovariectomy caused reduced bone mass to the same degree in these mice as in normal mice (55). We also could show that estrogen treatment increased bone mass to the same degree in mice with deletion of the Wnt16 gene as compared to normal mice. These observations demonstrate that the bone-sparing effect of estrogen and WNT16 are independent of each other, but WNT16-targeted therapies might still be useful for treatment of post-menopausal osteoporosis. This strategy might also be useful for treatment of glucocorticoid-induced osteoporosis since we have shown that prednisolone-induced bone loss in mice can be prevented by overexpression of Wnt16 in osteoblasts (56).
Clinical evidence for the importance of WNT signalling in regulation of bone mass
In addition to the observations that gain-of-function and loss-of-function mutations in the LRP5 gene are the cause of high bone mass in humans and the osteoporosis pseudoglioma syndrome, respectively, loss-of-function mutations in the LRP6 gene have also been found to be associated with low bone mineral density and hip fractures early in life(57). Also LRP4 seems to be important for LRP/WNT/FRIZZLED signalling since it has been reported that two mutations of the LRP4 gene are associated with high bone density (31). We described an unusual case of the association of a high bone mass phenotype with primary hyperparathyroidism and resistance to the catabolic action of PTH on the bone. In spite of the negative result of Lrp5 testing, other genes of the Wnt pathway might be responsible (58).
The importance of the different inhibitors of LRP/WNT/FRIZZLED is not only demonstrated by the fact that loss-of-function mutations in the SOST gene (encoding sclerostin) result in high bone mass in patients with van Buchem´s disease and sclerosteosis, but also by reports showing that mutations in the DKK1 gene and in the SFRP4 gene are associated with skeletal pathologies in humans. Thus, a mutation in the DKK1 gene has been reported to be associated with childhood-onset low bone mineral density and increased fracture susceptibility not related to osteogenesis imperfecta (59). As mentioned above, patients with Pyle´s disease suffer from increased fracture susceptibility due to decreased cortical thickness. This disease is caused by mutations in the SFRP4 gene (23).
A family with dominantly inherited, severe, early-onset osteoporosis has been found to have a missense mutation in the WNT1 gene (60). Mutations in the same gene have also been found in two siblings with a skeletal disease thought to be a variant of osteogenesis imperfecta (60). In another study it is reported that mutations in the WNT1 gene have been found in one family with autosomal-recessive osteogenesis imperfecta and in another family with dominantly inherited early-onset osteoporosis (34). WNT1 mutations have also been reported in another unrelated family with recessive severe bone fragility(61). Mutations in the WNT3a gene have been demonstrated in a family with early-onset osteoporosis (59). These findings are in agreement with experimental studies showing that WNT1 and WNT3a can stimulate osteoblastic activities through canonical signalling.
Associations with bone mineral density and genes encoding proteins involved in LRP/WNT/FRIZZLED signalling have also been observed in genome-wide analysis. Thus, single nucleotide polymorphisms in the WLS, LRP5, SOST, DKK1, SFRP4, WNT3, WNT4, WNT5b and WNT16 genes have all been associated with low bone mineral density (62, 63). Of particular interest are the WNT4, WNT16 and SOST loci which also have been found to be associated with low-trauma fractures (62, 64, 65).
WNT signalling as therapeutical targets
The many clinical and experimental studies demonstrating the importance of proteins involved in LRP/WNT/FRIZZED signalling for bone mass have made them interesting candidates for drug development. Several pathways in this signalling system increase bone mass by enhancing bone formation and since the most frequently osteoporosis drugs currently used inhibit bone resorption to prevent further bone loss in the patients, it is of interest to develop drugs acting to enhance bone mass by inducing new bone formation. The bisphosphonates, acting by killing mature osteoclasts, and denosumab, which is an antibody neutralizing RANKL inhibiting differentiation of mature osteoclasts from osteoclast progenitors cells, have both been found to decrease fractures mainly in vertebrae made up by trabecular bone. It is, therefore, a need to develop drugs which can enhance cortical bone since most fractures in elderly patients are caused by cortical-bone fragility. The fact that some WNT signalling molecules have been associated preferentially with cortical bone makes drug development based upon these molecules of special interest. WNT signalling, however, is important for differentiation and function of a large variety of tissues and organs as well as in tumour cell biology. It is, therefore, a large challenge to develop drugs that specifically target the skeleton. One such possibility is sclerostin which has been found to be mainly expressed in osteocytes. Preclinical studies found that treatment with antibodies neutralizing sclerostin enhanced trabecular bone mass and inhibited bone loss induced by ovariectomy and inflammation (66-68).
Prompted by the preclinical studies, antibodies neutralizing human sclerostin were developed. A phase I study found that a single subcutaneous or intravenous injection of anti-sclerostin resulted in increased bone mineral density in lumbar spine and total hip (69). The phase II study reported that monthly subcutaneous injections, or injections every third month, of romosozumab (anti-sclerostin antibody) increased bone mineral density after 12 months treatment in lumbar spine (11.3%), significantly more than alendronate (4.1%) or teriparatide (7.1%) (70). Also bone mineral density in total hip and femoral neck was enhanced more effectively by romosozumab. In line with an increased activation of canonical WNT pathway, serum levels of bone formation markers increased and markers for resorption decreased (due to increased expression of OPG). However, the formation markers returned to baseline levels after 6 months indicating that long-term treatment may not be effective. Two phase III studies have been published also demonstrating the promising effects by romosozumab. In the FRAME study, 7.180 postmenopausal women were treated with monthly subcutaneous injections with romosozumab for 12 months followed by 12 months with subcutaneous injections with denosumab every 6 months and compared to a group given placebo for 12 months followed by 12 months with denosumab (71). At 12 months, vertebral fractures were seen in 16/3321 patients in the romosozumab group compared to 59/3322 in the control group. At 24 months, 21/3325 patients who had received romosozumab followed by denosumab had experienced vertebral fractures compared to 84/3327 in the control group. Thus, the risk for vertebral fractures was 75% lower in the romosozumab group at both time points. There was, however, no statistically significant effect on non-vertebral fractures, which may be due to an unexpected low rate of non-vertebral fractures in the control group made up of many patients from Latin America. In the ARCH study, 4.093 postmenopausal women were given either romosozumab for 12 months and alendronate for the following 12 months or alendronate during 24 months (72). In the romosozumab group, 127 vertebral fractures in 2046 patients were observed compared to 243/2047 in the alendronate group during the 24 months treating period, resulting in a 48% reduction of new vertebral fractures. There was also a significant 19% reduction of non-vertebral fractures (P=0.04) and hip fractures (P=0.02). These studies demonstrate that targeting sclerostin is clearly a more effective treatment for osteoporosis than targeting bone resorption.
In the FRAME study, no differences in adverse side effect were observed in the romosozumab group. In the ARCH study, however, serious cardiovascular adverse events were more often observed during the first year in the romosozumab group (50/2040 patients) than in the alendronate group (38/2014 patients). It is unclear why these side effects were observed in the ARCH study but not in the FRAME study. One possibility might be that alendronate has a cardioprotective effect but that has not been possible to demonstrate. The difference might be related to the fact that patients were older in the ARCH study and therefore were less healthy with higher risk for cardiovascular disease (73,74). It should also be noted that WNT signalling has been shown in vessel walls and that sclerostin has been found to be expressed in arteries in patients with calcifications. It remains to be seen if romsozumab will be approved by FDA and, if so, if physicians will prescribe this drug and if patients will accept the treatment given the potential risk of increased risk for cardiovascular side effects (74).
In conclusion, several components of the WNT signalling system have been shown in preclinical and clinical studies to have robust effects on bone mass through direct effects on osteoblasts and through both direct and indirect effects on osteoclasts. Most attention has been paid to the direct effects on osteoblasts prompting efforts to develop true osteoanabolic drugs. The large challenge is to find specificity for the skeleton since WNT signalling is involved in many cellular physiological and pathological activities outside the skeleton, including proliferation of tumor cells. Based upon the observations that the WNT inhibitor sclerostin seems to be expressed exclusively in osteocytes and that patients with loss-of-function mutations in the SOST gene encoding sclerostin have high bone mass, neutralizing antibodies to sclerostin have been developed and gone through phase III clinical studies. Two studies have shown that romosozumab (anti-sclerostin antibody) given subcutaneously monthly to postmenopausal women is more effective to prevent vertebral fractures than anti-resorptive therapy. Similar to the anti-resorptive drugs, romosozumab is less effective to prevent non-vertebral fractures, demonstrating the need for drugs that more effectively target cortical bone. Such drugs could potentially be developed by increasing WNT16 which has been shown to have a unique protective role for cortical bone.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgement
The studies performed in the author´s (UHL) laboratory have been supported by the Swedish Research Council, the Swedish Foundation for Strategic Research, COMBINE, the ALF/LUA research grant in Gothenburg, the Lundberg Foundation, the County Council of Västerbotten, the Swedish Dental Society, the Swedish Rheumatism Association, and the Royal 80 Year Fund of King Gustav V.
References
- 1.Nusslein-Volhard C., Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287(5785):795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 2.Nusse R., Varmus H. E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31(1):99–109. doi: 10.1016/0092-8674(82)90409-3. [DOI] [PubMed] [Google Scholar]
- 3.Rijsewijk F., Schuermann M., Wagenaar E., Parren P., Weigel D., Nusse R. The Drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell. 1987;50(4):649–657. doi: 10.1016/0092-8674(87)90038-9. [DOI] [PubMed] [Google Scholar]
- 4.Nusse R., Varmus H. E. Wnt genes. Cell. 1992;69(7):1073–1087. doi: 10.1016/0092-8674(92)90630-u. [DOI] [PubMed] [Google Scholar]
- 5.Gong Y, Vikkula M, Boon L, Liu J, Beighton P, Ramesar R, Peltonen L, Somer H, Hirose T, Dallapiccola B, De Paepe A, Swoboda W, Zabel B, Superti-Furga A, Steinmann B, Brunner H G, Jans A, Boles R G, Adkins W, van den Boogaard M J, Olsen B R, Warman M L. Osteoporosis-pseudoglioma syndrome, a disorder affecting skeletal strength and vision, is assigned to chromosome region 11q12-13. Am J Hum Genet. 1996;59(1):146–151. [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson M. L., Gong G., Kimberling W., Recker S. M., Kimmel D. B., Recker R. B. Linkage of a gene causing high bone mass to human chromosome 11 (11q12-13) Am J Hum Genet. 1997;60(6):1326–1332. doi: 10.1086/515470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heaney C., Shalev H., Elbedour K., Carmi R., Staack J. B., Sheffield V. C., Beier D. R. Human autosomal recessive osteopetrosis maps to 11q13, a position predicted by comparative mapping of the murine osteosclerosis (oc) mutation. Human molecular genetics. 1998;7(9):1407–1410. doi: 10.1093/hmg/7.9.1407. [DOI] [PubMed] [Google Scholar]
- 8.Gong Y., Slee R. B., Fukai N., Rawadi G., Roman-Roman S., Reginato A. M., Wang H., Cundy T., Glorieux F. H., Lev D., Zacharin M., Oexle K., Marcelino J., Suwairi W., Heeger S., Sabatakos G., Apte S., Adkins W. N., Allgrove J., Arslan-Kirchner M., Batch J. A., Beighton P., Black G. C., Boles R. G., Boon L. M., Borrone C., Brunner H. G., Carle G. F., Dallapiccola B., De Paepe A., Floege B., Halfhide M. L., Hall B., Hennekam R. C., Hirose T., Jans A., Juppner H., Kim C. A., Keppler-Noreuil K., Kohlschuetter A., LaCombe D., Lambert M., Lemyre E., Letteboer T., Peltonen L., Ramesar R. S., Romanengo M., Somer H., Steichen-Gersdorf E., Steinmann B., Sullivan B., Superti-Furga A., Swoboda W., van den Boogaard M. J., Van Hul W., Vikkula M., Votruba M., Zabel B., Garcia T., Baron R., Olsen B. R., Warman M. L. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107(4):513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 9.Little R. D., Carulli J. P., Del Mastro R. G., Dupuis J., Osborne M., Folz C., Manning S. P., Swain P. M., Zhao S. C., Eustace B., Lappe M. M., Spitzer L., Zweier S., Braunschweiger K., Benchekroun Y., Hu X., Adair R., Chee L., FitzGerald M. G., Tulig C., Caruso A., Tzellas N., Bawa A., Franklin B., McGuire S., Nogues X., Gong G., Allen K. M., Anisowicz A., Morales A. J., Lomedico P. T., Recker S. M., Van Eerdewegh P., Recker R. R., Johnson M. L. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70(1):11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyden L. M., Mao J., Belsky J., Mitzner L., Farhi A., Mitnick M. A., Wu D., Insogna K., Lifton R. P. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346(20):1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 11.Clevers H., Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 12.MacDonald B. T., Tamai K., He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Developmental cell. 2009;17(1):9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baron R., Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179–192. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- 14.Lerner U. H., Ohlsson C. The WNT system: background and its role in bone. J Int Med. 2015;277(6):630–649. doi: 10.1111/joim.12368. [DOI] [PubMed] [Google Scholar]
- 15.Balemans W., Patel N., Ebeling M., Van Hul E., Wuyts W., Lacza C., Dioszegi M., Dikkers F. G., Hildering P., Willems P. J., Verheij J. B., Lindpaintner K., Vickery B., Foernzler D., Van Hul W. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39(2):91–97. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balemans W., Ebeling M., Patel N., Van Hul E., Olson P., Dioszegi M., Lacza C., Wuyts W., Van Den Ende J., Willems P., Paes-Alves A. F., Hill S., Bueno M., Ramos F. J., Tacconi P., Dikkers F. G., Stratakis C., Lindpaintner K., Vickery B., Foernzler D., Van Hul W. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Human molecular genetics. 2001;10(5):537–543. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 17.Kim S. J., Bieganski T., Sohn Y. B., Kozlowski K., Semenov M., Okamoto N., Kim C. H., Ko A. R., Ahn G. H., Choi Y. L., Park S. W., Ki C. S., Kim O. H., Nishimura G., Unger S., Superti-Furga A., Jin D. K. Identification of signal peptide domain SOST mutations in autosomal dominant craniodiaphyseal dysplasia. Human genetics. 2011;129(5):497–502. doi: 10.1007/s00439-011-0947-3. [DOI] [PubMed] [Google Scholar]
- 18.van Bezooijen R. L., Roelen B. A., Visser A., van der Wee-Pals L., de Wilt E., Karperien M., Hamersma H., Papapoulos S. E., ten Dijke P., Lowik C. W. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. The Journal of experimental medicine. 2004;199(6):805–814. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robling A. G., Niziolek P. J., Baldridge L. A., Condon K. W., Allen M. R., Alam I., Mantila S. M., Gluhak-Heinrich J., Bellido T. M., Harris S. E., Turner C. H. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283(9):5866–5875. doi: 10.1074/jbc.M705092200. [DOI] [PubMed] [Google Scholar]
- 20.Keller H., Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37(2):148–158. doi: 10.1016/j.bone.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 21.Morvan F., Boulukos K., Clement-Lacroix P., Roman Roman S., Suc-Royer I., Vayssiere B., Ammann P., Martin P., Pinho S., Pognonec P., Mollat P., Niehrs C., Baron R., Rawadi G. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res. 2006;21(6):934–945. doi: 10.1359/jbmr.060311. [DOI] [PubMed] [Google Scholar]
- 22.Pinzone J. J., Hall B. M., Thudi N. K., Vonau M., Qiang Y. W., Rosol T. J., Shaughnessy J. D., Jr. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood. 2009;113(3):517–525. doi: 10.1182/blood-2008-03-145169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiper P. O. S., Saito H., Gori F., Unger S., Hesse E., Yamana K., Kiviranta R., Solban N., Liu J., Brommage R., Boduroglu K., Bonafe L., Campos-Xavier B., Dikoglu E., Eastell R., Gossiel F., Harshman K., Nishimura G., Girisha K. M., Stevenson B. J., Takita H., Rivolta C., Superti-Furga A., Baron R. Cortical-Bone Fragility--Insights from sFRP4 Deficiency in Pyle's Disease. N Engl J Med. 2016;374(26):2553–2562. doi: 10.1056/NEJMoa1509342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato M., Patel M. S., Levasseur R., Lobov I., Chang B. H., Glass D. A., 2nd, Hartmann C., Li L., Hwang T. H., Brayton C. F., Lang R. A., Karsenty G., Chan L. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. The Journal of cell biology. 2002;157(2):303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Babij P., Zhao W., Small C., Kharode Y., Yaworsky P. J., Bouxsein M. L., Reddy P. S., Bodine P. V., Robinson J. A., Bhat B., Marzolf J., Moran R. A., Bex F. High bone mass in mice expressing a mutant LRP5 gene. J Bone Miner Res. 2003;18(6):960–974. doi: 10.1359/jbmr.2003.18.6.960. [DOI] [PubMed] [Google Scholar]
- 26.Cui Y., Niziolek P. J., MacDonald B. T., Zylstra C. R., Alenina N., Robinson D. R., Zhong Z., Matthes S., Jacobsen C. M., Conlon R. A., Brommage R., Liu Q., Mseeh F., Powell D. R., Yang Q. M., Zambrowicz B., Gerrits H., Gossen J. A., He X., Bader M., Williams B. O., Warman M. L., Robling A. G. Lrp5 functions in bone to regulate bone mass. Nat Med. 2011;17(6):684–691. doi: 10.1038/nm.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ai M., Holmen S. L., Van Hul W., Williams B. O., Warman M. L. Reduced affinity to and inhibition by DKK1 form a common mechanism by which high bone mass-associated missense mutations in LRP5 affect canonical Wnt signaling. Molecular and cellular biology. 2005;25(12):4946–4955. doi: 10.1128/MCB.25.12.4946-4955.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Semenov M. V., He X. LRP5 mutations linked to high bone mass diseases cause reduced LRP5 binding and inhibition by SOST. J Biol Chem. 2006;281(50):38276–38284. doi: 10.1074/jbc.M609509200. [DOI] [PubMed] [Google Scholar]
- 29.Holmen S. L., Giambernardi T. A., Zylstra C. R., Buckner-Berghuis B. D., Resau J. H., Hess J. F., Glatt V., Bouxsein M. L., Ai M., Warman M. L., Williams B. O. Decreased BMD and limb deformities in mice carrying mutations in both Lrp5 and Lrp6. J Bone Miner Res. 2004;19(12):2033–2040. doi: 10.1359/JBMR.040907. [DOI] [PubMed] [Google Scholar]
- 30.Kubota T., Michigami T., Sakaguchi N., Kokubu C., Suzuki A., Namba N., Sakai N., Nakajima S., Imai K., Ozono K. Lrp6 hypomorphic mutation affects bone mass through bone resorption in mice and impairs interaction with Mesd. J Bone Miner Res. 2008;23(10):1661–1671. doi: 10.1359/jbmr.080512. [DOI] [PubMed] [Google Scholar]
- 31.Leupin O., Piters E., Halleux C., Hu S., Kramer I., Morvan F., Bouwmeester T., Schirle M., Bueno-Lozano M., Fuentes F. J., Itin P. H., Boudin E., de Freitas F., Jennes K., Brannetti B., Charara N., Ebersbach H., Geiss S., Lu C. X., Bauer A., Van Hul W., Kneissel M. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286(22):19489–19500. doi: 10.1074/jbc.M110.190330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banziger C., Soldini D., Schutt C., Zipperlen P., Hausmann G., Basler K. Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell. 2006;125(3):509–522. doi: 10.1016/j.cell.2006.02.049. [DOI] [PubMed] [Google Scholar]
- 33.Zhong Z., Zylstra-Diegel C. R., Schumacher C. A., Baker J. J., Carpenter A. C., Rao S., Yao W., Guan M., Helms J. A., Lane N. E., Lang R. A., Williams B. O. Wntless functions in mature osteoblasts to regulate bone mass. Proc Natl Acad Sci USA. 2012;109(33):E2197–2204. doi: 10.1073/pnas.1120407109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keupp K., Beleggia F., Kayserili H., Barnes A. M., Steiner M., Semler O., Fischer B., Yigit G., Janda C. Y., Becker J., Breer S., Altunoglu U., Grunhagen J., Krawitz P., Hecht J., Schinke T., Makareeva E., Lausch E., Cankaya T., Caparros-Martin J. A., Lapunzina P., Temtamy S., Aglan M., Zabel B., Eysel P., Koerber F., Leikin S., Garcia K. C., Netzer C., Schonau E., Ruiz-Perez V. L., Mundlos S., Amling M., Kornak U., Marini J., Wollnik B. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet. 2013;92(4):565–574. doi: 10.1016/j.ajhg.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boland G. M., Perkins G., Hall D. J., Tuan R. S. Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. Journal of cellular biochemistry. 2004;93(6):1210–1230. doi: 10.1002/jcb.20284. [DOI] [PubMed] [Google Scholar]
- 36.Shin H. R., Islam R., Yoon W. J., Lee T., Cho Y. D., Bae H. S., Kim B. S., Woo K. M., Baek J. H., Ryoo H. M. Pin1-mediated Modification Prolongs the Nuclear Retention of beta-Catenin in Wnt3a-induced Osteoblast Differentiation. J Biol Chem. 2016;291(11):5555–5565. doi: 10.1074/jbc.M115.698563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiang Y. W., Shaughnessy J. D., Jr., Yaccoby S. Wnt3a signaling within bone inhibits multiple myeloma bone disease and tumor growth. Blood. 2008;112(2):374–382. doi: 10.1182/blood-2007-10-120253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tu X., Joeng K. S., Nakayama K. I., Nakayama K., Rajagopal J., Carroll T. J., McMahon A. P., Long F. Noncanonical Wnt signaling through G protein-linked PKCdelta activation promotes bone formation. Developmental cell. 2007;12(1):113–127. doi: 10.1016/j.devcel.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cawthorn W. P., Bree A. J., Yao Y., Du B., Hemati N., Martinez-Santibanez G., MacDougald O. A. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a beta-catenin-dependent mechanism. Bone. 2012;50(2):477–489. doi: 10.1016/j.bone.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bennett C. N., Longo K. A., Wright W. S., Suva L. J., Lane T. F., Hankenson K. D., MacDougald O. A. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci U S A. 2005;102(9):3324–3329. doi: 10.1073/pnas.0408742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bennett C. N., Ouyang H., Ma Y. L., Zeng Q., Gerin I., Sousa K. M., Lane T. F., Krishnan V., Hankenson K. D., MacDougald O. A. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J Bone Miner Res. 2007;22(12):1924–1932. doi: 10.1359/jbmr.070810. [DOI] [PubMed] [Google Scholar]
- 42.Stevens J. R., Miranda-Carboni G. A., Singer M. A., Brugger S. M., Lyons K. M., Lane T. F. Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells. J Bone Miner Res. 2010;25(10):2138–2147. doi: 10.1002/jbmr.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang J., Sonoyama W., Wang Z., Jin Q., Zhang C., Krebsbach P. H., Giannobile W., Shi S., Wang C. Y. Noncanonical Wnt-4 signaling enhances bone regeneration of mesenchymal stem cells in craniofacial defects through activation of p38 MAPK. J Biol Chem. 2007;282(42):30938–30948. doi: 10.1074/jbc.M702391200. [DOI] [PubMed] [Google Scholar]
- 44.Yu B., Chang J., Liu Y., Li J., Kevork K., Al-Hezaimi K., Graves D. T., Park N. H., Wang C. Y. Wnt4 signaling prevents skeletal aging and inflammation by inhibiting nuclear factor-kappaB. Nat Med. 2014;20(9):1009–1017. doi: 10.1038/nm.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maeda K., Kobayashi Y., Udagawa N., Uehara S., Ishihara A., Mizoguchi T., Kikuchi Y., Takada I., Kato S., Kani S., Nishita M., Marumo K., Martin T. J., Minami Y., Takahashi N. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med. 2012;18(3):405–412. doi: 10.1038/nm.2653. [DOI] [PubMed] [Google Scholar]
- 46.Hu H., Hilton M. J., Tu X., Yu K., Ornitz D. M., Long F. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 2005;132(19):49–60. doi: 10.1242/dev.01564. [DOI] [PubMed] [Google Scholar]
- 47.Chen J., Tu X., Esen E., Joeng K. S., Lin C., Arbeit J. M., Ruegg M. A., Hall M. N., Ma L., Long F. WNT7B promotes bone formation in part through mTORC1. PLoS Genet. 2014;10(1) doi: 10.1371/journal.pgen.1004145. e1004145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Souza P. P., Lerner U. H. The role of cytokines in inflammatory bone loss. Immunological investigations. 2013;42(7):555–622. doi: 10.3109/08820139.2013.822766. [DOI] [PubMed] [Google Scholar]
- 49.Takayanagi H. New developments in osteoimmunology. Rheumatology. 2012;8(11):684–689. doi: 10.1038/nrrheum.2012.167. Nature reviews. [DOI] [PubMed] [Google Scholar]
- 50.Glass D. A., Bialek P., Ahn J. D., Starbuc M., Patel M. S., Clevers H., Taketo M. M., Long F., McMahon A. P., Lang R. A., Karsenty G. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Developmental cell. 2005;8(5):751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 51.Holmen S. L., Zylstra C. R., Mukherjee A., Sigler R. E., Faugere M. C., Bouxsein M. L., Deng L., Clemens T. L., Williams B. O. Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem. 2005;280(22):21162–21168. doi: 10.1074/jbc.M501900200. [DOI] [PubMed] [Google Scholar]
- 52.Kramer I., Halleux C., Keller H., Pegurri M., Gooi J. H., Weber P. B., Feng J. Q., Bonewald L. F., Kneissel M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Molecular and cellular biology. 2010;30(12):3071–3085. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moverare-Skrtic S., Henning P., Liu X., Nagano K., Saito H., Borjesson A. E., Sjogren K., Windahl S. H., Farman H., Kindlund B., Engdahl C., Koskela A., Zhang F. P., Eriksson E. E., Zaman F., Hammarstedt A., Isaksson H., Bally M., Kassem A., Lindholm C., Sandberg O., Aspenberg P., Savendahl L., Feng J. Q., Tuckermann J., Tuukkanen J., Poutanen M., Baron R., Lerner U. H., Gori F., Ohlsson C. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat Med. 2014;20(11):1279–1288. doi: 10.1038/nm.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohlsson C., Henning P., Nilsson K. H., Wu J., Gustafsson K. L., Sjogren K., Tornqvist A. E., Koskela A., Zhang F. P., Lagerquist M. K., Poutanen M., Tuukkanen J., Lerner U. H., Moverare-Skrtic S. Inducible Wnt16 inactivation: WNT16 regulates cortical bone thickness in adult mice. J Endocrinol. 2018 doi: 10.1530/JOE-18-0020. pii: JOE-18-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moverare-Skrtic S., Henning P., Gustafsson K. L., Sjogren K., Windahl S. H., Koskela A., Tuukkanen J., Borjesson A. E., Lagerquist M. K., Lerner U. H., Zhang F. P., Gustafsson J. A., Poutanen M., Ohlsson C. The bone-sparing effects of estrogen and WNT16 are independent of each other. Proc Natl Acad Sci U S A. 2015;112(48):14972–14977. doi: 10.1073/pnas.1520408112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohlsson C., Nilsson K. H., Henning P., Wu J., Gustafsson K. L., Poutanen M., Lerner U. H., Moverare-Skrtic S. WNT16 Overexpression Partly Protects Against Glucocorticoid-induced Bone Loss. Am J Physiol Endocrinol Metab. 2018 doi: 10.1152/ajpendo.00292.2017. [DOI] [PubMed] [Google Scholar]
- 57.Mani A., Radhakrishnan J., Wang H., Mani A., Mani M. A., Nelson-Williams C., Carew K. S., Mane S., Najmabadi H., Wu D., Lifton R. P. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007;315(5816):1278–1282. doi: 10.1126/science.1136370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grigorie D, Constantini A, Sucaliuc A. Suspected non-LRP5 mutation associated with high bone mass unaltered by concurrent symptomatic primary hyperparathyroidism of long duration. Acta Endocrinologica (Buc) 2016;12(4):461–464. doi: 10.4183/aeb.2016.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Korvala J., Loija M., Makitie O., Sochett E., Juppner H., Schnabel D., Mora S., Cole W. G., Ala-Kokko L., Mannikko M. Rare variations in WNT3A and DKK1 may predispose carriers to primary osteoporosis. European journal of medical genetics. 2012;55(10):515–519. doi: 10.1016/j.ejmg.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 60.Laine C. M., Joeng K. S., Campeau P. M., Kiviranta R., Tarkkonen K., Grover M., Lu J. T., Pekkinen M., Wessman M., Heino\ T. J., Nieminen-Pihala\ V., Aronen M., Laine T., Kroger H., Cole W. G., Lehesjoki A. E., Nevarez L., Krakow D., Curry C. J., Cohn D. H., Gibbs R. A., Lee B. H., Makitie O. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med. 2013;368(19):1809–1816. doi: 10.1056/NEJMoa1215458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palomo T., Al-Jallad H., Moffatt P., Glorieux F. H., Lentle B., Roschger P., Klaushofer K., Rauch F. Skeletal characteristics associated with homozygous and heterozygous WNT1 mutations. Bone. 2014;67(1):63–70. doi: 10.1016/j.bone.2014.06.041. [DOI] [PubMed] [Google Scholar]
- 62.Estrada K, Styrkarsdottir U, Evangelou E, Hsu Y H, Duncan E L, Ntzani E E, Oei L, Albagha O M, Amin N, Kemp J P, Koller D L, Li G, Liu C T, Minster R L, Moayyeri A, Vandenput L, Willner D, Xiao S M, Yerges-Armstrong L M, Zheng H F, Alonso N, Eriksson J, Kammerer C M, Kaptoge S K, Leo P J, Thorleifsson G, Wilson S G, Wilson J F, Aalto\ V, Alen M, Aragaki A K, Aspelund T, Center J R, Dailiana Z, Duggan D J, Garcia M, Garcia-Giralt N, Giroux S, Hallmans G, Hocking L J, Husted L B, Jameson K A, Khusainova R, Kim G S, Kooperberg C, Koromila T, Kruk M, Laaksonen M, Lacroix A Z, Lee S H, Leung P C, Lewis J R, Masi L, Mencej-Bedrac S, Nguyen T V, Nogues X, Patel M S, Prezelj J, Rose L M, Scollen S, Siggeirsdottir K, Smith A V, Svensson O, Trompet S, Trummer O, van Schoor N M, Woo J, Zhu K, Balcells S, Brandi M L, Buckley B M, Cheng S, Christiansen C, Cooper C, Dedoussis G, Ford I, Frost M, Goltzman D, Gonzalez-Macias J, Kahonen M, Karlsson M, Khusnutdinova E, Koh J M, Kollia P, Langdahl B L, Leslie W D, Lips P, Ljunggren O, Lorenc R S, Marc J, Mellstrom D, Obermayer-Pietsch B, Olmos J M, Pettersson-Kymmer U, Reid D M, Riancho J A, Ridker P M, Rousseau F, Slagboom P E, Tang N L, Urreizti R, Van Hul W, Viikari J, Zarrabeitia M T, Aulchenko Y S, Castano-Betancourt M, Grundberg E, Herrera L, Ingvarsson T, Johannsdottir H, Kwan T, Li R, Luben R, Medina-Gomez C, Palsson S T, Reppe S, Rotter J I, Sigurdsson G, van Meurs J B, Verlaan D, Williams F M, Wood A R, Zhou Y, Gautvik K M, Pastinen T, Raychaudhuri S, Cauley J A, Chasman D I, Clark G R, Cummings S R, Danoy P, Dennison E M, Eastell R, Eisman J A, Gudnason V, Hofman A, Jackson R D, Jones G, Jukema J W, Khaw K T, Lehtimaki T, Liu Y, Lorentzon M, McCloskey E, Mitchell B D, Nandakumar K, Nicholson G C, Oostra B A, Peacock M, Pols H A, Prince R L, Raitakari O, Reid I R, Robbins J, Sambrook P N, Sham P C, Shuldiner A R, Tylavsky F A, van Duijn C M, Wareham N J, Cupples L A, Econs M J, Evans D M, Harris T B, Kung A W, Psaty B M, Reeve J, Spector T D, Streeten E A, Zillikens M C, Thorsteinsdottir U, Ohlsson C, Karasik D, Richards J B, Brown M A, Stefansson K, Uitterlinden A G, Ralston S H, Ioannidis J P, Kiel D P, Rivadeneira F. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. 2012;44(5):491–501. doi: 10.1038/ng.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hsu YH., Kiel DP. Clinical review: Genome-wide association studies of skeletal phenotypes: what we have learned and where we are headed. J Clin Endocrinol Metab. 2012;97(10):E1958–1977. doi: 10.1210/jc.2012-1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zheng H. F., Tobias J. H., Duncan E., Evans D. M., Eriksson J., Paternoster L., Yerges-Armstrong L. M., Lehtimaki T., Bergstrom U., Kahonen M., Leo P. J., Raitakari O., Laaksonen M., Nicholson G. C., Viikari J., Ladouceur M., Lyytikainen L. P., Medina-Gomez C., Rivadeneira F., Prince R. L., Sievanen H., Leslie W. D., Mellstrom D., Eisman J. A., Moverare-Skrtic S., Goltzman D., Hanley D. A., Jones G., St Pourcain B., Xiao Y., Timpson N. J., Smith G. D., Reid I. R., Ring S. M., Sambrook P. N., Karlsson M., Dennison E. M., Kemp J. P., Danoy P., Sayers A., Wilson S. G., Nethander M., McCloskey E., Vandenput L., Eastell R., Liu J., Spector T., Mitchell B. D., Streeten E. A., Brommage R., Pettersson-Kymmer U., Brown M. A., Ohlsson C., Richards J. B., Lorentzon M. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet. 2012;8(7) doi: 10.1371/journal.pgen.1002745. e1002745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garcia-Ibarbia C., Perez-Nunez M. I., Olmos J. M., Valero C., Perez-Aguilar M. D., Hernandez J. L., Zarrabeitia M. T., Gonzalez-Macias J., Riancho J. A. Missense polymorphisms of the WNT16 gene are associated with bone mass, hip geometry and fractures. Osteoporos Int. 2013;24(9):2449–2454. doi: 10.1007/s00198-013-2302-0. [DOI] [PubMed] [Google Scholar]
- 66.Li X., Ominsky M. S., Warmington K. S., Morony S., Gong J., Cao J., Gao Y., Shalhoub V., Tipton B., Haldankar R., Chen Q., Winters A., Boone T., Geng Z., Niu Q. T., Ke H. Z., Kostenuik P. J., Simonet W. S., Lacey D. L., Paszty C. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009;24(4):578–588. doi: 10.1359/jbmr.081206. [DOI] [PubMed] [Google Scholar]
- 67.Eddleston A., Marenzana M., Moore A. R., Stephens P., Muzylak M., Marshall D., Robinson M. K. A short treatment with an antibody to sclerostin can inhibit bone loss in an ongoing model of colitis. J Bone Miner Res. 2009;24(10):1662–1671. doi: 10.1359/jbmr.090403. [DOI] [PubMed] [Google Scholar]
- 68.Taut A. D., Jin Q., Chung J. H., Galindo-Moreno P., Yi E. S., Sugai J. V., Ke H. Z., Liu M., Giannobile W. V. Sclerostin antibody stimulates bone regeneration after experimental periodontitis. J Bone Miner Res. 2013;28(11):2347–2356. doi: 10.1002/jbmr.1984. [DOI] [PubMed] [Google Scholar]
- 69.Padhi D., Jang G., Stouch B., Fang L., Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26(1):19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 70.McClung M. R., Grauer A., Boonen S., Bolognese M. A., Brown J. P., Diez-Perez A., Langdahl B. L., Reginster J. Y., Zanchetta J. R., Wasserman S. M., Katz L., Maddox J., Yang Y. C., Libanati C., Bone H. G. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. 2014;370(5):412–420. doi: 10.1056/NEJMoa1305224. [DOI] [PubMed] [Google Scholar]
- 71.Cosman F., Crittenden D. B., Adachi J. D., Binkley N., Czerwinski E., Ferrari S., Hofbauer L. C., Lau E., Lewiecki E. M., Miyauchi A., Zerbini C. A., Milmont C. E., Chen L., Maddox J., Meisner P. D., Libanati C., Grauer A. Romosozumab Treatment in Postmenopausal Women with Osteoporosis. N Engl J Med. 2016;375(16):1532–1543. doi: 10.1056/NEJMoa1607948. [DOI] [PubMed] [Google Scholar]
- 72.Saag K. G., Petersen J., Brandi M. L., Karaplis A. C., Lorentzon M., Thomas T., Maddox J., Fan M., Meisner P. D., Grauer A. Romosozumab or Alendronate for Fracture Prevention in Women with Osteoporosis. N Engl J Med. 2017;377(15):1417–1427. doi: 10.1056/NEJMoa1708322. [DOI] [PubMed] [Google Scholar]
- 73.Ferrari S. L. Osteoporosis: Romosozumab to rebuild the foundations of bone strength. Rheumatology. 2018;14(3):128. doi: 10.1038/nrrheum.2018.5. Nature reviews. [DOI] [PubMed] [Google Scholar]
- 74.Khosla S. Bone diseases: Romosozumab - on track or derailed? Endocrinology. 2017;13(12):697–698. doi: 10.1038/nrendo.2017.136. Nature reviews. [DOI] [PMC free article] [PubMed] [Google Scholar]