

Abstract
Purpose of review
This article discusses recent advances in the understanding of clinical and genetic aspects of primary ataxias, including congenital, autosomal recessive, autosomal dominant, episodic, X-linked, and mitochondrial ataxias, as well as idiopathic degenerative and secondary ataxias.
Recent findings
Many important observations have been published in recent years in connection with primary ataxias, particularly new loci and genes. The most commonly inherited ataxias may present with typical and atypical phenotypes. In the group of idiopathic degenerative ataxias, genes have been found in patients with multiple system atrophy type C. Secondary ataxias represent an important group of sporadic, cerebellar, and afferent/sensory ataxias.
Summary
Knowledge of primary ataxias has been growing rapidly in recent years. Here we review different forms of primary ataxia, including inherited forms, which are subdivided into congenital, autosomal recessive cerebellar ataxias, autosomal dominant cerebellar ataxias, episodic ataxias, X-linked ataxias, and mitochondrial ataxias, as well as sporadic ataxias and idiopathic degenerative ataxias. Secondary or acquired ataxias are also reviewed and the most common causes are discussed.
Keywords: autosomal recessive cerebellar ataxias, congenital ataxias, secondary ataxias, spinocerebellar ataxias, sporadic ataxias
INTRODUCTION
Ataxia is a disorder of balance and coordination [1▪]. The commonest forms are cerebellar ataxia, in which the cerebellum and its afferent or efferent projections are affected, and afferent/sensory ataxia, in which the proprioceptive pathways are affected [1▪,2]. Cerebellar ataxia is a syndrome that includes several signs and symptoms, such as gait ataxia, dysarthria, nystagmus, tremor, and cognitive dysfunction [1▪,2–4]. Ataxias can be classified as primary or secondary, as well as hereditary or sporadic [1▪,2–4].
PRIMARY ATAXIAS
Primary cerebellar ataxias are further subdivided into sporadic and hereditary ataxias. The latter include autosomal recessive cerebellar ataxias (ARCAs), autosomal dominant cerebellar ataxias, currently known as spinocerebellar ataxias (SCAs), episodic ataxias, X-linked cerebellar ataxias, and mitochondrial ataxias. Idiopathic degenerative cerebellar ataxias include the cerebellar form of multiple system atrophy (MSA-C) and idiopathic late-onset cerebellar ataxias [3–9].
CONGENITAL ATAXIAS
Congenital ataxias can be caused by cerebellar mal-formations or pontocerebellar hypoplasia and present with cerebellar ataxia. The most widely known form is Joubert’s syndrome, a rare autosomal recessive disease characterized by a congenital hind-brain malformation, which can be identified on MRI as the ‘molar tooth sign’ [10,11] (Fig. 1). The clinical picture includes neonatal hypotonia, cerebellar ataxia, ocular motor apraxia, breathing dysregulation, and multiple organ involvement. To date, more than 20 causative genes have been identified, most of which encode proteins of the primary cilium, a subcellular organelle involved in crucial cellular functions. Joubert’s syndrome is part of an emerging class of genetic disorders known as ciliopathies [10,11].
FIGURE 1.
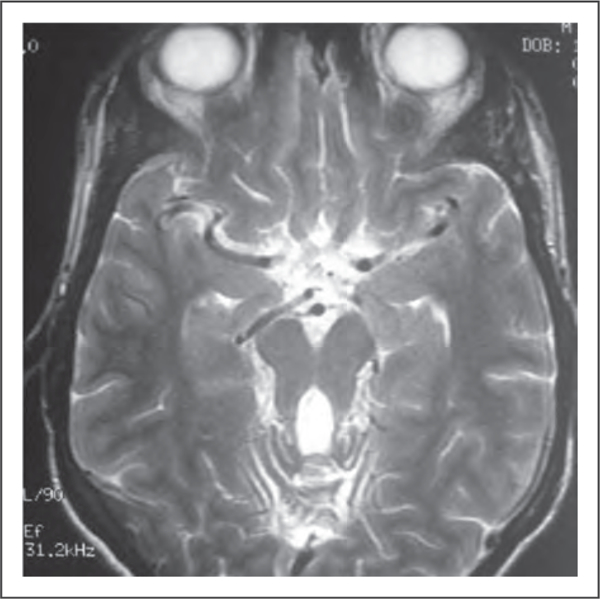
Brain MRI, T2-weighted, axial view. Molar tooth sign, Joubert’s syndrome. Adapted with permission.
INHERITED ATAXIAS
Inherited cerebellar ataxias constitute an extensive group of clinically and genetically heterogeneous, complex neurodegenerative disorders caused by a large number of genetic mutations. Among the hereditary cerebellar ataxias, there are almost 40 forms of SCAs, more than 30 ARCAs, two X-linked ataxias, and several forms of mitochondrial ataxias [7-9,12,13▪].
SCAs are predominantly caused by unstable repeat expansions in either coding (SCAs types 1, 2, 3, 6, 7, and 17, and dentatorubral-pallidoluysian atrophy) or noncoding (SCAs types 8,10, 12, 31, and 36) regions of the relevant genes [3,4,9,12,13▪]. More rarely, they are caused by conventional mutations, such as missense mutations, insertions, and deletions (SCAs types 5, 11, 13, 14, 15, 20, 23, 27, 28, and 35). SCAs caused by unstable repeat expansions in the coding area of the relevant genes have a trinucleotide (CAG) repeat, which causes cells to produce expanded polyglutamine tracts. These diseases are known as polyglutamine disorders [9,12,13▪,14]. Another group of inherited ataxias are episodic ataxias due to ion channel mutations [15]. Among the ARCAs, Friedreich’s ataxia (FRDA) is associated with homozygous triplet GAA expansions in the FXN gene [7,8,16]. The ataxia telangiectasia gene, known as ATM, is located on chromosome 11q22–23 [7,8,14,17▪]. Other ARCAs are associated with different, conventional mutations. Some, such as fragile X-associated tremor/ataxia syndrome (FXTAS), are X-linked, whereas others are mitochondrial ataxias [polymerase γ (POLG) ataxia] [18▪,19].
AUTOSOMAL RECESSIVE CEREBELLAR ATAXIAS
ARCAs are included in the heterogeneous group of inherited ataxias. They are typically characterized by cerebellar and spinal cord degeneration and have a relatively early age of onset [3,4,7,8]. The most common forms of ARCAs that have been genetically defined are shown in Table 1. In white children, the most common form is FRDA, followed by ataxia telangiectasia [7,8,16,17▪].
Table 1.
Autosomal recessive cerebellar ataxias – most common forms with genetic definition
| ARCAs | Locus | Gene | Protein |
|---|---|---|---|
| FRDA | 9q13 | FXN | Frataxin |
| AVED | 8q12.3 | TTPA | α-Tocopherol transfer protein |
| ARSACS | 13q12 | SACS | Sacsin |
| AT | 11q22.3 | ATM | Serine protein kinase |
| ATLD | 11q21 | MRE11 | Meiotic recombination 11 |
| AOA1 | 9p13 | APTX | Aprataxin |
| AOA2 | 9q34 | SETX | Senataxin |
| MIRAS/SANDO | 15q25 | POLG1 | DNA polymerase subunit g-1 |
| MSS | 5q31 | SIL1 | Nucleotide exchange factor SIL1 |
| ARCA1 | 6q25 | SYNE1 | Nesprin-1 |
| ARCA2 | 1q42.2 | CABC1 | Chaperone activity of bc1 complex like |
| ARCA3 | 3p22.1 | ANO10 | Anoctamin-10 |
AOA1, ataxia with oculomotor apraxia type 1; AOA2, ataxia with oculomotor apraxia type 2; ARCA1, autosomal recessive cerebellar ataxia type 1; ARCA2, autosomal recessive cerebellar ataxia type 2, with coenzyme Q10 deficiency; ARCA3, autosomal recessive cerebellar ataxia type 3 caused by mutations in ANO10; ARSACS, autosomal recessive spastic cerebellar ataxia of Charlevoix-Saguenay; AT, ataxia telangiectasia; ATLD, ataxia telangiectasia-like disorder; AVED, ataxia with vitamin E deficiency; FRDA, Friedreich’s ataxia; MIRAS, mitochondrial recessive ataxia syndrome; MSS, Marinesco-Sjogren syndrome; SANDO, sensory ataxic neuropathy, dysarthria, and ophathalmoparesis
FRIEDREICH’S ATAXIA
Since the identification of the FRDA gene and the GAA trinucleotide expansion that leads to FRDA, phenotypic variants of this ataxia have frequently been reported in individuals carrying pathogenic mutations, some of which do not fit the classic descriptions of this condition [16,20]. Atypical phenotypes include late-onset and very-late-onset ataxia, with small GAA expansions, retained reflexes, pyramidal signs, and movement disorders [21,22]. FRDA is predominantly an afferent/sensory ataxia; however, the presence of a cerebellar component was confirmed in neuropathological studies and recently in neuroimaging studies (Fig. 2) [16,20,23]. Although there is no consensus regarding treatment, antioxidants such as coenzyme Q10 and its derivatives, including idebenone, have been used. Idebenone has shown significant benefits for hypertrophic cardiomyopathy but is ineffective for neurological conditions [24,25]. More recently, new drugs have been tested, including deferiprone, as well as epigenetic therapy [26,27].
FIGURE 2.
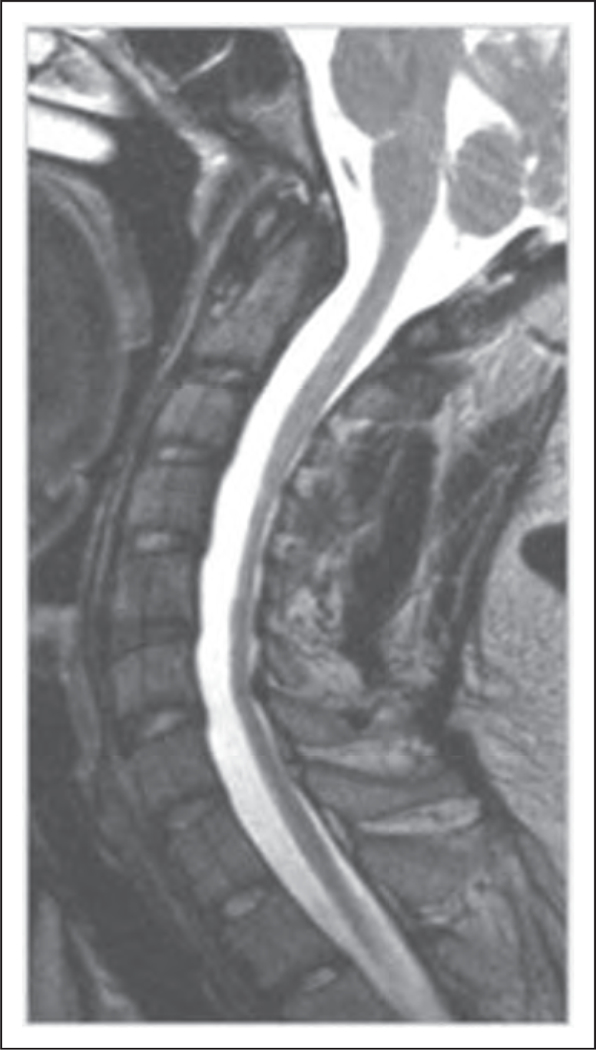
Spinal cord MRI, T2-weighted, sagittal view.Cervical spinal cord atrophy in a patient with Friedreich’s ataxia. Adapted with permission.
ATAXIA TELANGIECTASIA
Since the ATM gene was first described, over 200 potentially pathogenic mutations involving almost all coding exons of this gene have been reported [28]. In addition to the classical phenotype, with cerebellar ataxia and oculocutaneous telangiectasia (Fig. 3), many cases of ataxia telangiectasia with milder phenotypes have been described [28]. These phenotypes include later disease onset; slower progression; longer life expectancy; a predominance of movement disorders, such as dystonia, myoclonus, and chorea, instead of cerebellar ataxia; the absence of ocular telangiectasia; and lower levels of chromosomal instability and cellular radiosensitivity [17▪,28]. In fact, ataxia telangiectasia represents a multisystem entity with variable neurological and systemic manifestations. ATM syndrome has been proposed as a more adequate designation for this entity (H.A.G. Teive, unpublished).
FIGURE 3.
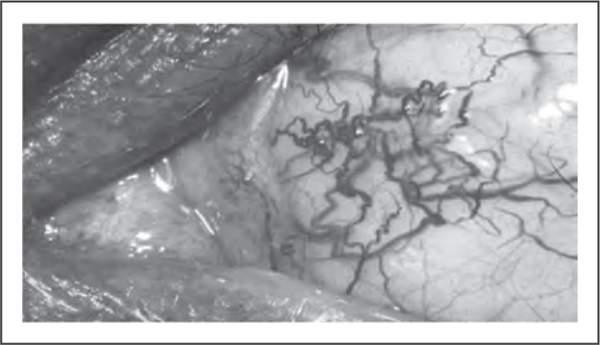
Conjunctival telangiectasia in a patient with ataxia telangiectasia. Adapted with permission.
OTHER AUTOSOMAL RECESSIVE CEREBELLAR ATAXIAS
Gordon Holmes’ syndrome, a peculiar form of ARCA, is characterized by hypogonadotropic hypogonadism, and different mutations have been found in patients with this form of ataxia, including STUB1, RNF216, OTUD4, and PNPLA6 gene mutations [29▪,30]. PNPLA6 mutations also cause Boucher-Neuhauser syndrome, a combination of ARCA and hypogonadotropic hypogonadism, which is also associated with chorioretinal dystrophy and hypersegmented neutrophils [29▪,31–33]. Another form of early-onset ARCA associated with retinal dystrophy is caused by a homozygous GRID2 deletion [34]. Mutations in the SNX-14 gene cause a rare form of ARCA associated with sensorineural hearing loss and intellectual disability [35]. Childhood-onset progressive myoclonic ataxia (Ramsay Hunt syndrome) is associated with a novel mutation in the GOSR2 gene [36]. Although significant progress has been made in the identification of ARCA genes, the genetic cause of disease remains undetermined in about 40–50% of ARCAs [30,37,38▪,39–43]. There is no treatment for these ataxias, with the exception of ataxia due to vitamin E deficiency and a group of ataxias associated with coenzyme Q10 deficiency [3,4,37,38▪].
SPINOCEREBELLAR ATAXIAS
SCAs constitute a large, complex group of heterogeneous autosomal dominant degenerative diseases characterized by progressive degeneration of the cerebellum and its afferent and efferent connections, as well as other nervous system structures [3–6,9,12,13▪,44▪▪,45▪▪]. Table 2 shows the main types of SCAs currently known (from SCA type 1 to SCA type 40) and gives the genetic loci, mutations, and proteins associated with each disease. SCA type 3 is the commonest form of the disease worldwide; types 1, 2, 6, and 7 have greatly varying prevalences depending on the ethnic background of the population [3–6,9,12,13▪].
Table 2.
Spinocerebellar ataxias – summary of genetic characteristics
| SCA | Locus | Gene | Mutation | Potentially distinguishable clinical features |
|---|---|---|---|---|
| SCA1 | 6p22–23 | ATXN1 | CAG | |
| SCA2 | 12q23–24.1 | ATXN2 | CAG | Slow saccades, areflexia |
| SCA3 | 14q32.1 | ATXN3/DMJ | CAG | Bulging eyes, fasciculations |
| SCA4 | 16q22.1 | SCA4 | – | |
| SCA5 | 11q13.2 | SPTBN2 | – | Down-beat nystagmus |
| SCA6 | 19p13 | CACNA1A | CAG | Coarse nystagmus and saccadic intrusion |
| SCA7 | 3p21.1-p12 | ATXN7 | CAG | Visual loss |
| SCA8 | 13q21 | ATXN8OS | CAG/CTG | Reduced penetrance |
| SCA10 | 22q13.31 | ATXN10 | ATTCT | Epilepsy |
| SCA11 | 15q15.2 | TTBK2 | – | |
| SCA12 | 5q32 | PPP2R2B | CAG | Arm tremor |
| SCA13 | 19q13.33 | KCNC3 | – | Absent eye findings |
| SCA14 | 19q13.4 | PRKCG | – | Tremor, myoclonus, facial myokymia |
| SCA15 | 3p26.1 | ITPR1 | Allelic to SCA16, 29 | |
| SCA16 | 3p26.1 | ITPR1 | Allelic to SCA15, 29 | |
| SCA17 | 6q27 | TBP | CAG | Huntington disease like |
| SCA18 | 7q22-q32 | IFRD1 | – | |
| SCA19 | 1p13.2 | KCND3 | Allelic to SCA22 | |
| SCA20 | 11p11.2-q13.3 | SCA20 | Multiple gene duplication | Spasmodic dysphonia |
| SCA21 | 7p21.3-p15.1 | SCA21 | – | |
| SCA22 | 1p13.2 | KCND3 | Allelic to SCA19 | |
| SCA23 | 20p13 | PDYN | – | |
| SCA25 | 2p21-p15 | SCA25 | – | |
| SCA26 | 19p13.3 | EEEF2 | ||
| SCA27 | 13q34 | FGF14 | – | Mental retardation, tremor |
| SCA28 | 18p11 | AFG3L2 | – | |
| SCA29 | 3p26.1 | ITPR1 | Allelic to SCA15, 16 | Congenital nonprogressive ataxia |
| SCA30 | 4q34.3-q35.1 | SCA30 | – | |
| SCA31 | 16q22 | SCA31 | TGGAA | |
| SCA32 | 7q32-q33 | SCA32 | – | |
| SCA34 | 6q12.3-q16.1 | ELOVL4 | – | Erythrokeratodermia |
| SCA35 | 20p13 | TGM6 | – | |
| SCA36 | 20p13 | NOP56 | GGCCTG | ALS/FTD like |
| SCA37 | 1p32 | SCA37 | ||
| SCA38 | 6p12.1 | ELOVLE5 | ||
| SCA40 | 14q32 | CCDC88C | ||
| DRPLA | 12p13.31 | ATN1 | CAG | Huntington’s disease like |
ALS, amyotrophic lateral sclerosis; FTD, frontotemporal dementia; SCA, spinocerebellar ataxia.
SPINOCEREBELLAR ATAXIA TYPE 3 (MACHADO-JOSEPH DISEASE)
The clinical picture of SCA type 3 is pleomorphic, with cerebellar ataxia in association with pyramidal signs; peripheral amyotrophy; nystagmus, ophthalmoparesis, and bulging eyes (Fig. 4); fasciculations of the face, tongue, and, occasionally, the limbs; and dystonia and parkinsonism [3,4,12,13▪,46,47]. Non-motor and extracerebellar features are common in patients with SCA type 3, particularly sleep disorders, cognitive and affective disturbances, movement disorders, psychiatric symptoms, olfactory dysfunction, peripheral neuropathy, pain, cramps, fatigue, nutritional problems, and dysautonomia [48–51].
FIGURE 4.

‘Bulging eyes’ in a patient with spinocerebellar ataxia type 3. Adapted with permission.
OTHER SPINOCEREBELLAR ATAXIAS
Other SCAs encompass a broad spectrum of clinical features and include SCA type 10 (caused by an expansion of an ATTCT pentanucleotide repeat), which is the second most common SCA in Mexico and the South of Brazil (Fig. 5) [52]. In the latter, the main phenotype observed is pure cerebellar ataxia, unlike the typical phenotype, which consists of cerebellar ataxia and epilepsy [52]. More recently, several new forms of SCAs with new loci and gene mutations have been described. These include SCA types 31, 34, 35, 36, 37, 38 and 40 (Fig. 6) [53–60]. In spite of the very large number of mutations described in individuals with SCA, many patients (30–40%) remain without a genetic/molecular diagnosis [12,13▪,14].
FIGURE 5.
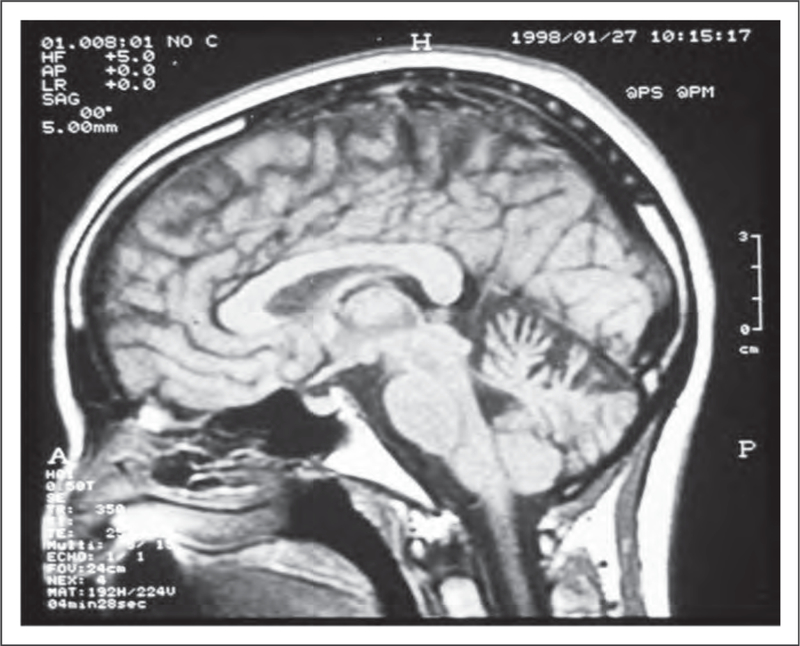
Brain MRI, T1-weighted, sagittal view. Cerebellar atrophy in a patient with spinocerebellar ataxia type 10. Adapted with permission.
FIGURE 6.
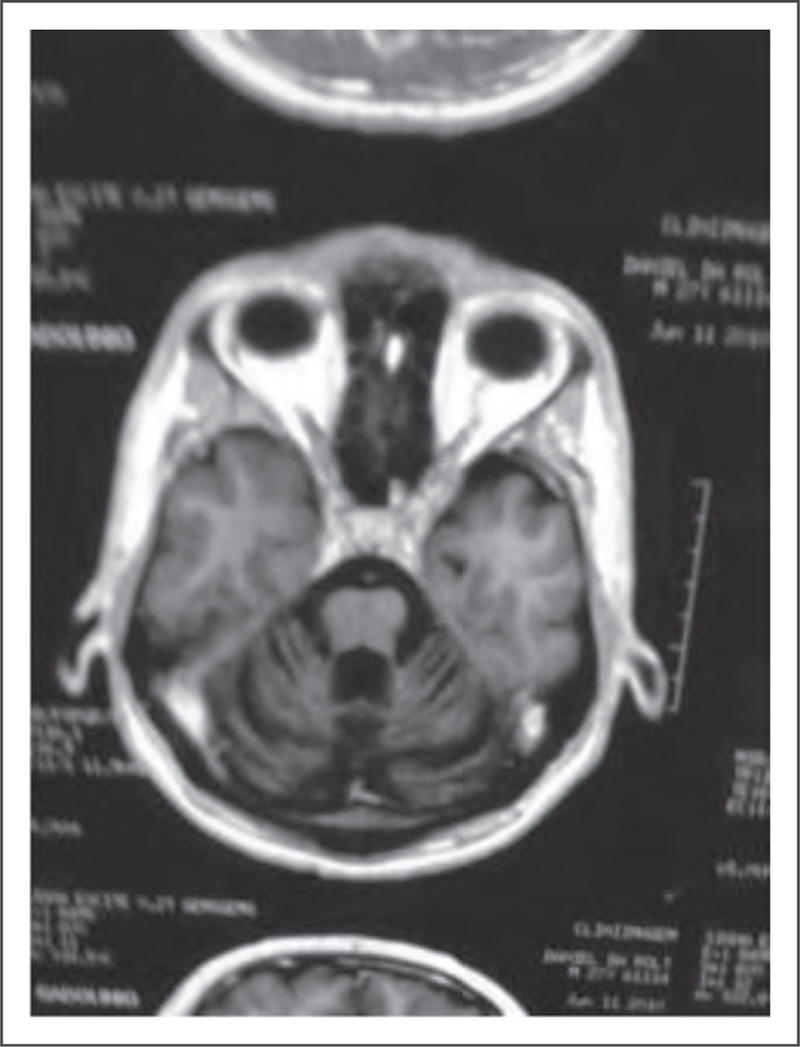
Spinocerebellar ataxia type 34. Brain MRI, axial view, T1-weighted, showing cerebellar atrophy. Adapted with permission.
SPINOCEREBELLAR ATAXIAS – TREATMENT
To date, there is no effective treatment for SCAs [62]. Neurorehabilitation is mandatory, and different studies using motor training, including physiotherapy or whole-body controlled videogames (‘exer-games’), have shown significant benefits for patients with SCAs [63,64▪]. New treatment options, including RNA interference therapy, and mesenchymal and cerebellar neural stem-cell transplantations, have been tested [65–68]. Treatment of SCA type 3 mouse models with the Hsp90 inhibitor 17-DMAG or overexpression of beclinl (an autophagy-related protein) was shown to mitigate motor and neuropathological deficits in SCA type 3 [69,70].
HEREDITARY EPISODIC ATAXIAS
Hereditary episodic ataxias are characterized by recurrent episodes of ataxia and vertigo, as well as progressive cerebellar ataxia [15,71,72▪]. To date, seven forms of episodic ataxia have been identified, the most common being types 1 and 2. Both are secondary to mutations in genes coding for ion channels and transport proteins [15,71,72▪]. Recently, mutations in the PRRT2 gene that cause autosomal dominant paroxysmal kinesigenic dyskinesia were associated to episodic ataxia [73] (Table 3). Acetazolamide, a carbonic-anhydrase inhibitor, may reduce the frequency and severity of attacks [15,71,72▪].
Table 3.
Episodic ataxias
| EA | Chromosome | Gene | Additional clinical features | Duration of ataxia attacks |
|---|---|---|---|---|
| EA-1 | 12p13 | KCNA1 | Facial myokimia/neuromyotonia | Seconds/up to 2 min |
| EA-2 | 19p13.2 | CAGNAIA | Nystagmus | Hours/days |
| EA-3 | 1q42 | – | Tinnitus/headache | – |
| EA-4 | – | – | Diplopia | Minutes/hours |
| EA-5 | 2q22–23 | CACNB4 | Epilepsy, vertigo | – |
| EA-6 | 5p13 | SLCA3 | Headache/hemiplegia | 2–3 h |
| EA-7 | 19q13 | – | Weakness | – |
| EA-Other | – | UBR4 | Weakness | – |
| EA-Other | – | PPRT2 | Paroxysmal dyskinesias | – |
EA, episodic ataxia; EA-Other, new loci of episodic ataxia, identified but still not confirmed.
X-LINKED ATAXIAS
X-linked SCAs are very rare forms of ataxia caused by X-linked recessive mutations [18▪]. At present, the most clinically relevant and common form is FXTAS [18▪]. FXTAS occurs predominantly in males over 50 years of age and is characterized by action tremor with an important kinetic component, cerebellar ataxia, cognitive dysfunction, and, occasionally, parkinsonism and autonomic dysfunction. It is caused by an intermediate CGG expansion (between 55 and 200 repeats) in the fragile X mental retardation 1 gene [18▪].
MITOCHONDRIAL ATAXIAS
In mitochondrial ataxias, cerebellar and sensory ataxias are usually combined with other features and are the result of abnormalities in mitochondrial DNA [19]. These forms of ataxia include maternally inherited heredoataxias due to point mutations in genes coding for RNAs and respiratory chain subunits or deletions/duplications of the mitochondrial DNA [19,74]. Mitochondrial recessive ataxia syndrome is caused by a mutation in the mitochondrial DNA POLG gene [19,74]. POLG-related ataxia is a mixed ataxia (with cerebellar and afferent/sensory ataxia) and presents with a large number of nonataxia features, such as sensory neuropathy, external ophthalmoplegia, ptosis, epilepsy, and hyperkinetic movement disorders [19,74].
IDIOPATHIC DEGENERATIVE ATAXIAS
Idiopathic cerebellar degeneration includes a group of disorders of unknown cause, such as MSA-C and idiopathic late-onset cerebellar ataxia, also known as sporadic adult-onset ataxia of unknown cause (SAOA) or even idiopathic sporadic cerebellar ataxia [3–5,75▪,76].
MULTIPLE SYSTEM ATROPHY
MSA is a sporadic and progressive neurodegenerative disease characterized by parkinsonism, cerebellar ataxia, and autonomic failure [75▪] (Fig. 7). In most Western populations, the clinical picture is dominated by parkinsonian features (defined as MSA-P), but cerebellar ataxia represents the most important motor feature in one-third of patients (defined as MSA-C). However, in Japan, there is a predominance of MSA-C (83.8%) [3,4,75▪]. Recently, homozygous and compound heterozygous mutations in the coenzyme Q2 gene (COQ2) were identified in Japanese patients with familial and sporadic MSA [77]. Although the frequency of mutations in COQ2 in a group of Chinese patients with MSA was 1.28%, [78] other studies failed to confirm the association between mutant COQ2 and MSA [79]. MSA and amyotrophic lateral sclerosis have been associated with a hexanucleotide repeat expansion in the protein C9orf72. This mutation has very diverse clinical presentations, including amyotrophic lateral sclerosis and frontotemporal dementia [80].
FIGURE 7.
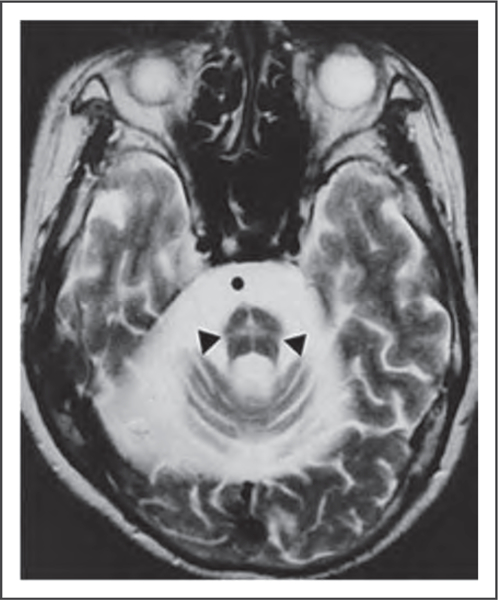
Hot cross bun sign of cerebellar form of multiple system atrophy. Adapted with permission from [61].
SPORADIC ADULT-ONSET ATAXIA OF UNKNOWN CAUSE
SAOA is an idiopathic neurodegenerative disorder previously defined as cerebello-olivary degeneration or pure cerebello-olivary degeneration of Marie, Foix, and Alajouanine [81]. Clinically, it presents as slowly progressive cerebellar ataxia, with onset after the age of 50 years and noncerebellar signs, such as chorea, and pyramidal and sensory signs [3,4,81]. The diagnosis is one of exclusion after secondary inherited ataxias and MSA have been eliminated [3,4]. In a Brazilian case series of eight elderly patients with SAOA, mild cognitive impairment and visual loss due to macular degeneration were observed in 50% of cases in addition to slowly progressive gait ataxia and cerebellar atrophy. Chorea was found concomitantly in three patients (H.A.G. Teive, unpublished).
WHOLE-EXOME SEQUENCING IN INHERITED AND SPORADIC ATAXIAS
Investigation of inherited ataxias is a challenge for neurologists because there is great clinical and genetic heterogeneity and a genetic diagnosis can only be made in 60% of cases [82]. Many patients therefore fail to receive a genetic diagnosis, limiting genetic counseling and prenatal diagnosis [83]. A powerful new tool that can be used to investigate these patients with undiagnosed inherited and sporadic ataxias is exome sequencing. Several studies have recently been published confirming that this is a very useful technique for evaluating this group of patients with primary ataxia [83,84▪].
SECONDARY ATAXIAS
Secondary or acquired ataxias include ataxias because of exogenous or endogenous nongenetic causes, including those of a toxic, paraneoplastic, immune-mediated, nutritional, and infectious nature, as well as focal injury to the cerebellum [3,4]. In this setting, neuroimaging studies are of capital importance in defining focal lesions in the cerebellum and its connections caused by, for example, neoplastic, inflammatory, demyelinating, and vascular disorders [3,4]. Ataxia can also be the result of the adverse effects of different drugs [85▪]. Drug-induced cerebellar ataxia is most commonly due to antiepileptic medicines (including oxcarbazepine, lamotrigine, and phenytoin), benzodiazepines (nitrazepam and triazolam), and antineoplastic/immunosuppressive drugs (cytarabine, tacrolimus, and cyclosporine) [85▪]. Chemicals such as alcohol, lithium, and toluene are also known to cause ataxia [85▪,86]. Several infectious disorders, such as syphilis and Whipple’s disease, mumps, and infectious mononucleosis, can lead to cerebellitis with cerebellar ataxia [3,4]. Endocrine abnormalities, particularly hypothyroidism, may present with cerebellar ataxia. Steroid-responsive encephalopathy associated with autoimmune thyroiditis, also known as Hashimoto’s encephalopathy, is defined by the presence of cerebellar ataxia, tremor, and myoclonus, as well as cognitive disorders and high serum levels of thyroperoxidase antibodies. As its name suggests, steroid-responsive encephalopathy associated with autoimmune thyroiditis responds well to steroid treatment [3,4]. Cerebellar and afferent/sensory ataxias can occur in individuals with a deficiency of vitamins such as thiamine, tocopherol, and cobalamine [3,4]. Paraneoplastic cerebellar degeneration is an immune-mediated cerebellar disorder associated with malignancy, particularly small-cell lung, breast, and ovary carcinomas, and Hodgkin’s lymphoma [87]. In paraneoplastic cerebellar degeneration, several types of autoantibodies are directed against neuronal antigens, the most common being anti-Yo, anti-Hu, and anti-Tr [3,4,87]. Cerebellar ataxia may occur in association with antibodies to glutamic acid decarboxylase (GAD), originally described in patients with stiff-person syndrome [3,4,87,88]. Anti-GAD ataxia is more common among women and can co-occur with insulin-dependent diabetes mellitus and thyroid diseases. Anti-GAD ataxia is variably responsive to intravenous immunoglobulins and steroids [3,4,87,88]. Gluten ataxia is another immune-mediated disorder caused by ingestion of gluten in patients who are genetically susceptible to this protein composite [89]. Gluten ataxia is characterized by progressive adult-onset gait ataxia with gaze-evoked nystagmus associated with signs of peripheral neuropathy. The antigliadin antibody is positive in 100% of patients. A gluten-free diet can improve gluten ataxia [89]. However, the relationship between cerebellar ataxia and antigliadin antibodies is currently very controversial, and several studies failed to confirm this association [90]. Finally, Miller Fisher syndrome, a Guillain-Barre syndrome variant, should also be considered in the differential diagnosis of cases with acute sensory ataxia [3,4].
CONCLUSION
Primary ataxias represent a very large group of neurodegenerative diseases and include sporadic and inherited forms, which are subdivided into congenital ataxias, ARCAs, SCAs, episodic ataxias, X-linked ataxias, mitochondrial ataxias, and idiopathic degenerative ataxias. Despite the increase in the number of defined genetic causes of primary ataxias, around 30–40% of patients remain without a diagnosis. Furthermore, to date, there is no cure for the majority of primary ataxias. Secondary ataxias are an important group of sporadic diseases and include immune-mediated ataxias and ataxias due to toxic agents and vitamin deficiencies among other causes.
KEY POINTS.
ARCAs are a group of primary ataxias; more than 30 forms have been described recently.
Autosomal dominant cerebellar ataxias or SCAs are a very large group of clinically and genetically very heterogeneous degenerative ataxias.
Other peculiar forms of primary ataxias are episodic ataxias, X-linked ataxias, and mitochondrial ataxias.
MSA-C and SAOA of unknown cause are idiopathic, degenerative forms of primary ataxias.
Secondary or acquired ataxias are immune-mediated ataxias or ataxias caused by toxic agents, particularly drugs and alcohol, and vitamin deficiencies.
Acknowledgements
The authors would like to thank the patients with ataxias for their permission to publish their photographs and MRI images.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
- 1.▪.Akbar U, Ashizawa T. Ataxia. Neurol Clin 2015; 33:225–248.Very nice overview about ataxia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pandolfo M, Manto M. Cerebellar and afferent ataxias. Continuum (Minneap Minn) 2013; 19:1312–1343. [DOI] [PubMed] [Google Scholar]
- 3.Klockgether T Acquired cerebellarataxiasand differential diagnosis In: Brice A, Pulst S-M, editors. Spinocerebellar degenerations. The ataxias and spastic paraplegias. Philadelphia: Butterworth Heinemann/Elsevier; 2007. pp. 61–77. [Google Scholar]
- 4.Teive HAG, Munhoz RP, Ashizawa T. Inherited and sporadic ataxias In: Albanese A, Jankovic J, editors. Hyperkinetic movementdisorders. Differential diagnosis and treatment. Oxford, UK: Wiley-Blackwell; 2012. pp. 279–295. [Google Scholar]
- 5.Klockgether T Update on degenerative ataxias. Curr Opin Neurol 2011; 24:339–345. [DOI] [PubMed] [Google Scholar]
- 6.Perlman SL. Spinocerebellar degenerations. Handb Clin Neurol 2011; 100:113–140. [DOI] [PubMed] [Google Scholar]
- 7.Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N Engl J Med 2012; 366:636–646. [DOI] [PubMed] [Google Scholar]
- 8.Vermeer S, van de Warrenburg BPC, Willemsen MAAP, et al. Autosomal recessive cerebellar ataxias: the current state of affairs. J Med Genet 2011; 48:651–659. [DOI] [PubMed] [Google Scholar]
- 9.DurrA.Autosomal dominant cerebellarataxias: polyglutamine expansions and beyond. Lancet Neurol 2010; 9:885–894. [DOI] [PubMed] [Google Scholar]
- 10.Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet 2009; 151C:326–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol 2013; 12:894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shakkottai VG, Fogel BL. Clinical neurogenetics: autosomal dominant spinocerebellar ataxia. Neurol Clin 2013; 31:987–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.▪.Rossi M, Perez-Lloret S, Doldan L, et al. Autosomal dominant cerebellar ataxias: a systematic review of clinical features. Eur J Neurol 2014; 21:607–615.Important and updated review about SCAs. [DOI] [PubMed] [Google Scholar]
- 14.Hersheson J, Haworth A, Houlden H. The inherited ataxias: genetic heterogeneity, mutation databases, and future directions in research and clinical diagnostics. Hum Mutat 2012; 33:1324–1332. [DOI] [PubMed] [Google Scholar]
- 15.Jen JC, Graves TD, Hess EJ, et al. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 2007; 130:2484–2493. [DOI] [PubMed] [Google Scholar]
- 16.Koeppen AH. Friedreich’s ataxia: pathology, pathogenesis, and molecular genetics. J Neurol Sci 2011; 303:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.▪.Méneret A,Ahmar-Beaugendre Y, Rieunier G, et al. The pleiotropic movement disorders phenotype of adult ataxia-telangiectasia. Neurology 2014; 83:1087–1095.Very interesting article about ataxia telangiectasia and its pleiotropic movement disorders. [DOI] [PubMed] [Google Scholar]
- 18.▪.Hagerman PJ, Hagerman RJ. Fragile X-associated tremor/ataxia syndrome. AnnNY Acad Sci 2015; 1338:58–70.Seminal review about FXTAS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horvath R, Hudson G, Ferrari G, et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain 2007; 129:1674–1684. [DOI] [PubMed] [Google Scholar]
- 20.Koeppen AH, Mazurkiewicz JE. Friedreich ataxia: neuropathology revised. J Neuropathol Exp Neurol 2013; 72:78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parkinson MH, Boesch S, NachbauerW, et al. Clinical features of Friedreich’s ataxia: classical and atypical phenotypes. J Neurochem 2013; 126:103–117. [DOI] [PubMed] [Google Scholar]
- 22.Hou JG, Jankovic J. Movement disorders in Friedreich’s ataxia. J Neurol Sci 2003; 206:59–64. [DOI] [PubMed] [Google Scholar]
- 23.Solbach K, Kraff O, Minnerop M, et al. Cerebellar pathology in Friedreich’s ataxia: atrophied dentate nuclei with normal iron content. Neuroimage Clin 2014; 6:93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rinaldi C, Tucci T, Maione S, et al. Low-dose idebenone treatment in Friedreich’s ataxia with and without cardiac hypertrophy. J Neurol 2009; 256:1434–1437. [DOI] [PubMed] [Google Scholar]
- 25.Lynch DR, Perlman SL, Meier T. A phase 3, double-blind, placebo-controlled trial of idebenone in Friedreich ataxia. Arch Neurol 2010; 67:941–947. [DOI] [PubMed] [Google Scholar]
- 26.Pandolfo M, Arpa J, Delatycki MB, et al. Deferiprone in Friedreich ataxia: a 6-month randomized controlled trial. Ann Neurol 2014; 76:509–521. [DOI] [PubMed] [Google Scholar]
- 27.Soragni E, Miao W, Ludicello M, et al. Epigenetic therapy for Friedeich ataxia. Ann Neurol 2014; 76:489–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verhagen MM, Abdo WF, Willemsen MA, et al. Clinical spectrum of ataxia- telangiectasia in adulthood. Neurology 2009; 73:430–437. [DOI] [PubMed] [Google Scholar]
- 29.▪.Synofzik M, Gonzalez MA, Lourenco CM, et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegeneration spectrum. Brain 2014; 137:69–77.Interesting article about PNPLA6 mutations and their phenotypes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heimdal K, Sanchez-Guixé M, Aukrust I, et al. STUB1 mutations in autosomal recessive ataxias - evidence for mutation-specific clinical heterogeneity. Orphanet J Rare Dis 2014; 9:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deik A, Johannes B, Rucker JC, et al. Compound heterozygous PNPLA6 mutations cause Boucher-Neuhauser syndrome with late-onset ataxia. J Neurol 2014; 261:2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Synofzik M, Kernstock C, Haack TB, Schols L. Ataxia meets chorioretinal dystrophy and hypogonadism: Boucher-Neuhauser syndrome due to PNPLA6 mutations. J Neurol Neurosurg Psychiatry 2015; 86:580–581. [DOI] [PubMed] [Google Scholar]
- 33.Tarnutzer AA, Gerth-Kahlert C, Timmann D, et al. Boucher-Neuhauser syndrome: cerebellar degeneration, chorioretinal dystrophy and hypogonadotropic hypogonadism: two novel cases and a review of 40 cases from the literature. J Neurol 2015; 262:194–202. [DOI] [PubMed] [Google Scholar]
- 34.Van Schil K, Meire F, Karlstetter M, et al. Early-onset autosomal recessive cerebellara ataxia associated with retinal dystrophy: new human hotfoot phenotype caused by homozygous GRID2 deletion. Genet Med 2015; 17:291–299. [DOI] [PubMed] [Google Scholar]
- 35.Thomas AC, Williams H, Setó-Salvia N, et al. Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am J Hum Genet 2014; 95:611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Egmond ME, Verschuuren-Bemelmans CC, Nibbeling EA, et al. Ramsay-Hunt syndrome: clinical characterization of progressive myoclonus ataxia caused by GOSR2 mutation. Mov Disord 2014; 29:139–143. [DOI] [PubMed] [Google Scholar]
- 37.Balreira A, Boczonadi V, Barca E, et al. ANO10 mutations cause ataxia and coenzyme Q10 deficiency. J Neurol 2014; 261:2192–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.▪.Renaud M, Anheim M, Kamsteeg EJ, et al. Autosomal recessive cerebellar ataxia type 3 due to ANO10 mutations: delineation and genotype-phenotype correlation study. JAMA Neurol 2014; 71:1305–1310.Important article about ANO10 mutations. [DOI] [PubMed] [Google Scholar]
- 39.Noreau A, Bourassa CV, Szuto A, et al. SYNE1 mutations in autosomal recessive cerebellar ataxia. JAMA Neurol 2013; 70:1296–1301. [DOI] [PubMed] [Google Scholar]
- 40.Nanetti L, Cavalieri S, Pensato V, et al. SETX mutations are a frequent genetic cause of juvenile and adult onset cerebellar ataxia with neuropathy and elevated serum alpha-fetoprotein. Orphanet J Rare Dis 2013; 8:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu YT, Hersheson J, Plagnol V, et al. Autosomal-recessive cerebellar ataxia caused by a novel ADCK3 mutation that elongates the protein: clinical, genetic and biochemical characterization. J Neurol Neurosurg Psychiatry 2014; 85:493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Demos MK, van Karnebeek CD, Ross CJ, et al. A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet J Rare Dis 2014; 9:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Synofzik M, Soehn AS, Gburek-Augustat J, et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum. Orphanet J Rare Dis 2013; 8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.▪▪.Hekman KE, Gomez CM. The autosomal dominant spinocerebellar ataxias: emerging mechanistic themes suggest pervasive Purkinje cell vulnerability. J Neurol Neurosurg Psychiatry 2015; 86:554–561.Very nice article about etiopathogenesis of the SCAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.▪▪.Matilla-Duenas A, Ashizawa T, Brice A, et al. Consensus paper: pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias. Cerebellum 2014; 13:269–302.Amazing article about neurodegeneration in SCAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moro A, Munhoz RP, Arruda WO, et al. Clinical relevance of ‘bulging eyes’ for the differential diagnosis of spinocerebellar ataxias. Arq Neuropsiquiatr 2013; 71:428–430. [DOI] [PubMed] [Google Scholar]
- 47.Teive HAG, Munhoz RP, Arruda WO, et al. Spinocerebellar ataxia type 3: sub¬phenotypes in a cohort of Brazilian patients. Arq Neuropsiquiatr 2014; 72:659–662. [DOI] [PubMed] [Google Scholar]
- 48.Pedroso JL, Franca MC Jr, Braga-Neto P, et al. Nonmotor and extracerebellar features in Machado-Joseph disease: a review. Mov Disord 2013; 28:1200–1208. [DOI] [PubMed] [Google Scholar]
- 49.van Gaalen J, Giunti P, van de Warrenburg BP. Movement disorders in spinocerebellar ataxias. Mov Disord 2011; 26:792–800. [DOI] [PubMed] [Google Scholar]
- 50.Moro A, Munhoz RP, Moscovich M, et al. Movement disorders in spinocerebellar ataxias in a cohort of Brazilian patients. Eur Neurol 2014; 72:360362. [DOI] [PubMed] [Google Scholar]
- 51.Moscovich M, Munhoz RP, Teive HAG, et al. Olfactory impairment in familial ataxias. J Neurol Neurosurg Psychiatry 2012; 83:97–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teive HAG, Munhoz RP, Arruda WO, et al. Spinocerebellar ataxia type 10: a review. Parkinsonism Relat Disord 2011; 17:655–661. [DOI] [PubMed] [Google Scholar]
- 53.Serrano-Munuera C, Corral-Juan M, Stevanin G, et al. New subtype of spinocerebellar ataxia with altered vertical eye movements mapping to chromosome 1p32. JAMA Neurol 2013; 70:764–771. [DOI] [PubMed] [Google Scholar]
- 54.Kobayashi H, Abe K, Matsuura T, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 2011; 89:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ikeda Y, Ohta Y, Kobayashi H, et al. Clinical features of SCA36. A novel spinocerebellar ataxia with motor neuron involvement (Asidan). Neurology 2012; 79:333–341. [DOI] [PubMed] [Google Scholar]
- 56.Garcia-Murias M, Quintáns B, Arias M, et al. Costa da morte’ ataxia is spinocerebellar ataxia 36: clinical and genetic characterization. Brain 2012; 135:1423–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Obayashi M, Stevanin G, Synofzik M, et al. Spinocerebellar ataxia type 36 exists in diverse populations and can be caused by a short hexanucleotide GGCCTG repeat expansion. J Neurol Neurosurg Psychiatry 2014. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 58.Guo YC, Lin JJ, Liao YC, et al. Spinocerebellar ataxia 35. Novel mutations in TGM6 with clinical and genetic characterization. Neurology 2014; 83:1554–1561. [DOI] [PubMed] [Google Scholar]
- 59.Cadieux-Dion M, Turcotte-Gauthier M, Noreau A, et al. Expanding the clinical phenotype associated with ELOVL4 mutation. Study of a large French-Canadian family with autosomal dominant spinocerebellar ataxia and erythro-keratodermia. JAMA Neurol 2014; 71:470–475. [DOI] [PubMed] [Google Scholar]
- 60.Di Gregorio E, Borroni B, Giorgio E, et al. ELOVL5 mutations cause spinocerebellar ataxia 38. Am J Hum Genet 2014; 95:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsoi H, Yu ACS, Chen ZS, et al. A novel missense mutation in CCDC88C activates the JNK pathway and causes a dominant form of spinocerebellar ataxia. J Med Genet 2014; 51:590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bushara K We cannot cure ataxia, we can only eradicate it. JAMA Neurol 2013; 70:1099. [DOI] [PubMed] [Google Scholar]
- 63.Synofzik M, Ilg W. Motor training in degenerative spinocerebellar disease: ataxia-specific improvements by intensive physiotherapy and exergames. Biomed Res Int 2014; 2014:583507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.▪.Ilg W, Bastian AJ, Boesch S, et al. Consensus paper: management of degenerative cerebellar disorders. Cerebellum 2014; 13:248–268.Excellent review about management of cerebellar ataxia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Costa Mdo C, Luna-Cancalon K, Fischer S, et al. Toward RNAi therapy for the polyglutamine disease Machado-Joseph disease. Mol Ther 2013; 21:1898–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nobrega C, Nascimento-Ferreira I, Onofre I, et al. RNA interference mitigates motor and neuropathological deficits in a cerebellar mouse model of Macha-do-Joseph disease. Plos One 2014; 9:e100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nakamura K, Mieda T, Suto N, et al. Mesenchymal stem cells as a potential therapeutic tool for spinocerebellar ataxia. Cerebellum 2014; 14:165–170. [DOI] [PubMed] [Google Scholar]
- 68.Mendonca LS, Nobrega C, Hirai H, et al. Transplantation of cerebellar neural stem cells improves motor coordination and neuropathology in Machado-Joseph disease mice. Brain 2015; 138:320–335. [DOI] [PubMed] [Google Scholar]
- 69.Silva-Fernandes A, Duarte-Silva S, Neves-Carvalho A, et al. Chronic treatment with 17-DMAG improves balance and coordination in a new mouse model of Machado-Joseph disease. Neurotherapeutics 2014; 11:433–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nascimento-Ferreira I, Nobrega C, Vasconcelos-Ferreira A, et al. Beclin 1 mitigates motor and neuropathological deficits in genetic mouse models of Machado-Joseph disease. Brain 2013; 136:2173–2188. [DOI] [PubMed] [Google Scholar]
- 71.Riant F, Vahedi K, Tournier-Lasserve E. Hereditary episodic ataxia. Rev Neurol 2011; 167:401–4407. [DOI] [PubMed] [Google Scholar]
- 72.▪.Waln O, Jankovic J. Paroxysmal movement disorders. Neurol Clin 2015; & 33:137–152.Very nice review about paroxysmal movement disorders, particularly about episodic ataxias. [DOI] [PubMed] [Google Scholar]
- 73.Gardiner AR, Bhatia KP, Stamelou M, et al. PRRT2 gene mutations: from paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine. Neurology 2012; 79:2115–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Synofzik M, Srulijes K, Godau J, et al. Characterizing POLG ataxia: clinics, electrophysiology and imaging. Cerebellum 2012; 11:1002–1011. [DOI] [PubMed] [Google Scholar]
- 75.▪.Lin DJ, Hermann KL, Schmahmann JD. Multiple system atrophy of the “ cerebellar type: clinical state of the art. Mov Disord 2014; 29:294–304.Very relevant article about MSA-C. [DOI] [PubMed] [Google Scholar]
- 76.Klockgether T Sporadic adult-onset ataxia of unknown etiology. Hand Clin Neurol 2012; 103:253–262. [DOI] [PubMed] [Google Scholar]
- 77.The Multiple-System Atrophy Research Collaboration. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med 2013; 369:233–244. [DOI] [PubMed] [Google Scholar]
- 78.Chen YP, Zhao B, Cao B, et al. Mutation of the COQ2 gene in ethnic Chinese patients with multiple-system atrophy. Neurobiol Aging 2015; 36:1222.e7–1222.e11. [DOI] [PubMed] [Google Scholar]
- 79.Schottlaender LV, Houlden H. Multiple-system atrophy (MSA) Brain Bank Collaboration. N Engl J Med 2014; 371:81. [DOI] [PubMed] [Google Scholar]
- 80.Goldman JS, Quinzii C, Dunning-Broadbent J, et al. Multiple system atrophy and amyotrophic lateral sclerosis in a family with hexanucleotide repeat expansions in C9orf72. JAMA Neurol 2014; 71:771–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fox SH, Nieves A, Bergeron C, Lang AE. Pure cerebello-olivary degeneration of Marie, Foix, and Alajouanine presenting with progressive cerebellar ataxia, cognitive decline, and chorea. Mov Disord 2003; 18:1550–1554. [DOI] [PubMed] [Google Scholar]
- 82.Nemeth AH, Kwasniewska AC, Lise S, et al. Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain 2013; 136:3106–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fogel BL, Lee H, Deignan JL, et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol 2014; 71:1237–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.▪.Pyle A, Smertenko T, Bargiela D, et al. Exome sequencing in underdiagnosed & inherited and sporadic ataxias. Brain 2015; 138:276–283.Very interesting update about whole-exome sequencing in SCAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.▪.van Gaalen J, Kerstens FG, Maas RP, et al. Drug-induced cerebellar ataxia: a & systematic review. CNS Drugs 2014; 28:1139–1153.Interesting article about drug-induced cerebellar ataxias. [DOI] [PubMed] [Google Scholar]
- 86.Manto M Toxic agents causing cerebellar ataxias. Handb Clin Neurol 2012; 103:201–213. [DOI] [PubMed] [Google Scholar]
- 87.Demarquay G, Honnorat J. Clinical presentation of immune-mediated cere¬bellar ataxia. Rev Neurol 2011; 167:408–417. [DOI] [PubMed] [Google Scholar]
- 88.Arino H, Gresa-Arribas N, Blanco Y, et al. Cerebellar ataxia and glutamic acid decarboxylase antibodies: immunologic profile and long-term effect of immunotherapy. JAMA Neurol 2014; 71:1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hadjivassiliou M, Aeschlimann P, Sanders DS, et al. Transgutaminase 6 antibodies in the diagnosis of gluten ataxia. Neurology 2013; 80:1740–1745. [DOI] [PubMed] [Google Scholar]
- 90.McKeon A, Lennon VA, Pittock SJ, et al. The neurologic significance of celiac disease biomarkers. Neurology 2014; 83:1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]