

Abstract
Solution pH plays an important role in structure and dynamics of biomolecular systems; however, pH effects cannot be accurately accounted for in conventional molecular dynamics simulations based on fixed protonation states. Continuous constant pH molecular dynamics (CpHMD) based on the λ-dynamics framework calculates protonation states on the fly during dynamical simulation at a specified pH condition. Here we report the CPU-based implementation of the CpHMD method based on the GBNeck2 generalized Born (GB) implicit-solvent model in the pmemd engine of the Amber molecular dynamics package. The performance of the method was tested using pH replica-exchange titration simulations of Asp, Glu and His sidechains in 4 miniproteins and 7 enzymes with experimentally known pKa’s, some of which are significantly shifted from the model values. The added computational cost due to CpHMD titration ranges from 11 to 33% for the data set and scales roughly linearly as the ratio between the titrable sites and number of solute atoms. Comparison of the experimental and calculated pKa’s using 2 ns per replica sampling yielded a mean unsigned error of 0.65, a root-mean-squared error of 0.90, and a linear correlation coefficient of 0.78. While this level of accuracy is similar to the GBSW-based CpHMD in CHARMM, in contrast to the latter, the current implementation was able to reproduce the experimental orders of the pKa’s of the coupled carboxylic dyads. We quantified the sampling errors, which revealed that prolonged simulation is needed to converge pKa’s of several titratable groups involved in salt-bridge-like interactions or deeply buried in the protein interior. Our benchmark data demonstrate that GBNeck2-CpHMD is an attractive tool for protein pKa predictions.
Graphical Abstract
INTRODUCTION
Solution pH is an important environmental factor that modulates the energetics and conformational dynamics of biomolecular systems. Many biological processes, such as protein folding,1 enzyme catalysis2 and ion/substrate transport3 are controlled by pH. However, pH effects are largely overlooked in the molecular dynamics (MD) community, and MD simulations are performed with titratable sites fixed in the protonated or the deprotonated state based on the solution or model pKa values. Over the past decade, two types of techniques have been developed to control solution pH in MD simulations. In the discrete constant pH molecular dynamics (CpHMD) technique, which is also known as the stochastic titration method,4 molecular dynamics is periodically interrupted by Monte-Carlo sampling of protonation states. This technique, using implicit-, explicit- or mixed-solvent schemes, has been implemented in widely used MD packages, such as Amber,5,6 CHARMM7,8 and Gromacs.4 In the continuous CpHMD technique, which has roots in the λ-dynamics method for free energy calculations,9 a set of fictitious λ coordinates, whose end points represent the protonated and deprotonated states, are propagated at the same time as the spatial coordinates.10 The continuous CpHMD technique, with the aforementioned three solvent schemes, has been implemented in CHARMM10–15 and recently in Gromacs.16 The development and application of both discrete and continuous CpHMD techniques have been recently reviewed.17 Here we focus on continuous CpHMD and for convenience, we will drop the word continuous in the remainder of the paper.
The original CpHMD method, which was implemented in CHARMM, utilizes the generalized Born (GB) implicit-solvent model, Generalized Born using molecular volume (GBMV)18 or Generalized Born with simple switching (GBSW),19 to sample conformational and protonation states.10,11 Making use of the temperature replica-exchange protocol to enhance sampling and ensure convergence,20 the GBSW-based CpHMD method was applied to understand protein folding mechanisms1,21,22 and blind prediction of protein pKa values.23 The latter study, which used 500 ps per replica sampling at a single pH condition to blindly predict the abnormally shifted pKa’s of 87 titratable sites, showed that GBSW-CpHMD was one of the most accurate and robust pKa calculation tools.24 Nevertheless, in the post-prediction analysis23 and other studies12,25 two issues that affect the accuracy of pKa calculations were identified. First, the effective Born radii of buried atoms given by the GBSW model19 are relatively insensitive to the degree of burial, which resulted in a systematic underestimation of the desolvation energies and consequently absolute pKa shifts for deeply buried sites.23 Second, small errors in describing the conformational environment and dynamics by the GBSW model, for example, partial loss of native structure,12 overcompaction22 and reduced mobility of hydrophobic cluster,12,25 and inability to represent solvent-mediated ion-pair interactions due to the lack of solvent granularity,12 can lead to pKa shifts in either directions.12 These GBSW-related errors are sometimes compounded by the force field bias, for example, overstabilization of α helix in the folded state.21,22 Thus, it appears that the accuracy of GB-based CpHMD methods may significantly benefit from the improved GB models and force fields. Further development of GB-based CpHMD is particularly desirable for pKa predictions, as it has an unmatched convergence speed among all constant pH methods.12
Over the past decade, the GB models in Amber molecular dynamics package26 have undergone considerable development. In the GBNeck model by Mongan et al., a so-called neck correction was introduced to account for the finite size of water molecules.27 In the most recent GBNeck2 model by Nguyen et al., the parameter set of GBNeck was significantly improved to better reproduce Poisson-Boltzmann solvation free energies and more importantly, the experimental structure, stability, and salt-bridge profiles of model peptides.28 Excitingly, the GBNeck2 model, when used with the newest ff14SB force field (sidechain only version)29 folded 16 proteins of various sizes and topologies to the native structures.30 These GB simulations also took advantage of GPU acceleration,31 which provided trajectories of 1 μs per day per GPU.
Motivated by the above developments and the desire to offer the community an accurate and fast pKa prediction tool, we implemented the GBNeck2-based CpHMD method in Amber (abbreviated as GBNeck2-CpHMD henceforth). To accelerate the convergence of pKa estimates,12 the existing pH replica-exchange (pH-REX) method in Amber32 was adapted. Following a brief description of methods and implementation details, we report pKa calculations for 4 mini-proteins and 7 enzymes that exhibit large pKa shifts and have been previously used to benchmark the CHARMM CpHMD methods.12,20 The method scales with the number of CPUs and has low computational cost. The accuracy of the calculated pKa’s will be compared to the GBSW-CpHMD in CHARMM.12,20 A detailed analysis of the deviations between calculated and experimental pKa’s will be presented, with a special emphasis on residues deeply buried in the protein interior and those involved in salt-bridge-like interactions, as they are the most challenging sites for accurate pKa predictions and showed the largest errors in the CHARMM CpHMD methods.12,20 We will also quantify sampling errors and compare results obtained with different force fields and solvent models before drawing conclusions.
METHODS AND IMPLEMENTATION
Continuous constant pH molecular dynamics
Based on the λ dynamics method for free energy calculations,9 the continuous CpHMD method makes use of an extended Hamiltonian to simultaneously propagate the atomic positions and the coordinates of the fictitious λ particles representing protonation/deprotonation of titratable sites.10 Expressing λ as sin2θ, the λ coordinate is continuous and bound between 0 and 1. Thus, the discretized version of λ can be used to represent the protonation state.
| (1) |
where cut1 and cut0 are the two cutoffs chosen to be 0.8 and 0.2, respectively. The total Hamiltionian that accounts for a set of deprotonation equilibria of ionizable sidechains in proteins, , becomes
| (2) |
In the above equation, the first and forth terms express the kinetic energies of the real atoms and fictitious λ particles, respectively. The second term represents the λ-independent bonded energies. The third term represents the λ-dependent electrostatic and van der Waals energies. For electrostatics, the atomic partial charges on a titratable site are linearly interpolated between the protonated (λ=0) and deprotonated (λ=1) states,
| (3) |
which enables the λ-dependent modulation of electrostatic energies, including the Coulomb energy as well as the GB solvation free energy,
| (4) |
Here rab is the distance between atom a and atom b, qa, qb are the respective partial charges, αa, αb are the respective effective Born radii, ϵp (1), ϵw (80) are the dielectric constants for the protein and water, respectively. κ is the inverse Debye length, κ2 = 8πq2I/ekBT, where I is the ionic strength, q is the charge of the salt ion, e is the elementary charge unit, and kB is the Boltzmann constant. Similarly, the van der Waals interactions involving titratable hydrogens are linearly scaled by λ.
The final term in Eq. 2 contains three biasing potential energies dependent on λ only,
| (5) |
Here Umod is a potential of mean force (PMF) function for titrating a model compound in solution, typically a blocked single amino acid. Subtraction of Umod from the total Hamiltonian allows the calculation of the relative deprotonation free energy of a titratable site in the protein environment (ΔGprotein) with respect to solution (ΔGmod). Umod is a quadratic function of λ
| (6) |
where Aj and Bj are the two parameters which can be determined using a fitting procedure (see Simulation Protocol).
The next biasing potential UpH allows the calculation of the deprotonation free energy change due to a change in solution pH,
| (7) |
where kB is Boltzmann constant, T is temperature, and pKamod is the model pKa, which can be taken from experiment. UpH(λj) is zero in the CpHMD simulation of a model compound at a solution pH identical to the model pKa, i.e., the protonated and deprotonated states are sampled with equal probability.
The last biasing potential Ubarr is a harmonic potential designed to decrease the probability of λ in the unphysical intermediate state.
| (8) |
where β is the height of the energy barrier. Finally, we note that the equations presented are used for single-site titration, e.g., Lys or Cys. The formalism for double-site titration, e.g., carboxyl and histidine residues can be found in ref.11
Implementation details
A new module (phmd) that implements the GB-based continuous CpHMD method10,11 was added in the pmemd molecular dynamics engine in Amber.26 Currently, the GBNeck2 model is used for the propagation of both spatial and titration coordinates, but other GB models can be readily adopted. The pH replica-exchange method implemented for the Monte Carlo based discrete constant pH method32 was adapted to work with CpHMD.
To mimic protonation/deprotonation events, CpHMD methods adopt a single topology approach, i.e., dummy hydrogens attached to the titration sites lose their partial charges and van der Waals interactions upon deprotonation. Additionally, a protonation state change is represented by the switch between two sets of sidechain atomic charges. With the CHARMM force fields, the implementation is straightforward, as only the sidechain charges are affected by the change in protonation state. However, with the Amber force fields, the backbone charges are dependent on the sidechain protonation state, which coupled with the fact that the backbone atoms can interact with sidechain atoms of neighboring residues due to 1–4 electrostatic interactions, presents a problem for CpHMD methods (both discrete and continuous) that rely on a single reference state.5 To circumvent this problem, a pragmatic approach similar to that used by Mongan et al.5 was adopted: the partial charges on the backbone are fixed to the values of a single protonation state (charged Asp/Glu and neutral His), and the residual charge (ranging from 0.10 to 0.14 e for Asp, Glu, and His) is absorbed onto the C-β atom. In order to avoid introducing potential artifacts to conformational dynamics, such a scheme is only deployed for titration dynamics. For conformational dynamics, the partial charges are unmodified and the charge interpolation between the protonated and deprotonated states is made to both backbone and sidechain atoms.
The dummy hydrogens on Asp and Glu require special attention. The dummy hydrogen can be on the same (syn) or opposite (anti) sides of the second carboxyl oxygen. Previous work showed that once an uncharged dummy hydrogen rotates to the latter position, it can no longer gain charge, i.e., titrate.5,10 Thus, following the CHARMM implementation,11 the dummy hydrogens were placed in the syn position at the beginning of the simulation, and the rotation barrier around the C-O bond was raised to 6 kcal/mol, to prevent the transition to the anti position. Although fixing the dummy hydrogens to the syn position is justified by its stability over the anti position based on the NMR experiment and quantum calculations of carboxylic acids,11 it is a topic that deserves further examination in the future.
In contrast to the CHARMM GB models (e.g., GBSW), hydrogen atoms have nonzero intrinsic Born radii in the Amber GB models. In contrast to the CHARMM GB models (e.g., GBSW), hydrogen atoms have nonzero intrinsic Born radii in the Amber GB models. As such, the intrinsic Born radius of the titratable hydrogen on a carboxylic group contributes to the solvation free energy of the protonated state. Consequently, the derivative of UGB (Eq. 4) with respect to λ should contain a term to account for the change in the effective Born radii due to protonation. The contribution of such a term is expected to be much smaller than that due to the λ-dependent charge perturbation. Additionally, the implementation would be computationally inefficient, given the complexity of how the implicit Born radii enter into the GB solvation free energy. Thus, we made the following approximation. The contributions of the titratable hydrogens of carboxylic residues (e.g., Asp and Glu) to the Born radii were excluded from the calculation in both the spatial and titration dynamics, and the intrinsic Born radii of the carboxyl oxygens were set to 1.4 Å regardless of the protonation state. For histidines, the intrinsic Born radius of the imidazole hydrogens was set to 1.17 Å regardless of the protonation state. Note, the default radius for the imidazole hydrogens is 1.3 Å. We reduced it to compensate for the overstabilized salt-bridge-like interactions, following the radius reduction of the guanodinium hydrogens on arginines by Nguyen, Roe, and Simmerling.28 Further details on the implementation and usage are available in Amber18 Reference Manual (http://ambermd.org/).
SIMULATION PROTOCOL
Structure preparation.
Blocked Asp, Glu and His (ACE-X-NH2) were built using the LEaP facility in Amber.26 The crystal structures or NMR models of the following proteins were retrieved from Protein Data Bank: the 36-residue villin headpiece subdomain (HP36, pdbid 1VII),33 45-residue binding domain of 2-oxoglutarate dehydrogenase multienzyme complex (BBL, pdbid 1W4H),34 56-residue N-terminal domain of ribosomal protein L9 (NTL9, pdbid 1CQU),35 56-residue turkey ovomucoid third domain (OMTKY, pdbid 1OMU),36 105-residue reduced form of human thioredoxin (pdbid 1ERT),37 129-residue hen egg-white lysozyme (HEWL, pdbid 2LZT),38 143-residue hyperstable Δ+PHS variant of staphylococcal nuclease (SNase, pdbid 3BDC),39 124-residue ribonuclease A (RNase A, pdbid 7RSA),40 155-residue E. coli ribonuclease H (RNase HI, pdbid 2RN2),41 185-residue oxidized form of Bacillus circulans xylanase (xylanase, pdbid 1BCX),42 and 389-residue unbound β-secretase 1 catalytic domain (BACE1, pdbid 1SGZ). If applicable, the first chain in the crystal structure or the first entry in the NMR models was used; the hydrogen atoms as well as crystal waters were removed.
The following steps were performed with CHARMM (version c42a1).43 The PDB structures were acetylated at N terminus and amidated at C terminus. Disulfide bonds were added as needed. Missing hydrogens were added with protonated His/Lys/Arg/Cys/Tyr and deprotonated Asp/Glu sidechains. The hydrogen positions were relaxed using 50 steps of steepest descent energy minimization in GBSW implicit solvent19 (default setting, see CHARMM documentation) where a harmonic force constant of 50 kcal/mol/Å2 was placed on the heavy atoms. Next, dummy hydrogens in syn orientation were added to Asp/Glu sidechains, followed by 10 steps of steepest descent and 10 steps of adopted basis Newton Raphson minimization in GB solvent. The resulting pdb files were converted into Amber parameter and coordinate files in LEaP.
Derivation of model parameters.
Following previous work,11 the average forces ⟨∂U/∂θ⟩ at fixed θx and ⟨∂U/∂θx⟩ at fixed θ were obtained for blocked model compounds, Ace-X-NH2, where X=Asp, Glu and His, using GB simulations of 5 ns per window. The ionic strength was 0.1 M in accordance with the experimental model pKa determination.44 For Asp/Glu, θ/θx values of 0.2, 0.4, 0.7854, 1.0, 1.2 and 1.4 were used, corresponding to λ/x values of 0.04, 0.15, 0.5, 0.71, 0.87 and 0.97, respectively. ⟨∂U/∂θ⟩ and ⟨dU/dθx⟩ values were fit to the model potential of mean force (PMF) function, Umodel(λ, x) = (a1λ2+a2λ+a3)(x+a4)2+a5λ2+a6λ, where a1, ...a6 are the fitting parameters. For His, fewer data points were needed.11 ⟨∂U/∂θ⟩ was determined with θx fixed at 0 and 1.57 (x = 0 or 1), and ⟨∂U/∂θx⟩ was determined with θ fixed at 1.57 (λ = 1). The force values were then fit to the following PMF function with five parameters: Umodel(λ, x) = λ2(a1×2 + a2x + a3) + λ(a4x + a5).
Molecular dynamics protocol.
Molecular dynamics was performed using the modified version of the pmemd molecular dynamics engine in Amber.26 Proteins were represented by the ff14SB Amber force field.29 Solvent was represented by the GBNeck2 implicit-solvent model (igb=8) with mbondi3 intrinsic Born radii.28 A 2-fs time step was used with bonds involving hydrogen atoms constrained with the SHAKE algorithm.45 In the GB calculations, a 0.15 M ionic strength was used.
pH replica-exchange CpHMD titration of blocked amino acids and pentapeptides
Five sets of titration simulations were run for each model compound and pentapeptide. Each REX simulation contained six pH replicas, placed in the pH range encompassing 1.5 pH unit below and 1 pH unit above the model pKa, with 0.5-pH unit interval. Exchanges were attempted every 1000 MD steps (2 ps), and each replica was run for 1 ns. The model pKa’s from Thurlkill et al. were used (Table 1).
Table 1:
Calculated pKa’s of blocked model compounds and alanine pentapeptides in solution
| Residue | Compounda | Peptidea | Thurlkillb | Nozakic | Castanẽdad |
|---|---|---|---|---|---|
| Asp | 3.7±0.07 | 3.8±0.05 | 3.67±0.04 | 4.0 | 3.90±0.01 |
| Glu | 4.2±0.11 | 4.1±0.05 | 4.25±0.05 | 4.4 | 4.36±0.01 |
| His | 6.4±0.03 | 6.4±0.03 | 6.54±0.04 | 6.3 | n/d |
| HID | 7.0±0.04 | 7.0±0.05 | n/d | 6.92e | n/d |
| HIE | 6.5±0.05 | 6.5 0.02 | n/d | 6.53e | n/d |
Error bars of the calculated pKa’s are the standard deviations based on five independent sets of REX titration runs, each lasting 1 ns per replica. Blocked model compound refers to Ac- X-NH2, while pentapeptide refers to Ac-Ala-Ala-X-Ala-Ala-NH2, where X is the titratable residue Asp, Glu or His.
pKa’s of pentapeptides in 0.1 M salt solution determined by Thurlkill et al. using NMR.44 These pKa’s were used as the model pKa’s in our simulations.
pKa’s of blocked amino acids given by Nozaki and Tanford46 using potentiometric titration. Ionic strength is unclear.
pKa’s of tripeptides Ac-Ala-X-Ala-NH2 in 0.1 M salt solution determined by Castañeda et al. using NMR.39
HID refers to the titration HID ⇌ HIP. HIE refers to the titration HIP ⇌ HIE.
pKa’s of model compounds measured by Tanokura using NMR titration.47
pH replica-exchange CpHMD titration of proteins.
One set of pH REX simulation was run for each protein. For proteins containing only acidic residues (HP36 and NTL9), the replicas were placed in the pH range 0–7.5 with 0.5-pH unit intervals. For other proteins, the replicas were placed in the pH range 0–9.5, with the same pH interval. Exchanges were attempted every 250 steps (0.5 ps), and each replica was run for 2 ns (unless otherwise stated).
pKa calculations and error analysis
We first calculated the unprotonated fractions (S) using the following definition: λ < 0.2 for the protonated, and λ > 0.8 for the deprotonated state. The residue-specific pKa’s were calculated by fitting S at the simulation pH conditions to the Hill equation,
| (9) |
where n is the Hill coefficient. Note, the pKa’s are insensitive to the cutoffs used to defined the protonation states. Tests using λ < 0.1 for the protonated and λ > 0.9 for the deprotonated state resulted in the same pKa’s.
There are a number of ways to estimate the errors in the calculated pKa’s. Here we present the one based on error propagation from the errors in the unprotonated fractions at different pH, δSi.
| (10) |
where index i refers to a pH condition. The derivation for ∂pKa/∂Si is presented in SI. δSi can be calculated using the block standard error formula
| (11) |
where σn is the standard deviation of Si, N is the total number of data points in the time series of λ, and n is the number of data points in each block. We chose the minimum block length to be the correlation time τ, which can be estimated using the autocorrelation function (ACF),
| (12) |
where ⟨. . . ⟩ indicates an average over all Δt intervals considered, Δt is the lag time, is the discretized version of λ for representing the protonation state (Eq. 1). The ACF tends toward zero, as Δt increases and looses memory. τ was estimated as the value of t for which the ACF first crosses zero. We note, this definition worked well for most residues, but not so for residues that have S values that are not converged (see later discussion).
RESULTS AND DISCUSSION
Titration of model compounds and alanine pentapeptides
To verify the accuracy of the model PMF parameters, titration simulations were performed on two sets of model systems, the blocked amino acids (model compounds), Ac-X-NH2, and alanine pentapeptides, Ac-Ala-Ala-X-Ala-Ala-NH2, in solution. If the model PMF parameters were accurate, the input model pKa’s would be reproduced exactly. We also expect identical pKa’s for the blocked amino acid and pentapeptide containing the same titratable group, as it is completely exposed to solvent. Table 1 lists the calculated pKa’s of Asp, Glu, and His in the two model systems and the experimental values reported by Thurlkill et al. (pentapeptides44), which were used as the input model values in the current work. Table 1 also gives the old reference set reported by Nozaki and Tanford,46 which were used in the CpHMD implementations in CHARMM,12,15,20 and the pKa’s measured by Castañeda et al. for Asp and Glu in alanine tripeptides Ac-Ala-X-Ala-NH2.39 The calculated pKa’s, based on five independent sets of pH-REX CpHMD titration simulations (1 ns per replica), show a standard deviation of about 0.05. The deviation between the pKa’s in the model compound and pentapeptide is below 0.1 pH units. Thus, in addition to the convergence of the unprotonated fractions and individual pKa’s (Fig. S1–S3), these data offer assurance that the sampling for the model systems is complete. The calculated pKa’s are within 0.1 pH units from the intended model values (from Thurlkill et al.44), indicating that the model PMF functions are accurate. We note that the deviations are smaller than the differences (up to 0.2–0.3 pH units) among the experimental values by Nozaki, Thurlkill, and Castañeda.
Overall convergence and CPU scaling for protein titration
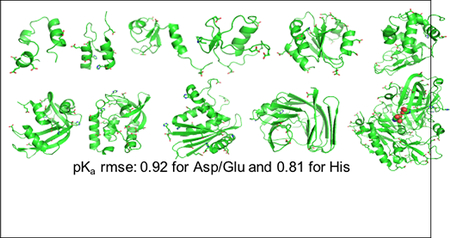
To benchmark the performance of the current implementation, we carried out titration simulations on 4 small proteins and 7 enzymes which show large pKa’s shifts relative to the model values (Fig. 1), making them ideal test systems for evaluating the accuracy of pKa calculations.48 Except for BACE1, these proteins have been previously used to benchmark the GBSW-CpHMD method in CHARMM.12,20 We titrated BACE1, which is a much larger enzyme and contains a hydrogen-bonded catalytic aspartic dyad, for which the pKa’s are experimentally known but very challenging to predict.2,49
Figure 1: Proteins used in the current benchmark set.

aa/titr refers to the number of amino acids/number of titratable groups. Titratable groups include Asp, Glu and His sidechains shown in the stick model. mue refers to the mean unsigned error with respect to the experimental pKa’s. For BACE1, mue calculation includes only the pKa’s of the catalytic dyad (highlighted in the van der Waals representation), as they were the only ones with experimental data.
We first examine the convergence behavior. Insufficient sampling and poor convergence of pKa’s could lead to poor fitting (of fraction of deprotonation vs. pH) to the generalized Henderson-Hasselbalch (or Hill) equation.5,20,32,50 No obviously bad fitting could be spotted in the titration plots (Fig. S4–S13). We then compared the pKa’s from the 2-ns and 1-ns simulation times (Table 3). The differences are all within 0.1 pH units, except for five residues showing pKa changes around 0.3 units (see later discussion). We also examined the time trace of the fraction of deprotonation S at all pH conditions. Except for the aforementioned five residues showing continued drift in the S values, all S curves converge within 1 ns (Fig. S14–S24). These data demonstrate that, for most residues, the pKa’s converge with 1 ns sampling per replica, which is on par with the GBSW-based CpHMD in CHARMM (note, the early data were obtained using the temperature-based replica exchange).12,20 We will come back to the pKa’s that are not converged.
Table 3:
Calculated and experimental pKa’s for 4 small proteins and 7 enzymes
| Residue | Expt | Calc | Residue | Expt | Calc | Residue | Expt | Calc | Residue | Expt | Calc |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BBL | HP36 | OMTKY | HEWL | ||||||||
| Asp129 | 3.9 | 2.8 (2.8) | Asp44 | 3.1 | 2.1 (2.1) | Asp8 | 2.7 | 2.5 (2.3) | Glu7 | 2.6 | 3.5 (3.5) |
| Glu141 | 4.5 | 4.1 (4.1) | Glu45 | 4.0 | 3.7 (3.7) | Glu11 | 4.1 | 3.9 (3.9) | His15 | 5.5 | 6.5 (6.6) |
| His142 | 6.5 | 6.9 (7.0) | Asp46 | 3.5 | 3.6 (3.5) | Glu20 | 3.2 | 3.7 (3.7) | Asp18 | 2.8 | 1.1 (1.1) |
| Asp145 | 3.7 | 2.6*(2.2) | Glu72 | 4.4 | 4.1 (4.2) | Asp28 | 2.3 | 3.6 (3.6) | Glu35 | 6.1 | 4.6 (4.7) |
| Glu161 | 3.7 | 3.3 (3.3) | Glu44 | 4.8 | 4.6 (4.6) | Asp48 | 1.4 | 1.8 (1.6) | |||
| Asp162 | 3.2 | 3.2 (3.2) | RNase A | His53 | 7.5 | 6.6 (6.7) | Asp52 | 3.6 | 3.3 (3.2) | ||
| Glu164 | 4.5 | 4.0 (4.1) | Glu2 | 2.8 | 3.2 (3.2) | Asp66 | 1.2 | 2.1 (2.2) | |||
| His166 | 5.4 | 6.0 (6.0) | Glu9 | 4.0 | 3.4 (3.3) | RNase H | Asp87 | 2.2 | 1.8*(1.4) | ||
| His12 | 6.2 | 6.4*(6.8) | Glu6 | 4.5 | 4.3*(4.1) | Asp101 | 4.5 | 4.8 (4.9) | |||
| NTL9 | Asp14 | 2.0 | 2.4 (2.3) | Asp10 | 6.1 | 3.4 (3.4) | Asp119 | 3.5 | 2.4 (2.2) | ||
| Asp8 | 3.0 | 2.1 (2.1) | Asp38 | 3.5 | 2.8 (2.9) | Glu32 | 3.6 | 3.2 (3.2) | |||
| Glu17 | 3.6 | 3.4 (3.5) | His48 | 6.0 | 7.2 (7.1) | Glu48 | 4.4 | 2.5 (2.5) | SNase | ||
| Asp23 | 3.1 | 2.9 (3.0) | Glu49 | 4.7 | 2.6 (2.4) | Glu57 | 3.2 | 4.1 (4.0) | His8 | 6.5 | 6.5 (6.5) |
| Glu38 | 4.0 | 3.6 (3.5) | Asp53 | 3.9 | 4.3 (4.2) | Glu61 | 3.9 | 2.8 (2.8) | Glu10 | 2.8 | 3.7 (3.7) |
| Glu48 | 4.2 | 3.8 (3.8) | Asp83 | 3.5 | 2.9 (2.9) | His62 | 7.0 | 6.9 (6.9) | Asp19 | 2.2 | 2.3 (2.0) |
| Glu54 | 4.2 | 3.8 (3.8) | Glu86 | 4.1 | 3.5 (3.4) | Glu64 | 4.4 | 3.1 (2.9) | Asp21 | 6.5 | 3.7 (4.0) |
| His105 | 6.7 | 6.3 (6.3) | Asp70 | 2.6 | 4.1 (4.1) | Asp40 | 3.9 | 2.8 (2.8) | |||
| Thioredoxin | Glu111 | 3.5 | 3.5 (3.5) | His83 | 5.5 | 6.2 (6.0) | Glu43 | 4.3 | 3.7 (3.9) | ||
| Glu6 | 4.8 | 3.9 (4.0) | His119 | 6.1 | 6.1 (6.0) | Asp94 | 3.2 | 3.2 (3.1) | Glu52 | 3.9 | 3.9 (3.9) |
| Glu13 | 4.4 | 4.4 (4.4) | Asp121 | 3.1 | 3.5 (3.5) | Asp102 | <2.0 | 3.4 (3.2) | Glu57 | 3.5 | 3.4 (3.4) |
| Asp16 | 4.0 | 4.0 (4.0) | Asp108 | 3.2 | 3.1 (3.0) | Glu67 | 3.8 | 4.5 (4.6) | |||
| Asp20 | 3.8 | 2.9 (2.9) | Xylanase | His114 | <5.0 | 7.0 (6.9) | Glu63 | 3.3 | 3.9 (3.9) | ||
| Asp26 | 9.9 | 6.2 (6.3) | Asp5 | 3.0 | 3.3 (3.1) | Glu119 | 4.1 | 3.9 (3.8) | Glu75 | 3.3 | 2.6 (2.5) |
| His43 | n/d | 6.1 (6.1) | Asp12 | 2.5 | 2.5 (2.4) | His124 | 7.1 | 6.2 (6.2) | Asp77 | <2.2 | 1.9 (1.6) |
| Glu47 | 4.1 | 4.3 (4.5) | Glu79 | 4.6 | 5.1*(5.4) | His127 | 7.9 | 6.6 (6.8) | Asp83 | <2.2 | 2.1*(1.8) |
| Glu56 | 3.3 | 4.5 (4.6) | Asp84 | <2.0 | 3.4 (3.3) | Glu129 | 3.6 | 4.6 (4.6) | Asp95 | 2.2 | 4.3 (4.3) |
| Asp58 | 5.3 | 3.8 (3.6) | Asp102 | <2.0 | 3.3 (3.4) | Glu131 | 4.3 | 4.3 (4.4) | Glu101 | 3.8 | 3.5 (3.8) |
| Asp60 | 2.8 | 3.6 (3.6) | Asp107 | 2.7 | 3.1 (3.2) | Asp134 | 4.1 | 4.3 (4.2) | His121 | 5.2 | 6.8 (6.9) |
| Asp61 | 4.2 | 4.6 (4.5) | Asp120 | 3.2 | 4.0 (4.0) | Glu135 | 4.3 | 4.2 (4.3) | Glu122 | 3.9 | 3.0 (2.9) |
| Asp64 | 3.2 | 3.1 (3.3) | Asp122 | 3.6 | 3.4 (3.6) | Glu147 | 4.2 | 3.9 (3.8) | Glu129 | 3.8 | 4.5 (4.5) |
| Glu68 | 4.9 | 4.3 (4.3) | His150 | <2.3 | 4.8 (4.8) | Asp148 | <2.0 | 2.4 (2.1) | Glu135 | 3.8 | 4.2 (4.1) |
| Glu70 | 4.6 | 5.0 (5.0) | His157 | 6.5 | 7.3 (7.3) | Glu154 | 4.4 | 3.8 (3.8) | |||
| Glu88 | 3.7 | 3.8 (3.8) | Glu173 | 6.7 | 7.0 (6.8) | ||||||
| Glu95 | 4.1 | 3.5 (3.5) | BACE1 | max | 3.7 | ||||||
| Glu98 | 3.9 | 3.9 (3.8) | Asp32 | 5.2 | 3.9 (3.9) | mue | 0.65 | ||||
| Glu103 | 4.4 | 4.7 (4.6) | Asp228 | 3.5 | 2.7 (2.8) | rmse | 0.90 |
In parentheses are the pKa’s calculated from the first half of the simulations (1 ns per replica). pKa’s with large uncertainties are indicated by an asterisk. Experimental data were obtained from the NMR titration of HP36,52 BBL,53,54 NTL9,55 OMTKY,56,57 thioredoxin,58 HEWL,59 SNase39,60 RNase A,61 RNase H,62,63 xylanase,64,65 and BACE1.66 The maximum unsigned error (max), mean unsigned error (mue), and root-mean-square error (rmse) are listed. BACE1 pKa’s are excluded to facilitate comparison with CHARMM (see Fig. 3 and Table 2).
For CpHMD to become a practical tool, it must scale well with number of processors and not have high computational overhead. Since the major CPU time for CpHMD titration is spent on evaluating the forces on λ particles, which involves calculations of the interactions between the real atoms and λ particles, it should increase with the number of atoms multiplied by the number of titratable sites, Natm × Ntitr. This is indeed the case for the 11 proteins that were titrated here, which span a range of 35–389 residues with 4–50 titration sites (Fig. 2a). Thus, CpHMD titration scales linearly with the system size, with a prefactor which is the number of titration sites.
Figure 2: Timing statistics of the current GBNeck2-CpHMD implementation.

a) CPU time for CpHMD titration scales linearly as the product of number of atoms (Natm) and number of titratable sites (Ntitr). The regression line is shown in green. For clarity, the data point for BACE1 is hidden. b) Percentage CPU time for the CpHMD titration relative to the GB calculation scales approximately linearly as the ratio between Ntitr and . Timing was based on the 5-ps single pH CpHMD runs using 2 cores on a workstation.
Now we come to the added CPU cost due to CpHMD titration. Given the above scaling behavior and the fact that the current GB calculation scales approximately as (slightly better than the expected quadratic scaling), we reasoned that the percentage CPU time used by CpHMD titration relative to the GB calculations should increase linearly as the ratio between Ntitr and . Regression of the actual CPU usage data returned a correlation coefficient of 0.88, thus confirming this conjecture (Fig. 2b). Thus, the percentage CPU usage for CpHMD titration scales approximately linearly as the fraction of titratable atoms. For the current data set, the CPU cost is 11– 33% of the GB calculations.
Overall accuracy of the protein pKa calculations
To assess the accuracy of the pKa calculations, we compare the calculated and experimental pKa’s as well as their pKa shifts with respect to Thurlkill’s model values. The pKa comparison returns a mean unsigned error (mue) of 0.70, a root-mean-square error (rmse) of 0.91, and a linear correlation coefficient R of 0.78 (Fig. 3a). Comparing the calculated and experimental pKa shifts yields an R of 0.56 (Fig. 3b). The worse correlation for the latter confirms that predicting pKa shifts is a more stringent test of pKa prediction techniques, as pointed out by Warshel and coworkers two decades ago.48
Figure 3: Comparison of the pKa calculations using the current GBNeck2-CpHMD in Amber and GBSW-CpHMD in CHARMM.

Left panel. Correlation between the experimental and calculated pKa’s from the Amber (a) and previously published CHARMM (d) CpHMD simulations. Middle panel. Correlation between the experimental and calculated pKa shifts from the Amber (b) and CHARMM (e) CpHMD simulations. Model pKa’s obtained by Thulkill et al. were used for the Amber CpHMD and those reported by Nozaki and Tanford were used for the CHARMM CpHMD simulations (Table 1). Data for Asp/Glu are shown in red while those for His are shown in blue. For clarity, the data points for Asp99 in thioredoxin are hidden in a, b, d, and e. The linear correlation coefficients R for the entire data set are given. The best fit lines are shown in green. The black lines indicate y = x. Right panel. (c) and (f): Histograms for the deviations between the calculated and experimental pKa’s for Asp/Glu (red) and His (blue). Bin size is 0.2 pH units. The histograms for His are inverted for better viewing. The CHARMM data were generated using the c22 force field51 and 1 ns per replica. For the CHARMM data, pH replica-exchange was used for HP36, BBL, NLT9, HEWL and SNase,12 and temperature replica-exchange was used for OMTKY, RNase A, RNase H, xylanase and thioredoxin.20
Examining the calculation errors for carboxylic groups and histidines separately is instructive. For the carboxylic groups, the mue, rmse, and R are 0.65, 0.92, and 0.62 respectively, while for His, they are 0.67, 0.81, and 0.16, respectively (Table 2). Although the rmse for histidines is only slightly lower than that for the carboxylic groups, the correlation is much worse, which may be related to the smaller range of the pKa values (about 3 for His vs. over 7 for carboxylic groups). We plotted the histograms of the pKa errors (Fig. 3c). For carboxylic groups, the highest peak is at zero, and the histogram is slightly skewed to the right, indicating a slight systematic overestimation of Asp/Glu pKa’s. For histidines, the peak is at zero; the error has a slightly broader range but no obvious skewness; however, no reliable conclusion can be drawn here due to the very limited data set.
Table 2:
Comparison of the pKa calculations for carboxylic groups and histidines with GBNeck2-CpHMD in Amber and GBSW-CpHMD in CHARMMa
| Amber | CHARMM | ||||||
|---|---|---|---|---|---|---|---|
| residue | N | mue | rmse | R | mue | rmse | R |
| Asp/Glu | 98 | 0.65 | 0.92 | 0.62 | 0.59 | 0.86 | 0.68 |
| His | 15 | 0.67 | 0.81 | 0.16 | 1.1 | 1.3 | 0.00 |
Listed are the total number of residues (N), the mean unsigned error (mue), root-mean- square error (rmse), and linear correlation coefficient (R) from comparing the calculated and experimental pKa’s.
The overall rmse and correlation with respect to the experimental pKa’s and pKa shifts of the current implementation are similar to that of the GBSW-based CpHMD in CHARMM (Fig. 3b and e). For carboxylic groups, GBSW-CpHMD gives a slightly better correlation (R of 0.68 vs. 0.62) and slightly smaller error (rmse of 0.86 vs. 0.92, see Table 2). The histograms of the pKa errors for carboxylic groups are clearly left skewed (Fig. 3c and f). For histidines, GBSW-CpHMD gives much larger errors (rmse of 1.3 vs. 0.81, Table 2), and there is no correlation between calculation and experiment (R of 0.0 vs. 0.16, Table 2), although the latter conclusion may not be reliable due to the small number of histidine pKa’s.
A useful application of a pKa calculation tool is to predict the acid and nucleophile components in catalytic dyads.49 For this purpose, the pKa order is the quantity of interest. The current GBNeck2-CpHMD correctly predicted the pKa order for Glu35/Asp52 in HEWL, Asp19/Asp21 in SNase, and Asp32/Asp228 in BACE1, although the pKa splitting is consistently smaller than experiment. In contrast, the GBSW-CpHMD in CHARMM cannot distinguish the pKa’s of coupled dyads.12 The latter was attributed to the distortion of the conformational environment of the buried dyad residues.12 Our previous work showed that the hybrid-solvent CpHMD in CHARMM, which uses explicit solvent for conformational sampling, can accurately predict the pKa splitting of the dyad residues because it more faithfully captures the protein conformational dynamics.12 Specifically, these simulations demonstrated that the nucleophile forms more hydrogen bonds than the proton donor (acid), while having similar or slightly higher solvent exposure.49 Remarkably, these local conformational features are reproduced in the GBNeck2-CpHMD simulations (Fig. S26), allowing better predictions than the GBSW-CpHMD in CHARMM.
Correlating the pKa errors with the degree of solvent exposure
The pKa’s of buried residues are the most challenging to predict.24 Previous work showed that pKa shifts due to desolvation penalty are insufficiently reproduced in the GBSW-based CpHMD.11,20,23 As a result, the pKa’s of buried carboxylic groups and histidines are systematically under- and overestimated, respectively.23 To determine whether desolvation is also underestimated in GBNeck2 and if so, whether it is a major source of pKa errors in GBNeck2-CpHMD titration, we plotted the pKa errors against fSASA, the ratio between the solvent accessible surface area of the titratable site in the protein and in solution. To isolate the error in estimating the desolvation penalty, we excluded the residues that have the carboxylic oxygen or histidine nitrogen within a 5 Å distance from the carboxylic oxygen in Asp/Glu or nitrogen in His/Lys/Arg in the crystal structure; the pKa’s of these residues may be subject to another source of error, namely, inaccurate representation of strong electrostatic interactions.12,23
Fig. 4a shows that the pKa errors of carboxylic groups not involved in strong electrostatic interactions are mostly negative. As fSASA decreases, there is a tendency for the error to become larger in magnitude (linear correlation coefficient of 0.54). The two largest errors (−3.7 for Asp26 of thioredoxin and −1.9 for Glu48 of RNase H) are associated with fSASA of 8% and 28%, respectively. These data suggest that the desolvation penalties of buried, and particularly deeply buried carboxylic groups are underestimated in the GBNeck2 model, consistent with the fact that the effective Born radii for these buried sites are too small as compared to the Poisson-Boltzmann radii.27 Performing the same analysis for histidines, we found no clear correlation between fSASA and pKa error, although the data set may be too small to draw a reliable conclusion (Fig. 4b).
Figure 4: Correlating the pKa errors with the degree of solvent exposure.

Upper panel. pKa errors vs. fSASA for carboxylic groups (a) and histidines (b) not involved in strong electrostatic interactions. fSASA refers to the ratio between the solvent accessible surface area (SASA) of the sidechain in protein and in the model. The average fSASA over all pH conditions are used. To guide the eye, a green regression line is drawn for the data in a). The correlation coefficient R is given. For Asp/Glu, SASA includes the carboxylic group (COO) and dummy hydrogens. For His, SASA includes all imidazole ring atoms. Lower panel. pKa errors vs. fSASA for carboxylic groups (c) and histidines (d) involved in strong electrostatic interactions. In all plots, data for carboxylic groups are colored red, and those for histidines are colored blue.
Residues involved in strong attractive electrostatic interactions
Overstabilization of salt-bridge interactions, a known limitation of the GB models,67 was a major source of error for GBSW-CpHMD in CHARMM.11,12,20 Thus, we examined the pKa errors of residues involved in strong electrostatic interactions, which, for simplicity, were defined as those with a distance below 5 Å between a carboxylic oxygen and nitrogen in His/Lys/Arg in the crystal structure. We divide the residues into two groups based on their fSASA values. For those with fSASA≥50%, the pKa errors are more or less symmetrically distributed around zero (Fig. 4c), suggesting no systematic error. Since the desolvation effect is small for these groups, it indicates that solvent-exposed salt bridges involving carboxylic groups are not overly strong, which is not surprising, as in GBNeck2, the intrinsic Born radii for Asp/Glu and Lys/Arg were optimized to closely match the explicit-solvent salt-bridge profiles.28
We next examine the carboxylic groups with fSASA less than 50%, for which the pKa’s are subject to both desolvation and salt-bridge-like interactions. Both factors, desolvation, which shifts the pKa up, and salt-bridge-like interactions, which shift the pKa down, may be underestimated, leading to error compensation, although the extent of the compensation is unclear. We note the latter, underestimation of salt-bridge interactions (“too wet”), which was observed in the GBSW-based CpHMD simulations,12,49 may be reduced here, as the Neck model approximately accounts for the interstitial water.27 Indeed, Fig. 4c shows that most of the pKa errors are small, except for Asp21 of SNase and Asp10 of RNase H, for which the pKa’s are underestimated by about 2.7 units (Table 3). Both residues are deeply buried (about 20% solvent exposure) and hydrogen bonded to another Asp in the enzyme active site.39,62 Considering our previous discussion regarding desolvation errors (Fig. 4a), we suggest that the underestimation of desolvation is much larger than the underestimation of attractive electrostatic interactions, leading to the negative pKa errors.
We turn to the histidines involved in strong electrostatic interactions. Due to the small number of data points, a general trend is hard to deduce. Therefore, we focus our attention on two extreme cases: the fully exposed His127 of RNase H, which has a calculated pKa of 6.6 (1.3 units lower than experiment), and the fully buried His48 of RNase A, which has a calculated pKa of 7.2 (1.1 units higher than experiment). In RNase H, the crystal structure shows a salt bridge between His127 and Glu119, which justifies the large experimental pKa shift of His127 (1.4 units). However, the salt bridge contradicts the small experimental pKa shift of Glu119 (−0.2 units). In the simulation, the salt bridge is formed for a small fraction of time (11 % at pH 5, see SI), consistent with the small pKa shifts for both His127 (0.1 units) and Glu119 (−0.4 units). Interestingly, the current results are consistent with the previous GBSW-CpHMD simulation.20 Thus, we could not find an explanation for the discrepancy between the calculated and experimental pKa of His127 in RNase H. We will defer further discussion to future studies using titration in explicit solvent.
His48 of RNase A is nearly completely buried (3% solvent exposure) and forms a salt bridge with Asp14. Thus, we believe the positive pKa error of 1.1 can be understood similarly as for Asp21 of SNase and Asp10 of RNase H. That is, the underestimation of desolvation (which shifts the histidine pKa down) overwhelms the underestimation of buried salt-bridge interaction (which shifts the histidine pKa up), leading to a positive pKa error for His48.
Analysis of sampling errors and convergence
To quantify sampling noise, we performed error analysis for the calculated pKa’s based on error propagation from the block standard error of the fraction of deprotonation (S) at different pH (see Methods). Most of the pKa’s have “sampling” errors below 0.1; however, six of them have errors in the range 0.2–0.3, as indicated by asterisks in Table 3 (actual data not shown). Not surprisingly, the large errors correspond to the aforementioned titratable groups with significant drift in the time trace of S values and 0.3–0.4 units changes in the pKa’s based on the first and second ns of the simulations. Thus, while the sampling noise is small for converged pKa’s, large errors are flags for incomplete protonation state sampling.
We examine the correlation time to better understand the convergence behavior. Fig. 5a shows that for Glu7, the autocorrelation function (ACF) of (Eq. 1) decays to zero after about 15 ps and oscillates around zero for the remainder of the simulation. This behavior is typical for pKa’s with small sampling errors, and can be attributed to fast convergence of the S value (Fig. 5b). In contrast, for Asp87 of HEWL, the ACF does not reach zero until about 350 ps and keeps decreasing for the remainder of the simulation (Fig. 5c). This behavior is typical for pKa’s with large sampling errors, and can be attributed to the incomplete convergence of the S value, which shows a decreasing trend toward the end of the simulation (Fig. 5d), indicating that the pKa would be higher and closer to the experimental value (Table 3) if the simulation was to continue. A closer look at the trajectory showed that the Asp87 forms a persistent salt-bridge-like interaction with His15 in the first half of the simulation, while in the second half of the simulation, the interaction is largely disrupted (data not shown). Persistent attractive interaction leads to prolonged correlation time, and is also the underlying reason for the large sampling errors in the pKa’s of Asp145 in BBL and Glu6 in RNaseH. The other two pKa’s with large sampling errors are from buried residues, His12 in RNase A and Glu79 in Xylanase. Consistently, the time trace of S shows a trend that decreases the deviation from the experimental pKa. The above analysis shows that the large “sampling” errors are due to insufficient convergence of the protonation-state sampling. Indeed, extending the simulations to 4 ns per replica improved the convergence of the unprotonated fractions S (Fig. S25) and reduced the errors for all pKa’s (Table 4), although Asp87 of HEWL and Glu79 of HEWL require even more sampling, as the errors are around 0.2, and the unprotonated fractions have not fully plateaued (Fig. S25). Encouragingly, however, with the prolonged sampling the changes of the pKa’s are mostly in the direction of the experimental values (Table 4).
Figure 5: Analysis of correlation time and protonation state sampling.

(a) and (c): Autocorrelation functions of (defined in Eq. 1) for Glu7 and Asp87 in HEWL, respectively. (b) and (d): λ (black) and S (green) values as functions of simulation time. The pH 3.5 replica for Glu7 and pH 2 replica for Asp87 were used.
Table 4:
Prolonged simulations improve slowly converging pKa’s.
| Protein | Residue | Expt | 2 ns | 4 ns |
|---|---|---|---|---|
| BBL | Asp145 | 3.7 | 2.6 (0.3) | 2.8 (0.1) |
| HEWL | Asp87 | 2.2 | 1.8 (0.2) | 1.9 (0.2) |
| RNase A | His12 | 6.2 | 6.4 (0.3) | 6.4 (0.1) |
| SNase | Asp83 | <2.2 | 2.1 (0.3) | 1.8 (0.1) |
| RNase H | Glu6 | 4.5 | 4.3 (0.2) | 4.3 (0.1) |
| Xylanase | Glu79 | 4.6 | 5.1 (0.3) | 4.4 (0.2) |
In parentheses are the sampling errors calculated using error propagation (Eq. 10).
Dependence on the force field and solvent model
To test the dependence on the force field, we performed an additional titration simulation for HEWL using GBNeck2 and the CHARMM c22 force field in Amber (Table 5). Since GBNeck2 is optimized for the Amber and not CHARMM force field, we avoid comparing to experimental values and instead focus on the pKa differences. The largest pKa differences are for the catalytic residues Glu35 and Asp52. With the ff14sb force field, the experimental pKa order is reproduced; however, with the c22 force field, the two pKa’s are nearly identical and higher than the ff14sb results by 1.6 and 3 units, respectively. Considering the desolvation penalty may be similar given the same Neck2 model, we suggest the large difference may come from the vacuum hydrogen bond strength in the two force fields.
Table 5:
Calculated pKa’s of HEWL using different force fields and solvent models
| Residue | Expta | Neck2-ff14sb | Neck2-c22c | SW-c22d |
|---|---|---|---|---|
| Glu7 | 2.6 | 3.5 | 3.5 | 2.6 |
| His15 | 5.5 | 6.5 | 7.6 | 5.3 |
| Asp18 | 2.8 | 1.1 | 3.2 | 2.9 |
| Glu35 | 6.1 | 4.6 | 6.2 | 4.4 |
| Asp48 | 1.4 | 1.8 | 1.3 | 2.8 |
| Asp52 | 3.6 | 3.3 | 6.3 | 4.6 |
| Asp66 | 1.2 | 2.1 | 2.3 | 1.2 |
| Asp87 | 2.2 | 1.8 | 2.2 | 2.0 |
| Asp101 | 4.5 | 4.8 | 3.9 | 3.3 |
| Asp119 | 3.5 | 2.4 | 2.6 | 2.5 |
Experimental data taken from Webb et al.59
Neck2-ff14sb denotes the GBNeck2-CpHMD simulation with the Amber ff14sb force field.
Neck2-c22 denotes the GBNeck2-CpHMD simulation with the CHARMM c22 force field51 in Amber.
SW-c22 denotes the previously published GBSW-CpHMD simulation with the GBSW model and CHARMM c22 force field.68
Next, we considered results with the c22 force field and the published GBSW-CpHMD data. The largest pKa differences are again Glu35 and Asp52 in addition to His15. The two pKa’s are nearly identical but lower than the Neck2-c22 results by 1.8 and 1.7 units, respectively. The lowered pKa values demonstrates that the desolvation induced pKa upshift is much smaller with the GBSW model, consistent with the previous observation that the GBSW model significantly underestimates the pKa’s of buried carboxylic groups.11,20,23
CONCLUDING REMARKS
We presented the implementation of the generalized Born (GB) implicit-solvent based continuous constant pH MD (CpHMD) method in Amber. The implementation was tested using pH replica-exchange titration of 2 ns per replica on 11 proteins, ranging from the 36-residue mini-protein HP36 to the 389-residue BACE1 enzyme. The simulations utilized the most recent GBNeck2 model28 and ff14sb force field.29 The CPU cost relative to the fixed-protonation-state GB calculations scales linearly as the ratio of titratable and total number of residues. For the present test set, where all carboxylic groups and histidines were allowed to titrate, the added computational cost was 11–33 %. Comparison of the calculated and experimental pKa’s of 96 carboxylic groups and 15 histidines gave a mean unsigned error of 0.65, a root-mean-squared error of 0.90, and a linear correlation coefficient of 0.78. Comparison of the calculated and experimental pKa’s shifts relative to the model values gave a linear correlation coefficient of 0.56. This level of accuracy is similar to the GBSW-based19 CpHMD implementation11,20 with the c22 force field51 in CHARMM.43 We found that the accuracy is slightly worse for carboxylic groups (a total of 97) but much better for histidines compared to the GBSW-CpHMD in CHARMM, although the number of histidines (15) is rather small. However, surprisingly, the current implementation correctly predicted the experimental orders of the pKa’s of the buried carboxylic dyads in hen egg white lysozyme, staphylococcal nuclease, and β-secretase 1, while GBSW-CpHMD could not distinguish them. Our analysis showed that the GBNeck2 simulations preserved the local hydrogen bonding environment and solvent exposure of the titratable sidechain, which are the molecular determinants for the pKa order. A systematic error in the GBNeck2-CpHMD titration is the underestimation of pKa shifts for deeply buried carboxylic groups, which can be largely attributed to the underestimation of the desolvation penalties, as was observed for the GBSW model. As to the pKa calculations for histidines, no definitive conclusions could be drawn due to the small range of pKa values and too few data points. We note that buried residues can become kinetically trapped in a conformational substate more readily than solvent-exposed residues. Thus, lack of conformational sampling is likely another contributor to the pKa errors. In principle, one can probe the extent of such errors by extensive conformational sampling to uncover all substates, as demonstrated in the CHARMM hybridsolvent CpHMD titration of a deeply buried lysine, for which both closed (crystal structure) and open states needed to be sampled to reproduce the experimental macroscopic pKa.69 This type of study was not pursued here, because prolonged conformational sampling in GB solvent likely leads to larger deviations from the native state and consequently larger pKa errors.6
We also conducted analysis of random sampling errors in the calculated pKa’s. With the simulation time of 2 ns per replica, the errors are below 0.1 pH units for all but 6 pKa’s which were not converged due to incomplete sampling of salt-bridge-like interactions and/or hydrophobic burial. Prolonging the simulations to 4 ns per replica demonstrated significant improvement in convergence and agreement with experimental data. Finally, we tested the dependence on the force field and solvent model using HEWL. The pKa’s of the catalytic dyad are most sensitive, which could be attributed to the differences in the hydrogen bond strength between the ff14sb and c22 force fields and the more severe underestimation of desolvation penalty by the GBSW model.
The current work points toward several directions of future improvement. The GB models in the Amber package are under active development,70 and improved GB models could be easily incorporated in CpHMD to potentially improve accuracy. The present method uses GB solvent for the propagation of both conformational and protonation degrees of freedom; however, the hybrid-solvent and all-atom CpHMD in CHARMM have demonstrated significantly improved accuracy relative to the GB-based version. Currently, extensions to use a hybrid-solvent scheme12 or fully explicit solvent12 are underway. To accelerate the computational speed, a GPU implementation will be pursued in the near future.
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge National Institutes of Health (R01GM098818 and R01GM118772) and National Science Foundation (CBET143595) for funding.
Footnotes
SOFTWARE AVAILABILITY
The GBNeck2-based CpHMD method along with the associated parameters and analysis tools will be distributed as a new functionality in pmemd of Amber18.
Supporting Information Available
Titration curves, analysis of the convergence of the pKa estimates, and an analysis of the environment surrounding the dyad residues.
References
- (1).Khandogin J; Chen J; Brooks CL III Exploring atomistic details of pH-dependent peptide folding. Proc. Natl. Acad. Sci. USA 2006, 103, 18546–18550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ellis CR; Shen J pH-Dependent Population Shift Regulates BACE1 Activity and Inhibition. J. Am. Chem. Soc 2015, 137, 9543–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Huang Y; Chen W; Dotson DL; Beckstein O; Shen J Mechanism of pH-dependent activation of the sodium-proton antiporter NhaA. Nat. Commun 2016, 7, 12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Baptista AM; Teixeira VH; Soares CM Constant-pH molecular dynamics using stochastic titration. J. Chem. Phys 2002, 117, 4184–4200. [Google Scholar]
- (5).Mongan J; Case DA; McCammon JA Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem 2004, 25, 2038–2048. [DOI] [PubMed] [Google Scholar]
- (6).Swails JM; York DM; Roitberg AE Constant pH Replica Exchange Molecular Dynamics in Explicit Solvent Using Discrete Protonation States: Implementation, Testing, and Validation. J. Chem. Theory Comput 2014, 10, 1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lee J; Miller BT; Damjanović A; Brooks BR Constant pH molecular dynamics in explicit solvent with enveloping distribution sampling and Hamiltonian exchange. J. Chem. Theory Comput 2014, 10, 2738–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Radak BK; Chipot C; Suh D; Jo S; Jiang W; Phillips JC; Schulten K; Roux B Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems. J. Chem. Theory Comput 2017, 13, 5933–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kong X; Brooks CL III λ-dynamics: A new approach to free energy calculations. J. Chem. Phys 1996, 105, 2414–2423. [Google Scholar]
- (10).Lee MS; Salsbury FR Jr.; Brooks CL III Constant-pH molecular dynamics using continuous titration coordinates. Proteins 2004, 56, 738–752. [DOI] [PubMed] [Google Scholar]
- (11).Khandogin J; Brooks CL III Constant pH molecular dynamics with proton tautomerism. Biophys. J 2005, 89, 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wallace JA; Shen JK Continuous constant pH molecular dynamics in explicit solvent with pH-based replica exchange. J. Chem. Theory Comput 2011, 7, 2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wallace JA; Shen JK Charge-leveling and proper treatment of long-range electrostatics in all-atom molecular dynamics at constant pH. J. Chem. Phys 2012, 137, 184105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Goh GB; Knight JL; Brooks CL III Constant pH molecular dynamics simulations of nucleic acids in explicit solvent. J. Chem. Theory Comput 2012, 8, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Huang Y; Chen W; Wallace JA; Shen J All-Atom Continuous Constant pH Molecular Dynamics With Particle Mesh Ewald and Titratable Water. J. Chem. Theory Comput 2016, 12, 5411–5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Donnini S; Tegeler F; Groenhof G; Grubmu¨ller H Constant pH molecular dynamics in explicit solvent with λ-dynamics. J. Chem. Theory Comput 2011, 7, 1962–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen W; Morrow BH; Shi C; Shen JK Recent development and application of constant pH molecular dynamics. Mol. Simul 2014, 40, 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lee MS; Feig M; Salsbury FR Jr.; Brooks CL III New analytic approximation to the standard molecular volume definition and its application to generalized Born calculations. J. Comput. Chem 2003, 24, 1348–1356. [DOI] [PubMed] [Google Scholar]
- (19).Im W; Lee MS; Brooks CL III Generalized Born model with a simple smoothing function. J. Comput. Chem 2003, 24, 1691–1702. [DOI] [PubMed] [Google Scholar]
- (20).Khandogin J; Brooks CL III Toward the accurate first-principles prediction of ionization equilibria in proteins. Biochemistry 2006, 45, 9363–9373. [DOI] [PubMed] [Google Scholar]
- (21).Khandogin J; Brooks CL III Linking folding with aggregation in Alzheimer’s beta amyloid peptides. Proc. Natl. Acad. Sci. USA 2007, 104, 16880–16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shen JK Uncovering specific electrostatic interactions in the denatured states of proteins. Biophys. J 2010, 99, 924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wallace JA; Wang Y; Shi C; Pastoor KJ; Nguyen B-L; Xia K; Shen JK Toward accurate prediction of pKa values for internal protein residues: the importance of conformational relaxation and desolvation energy. Proteins 2011, 79, 3364–3373. [DOI] [PubMed] [Google Scholar]
- (24).Alexov E; Mehler EL; Baker N; Baptista AM; Huang Y; Milletti F; Nielsen JE; Farrell D; Carstensen T; Olsson MHM; Shen JK; Warwicker J; Williams S; Word JM Progress in the prediction of pKa values in proteins. Proteins 2011, 79, 3260–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Morrow BH; Wang Y; Wallace JA; Koenig PH; Shen JK Simulating pH titration of a single surfactant in ionic and nonionic surfactant micelles. J. Phys. Chem. B 2011, 115, 14980–14990. [DOI] [PubMed] [Google Scholar]
- (26).Case D; Betz R; Botello-Smith W; Cerutti D; Cheatham T III; Darden T; Duke R; Giese T; Gohlke H; Goetz A; Homeyer N; Izadi S; Janowski P; Kaus J; Kovalenko A; Lee T; LeGrand S; Li P; Lin C; Luchko T; Luo R; Madej B; Mermelstein D; Merz K; Monard G; Nguyen H; Nguyen H; Omelyan I; Onufriev A; Roe D; Roitberg A; Sagui C; Simmerling C; Swails J; Walker R; Wang J; Wolf R; Wu X; Xiao L; York D; Kollman P AMBER 2016. 2016. [Google Scholar]
- (27).Mongan J; Simmerling C; McCammon JA; Case DA; Onufriev A Generalized Born Model with a Simple, Robust Molecular Volume Correction. J. Chem. Theory Comput 2007, 3, 156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Nguyen H; Roe DR; Simmerling C Improved Generalized Born Solvent Model Parameters for Protein Simulations. J. Chem. Theory Comput 2013, 9, 2020–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nguyen H; Maier J; Huang H; Perrone V; Simmerling C Folding Simulations for Proteins with Diverse Topologies Are Accessible in Days with a Physics-Based Force Field and Implicit Solvent. J. Am. Chem. Soc 2014, 136, 13959–13962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Götz AW; Williamson MJ; Xu D; Poole D; Grand SL; Walker RC Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput 2012, 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Meng Y; Roitberg AE Constant pH replica exchange molecular dynamics in biomolecules using a discrete protonation model. J. Chem. Theory Comput 2010, 6, 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).McKnight1 CJ; Matsudaira PT; Kim PS NMR structure of the 35-residue villin headpiece subdomain. Nat. Struct. Biol 1997, 4, 180–184. [DOI] [PubMed] [Google Scholar]
- (34).Ferguson N; Sharpe TD; Schartau PJ; Sato S; Allen MD; Johnson CM; Rutherford TJ; Fersht AR Ultra-fast barrier-limited folding in the peripheral subunit-binding domain family. J. Mol. Biol 2005, 353, 427–446. [DOI] [PubMed] [Google Scholar]
- (35).Luisi DL; Kuhlman B; Sideras K; Evans PA; Raleigh DP Effects of varying the local propensity to form secondary structure on the stability and folding kinetics of a rapid folding mixed α/β protein: characterization of a truncation mutant of the N-terminal domain of the ribosomal protein L9. J. Mol. Biol 1999, 289, 167–174. [DOI] [PubMed] [Google Scholar]
- (36).Hoogstraten CG; Choe S; Westler WM; Markley JL Comparison of the accuracy of protein solution structures derived from conventional and network-edited NOESY data. Protein Sci. 1995, 4, 2289–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Weichsel A; Gasdaska JR; Powis G; Montfort WR Crystal structures of reduced, oxidized, and mutated human thioredoxins: evidence for a regulatory homodimer. Structure 1996, 4, 735–751. [DOI] [PubMed] [Google Scholar]
- (38).Ramanadham M; Sieker LC; Jensen LH Refinement of triclinic lysozyme: II. The method of stereochemically restrained least squares. Acta Crystallogr. B 1990, 46, 63–69. [DOI] [PubMed] [Google Scholar]
- (39).Castañeda CA; Fitch CA; Majumdar A; Khangulov V; Schlessman JL; García-Moreno E, Molecular B determinants of the pKa values of Asp and Glu residues in staphylococcal nuclease. Proteins 2009, 77, 570–588. [DOI] [PubMed] [Google Scholar]
- (40).Wlodawer A; Svensson LA; Sjölin L; Gilliland GL Structure of phosphate-free ribonuclease A refined at 1.26 Angstrom. Biochemistry 1988, 27, 2705–2717. [DOI] [PubMed] [Google Scholar]
- (41).Katayangi K; Miyagawa M; Matsushima M; Ishikawa M; Kanaya S; Nakamura H; Ikehara M; Matsuzaki T; Morikawa K Structural details of ribonuclease H from Escherichia coli as refined to an atomic resolution. J. Mol. Biol 1992, 223, 1029–1052. [DOI] [PubMed] [Google Scholar]
- (42).Wakarchuk WW; Campbell RL; Sung WL; Davoodi J; Yaguchi M Mutational and crystallographic analyses of the active site residues of the bacillus circulans xylanase. Protein Sci. 1994, 3, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Brooks BR; Brooks CL III; Mackerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner AR; Feig M; Fischer S; Gao J; Hodoscek M; Im, W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor RW; Post CB; Pu JZ; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York DM; Karplus M CHARMM: the biomolecular simulation program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Thurlkill RL; Grimsley GR; Scholtz JM; Pace CN pK values of the ionizable groups of proteins. Protein Sci. 2006, 15, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ryckaert JP; Ciccotti G; Berendsen HJC Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- (46).Nozaki Y; Tanford C Examination of titration behavior. Methods Enzymol. 1967, 11, 715–734. [Google Scholar]
- 47.() Tanokura M 1H-NMR study on the tautomerism of the imidazole ring of histidine residues: I. Microscopic pK values and molar ratios of tautomers in histidine-containing peptides. Biochim. Biophys. Acta 1983, 742, 576–585. [DOI] [PubMed] [Google Scholar]
- (48).Schutz CN; Warshel A What are the dielectric constants of proteins and how to validate electrostatic models? Proteins 2001, 44, 400–417. [DOI] [PubMed] [Google Scholar]
- (49).Huang Y; Yue Z; Tsai C-C; Henderson JA; Shen J Predicting Catalytic Proton Donors and Nucleophiles in Enzymes: How Adding Dynamics Helps Elucidate the Structure-Function Relationships. J. Phys. Chem. Lett 2018, 9, 1179–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Swails JM; Roitberg AE Enhancing conformation and protonation state sampling of hen egg white lysozyme using pH replica exchange molecular dynamics. J. Chem. Theory Comput 2012, 8, 4393–4404. [DOI] [PubMed] [Google Scholar]
- (51).MacKerell AD Jr.; Bashford D; Bellott M; Dunbrack RL Jr.; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S; Joseph-McCarthy D; Kuchnir L; Kuczera K; Lau FTK; Mattos C; Michnick S; Ngo T; Nguyen DT; Prodhom B; Reiher WE III; Roux B; Schlenkrich M; Smith JC; Stote R; Straub J; Watanabe M; Wiórkiewicz-Kuczera J; Yin D; Karplus M All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (52).Xiao S; Patsalo V; Shan B; Bi Y; Green DF; Raleigh DP Rational modification of protein stability by targeting surface sites leads to complicated results. Proc. Natl. Acad. Sci. USA 2013, 110, 11337–11342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Arbely E; Rutherford TJ; Sharpe TD; Ferguson N; Fersht AR Downhill versus barrier-limited folding of BBL 1: energetic and structural pertubation effects upon protonation of a histidine of unusually low pKa. J. Mol. Biol 2009, 387, 986–992. [DOI] [PubMed] [Google Scholar]
- (54).Arbely E; Rutherford TJ; Neuweiler H; Sharpe TD; Ferguson N; Fersht AR Carboxyl pKa values and acid denaturation of BBL. J. Mol. Biol 2010, 403, 313–327. [DOI] [PubMed] [Google Scholar]
- (55).Kuhlman B; Luisi DL; Young P; Raleigh DP pKa values and the pH dependent stability of the N-terminal domain of L9 as probes of electrostatic interactions in the denatured state. Differentiation between local and nonlocal interactions. Biochemistry 1999, 38, 4896–4903. [DOI] [PubMed] [Google Scholar]
- (56).Schaller W; Robertson AD pH, ionic strength, and temperature dependences of ionization equilibria for the carboxyl groups in turkey ovomucoid third domain. Biochemistry 1995, 34, 4714–4723. [DOI] [PubMed] [Google Scholar]
- (57).Forsyth WR; Gilson MK; Antosiewicz J; Jaren OR; Robertson AD Theoretical and experimental analysis of ionization equilibria in ovomucoid third domain. Biochemistry 1998, 37, 8643–8652. [DOI] [PubMed] [Google Scholar]
- (58).Qin J; Clore GM; Gronenborn AM Ionization equilibria for side-chain carboxyl groups in oxidized and reduced human thioredoxin and in the complex with its target peptide from the transcription factor nfκB. Biochemistry 1996, 35, 7–13. [DOI] [PubMed] [Google Scholar]
- (59).Webb H; Tynan-Connolly BM; Lee GM; Farrell D; O’Meara F; Søndergaard CR; Teilum K; Hewage C; McIntosh LP; Nielsen JE Remeasuring HEWL pKa values by NMR spectroscopy: methods, analysis, accuracy, and implications for theoretical pKa calculations. Proteins 2011, 79, 685–702. [DOI] [PubMed] [Google Scholar]
- (60).Fitch CA; Karp DA; Lee KK; Stites WE; Lattman EE; E. BG-M Experimental pKa values of buried residues: analysis with continuum methods and role of water penetration. Biophys. J 2002, 82, 3289–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Baker WR; Kintanar A Characterization of the pH titration shifts of ribonuclease A by one- and two-dimensional nuclear magnetic resonance spectroscopy. Arch. Biochem. Biophys 1996, 327, 189–199. [DOI] [PubMed] [Google Scholar]
- (62).Oda Y; Yoshida M; Kanaya S Role of histidine 124 in the catalytic function of ribonuclease HI from Escherichia coli. J. Biol. Chem 1993, 268, 88–92. [PubMed] [Google Scholar]
- (63).Oda Y; Yamazaki T; Nagayama K; Kanaya S; Kuroda Y; Nakamura H Individual ionization constants of all the carboxyl groups in ribonuclease HI from escherichia coli determined by NMR. Biochemistry 1994, 33, 5275–5284. [DOI] [PubMed] [Google Scholar]
- (64).Plesniak LA; Connelly GP; Wakarchuk WW; Mcintosh LP Characterization of a buried neutral histidine residue in Bacillus circulans xylanase: NMR assignments, pH titration, and hydrogen exchange. Protein Sci. 1996, 5, 2319–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Joshi MD; Hedberg A; Mcintosh LP Complete measurement of the pKa values of the carboxyl and imidazole groups in Bacillus circulans xylanase. Protein Sci. 1997, 6, 2667–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Toulokhonova L; Metzler WJ; Witmer MR; Copeland RA; Marcinkeviciene J Kinetic studies on β-site amyloid precursor protein-cleaving enzyme (BACE). J. Biol. Chem 2003, 278, 4582–4589. [DOI] [PubMed] [Google Scholar]
- (67).Geney R; Layten M; Gomperts R; Hornak V; Simmerling C Investigation of salt bridge stability in a Generalized Born solvent model. J. Chem. Theory Comput 2006, 2, 115–127. [DOI] [PubMed] [Google Scholar]
- (68).Chen W; Wallace J; Yue Z; Shen J Introducing titratable water to all-atom molecular dynamics at constant pH. Biophys. J 2013, 105, L15–L17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Shi C; Wallace JA; Shen JK Thermodynamic coupling of protonation and conformational equilibria in proteins: theory and simulation. Biophys. J 2012, 102, 1590–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Forouzesh N; Izadi S; Onufriev AV Grid-Based Surface Generalized Born Model for Calculation of Electrostatic Binding Free Energies. J. Chem. Inf. Model 2017, 57, 2505–2513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.