

Abstract
Background
Cystic fibrosis is a genetic disorder in which abnormal mucus in the lungs is associated with susceptibility to persistent infection. Pulmonary exacerbations are when symptoms of infection become more severe. Antibiotics are an essential part of treatment for exacerbations and inhaled antibiotics may be used alone or in conjunction with oral antibiotics for milder exacerbations or with intravenous antibiotics for more severe infections. Inhaled antibiotics do not cause the same adverse effects as intravenous antibiotics and may prove an alternative in people with poor access to their veins. This is an update of a previously published review.
Objectives
To determine if treatment of pulmonary exacerbations with inhaled antibiotics in people with cystic fibrosis improves their quality of life, reduces time off school or work and improves their long‐term survival.
Search methods
We searched the Cochrane Cystic Fibrosis Group's Cystic Fibrosis Trials Register. Date of the last search: 03 October 2018.
We searched ClinicalTrials.gov, the Australia and New Zealand Clinical Trials Registry and WHO ICTRP for relevant trials. Date of last search: 09 October 2018.
Selection criteria
Randomised controlled trials in people with cystic fibrosis with a pulmonary exacerbation in whom treatment with inhaled antibiotics was compared to placebo, standard treatment or another inhaled antibiotic for between one and four weeks.
Data collection and analysis
Two review authors independently selected eligible trials, assessed the risk of bias in each trial and extracted data. They assessed the quality of the evidence using the GRADE criteria. Authors of the included trials were contacted for more information.
Main results
Four trials with 167 participants are included in the review. Two trials (77 participants) compared inhaled antibiotics alone to intravenous antibiotics alone and two trials (90 participants) compared a combination of inhaled and intravenous antibiotics to intravenous antibiotics alone. Trials were heterogenous in design and two were only available in abstract form. Risk of bias was difficult to assess in most trials, but for all trials we judged there to be a high risk from lack of blinding and an unclear risk with regards to randomisation. Results were not fully reported and only limited data were available for analysis.
Inhaled antibiotics alone versus intravenous antibiotics alone
Only one trial (n = 18) reported a perceived improvement in lifestyle (quality of life) in both groups (very low‐quality of evidence). Neither trial reported on time off work or school. Both trials measured lung function, but there was no difference reported between treatment groups (very low‐quality evidence). With regards to our secondary outcomes, one trial (n = 18) reported no difference in the need for additional antibiotics and the second trial (n = 59) reported on the time to next exacerbation. In neither case was a difference between treatments identified (both very low‐quality evidence). The single trial (n = 18) measuring adverse events and sputum microbiology did not observe any in either treatment group for either outcome (very low‐quality evidence).
Inhaled antibiotics plus intravenous antibiotics versus intravenous antibiotics alone
Neither trial reported on quality of life or time off work or school. Both trials measured lung function, but found no difference between groups in forced expiratory volume in one second (one trial, n = 28, very low‐quality evidence) or vital capacity (one trial, n = 62). Neither trial reported on the need for additional antibiotics or the time to the next exacerbation; however, one trial (n = 28) reported on hospital admissions and found no difference between groups. Both trials reported no difference between groups in adverse events (very low‐quality evidence) and one trial (n = 62) reported no difference in the emergence of antibiotic‐resistant organisms (very low‐quality evidence).
Authors' conclusions
There is little useful high‐level evidence to judge the effectiveness of inhaled antibiotics for the treatment of pulmonary exacerbations in people with cystic fibrosis. The included trials were not sufficiently powered to achieve their goals. Hence, we are unable to demonstrate whether one treatment was superior to the other or not. Further research is needed to establish whether inhaled tobramycin may be used as an alternative to intravenous tobramycin for some pulmonary exacerbations.
Plain language summary
Inhaling antibiotics to treat temporary worsening of lung infection in people with cystic fibrosis
Review question
We reviewed the evidence for inhaling antibiotics to treat exacerbations (flare ups) of lung infection in people with cystic fibrosis.
Background
Cystic fibrosis is a serious genetic disorder that results in abnormally sticky mucus in several parts of the body. In the lungs the sticky mucus can lead to repeated infection. An exacerbation makes the symptoms more severe. Antibiotics are an essential part of treatment and may be given by mouth, by needle into the blood stream or by inhaling the drug. We wanted to learn if inhaling antibiotics improved general health compared to the other methods.
This might mean that people with cystic fibrosis could avoid hospitalisation for intravenous antibiotics and some side effects. Inhaling the antibiotics would also be easier for people who have difficulty with access to their veins. This is an updated version of the review.
Search date
The evidence is current to: 09 October 2018.
Study characteristics
We found four trials with a total of 167 participants, two of which compared inhaled antibiotics alone to intravenous antibiotics alone (77 participants) and two which compared a combination of inhaled and intravenous antibiotics to intravenous antibiotics alone (90 participants) for treating exacerbations in people with cystic fibrosis. In all trials the inhaled antibiotics were compared to the same antibiotics given intravenously. The numbers of participants in each trial ranged from 18 to 62.
Key results
Inhaled antibiotics alone versus intravenous antibiotics alone
One trial (18 participants) reported a perceived improvement in lifestyle in both groups but neither trial reported on time off work or school. Both trials measured lung function, but neither reported any difference between treatment groups. One trial (18 participants) reported no difference in the need for additional antibiotics and the second trial (59 participants) reported on the time to next exacerbation ‐ there was no difference between inhaled or intravenous antibiotics for either outcome. Only one trial (18 participants) measured adverse events and sputum microbiology, but did not find any difference between treatments for either outcome.
Inhaled antibiotics plus intravenous antibiotics versus intravenous antibiotics alone
Neither trial reported on quality of life or time off work or school. Both trials reported lung function, but found no difference between groups. Neither trial reported on the need for additional antibiotics or the time to the next exacerbation; however, one trial (28 participants) reported on hospital admissions and found no difference between groups. Both trials reported no difference between groups in adverse events and one trial (62 participants) reported no difference in the emergence of antibiotic‐resistant organisms.
Quality of the evidence
We graded the quality of the evidence as very low. We had concerns since none of the trials stated how the participants were diagnosed with CF or how they defined an exacerbation. It was not possible to keep the treatment group secret from the participants as the trials compared different ways of giving the antibiotics and we thought this would likely influence some of the results. We were not sure whether the participants were put into the different groups truly at random and we do not know how this might affect the results.
Summary of findings
Background
Description of the condition
Pulmonary manifestations of cystic fibrosis (CF) are characterised by abnormal airway secretions associated with infection and inflammation leading to bronchiectasis. In almost all people with CF, there is an inevitable progression in the severity of lung disease resulting in more severe airway damage, progressive airflow obstruction and premature death due to respiratory failure (Gibson 2003a).
It is well‐recognised that there are periodic increases in the severity of lung disease, which are referred to as pulmonary exacerbations (Elborn 2007). Exacerbations are one of the most important clinical events in the course of the disease for people with CF because of the increased symptoms, the acceleration in the rate of decline in lung function, and the need for increased treatment (de Boer 2011; Sanders 2010; Sanders 2011). Severe exacerbations have been associated with CF‐related diabetes (Marshall 2005), sleep disturbances (Dobbin 2005) and may lead to reduced survival (de Boer 2011). Multiple factors play a role in the pathogenesis of pulmonary exacerbations, such as respiratory microbiome, host defences and environmental exposures (Goss 2007).
Defining pulmonary exacerbations in CF is challenging due to the variability and the subjective nature of presenting symptoms. A combination of symptoms, physical signs and laboratory findings have been used to help with diagnosis and grade severity (Gibson 2003a); the major criteria for diagnosing a pulmonary exacerbation are changes in an individual's symptoms (Goss 2007). The Fuchs criteria have been used in many clinical trials to define an exacerbation (Bilton 2011). It relies on treatment with intravenous antibiotics for four of the following respiratory signs or symptoms: new or increased haemoptysis; a change in sputum; increased cough; increased dyspnoea; fever above 38°C; fatigue; malaise; weight loss; sinus pain or pressure; change in sinus drainage; change in lung auscultation; a drop in FEV1 of at least 10% from baseline; or new radiographic findings to identify an exacerbation state (Fuchs 1994). However, no consensus diagnostic criteria exist (BMJ 2018).
Description of the intervention
Antibacterial drugs are an essential treatment for pulmonary exacerbations. The selection of antibiotic treatment will depend on the organism(s) usually found in respiratory secretions, as well as the precipitating factor or identification of a new infection, or both. Pseudomonas aeruginosa is the usual organism, particularly in adults (Smyth 2006), although methicillin‐resistant Staphylococcus aureus (MRSA) has recently emerged as an important pathogen in people with CF, with a 10‐fold increase in prevalence between 1996 and 2014 (Jennings 2017).
Most inhaled antibiotics are delivered as an aerosol, generated from a nebuliser. Administration takes between five and 20 minutes, generally twice each day; the shorter delivery times reflect relatively recent developments in nebuliser technology. The nebulisers which are used to administer the antibiotics are relatively expensive and require training of the patient or caregiver to use them correctly. There are dry powder devices becoming available which should be more convenient than nebulisers. The type of nebulisers used to administer drugs in CF is the subject of a further Cochrane Review (Daniels 2013).
How the intervention might work
Treatment of exacerbations using antibiotics with activity against P aeruginosa reduces symptoms and improves lung function (Gold 1987; Wientzen 1980). A Cochrane Review of nebulised anti‐pseudomonal antibiotics for maintenance treatment of individuals with CF and P aeruginosa infection have shown them to be associated with an improvement in lung function and a reduction in the frequency of exacerbations requiring additional antibiotic treatment (Smith 2018). In people with stable disease, inhaled antibiotics have been shown to reduce concentrations of P aeruginosa in sputum and to increase forced expiratory volume at one second (FEV1) two weeks after onset of treatment suggesting their usefulness for treating exacerbations (Ramsey 1993).
Why it is important to do this review
In practice, inhaled antibiotics are almost certainly used to treat pulmonary exacerbations. The global frequency of such use is not known, but an article published by the Epidemiologic Study of Cystic Fibrosis (ESCF) from the USA and Canada reported that 24% of pulmonary exacerbations were treated with inhaled antibiotics (Wagener 2012). While their use has been further noted in earlier papers (Moskowitz 2008; Smyth 2008) and four articles reviewing treatment of pulmonary exacerbations mention the use of inhaled antibiotics (Flume 2009; Gibson 2003a; Smyth 2006; Smyth 2008), there are no firm recommendations and no evidence is cited. The use of inhaled antibiotics for P aeruginosa was recommended in the recent National Institute for Health and Care Excellence (NICE) guidance in conjunction with intravenous (IV) antibiotics in individuals experiencing new P aeruginosa infection to facilitate eradication; however, there is no strong evidence to support this recommendation (NICE 2017).
New inhaled antibiotics are being developed, as are devices for administering them, and these will probably increase inhaled antibiotic use for treating pulmonary exacerbations.
The use of inhaled antibiotics to treat pulmonary exacerbations has advantages compared to IV administration. Inhaled antibiotics can augment oral therapy for milder exacerbations and avoid hospitalisation and IV access. Furthermore, inhaled aminoglycosides can reduce the risk of kidney damage and loss of hearing that is associated with IV aminoglycosides. Finally, inhaled antibiotics could provide another treatment option to IV antibiotics for those individuals with difficult venous access.
Hence, there is a need to establish whether there is evidence of an effect of inhaled antibiotics for treating pulmonary exacerbations in CF.
Objectives
To determine if treatment of pulmonary exacerbations with inhaled antibiotics in people with CF improves their quality of life (QoL), reduces time off school or work and improves their long‐term lung function.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials.
Types of participants
Children and adults with CF who are diagnosed with having a pulmonary exacerbation. Diagnosis of CF to be made using clinical criteria and confirmed by sweat testing or genetic analysis. A pulmonary exacerbation was taken as defined by the individual trial protocol.
Types of interventions
We will consider any inhaled antibiotic, at any dose, using any method of aerosol delivery. Duration of treatment will be between one and four weeks. The inhaled antibiotic will be administered alone or in addition to the usual treatment for pulmonary exacerbations and compared to either placebo or other antibiotic treatment.
Types of outcome measures
Primary outcomes
QoL (as measured by a validated tool such as CFQoL (Gee 2000) or CFQ‐R (Quittner 2009))
Time off work or school
-
Lung function (spirometry)
FEV1 (litre or per cent (%) predicted) absolute values or change values
FVC (litre or % predicted) absolute values or change values
Annual change in FEV1
Secondary outcomes
Need for hospital admission (in the short term (up to four weeks))
-
Need for additional antibiotics
IV
oral
Time to next pulmonary exacerbation
Weight (kg)
-
Adverse effects
mild ‐ resulting in no change in treatment (e.g. cough, bronchospasm)
moderate ‐ resulting in change in treatment (e.g. loss of hearing, nephrotoxicity)
severe ‐ needs hospital admission or is life‐threatening (e.g. anaphylactic reactions, nephrotoxicity)
-
Microbiology
emergence of new organisms
emergence of resistant organisms
Search methods for identification of studies
There are no restrictions regarding language or publication status.
Electronic searches
We identified relevant trials from the Group's Cystic Fibrosis Trials Register using the terms: antibiotics AND (acute treatment OR unknown) AND (inhaled OR not stated).
The Cystic Fibrosis Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of The Cochrane Library), weekly searches of MEDLINE, a search of EMBASE to 1995 and the prospective handsearching of two journals ‐ Pediatric Pulmonology and the Journal of Cystic Fibrosis. Unpublished work is identified by searching the abstract books of three major cystic fibrosis conferences: the International Cystic Fibrosis Conference; the European Cystic Fibrosis Conference and the North American Cystic Fibrosis Conference. For full details of all searching activities for the register, please see the relevant sections of the Cystic Fibrosis and Genetic Disorders Group's website.
Date of latest search of CF Trials Register: 03 October 2018.
We also searched for ongoing trials using ClinicalTrials.gov (using the terms cystic fibrosis AND inhaled antibiotics), the World Health Organisation International Clinical Trials Registry Platform (WHO ICTRP) (using the terms cystic fibrosis AND inhaled antibiotics) and the Australia and New Zealand Clinical Trials Registry (using the terms cystic fibrosis AND inhaled antibiotics).
Date searched: 09 October 2018.
Data collection and analysis
For any of the methods stated below, which we have not been able to undertake in this version of the review, we plan to do so if data become available for a future update.
Selection of studies
Two review authors independently screened titles and abstracts of the citations retrieved from the searches (SS and EC). The same two authors read the full text articles identified from the title and abstract screening to select trials that met the inclusion criteria (for earlier versions of the review NJ and TR, for this update two of the three authors SS and EC). Where there was disagreement between the authors on the trials selected, we attempted to reach a decision by consensus or by involving the third author to arbitrate (NR). Full text articles from the previous version of the review were not re‐assessed.
Data extraction and management
Two authors recorded details of trial design, participant characteristics, interventions, quality assessment and the relevant outcome data using a customised data extraction form (for previous versions of the review NJ and TR, from 2018 SS and EC). We settled any disagreement by consensus. We have reported the following characteristics in the 'Characteristics of included studies' table where data were available from the papers:
criteria for diagnosis of CF;
definition of pulmonary exacerbation;
type of infection;
trial design;
inhaled antibiotic and dose;
aerosol delivery method;
duration of treatment;
comparison intervention;
other treatments (e.g. IV or oral antibiotics and setting (inpatient or outpatient));and
severity of exacerbation using baseline FEV1 (% predicted).
We have presented separate comparisons for inhaled antibiotics alone versus IV antibiotics and for combination inhaled and IV antibiotics versus IV antibiotics alone. As planned, we have reported outcomes at up to one week, between one and two weeks, more than two weeks to three weeks, more than three weeks to four weeks. We have also considered additional follow‐up data recorded at other time periods.
It is recognised that lung function is an indicator of morbidity and mortality, so, if in future we obtain data for annual change in FEV1 we will report this as a surrogate marker for long‐term survival.
Assessment of risk of bias in included studies
Two authors (for previous versions of the review NJ and TR, from 2018 SS and EC) assessed the risk of bias of the selected trials using the domain‐based evaluation as described in theCochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We assessed the following domains as having either a low, unclear or high risk of bias:
randomisation (low risk ‐ random number table, computer‐generated lists or similar methods; unclear risk ‐ described as randomised, but no details given; high risk ‐ e.g. alternation, the use of case record numbers, and dates of birth or day of the week);
concealment of allocation (low risk ‐ e.g. list from a central independent unit, on‐site locked computer, identically appearing numbered drug bottles or containers prepared by an independent pharmacist or investigator, or sealed opaque envelopes; unclear risk ‐ not described; high risk ‐ if allocation sequence was known to, or could be deciphered by the investigators who assigned participants or if the trial was quasi‐randomised);
blinding (of participants, personnel and outcome assessors);
incomplete outcome data (whether investigators used an intention‐to‐treat analysis);
selective outcome reporting;
other potential sources of bias.
We also noted whether the included trials reported any sample size calculations.
Where there was disagreement between the authors on a trial's evaluation, we reached a decision by consensus or by mediation by the contact editor. Results of the risk of bias assessment are presented in the 'Risk of bias' tables (Characteristics of included studies). The new author team did not re‐assess the judgements for trials previously included in the review.
Measures of treatment effect
For binary outcome measures, we calculated a pooled estimate of the treatment effect for each outcome across trials using risk ratio (RR) and 95% confidence intervals (CIs) where appropriate.
If other types of data are included in future updates of the review, we will analyse these as follows. For continuous outcomes, we will record either mean relative change from baseline for each group or mean post‐treatment or post‐intervention values and the standard deviation (SD). If the papers report standard errors (SE) (and if it is possible) we will convert these to SDs. We will present a pooled estimate of treatment effect by calculating the mean difference and 95% CIs. If we become aware that some data are skewed and therefore we are not able to enter and analyse these within RevMan, we will report these narratively (RevMan 2014).
We will analyse any count data using a rate ratio (or narratively if this is not possible).
For any time‐to‐event outcomes included in the review, we plan to obtain a mixture of logrank and Cox model estimates from the trials; we aim to combine all results using the generic inverse variance method as we hope to be able to convert the logrank estimates into log hazard ratios and SEs as detailed in theCochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
We will analyse longitudinal data using the most appropriate method available (Jones 2009).
Unit of analysis issues
An important factor in the validity of cross‐over trials is that the severity of the disease is stable, so that disease status at the start point of each period is similar. This is very unlikely to be the case for pulmonary exacerbations in CF and therefore we will only include first‐arm data from any eligible cross‐over trials; where these data are unavailable we have excluded these trials from the review.
Dealing with missing data
Where possible, we have reported the numbers and reasons for dropouts and withdrawals in all intervention groups. We have also stated whether the papers specify that there were no dropouts or withdrawals. We have contacted authors for clarification on some missing information.
In order to allow an intention‐to‐treat analysis, we sought data on the number of participants with each outcome event, by allocated treated group, irrespective of compliance and whether or not the individual was later thought to be ineligible or otherwise excluded from treatment or follow‐up.
Assessment of heterogeneity
If sufficient data had been available, we planned to assess the degree of heterogeneity between trials through visual examination of the combined data presented in the forest plots, and by considering the I² statistic (Higgins 2003) together with Chi² values (Deeks 2011) and their confidence intervals. The I² statistic is a measure which describes the percentage of total variation across trials that are due to heterogeneity rather than by chance (Higgins 2003). The values of I² lie between 0% and 100%, and a simplified categorization of heterogeneity that we plan to use is of low (I² value of approximately 25%), moderate (I² value of approximately 50%), and high (I² value of approximately 75%) (Higgins 2003).
Assessment of reporting biases
In the tables below, we reported when the primary investigators took measurements during the trial, what measurements were reported within the published paper and what data we report in the review (Characteristics of included studies). For the two full papers included, we compared the methods sections to the results sections to identify any potential selective reporting. We also used knowledge of the clinical background to identify standard outcome measures that are used, but may not have been reported by the investigators. We further attempted to assess the impact of the reporting of several surrogate outcomes that are not directly relevant.
If, in future updates, we are able to include a sufficient number of trials, we will attempt to assess whether our review is subject to publication bias by using a funnel plot. If asymmetry is detected, causes other than publication bias will be explored.
Data synthesis
If, in future updates of this review, we identify moderate to high levels of heterogeneity (as defined above), we will present pooled estimates of the treatment effect using a random‐effects model. If this level of heterogeneity is not identified, we will compute pooled estimates of the treatment effect for each outcome under a fixed‐effect model.
Subgroup analysis and investigation of heterogeneity
In future, if we find moderate to high heterogeneity (over 50%) and a sufficient number of trials are included, we will investigate the possible causes further by performing subgroup analyses based on the the dose and the inhaled antibiotic, the pathogen causing the exacerbation, the severity of the pulmonary exacerbation, the severity of respiratory disease prior to the exacerbation (FEV1 % predicted*), age of participants (children versus adults) and the duration and setting of treatment.
We plan to use the definitions of disease severity presented by the American Thoracic Society (Pellegrino 2005):
| Degree of severity | FEV1 % predicted |
| Mild | over 70% |
| Moderate | 60% to 69% |
| Moderately severe | 50% to 59% |
| Severe | 35% to 49% |
| Very severe | less than 35% |
Sensitivity analysis
If we are able to include a sufficient number of trials in a future update of the review, we plan to perform a sensitivity analysis based on the risk of bias of the trials (e.g. including and excluding trials assessed as having a high risk of bias).
Summary of findings
In accordance with current Cochrane guidance we have included a summary of findings table for each comparison in the review. The two comparisons are as follows:
inhaled antibiotics compared to IV antibiotics;
an inhaled antibiotic in addition to IV antibiotics compared to IV antibiotics.
We have selected the following seven outcomes, which we consider to be the most important, to include in the tables.
QoL (as measured by a validated tool such as CFQoL (Gee 2000) or CFQ‐R (Quittner 2009))
Time off work or school
Lung function (spirometry)
Need for additional antibiotics (IV or oral)
Time to next pulmonary exacerbation
Adverse events
Microbiology ‐ emergence of new or resistant strains
We used the GRADE approach to assess the quality of the evidence for each outcome based on the risk of bias within the trials, relevance to our population of interest (indirectness), unexplained heterogeneity or inconsistency, imprecision of the results or high risk of publication bias. We downgraded the evidence once if the risk was serious and twice if the risk was deemed to be very serious.
Results
Description of studies
Results of the search
The searches identified a total of 211 separate trials for consideration. We included four trials in the review with a total of 167 participants and excluded 203 trials (115 on the basis of title alone, and 88 which are listed in 'Excluded studies'). One trial was previously listed in ongoing trials (Soulsby 2010) and is now listed in Studies awaiting classification. This trial is now complete and we have obtained study data from the author. However, it is a small, cross‐over trial for which separate first‐arm data have not yet been provided. We have contacted the author again (18 April 2018) for additional information and we will include this in a future update, if these data become available.Two further trials are currently awaiting classification (Postnikov 2007; Semykin 2010) and one new trial is listed in Ongoing studies. A flow chart showing the process of trial selection is presented in the figures (Figure 1).
1.
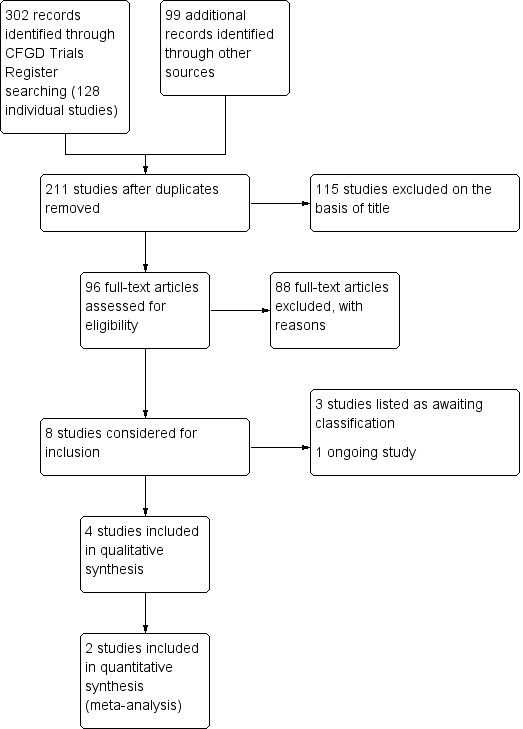
PRISMA flow diagram
Included studies
Inhaled antibiotics alone versus IV antibiotics
Trial design
Two trials (77 participants) of parallel design were included in the review (Cooper 1985; Shatunov 2001). Duration of treatment was described as 14 days in one trial (Shatunov 2001) and not stated in the second trial (Cooper 1985). One trial specifically stated that participants were admitted to hospital for pulmonary exacerbations and treated as inpatients (Cooper 1985). In the remaining trial it is not clear if participants were treated as inpatients or outpatients (Shatunov 2001).
Participants
The number of participants in each trial was 18 (Cooper 1985) and 59 (Shatunov 2001). Neither trial defined their criteria for the diagnosis of CF or gave a definition for a pulmonary exacerbation. One trial reports "pseudomonal‐related" pulmonary deterioration (Cooper 1985), and the second trial describes participants as being chronically colonised or infected with P aeruginosa and having pulmonary exacerbations (Shatunov 2001).
Interventions
Both trials compared inhaled antibiotic treatment to the same antibiotics given intravenously (Cooper 1985; Shatunov 2001). The drugs used by inhalation were tobramycin and carbenicillin (Cooper 1985) and ceftazidime (Shatunov 2001).
| Study ID | Active treatment | Comparator treatment(s) |
| Cooper 1985 | Inhaled tobramycin + inhaled carbenicillin (dose not specified) | IV ticarcillin + IV tobramycin (dose not specified) |
| Shatunov 2001 | Inhaled ceftazidime (1500 mg 1x daily) | IV ceftazidime (150 mg/kg/day in 3 divided doses) or IV ceftazidime (150 mg/kg/day in 2 divided doses) |
Outcomes
Both trials reported on lung function; FEV1 was reported in one trial (Cooper 1985) and the second trial described measuring "ventilation parameters and peakflowmetry" (Shatunov 2001). Other reported outcomes were the time until the next pulmonary exacerbation (Shatunov 2001), adverse effects (Cooper 1985), microbiological outcomes (Shatunov 2001), QoL (Cooper 1985) and need for additional IV antibiotics (Cooper 1985).
Both trials stated explicitly that they had measured outcomes at baseline and end of treatment (Cooper 1985; Shatunov 2001).
Inhaled antibiotics plus IV antibiotics versus IV antibiotics alone
Trial design
Two trials (90 participants) of parallel design were included in the review (Schaad 1987; Stephens 1983). Duration of treatment in both was described as 14 days or two weeks (Schaad 1987; Stephens 1983) and both trials specifically state that participants were admitted to hospital for pulmonary exacerbations and treated as inpatients (Schaad 1987; Stephens 1983).
Participants
The number of participants in each trial were 62 (Schaad 1987) and 28 (Stephens 1983). Neither trial defined their criteria for the diagnosis of CF or gave a definition for a pulmonary exacerbation. One trial gave baseline FEV1 data to indicate the severity of the exacerbation (Stephens 1983). With regards to the pathogen causing the pulmonary exacerbation, there are no consistent inclusion criteria. In one trial the isolation ofP aeruginosa from sputum was a definite inclusion criteria (Schaad 1987). The second trial does not discuss possible causes of infection, although it is stated within the trial report that at baseline sputum colony counts of P aeruginosa were comparable between treatment and control groups (Stephens 1983).
Interventions
Both trials compared the addition of an inhaled antibiotic to an IV combination therapy that included the same drug given intravenously (Schaad 1987; Stephens 1983). The inhaled antibiotics were amikacin (Schaad 1987) and tobramycin (Stephens 1983).
| Study ID | Active treatment | Comparator treatment(s) |
| Schaad 1987 | Inhaled amikacin (100 mg 2x daily) + IV ceftazidime (250 mg/kg/day in 4 divided doses) + IV amikacin (33 mg/kg/day in 3 divided doses) | IV ceftazidime (250 mg/kg/day in 4 divided doses) + IV amikacin (33 mg/kg/day in 3 divided doses) |
| Stephens 1983 | Inhaled tobramycin (80 mg) mixed with salbutamol (1 mL) plus buffering nebulising solution (2 ml) 3x daily + IV tobramycin (10 mg/kg/day in 3 divided doses) + IV ticarcillin (300 mg/kg/day in 4 divided doses) | IV tobramycin (10 mg/kg/day in 3 divided doses) + IV ticarcillin (300 mg/kg/day in 4 divided doses) |
Outcomes
Both trials reported on lung function; FEV1 was reported in one trial (Stephens 1983) and the second trial measured lung volumes and airway resistance during quiet breathing (Schaad 1987). Other reported outcomes were the time until the next pulmonary exacerbation (Stephens 1983), adverse effects (Schaad 1987; Stephens 1983) and microbiological outcomes (Schaad 1987; Stephens 1983), weight (Schaad 1987; Stephens 1983) and need for hospital admission (Stephens 1983).
Both trials stated explicitly that they had measured outcomes at baseline and end of treatment; and one further reported follow‐up data at four to six weeks after end of treatment (Schaad 1987).
Excluded studies
A total of 115 trials were excluded on the basis of title alone, further details of these trials are not presented. Papers were obtained for the remaining identified trials and a total of 88 are listed in the review as excluded trials; reasons for exclusion are given in the tables (Characteristics of excluded studies). Most trials were excluded as they were in people with chronic disease (n = 45); some trials stated that participants were clinically stable (n = 9) or that they were undergoing maintenance treatment (n = 4); and two trials were single‐dose trials. Seven trials looked at eradication treatment and two at prophylaxis. Two trials looked at the correct intervention in the correct population, but were of cross‐over design and first‐arm data were not available. In two trials, the intervention was not an inhaled antibiotic. In the remaining 15 trials participants were not included if they had pulmonary exacerbations.
Studies awaiting classification
There are three trials awaiting classification (Postnikov 2007; Semykin 2010; Soulsby 2010).
One trial is a two‐arm cross‐over trial which lasted two weeks and recruited 15 children aged between seven and 17 years of age (Postnikov 2007). Participants received either once‐daily or twice‐daily amikacin at a dose of between 15 mg/kg/day and 20 mg/kg/day in combination with ceftazidime or meropenem. The investigators planned to report on lung function, microbiology and adverse events at baseline and at day 14. It is currently not clear from the abstract whether the amikacin was inhaled or IV (Postnikov 2007).
One trial is a three arm of parallel trial in 108 participants aged between four and 17 years with chronic P aeruginosa infection experiencing an exacerbation. Treatments compared were twice‐daily TOBI® 300 mg or twice‐daily Bramitob® 300 mg (both in combination with IV ceftazidime and oral ciprofloxacin) versus IV cefepime plus IV amikacin versus IV meropemem plus IV amikacin. Investigators measured clinical symptoms, lung function and microbiology. To date this trial has only been published as an abstract (Semykin 2010).
The third trial currently awaiting classification is an open‐label cross‐over RCT in 24 people older than six years of age and with chronic P aeruginosa infection experiencing an exacerbation. Investigators compared IV tobramycin at the dose they received on their last admission (usually 7 ‐ 10 mg/kg) once daily for 14 days versus inhaled tobramycin at a dose of 300 mg twice daily for 14 days. Outcomes include lung function, time to next exacerbation, adverse events (renal function and antibiotic resistance), weight and QoL (Soulsby 2010).
Ongoing studies
One cross‐over trial is ongoing (Prevotat 2018). Investigators are recruiting participants of at least eight years of age with chronic P aeruginosa infection experiencing an exacerbation. The treatment comparison is a 'short cure' (14 days of IV Nebcin plus five days of IV tobramycin followed by nine days of inhaled tobramycin (Tobi Inhalant Product) 300 mg twice daily) versus 'standard' treatment (14 days IV Nebcin plus 14 days of IV tobramycin).
Risk of bias in included studies
Summary figures for the risk of bias are presented in the figures (Figure 2; Figure 3).
2.
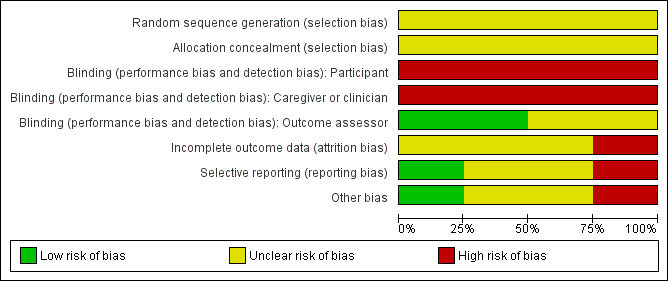
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Generation of sequence
All of the trials were judged to have an unclear risk of bias. Three of the trials described participants as being randomly allocated to groups but gave no description of the randomisation process (Cooper 1985; Schaad 1987; Stephens 1983). One trial stated that participants were "divided into three groups" but again, no details of the process were given (Shatunov 2001).
Concealment of allocation
None of the included trials discuss allocation concealment and hence were judged to have an unclear risk of bias (Cooper 1985; Schaad 1987; Shatunov 2001; Stephens 1983).
Blinding
The risk of bias from blinding of participants, of caregivers or clinicians and of outcome assessors was assessed separately. Given that the interventions being compared were either inhaled antibiotics or a combination of inhaled and IV antibiotics versus IV antibiotics and there were no placebo interventions, it was not possible to blind either the participants or the caregivers to the treatment arm. This has resulted in all trials being judged to have a high risk of bias for blinding in the groups 'Participant' and 'Caregiver or clinician'. Given that the majority of the outcomes in this review are not subjective, it is unclear how this risk of bias will impact on the results. However, the blinding of outcomes assessors would have been possible. Two trials report that outcome assessors were blinded to the treatment group (Schaad 1987; Stephens 1983). Schaad reports that clinical evaluations, radiographs and sputum analysis were all assessed by individuals who had no knowledge of treatment group (Schaad 1987). Stephens only reports that the technician performing the lung function tests was not aware of the treatment group and does not give any details for other outcomes (Stephens 1983). We have judged these two trials to have a low risk of bias (Schaad 1987; Stephens 1983). The remaining two trials did not discuss blinding of outcome assessors and are judged to have an unclear risk of bias (Cooper 1985; Shatunov 2001).
Incomplete outcome data
Three trials were judged to have an unclear risk of bias as withdrawals were not discussed (Cooper 1985; Shatunov 2001; Stephens 1983).
We regarded the trial by Schaad to have a high risk of bias, this was because not all outcomes were reported for all of the participants, and reasons for this were not given (Schaad 1987).
Selective reporting
Two of the included trials were only published as abstracts and so it is not clear from the available information if all the outcomes that were planned to be measured were indeed reported (Cooper 1985; Shatunov 2001). We therefore judged both of these trials to have an unclear risk of bias.
Two trials have been published as full papers (Schaad 1987; Stephens 1983). The Stephens paper explicitly states that FVC would be measured, but no data were reported on this outcome. Also, outcomes were measured on days 1, 5 and 12, but the table in the paper only presents clinical status data for the start and end of treatment (Stephens 1983). It is unclear whether the trial investigators have selectively reported data by time‐point. The Schaad paper appears to be consistent between those outcomes stated as being measured and those mentioned in the results section, giving this a low risk of bias (Schaad 1987).
We would also like to note that for the outcome 'Time to next pulmonary exacerbation' the trial where this was reported presented data in a manner which did not allow us to undertake our planned analysis (hazard ratios), which we regard as a potential form of selective reporting (Shatunov 2001).
Other potential sources of bias
We judged one trial as having a high risk of bias for this section (Schaad 1987). Schaad reports that 13 participants enrolled twice and six participants participated three times (Schaad 1987).
For one trial we did not identify any other potential source of bias, and was thus judged as having a low risk of bias (Stephens 1983).
The remaining two trials were both only published in abstract form and so, due to limited information, we were not able to identify any other potential sources of bias; we have therefore judged these to have an unclear risk of bias (Cooper 1985; Shatunov 2001).
Effects of interventions
Summary of findings for the main comparison. Summary of findings: inhaled antibiotics compared with IV antibiotics.
| Inhaled antibiotics compared with IV antibiotics for pulmonary exacerbations in CF | ||||||
|
Patient or population: children and adults with CF and an acute exacerbation Settings: inpatient in hospital Intervention: inhaled antibiotics Comparison: IV antibiotics | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| IV antibiotics | Inhaled antibiotics | |||||
|
Quality of life perceived change in lifestyle after treatment Follow‐up: 4 weeks |
There was a perceived improvement in lifestyle after treatment in both groups. | 18 (1) | ⊕⊝⊝⊝ very low1,2 | No data provided. | ||
| Time off work or school | This outcome was not reported. | |||||
|
Lung function change in FEV1 after treatment Follow‐up: 4 weeks after completion of therapy |
There was improvement in FEV1 after treatment in both groups but this was not significant. | 18 (1) | ⊕⊝⊝⊝ very low1,2 | Not enough data to enter into analysis. A second 3‐arm trial also measured lung function but the specific measurements were not described; there was no difference reported in treatment with twice‐daily IV ceftazidine or once‐daily inhaled ceftazidine, but both of these treatments were better than 3‐times daily IV ceftazidine. |
||
|
Need for additional IV or oral antibiotics Follow‐up: 4 weeks after completion of therapy |
No events were seen in the control group. | RR 6.11 (0.33 to 111.71) | 18 (1) | ⊕⊝⊝⊝ very low1,2 | Very little data reported. | |
|
Time to next pulmonary exacerbation Follow‐up: 4 weeks after completion of therapy |
The time to the next exacerbation was longest in the once‐daily inhaled antibiotic group, less in the twice‐daily IV antibiotic group, but both longer than in the group receiving IV antibiotics three‐times‐daily. | 59 (1) | ⊕⊝⊝⊝ very low1,2 | Very little data reported. | ||
|
Adverse events Follow‐up: 4 weeks after completion of therapy |
No adverse events were observed in either group. | 18 (1) | ⊕⊝⊝⊝ very low1,2 | |||
|
Microbiology emergence of resistant organisms Follow‐up: not stated |
No results reported for this outcome. | 59 (1) | ⊕⊝⊝⊝ very low1,2 | No results reported. This outcome was stated to be measured in the Shatunov trial, but no results were reported (Shatunov 2001) |
||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CF: cystic fibrosis; CI: confidence interval; FEV1: forced expiratory volume at 1 second; RR: risk ratio; IV: intravenous. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. Downgraded twice for unclear or high risk of bias within the trial.
2. Downgraded once due to imprecision ‐ very low participant numbers.
Summary of findings 2. Summary of findings: inhaled antibiotics plus IV antibiotics compared with IV antibiotics only.
| Inhaled plus IV antibiotics compared with IV antibiotics only for pulmonary exacerbation in CF | ||||||
|
Patient or population: children and adults with pulmonary exacerbation in CF Settings: inpatient in hospital Intervention: inhaled antibiotics plus IV antibiotics Comparison: IV antibiotics | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| IV antibiotics alone | IV + inhaled antibiotics | |||||
| Quality of life | This outcome was not reported. | |||||
| Time off work or school | This outcome was not reported. | |||||
|
Lung function change in FEV1 % predicted from baseline Follow‐up: end of treatment (discharge from hospital which ranged from 14 days to 26 days |
No significant difference was found between groups in change in FEV1 (intervention group 6.7%, comparison group 3.9%). | 28 (1) | ⊕⊝⊝⊝ very low1,2 | |||
| Need for additional IV or oral antibiotics | This outcome was not reported. | |||||
| Time to next pulmonary exacerbation | This outcome was not reported. | |||||
|
Adverse events moderate adverse events experienced during and after treatment Follow‐up: 4 ‐ 6 weeks after the end of treatment (2 weeks) |
No reports of renal toxicity in either group. Serum creatinine levels remained at less than 1.0 mg/dL and did not increase by more than 0.5 mg/dL in any participant; no proteinuria or cylinduria observed in either group. | 28 (1) | ⊕⊝⊝⊝ very low1,2 | No data were available for analysis so narrative text has been used. A further study reported this outcome but did not state which group the adverse events occurred in and therefore provides no evidence. |
||
|
Microbiology emergence of resistant organisms Follow‐up: 1 to 2 weeks |
83 per 1000 | 187 per 1000 (22 to 1000) | RR 2.25 (0.27 to 19.04) | 28 (1) | ⊕⊝⊝⊝ very low1,2 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CF: cystic fibrosis; CI: confidence interval; FEV1: forced expiratory volume at 1 second; RR: risk ratio; IV: intravenous. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. Downgraded twice due to high or unclear risk of bias in the one trial included for this outcome and possible selective reporting of some outcomes..
2. Downgraded once due to imprecision as it is a small study with a small number of participants.
In the summary of findings tables, the quality of the evidence has been graded for pre‐defined outcomes (see above) and definitions of these gradings provided.
Inhaled antibiotics alone versus IV antibiotics
Two trials (n = 77) reported on this comparison (Cooper 1985; Shatunov 2001) and results are summarised in the tables (Table 1).
Primary outcomes
1. QoL
One abstract stated 'perceived improvement in lifestyle' as an outcome in the trial; however, no results were reported (Cooper 1985). The quality of this evidence was deemed to be very low.
2. Time off work or school
Neither trial reported on this outcome.
3. Lung function
Both trials stated that lung function was an outcome measure. One trial did not specify which 'ventilatory parameters' were measured (Shatunov 2001). The abstract reports no difference in treatment with twice‐daily IV ceftazidine or once‐daily inhaled ceftazidine, but that both of these treatment regimens were better than treatment with three‐times daily IV ceftazidine (P < 0.05) (Shatunov 2001).
a. FEV1
Cooper reported FEV1 % predicted was 42% before treatment and 55% post‐treatment in the inhaled tobramycin and carbenicillin group and 39% before treatment and 52% after treatment in the IV antibiotic group (same combination of drugs given intravenously); there was no statistically significant difference in FEV1 at end of treatment between treatment or control. There was insufficient information for meta‐analysis (Cooper 1985).
The GRADE evidence for this outcome was deemed to be very low (Table 1).
b. FVC
Only Cooper reported the mean FVC % predicted at the end of treatment. In the group on inhaled antibiotics (n = 8) this was 73% predicted compared to 68% predicted in the group receiving the same antibiotics intravenously (n = 10) (Cooper 1985).
c. Annual change in FEV1
Neither trial reported on this outcome.
Secondary outcomes
1. Need for hospital admission
Neither trial reported on this outcome.
2. Need for additional antibiotics
a. IV
One trial reported that two participants from the inhaled group needed additional IV antibiotics, RR 6.11 (95% CI 0.33 to 111.71) (Analysis 1.1) (Cooper 1985). The GRADE evidence was again deemed to be very low.
1.1. Analysis.
Comparison 1 Inhaled antibiotics versus intravenous antibiotics, Outcome 1 Need for additional IV antibiotics.
b. oral
Neither trial reported on the need for additional oral antibiotics.
3. Time to next pulmonary exacerbation
Only one trial reported information on the time to the next pulmonary exacerbation, but not in a format that allowed a meta‐analysis (Shatunov 2001). Our original planned analysis was not possible as this trial did not report hazard ratios.
Shatunov reported that the time to next exacerbation was maximal in the once‐daily inhaled antibiotic group, less in the twice‐daily IV antibiotic group, but both longer than in the group receiving IV antibiotics three‐times‐daily (Shatunov 2001).
4. Weight
Neither trial reported on weight.
5. Adverse effects
a. Mild
Neither trial reported on this outcome.
b. Moderate
Cooper monitored renal and auditory changes and reported no adverse effects in either the inhaled or the IV antibiotic groups (Cooper 1985).
c. Severe
Neither trial reported on this outcome.
6. Microbiology
Both trials measured this outcome (Cooper 1985; Shatunov 2001). Cooper stated that there were no adverse effects seen in sputum microbiology (Cooper 1985).
a. Emergence of new organisms
No trials reported on the emergence of new organisms.
b. Emergence of resistant organisms
This outcome was stated to be measured in the Shatunov trial, but no results were reported (Shatunov 2001).
Inhaled antibiotics plus IV antibiotics versus IV antibiotics alone
Two trials reported on this comparison (n = 90) (Schaad 1987; Stephens 1983). Due to multiple enrolments in the Schaad trial, data were reported on episodes, rather than on participants (therefore not independent), and thus data cannot be entered into the meta‐analysis for any of the outcomes listed below (Schaad 1987). Results are summarised in the tables (Table 2).
Primary outcomes
1. QoL
Neither trial reported on this outcome.
2. Time off work or school
Neither trial reported on this outcome.
3. Lung function
Both trials stated that lung function was an outcome measure (Schaad 1987; Stephens 1983).
a. FEV1
One trial reported the change from baseline in FEV1 % predicted, but there was insufficient information for meta‐analysis (Stephens 1983). There was a 6.7% change in the group receiving the combination of inhaled and IV antibiotics and a 3.9% change in the IV antibiotics only group; the difference between groups was described as not statistically significant.
The GRADE evidence for this outcome was deemed to be very low.
b. FVC
Neither trial reported FVC, but Schaad reported results of vital capacity (VC) measured during quiet breathing (Schaad 1987). At the end of the two‐week treatment, the mean (SD) VC % predicted for the inhaled amikacin plus IV amikacin and ceftazidine group (n = 30) was 57% (16%) and for the group (n = 24) given both drugs IV was 62% (16%). At follow‐up four to six weeks later, the inhaled plus IV group (n = 12) had a mean (SD) VC % predicted of 51% (20%) and the IV alone group (n = 14) of 57% (17%).
c. Annual change in FEV1
Neither trial reported on this outcome.
Secondary outcomes
1. Need for hospital admission
One trial reported on the need for hospital admission (Stephens 1983); the results were not statistically significant, RR 1.50 (95% CI 0.15 to 14.68) (Analysis 2.1).
2.1. Analysis.
Comparison 2 Inhaled antibiotics plus intravenous antibiotics versus intravenous antibiotics, Outcome 1 Need for hospital admission.
2. Need for additional antibiotics
Neither trial reported on the need for either additional IV or additional oral antibiotics.
3. Time to next pulmonary exacerbation
Neither trial reported on this outcome.
4. Weight
Both trials reported data for weight, although we were not able to enter data from either trial into a meta‐analysis (Schaad 1987; Stephens 1983).
Schaad reported on weight as degree of underweight (%) at the end of treatment and at follow‐up (Schaad 1987). At the end of treatment Schadd reported in the IV plus inhaled group (n = 43) a mean (SD) % of underweight as 13.1% (7.1%) compared to the IV alone group (n = 44) where mean (SD) was 13.5% (7.3%). At follow‐up (four to six weeks), in the IV plus inhaled group (n = 36) the mean (SD) % of underweight was 14.8% (7.7%) compared to the IV alone group where the mean (SD) was 14.9% (7.8%) (Schaad 1987).
Stephens reported that the mean change in weight for the inhaled group (n = 16) was 1.7 and in the control group (n = 12) was 2.2 (Stephens 1983). The units of weight were not specified and no variance measures were reported. It was reported within the paper that the difference was not significant.
5. Adverse effects
a. Mild
One trial reported mild adverse events, but we are not able to present these in the analysis (Schaad 1987). Schaad reported mild adverse effects but did not state in which of the treatment groups these events had occurred (Schaad 1987). Phlebitis was reported in five participants (6%) and urticarial rash in a further four (5%) participants. Furthermore Schaad reported no significant changes in blood urea nitrogen, creatinine and urine analysis (Schaad 1987).
b. Moderate
Both trials reported on moderate adverse events (Schaad 1987; Stephens 1983).
Schaad reported on moderate adverse events, but as before did not break these down by treatment group precluding inclusion in the analysis (Schaad 1987). Pre‐ and post‐treatment audiograms were undertaken in 81 (93%) out of 87 participants and interpreted as unchanged in all. While a more than three‐fold increase in aspartate transaminase (SGOT) and alanine aminotransferase (SGPT) activities were noted in five out of 87 participants at end of therapy, this was not the case at follow‐up. Further transient haematologic abnormalities were noted in eight participants as follows: four developed eosinophilia; three developed neutropenia; and one developed thrombocytopenia (Schaad 1987).
Stephens reported that there were no reports of renal toxicity in either group (Analysis 2.2). Furthermore, serum creatinine levels remained at less than 1.0 mg/dL and did not increase by more than 0.5 mg/dL in any participant; no proteinuria or cylinduria observed in either group (Stephens 1983). The GRADE assessment for moderate adverse events was very low.
2.2. Analysis.
Comparison 2 Inhaled antibiotics plus intravenous antibiotics versus intravenous antibiotics, Outcome 2 Adverse events.
c. Severe
Schaad reported that there were no significant adverse events observed in either group (Schaad 1987).
6. Microbiology
Both trials reported on this outcome (Schaad 1987; Stephens 1983).
a. Emergence of new organisms
Neither trial reported on the emergence of new organisms.
b. Emergence of resistant organisms
Schaad reported that at the end of treatment, two out of 39 strains were resistant to ceftazidime and two were resistant to amikacin in the group using inhaled amikacin combined with IV amikacin and ceftazidime. In the group treated with the same IV antibiotics, three out of 39 strains were resistant to ceftazidime and one to amikacin (Schaad 1987). Stephens reported that there was no difference in the rate of P aeruginosa isolates being resistant to inhaled tobramycin, RR 2.25 (95% CI 0.27 to 19.04) (Analysis 2.3) (Stephens 1983). The GRADE assessment found the evidence to be of very low quality.
2.3. Analysis.
Comparison 2 Inhaled antibiotics plus intravenous antibiotics versus intravenous antibiotics, Outcome 3 Development of resistant organisms.
Discussion
Pulmonary exacerbations associated with chronic P aeruginosa infection are well recognised in people with CF, although there is no agreed definition and pathophysiology is not well understood. Exacerbations are associated with both short‐ and long‐term effects on health (Sanders 2010). The usual treatment of exacerbations is a combination of two antibiotics delivered intravenously and more airway clearance for two to three weeks, frequently in hospital (Flume 2009; Gibson 2003a). In some instances oral antibiotic treatment (ciprofloxacin) is the first antibiotic used. There has been limited research on the effect of inhaled antibiotics for treatment of pulmonary exacerbations in people with CF. The rationale to undertake these trials is to provide people with CF with therapeutic options that are less invasive and potentially less toxic but equally effective as compared to other treatment options (e.g. IV antibiotics).
Summary of main results
We identified four RCTs with 167 participants from our search of the literature. Two trials compared inhaled antibiotics alone to IV antibiotics alone (n = 77), while two compared a combination of inhaled and IV antibiotics to IV antibiotics alone (n = 90). Outcome measures of interest were either not reported or reported in a style that was not suitable for meta‐analysis. It is of note that in the two trials that reported data for FEV1 (one from each comparison), no differences were found between the groups. In one of the trials comparing inhaled antibiotics alone to IV antibiotics alone the time to the next exacerbation was not found to be different (Shatunov 2001). No significant adverse events were reported in any of the trials in either comparison. Larger trials are needed to confirm these findings.
Overall completeness and applicability of evidence
Two of the included trials used inhaled tobramycin (one of these combined it with inhaled carbenicillin and the second with IV tobramycin and IV ticarcillin) which is the most commonly‐used inhaled antibiotic. The dose used was not stated in one of these trials. Trials were varied in design and specific intervention and comparator, making meta‐analyses of the results not appropriate.
The trials included in this review were designed to show that inhaled antibiotics are equally as effective as other treatment options. Such equivalence trials require large numbers of participants to ensure confidence in the results. The included trials all had a small sample size, which would make them prone to a Type II error, indeed in the two trials which provided data on lung function there were only 41 participants and a difference could have been missed.
The only trials we were able to include in the review were relatively old and in a fast‐moving area of treatment development it brings into question the applicability of any results to the current domain. Our primary outcome was change in QoL, but this was only measured in one of the four trials and the quality of the evidence was very low.
There is currently one ongoing trial which is recruiting participants; we will report on this trial at the next update of the review (Prevotat 2018).
Quality of the evidence
The overall quality of the evidence was either low or very low across all outcomes reported in the trials included in both comparisons. The risk of bias was unclear overall for all of the trials, largely due to the fact there was a lack of information provided (two trials were only published in abstract form). Given the obvious inability to blind participants and caregivers to mode of delivery (IV versus inhaled) there is a potential risk of bias for subjective outcomes, such as QoL (our primary outcome). However, given the lack of data included in the review for subjective outcomes, this potential risk of bias currently has no bearing on our conclusions.
We also downgraded the evidence because of low participant numbers within the trials causing imprecision.
Potential biases in the review process
Although few trials were identified, we are confident that all eligible trials have been identified by the comprehensive search undertaken.
We have contacted authors for clarification on some missing information. However, we have received no additional unpublished data. We are unable to comment on the potential risk of bias this poses.
Given the limited data available there is a low risk of bias due to data handling errors.
Agreements and disagreements with other studies or reviews
Our findings concur with those published in the American CF pulmonary guidelines, in that these also highlight a lack of evidence for the use of inhaled antibiotics during the treatment of a pulmonary exacerbation (Flume 2009). In the UK, CF Trust guidelines state that there is no evidence that inhaled antibiotics are suitable alternatives to IV antibiotics for pulmonary exacerbations, or that there is clinical benefit when used in addition to IV antibiotics in this setting (CF Trust 2009). Most recently, the newly published NICE guidelines recommend the use of inhaled antibiotics in conjunction with oral antibiotics for mild exacerbations but there is little evidence to support this (NICE 2017).
Authors' conclusions
Implications for practice.
The evidence of benefit to people with cystic fibrosis (CF) from use of inhaled antibiotics as long‐term suppression of respiratory infection suggests there might also be benefit for treatment of exacerbations (Smith 2018). The evidence is strongest for inhaled tobramycin. A number of authors have advocated the use of inhaled antibiotics to treat exacerbations without citing evidence of effect (Gibson 2003a; Moskowitz 2008). This review has found no high level evidence to inform the use of inhaled antibiotics for exacerbations. There were only four trials comparing inhaled antibiotics (either alone or in combination with intravenous (IV) antibiotics) with IV antibiotics for exacerbations and these were inadequate for a valid analysis.
When considering the use of antibiotics to treat pulmonary exacerbations, there are situations when IV antibiotic therapy is challenging, e.g. where IV access is difficult; where an admission or home treatment with IV antibiotics cannot be arranged for social reasons; or where the clinician wants to avoid the adverse effects of systemic therapy. An inhaled aminoglycoside may be useful when an IV aminoglycoside is contra‐indicated because of renal impairment or a risk of drug‐induced hearing loss. In such circumstances an oral quinolone (such as ciprofloxacin) and a nebulised antibiotic may be an alternative strategy (Smyth 2008). However, evidence for the effectiveness of these strategies is absent.
Implications for research.
Consumer input into this review has highlighted how important this topic is to people with CF and how disappointing it is that the included trials reported outcomes that were not relevant to people with CF. Early detection and treatment of pulmonary exacerbations is central to CF care and other options for antibiotic delivery in these circumstances (apart from IV treatment) should be evaluated. However, we were only able to identify a single ongoing trial of nebulised antibiotics for the management of pulmonary exacerbations (Prevotat 2018).
One of the difficulties when describing the effects of treatments for exacerbations is the lack of a consensus definition of what constitutes a mild, moderate or severe exacerbation. Research would benefit from a move towards such a consensus definition.
Randomised controlled trials are required in order to answer the following questions in people with CF experiencing pulmonary exacerbations.
For mild pulmonary exacerbations, does inhaled tobramycin (or another inhaled antibiotic) added to oral ciprofloxacin improve outcomes compared to ciprofloxacin alone? The outcomes of interest are rate of return to health (quality of life, time off work or school), lung function (e.g. forced expiratory volume in one second (FEV1)), weight, need for a rescue course of IV antibiotics and time to next exacerbation. Safety outcomes (such as known adverse effects of tobramycin) should also be studied.
For more severe pulmonary exacerbations, is inhaled tobramycin as effective as IV tobramycin when either one is added to other IV antibiotics?
We feel it is important that outcomes such as quality of life or time off work or school are recorded.
What's new
| Date | Event | Description |
|---|---|---|
| 22 October 2018 | New search has been performed | 143 references to 40 trials were identified in the searches. We excluded 25 references immediately on title and did not list these in the review. Excluded studies We identified 85 additional references to 25 trials previously listed as excluded in the review (Al‐Aloul 2004; Clancy 2013; Coates 2011; Dorkin 2011; Elborn 2015; Geller 2011; Goss 2009; Griffith 2008; Konstan 2010; Konstan 2011; Mainz 2014; Mazurek 2011; McCoy 2008; Noah 2010; Proesmans 2013; Ratjen 2010; Retsch‐Bogart 2007; Rietschel 2009; Schuster 2013; Stass 2008; Taccetti 2012; Tramper‐Stranders 2009; Trapnell 2010; Treggiari 2011; Wainwright 2002). We identified 13 new trials (30 references) which we have listed as excluded studies (Dasenbrook 2015; Day 1988; Einarsson 2017; Eisenberg 1997; Elborn 2015; Flume 2014; Flume 2015a; Flume 2016; Geller 2004; Herrmann 2017; Kapranov 1995; Nasr 2006; Ruddy 2013). Ongoing studies One trial was identified from clinicaltrials.gov, but is at the recruitment stage so has been listed as an ongoing study (Prevotat 2018). Studies awaiting classification One trial, originally listed as ongoing, has now been completed; but it is a cross‐over trial and no first‐arm data are currently available. The author has been contacted and we have moved the trial to awaiting classification pending a response (Soulsby 2010). One reference to one newly identified trial is listed as awaiting classification (Postnikov 2007). The 'Plain language summary' has been updated in accordance with the latest guidelines and summary of findings tables added. |
| 22 October 2018 | New citation required but conclusions have not changed | The previous author team have stepped down and a new review team has taken this review on. |
Acknowledgements
We would like to thank the contact editor for this review, Dr Kevin Southern, and the peer reviewers for their helpful and constructive comments. We would also like to thank Natalie Soulsby for providing us with trial data.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Data and analyses
Comparison 1. Inhaled antibiotics versus intravenous antibiotics.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Need for additional IV antibiotics | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 At 1 to 2 weeks | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Renal toxicity | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
1.2. Analysis.
Comparison 1 Inhaled antibiotics versus intravenous antibiotics, Outcome 2 Adverse events.
Comparison 2. Inhaled antibiotics plus intravenous antibiotics versus intravenous antibiotics.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Need for hospital admission | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 At 1 to 2 weeks | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Renal toxicity | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Development of resistant organisms | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 At 1 to 2 weeks | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Cooper 1985.
| Methods | Randomised controlled trial. Parallel design. Single centre in Canada. Duration: 4 weeks. |
|
| Participants | 18 participants who had CF (method of CF diagnosis not stated) and acute pseudomonas‐related pulmonary deterioration (no definition given) requiring intensive treatment and admission to hospital. Inhaled group: n = 8; IV group: n = 10. Age and sex of participants not stated. |
|
| Interventions | High‐dose inhaled: carbenicillin and tobramycin. High‐dose IV: ticarcillin and tobramycin. |
|
| Outcomes | QoL, lung function (FEV1, FVC), need for additional IV antibiotics, adverse effects. | |
| Notes | Single abstract only published. Mean values given for lung function data, but no SDs. Sample size calculation not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Abstract states ‘randomly allocated’, no details of method given. |
| Allocation concealment (selection bias) | Unclear risk | Not discussed. |
| Blinding (performance bias and detection bias) Participant | High risk | Not possible, inhaled versus IV treatment. |
| Blinding (performance bias and detection bias) Caregiver or clinician | High risk | Not possible, inhaled versus IV treatment. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Not discussed. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Appear to be no withdrawals. |
| Selective reporting (reporting bias) | Unclear risk | No access to protocol, this is only an abstract so no complete methods and results sections to compare. |
| Other bias | Unclear risk | Not clear, only abstract with limited information. |
Schaad 1987.
| Methods | Randomised controlled trial. Parallel design. Single centre in Switzerland (Berne). Duration: 2 weeks (average 15 days). |
|
| Participants | Participants with CF (no information on how CF diagnosed) admitted for exacerbation of pulmonary symptoms (no further details given). Inclusion criteria: isolation of Pa from sputum on admission; absence of life‐threatening illness, renal or hepatic failure and history of drug allergy; at least 6 months since last hospital admission. 87 data sets (62 participants, but 87 episodes ‐ 13 enrolled twice (possibly in different arms) and 6 enrolled 3 times). Mean age 15 years, range 3 ‐ 24 years. Baseline data not presented: “two therapy groups comparable in sex, age, clinical/radiographic score and IV anti‐pseudomonal therapy”. |
|
| Interventions |
IV alone: ceftazidime (250 mg/kg/day in 4 divided doses) plus amikacin (33 mg/kg/day in 3 divided doses) administered over 5 min. IV + inhaled: ceftazidime (250 mg/kg/day in 4 divided doses) plus amikacin (33 mg/kg/day in 3 divided doses) administered over 5 min plus aerosolized amikacin (1x 100 mg vial amikacin sulphate in 2 ml aqueous solution) 2x daily via Pari Jet nebuliser after chest physio. IV (ceftazidime plus amikacin) (44 episodes) versus IV (ceftazidime plus amikacin) plus inhaled amikacin (43 episodes). |
|
| Outcomes | Lung function (VC, RV, FRC and airway resistance), weight, adverse effects, microbiology, serum and sputum concentrations, radiologic score, clinical or radiographic score, erythrocyte sedimentation rate, leukocytes, band neutrophils. Reported at baseline, end of therapy (2 weeks) and 4 ‐ 6 weeks later. |
|
| Notes | States on page 600 “87 were randomly allocated..” but on page 601 “62 patients were admitted to the study and received 87 course of therapy by random assignment. 13 patients were enrolled twice, 6 participated 3 times”. Number of participants analysed differs for different outcomes, but at end of therapy ranges from 54 to 87, at follow‐up 26 to 68. Sample size calculation not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Paper states “87 patients randomly allocated ...” but not details given of method. |
| Allocation concealment (selection bias) | Unclear risk | Not discussed. |
| Blinding (performance bias and detection bias) Participant | High risk | Not possible, comparison of IV versus IV + inhaled antibiotics. |
| Blinding (performance bias and detection bias) Caregiver or clinician | High risk | Not possible, comparison of IV versus IV + inhaled antibiotics. |
| Blinding (performance bias and detection bias) Outcome assessor | Low risk | Paper states: ‐ “ clinical evaluations done by the same investigator (JWK) who was not aware of the type of anti‐microbial therapy” ‐ “radiographs graded ... by a pediatric radiologist who had no knowledge of a patient’s treatment” ‐ “Sputum studies ... processed by the Institute of Microbiology, University of Berne for culture and antibiotic susceptibility testing, by the Pulmonary Division Laboratory, University of Berne for microscopic analysis and by the Division of Paediatric Infectious Diseases, University of Geneva, for measurement of elastolytic activity and total protein concentration.” |
| Incomplete outcome data (attrition bias) All outcomes | High risk | All 87 courses completed (62 participants; 13 enrolled twice and 6 enrolled 3 times), but not all outcomes reported for all of the participants, and reason for this not given. Only 68 participants available for follow‐up (IV = 32; IV + inhaled = 36) not sure if any of these were multiple enrolments. Number of participants analysed differs for different outcomes, but at end of therapy ranges from 54 to 87, at follow‐up 26 to 68. |
| Selective reporting (reporting bias) | Low risk | Consistency between outcomes stated as being measured and those mentioned in the results section. |
| Other bias | High risk | Some participants appear in trial on multiple counts: States on page 601 "62 patients were admitted to the study and received 87 course of therapy by random assignment. 13 patients were enrolled twice, six participated 3 times". |
Shatunov 2001.
| Methods | Parallel design. Single centre in Russia. Duration: 2 weeks. |
|
| Participants | 59 children with CF infected with P aeruginosa and in a period of moderate pulmonary exacerbation (no further information provided). Not clear how many in each intervention group. Age range 5 ‐ 15 years, gender split not described. Method of CF diagnosis not stated. |
|
| Interventions | 3 groups: A: ceftazidime 150 mg/kg/day IV every 8 hours. B: ceftazidime 150 mg/kg/day IV every 12 hours. C: ceftazidime inhalation 1500 mg once daily via Pari28® and Portaneb® jet nebulisers. |
|
| Outcomes | Peakflowmetry (but not clear which lung function tests used), oxygen saturation, chest X‐ray, time to next pulmonary exacerbation, duration of P aeruginosa eradication, emergence of new or resistant organisms. Measured at baseline and end of treatment. |
|
| Notes | Abstract only. No data presented to enter into meta‐analysis. Sample size calculation not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Abstract states ‘divided into 3 groups’ but does not state 'randomised' or describe process. Authors contacted and awaiting response. |
| Allocation concealment (selection bias) | Unclear risk | Not discussed. |
| Blinding (performance bias and detection bias) Participant | High risk | Not possible: IV twice‐daily versus IV 3‐times‐daily versus inhaled. |
| Blinding (performance bias and detection bias) Caregiver or clinician | High risk | Not possible: IV twice‐daily versus IV 3‐times‐daily versus inhaled once‐daily. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Not discussed. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not clear if all randomised participants were treated or if there were any withdrawals. |
| Selective reporting (reporting bias) | Unclear risk | Abstract only ‐ results described in general terms only. States: Ventilation parameters, peak‐flowmetry, oxygen saturation, chest X‐ray, sputum bacteriology were done before and after. Sputum and serum samples for CTZ concentrations were collected after the first administration and for steady‐state concentrations. Frequency and duration of P aeruginosa eradication and time until next exacerbation were analysed too. |
| Other bias | Unclear risk | Not clear, only abstract with limited information. |
Stephens 1983.
| Methods | Randomised controlled trial. Parallel design. Single centre in Canada. Duration: 14 days. |
|
| Participants | 28 people with CF (no information on how CF diagnosed) admitted to hospital for treatment of pulmonary exacerbation. IV + inhaled group: n = 16; IV alone group: n = 12. Age: IV + inhaled: mean (SD) 15.3 (3.5) years; IV: mean (SD) 15.1 (4.7) years. Age range for both groups 7 ‐ 22 years. Gender: IV + inhaled: 9 males, 7 females; IV: 9 males, 3 females. Clinical status: admitted to hospital for treatment of exacerbation (no definition given). Groups comparable in age and details listed below. Mean (SD) Schwachman score: IV + inhaled: 53.4 (9.5); IV: 55.4 (9.0). Mean (SD) FEV1 % predicted: IV + inhaled: 64.3 (11.8); IV: 72.2 (10.9). Mean (SD) FEF25‐75 % predicted: IV + inhaled: 30.6 (26.5); IV: 36.6 (21.0). Mean (SD) log10 sputum colony count of P aeruginosa: IV + inhaled: 6.9 (1.6); IV: (7.4 (1.2). |
|
| Interventions |
IV + inhaled: IV (tobramycin 10 mg/kg/day in 3 divided doses plus ticarcillin 300 mg/kg/day in 4 divided doses, each dose infused over 30 minutes) plus 80 mg aerosolized tobramycin mixed with 1 ml salbutamol and 2 ml buffering nebulising solution 3x daily via a Bennet twin jet nebulizer. IV alone: tobramycin 10 mg/kg/day in 3 divided doses plus ticarcillin 300 mg/kg/day in 4 divided doses, each dose infused over 30 minutes. |
|
| Outcomes | FEV1, FVC, FEF25‐75, need for hospital admission, weight, adverse effects, emergence of resistant organisms, serum concentrations, sleeping pulse and respiratory rates, temperature, Schwachman score, sputum culture, antimicrobial sensitivities. Data reported at start of trial and discharge from hospital (25 participants discharged after 14 days, 2 from treatment and 1 from control group discharged at 21 to 26 days). Serum concentrations for adjustment of IV tobramycin measured on days 1, 5 and 12 ‐ day 5 not reported in paper. |
|
| Notes | All P aeruginosa strains sensitive to ≤8 µg/ml tobramycin and ≤100 µg/ml ticarcillin. For FEV1 and weight, paper reports means, but no SDs, so data can not be entered into RevMan. Paper also reports that "Eradication of Pa transient and all children re‐colonized within 1 to 2 months after discharge." Sample size calculation not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Paper states "allocated randomly to either the control or treatment groups" but process not described. |
| Allocation concealment (selection bias) | Unclear risk | Not discussed. |
| Blinding (performance bias and detection bias) Participant | High risk | Not possible, IV + inhaled versus IV alone. |
| Blinding (performance bias and detection bias) Caregiver or clinician | High risk | Not possible, IV + inhaled versus IV alone. |
| Blinding (performance bias and detection bias) Outcome assessor | Low risk | For lung function tests paper states "technician performing the tests was not aware of the treatment", but no details given for other outcome measures. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Appear to be no withdrawals. |
| Selective reporting (reporting bias) | High risk | Data for FVC not reported. Furthermore, paper states that "Serum concentrations were obtained prior to and 30 minutes after completion of the 30‐min infusion. Serum concentrations of tobramycin and ticarcillin were measured on day 1, 5 and 12. Sleeping pulse & respiratory rates recorded daily and body temp (oral) 4 times a day. Schwachman score, quantitative sputum culture & antimicrobial sensitivities pulmonary function tests were performed at entry and at end of 2 weeks of treatment. Sputum obtained prior to 1st morning dose of inhaled tobramycin." Table 2 in the paper reports these clinical status data for start and at end of treatment, but no data are given for day 5 ‐ unclear if this is selectively reported or not. |
| Other bias | Low risk | No other bias identified. |
BMI: body mass index CF: cystic fibrosis FEF25‐75: mid‐expiratory flow FEV1: forced expiratory volume at one second FRC: functional residual capacity FVC: forced vital capacity IV: intravenous P aeruginosa: Pseudomonas aeruginosa PEG: percutaneous endoscopic gastrostomy QoL: quality of life RV: residual volume SD: standard deviation VC: vital capacity
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adeboyeku 2002 | Not pulmonary exacerbation. |
| Al‐Aloul 2004 | Cross‐over trial. First‐arm data unavailable. |
| Alothman 2002 | Not pulmonary exacerbation. |
| Bresnik 2008 | Not pulmonary exacerbation. |
| Bruinenberg 2008 | Chronic infection. |
| Carswell 1987 | Chronic infection. |
| Chua 1990 | Not pulmonary exacerbation. |
| Chuchalin 2007 | Chronic infection. |
| Clancy 2013 | Chronic infection. |
| Coates 2011 | Trial of nebuliser device in patients with stable disease. |
| Colin 2003 | Chronic infection. |
| Dasenbrook 2015 | Chronic infection |
| Davies 2004 | Chronic infection. |
| Day 1988 | Chronic infection. Duration of treatment longer than 4 weeks |
| Dodd 1997 | Participants clinically stable. |
| Dodd 1998 | Not pulmonary exacerbation. |
| Dorkin 2011 | Chronic infection. |
| Einarsson 2017 | Wrong intervention ‐ not inhaled antibiotics |
| Eisenberg 1997 | Study of nebuliser, participants do not appear to have Pex, single administration of each intervention. |
| Elborn 2015 | Clinically stable people with CF with chronic P. aeruginosa. |
| Flume 2014 | Chronic infection |
| Flume 2015a | Chronic infection |
| Flume 2016 | Chronic infection in stable patients |
| Frederiksen 1997 | Maintenance treatment. |
| Geborek 2003 | Cross‐over trial. First‐arm data unavailable. |
| Geller 2004 | Single dose study of inhaled antibiotic. |
| Geller 2007 | Participants clinically stable. |
| Geller 2011 | Patients clinically stable. |
| Gibson 2003b | Chronic infection. |
| Gibson 2004 | Patients clinically stable. |
| Goss 2009 | Chronic infection. |
| Griffith 2008 | Patients clinically stable. |
| Gulliver 2003 | Not pulmonary exacerbation. |
| Herrmann 2017 | Chronic infection. Duration of intervention longer than 4 weeks |
| Hodson 2002 | Chronic infection. |
| Huls 2000 | Not pulmonary exacerbation. |
| Jenkins 1985 | Chronic infection. |
| Jensen 1987 | Chronic infection. |
| Kapranov 1995 | Wrong intervention. Not an inhaled antibiotic |
| Kenny 2009 | Eradication, not pulmonary exacerbation. |
| Konstan 2010 | Chronic infection. |
| Konstan 2011 | Chronic infection. |
| Kun 1984 | Not pulmonary exacerbation. |
| Ledson 2002 | Chronic infection. |
| Lenoir 2007 | Chronic infection. |
| Mainz 2014 | Not pulmonary exacerbation, pulmonary exacerbation given as reason for exclusion from trial. |
| Mazurek 2011 | Chronic infection. |
| McCoy 2008 | Chronic infection. |
| Nasr 2006 | Participants had chronic disease, treatment for 28 days. |
| Nathanson 1985 | Chronic infection. |
| Nikolaizik 1996 | Chronic infection. |
| Nikolaizik 2005 | Chronic infection. |
| Nikonova 2010 | Chronic infection. |
| Noah 2010 | Patients clinically stable. |
| Nolan 1982 | Prophylaxis, not pulmonary exacerbation. |
| Novartis 2010 | Eradication, not pulmonary exacerbation. |
| Oermann 2009 | Chronic infection |
| Poli 2005 | Chronic infection. |
| Proesmans 2013 | Eradication, not pulmonary exacerbation. |
| Ramsey 1993 | Maintenance treatment. |
| Ramsey 1999 | Chronic infection. |
| Ratjen 2010 | Eradication, not pulmonary exacerbation. |
| Regelmann 1990 | Stable disease. |
| Retsch‐Bogart 2007 | Chronic infection. |
| Retsch‐Bogart 2008 | Chronic infection. |
| Rietschel 2009 | Chronic infection. |
| Rosenfeld 2006 | Not pulmonary exacerbation. |
| Ruddy 2013 | Participants had chronic disease, treatment for 28 days. |
| Schaad 1997 | Maintenance treatment. |
| Schelstraete 2009 | Eradication, not pulmonary exacerbation. |
| Schuster 2013 | Maintenance treatment. |
| Smith 1994 | Not pulmonary exacerbation. |
| Stass 2008 | Single‐dose pharmacokinetic trial, not pulmonary exacerbation. |
| Stass 2009 | Chronic infection |
| Stead 1987 | Chronic infection. |
| Stroobant 1985 | Not pulmonary exacerbation. |
| Taccetti 2012 | Eradication, not pulmonary exacerbation. |
| Tramper‐Stranders 2009 | Prophylaxis. |
| Trapnell 2010 | Chronic infection. |
| Treggiari 2011 | Chronic infection. |
| Tullis 2014 | Not pulmonary exacerbation, trial duration up to 48 weeks. |
| Valerius 1991 | Chronic infection. |
| Wainwright 2002 | Eradication, not pulmonary exacerbation. |
| Wainwright 2011 | Chronic infection. |
| Westerman 2003 | Chronic infection. |
| Westerman 2005 | Chronic disease. |
| Wiesemann 1998 | Chronic infection. |
| Yasmin 1974 | Not pulmonary exacerbation. |
CF: cystic fibrosis P. aeruginosa: Pseudomonas aeruginosa
Characteristics of studies awaiting assessment [ordered by study ID]
Postnikov 2007.
| Methods | 2‐arm cross‐over design. Duration: 14 days. |
| Participants | 15 children with CF aged 7 ‐ 17 years. |
| Interventions | Twice‐daily versus once‐daily amikacin. Dose: 15 mg/kg/day ‐ 20 mg/kg/day in combination with ceftazidime or meropenem. |
| Outcomes | Lung function (FVC, FEV1), P aeruginosa colonies, nephrotoxicity, ototoxicity, serum levels of amikacin. Measured on day 1 and day 14. |
| Notes | Not clear if inhaled or IV. |
Semykin 2010.
| Methods | Randomised controlled trial. 3‐arm parallel design. |
| Participants | 108 participants with chronic P aeruginosa. Age range: 4 ‐ 17 years. |
| Interventions | Group A: (n = 32, age range 4‐16 years) TOBI® (n = 16) 300 mg bid or Bramitob® (n = 16) 300 mg bid + IV ceftazidime plus oral ciprofloxacin. Group B: (n = 39, age range 6 ‐ 17 years) IV cefepime + IV amikacin. Group C: (n = 37, age 4 ‐ 17 years) IV meropemem + IV amikacin. |
| Outcomes | Clinical symptoms, lung function (FVC, FEV1), sputum P aeruginosa density. |
| Notes |
Soulsby 2010.
| Methods | Randomised controlled trial. Randomisation order generation using the toss of a coin. No blinding. Open (masking not used). Phase IV trial. |
| Participants | Diagnosed with CF. Chronically colonised with P aeruginosa. Exacerbation of a lung infection. Male and female. Minimum age: 6 years old. Target sample size: 24. |
| Interventions | IV tobramycin at the dose they received on their last admission (usually 7 ‐ 10 mg/kg) once daily for 14 days versus inhaled tobramycin at a dose of 300 mg twice daily for 14 days. Cross‐over to other treatment arm at next admission (at least 6 weeks apart). |
| Outcomes |
Primary outcomes 1. FEV1 % predicted (measured at admission, on last day of treatment (day 14) and next clinical appointment ‐ usually 6 weeks after end of treatment). 2. Time to next admission for an exacerbation (measured in weeks). 3. Change in renal function (measured on day 1 and 8 using blood samples also measured using urine samples on day 1, 14 and at next clinic visit ‐ usually 6 weeks after end of treatment). Secondary outcomes 1. Weight measured (measured at admission, on last day of treatment (day 14) and next clinical appointment ‐ usually 6 weeks after end of treatment). 2. QoL questionnaire (measured at day 1 and 14). 3. Antibiotic resistance to P aeruginosa using sputum samples (day 1, 14 and next clinical appointment ‐ usually 6 weeks after end of treatment). |
| Notes | Funding source: The Society of Hospital Pharmacists of Australia, PO Box 1774, Collingwood, VIC 3066, Australia. We have no first‐arm data for this small cross over trial; author contacted to see if we can obtain the data we need to include it. |
FEV1: forced expiratory volume at 1 second FVC: forced vital capacity IV: intravenous P aeruginosa: Pseudomonas aeruginosa QoL: quality of life
Characteristics of ongoing studies [ordered by study ID]
Prevotat 2018.
| Trial name or title | Evaluation of short antibiotic combination courses followed by aerosols in cystic fibrosis (TOBRAMUC). |
| Methods | Randomised cross‐over trial. |
| Participants | Estimated enrolment: 97 participants. Inclusion criteria: 8 years and older; CF confirmed by sweat or genetic test; clinical signs of an exacerbation (increased cough, sputum (abundance, purulence), fever, anorexia, weight loss and FEV1) or acute exacerbations (defined at the clinician's discretion); FEV1 ≥ 25 %; P aeruginosa chronic carriers; received at least 1 IV course of antibiotics in the year prior to inclusion. |
| Interventions | Intervention group (short cure): 14 days IV Nebcin plus 5 days of IV tobramycin followed by 9 days of inhaled tobramycin (Tobi Inhalant Product) 300 mg 2x daily. Control group (standard): 14 days IV Nebcin plus 14 days of IV tobramycin. |
| Outcomes |
Primary outcome FEV1 (up to 18 months post treatment) measure of dyspnoea. Secondary outcomes FEV1 (change from baseline at day 16 or day 22) measure of dyspnoea. Visual analogue scale (change from baseline at day 16 or day 22) measure of dyspnoea, condition of patient, bronchial congestion. Number of participants with bronchial congestion (change from baseline at day 16 or day 22). Sputum sample culture (change from baseline at day 16 or day 22) (a descriptive analysis of P aeruginosa, and the other bacteria in the bacterial flora of sputum). Time to first exacerbation post treatment (up to 18 months). Number of pulmonary exacerbations and those leading to hospitalisation (up to 18 months post treatment). |
| Starting date | 13 June 2017 |
| Contact information | Anne Prévotat, MD anne.prevotat@chru‐lille.fr |
| Notes |
CF: cystic fibrosis FEV1: forced expiratory volume at 1 second IV: intravenous P aeruginosa: Pseudomonas aeruginosa
Differences between protocol and review
Unit of analysis issues
Previously we proposed excluding all cross‐over trials from this review. However, on revisiting the issue of the inclusion of such trials, all three authors agreed that the inclusion of first‐arm data from these trials would be appropriate.
Summary of findings tables have been included in line with updated Cochrane guidance.
Contributions of authors
Up to 2018
Gerard Ryan conceived the protocol and all three authors worked on drafting the protocol.
NIkki Jahnke and Tracey Remmington undertook trial selection and data extraction. Nikki Janke entered data and with Gerard Ryan drafted the first version of the review. All authors worked on finalising the review following peer reviewer comments and editorial input.
From 2018
Sherie Smith and Edward Charbek undertook trial selection and data extraction. Nicola Rowbotham arbitrated on discrepancies in inclusion decisions and added clinical expertise.
All authors contributed to updating the text of the review.
Sources of support
Internal sources
NIHR National Programme (Department of Health), UK.
External sources
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
No conflicts of interest were reported by Sherie Smith or Ed Charbek
Nicola J Rowbotham is an NIHR academic clinical fellow. She has received non‐financial support (travel and accommodation) for conference attendance from TEVA.
Previous author team
Dr Gerard Ryan received funds for research for a multicentre industry‐sponsored trial of aztreonam. Dr Ryan worked in a CF centre that contributed participants to the trial with funding going to the Lung Institute of Western Australia (LIWA) for research staff. No funding went to the CF centre.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Cooper 1985 {published data only}
- Cooper DM, Harris M, Mitchell I. Comparison of intravenous and inhalation antibiotic therapy in acute pulmonary deterioration in cystic fibrosis. American Review of Respiratory Disease 1985;131:A242. [CFGD Register: PI129] [Google Scholar]
Schaad 1987 {published data only}
- Schaad UB, Wedgwood Krucko J, Suter S, Kraemer R. Efficacy of inhaled amikacin as adjunct to intravenous combination therapy (ceftazidime and amikacin) in cystic fibrosis. Journal of Pediatrics 1987;111(4):599‐605. [CFGD Register: PI56] [DOI] [PubMed] [Google Scholar]
Shatunov 2001 {published data only}
- Shatunov SM. Comparative efficacy of different methods of ceftazidime administration in children with cystic fibrosis. Proceedings of the 11th European Respiratory Society Annual Congress; 2001 Sept 22‐26; Berlin. 2001:860. [CFGD Registerer: PI164]
Stephens 1983 {published data only}
- Stephens D, Garey N, Isles A, Levison H, Gold R. Efficacy of inhaled tobramycin in the treatment of pulmonary exacerbations in children with cystic fibrosis. Pediatric Infectious Disease 1983;2(3):209‐11. [CFGD Register: PI105] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Adeboyeku 2002 {published data only}
- Adeboyeku DU, Agent P, Jackson V, Hodson M. A double blind randomised study to compare the safety and tolerance of differing concentrations of nebulised colistin administered using HaloLite in cystic fibrosis (CF) patients. Pediatric Pulmonology 2001;32(Suppl 22):288. [CFGD Register: PI165] [Google Scholar]
Al‐Aloul 2004 {published data only}
- Al‐Aloul M, Miller H, Browning P, Ledson MJ, Walshaw MJ. A randomised cross over trial of TOBI® vs IV tobramycin in acute pulmonary exacerbations in CF. Pediatric Pulmonology 2004;38(Suppl 27):249. [CFGD Register: PI186a] [Google Scholar]
- Al‐Aloul M, Miller H, Ledson MJ, Walshaw MJ. Tobramycin nebuliser solution (TOBI): a renal sparing alternative to intravenous (IV) tobramycin in acute pulmonary exacerbations in CF. Thorax 2004;59(Suppl II):ii79. [CFGD Register: PI186d] [Google Scholar]
- Al‐Aloul M, Miller H, Ledson MJ, Walshaw MJ. Tobramycin nebuliser solution in the treatment of cystic fibrosis pulmonary exacerbations: effect on sputum pseudomonas aeruginosa density. Thorax 2005;2(Suppl 2):ii92. [CFGD Register: PI186b] [Google Scholar]
- Al‐Aloul M, Nazareth D, Walshaw M. Nebulized tobramycin in the treatment of adult CF pulmonary exacerbations. Journal of Aerosol Medicine and Pulmonary Drug Delivery 2014;27(4):299‐305. [CFGD Register: PI186c] [DOI] [PubMed] [Google Scholar]
Alothman 2002 {published data only}
- Alothman GA, Alsaadi MM, Ho BL, Ho SL, Dupuis A, Corey M, et al. Evaluation of bronchial constriction in children with cystic fibrosis after inhaling two different preparations of tobramycin. Chest 2002;122(3):930‐4. [CFGD Register: PI157b] [DOI] [PubMed] [Google Scholar]
- Alothman GA, Coates AL, Corey M, Dupuis A, Ho SL, Ho BL, et al. In cystic fibrosis (CF) patients, does the inhalation of an intravenous tobramycin preparation result in more bronchospasm than a preservative free tobramycin preparation?. Pediatric Pulmonology 2000;30(Suppl 20):298‐9. [CFGD Register: PI157a] [Google Scholar]
Bresnik 2008 {published data only}
- Bresnik M. Open‐label, randomized, Phase 3 trial to evaluate the efficacy and safety of AZLI versus TOBI® in an intermittent aerosolized antibiotic regimen in patients With CF (US and EU), followed by an open‐label, single‐arm extension (EU). ClinicalTrials.gov (accessed 08 December 2010). [NCT00757237]
Bruinenberg 2008 {published data only}
- Bruinenberg P, Otulana B, Blanchard J, Morishige R, Cipolla D, Wilson J, et al. The effect of once‐a day inhaled liposomal ciprofloxacin hydrochloride on sputum bacterial density in cystic fibrosis patients with chronic pulmonary P. aeruginosa colonization. Pediatric Pulmonology 2008;43(Suppl 31):344. [CFGD Register: PI216] [Google Scholar]
Carswell 1987 {published data only}
- Carswell F, Ward C, Cook DA, Speller DC. A controlled trial of nebulized aminoglycoside and oral flucloxacillin versus placebo in the outpatient management of children with cystic fibrosis. British Journal of Diseases of the Chest 1987;81(4):356‐60. [CFGD Register: PI54] [DOI] [PubMed] [Google Scholar]
Chua 1990 {published data only}
- Chua H, Collis G, Souef P. Bronchial response of children with cystic fibrosis to nebulised antibiotics. Australian and New Zealand Journal of Medicine 1990;20:537. [CFGD Register: PI66b] [Google Scholar]
- Chua HL, Collis GG, Souef PN. Bronchial response to nebulized antibiotics in children with cystic fibrosis. European Respiratory Journal 1990;3(10):1114‐6. [CFGD Register: PI66a] [PubMed] [Google Scholar]
Chuchalin 2007 {published data only}
- Chuchalin A, Csiszer E, Gyurkovics K, Bartnicka MT, Sands D, Kapranov N, et al. A formulation of aerosolized tobramycin (Bramitob) in the treatment of patients with cystic fibrosis and Pseudomonas aeruginosa infection: a double‐blind, placebo‐controlled, multicenter study. Paediatric Drugs 2007;9(Suppl 1):21‐31. [CFGD Register: PI201c] [DOI] [PubMed] [Google Scholar]
- Chuchalin A, Gyurkovics K, Mazurek H, Varoli G, Monici Preti P. Long‐term administration of aerosolised tobramycin in patients with cystic fibrosis. European Respiratory Journal 2005;26 Suppl 49:619s. [CFGD Register: PI201a] [Google Scholar]
- Chuchalin A, Gyurkovics K, Mazurek HVG, Monici Preti PA. Long‐term administration of nebulised tobramycin in patients with cystic fibrosis. Journal of Cystic Fibrosis 2006;5 Suppl:S42. [CFGD Register: PI201b] [Google Scholar]
Clancy 2013 {published data only}
- Clancy JP, Dupont L, Konstan MW, Billings J, Fustik S, Goss CH, et al. Phase II studies of nebulised Arikace in CF patients with Pseudomonas aeruginosa infection. Thorax 2013;68(9):818‐25. [CENTRAL: 876398; CFGD Register: PI207e // PI222c; CRS: 5500050000000084; PUBMED: 23749840] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy JP, Dupont L, Konstan MW, Billings J, Fustik S, Goss CH, et al. Phase II studies of nebulised Arikace in CF patients with Pseudomonas aeruginosa infection. Thorax 2013;68(9):818‐25. Online supplementary data. [CFGD Register: PI222d // PI207f] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy JP, Minic P, Dupont L, Goss CH, Quittner AL, Lymp JF, et al. Full analysis of data from two phase II blinded & placebo‐controlled studies of nebulized liposomal amikacin for inhalation (Arikace®) in the treatment of CF patients with pseudomonas aeruginosa lung infection. Pediatric Pulmonology 2010;45 Suppl 33:299. [Abstract no.: 227; CENTRAL: 848916; CFGD Register: PI207d // PI222b ; CRS: 5500100000010628] [Google Scholar]
- Dupont L, Minic P, Fustic S, Mazurek H, Solyom E, Feketova A, et al. A randomised placebo‐controlled study of nebulized liposomal amikacin (Arikace™) in the treatment of cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. Journal of Cystic Fibrosis 2008;7 Suppl 2:S26. [CFGD Register: PI207a] [Google Scholar]
- Dupont LJ, Clancy JP, Minic P, Goss CH, Fustic S, Mazurek H, et al. Evaluation of two phase II blinded and placebo‐controlled studies of nebulized liposomal amikacin (arikace") in the treatment of cystic fibrosis patients with pseudomonas aeruginosa lung infection. American Journal of Respiratory and Critical Care Medicine 2010;181(Meeting Abstracts):A1836. [CFGD Register: PI222e // PI207g] [Google Scholar]
- Dupont LJ, Minic P, Fustik S, Mazurek H, Solyom E, Feketeova A, et al. A randomized placebo‐controlled study of nebulized liposomal amikacin (Arikace™) in the treatment of cystic fibrosis patients with chronic Pseudomonas Aeruginosa lung infection. Pediatric Pulmonology 2008;43 Suppl 31:301. [CFGD Register: PI207b] [Google Scholar]
- Gupta R, Dupont L, Minin P, Fustik S, Mazurek H, Solyom E. A randomized placebo‐controlled study of nebulized liposomal amikacin (Arikace™) in the treatment of cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection (protocol TR02‐105). American Journal of Respiratory and Critical Care Medicine 2009;179:no pagination. [Abstract no.: A1199; CFGD Register: PI207c] [Google Scholar]
Coates 2011 {published data only}
- Coates AL, Denk O, Leung K, Ribeiro N, Chan J, Green M, et al. Higher tobramycin concentration and vibrating mesh technology can shorten antibiotic treatment time in cystic fibrosis. Pediatric Pulmonology 2011;46(4):401‐8. [CENTRAL: 786190; CFGD Register: PI241b ; CRS: 5500100000006333] [DOI] [PubMed] [Google Scholar]
- Denk O, Coates AL, Keller M, Leung K, Green M, Chan J, et al. Lung delivery of a new tobramycin nebuliser solution (150mg/1.5ml) by an investigational eFlow® nebuliser is equivalent to TOBI® but in a fraction of time. Journal of Cystic Fibrosis 2009;8 Suppl 2:S66. [Abstract no.: 264; CENTRAL: 794467; CFGD Register: PI241c; CRS: 5500100000003576] [Google Scholar]
- Keller M, Coates AL, Griese M, Denk O, Schierholz J, Knoch M. In‐vivo data support equivalent therapeutic efficacy of a new tobramycin inhalation solution (150mg/1.5ml) administered by the eFlow® electronic nebuliser compared to TOBI® in the PARI LC PLUS®. Journal of Cystic Fibrosis 2010;9 Suppl 1:S22. [Abstract no.: 84; CENTRAL: 794286; CFGD Register: PI241a ; CRS: 5500100000003569] [Google Scholar]
Colin 2003 {published data only}
- Anbar RD, Yu X, Colin AA. Reduction of pulmonary hospitalizations during a randomized, controlled, open‐label study of tobramycin solution for inhalation in young CF patients with mild lung disease. Pediatric Pulmonology 2003;36(Suppl 25):296. [CFGD Register: PI175b] [Google Scholar]
- Colin AA, Anbar RD, Yu X. Reduction in pulmonary hospitalizations during a randomized, controlled, open‐label study of tobramycin solution for inhalation in young CF patients with mild lung disease. Journal of Cystic Fibrosis 2003;2(Suppl 1):S22. [CFGD Register: PI175a] [Google Scholar]
- Murphy TD, Anbar RD, Lester LA, Nasr SZ, Nickerson B, VanDevanter DR, et al. Treatment with tobramycin solution for inhalation reduces hospitalizations in young CF subjects with mild lung disease. Pediatric Pulmonology 2004;38(4):314‐20. [CFGD Register: PI175c] [DOI] [PubMed] [Google Scholar]
Dasenbrook 2015 {published data only}
- Dasenbrook EC. Emerging therapies in cystic fibrosis: aerovanc for the treatment of chronic MRSA. Pediatric Pulmonology 2015;50 Suppl 41:149. [Abstract no.: S11.4; CFGD Register: PI289a] [Google Scholar]
- Marich C, Lord J, Dasenbrook EC, Flume PA, Jouhikainen T. Pharmacokinetics of vancomycin in plasma and sputum following pulmonary administration in cystic fibrosis patients with persistent methicillin‐resistant staphylococcus aureus infection. Pediatric Pulmonology 2016;51 Suppl 45:298‐99. [Abstract no.: 282; CFGD Register: PI289b] [Google Scholar]
Davies 2004 {published data only}
- Conway SP, Davies JC, Etherington C, Goldman HH, Howard E, Jaffe A, et al. Piloting the use of the cystic fibrosis questionnaire (CFQ) in CF patients changing to dry powder inhaled colistimethate. Journal of Cystic Fibrosis 2007;6(Suppl 1):S76. [CFGD Register: PI189b] [Google Scholar]
- Davies JC, Hall P, Francis J, Scott S, Geddes DM, Conway S, et al. A dry powder formulation of colistimethate sodium is safe and well tolerated in adults and children with CF. Pediatric Pulmonology 2004;38(Suppl 27):283. [CFGD Register: BD188c // BD189a // BD165p] [Google Scholar]
- Goldman MH, Howard E. FEV1% predicted may not be a simple end point for CF studies. Journal of Cystic Fibrosis 2007;6(Suppl 1):S34. [CFGD Register: PI189c] [Google Scholar]
Day 1988 {published data only}
- Day AJ, Williams J, McKeown C, Bruton A, Weller PH. Evaluation of inhaled colomycin in children with cystic fibrosis. Excerpta Medica, Asia Pacific Congress Series 1988;74:R(c)3. [CENTRAL: 291275; CFGD Register: PI85; CRS: 5500100000001339] [Google Scholar]
Dodd 1997 {published data only}
- Dodd M, Maddison J, Abbott J, Webb AK. The effect of the tonicity of nebulised colistin on lung function in adults with cystic fibrosis. Proceedings of 18th European Cystic Fibrosis Conference; 1993 May 21‐26; Madrid, Spain. 1993:121. [CFGD Register: PI100a]
- Dodd ME, Abbott J, Maddison J, Moorcroft AJ, Webb AK. Effect of tonicity of nebulised colistin on chest tightness and pulmonary function in adults with cystic fibrosis. Thorax 1997;52(7):656‐8. [CFGD Register: PI100b] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd ME, Maddison J, Abbot J, Webb AR. The effect of the tonicity of nebulised colistin on chest tightness and lung function in adults with cystic fibrosis. European Respiratory Journal 1993;6(Suppl 17):515s. [CFGD Register: PI100c] [Google Scholar]
Dodd 1998 {published data only}
- Dodd ME, Haworth CS, Moorcroft AJ, Miles J, Webb AK. Is medicine evidence‐based when there is discrepancy between patient reported and objective measures of compliance in clinical trials?. Pediatric Pulmonology 1998;26(Suppl 17):389. [CFGD Register: PI237] [Google Scholar]
Dorkin 2011 {published data only}
- Bayer. Study to evaluate the safety and efficacy of ciprofloxacin (inhaled) in patients with cystic fibrosis. www.clinicaltrials.gov (accessed 17 Feb 2010):ClinicalTrials.gov identifier: NCT00645788. [CENTRAL: 744140; CFGD Register: PI261b ; CRS: 5500100000003466]
- Cystic Fibrosis Foundation. Inhaled ciprofloxacin. Www.cff.org (www.cff.org/clinicaltrials) (accessed 17 Feb 2010) 2010. [CENTRAL: 744141; CFGDK Register: PI261c; CRS: 5500100000003467]
- Dorkin H, Criollo M, Reimnitz P, Alder J, Hampel B. Randomized, double‐blind, placebo‐controlled, multicenter study to evaluate the safety and efficacy of inhaled ciprofloxacin compared with placebo in patients with cystic fibrosis‐ a phase IIB study of ciprofloxacin dry powder for inhalation (DPI). Pediatric Pulmonology 2011;46 Suppl 34:296. [Abstract no.: 235; CFGD Register: PI261a] [Google Scholar]
Einarsson 2017 {published data only}
- Einarsson G, Flanagan E, Lee A, Elborn JS, Tunney M, Plant BJ. Longitudinal airway microbiota profiling in cystic fibrosis patients enrolled in the CFMATTERS clinical trial. Journal of Cystic Fibrosis 2017;16(Supplement 1):S4. [Abstract no.: WS03.1; CFGD Register: PI295] [Google Scholar]
Eisenberg 1997 {published data only}
- Eisenberg J, Pepe M, Williams Warren J, Vasiliev M, Montgomery AB, Smith AL, et al. A comparison of peak sputum tobramycin concentration in patients with cystic fibrosis using jet and ultrasonic nebulizer systems. Aerosolized Tobramycin Study Group. Chest 1997;111(4):955‐62. [CENTRAL: 138692; CRS: 5500100000000815; EMBASE: 1997120219; PI117 ; PUBMED: 9106575] [DOI] [PubMed] [Google Scholar]
Elborn 2015 {published data only}
- Elborn JS, Flume PA, Cohen F, Loutit J, VanDevanter DR. Safety and efficacy of prolonged levofloxacin inhalation solution (APT‐1026). Journal of Cystic Fibrosis 2016;15(5):634‐40. [CFGD Register: PI262d] [DOI] [PubMed] [Google Scholar]
- Elborn JS, Flume PA, Loutit J, Cohen F. Prolonged improvement in lung function and quality of life in cystic fibrosis: a 24‐week extension study of levofloxacin nebulization solution (APT‐1026) versus tobramycin nebulization solution in stable CF patients with chronic pseudomonas aeruginosa infection. Journal of Cystic Fibrosis 2014;13 Suppl 2:S16. [Abstract no.: WS7.5; CENTRAL: 1000055; CFGD Register: PI262b; CRS: 5500131000000008] [Google Scholar]
- Elborn JS, Geller D, Conrad D, Aaron S, Smyth AR, Fischer R, et al. Phase 3 trial of inhaled levofloxacin (Aeruquinâ™, MP‐376, APT‐1026) vs. tobramycin inhalation solution (TIS) in intensively treated CF patients over 6 months. Journal of Cystic Fibrosis 2013;12 Suppl 1:S35. [Abstract no.: WS17.6; CENTRAL: 867323; CFGD Register: PI262a; CRS: 5500100000011291] [Google Scholar]
- Elborn JS, Geller DE, Conrad D, Aaron SD, Smyth AR, Fischer R, et al. A phase 3, open‐label, randomized trial to evaluate the safety and efficacy of levofloxacin inhalation solution (APT‐1026) versus tobramycin inhalation solution in stable cystic fibrosis patients. Journal of Cystic Fibrosis 2015;14(4):507‐14. [CENTRAL: 1038490; CFGD Register: PI262c; CRS: 5500131000000327; JID:: 101128966; PUBMED: 25592656] [DOI] [PubMed] [Google Scholar]
- Elborn JS, Geller DE, Conrad D, Aaron SD, Smyth AR, Fischer R, et al. A phase 3, open‐label, randomized trial to evaluate the safety and efficacy of levofloxacin inhalation solution (APT‐1026) versus tobramycin inhalation solution in stable cystic fibrosis patients. Journal of Cystic Fibrosis 2015;14:507‐14. Online supplementary material. [CFGD Register: PI262e] [DOI] [PubMed] [Google Scholar]
- Devanter DR, Elborn JS, Flume P, Polu K, LiPuma JJ. Microbiologic changes observed over 6 months in a randomized, open‐label comparison of inhaled levofloxacin and inhaled tobramycin in persons with cystic fibrosis and chronic P. aeruginosa (Pa) airway infection. Journal of Cystic Fibrosis 2016;15:S60‐1. [CFGD Register: PI262f] [Google Scholar]
Flume 2014 {published data only}
- Fischer R, Flume PA, Devanter DR, Polu K, Pecoraro M, Bhatt N, et al. Pulmonary exacerbations and changes in lung function in CF adults with P. aeruginosa treated with inhaled levofloxacin (Quinsair®) or tobramycin. Pediatric Pulmonology 2016;51 Suppl 45:359. [Abstract no.: 436; CFGD Register: PI283d] [Google Scholar]
- Flume P. Trial of aeroquin versus tobramycin inhalation solution (TIS) in cystic fibrosis (CF) patients. clinicaltrials.gov/ct2/show/NCT01270347 (first received 05 January 2011). [CFGD Register: PI283a; clinicaltrials.gov: NCT01270347]
- Flume P, Elborn JS, Polu K, Llorens L, Pecoraro ML, Bhatt N, et al. History of pulmonary exacerbations (pex) AS a predictor of response to nebulized levofloxacin compared with nebulized tobramycin. Journal of Cystic Fibrosis 2016;2015 Suppl 1:S61. [Abstract no.: 38; CFGD Register: PI283e] [Google Scholar]
- Flume P, VanDevanter DR, Cohen F, Fleming R, Elborn JS. Safety profile of levofloxacin inhalation solution from 3 controlled cystic fibrosis trials. Journal of Cystic Fibrosis 2015;14 Suppl 1:S87. [Abstract no.: 117; CFGD Register: PI240f // PI283c // PI284c] [Google Scholar]
- NCT01180634. A Phase 3, multi‐center, multinational, randomized, double‐blind, placebo‐controlled study to evaluate the efficacy and safety of MP‐376 (Levofloxacin Inhalation Solution; Aeroquin™) in stable cystic fibrosis patients. clinicaltrials.gov/show/nct01180634 (first received 12 August 2010). [CFGD Register: PI284a]
- Schwarz C, Spinola M, Flume PA, Elborn JS. Potential taste disturbance is largely mild and diminishes with continuing treatment in cystic fibrosis patients treated with levofloxacin inhaled solution. Journal of Cystic Fibrosis 2018;17(Suppl 3):S81‐2. [CFGD Register: PI283f] [Google Scholar]
Flume 2015a {published data only}
- Flume PA, Clancy JP, Retsch‐Bogart GZ, Tullis DE, Bresnik M, Derchak PA, et al. Continuous alternating inhaled antibiotics for chronic pseudomonal infection in cystic fibrosis. Journal of Cystic Fibrosis 2016;15(6):809‐15. [CFGD Register: PI288b] [DOI] [PubMed] [Google Scholar]
- Flume PA, Clancy JP, Retsch‐Bogart GZ, Tullis E, Bresnik M, Derchak PA, et al. Aztreonam for inhalation solution (AZLI) and tobramycin inhalation solution (TIS) continuous alternating therapy (CAT) for cystic fibrosis (CF) patients with chronic pseudomonas aeruginosa (PA) infection: a randomized, double‐blind, placebo‐controlled trial. Pediatric Pulmonology 2015;50 Suppl 41:352. [Abstract no.: 428; CFGD Register: PI288a] [Google Scholar]
- Flume PA, VanDevanter DR, Morgan EE, Dudley MN, Loutit JS, Bell SC, et al. A phase 3, multi‐center, multinational, randomized, double‐blind, placebo‐controlled study to evaluate the efficacy and safety of levofloxacin inhalation solution (APT‐1026) in stable cystic fibrosis patients. Journal of Cystic Fibrosis 2016;15(4):495‐502. Online supplement. [CFGD Register: PI284e] [DOI] [PubMed] [Google Scholar]
Flume 2016 {published data only}
- Flume P, VanDevanter DR, Cohen F, Fleming R, Elborn JS. Safety profile of levofloxacin inhalation solution from 3 controlled cystic fibrosis trials. Journal of Cystic Fibrosis 2015;14 Suppl 1:S87. [Abstract no.: 117; CFGD Register: PI240f // PI283c // PI284c] [Google Scholar]
- Flume PA, VanDevanter DR, Morgan EE, Dudley MN, Loutit JS, Bell SC, et al. A phase 3, multi‐center, multinational, randomized, double‐blind, placebo‐controlled study to evaluate the efficacy and safety of levofloxacin inhalation solution (APT‐1026) in stable cystic fibrosis patients. Journal of Cystic Fibrosis 2016;15(4):495‐502. Online supplement. [CFGD Register: PI284e] [DOI] [PubMed] [Google Scholar]
- NCT01180634. A Phase 3, multi‐center, multinational, randomized, double‐blind, placebo‐controlled study to evaluate the efficacy and safety of MP‐376 (Levofloxacin Inhalation Solution; Aeroquin™) in stable cystic fibrosis patients. clinicaltrials.gov/show/nct01180634 (first received 12 August 2010). [CFGD Register: PI284a]
Frederiksen 1997 {published data only}
- Frederiksen B, Hansen A, Koch C, Hoiby N. Delay of recurrence of Pseudomonas aeruginosa in patients with cystic fibrosis with inhaled colistin and oral ciproxin: a comparison between 3 weeks and 3 months of treatment. Pediatric Pulmonology 1997;24(Suppl 14):288. [CFGD Register: PI118a] [Google Scholar]
- Frederiksen B, Pressler T, Koch C, Hoiby N. Endpoints for evaluating early anti‐pseudomonal treatment: changes in pseudomonas prevalence and in pulmonary function. Pediatric Pulmonology 2003;36(Suppl 25):334. [CFGD Register: PI118b] [Google Scholar]
Geborek 2003 {published data only}
- Geborek A, Hjelte L, Lindblad A, Mared L, Eriksson L, Johannesson M, et al. Cross‐over study of TOBI® vs. intravenous tobramycin in combination treatment of pulmonary exacerbations in cystic fibrosis patients. Journal of Cystic Fibrosis 2003;2(Suppl 1):S22. [CFGD Register: PI176] [Google Scholar]
Geller 2004 {published data only}
- Geller DE, Rodriguez CA, Howenstine M, Murphy T, Voter K, Nickerson B, et al. The effects of doubling concentration of tobramycin solution for inhalation on pharmacokinetics (PK), safety and delivery time in patients with cystic fibrosis (CF). American Journal of American Journal of Respiratory and Critical Care Medicine 2004;169(7):A391. [CENTRAL: 486943; CFGD Register: PI183a; CRS: 5500100000002627] [Google Scholar]
- Rosenfeld M, Geller DE, Rodriguez CA, Howenstine M, Konstan M, Ordonez C, et al. Serum pharmacokinetics of two preparations of tobramycin solution for inhalation in young cystic fibrosis patients. American Journal of Respiratory and Critical Care Medicine 2004;169(7):A386. [CENTRAL: 495351; CFGD Register: PI183b; CRS: 5500100000002644] [Google Scholar]
Geller 2007 {published data only}
- Geller DE, Flume P, Schwab R, Fornos P, Conrad DJ, Morgan E, et al. A phase 1 safety, tolerability and pharmacokinetic (PK) study of MP‐376 (levofloxacin solution for inhalation) in stable cystic fibrosis (CF) patients. Pediatric Pulmonology 2008;43 Suppl 31:315. [Abstract no.: 321; CFGD Register: PI187a] [Google Scholar]
- Geller DE, Konstan MW, Smith J, Noonberg SB, Conrad C. Novel tobramycin inhalation powder in cystic fibrosis subjects: Pharmacokinetics and safety. Pediatric Pulmonology 2007;42(4):307‐13. [CFGD Register: PI187c] [DOI] [PubMed] [Google Scholar]
- Rodriguez CA, Shrewsbury SB, Potter SN, Nardella P, Geller DE. Single dose pharmacokinetics of tobramycin after administration of a novel dry powder formulation (TPI) in subjects with cystic fibrosis (cf). Pediatric Pulmonology 2004;38(Suppl 27):250. [CFGD Register: PI187b] [Google Scholar]
Geller 2011 {published data only}
- Conrad D, Flume P, Sindel L, Andrews S, Morgan L, Loutit J, et al. Phase 2b study of inhaled MP‐376 (Aeroquin, levofloxacin inhalation solution) in stable cystic fibrosis (CF) patients with chronic Pseudomonas Aeruginosa (PA) lung infection. American Journal of Respiratory and Critical Care Medicine 2010;181(Meeting Abstracts):no pagination. [CFGD Register: PI240g] [Google Scholar]
- Flume P, Geller DE, Sindel L, Staab D, Fischer R, Rietmüller J, et al. Effects of inhaled MP‐376 (aeroquin, levofloxacin inhalation solution) on lung function in stable cystic fibrosis (CF) patients with chronic Pseudomonas aeruginosa (PA) lung infection. Journal of Cystic Fibrosis 2010;9(Suppl 1):S23. [Abstract no.: 86; CFGD Register: PI240a] [Google Scholar]
- Flume P, Morgan EE, Loutit J. A Phase 3, multi‐center, multinational, randomized, double‐blind, placebo‐controlled study to evaluate the efficacy and safety of MP‐376 (levofloxacin inhalation solution; Aeroquin™) in stable cystic fibrosis patients. ClinicalTrials.gov (accessed 08 December 2010). [CTG: NCT01180634] [DOI] [PubMed]
- Flume PA, Geller DE, Loutit JS, Dudly MN, Conrad D, Mpex 204 Study Group. Effects of inhaled MP‐376 (Aeroquin™ levofloxacin inhalation solution) on cystic fibrosis patients with both Staphylococcus aureus (SA) and Pseudomonas aeruginosa (PA) lung infection. Journal of Cystic Fibrosis 2011;10 Suppl 1:S22. [Abstract no.: 87; CFGD Register : PI240b] [Google Scholar]
- Geller DE, Flume PA, Staab D, Fischer R, Loutit JS, Conrad DJ. Levofloxacin inhalation solution (MP‐376) in patients with cystic fibrosis with Pseudomonas aeruginosa. American Journal of Respiratory and Critical Care Medicine 2011;183(11):1510‐6. [CFGD Register: PI240d] [DOI] [PubMed] [Google Scholar]
- Geller DE, Flume PA, Staab D, Fischer R, Loutit JS, Conrad DJ. Online supplemental methods to "Levofloxacin inhalation solution (MP‐376) in patients with cystic fibrosis with Pseudomonas aeruginosa". American Journal of Respiratory and Critical Care Medicine 2011;183(11):1510‐6. Online supplemental methods. [CFGD Register: PI240e] [DOI] [PubMed] [Google Scholar]
- Geller DFPA, Sindel L, Staab D, Fischer R, Loutit J, Conrad D. Effects of inhaled MP‐376 (aeroquin, levofloxacin inhalation solution) on the need for other anti‐pseudomonal antimicrobials in stable CF patients with chronic pseudomonas aeruginosa lung infection. Pediatric Pulmonology 2010;45 Suppl 33:301. [Abstract no.: 232; CFGD Register: PI240c] [Google Scholar]
Gibson 2003b {published data only}
- Gibson RL, Emerson J, McNamara S. A randomized controlled trial of inhaled tobramycin in young children with cystic fibrosis: eradication of Pseudomonas from the lower airway. Pediatric Pulmonology 2002;Suppl 24:300. [CFGD Register: PI151c] [Google Scholar]
- Gibson RL, Emerson J, McNamara S, Burns JL, Rosenfeld M, Yunker A, et al. Online Supplement to 'Significant microbiological effect of inhaled tobramycin in young children with cystic fibrosis' [online]. American Journal of Respiratory and Critical Care Medicine 2003;167(6):841 Online. [CFGD Register: PI151e] [DOI] [PubMed] [Google Scholar]
- Gibson RL, Emerson J, McNamara S, Burns JL, Rosenfeld M, Yunker A, et al. Significant microbiological effect of inhaled tobramycin in young children with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 2003;167(6):841‐9. [CFGD Register: PI151d] [DOI] [PubMed] [Google Scholar]
- Rosenfeld M. Serum and lower respiratory tract tobramycin concentrations produced by inhaled tobramycin (TOBI) in young children with cystic fibrosis. Pediatric Pulmonology 1999;28(Suppl 19):106‐8. [CFGD Register: PI151b] [Google Scholar]
- Rosenfeld M, Borowitz D, Emerson J, Gibson R, McCoy K, McNamara S, et al. Serum pharmacokinetics and safety of inhaled tobramycin in very young CF patients. Pediatric Pulmonology 1999;28(Suppl 19):262. [CFGD Register: PI151a] [Google Scholar]
Gibson 2004 {published data only}
- Gibson RL, Retsch‐Bogart G, Ahrens R, Clancy J, Daines C, Milla C, et al. Safety and tolerability of aztreonam for inhalation (AI) in cystic fibrosis patients. Pediatric Pulmonology 2004;38(Suppl 27):253. [CFGD Register: PI188a] [Google Scholar]
- Gibson RL, Retsch‐Bogart GZ, Oermann C, Milla C, Pilewski J, Daines C, et al. Microbiology, safety, and pharmacokinetics of aztreonam lysinate for inhalation in patients with cystic fibrosis. Pediatric Pulmonology 2006;41(7):656‐65. [CFGD Register: PI188b] [DOI] [PubMed] [Google Scholar]
Goss 2009 {published data only}
- Clancy JP, Dupont L, Konstan MW, Billings J, Fustik S, Goss CH, et al. Phase II studies of nebulised Arikace in CF patients with Pseudomonas aeruginosa infection. Thorax 2013;68(9):818‐25. [CENTRAL: 876398; CFGD Register: PI207e // PI222c; CRS: 5500050000000084; PUBMED: 23749840] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy JP, Minic P, Dupont L, Goss CH, Quittner AL, Lymp JF, et al. Full analysis of data from two phase II blinded & placebo‐controlled studies of nebulized liposomal amikacin for inhalation (Arikace®) in the treatment of CF patients with pseudomonas aeruginosa lung infection. Pediatric Pulmonology 2010;45 Suppl 33:299. [Abstract no.: 227; CENTRAL: 848916; CFGD Register: PI207d // PI222b ; CRS: 5500100000010628] [Google Scholar]
- Goss CH, Clancy JP, Nick JA, Billings J, Rubenstein RC, Young KR, et al. A phase 2 blinded and placebo‐controlled study of nebulized liposomal amikacin (arikace™) in the treatment of CF patients with pseudomonas aeruginosa lung infection. Pediatric Pulmonology 2009;44 Suppl 32:295. [Abstract no.: 239; CFGD Register: PI222a] [Google Scholar]
Griffith 2008 {published data only}
- Geller DE, Flume P, Schwab R, Fornos P, Conrad DJ, Morgan E, et al. A phase safety, tolerability and pharmacokinetic (PK) study of MP‐376 (levofloxacin solution for inhalation) in stable cystic fibrosis (CF) patients. Pediatric Pulmonology 2008;43(Suppl 1):315. [Abstract no.: 321; CFGD Register: PI210b] [Google Scholar]
- Griffith DC, Hansen C, Pressler T, Balchen T, Jensen TJ, Geller DE, et al. Single‐dose pharmacokinetics of aerosol MP‐376 (levofloxacin solution for inhalation) in cystic fibrosis patients: PK‐PD implications. Journal of Cystic Fibrosis 2008;7(Suppl 2):S26. [CENTRAL: 643120; CFGD Register: PI210a; CRS: 5500100000003230] [Google Scholar]
- Kearns GL, Rubino CM, Griffith DC, Geller DE, Forrest A, Bhavnani SM, et al. Levofloxacin pharmacokinetics (PK) after administration of MP‐376 (Levofloxacin inhalation solution; Aeroquin) in children with cystic fibrosis. Journal of Cystic Fibrosis 2011;10 Suppl 1:S23. [Abstract no.: 88; CFGD Register: PI210d] [Google Scholar]
- Morgan EE, Dudley MN. Phase I, single and multi‐dose, placebo controlled, randomized, dose‐escalation study to evaluate the safety, tolerability and PK profile of MP‐376 using the PARI eFlow nebulizer for 14 days to CF patients. ClinicalTrials.gov (accessed 08 December 2010). [Clinicaltrials.gov: NCT00503490]
- Stockmann C, Hillyard B, Ampofo K, Spigarelli MG, Sherwin CM. Levofloxacin inhalation solution for the treatment of chronic Pseudomonas. Expert Review of Respiratory Medicine 2015;9(1):13‐22. [CFGD Register: PI210c] [DOI] [PubMed] [Google Scholar]
Gulliver 2003 {published data only}
- Gulliver T, Wilson S, Williams G, Harris M, Cooper D. Nebulized tobramycin (intravenous solution) is tolerated without inducing cough and wheeze in cystic fibrosis patients [abstract]. Proceedings of the Thoracic Society of Australia & New Zealand Annual Scientific Meeting; 2003 April 4‐9; Adelaide, Australia. 2003:Abst P139. [CFGD Register: PI184]
Herrmann 2017 {published data only}
- Herrmann G, Freitag E, Kiefer L, Bender V, Adams C, Graepler‐Mainka U, et al. Combined dry powder tobramycin and nebulized colistin versus colistin inhalation in CF patients ‐ a randomised, open label phase III clinical study. Journal of Cystic Fibrosis 2017;16 Suppl 1:S54. [CFGD Register: PI294] [Google Scholar]
Hodson 2002 {published data only}
- Govan JR. Insights into cystic fibrosis microbiology from the European tobramycin trial in cystic fibrosis. Journal of Cystic Fibrosis 2002;1 Suppl 2:203‐8. [CFGD Register: PI153e] [DOI] [PubMed] [Google Scholar]
- Hodson ME, Gallagher CG. New clinical evidence from the European tobramycin trial in cystic fibrosis. Journal of Cystic Fibrosis 2002;1 Suppl 2:S199‐202. [CFGD Register: PI153d] [DOI] [PubMed] [Google Scholar]
- Hodson ME, Gallagher CG, Govan JRW. A randomised clinical trial of nebulised tobramycin or colistin in cystic fibrosis. European Respiratory Journal 2002;20(3):658‐64. [CFGD Register: PI153c] [DOI] [PubMed] [Google Scholar]
- Hodson ME, Gallagher CG, Govan JRW, PL TNDS101CTG. Randomised UK / Eire clinical trial of the efficacy and safety of tobramycin 300 mg/5 ml nebuliser solution or nebulised colistin in CF patients [abstract]. Pediatric Pulmonology 2000;Suppl 20:248‐9. [CFGD Register: PI153b] [Google Scholar]
- Hodson ME, Gallagher CG, Govan JRW, the PL‐TNDS‐101CTG. Randomised UK/Eire clinical trial of the efficacy and safety of tobramycin 300 mg/5 mL nebuliser solution or nebulised colistin. Proceedings of the 13th International Cystic Fibrosis Congress; 2000 June 4‐8; Stockholm. 2000:145. [CFGD Register: PI153a]
Huls 2000 {published data only}
- App EM, Huls G, Bittner‐Dersch P, Stolz S, Lindemann H, Matthys H. Impaired lung function influences the serum concentration of inhaled drugs in cystic fibrosis. Pediatric Pulmonology 2000;30(Suppl 20):279‐80. [CFGD Register: PI156b] [Google Scholar]
- Huls G, App EM, Bittner‐Dersch P, Stolz S, Lindemann H. Impaired lung function influences the serum concentration of inhaled drugs in cystic fibrosis. Proceedings of the 13th International Cystic Fibrosis Congress, 4‐8 June 2000, Stockholm, Sweden. 2000:177. [CFGD Register: PI156a]
Jenkins 1985 {published data only}
- Jenkins SG, Kelly WC, Mason WG, Peele JD, Cruse MA, Coludro EO, et al. Aerosolized amikacin administration to cystic fibrosis patients chronically infected with pseudomonas aeruginosa. Cystic Fibrosis Club Abstracts. 1985:147. [CFGD Register: PI131]
Jensen 1987 {published data only}
- Jensen T, Pedersen SS, Garne S, Heilmann C, Hoiby N, Koch C. Colistin inhalation therapy in cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. Journal of Antimicrobial Chemotherapy 1987;19(6):831‐8. [CFGD Register: PI51] [DOI] [PubMed] [Google Scholar]
Kapranov 1995 {published data only}
- Kapranov NI, Belousov YB, Kashyrskaya NY, Smirnova EY. Quinoline therapy in children with cystic fibrosis. Proceedings of the 20th European Cystic Fibrosis Conference; 1995 June 18‐21; Brussels, Belgium. 1995:P19. [CENTRAL: 291377; CFGD Register: PI104; CRS: 5500100000001422]
Kenny 2009 {published data only}
- Kenny S, Hall V, Goldsmith C, Moore J, Rendall JC, Elborn JS. Eradication of new Pseudomonas aeruginosa in adults with CF. Journal of Cystic Fibrosis 2009;8(Suppl 2):S39. [CFGD Register: PI229] [Google Scholar]
Konstan 2010 {published data only}
- Chiron R, Geller DE, Angyalosi G, Debonnett L, Yadao A, Bader G, et al. Tobramycin powder for inhalation is effective in advanced stage CF lung disease: the EAGER trial. Journal of Cystic Fibrosis 2014;13 Suppl 2:S57. [Abstract no.: 42; CENTRAL: 996576; CFGD Register: PI239k; CRS: 5500129000000011] [Google Scholar]
- Geller DE, Flume PA, Brockhaus F, Zhang J, Angylosi G, He E, et al. Treatment convenience and satisfaction of tobramycin inhalation powder (TIP) versus TOBI in cystic fibrosis (CF) patients. Journal of Cystic Fibrosis 2010;9(Suppl 1):S22. [Abstract no.: 82; CFGD Register: PI239b] [Google Scholar]
- Geller DE, Flume PA, Konstan M, Angyalosi G, Higgins M. Microbiological and clinical response to tobramycin inhalation powder (TIP™) in cystic fibrosis patients with chronic Pseudomonas aeruginosa (Pa) infection. Journal of Cystic Fibrosis 2011;10 Suppl 1:S21. [Abstract no.: 82; CFGD Register: PI239g] [Google Scholar]
- Geller DE, Nasr SZ, Piggott S, He E, Angyalosi G, Higgins M. Tobramycin inhalation powder in cystic fibrosis patients: response by age group. Respiratory Care 2014;59(3):388‐98. [CFGD Register: PI239l] [DOI] [PubMed] [Google Scholar]
- Konstan M. A randomised open‐label, multicentre Phase 3 Trial to assess the safety of tobramycin inhalation powder compared to tobramycin solution for inhalation in cystic fibrosis subjects. ClinicalTrials.gov (accessed 08 December 2010). [Clinicaltrials.gov: NCT00388505]
- Konstan M, Flume PA, Brockhaus F, Angyalosi G, He, E, Geller D. Safety and efficacy of tobramycin inhalation powder (TIP) in treating CF patients infected with Pseudomonas aeruginosa (Pa) [abstract]. Journal of Cystic Fibrosis 2010;9(Suppl 1):S22. [CFDG Register: PI239a] [Google Scholar]
- Konstan MW, Flume PA, Brockhaus F, Angyalosi G, He E, Zhang J, et al. Tobramycin inhalation powder (TIP) versus tobramycin inhalation solution (TOBI®): the EAGER trial. Pediatric Pulmonology 2010;45(Suppl 33):303. [Abstract no.: 235; CFGD Register: PI239c] [Google Scholar]
- Konstan MW, Flume PA, Kappler M, Chiron R, Higgins M, Brockhaus F, et al. Safety, efficacy and convenience of tobramycin inhalation powder in cystic fibrosis patients: The EAGER trial. Journal of Cystic Fibrosis 2011;10(1):54‐61. [CFGD Register: PI239e] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColley S, Rietschel E, Brockhaus F, Angyalosi G, Higgins M. Safety of inhaled tobramycin in patients with cystic fibrosis. Pediatric Pulmonology 2011;46 Suppl 34:344. [Abstract no.: 365; CFGD Register: PI239h // PI227d] [Google Scholar]
- Nasr S, Nick J, Ezzet N, Gallo P, Debonnett L, Angyalosi G, et al. Reduced administration time for inhaled tobramycin in cystic fibrosis patients: results from the EAGER trial. Pediatric Pulmonology 2013;48 Suppl 36:283. [Abstract no.: 219; CENTRAL: 921693; CFGD Register: PI239j; CRS: 5500125000000404] [Google Scholar]
- Regnault A, Balp M M, Kulich K, Esteve L, Viala DM. Validation of the treatment satisfaction questionnaire for medication (TSQM) in cystic fibrosis. Journal of Cystic Fibrosis 2011;10 Suppl 1:S85. [Abstract no.: 332; CFGD Register: PI239f] [DOI] [PubMed] [Google Scholar]
- Regnault A, Balp MM, Kulich K, Esteve L, Viala‐Danten M. Association of treatment satisfaction and compliance of cystic fibrosis (CF) patients using inhaled tobramycin treatment in the EAGER study. Journal of Cystic Fibrosis 2011;10(Suppl 1):S82. [Abstract no.: 323; CFGD Register: PI239d] [Google Scholar]
- Regnault A, Balp MM, Kulich K, Viala‐Danten M. Validation of the Treatment Satisfaction Questionnaire for Medication in patients with cystic fibrosis. Journal of Cystic Fibrosis 2012;11(6):494‐501. [CENTRAL: 867326; CFGD Register: PI239i; CRS: 5500100000011297; PUBMED: 22583743] [DOI] [PubMed] [Google Scholar]
Konstan 2011 {published data only}
- Konstan MW, Geller DE, Brockhaus F, Zhang J, Angyalosi G. Tobramycin inhalation powder is effective and safe in the treatment of chronic pulmonary Pseudomonas aeruginosa (Pa) infection in patients with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 2009;179:no pagination. [Abstract no.: A1186; CFGD Register: PI227a] [Google Scholar]
- Konstan MW, Geller DE, Minic P, Brockhaus F, Zhang J, Angyalosi G. Effective treatment of chronic Pseudomonas aeruginosa (Pa) infection with tobramycin inhalation powder in CF patients. Journal of Cystic Fibrosis 2009;8 Suppl 2:S27. [Abstract no.: 105; CFGD Register: PI227b] [Google Scholar]
- Konstan MW, Geller DE, Minic P, Brockhaus F, Zhang J, Angyalosi G. Tobramycin inhalation powder for P. aeruginosa infection in cystic fibrosis: The EVOLVE trial. Pediatric Pulmonology 2011;46(3):230‐8. [CFGD Register: PI227e] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColley S, Rietschel E, Brockhaus F, Angyalosi G, Higgins M. Safety of inhaled tobramycin in patients with cystic fibrosis. Pediatric Pulmonology 2011;46 Suppl 34:344. [Abstract no.: 365; CFGD Register: PI239h // PI227d] [Google Scholar]
- Novartis. A randomised double‐blind, placebo‐controlled, multicenter, Phase 3 Trial to assess the efficacy and safety of tobramycin inhalation powder (TIP) in cystic fibrosis (CF) subjects. clinicaltrials.gov (accessed 08 December 2010). [CFGD Register: PI227c; NCT00125346]
Kun 1984 {published data only}
- Kun P, Landau LI, Phelan PD. Nebulized gentamicin in children and adolescents with cystic fibrosis. Australian Paediatric Journal 1984;20:43‐5. [CFGD Register: PI106] [DOI] [PubMed] [Google Scholar]
Ledson 2002 {published data only}
- Ledson MJ, Gallagher MJ, Cowperthwaite C, Robinson M, Convery RP, Walshaw MJ. A randomised double blind placebo controlled crossover trial of nebulised taurolidine in adult CF patients colonised with B Cepacia. Proceedings of 22nd European Cystic Fibrosis Conference; 1998 June 13‐19; Berlin, Germany. 1998:120. [CFGD Register: PI128a]
- Ledson MJ, Gallagher MJ, Robinson M, Cowperthwaite C, Williets T, Hart CA, et al. A randomized double‐blinded placebo‐controlled crossover trial of nebulized taurolidine in adult cystic fibrosis patients infected with Burkholderia cepacia. Journal of Aerosol Medicine 2002;15(1):51‐7. [CFGD Register: PI128b] [DOI] [PubMed] [Google Scholar]
Lenoir 2007 {published data only}
- Lenoir G, Antypkin YG, Miano A, Moretti P, Zanda M, Varoli G, et al. Efficacy, safety, and local pharmacokinetics of highly concentrated nebulized tobramycin in patients with cystic fibrosis colonized with Pseudomonas aeruginosa. Paediatric Drugs 2007;9 Suppl 1:11‐20. [CFGD Register: PI196c] [DOI] [PubMed] [Google Scholar]
- Lenoir G, Aryayev N, Varoli G, Monici Preti P. Aerosolized tobramycin in the treatment of patients with cystic fibrosis and pseudomonas aeruginosa infection. Journal of Cystic Fibrosis 2006;5 Suppl:S42. [CFGD Register: PI196b] [Google Scholar]
- Lenoir G, Aryayev N, Varoli G, Monici Preti P. Highly concentrated aerosolized tobramycin in the treatment of patients with cystic fibrosis and Pseudomonas aeruginosa infection. European Respiratory Journal 2005;26 Suppl 49:620s. [CFGD Register: PI196a] [Google Scholar]
Mainz 2014 {published data only}
- Mainz J. Nasal inhalation of tobramycin by the Pari sinus nebuliser in patients with cystic fibrosis and Pseudomonas aeruginosa colonization in the upper airways. ClinicalTrials.gov (accessed 08 December 2010). [Clinicaltrials.gov: NCT00774072]
- Mainz JG, Schadlich K, Schien C, Michl R, Schelhorn‐Neise P, Koitschev A, et al. Sinonasal inhalation of tobramycin vibrating aerosol in cystic fibrosis patients with upper airway Pseudomonas aeruginosa colonization: results of a randomized, double‐blind, placebo‐controlled pilot study. Drug Design, Development and Therapy 2014;8:209‐17. [CENTRAL: 981442; CFGD Register: PI248b; CRS: 5500125000000721; PUBMED: 24596456] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainz JG, Schien C, Schadlich K, Pfister W, Schelhorn‐Neise P, Koitschev A, et al. Sinonasal inhalation of tobramycin in cystic fibrosis patients with P. aeruginosa colonization of the upper airways ‐ results of a multicentric placebo‐controlled pilot study. Journal of Cystic Fibrosis 2011;10 Suppl 1:S21. [Abstract no.: 83; CENTRAL: 848866; CFGD Register: PI248a; CRS: 5500100000010543] [Google Scholar]
- Mainz JG, Schiller I, Ritschel C, Mentzel HJ, Riethmuller J, Koitschev A, et al. Sinonasal inhalation of dornase alfa in CF: A double‐blind placebo‐controlled cross‐over pilot trial. Auris, Nasus, Larynx 2011;38(2):220‐7. [CFGD Register: PI248c] [DOI] [PubMed] [Google Scholar]
Mazurek 2011 {published data only}
- Cicirello H, Mazurek H, Chiron R, Pelikan L, Geidel C, Bolbas K, et al. Efficacy and safety of two inhaled tobramycin solutions in patients with cystic fibrosis and chronic pseudomonas aeruginosa infection: Results from a head to head comparison. American Journal of Respiratory and Critical Care Medicine 2011;183(1 Meeting Abstracts):no pagination. [CFGD Register: PI249e] [Google Scholar]
- Mazurek H, Chiron R, Kucerova T, Geidel C, Bolbas K, Chuchalin A, et al. Long‐term efficacy and safety of aerosolized tobramycin 300 mg/4 ml in cystic fibrosis. Pediatric Pulmonology 2014;49(11):1076‐89. [CENTRAL: 1015252; CFGD Register: PI249d; CRS: 5500131000000251; JID: 8510590; PUBMED: 24464974] [DOI] [PubMed] [Google Scholar]
- Mazurek H, Chiron R, Pelikan L, Geidel C, Bolbas K, Antipkin Y, et al. Comparison of two inhaled tobramycin solutions in cystic fibrosis patients with chronic pseudomonas aeruginosa infection: results in different age subgroups. Journal of Cystic Fibrosis 2011;10(Suppl 1):S28. [CFGD Register: PI249b] [Google Scholar]
- Mazurek H, Chiron R, Varoli G, Santoro D, Cicirello H, Antipkin Y. Efficacy on lung function and safety of multiple courses of tobramycin 300mg/4 ml nebuliser solution (Bramitob) in patients with cystic fibrosis and chronic pseudomonas aeruginosa infection: results from a 48‐week extension phase. Journal of Cystic Fibrosis 2012;11 Suppl1:S74. [Abstract no.: 69; CENTRAL: 867264; CFGD Register: PI249c; CRS: 5500100000011290] [Google Scholar]
- Mazurek H, Lenoir G, Pelikan L, Geidel C, Bolbas K, Antipkin Y, et al. Head‐to‐head comparison of two inhaled tobramycin solutions in cystic fibrosis (CF) patients with chronic pseudomonas aeruginosa (Pa) infection. Journal of Cystic Fibrosis 2011;10(Suppl 1):S28. [CFGD Register: PI249a] [Google Scholar]
McCoy 2008 {published data only}
- McCoy K, Retsch‐Bogart G, Gibson RL, Oermann C, Braff M, Montgomery AB. Investigation of susceptibility breakpoints for inhaled antibiotic therapies in cystic fibrosis. Pediatric Pulmonology 2010;45 Suppl 33:341. [Abstract no.: 340; CENTRAL: 848913; CFGD Register: PI212g // PI213i; CRS: 5500100000010623] [Google Scholar]
- McCoy K, Retsch‐Bogart G, Oermann C, Gibson R, Montgomery AB. Aztreonam lysine for inhalation (AZLI) for CF patients with P. aeruginosa (PA) infection. Journal of Cystic Fibrosis 2007;6 Suppl 1:S10. [CFGD Register: PI212a] [Google Scholar]
- McCoy KS, Quittner AL, Oermann CM, Gibson RL, Retsch‐Bogart GZ, Montgomery AB. Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis. American Journal of Respiratory Critical Care Medicine 2008;178:921‐8. [CFGD Register: PI212c] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy KS, Quittner AL, Oermann CM, Gibson RL, Retsch‐Bogart GZ, Montgomery AB. Online Data Supplement to 'Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis' [online]. American Journal of Respiratory and Critical Care Medicine 2008;178(9):921‐8. [CFGD Register: PI212d] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy KS, Retsch‐Boagrt GZ, Gibson RL, Oermann CM, McKevitt M, Montgomery AB. Efficacy of Aztreonam Lysine for inhalation (AZLI) in patients with cystic fibrosis and drug resistant P. aeruginosa (DRPA). Journal of Cystic Fibrosis 2009;8 Suppl 2:S28. [CFGD Register: PI212e // PI213e] [Google Scholar]
- McCoy KS, Retsch‐Bogart GZ, Gibson R, Oermann C, Braff MH, Montgomery AB. Relevance of established susceptibility breakpoints to clinical efficacy of inhaled antibiotic therapies in cystic fibrosis. Pediatric Pulmonology 2008;43 Suppl 31:351. [CFGD Register: PI212b // PI213c] [Google Scholar]
- Plosker GL. Aztreonam lysine for inhalation solution: in cystic fibrosis. Drugs 2010;70(14):1843‐55. [CFGD Register: PI212f // PI213h] [DOI] [PubMed] [Google Scholar]
- Quittner AL, Henig NR, Lewis S, Derchak PA, McCoy KS, Oermann CM, et al. Effects of chronic intermittent aztreonam for inhalation solution (AZLI) on health‐related quality of life (HRQOL) in persons with cystic fibrosis (CF) and P. aeruginosa. Pediatric Pulmonology 2011;46 Suppl 34:299. [Abstract no.: 240; CENTRAL: 867260; CFGD Register: PI213j // PI220f ; CRS: 5500100000011261] [Google Scholar]
Nasr 2006 {published data only}
- Nasr SZ, Gordon D, Sakmar E, Eckhardt BP, Strouse PJ. Comparison of high resolution computerized tomography (HRCT) of the chest and pulmonary function testing in evaluating the effect of tobramycin solution for inhalation in cystic fibrosis (CF) patients. European Respiratory Journal 2004;24(Suppl 48):P2403. [CENTRAL: 518355; CFGD Register: DG2a ; CRS: 5500100000002727] [Google Scholar]
- Nasr SZ, Gordon D, Sakmar E, Yu X, Christodoulou E, Eckhardt BP, et al. High resolution computerized tomography of the chest and pulmonary function testing in evaluating the effect of tobramycin solution for inhalation in cystic fibrosis patients. Pediatric Pulmonology 2006;41(12):1129‐37. [CENTRAL: 573172; CFGD Register: DG2c; CRS: 5500100000002876; EMBASE: 2006604093; PUBMED: 17068818] [DOI] [PubMed] [Google Scholar]
- Nasr SZ, Gordon D, Yu X, Sakmar E, Eckhardt BP, Strause P. Comparison of high resolution computerized tomography (HRCT) of the chest and pulmonary function testing in evaluating of the effect of tobramycin solution for inhalation (TSI) in cystic fibrosis (CF) subjects with mild lung disease. Pediatric Pulmonology 2004;38(Suppl 27):299. [CENTRAL: 526252; CFGD Register: DG2b; CRS: 5500100000002752] [Google Scholar]
- Nasr SZ, Sakmar E, Christodoulou E, Eckhardt BP, Streetman DS, Strouse PJ. The use of high resolution computerized tomography (HRCT) of the chest in evaluating the effect of tobramycin solution for inhalation in cystic fibrosis lung disease. Pediatric Pulmonology 2010;45(5):440‐9. [CENTRAL: 752249; CFGD Register: DG2e; CRS: 5500125000000357; PUBMED: 20425851] [DOI] [PubMed] [Google Scholar]
- Nasr SZ, Sakmar E, Eckhardt BP, Strouse PJ. High resolution computerized tomography (HRCT) of the chest vs. pulmonary function testing utility in evaluating the effect of tobramycin solution for inhalation in cystic fibrosis. Pediatric Pulmonology 2008;43 Suppl 31:366. [CENTRAL: 690468; CFGD Register: DG2d; CRS: 5500100000003327] [Google Scholar]
Nathanson 1985 {published data only}
- Nathanson I, Cropp GJA, Li P, Neter E. Effectiveness of aerosolized gentamicin in cystic fibrosis (CF). Cystic Fibrosis Club Abstracts; 1985. 1985; Vol. 28:145. [CENTRAL: 291475; CFGD Register: PI130; CRS: 5500100000001502]
Nikolaizik 1996 {published data only}
- Nikolaizik WH, Jenni‐Galovic V, Schoni MH. Bronchial constriction after nebulized tobramycin preparations and saline in patients with cystic fibrosis. European Journal of Pediatrics 1996;155(7):608‐11. [DOI] [PubMed] [Google Scholar]
Nikolaizik 2005 {published data only}
- Nikolaizik WH, Vietzke D, Ratjen F. A pilot study to compare tobramycin 80 mg injectable preparation with 300 mg solution for inhalation in cystic fibrosis patients. Canadian Respiratory Journal 2008;15(5):259‐62. [CFGD Register: PI191c] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaizik WH, Vietzke D, Ratjen F. Comparison of tobramycin 80 mg (IV‐ Preparation) and 300mg solution for inhalation in cystic fibrosis patients. European Respiratory Journal 2005;26(Suppl 49):620s. [CFGD Register: PI191b] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaizik WH, Vietzke D, Ratjen F. Comparison of tobramycin 80mg (IV‐preparation) and 300mg solution inhaled twice daily for chronic P. aeruginosa infection. Journal of Cystic Fibrosis 2005;4(Suppl):S53. [CFGD Register: PI191a] [Google Scholar]
Nikonova 2010 {published data only}
- Nikonova VS, Kashirskaya NY, Kapranov NI. Efficacy and safety of tobramycin and colistin for inhalation in children with cystic fibrosis from Moscow region. Journal of Cystic Fibrosis 2010;9 Suppl 1:S54. [Abstract no.: 210; CENTRAL: 774753; CFGD Register: PI242; CRS: 5500100000003505] [Google Scholar]
Noah 2010 {published data only}
- Noah T, Ivins S, Abode K, Harris W, Henry M, Leigh M. Comparison of antibiotics for early pseudomonas infection in CF: interim data analysis. Pediatric Pulmonology 2007;42(Suppl 30):332. [CFGD Register: PI205a] [Google Scholar]
- Noah TL, Ivins SS, Abode KA, Stewart PW, Michelson PH, Harris WT, et al. Inhaled versus systemic antibiotics and airway inflammation in children with cystic fibrosis and Pseudomonas. Pediatric Pulmonology 2010;45(3):281‐90. [CFGD Register: PI205b] [DOI] [PubMed] [Google Scholar]
Nolan 1982 {published data only}
- Nolan G, McIvor P, Levison H, Fleming PC, Corey M, Gold R. Antibiotic prophylaxis in cystic fibrosis: inhaled cephaloridine as an adjunct to oral cloxacillin. Journal of Pediatrics 1982;101(4):626‐30. [CFGD Register: PI110] [DOI] [PubMed] [Google Scholar]
Novartis 2010 {published data only}
- Novartis. A randomized, double‐blind, placebo‐controlled, crossover multi‐center study to assess the efficacy and safety of inhaled tobramycin nebuliser solution (TOBI®) for the treatment of early infections of P. aeruginosa in cystic fibrosis subjects aged from 3 months to less than 7 years. ClinicalTrials.gov (accessed 08 December 2010). [Clinicaltrials.gov: NCT01082367; EARLY trial]
Oermann 2009 {published data only}
- Oermann CM, McCoy KS, Retsch‐Bogart GZ, Gibson R, McKevitt M, Montgomery B. Antibiotic susceptibility in Pseudomonas Aeruginosa (PA) isolates following repeated exposure to aztreonam for inhalation solution (AZLI) in patients with cystic fibrosis. Pediatric Pulmonology 2009;44(Suppl 32):309. [Abstract no.: 278; CENTRAL: 735839; CRS: 5500100000003411] [Google Scholar]
- Oermann CM, McCoy KS, Retsch‐Bogart GZ, Gibson R, McKevitt M, Montgomery B. Effect of repeated exposure to aztreonam for inhalation solution (AZLI) therapy on cystic fibrosis respiratory pathogens. Pediatric Pulmonology 2009;44 Suppl 32:335. [Abstract no.: 353; CENTRAL: 744123; CRS: 5500100000003452] [Google Scholar]
- Oermann CM, McCoy KS, Retsch‐Bogart GZ, Gibson RL, Montgomery AB. Effect of multiple courses of Aztreonam Lysine for inhalation (AZLI) on FEV1 and weight in patients with cystic fibrosis (CF) and Pseudomonas aeruginosa (PA): analysis of 18 month data from CP‐AI‐006. Journal of Cystic Fibrosis 2009;8 Suppl 2:S28. [Abstract no.: 107; CENTRAL: 744122; CRS: 5500100000003451] [Google Scholar]
- Oermann CM, McCoy KS, Retsch‐Bogart GZ, Gibson RL, Quittner AL, Montgomery AB. Adherence over multiple courses of Aztreonam for inhalation (AZLI): effect on disease‐related endpoints in patients with cystic fibrosis (CF) and Pseudomonas aeruginosa (PA). Journal of Cystic Fibrosis 2009;8 Suppl 2:S28. [Abstract no.: 109; CENTRAL: 744121; CRS: 5500100000003450] [Google Scholar]
- Oermann CM, Retsch‐Bogart GZ, Quittner AL, Gibson RL, McCoy KS, Montgomery AB, et al. An 18‐month study of the safety and efficacy of repeated courses of inhaled aztreonam lysine in cystic fibrosis. Pediatric Pulmonology 2010;45(11):1121‐34. [CENTRAL: 770178; CRS: 5500100000003719] [DOI] [PMC free article] [PubMed] [Google Scholar]
Poli 2005 {published data only}
- Poli G, Acerbi D, Pennini R, Soliani Raschini A, Bianco F, Corrado M, et al. Clinical pharmacology study of a new tobramycin solution for nebulisation. Journal of Cystic Fibrosis 2006;5(Suppl):S43. [CFGD Register: PI195b] [Google Scholar]
- Poli G, Acerbi D, Pennini R, Soliani Raschini A, Corrado M, Eichler HG, et al. Clinical pharmacology study of a new tobramycin solution for nebulisation. European Respiratory Journal 2005;26(Suppl 49):729s. [CFGD Register: PI195a] [DOI] [PubMed] [Google Scholar]
- Poli G, Acerbi D, Pennini R, Soliani Raschini A, Corrado ME, Eichler HG, et al. Clinical pharmacology study of Bramitob, a tobramycin solution for nebulization, in comparison with Tobi. Paediatric Drugs 2007;9(Suppl 1):3‐9. [CFGD Register: PI195c] [DOI] [PubMed] [Google Scholar]
Proesmans 2013 {published data only}
- Proesmans M, Boulanger L, Vermeulen F, Boeck K. Eradication of recent Pseudomonas aeruginosa isolation: TOBI versus colistin/ ciprofloxacin. Journal of Cystic Fibrosis 2008;7(Suppl 2):S64. [CFGD Register: PI208a] [Google Scholar]
- Proesmans M, Boulanger L, Vermeulen F, Boeck K. Eradication of recent Pseudomonas aeruginosa isolation: TOBI versus colistin/ciprofloxacin. Pediatric Pulmonology 2009;44 Suppl 32:321. [Abstract no.: 311; CFGD Register: PI208b] [Google Scholar]
- Proesmans M, Boulanger L, Vermeulen F, Boeck K. Eradication of recent pseudomonas aeruginosa infection: TOBI versus Colistineb®/ ciprofloxacin. Journal of Cystic Fibrosis 2011;10 Suppl 1:S26, Abstract no: 102. [CFGD Register: PI208c] [Google Scholar]
- Proesmans M, Vermeulen F, Boulanger L, Verhaegen J, Boeck K. Comparison of two treatment regimens for eradication of pseudomonas aeruginosa infection in children with cystic fibrosis. Journal of Cystic Fibrosis 2013;12(1):29‐34. [CFGD Register: PI208d] [DOI] [PubMed] [Google Scholar]
Ramsey 1993 {published data only}
- Fiel SB. Aerosol delivery of antibiotics to the lower airways of patients with cystic fibrosis. Chest 1995;107(2 Suppl):61S‐4S. [CFGD Register: PI75b] [DOI] [PubMed] [Google Scholar]
- Ramsey BW, Dorkin HL, Eisenberg JD, Gibson RL, Harwood IR, Kravitz RM, et al. Efficacy of aerosolized tobramycin in patients with cystic fibrosis. New England Journal of Medicine 1993;328(24):1740‐6. [CFGD Register: PI75a] [DOI] [PubMed] [Google Scholar]
Ramsey 1999 {published data only}
- Birnbaum HG, Greenberg P, Finkelstein S, Berndt E, Otto KL, Montgomery AB, et al. Economic analysis of hospitalization and home IV anti‐pseudomonal antibiotic use in CF patients on tobramycin solution for inhalation (TOBI). Pediatric Pulmonology 1998;Suppl 17:273. [CFGD Register: PI120f] [Google Scholar]
- Bowman CM. The long‐term use of inhaled tobramycin in patients with cystic fibrosis. Journal of Cystic Fibrosis 2002;1 Suppl 2:S194‐8. [CFGD Register: PI120cc] [DOI] [PubMed] [Google Scholar]
- Burns JL, Dalfsen JM, Shawar RM, Otto KL, Garber RL, Quan JM, et al. Effect of chronic intermittent administration of inhaled tobramycin on respiratory microbial flora in patients with cystic fibrosis. Journal of Infectious Diseases 1999;179(5):1190‐6. [CFGD Register: PI120i] [DOI] [PubMed] [Google Scholar]
- Casey S, Ramsey B, Borowitz D. Nutritional benefits of chronic intermittent Pseudomonas aeruginosa suppression with tobramycin solution for inhalation in adolescents. 13th International Cystic Fibrosis Congress; 2000 June 4‐8; Stockholm, Sweden. 2000:172. [CFGD Register: PI120ff]
- Enger C, Rothman K, Kylstra JW. Mortality rates during two years of treatment with intermittent inhaled tobramycin (TOBI) in CF. Pediatric Pulmonology 1999;Suppl 19:339‐40. [CFGD Register: PI120m] [Google Scholar]
- Fiel S, VanDevanter DR, Yu X. Effect of tobramycin solution for inhalation on pulmonary function decline rates in exacerbating and stable CF patients. Abstracts of the 24th European Cystic Fibrosis Conference; 2001 June 6‐9; Vienna. 2001:P195. [CFGD Register: PI120s]
- Fiel SB. Long term effect of tobramycin solution for inhalation on reduction of hospitalization of CF patients. European Respiratory Journal 2000;16 Suppl 31:1545. [CFGD Register: PI120dd] [Google Scholar]
- Geller DE, Pitlick WH, Nardella PA, Tracewell WG, Ramsey BW. Pharmacokinetics and bioavailability of aerosolized tobramycin in cystic fibrosis. Chest 2002;122(1):219‐26. [CFGD Register: PI120bb] [DOI] [PubMed] [Google Scholar]
- Graff GR, Gordon DC, Van D, Burns JL. Epidemiology of Stenotrophomonas maltophilia culture in cystic fibrosis (CF) patients during the tobramycin solution for inhalation (TOBI) study. Pediatric Pulmonology 2000;Suppl 20:283. [CFGD Register: PI120p] [Google Scholar]
- Hazinski TA. Intermitent administration of inhaled tobramycin in patients with cystic fibrosis [comment]. Journal of Pediatrics 1999;135(1):130. [CFGD Register: PI120j] [PubMed] [Google Scholar]
- Klystra JW, Bowman CM, Meyer U, Montgomery AB, Schaeffler B, Stewart P, et al. Who benefits more? An age‐stratified analysis of lung function and weight gain in CF patients using inhaled tobramycin. Netherlands Journal of Medicine 1999;54 Suppl:S83. [CFGD Register: PI120h] [Google Scholar]
- Konstan MW, VanDevanter DR. Peripheral white blood cell count as a surrogate marker for progression of lung disease in cystic fibrosis. Pediatric Pulmonology 2001;Suppl 22:305. [CFGD Register: PI120v] [Google Scholar]
- LeLorier J, Perreault S, Birnbaum H, Greenberg P, Sheehy O. Savings in direct medical costs from the use of tobramycin solution for inhalation in patients with cystic fibrosis. Clinical Therapeutics 2000;22(1):140‐51. [CFGD Register: PI120o] [DOI] [PubMed] [Google Scholar]
- LiPuma JJ. Microbiological and immunologic considerations with aerosolized drug delivery. Chest 2001;120(Suppl 3):118S‐23S. [CFGD Register: PI120x] [DOI] [PubMed] [Google Scholar]
- MacLeod DL, Nelson LE, Shawar RM, Lin BB, Lockwood LG, Dirks JE, et al. Aminoglycoside‐resistance mechanisms for cystic fibrosis Pseudomonas aeruginosa isolates are unchanged by long‐term, intermittent, inhaled tobramycin treatment. Journal of Infectious Diseases 2000;181(3):1180‐4. [CFGD Register: PI120n] [DOI] [PubMed] [Google Scholar]
- Moss R, Klystra JW, Montgomery AB, Gibson R. Who benefits more? An analysis of FEV1 and weight in adolescents (age 13 ‐ <18) CF patients using inhaled tobramycin (TOBI). Pediatric Pulmonology 1999;Suppl 19:243. [CFGD Register: PI120k] [Google Scholar]
- Moss RB. Administration of aerosolized antibiotics in cystic fibrosis patients. Chest 2001;120 Suppl 3:107S‐13S. [CFGD Register: PI120y] [DOI] [PubMed] [Google Scholar]
- Moss RB. Long‐term benefits of inhaled tobramycin in adolescent patients with cystic fibrosis. Chest 2002;121(1):55‐63. [CFGD Register: PI120aa] [DOI] [PubMed] [Google Scholar]
- Nickerson B, Montgomery AB, Klystra JW, Ramsey BW. Safety and effectiveness of 2 years of treatment with intermittent inhaled tobramycin in CF patients. Pediatric Pulmonology 1999;Suppl 19:243‐4. [CFGD Register: PI120L] [Google Scholar]
- Otto KL, Montgomery AB, Lin A, Ramsey BW. Seasonality of hospitalisations for pulmonary exacerbations and lower respiratory tract infections in phase III tobramycin solution for inhalation trials. Pediatric Pulmonology 1998;4 Suppl 17:273. [CFGD Register: PI120e] [Google Scholar]
- Quan JM, Vasiljev M, Schaeffler B, Phelps C, Burrington C, Meyer U. Treatment for exacerbation only does not arrest progressive lung function decline in CF. The Netherlands Journal of Medicine 1999;54 Suppl:S84. [CFGD Register: PI120g] [Google Scholar]
- Quittner A, Gordon D, Yu X. Convergence of quality of life assessments and clinical outcomes in patients enrolled in the tobramycin solution for inhalation (TSI) trials. Abstracts of the 24th European Cystic Fibrosis Conference; 2001 June 6‐9; Vienna. 2001:P326. [CFGD Register: PI120t]
- Quittner AL, Buu A. Effects of tobramycin solution for inhalation on global ratings of quality of life in patients with cystic fibrosis and Pseudomonas aeruginosa infection. Pediatric Pulmonology 2002;33(4):269‐76. [CFGD Register: PI120z] [DOI] [PubMed] [Google Scholar]
- Quittner AL, Buu A, Gordon D. Longitudinal changes in global ratings of quality of life for patients in the tobramycin solution for inhalation (TSI) trials. Pediatric Pulmonology 2001;Suppl 22:349. [CFGD Register: PI120w] [Google Scholar]
- Ramsey B, Burns J, Smith A. Safety and efficacy of tobramycin solution for inhalation in patients with cystic fibrosis: the results of two phase III placebo controlled clinical trial. Pediatric Pulmonology 1997;Suppl 14:137‐8. [CFGD Register: PI120c] [Google Scholar]
- Ramsey BW, Bowman MC, Montgomery AB, Smith AR, TOBI SG. Design of Phase 3 aerosolized tobramycin studies for chronic intermittent therapy. Proceedings of the 21st European Cystic Fibrosis Conference; 1997 June 1‐6; Davos. 1997:144. [CFGD Register: PI120b]
- Ramsey BW, Bowman MC, Vasiljev‐K M, Smith AR, TOBI SG. Results of Phase 3 aerosolized tobramycin studies for chronic intermittent therapy. Proceedings of the 21st European Cystic Fibrosis Conference; 1997 June 1‐6; Davos. 1997:144. [CFGD Register: PI120a]
- Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams Warren J, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. New England Journal of Medicine 1999;340(1):23‐30. [CFGD Register: PI120d] [DOI] [PubMed] [Google Scholar]
- Rosenfeld M, Emerson J, Williams‐Warren J, Pepe M, Smith A, Montgomery AB, et al. Defining a pulmonary exacerbation in cystic fibrosis. Journal of Pediatrics 2001;139(3):359‐65. [CFGD Register: PI120ee] [DOI] [PubMed] [Google Scholar]
- Taylor CJ. Nebulised high dose Tobramycin (TOBI) in adolescents with cystic fibrosis. Pediatric Pulmonology 2001;Suppl 22:291. [CFGD Register: PI120u] [Google Scholar]
- VanDevanter D, Hou K, Yu X. Effect of tobramycin solution for inhalation (TOBI) on long term rates of lung function decline in Pseudomonas aeruginosa‐infected cystic fibrosis (CF) patients with mild to moderate lung disease. Pediatric Pulmonology 2000;Suppl 20:298. [CFGD Register: PI120q] [Google Scholar]
- VanDevanter DR, Yu X, Konstan MW. Effect of tobramycin solution for inhalation on circulating white blood cell counts in exacerbating and stable CF patients. Abstracts of the 24th European Cystic Fibrosis Conference; 2001 June 6‐9; Vienna. 2001:P172. [CFGD Register: PI120r]
- Wagener J, Moss R, Wilmott R, Konstan M, Zeitlin P, Walz D, et al. A study of the safety and efficacy of tobramycin for inhalation in young children with cystic fibrosis. A Phase II multicenter randomised trial of tobramycin for inhalation in young children with cystic fibrosis. ClinicalTrials.gov (accessed 08 December 2010). [NCT00006280]
Ratjen 2010 {published data only}
- Ratjen F, Munck A, Campello V. Inhaled tobramycin nebuliser solution for treatment of early Pseudomonas aeruginosa infection: first results from the Elite study. Pediatric Pulmonlogy 2006;41 Suppl 29:318. [CFGD Register: PI197b] [Google Scholar]
- Ratjen F, Munck A, Campello V. Safety of inhaled tobramycin nebuliser solution for treatment of early pseudomonas aeruginosa infection: first results from the ELITE study. Journal of Cystic Fibrosis 2006;5 Suppl:S22. [CFGD Register: PI197a] [Google Scholar]
- Ratjen F, Munck A, Kho P. Short and long‐term efficacy of inhaled tobramycin in early P. aeruginosa infection: the ELITE study. Pediatric Pulmonology 2008;43 Suppl 31:319. [CFGD Register: PI197d] [Google Scholar]
- Ratjen F, Munck A, Kho P, Angyalosi G. Treatment of early Pseudomonas aeruginosa infection in patients with cystic fibrosis: the ELITE trial. Thorax 2010;65(4):286‐91. [CFGD Register: PI197e] [DOI] [PubMed] [Google Scholar]
- Ratjen F, Stenglein S, Munck A. Inhaled tobramycin nebulizer solution for treatment of early Pseudomonas aeruginosa infection; the ELITE study. Journal of Cystic Fibrosis 2008;7 Suppl 2:S26. [CFGD Register: PI197c] [Google Scholar]
Regelmann 1990 {published data only}
- Regelmann WE, Elliott GR, Clawson CC, Warwick WJ. Reduction of sputum Ps. Aeruginosa density by antibiotics improves lung function in CF more than bronchodilators and chest physiotherapy alone. Pediatric Pulmonology 1988;5(Suppl 2):97. [CFGD Register: PI65a] [DOI] [PubMed] [Google Scholar]
- Regelmann WE, Elliott GR, Warwick WJ, Clawson CC. Reduction of sputum Pseudomonas aeruginosa density by antibiotics improves lung function in cystic fibrosis more than do bronchodilators and chest physiotherapy alone. American Review of Respiratory Disease 1990;141(4 Pt 1):914‐21. [CFGD Register: PI65b] [DOI] [PubMed] [Google Scholar]
Retsch‐Bogart 2007 {published data only}
- McCoy K, Retsch‐Bogart G, Gibson RL, Oermann C, Braff M, Montgomery AB. Investigation of susceptibility breakpoints for inhaled antibiotic therapies in cystic fibrosis. Pediatric Pulmonology 2010;45 Suppl 33:341. [Abstract no.: 340; CENTRAL: 848913; CFGD Register: PI212g // PI213i ; CRS: 5500100000010623] [Google Scholar]
- McCoy KS, Retsch‐Boagrt GZ, Gibson RL, Oermann CM, McKevitt M, Montgomery AB. Efficacy of Aztreonam Lysine for inhalation (AZLI) in patients with cystic fibrosis and drug resistant P. aeruginosa (DRPA). Journal of Cystic Fibrosis 2009;8 Suppl 2:S28. [CFGD Register: PI212e // PI213e] [Google Scholar]
- McCoy KS, Retsch‐Bogart GZ, Gibson R, Oermann C, Braff MH, Montgomery AB. Relevance of established susceptibility breakpoints to clinical efficacy of inhaled antibiotic therapies in cystic fibrosis. Pediatric Pulmonology 2008;43 Suppl 31:351. [CFGD Register: PI212b // PI213c] [Google Scholar]
- Plosker GL. Aztreonam lysine for inhalation solution: in cystic fibrosis. Drugs 2010;70(14):1843‐55. [CFGD Register: PI212f // PI213h] [DOI] [PubMed] [Google Scholar]
- Quittner AL, Retsch‐Bogart GZ, McCoy KS, Oermann CM, Gibson R, Lewis S. Effect of a 28‐day course of aztreonam for inhalation solution (AZLI) on responses to individual CFQ‐R respiratory symptoms score questions among patients with CF. Pediatric Pulmonology 2009;44 Suppl 32:305. [Abstract no.: 266; CFGD Register: PI213f] [Google Scholar]
- Retsch‐Bogart GZ, McCoy KS, Gibson RL, Oermann C, Montgomery AB. Source of improvements in lung function in patients with cystic fibrosis (CF) following treatment with aztreonam lysine for inhalation (AZLI). Pediatric Pulmonology 2008;42 Suppl 30:320. [CFGD Register: PI213d] [Google Scholar]
- Retsch‐Bogart GZ, McCoy KS, Gibson RL, Oermann CM, Braff MH, Montgomery AB. Sustained improvement in pulmonary function following a 28‐day course of 75 MG AZLI TID therapy. Pediatric Pulmonology 2008;43 Suppl 31:320. [CFGD Register: PI211d // PI213b] [Google Scholar]
- Retsch‐Bogart GZ, Montgomery B, Gibson R, McCoy K, Oermann CM, Cooper P. Phase 3 trial (AIR‐CF 1) measuring improvement in respiratory symptoms in patients with cystic fibrosis (CF) following treatment with aztreonam lysine for inhalation. Pediatric Pulmonology 2007;42 Suppl 30:310. [CFGD Register: PI213a] [Google Scholar]
- Retsch‐Bogart GZ, Quittner AL, Gibson RL, Oermann CM, McCoy KS, Montgomery AB, et al. Efficacy and safety of inhaled aztreonam lysine for airway pseudomonas in cystic fibrosis. Chest 2009;135(5):1223‐32. [CFGD Register: PI213g] [DOI] [PMC free article] [PubMed] [Google Scholar]
Retsch‐Bogart 2008 {published data only}
- Burns JL, Stapp J, Lofland D, AI P2SG. Microbiology results from a phase 2 clinical study of aztreonam lysinate for inhalation (AI): a new inhaled antibiotic to treat CF patients with Pseudomonas aeruginosa (PA). Journal of Cystic Fibrosis 2005;4 Suppl:S55. [CFGD Register: PI211a] [Google Scholar]
- Retsch‐Bogart GZ, Burns JL, Otto KL, Liou TG, McCoy K, Oermann C, et al. A phase 2 study of aztreonam lysine for inhalation to treat patients with cystic fibrosis and Pseudomonas aeruginosa infection. Pediatric Pulmonology 2008;43(1):47‐58. [CFGD Register: PI211c] [DOI] [PubMed] [Google Scholar]
- Retsch‐Bogart GZ, Gibson RL, AI P2SG. A phase 2 study of aztreonam lysinate for inhalation to treat cystic fibrosis patients with Pseudomonas aeruginosa infection. American Thoracic Society International Conference; 2005 May 20‐25; San Diego, USA. 2005:A576. [CFGD Register: PI211b]
- Retsch‐Bogart GZ, McCoy KS, Gibson RL, Oermann CM, Braff MH, Montgomery AB. Sustained improvement in pulmonary function following a 28‐day course of 75 MG AZLI TID therapy. Pediatric Pulmonology 2008;43 Suppl 31:320. [CFGD Register: PI211d // PI213b] [Google Scholar]
Rietschel 2009 {published data only}
- Rietschel E, Posselt HG, Heuer HE, Merkel N, Staab D. Pharmacokinetics of tobramycin (TOBITM) after 4 and 8 weeks of continuous once daily or twice daily inhalations. Journal of Cystic Fibrosis 2009;8(Suppl 2):S27. [CFGD Register: PI232a] [Google Scholar]
- Rietschel E, Staab D, Merkel N, Konigsbruggen S, Posselt H. Pharmacokinetics of continuous treatment with O.D. or B.I.D inhalation of tobramycin (TOBI™) via Pari Eflow™ Rapid. Pediatric Pulmonology 2010;45 Suppl 33:320. [Abstract no.: 283; CFGD Register: PI232c] [Google Scholar]
- Rietschel E, Staab D, von Konigsbruggen, Merkel N, Heuer HE, Posselt HG. Pharmacokinetics of continuous treatment with o.d. or b.i.d. inhalation of tobramycin (TOBITM). Journal of Cystic Fibrosis 2010;9(Suppl 1):S23. [CFGD Register: PI232b] [Google Scholar]
Rosenfeld 2006 {published data only}
- Rosenfeld M, Emerson J, Uh D, Anderson G, Genatossio A, McNamara S, et al. Does tobramycin accumulate in respiratory secretions with repeated aerosol administration: a pilot study. Pediatric Pulmonology 2006;41(Suppl 29):327. [CFGD Register: PI203] [Google Scholar]
Ruddy 2013 {published data only}
- Ruddy J, Emerson J, Moss R, Genatossio A, McNamara S, Burns JL, et al. Sputum tobramycin concentrations in cystic fibrosis patients with repeated administration of inhaled tobramycin. Journal of Aerosol Medicine and Pulmonary Drug Delivery 2013;26(2):69‐75. [5K23RR15529/RR/NCRR: NIH HHS/United States; CENTRAL: 880221; CFGD Register: PI277; CRS: 5500127000000010; GR:: 1UL1RR025744/RR/NCRR NIH HHS/United States; JID:: 101475057; K23: RR015529/RR/NCRR NIH HHS/United States; OID: [Other ID]: NLM: PMC3621259; PMCID:: PMC3621259; PUBMED: 22620494; UL1: TR000423/TR/NCATS NIH HHS/United States] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schaad 1997 {published data only}
- Schaad UB, Wedgwood J, Ruedeberg A, Kraemer R, Hampel B. Ciprofloxacin as antipseudomonal treatment in patients with cystic fibrosis. Pediatric Infectious Disease Journal 1997;16(1):106‐11. [CFGD Register: PI116; CRS: 5500100000010666] [DOI] [PubMed] [Google Scholar]
Schelstraete 2009 {published data only}
- Schelstraete P, Deschaght P, Daele S, Haerynck F, Simaey L, Vaneechoutte M, et al. Genotype based evaluation of eradication treatment of new P. aeruginosa infections in CF patients. Journal of Cystic Fibrosis 2009;8(Suppl 2):S39. [CFGD Register: PI228] [DOI] [PubMed] [Google Scholar]
Schuster 2013 {published data only}
- Goldman M, Schuster A, Halliburn C, Döring G, The Freedom Study Group. A randomised, open label phase 3 study to evaluate the efficacy and safety of a dry powder formulation of inhaled colistimethate sodium (Colobreathe®) versus tobramycin nebuliser solution (TNS) in cystic fibrosis subjects with chronic Pseudomonas aeruginosa lung infection. Journal of Cystic Fibrosis 2012;11 Suppl 1:S12. [Abstract no.: WS5.5; CFGD Register: PI214b] [Google Scholar]
- Goldman MH, Pitt T. Lack of emergence of antimicrobial resistance of Pseudomonas Aeruginosa after six months inhalation of dry powder colistimethate. Pediatric Pulmonology 2008;43(Suppl 31):331. [CFGD Register: PI214a] [Google Scholar]
- Goldman MH, Shuster A, Haliburn C, Doring G. A randomised, open label phase 3 study to evaluate the efficacy and safety of a dry powder formulation of colistimethate sodium (Colobreathe®) versus tobramycin nebuliser solution (TNS) in cystic fibrosis subjects with chronic Pseudomonas aeruginosa lung infection. Pediatric Pulmonology 2012;47 Suppl 35:353. [Abstract no.: 363; CFGD Register: PI214c] [Google Scholar]
- Goldman MH, Werner T, Schuster A. Does persistence with inhaled dry powder antibiotic treatment improve tolerability?. Pediatric Pulmonology 2013;48 Suppl 36:347. [Abstract no.: 389; CENTRAL: 921669; CFGD Register: PI214f; CRS: 5500125000000396] [Google Scholar]
- Schuster A, Haliburn C, Doring G, Goldman MH, FSG. Online Data Supplement to 'Safety, efficacy and convenience of colistimethate sodium dry powder for inhalation (Colobreathe DPI) in patients with cystic fibrosis: a randomised study [online]. Thorax 2013;68(4):344‐350 Online. [CENTRAL: 867327; CFGD Register: PI214e; CRS: 5500100000011299] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster A, Haliburn C, Doring G, Goldman MH, FSG. Safety, efficacy and convenience of colistimethate sodium dry powder for inhalation (Colobreathe DPI) in patients with cystic fibrosis: a randomised study. Thorax 2013;68(4):344‐50. [CENTRAL: 864435; CFGD Register: PI214d; CRS: 5500100000011298; PUBMED: 23135343] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster A, Pressler T, Zhu H, Haberman R. Efficacy and safety of colistimethate sodium in a paediatric population: results from the FREEDOM trial. Journal of Cystic Fibrosis 2015;14:S87. [CFGD Register: PI214g] [Google Scholar]
Smith 1994 {published data only}
- Smith A, Ramsey B. Aerosol antibiotic therapy. Proceedings of 19th European Cystic Fibrosis Conference; 1994 May 29‐June 3; Paris, France. 1994:L21. [CFGD Register: PI102]
Stass 2008 {published data only}
- Stass H, Delesen H, Nagelschmitz J, Staab D. Safety and pharmacokinetics of ciprofloxacin dry powder for inhalation in cystic fibrosis: a Phase I, randomized, single‐dose, dose‐escalation study. Journal of Aerosol Medicine and Pulmonary Drug Delivery 2015;28(2):106‐15. [CENTRAL: 1037796; CFGD Register: PI215c; CRS: 5500131000000317; JID:: 101475057; PUBMED: 25050456] [DOI] [PubMed] [Google Scholar]
- Stass H, Ludwig M, Nagelschmitz J, Stabb D. Safety and pharmacokinetics of inhaled dry powder ciprofloxacin after single and multiple inhalations in patients with cystic fibrosis. Pediatric Pulmonology 2008;43 Suppl 31:300. [CENTRAL: 671400; CFGD Register: PI215a; CRS: 5500100000003282] [Google Scholar]
- Stass H, Weimann B, Nagelschmitz J, Rolinck‐Werninghaus C, Staab D. Tolerability and pharmacokinetic properties of ciprofloxacin dry powder for inhalation in patients with cystic fibrosis: a phase I, randomized, dose‐escalation study. Clinical Therapeutics 2013;35(10):1571‐81. [CFGD Register: PI215b; CRS: 5500127000000012; JID: 7706726; PUBMED: 24054830] [DOI] [PubMed] [Google Scholar]
Stass 2009 {published data only}
- Stass H, Nagelschmitz J, Posselt H. Safety, tolerability and pharmacokinetics of ciprofloxacin dry powder for inhalation in adolescent patients with CF. Pediatric Pulmonology 2009;44 Suppl 32:302. [Abstract no.: 258; CENTRAL: 744139; CRS: 5500100000003465] [Google Scholar]
Stead 1987 {published data only}
- Stead RJ, Hodson ME, Batten JC. Inhaled ceftazidime compared with gentamicin and carbenicillin in older patients with cystic fibrosis infected with Pseudomonas aeruginosa. British Journal of Diseases of the Chest 1987;81(3):272‐9. [CFGD Register: PI53b] [DOI] [PubMed] [Google Scholar]
- Stead RJ, Hodson ME, Batten JC. Nebulised ceftazidime compared with gentamicin and carbenicillin in adults with cystic fibrosis infected with Ps. aeruginosa. Proceedings of 13th Annual Meeting of the European Working Group for Cystic Fibrosis; 1985 Nov 3‐8; Jerusalem, Israel. 1985:51. [CFGD Register: PI53a]
Stroobant 1985 {published data only}
- Heaf DP, Tyson S, Dinwiddie R, Matthew D. A comparison of inhaled therapies in children with cystic fibrosis. Proceedings of 9th International Cystic Fibrosis Congress; 1984 June 9‐15; Brighton, England. 1984:274. [CFGD Register: PI83a]
- Stroobant J, Heaf DP, Tyson S, Matthew DJ. Effect of inhaled azlocillin, mistabron and combination therapy in children with cystic fibrosis. Pediatric Research 1985;19:1099. [CFGD Register: PI83c] [Google Scholar]
- Stroobant J, Heaf DP, Tyson S, Matthew DJ. Effect of inhaled azlocillin, mistabron and combination therapy in children with cystic fibrosis. Proceedings of 13th Annual Meeting of the European Working Group for Cystic Fibrosis; 1985 Nov 3‐8; Jerusalem, Israel. 1985:47. [CFGD Register: PI83b]
Taccetti 2012 {published data only}
- Cariani L, Defilippi G, Costantini D, Claut L, Clarizia G, D'accico M, et al. Semi‐automated rep‐pcr genotyping of pseudomonas aeruginosa in Italian CF patients in eradication therapy. Pediatric Pulmonology 2010;45 Suppl 33:348. [Abstract no.: 360; CFGD Register: PI230c] [Google Scholar]
- Dolce D, Cariani L, Ravenni N, Mergni G, Biffi A, Colombo C, et al. Anti‐ P. aeruginosa antibodies and microbiological outcome in patients treated with early eradication therapy. Pediatric Pulmonology 2013;48 Suppl 36:288. [Abstract no.: 231; CENTRAL: 921692; CFGD Register: PI230i; CRS: 5500125000000403] [Google Scholar]
- Taccetti G, Bianchini E, Cariani L, Buzzetti R, Costantini D, Trevisan F, et al. Early antibiotic treatment for Pseudomonas aeruginosa eradication in patients with cystic fibrosis: a randomised multicentre study comparing two different protocols. Thorax 2012;67(10):853‐9. [CFGD Register: PI230g] [DOI] [PubMed] [Google Scholar]
- Taccetti G, Bianchini E, Zavataro L, Campana S, Defilippi G, Ravenni N, et al. Pseudomonas aeruginosa eradication in cystic fibrosis: preliminary data from a randomized multicenter study of two different early antibiotic treatment protocols. Pediatric Pulmonology 2010;45 Suppl 33:337. [Abstract no.: 332; CFGD Register: PI230d] [Google Scholar]
- Taccetti G, Bianchini E, Zavataro L, Campana S, Defilippi G, Ravenni N, et al. Pseudomonas aeruginosa microbiological status and emergence of other pathogens after early eradication treatment in cystic fibrosis: a post‐trial follow‐up. Pediatric Pulmonology 2011;46 Suppl 34:317. [Abstract no.: 292; CFGD Register: PI230e] [Google Scholar]
- Taccetti G, Bianchini E, Zavataro L, Campana S, Ravenni N, Boni V, et al. Early antibiotic treatment for Pseudomonas aeruginosa eradication in cystic fibrosis patients: a randomized multicenter study of two different protocols. Pediatric Pulmonology 2009;44(Suppl 32):354. [CFGD Register: PI230a] [Google Scholar]
- Taccetti G, Bianchini E, Zavataro L, Costantini D, Galici V, Campana S, et al. Pseudomonas aeruginosa eradication in cystic fibrosis: final results of a randomized multicenter study of two different early antibiotic treatment protocols. Pediatric Pulmonology 2011;46 Suppl 34:317. [Abstract no.: 291; CFGD Register: PI230f] [Google Scholar]
- Taccetti G, Cocchi P, Dolce D, Galici V, Mergni G, Gagliardini R, et al. Is early eradication treatment against P. aeruginosa associated with the emergence of other non‐fermenter gram negatives. Pediatric Pulmonology 2013;48 Suppl 36:328. [Abstract no.: 338; CENTRAL: 921671; CFGD Register: PI230h; CRS: 5500125000000398] [Google Scholar]
- Zavataro L, Taccetti G, Cariani L, Ravenni N, Braccini G, Bresci S, et al. Epidemiology of first/new Pseudomonas aeruginosa infection in cystic fibrosis patients. Journal of Cystic Fibrosis 2010;9 Suppl 1:S29. [Abstract no.: 110; CFGD Register: PI230b] [Google Scholar]
Tramper‐Stranders 2009 {published data only}
- Tramper‐Stranders G, Wolfs TFW, Aalderen W, Kouwenberg J, Nagelkerke A, Ent CK. Prevention of initial P. aeruginosa infection in children with cystic fibrosis: a multi‐centre double‐blind randomised controlled trial. Journal of Cystic Fibrosis 2009;8 Suppl 2:S37. [Abstract no.: 148; CFGD Register: PI231a] [Google Scholar]
- Tramper‐Stranders GA, Wolfs TF, Haren Noman S, Aalderen WM, Nagelkerke AF, Nuijsink M, et al. Controlled trial of cycled antibiotic prophylaxis to prevent initial Pseudomonas aeruginosa infection in children with cystic fibrosis. Thorax 2010;65(10):915‐20. [CENTRAL: 761942; CFGD Register: PI231b; CRS: 5500125000000359; PUBMED: 20729233] [DOI] [PubMed] [Google Scholar]
Trapnell 2010 {published data only}
- McColley SA, Trapnell BC, Kissner D, McKevitt M, Montgomery AB, Rosen J, for the FTI Study Group. Fosfomycon/tobramycin for inhalation (FTI): microbiological results of a phase 2 placebo‐controlled trial in patients with cystic fibrosis and pseudomonas aeruginosa. Pediatric Pulmonology 2010;45(Suppl 33):338. [CFGD Register: PI247c] [Google Scholar]
- Trapnell B. A Phase 2, double‐blind, multicenter, randomized, placebo‐controlled trial evaluating fosfomycin/tobramycin for inhalation in patients with cystic fibrosis and Pseudomonas Aeruginosa. ClinicalTrials.gov (accessed 08 December 2010). [NCT00794586]
- Trapnell BC, Kissner D, Montgomery AB, Newcomb T, Geller D. Fosfomycon/tobramycin for inhalation (FTI): safety results of a phase 2 placebo‐controlled trial in patients with cystic fibrosis and pseudomonas aeruginosa. Pediatric Pulmonology 2010;45(Suppl 33):302. [Abstract no.: 234; CFGD Register: PI247b] [Google Scholar]
- Trapnell BC, McColley SA, Kissner DG, Rolfe MW, Rosen JM, McKevitt M, et al. Fosfomycin/tobramycin for inhalation in patients with cystic fibrosis with pseudomonas airway infection. American Journal of Respiratory and Critical Care Medicine 2012;185(2):171‐8. [CFGD Register: PI247d] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell BC, Rolfe M, McColley S, Montgomery AB, Moorehead L, Geller D. Fosfomycon/tobramycin for inhalation (FTI): efficacy results of a phase 2 placebo‐controlled trial in patients with cystic fibrosis and pseudomonas aeruginosa. Pediatric Pulmonology 2010;45(Suppl 33):302. [Abstract no.: 233; CFGD Register: PI247a] [Google Scholar]
Treggiari 2011 {published data only}
- Anstead M, Heltshe SL, Khan U, Barbieri JT, Langkamp M, Doring G, et al. Pseudomonas aeruginosa serology and risk for re‐isolation in the EPIC trial. Journal of cystic fibrosis 2013;12(2):147‐53. [CFGD Register: PI202m] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstead M, Lymp J, Khan U, Barbieri J, Langkamp M, Doring G, et al. Pseudomonas aeruginosa serology predicts response to treatment and re‐infection in the EPIC clinical study. Pediatric Pulmonology 2011;46 Suppl 34:303. [Abstract no.: 254; CFGD Register: PI202g] [Google Scholar]
- Anstead M, Saiman L, Mayer‐Hamblett N, Lands LC, Kloster M, Goss CH, et al. Pulmonary exacerbations in CF patients with early lung disease. Journal of Cystic Fibrosis 2014;13(1):74‐9. [CENTRAL: 961723; CFGD Register: PI202L; CRS: 5500050000000096; EMBASE: 2013796396] [DOI] [PubMed] [Google Scholar]
- Hamblett NM, Retsch‐Bogart GZ, Treggiari M, Kronmal RA, Khan U, Williams J, Ramsey BW. Safety and efficacy of anti‐pseudomonal therapy for early eradication of Pseudomonas aeruginosa: the EPIC study. Pediatric Pulmonology 2009;44(Suppl 32):183. [CFGD Register: PI202b] [Google Scholar]
- Hoffman LR, Ramsey BW, Kulasekara HD, Retsch‐Bogart GZ, Wolter DJ, Pope CE, et al. Pseudomonas aeruginosa (PA) phenotypes associated with persistent early infection in CF patients in the EPIC Clinical Trial. Pediatric Pulmonology 2012;47 Suppl 35:317. [Abstract no.: 266; CFGD Register: PI202j] [Google Scholar]
- Jorth P, Hisert KB, Garudathri J, Wolter D, Hoffman L, Singh P. Studies on the effects of ciprofloxacin on pseudomonas aeruginosa evolution in cystic fibrosis patients. Pediatric Pulmonology 2014;49:349. [CFGD Register: PI202n] [Google Scholar]
- Jorth P, Rezayat A, Hisert KB, Garudathri J, Khan U, Hamblett NM, et al. Early evolution of pseudomonas aeruginosa during cystic fibrosis infection. Pediatric Pulmonology 2015;50 Suppl 41:304. [Abstract no.: 300; CFGD Register: PI202p] [Google Scholar]
- Khan U, Mayer‐Hamblett N, Retsch‐Bogart G, Treggiari M, Ramsey B. Association between baseline pseudomonas aeruginosa positivity in EPIC clinical trial participants & prior antibiotic exposure. Pediatric Pulmonology 2010;45 Suppl 33:335. [Abstract no.: 326; CFGD Register: PI202f] [Google Scholar]
- Mayer‐Hamblett N, Kloster M, Rosenfeld M, Gibson RL, Retsch‐Bogart GZ, Emerson J, et al. Impact of sustained eradication of new Pseudomonas aeruginosa infection on long‐term outcomes in cystic fibrosis. Clinical Infectious Diseases 2015;61(5):707‐15. [CFGD Register: PI202o] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer‐Hamblett N, Kronmal RA, Gibson RL, Rosenfeld M, Retsch‐Bogart G, Treggiari MM, et al. Initial Pseudomonas aeruginosa treatment failure is associated with exacerbations in cystic fibrosis. Pediatric Pulmonology 2012;47(2):125‐34. [CFGD Register: PI202i] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer‐Hamblett N, Rosenfeld M, Treggiari MM, Konstan MW, Retsch‐Bogart G, Morgan W, et al. Standard care versus protocol based therapy for new onset Pseudomonas aeruginosa in cystic fibrosis. Pediatric Pulmonology 2013;48(10):943‐53. [CENTRAL: 916577; CFGD Register: PI202k; CRS: 5500125000000727; PUBMED: 23818295] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey B. TOBI use in infants and children with early Pseudomonas Aeruginosa infection ‐ duration of effect and epic update [abstract]. Pediatric Pulmonology 2005;40(Suppl 28):146. [CFGD Register: PI202a] [Google Scholar]
- Treggiari M, Retsch‐Bogart G, Mayer‐Hamblett N, Khan U, Kronmal R, Ramsey B, et al. Comparative efficacy and safety of four randomized regimens to treat early Pseudomonas aeruginosa infection in children with cystic fibrosis. Journal of Cystic Fibrosis 2010;9 Suppl 1:S54. [Abstract no.: 209; CFGD Register: PI202e] [Google Scholar]
- Treggiari M, Retsch‐Bogart GZ, Mayer‐Hamblett N, Kronmal R, Khan U, Williams J, et al. Early anti‐pseudomonal infection in children with CF: study population and conduct of the "EPIC" clinical trial. Pediatric Pulmonology 2009;44 Suppl 32:316. [Abstract no.: 299; CFGD Register: PI202c] [Google Scholar]
- Treggiari MM, Retsch‐Bogart G, Mayer‐Hamblett N, Khan U, Kulich M, Kronmal R, et al. Comparative efficacy and safety of 4 randomized regimens to treat early Pseudomonas aeruginosa infection in children with cystic fibrosis. Archives of Pediatric & Adolescent Medicine 2011;165(9):847‐56. [CFGD Register: PI202h] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treggiari MM, Rosenfeld M, Mayer‐Hamblett N, Retsch‐Bogart G, Gibson RL, Williams J, et al. Early anti‐pseudomonal acquisition in young patients with cystic fibrosis: rationale and design of the EPIC clinical trial and observational study. Contempory Clinical Trials 2009;30(3):256‐68. [CFGD Register: PI202d] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tullis 2014 {published data only}
- Balfour‐Lynn IM. At last, Burkholderia spp. is one of the inclusion criteria‐‐a negative (but published) randomised controlled trial. Journal of Cystic Fibrosis 2014;13(3):241‐2. [CFGD Register: PI259i] [DOI] [PubMed] [Google Scholar]
- Burns J, LiPuma JJ, Retsch‐Bogart G, Bresnik M, Henig N, McKevitt M, et al. No antibiotic cross‐resistance after 1 year of continuous aztreonam for inhalation solution (AZLI) in cystic fibrosis (CF) patients (pts) with chronic Burkholderia (BURK) infection. Journal of Cystic Fibrosis 2012;11 Suppl 1:S71. [Abstract no.: 58; CFGD Register: PI259d] [Google Scholar]
- Burns JL, LiPuma J, McKevitt M, Lewis S, Retsch‐Bogart GZ, Bresnik M, et al. The effect of burkholderia colony morphology in Cystic Fibrosis (CF) patients with chronic burkholderia species in a randomized trial of Aztreonam For Inhalation Solution (AZLI). Pediatric Pulmonology 2012;47(S35):333. [Abstract no.: 307; CFGD Register: PI259g] [Google Scholar]
- Gilead Sciences. Phase 3b randomized, double‐blind, placebo‐controlled two‐part trial to assess the safety and efficacy of continuous aztreonam for inhalation solution (AZLI) in subjects with cystic fibrosis (CF) and chronic Burkholderia species infection. ClinicalTrials.gov (accessed 08 December 2010). [CTG: NCT01059565]
- Tullis DE, Burns JL, Retsch‐Bogart GZ, Bresnik M, Henig NR, Lewis SA, et al. Inhaled aztreonam for chronic Burkholderia infection in cystic fibrosis: a placebo‐controlled trial. Journal of Cystic Fibrosis 2014;13(3):296‐305. [CFGD Register: PI259h] [DOI] [PubMed] [Google Scholar]
- Tullis E, Burns J, Retsch‐Bogart G, Bresnik M, Henig N, Lewis S, et al. Aztreonam for inhalation solution (AZLI) in cystic fibrosis (CF) patients with chronic Burkholderia species (BURK) infection: final results from a randomized, placebo‐controlled trial. Journal of Cystic Fibrosis 2012;11 Suppl 1:S11. [Abstract no.: WS5.4; CFGD Register: PI259e] [Google Scholar]
- Tullis E, Burns JL, Retsch‐Bogart G, Bresnik M, Henig Nr, Lewis S, et al. Aztreonam for inhalation solution (AZLI) in cystic fibrosis (CF) patients with chronic burkholderia species (Burk) infection: initial results from a randomized, placebo‐controlled trial. Pediatric Pulmonology 2011;46 Suppl 34:296. [Abstract no.: 234; CFGD Register: PI259b] [Google Scholar]
- Tullis E, Burns JL, Retsch‐Bogart G, Bresnik M, Lewis S, Montgomery AB, et al. Aztreonam 75mg powder and solvent for nebuliser solution (AZLI) in cystic fibrosis (CF) patients with chronic Burkholderia species (Burk) infection: baseline demographics and microbiology from randomized , placebo‐controlled trial. Journal of Cystic Fibrosis 2011;10 Suppl 1:S22. [Abstract no.: 85; CFGD Register: PI259c] [Google Scholar]
- Tullis E, Burns JL, Retsch‐Bogart GZ, Bresnik M, Lewis S, LiPuma J. Lung function in Cystic Fibrosis (CF) patients with chronic burkholderia (BURK) species infection over the course of a prospective, randomized trial of aztreonam for inhalation solution (AZLI). Pediatric Pulmonology 2012;47(S35):334. [Abstract no.: 310; CFGD Register: PI259f] [Google Scholar]
- Tullis E, LiPuma JJ, Retsch‐Bogart G, Bresnik M, Henig N, McKevitt M, et al. Effects of continuous aztreonam for inhalation solution (AZLI) use on pathogens and antibiotic susceptibility in cystic fibrosis (CF) patients with chronic burkholderia species infection. Pediatric Pulmonology 2011;46 Suppl 34:305. [Abstract no.: 257; CFGD Register: PI259a] [Google Scholar]
Valerius 1991 {published data only}
- Valerius NH, Koch C, Høiby N. Prevention of Chronic colonization with Pseudomonas aeruginosa in patients with CF by early treatment with ciprofloxacin and colistin aerosol inhalations. Pediatric Pulmonology 1990;9(Suppl 5):219. [CFGD Register: PI70a] [Google Scholar]
- Valerius NH, Koch C, Høiby N. Prevention of chronic Pseudomonas aeruginosa colonisation in cystic fibrosis by early treatment. Lancet 1991;338(8769):725‐6. [CFGD Register: PI70b] [DOI] [PubMed] [Google Scholar]
Wainwright 2002 {published data only}
- Byrnes CA, Vidmar S, Cheney JL, Carlin JB, Armstrong DS, Cooper PJ, et al. Prospective evaluation of respiratory exacerbations in children with cystic fibrosis from newborn screening to 5 years of age. Thorax 2013;68(7):643‐51. [CENTRAL: 904924; CFGD Register: PE167i; CRS: 5500125000000477; PUBMED: 23345574] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheney J, Vidmar S, Grimwood K, Carlin JB, Wainwright C, on behalf of ACFBALStudy Group. Interim outcomes of Pseudomonas aeruginosa (Pa) eradication protocol in young children in the Australasian Cystic Fibrosis Bronchoalveolar. Journal of Cystic Fibrosis 2009;8 Suppl 2:S39. [Abstract no.: 157; CFGD Register: PE167c] [Google Scholar]
- Cheney J, Wainwright C, ACFBAL SG. Trials, tribulations and triumphs of a cystic fibrosis study ‐ a behind the scenes look at the workings of an international multi‐centre study. Journal of Cystic Fibrosis 2010;9 Suppl 1:S118. [Abstract no.: 452; CENTRAL: 790042; CFGD Register: PE167g; CRS: 5500125000000092] [Google Scholar]
- Kidd TJ, Ramsay KA, Vidmar S, Carlin JB, Bell SC, Wainwright CE, et al. Pseudomonas aeruginosa genotypes acquired by children with cystic fibrosis by age. Journal of cystic fibrosis 2015;14(3):361‐9. [CFGD Register: PE167k] [DOI] [PubMed] [Google Scholar]
- Moodie M, Lal A, Vidmar S, Armstrong DS, Byrnes CA, Carlin JB, et al. Costs of bronchoalveolar lavage‐directed therapy in the first 5 years of life for children with cystic fibrosis. Journal of Pediatrics 2014;165(3):564‐9. [CFGD Register: PE167j] [DOI] [PubMed] [Google Scholar]
- Wainwright C, Carlin J, Cooper P, Byrnes C, Martin J, Grimwood K, et al. Australasian cystic fibrosis BAL study interim analysis. Pediatric Pulmonology 2006;41 Suppl 29:317. [CFGD Register: PE167a] [Google Scholar]
- Wainwright CE, Carlin J, Cooper P, Byrnes C, Whitehead B, Martin J, et al. Early infection with pseudomonas aeruginosa can be cleared in young children with cystic fibrosis [abstract]. Pediatric Pulmonology 2002;34(Suppl 24):300‐1. [CFGD Register: PE167d] [Google Scholar]
- Wainwright CE, Grimwood K, Carlin JB, Vidmar S, Cooper PJ, Francis PW, et al. Safety of bronchoalveolar lavage in young children with cystic fibrosis. Pediatric Pulmonology 2008;43(10):965‐72. [CFGD Register: PE167b] [DOI] [PubMed] [Google Scholar]
- Wainwright CE, Kidd TJ, Ramsey KA, Bell SC, Grimwood K. Australasian CF bronchoalveolar lavage (ACFBAL) study: P.Aeruginosa (PA) genotypes in pre‐school CF children. Pediatric Pulmonology 2011;46 Suppl 34:320. [Abstract no.: 299; CENTRAL: 874662; CFGD Register: PE167h ; CRS: 5500125000000093] [Google Scholar]
- Wainwright CE, Vidmar S, Armstrong DS, Byrnes CA, Carlin JB, Cheney J, et al. Effect of bronchoalveolar lavage‐directed therapy on Pseudomonas aeruginosa infection and structural lung injury in children with cystic fibrosis: a randomized trial. JAMA 2011;306(2):163‐71. [CFGD Register: PE167e] [DOI] [PubMed] [Google Scholar]
- Wainwright CE, Vidmar S, Armstrong DS, Byrnes CA, Carlin JB, Cheney J, et al. Online Supplement to 'Effect of bronchoalveolar lavage‐directed therapy on Pseudomonas aeruginosa infection and structural lung injury in children with cystic fibrosis: a randomized trial' [online]. JAMA 2011;306(2):163‐171 Online. [CENTRAL: 788878; CFGD Register: PE167f; CRS: 5500100000011126; PUBMED: 21750293] [DOI] [PubMed] [Google Scholar]
Wainwright 2011 {published data only}
- A double‐blind, multicenter, multinational, randomized, placebo‐controlled trial evaluating aztreonam lysine for inhalation in patients with cystic fibrosis, mild lung disease, and P. Aeruginosa (AIR‐CF4). ClinicalTrials.gov (accessed 08 December 2010). [NCT00712166]
- Wainwright C, Nakamura C, Geller D, Montgomery AB. A double‐blind, multinational, randomized, placebo‐controlled trial evaluating aztreonam for inhalation solution (AZLI) in patients with cystic fibrosis (CF), mild lung disease and P. aeruginosa. Journal of Cystic Fibrosis 2010;9(Suppl 1):S22. [CFGD Register: PI243a] [DOI] [PubMed] [Google Scholar]
- Wainwright CE, Quittner AL, Geller DE, Nakamura C, Wooldridge JL, Gibson RL, et al. Aztreonam for inhalation solution (AZLI) in patients with cystic fibrosis, mild lung impairment, and P. aeruginosa. Journal of Cystic Fibrosis 2011;10(4):234‐42. [CENTRAL: 801029; CFGD Register: PI243b; CRS: 5500100000011273; PUBMED: 21441078] [DOI] [PubMed] [Google Scholar]
Westerman 2003 {published data only}
- Westerman EM, Brun PP, Touw DJ, Frijlink HW, Heijerman HG. Effect of nebulized colistin sulphate and colistin sulphomethate on lung function in patients with cystic fibrosis: a pilot study. Journal of Cystic Fibrosis 2004;3(1):23‐8. [CFGD Register: PI177b] [DOI] [PubMed] [Google Scholar]
- Westerman EM, Brun PPH, Frijlink HW, Heijerman HGM. Effect of nebulised colistin sulphate and colistin sulphomethate on lung function in patients with cystic fibrosis. Journal of Cystic Fibrosis 2003;2(Suppl 1):S55. [CFGD Register: PI177a] [DOI] [PubMed] [Google Scholar]
Westerman 2005 {published data only}
- Westerman EM, Boer AH, Brun PP, Touw DJ, Roldaan AC, Frijlink HW, et al. Dry powder inhalation of colistin in cystic fibrosis patients: a single dose pilot study. Journal of Cystic Fibrosis 2007;6(4):284‐92. [CFGD Register: PI193b] [DOI] [PubMed] [Google Scholar]
- Westerman EM, Boer AH, Brun PPH, Touw DJ, Frijlink HW, Heijerman HGM. Colistin dry powder inhalation in cystic fibrosis: novel Twincer® inhaler compared to nebulization: a pilot study [abstract]. Journal of Cystic Fibrosis 2005;4(Suppl 1):S64. [CFGD Register: PI193a] [Google Scholar]
Wiesemann 1998 {published data only}
- Ratjen F, Steinkamp G, Döring G, Bauernfeind A, Wiesemann HG, Hardt H. Prevention of chronic pseudomonas aeruginosa infection by early inhalation therapy with tobramycin. Pediatric Pulmonology 1994;18(Suppl 10):255. [CFGD Register: PI101a] [Google Scholar]
- Wiesemann HG, Steinkamp G, Ratjen F, Bauernfeind A, Przyklenk B, Doring G, et al. Placebo‐controlled, double‐blind, randomized study of aerosolized tobramycin for early treatment of Pseudomonas aeruginosa colonization in cystic fibrosis. Pediatric Pulmonology 1998;25:88‐92. [CFGD Register: PI101b] [DOI] [PubMed] [Google Scholar]
Yasmin 1974 {published data only}
- Yasmin N, Laraya‐Cuasay LR, Mueller S, Liberi P, Braverman S, Capitanio M, et al. A critical evaluation of antibiotic aerosol in patients with cystic fibrosis. Proceedings of 15th Annual Meeting Cystic Fibrosis Club Abstracts. 1974. [CFGD Register: PI79]
References to studies awaiting assessment
Postnikov 2007 {published data only}
- Postnikov SS, Semykin SY, Polikarpova SV, Dubovik LG, Gracheva LA, Sagatelyan RM. A prospective trial on the efficacy and tolerability of twice‐daily dosing (TDD) versus once‐daily dosing (ODD) amikacin in cystic fibrosis patients. Journal of Cystic Fibrosis 2007;6 Suppl 1:S34. [CENTRAL: 614340; CFGD Register: PI204; CRS: 5500100000003148] [Google Scholar]
Semykin 2010 {published data only}
- Semykin SY, Polikarpova SV, Dubovik LG, Kashirskaya NY. Efficiency of the inhalational tobramycin therapy in complex antibacterial therapy of lung exacerbation in cystic fibrosis children with chronic pseudomonas aeruginosa infection. Journal of Cystic Fibrosis 2010;9 Suppl 1:S55. [Abstract no.: 214; CFGD Register: PI246] [Google Scholar]
Soulsby 2010 {published data only}
- Soulsby N. A randomised cross over pilot study of inhaled tobramycin as a treatment option for hospitalised patients with cystic fibrosis versus standard treatment of intravenous tobramycin. www.anzctr.org.au [accessed 08 December 2010].
References to ongoing studies
Prevotat 2018 {published data only}
- NCT03066453. Evaluation of short antibiotic combination courses followed by aerosols in cystic fibrosis (TOBRAMUC). clinicaltrials.gov/ct2/show/NCT03066453 (first received 28 February 2017).
Additional references
Bilton 2011
- Bilton D, Canny G, Conway S, Dumcius S, Hjelte L, Proesmans M, et al. Pulmonary exacerbation: towards a definition for use in clinical trials. Report from the EuroCareCF Working Group on outcome parameters in clinical trials. Journal of Cystic Fibrosis 2011;10 Suppl 2:S79‐81. [DOI] [PubMed] [Google Scholar]
BMJ 2018
- BMJ Best Practice. Respiratory disease: acute pulmonary exacerbation. bestpractice.bmj.com Feb 28 2018.
CF Trust 2009
- UK Cystic Fibrosis Trust Antibiotic Working Group. Antibiotic treatment for cystic fibrosis. www.cftrust.org.uk/aboutcf/publications/consensusdoc/Antibiotic_treatment_for_Cystic_Fibrosis.pdf (accessed 07 October 2011).
Daniels 2013
- Daniels T, Mills N, Whitaker P. Nebuliser systems for drug delivery in cystic fibrosis. Cochrane Database of Systematic Reviews 2013, Issue 4. [DOI: 10.1002/14651858.CD007639.pub2] [DOI] [PubMed] [Google Scholar]
de Boer 2011
- de Boer K, Vandemheen KL, Tullis E, Doucette S, Fergusson D, Freitag A, et al. Exacerbation frequency and clinical outcomes in adult patients with cystic fibrosis. Thorax 2011;66(8):680‐5. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JP, Altman DG (editors) on behalf of the Cochrane Statistical Methods Group. Chapter 9 Analysing data and undertaking meta‐analysis. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Dobbin 2005
- Dobbin Catherine J, Bartlett Delwyn, Melehan Kerri, Grunstein Ronald R, Bye Peter T P. The Effect of Infective Exacerbations on Sleep and Neurobehavioral Function in Cystic Fibrosis. American Journal of Respiratory and Critical Care Medicine 2005;172(1):99‐104. [DOI] [PubMed] [Google Scholar]
Elborn 2007
- Elborn JS, Bell SC. Pulmonary exacerbations in cystic fibrosis and bronchiectasis. Thorax 2007;62(4):288‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Flume 2009
- Flume PA, Mogayzel PJ, Robinson KA, Goss CH, Rosenblatt RL, Kuhn RJ, et al. Cystic fibrosis pulmonary guidelines, treatment of pulmonary exacerbations. American Journal of Respiratory and Critical Care Medicine 2009;180(9):801‐8. [DOI] [PubMed] [Google Scholar]
Fuchs 1994
- Fuchs Henry J, Borowitz Drucy S, Christiansen David H, Morris Edward M, Nash Martha L, Ramsey Bonnie W, et al. Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symptoms and on Pulmonary Function in Patients with Cystic Fibrosis. New England Journal of Medicine 1994;331(10):637‐42. [DOI] [PubMed] [Google Scholar]
Gee 2000
- Gee L, Abbott J, Conway S, Etherington C, Webb A. Development of a disease specific health related quality of life measure for adults and adolescents with cystic fibrosis. Thorax 2000;55(11):946‐54. [DOI: 10.1136/thorax.55.11.946] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gibson 2003a
- Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. American Journal of Respiratory & Critical Care Medicine 2003;168(8):918‐51. [DOI] [PubMed] [Google Scholar]
Gold 1987
- Gold R, Carpenter S, Heurter H, Corey M, Levison H. Randomized trial of ceftazidime versus placebo in the management of acute respiratory exacerbations in patients with cystic fibrosis. Journal of Pediatrics 1987;111(6 Pt 1):907‐13. [PUBMED: 3316565 ] [DOI] [PubMed] [Google Scholar]
Goss 2007
- Goss CH, Burns JL. Exacerbations in cystic fibrosis. 1: Epidemiology and pathogenesis. Thorax 2007;62(4):360‐7. [PUBMED: 17387214] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Jennings 2017
- Jennings M T, Dasenbrook E C, Lechtzin N, Boyle M P, Merlo C A. Risk factors for persistent methicillin‐resistant Staphylococcus aureus infection in cystic fibrosis. Journal of Cystic Fibrosis 2017;16(6):681‐6. [DOI] [PubMed] [Google Scholar]
Jones 2009
- Jones AP, Riley RD, Williamson PR, Whitehead A. Meta‐analysis of individual patient data versus aggregate data from longitudinal clinical trials. Clinical Trials 2009;6(1):16‐27. [DOI] [PubMed] [Google Scholar]
Marshall 2005
- Marshall B C, Butler S M, Stoddard M, Moran A M, Liou T G, Morgan W J. Epidemiology of cystic fibrosis‐related diabetes. The Journal of Pediatrics 2005;146(5):681‐7. [DOI] [PubMed] [Google Scholar]
Moskowitz 2008
- Moskowitz SM, Silva SJ, Mayer‐Hamblett N, Pasta DJ, Mink DR, Mabie JA, et al. Shifting patterns of inhaled antibiotic use in cystic fibrosis. Pediatric Pulmonology 2008;43(9):874‐81. [DOI] [PubMed] [Google Scholar]
NICE 2017
- National Institute for Health and Care Excellence. Cystic fibrosis: diagnosis and management. https://www.nice.org.uk/guidance/ng78 October 2017:Accessed 21 June 2018. [PubMed]
Pellegrino 2005
- Pellegrino R, Viegi G, Brusasco V, Crapo RO, Burgos F, Casaburi R, et al. Interpretative strategies for lung function tests. European Respiratory Journal 2005;26(5):948–68. [DOI: 10.1183/09031936.05.00035205] [DOI] [PubMed] [Google Scholar]
Quittner 2009
- Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire‐Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 2009;135(6):1610‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Sanders 2010
- Sanders DB, Bittner RCL, Rosenfeld M, Hoffman LR, Redding GJ, Goss CH. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. American Journal of Respiratory and Critical Care Medicine 2010;182(5):627‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sanders 2011
- Sanders DB, Bittner RC, Rosenfeld M, Redding GJ, Goss CH. Pulmonary exacerbations are associated with subsequent FEV1 decline in both adults and children with cystic fibrosis. Pediatric Pulmonology 2011;46(4):393‐400. [DOI] [PubMed] [Google Scholar]
Smith 2018
- Smith S, Rowbotham N, Regan K. Inhaled anti‐pseudomonal antibiotics for long‐term therapy in cystic fibrosis. Cochrane Database of Systematic Reviews 2018, Issue 3. [DOI: 10.1002/14651858.CD001021.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Smyth 2006
- Smyth A. Update on treatment of pulmonary exacerbations in cystic fibrosis. Current Opinion in Pulmonary Medicine 2006;12(6):440‐4. [PUBMED: 17053495 ] [DOI] [PubMed] [Google Scholar]
Smyth 2008
- Smyth A, Elborn JS. Exacerbations in cystic fibrosis: 3 · Management. Thorax 2008;63(2):180‐4. [DOI] [PubMed] [Google Scholar]
Wagener 2012
- Wagener JS, Rasouliyan L, Vandevanter DR, Pasta DJ, Regelmann WE, Morgan WJ, et al. Oral, inhaled, and intravenous antibiotic choice for treating pulmonary exacerbations in cystic fibrosis. Pediatric Pulmonology 2012 Aug 8 [Epub ahead of print]. [DOI: 10.1002/ppul.22652] [DOI] [PMC free article] [PubMed]
Wientzen 1980
- Wientzen R, Prestidge CB, Kramer RI, McCracken GH, Nelson JD. Acute pulmonary exacerbations in cystic fibrosis. A double‐blind trial of tobramycin and placebo therapy. American Journal of Diseases of Children 1980;134(12):1134‐8. [PUBMED: 7004176] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Ryan 2012
- Ryan G, Jahnke N, Remmington T. Inhaled antibiotics for pulmonary exacerbations in cystic fibrosis. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.CD008319.pub2] [DOI] [PubMed] [Google Scholar]