

Abstract
Background
This is an updated version of the Cochrane Review previously published in 2013. This review is one in a series of Cochrane Reviews investigating pair‐wise monotherapy comparisons.
Epilepsy is a common neurological condition in which abnormal electrical discharges from the brain cause recurrent unprovoked seizures. It is believed that with effective drug treatment, up to 70% of individuals with active epilepsy have the potential to become seizure‐free and go into long‐term remission shortly after starting drug therapy with a single antiepileptic drug in monotherapy.
Worldwide, phenytoin is a commonly used antiepileptic drug. It is important to know how newer drugs, such as oxcarbazepine, compare with commonly used standard treatments.
Objectives
To review the time to treatment failure, remission and first seizure with oxcarbazepine compared to phenytoin, when used as monotherapy in people with focal onset seizures or generalised tonic‐clonic seizures (with or without other generalised seizure types).
Search methods
We searched the following databases on 20 August 2018: the Cochrane Register of Studies (CRS Web), which includes the Cochrane Epilepsy Group Specialized Register and the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE (Ovid, 1946 to 20 August 2018), ClinicalTrials.gov, and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP). We handsearched relevant journals and contacted pharmaceutical companies, original trial investigators and experts in the field.
Selection criteria
We included randomised controlled trials comparing monotherapy with either oxcarbazepine or phenytoin in children or adults with focal onset seizures or generalised onset tonic‐clonic seizures.
Data collection and analysis
This was an individual participant data (IPD) review. Our primary outcome was time to treatment failure and our secondary outcomes were time to first seizure post‐randomisation, time to six‐month and 12‐month remission, and incidence of adverse events. We used Cox proportional hazards regression models to obtain trial‐specific estimates of hazard ratios (HRs) with 95% confidence intervals (CIs), using the generic inverse variance method to obtain the overall pooled HR and 95% CI.
Main results
Individual participant data were available for 480 out of a total of 517 participants (93%), from two out of three included trials. For remission outcomes, a HR of less than one indicated an advantage for phenytoin; and for first seizure and treatment failure outcomes, a HR of less than one indicated an advantage for oxcarbazepine.
The results for time to treatment failure for any reason related to treatment showed a potential advantage of oxcarbazepine over phenytoin, but this was not statistically significant (pooled HR adjusted for epilepsy type: 0.78 95% CI 0.53 to 1.14, 476 participants, two trials, moderate‐quality evidence). Our analysis showed that treatment failure due to adverse events occurred later on with oxcarbazepine than phenytoin (pooled HR for all participants: 0.22 (95% CI 0.10 to 0.51, 480 participants, two trials, high‐quality evidence). Our analysis of time to treatment failure due to lack of efficacy showed no clear difference between the drugs (pooled HR for all participants: 1.17 (95% CI 0.31 to 4.35), 480 participants, two trials, moderate‐quality evidence).
We found no clear or statistically significant differences between drugs for any of the secondary outcomes of the review: time to first seizure post‐randomisation (pooled HR adjusted for epilepsy type: 0.97 95% CI 0.75 to 1.26, 468 participants, two trials, moderate‐quality evidence); time to 12‐month remission (pooled HR adjusted for epilepsy type 1.04 95% CI 0.77 to 1.41, 468 participants, two trials, moderate‐quality evidence) and time to six‐month remission (pooled HR adjusted for epilepsy type: 1.06 95% CI 0.82 to 1.36, 468 participants, two trials, moderate‐quality evidence).
The most common adverse events reported in more than 10% of participants on either drug were somnolence (28% of total participants, with similar rates for both drugs), headache (15% of total participants, with similar rates for both drugs), dizziness (14.5% of total participants, reported by slightly more participants on phenytoin (18%) than oxcarbazepine (11%)) and gum hyperplasia (reported by substantially more participants on phenytoin (18%) than oxcarbazepine (2%)).
The results of this review are applicable mainly to individuals with focal onset seizures; 70% of included individuals experienced seizures of this type at baseline. The two studies included in IPD meta‐analysis were generally of good methodological quality but the design of the studies may have biased the results for the secondary outcomes (time to first seizure post‐randomisation, time to six‐month and 12‐month remission) as seizure recurrence data were not collected following treatment failure or withdrawal from the study. In addition, misclassification of epilepsy type may have impacted on results, particularly for individuals with generalised onset seizures.
Authors' conclusions
High‐quality evidence provided by this review indicates that treatment failure due to adverse events occurs significantly later with oxcarbazepine than phenytoin. For individuals with focal onset seizures, moderate‐quality evidence suggests that oxcarbazepine may be superior to phenytoin in terms of treatment failure for any reason, seizure recurrence and seizure remission. Therefore, oxcarbazepine may be a preferable alternative treatment than phenytoin, particularly for individuals with focal onset seizures. The evidence in this review which relates to individuals with generalised onset seizures is of low quality and does not inform current treatment policy.
We recommend that future trials should be designed to the highest quality possible with regards to choice of population, classification of seizure type, duration of follow‐up (including continued follow‐up after failure or withdrawal of randomised treatment), choice of outcomes and analysis, and presentation of results.
Plain language summary
Oxcarbazepine versus phenytoin monotherapy (single medication treatment) for epilepsy
This is an updated version of the Cochrane Review first published in Issue 2, 2006 of the Cochrane Database of Systematic Reviews.
Background
Epilepsy is a common disorder in which abnormal electrical discharges from the brain cause recurrent seizures. We studied two types of epileptic seizures in this review: generalised onset seizures, in which electrical discharges begin in one part of the brain and move throughout the brain; and focal onset seizures, in which the seizure is generated in and affects one part of the brain (the whole hemisphere of the brain or part of a lobe of the brain). Focal seizures may become generalised (secondary generalisation) and move from one part of the brain throughout the brain. For around 70% of people with epilepsy, a single antiepileptic medication can control generalised onset or focal onset seizures.
Objective
Worldwide, phenytoin is a commonly used antiepileptic medication and oxcarbazepine is one of a newer generation of antiepileptic medications. The aim of this review was to compare how effective these medications are at controlling seizures, to find out if they are associated with side effects that may result in individuals stopping the medication, and to help people choose between these medications.
Methods
We assessed the evidence from three studies (specifically, randomised controlled trials) comparing oxcarbazepine with phenytoin. We were able to combine information for 480 people from two of the three trials. For the remaining 37 people from one trial, information was not available to use in this review. The evidence is current to 20 August 2018.
Results
The review found that people taking oxcarbazepine stop taking treatment because of side effects significantly later than people taking phenytoin. Our results also showed that people with focal onset seizures taking phenytoin may stop taking treatment for any reason earlier than people with focal onset seizures taking oxcarbazepine. The results also suggest that people with focal onset seizures taking oxcarbazepine may experience a repeat seizure later, and achieve freedom from seizures earlier, than people with focal onset seizures taking phenytoin. There was no clear difference between the drugs in terms of withdrawal from the treatment, seizure recurrence and seizure remission for individuals with generalised onset seizures.
Quality of the evidence
The two studies included in analysis were well designed but no information about seizures was recorded after people stopped taking their trial medication, which may have impacted on the results of the study.
Most people (70%) included in the studies within this review had focal onset seizures, so the results are mainly relevant to people with this epilepsy type. Also up to 30% of the people in the trials used in our results may have been wrongly classified as having generalised seizures, which may have impacted on the results.
For these reasons, we judged the quality of the evidence provided by this review to be of moderate quality for people with focal onset seizures, and low quality for people with generalised onset seizures.
Conclusions
For people with focal onset seizures, oxcarbazepine may be a preferable treatment to phenytoin, but more information is needed for people with generalised onset seizures to choose between these medications. We recommend that all future trials comparing these medications, or any other antiepileptic medications, should be designed using high‐quality methods. Seizure types of people included in trials should also be classified very carefully.
Summary of findings
Background
This is an updated version of the original Cochrane Review published in 2006 (Muller 2006), and updated in 2013 (Nolan 2013a).
Description of the condition
Epilepsy is a common neurological condition in which recurrent, unprovoked seizures are caused by abnormal electrical discharges from the brain. Epilepsy is a disorder of many heterogenous seizure types, with an estimated incidence of 33 to 57 per 100,000 person‐years worldwide (Annegers 1999; Hirtz 2007; MacDonald 2000; Olafsson 2005; Sander 1996), accounting for approximately 1% of the global burden of disease (Murray 1994). The lifetime risk of epilepsy onset is estimated to be 1300 to 4000 per 100,000 person years (Hauser 1993; Juul‐Jenson 1983) and the lifetime prevalence could be as much as 70 million people worldwide (Ngugi 2010). It is believed that with effective drug treatment, up to 70% of individuals with active epilepsy have the potential to become seizure free and go into long‐term remission shortly after starting drug therapy (Cockerell 1995; Hauser 1993; Sander 2004), and that around 70% of individuals can achieve seizure freedom using a single antiepileptic drug (AED) in monotherapy (Cockerell 1995). Current guidelines from the National Institute for Health and Care Excellence (NICE) recommend that both adults and children with epilepsy should be treated with monotherapy wherever possible (NICE 2012). The remaining 30% of individuals experience refractory or drug resistant seizures which often require treatment with combinations of antiepileptic drugs (AEDs) or alternative treatments such as epilepsy surgery (Kwan 2000).
We will study two seizure types in this review: generalised onset seizures, in which electrical discharges begin in one part of the brain and move throughout the brain; and focal onset seizures, in which the seizure is generated in and affects one part of the brain (the whole hemisphere of the brain or part of a lobe of the brain).
Description of the intervention
Oxcarbazepine is one of the newer antiepileptic drugs and has similar chemical properties to its parent compound carbamazepine. It is licensed in a number of countries for use as both monotherapy and add‐on (adjunctive) therapy.
When used as monotherapy, oxcarbazepine has been shown to be as effective in terms of seizure control as first‐line antiepileptic drugs carbamazepine (Dam 1989), phenytoin (Bill 1997; Guerreiro 1997) and sodium valproate (Christe 1997). Oxcarbazepine is generally well tolerated as monotherapy in adults (Beydoun 2000; Bill 1997; Dam 1989Christe 1997; Schachter 1999) and children (Guerreiro 1997). It has been shown to have a low incidence of cosmetic side effects and serious adverse events such as allergic reactions (Kwan 2003), resulting in significantly lower discontinuation rates compared to carbamazepine in adults (Dam 1989), and phenytoin in adults (Bill 1997) and children (Guerreiro 1997).
Common adverse events, reported in more than 5% of participants receiving oxcarbazepine monotherapy, are similar in adults and children and include somnolence, headache, dizziness, nausea, vomiting, fatigue and rash (Bang 2003; Kwan 2003; Wellington 2001). Oxcarbazepine has been shown to be better tolerated than phenytoin in adults (particularly in terms of gum hyperplasia, tremor, diplopia and nystagmus (Bill 1997)), and in children (particularly in terms of gum hyperplasia, nervousness, dizziness, hypertrichosis and ataxia (Guerreiro 1997)).
Worldwide, phenytoin is a commonly used antiepileptic drug for participants with focal onset seizures and generalised onset tonic‐clonic seizures. Although phenytoin is no longer considered as a first‐line treatment in Europe (Wallace 1997), it is more commonly used in the USA (Wilder 1995). Phenytoin is associated with long‐term cosmetic changes including gum hypertrophy, acne and coarsening of the facial features (Mattson 1985; Scheinfeld 2003), as well as low folic acid levels, predisposing participants to megaloblastic anaemia (Carl 1992). It can also cause a rash (Tennis 1997) in 5% to 10% of participants, which on rare occasions may be life threatening. It is also associated with congenital abnormalities (Gladstone 1992; Nulman 1997), where the risk is estimated to be two to three times that of the general population (Meador 2008). Phenytoin is also particularly associated with fetal hydantoin syndrome (Scheinfeld 2003).
How the intervention might work
Antiepileptic drugs suppress seizures by reducing neuronal excitability, hence reducing the probability that a seizure will occur.
Oxcarbazepine and phenytoin are broad spectrum treatments suitable for many seizure types, and both have an anticonvulsant mechanism through blocking ion channels, binding with neurotransmitter receptors or through inhibiting the metabolism or reuptake of neurotransmitters and the modulation of gamma‐aminobutyric acid‐A (GABA‐A) receptors (Faigle 1990; Granger 1995; Grant 1992; Ragsdale 1991; Willow 1985).
Why it is important to do this review
With evidence that up to 70% of individuals with a new epilepsy diagnosis enter a long‐term remission of seizures shortly after starting drug therapy (Cockerell 1995; Hauser 1993; Sander 2004), the correct choice of first‐line antiepileptic therapy for individuals with newly diagnosed seizures is of great importance. It is important to know how newer drugs, such as oxcarbazepine, compare with first‐line standard treatments. Our aim in this systematic review is to overview existing evidence for the comparative efficacy and tolerability of oxcarbazepine and phenytoin (one of the standard antiepileptic drugs) when used as monotherapy.
There are difficulties in undertaking a systematic review of epilepsy monotherapy trials as the important efficacy outcomes require analysis of time‐to‐event data (for example, time to first seizure after randomisation). Although methods have been developed to synthesise time‐to‐event data using summary information (Parmar 1998; Williamson 2002), the appropriate statistics are not commonly reported in published epilepsy trials (Nolan 2013b; Williamson 2000). Furthermore, although most epilepsy monotherapy trials collect seizure data, there has been no uniformity in the definition and reporting of outcomes. For example, trials may report time to 12‐month remission but not time to first seizure or vice versa, or some trials may define time to first seizure from the date of randomisation while others use the date of achieving maintenance dose. Trial investigators have also adopted differing approaches to the analysis, particularly with respect to the censoring of time‐to‐event data. For these reasons, we performed this review using individual participant data (IPD), which helps to overcome these problems. This review is one in a series of Cochrane IPD reviews investigating pair‐wise monotherapy comparisons (Marson 2000; Nevitt 2017b; Nevitt 2018a; Nevitt 2018b; Nolan 2013c; Nevitt 2018c; Nolan 2016b). These data have also been included in IPD network meta‐analyses of antiepileptic drug monotherapy (Tudur Smith 2007; Nevitt 2017a)
Objectives
To review the time to treatment failure, remission and first seizure with oxcarbazepine compared to phenytoin, when used as monotherapy in people with focal onset seizures or generalised tonic‐clonic seizures (with or without other generalised seizure types).
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) which used either an adequate method of allocation concealment (e.g. sealed opaque envelopes) or a 'quasi' method of randomisation (e.g. allocation by date of birth).
Studies may have been double‐blind, single‐blind, or unblinded.
Studies must have included a comparison of oxcarbazepine monotherapy versus phenytoin monotherapy in individuals with epilepsy.
Types of participants
We included children or adults with focal onset seizures (simple focal, complex focal or secondarily generalised tonic‐clonic seizures) or generalised onset tonic‐clonic seizures, with or without other generalised seizure types (in other words, those who had only generalised tonic‐clonic seizures and those who had both generalised onset tonic‐clonic seizures and generalised seizures of other types, e.g. absence, myoclonic, etc.).
We excluded individuals with other generalised seizure types alone without generalised tonic‐clonic seizures (e.g. those who had only absence seizures without any generalised tonic‐clonic seizures) due to differences in first‐line treatment guidelines for other generalised seizure types (NICE 2012).
We included individuals with a new diagnosis of epilepsy, or those who have had a relapse following withdrawal of antiepileptic monotherapy.
Types of interventions
Oxcarbazepine or phenytoin as monotherapy.
Types of outcome measures
Below is a list of outcomes investigated in this review. Reporting of these outcomes in the original trial report was not an eligibility requirement for inclusion in this review.
Primary outcomes
Time to treatment failure (retention time). This was a combined outcome reflecting both efficacy and tolerability, as the following may have lead to failure of treatment: continued seizures, side effects, non‐compliance or the initiation of add‐on treatment. This is an outcome to which the participant makes a contribution and is the primary outcome measure recommended by the Commission on Antiepileptic Drugs of the International League Against Epilepsy (ILAE 1998; ILAE 2006).
Time to treatment failure is considered according to three definitions:
time to treatment failure for any treatment related reason (continued seizures, side effects, non‐compliance or the initiation of add‐on treatment);
time to treatment failure due to adverse events (i.e. side effects);
time to treatment failure due to lack of efficacy (i.e. continued seizures).
Secondary outcomes
Time to first seizure post‐randomisation
Time to achieve 12‐month remission (seizure‐free period)
Time to achieve six‐month remission (seizure‐free period)
Incidence of adverse events (all reported whether related or unrelated to treatment)
Search methods for identification of studies
Electronic searches
Searches were run for the original review in 2006 and subsequent searches were run in April 2008, July 2010, November 2011, June 2012, January 2013, and February 2015.
For the latest update we searched the following databases on 20 August 2018, with no language restrictions.
The Cochrane Register of Studies (CRS Web), which includes the Cochrane Epilepsy Group Specialized Register and the Cochrane Central Register of Controlled Trials (CENTRAL), using the strategy outlined in Appendix 1.
MEDLINE (Ovid, 1946 to 20 August 2018), using the strategy outlined in Appendix 2.
ClinicalTrials.gov, using the strategy outlined in Appendix 3.
The World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP), using the strategy outlined in Appendix 4.
Searching other resources
In addition, we handsearched relevant journals, reviewed the reference lists of included studies to search for additional reports of relevant studies, contacted Novartis (manufacturers of oxcarbazepine) and Parke‐Davis (manufacturers of phenytoin) and researchers in the field to seek any ongoing or unpublished studies.
Data collection and analysis
Selection of studies
Two of the authors (MM and SJN) independently assessed all identified trials for inclusion. Any disagreements were resolved by mutual discussion.
Data extraction and management
We requested the following IPD for all trials meeting our inclusion criteria.
Trial methods
Method of generation of random list
Method of concealment of randomisation
Stratification factors
Blinding methods
Participant covariates
Gender
Age
Seizure types
Time between first seizure and randomisation
Number of seizures prior to randomisation (with dates)
Presence of neurological signs
Electroencephalographic (EEG) results
Computerised tomography/magnetic resonance imaging (CT/MRI) results
Follow‐up data
Treatment allocation
Date of randomisation
Dates of follow‐up
Dates of seizures post‐randomisation or seizure frequency data between follow‐up visits
Dates of treatment failure and reasons for treatment failure
Dose
Dates of dose changes
For each trial for which we did not obtain IPD, we carried out an assessment to see whether any relevant aggregate level data had been reported or could be indirectly estimated using the methods of Parmar 1998 and Williamson 2002.
For included trials with IPD provided (Bill 1997; Guerreiro 1997), seizure data were provided in terms of the mean number of seizures recorded per week in the titration period (first eight weeks) and the maintenance period (following 48 weeks) rather than specific dates of seizures. To enable time‐to‐event outcomes to be calculated, we applied linear interpolation to approximate the days on which seizures occurred. For example, if the mean number of seizures per week in the titration period was 0 and in the maintenance period it was 0.02115, and the participant started treatment on 28 September 1993 and ended treatment on 19 October 1994 (interval of 387 days), then the date of first seizure would be approximately 165.5 days after the start of the maintenance period and thus 221.5 days after the start of treatment. This allowed an estimate of the time to six‐ and 12‐month remission and the time to first seizure to be computed.
We calculated time to first seizure from the date of randomisation to the date that their first seizure was estimated to have occurred. If the mean number of seizures per week data were missing for the titration period (first eight weeks), the estimated time of the first seizure could not be calculated. Eight participants in total (five in Bill 1997, and three in Guerreiro 1997) had missing seizure data for the titration period (all eight also had missing seizure data for the maintenance period). The number of days on trial medication ranged between one and 36 days for these eight participants. We excluded them from analyses of time to first seizure, time to six‐month remission and time to 12‐month remission, but included them in the analysis of time to treatment failure.
We calculated time to six‐ and 12‐month remission from the date of randomisation to the date (or estimated date) the individual had first been free from seizures for six or 12 months respectively. If the participant had one or more seizures in the titration period, a six‐ or 12‐month seizure‐free period could also occur between the estimated date of the last seizure in the titration period and the estimated date of the first seizure in the maintenance period.
If the mean 'number of seizures per week' data were missing for the maintenance period (but not for the titration period), the values for six‐ and 12‐month remission would be censored at the end of the titration period (effectively excluding them from the analysis). These outcomes were also censored if the individual died or follow‐up ceased prior to the occurrence of the event of interest.
For both trials (Bill 1997; Guerreiro 1997), the date of and reason for the treatment failure were provided directly (see Table 3 for reasons of premature discontinuation of treatment). Time to treatment failure was calculated as date of randomisation to date of treatment failure. For the analysis of time‐to‐event, we defined an 'event' as treatment failure because of reasons related to the treatment (i.e. lack of efficacy, adverse events, or both lack of efficacy and adverse events), non‐compliance with the treatment regimen, withdrawal of consent from the trial, etc.). We censored the outcome if treatment failure or withdrawal of treatment was for reasons not related to the trial treatment, i.e. loss to follow‐up, death (not treatment or epilepsy related), withdrawal of treatment due to remission, etc. We also censored individuals who were still on allocated treatment at the date of the end of follow‐up. We considered documented reasons for treatment failure or treatment withdrawal on a case‐by‐case basis for relation to treatment; three of the review authors (SJN, MM and AGM) independently classified reasons for treatment failure as 'events' or 'censored' and resolved any disagreements by discussion. We extracted detail about the reason for the treatment failure from study case report forms when necessary, e.g. for death and protocol violation(s). Two deaths were recorded. One was classified as a censored value, because the cause of death was unrelated to the treatment or the condition. The other death was classified as an event: the participant died after experiencing an episode of status epilepticus, but had been non‐compliant and discontinued treatment before they died.
1. Reasons for premature discontinuation (treatment failure).
| Reason for early termination | Bill 1997 | Guerreiro 1997 | Total | ||||
| OXC | PHT | OXC | PHT | OXC | PHT | All | |
| Adverse events (Event)a | 5 | 18 | 2 | 14 | 7 | 32 | 39 |
| Lack of efficacy (Event) | 1 | 1 | 4 | 3 | 5 | 4 | 9 |
| Non‐compliance/protocol violation (Event) | 29 | 20 | 9 | 7 | 38 | 27 | 65 |
| Illness or death (not treatment related, censored)b | 2 | 5 | 1 | 0 | 3 | 5 | 8 |
| Lost to follow‐up (censored) | 19 | 17 | 8 | 9 | 27 | 26 | 53 |
| Other (censored)c | 0 | 0 | 0 | 1 | 0 | 1 | 1 |
| Completed the study (censored) | 87 | 83 | 73 | 62 | 160 | 145 | 305 |
| Total | 143 | 144 | 97 | 96 | 240 | 240 | 480 |
a. One participant on phenytoin (in Bill 1997) had an episode of status epilepticus following non‐compliance with treatment and died. This was classified as a serious adverse event ('Event') in the analysis of time to treatment failure.
b. One participant (in Bill 1997) died after getting caught up in political violence. This was deemed to not be related to treatment and was censored in the analysis of time to treatment failure.
c. One participant (in Guerreiro 1997) immediately withdrew from treatment at baseline (following randomisation). This was deemed to not be related to treatment and was censored in the analysis of time to treatment failure.
For the analysis of 'time to treatment failure due to adverse events', only treatment failures which were documented to be due to adverse events (either as a sole reason or due to both a lack of efficacy and adverse events) were classed as an 'event' within time‐to‐event analyses and all other reasons for treatment failure were censored. Similarly, for the analysis of 'time to treatment failure due to lack of efficacy' only treatment failures which were documented to be due to lack of efficacy (i.e. continued seizures, either as a sole reason or due to both a lack of efficacy and adverse events) were classed as an 'event' within time‐to‐event analyses and all other reasons for treatment failure were censored.
Assessment of risk of bias in included studies
Two review authors (SJN and MM) independently assessed the risk of bias for each trial using the Cochrane 'Risk of bias' tool, as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We rated each of the following six domains as low, unclear or high risk of bias: method of generating random sequence, allocation concealment, blinding methods (blinding of participants and personnel and blinding of outcome assessment), incomplete outcome data, selective outcome reporting and other sources of bias. Any discrepancies in 'Risk of bias' judgements of the two review authors were resolved by discussion. In the event of the presence of high risk of bias in included trials (due to inadequate allocation concealment or lack of blinding), we planned to perform sensitivity analyses excluding these trials.
Measures of treatment effect
We measured all outcomes in this review as time‐to‐event outcomes with the hazard ratio (HR) and 95% confidence interval (CI) used as the measure of treatment effect. We calculated outcomes from IPD provided, where possible, or extracted from published trials if possible.
Unit of analysis issues
We did not have any unit of analysis issues. The unit of allocation and analysis was the individual for all included trials; and no trials included in meta‐analyses were of a repeated measures (longitudinal) nature or of a cross‐over design.
Dealing with missing data
For each trial that supplied IPD, we reproduced results from trial results where possible and performed the following consistency checks.
We cross‐checked trial details against any published report of the trial and contacted original trial authors if we found missing data, errors or inconsistencies. If trial authors could not resolve inconsistencies between the IPD and the published data, depending on the extent of the inconsistencies, we planned to perform sensitivity analysis or we excluded the data from the meta‐analysis.
We reviewed the chronological randomisation sequence and checked the balance of prognostic factors, taking account of factors stratified for in the randomisation procedure.
Assessment of heterogeneity
We assessed heterogeneity statistically using the Q test (P < 0.10 for significance) and the I² statistic (where a value of greater than 50% indicates considerable heterogeneity; Higgins 2003), and visually by inspecting forest plots.
Assessment of reporting biases
Two review authors (SJN and MM) undertook a full quality and 'Risk of bias' assessment. In theory, a review using IPD should overcome issues of reporting biases as unpublished data can be provided and unpublished outcomes calculated.
Data synthesis
We aimed to carry out our analysis on an intention‐to‐treat basis (that is, where participants are analysed in the group to which they were randomised, irrespective of which treatment they actually received). However, in the two trials included in meta‐analysis, participants were not followed up after treatment failure or treatment withdrawal (see Table 3 for reasons for premature discontinuation of treatment). For most of these participants, the reason for treatment failure classed as an event for the analysis of time to treatment failure, and these participants had to be censored at the time of treatment failure for the seizure outcomes, which contravenes the principle of intention‐to‐treat.
For all outcomes, we investigated the relationship between the time‐to‐event and treatment effect of the antiepileptic drugs (AEDs). We used Cox proportional hazards regression models to obtain trial‐specific estimates of log (hazard ratio) or treatment effect and associated standard errors in Stata Statistical Software, version 14 (Stata 2015). The model assumes that the ratio of hazards (risks) between the two treatment groups is constant over time (i.e. hazards are proportional). We tested this proportional hazards assumption of the Cox regression model for each outcome of each trial by testing the statistical significance of a time varying covariate in the model. We evaluated overall pooled estimates of hazard ratios (with 95% confidence intervals) using the generic inverse variance method. We expressed results as hazard ratios (HRs) and 95% confidence interval (CIs).
By convention, a HR greater than one indicates that an event is more likely to occur earlier on oxcarbazepine than on phenytoin. Hence, for time to treatment failure or time to first seizure, a HR greater of less one indicates a clinical advantage for oxcarbazepine (e.g. HR = 0.8 would suggest a 20% reduction in hazard of treatment failure from oxcarbazepine compared to phenytoin), and for time to achieve six‐month and 12‐month remission, a HR of less than one indicates a clinical advantage for phenytoin.
We anticipated that adverse events may have been recorded using different methods and reported in different levels of detail across included studies, therefore we did not analyse incidence of adverse events and instead reported this data narratively.
Subgroup analysis and investigation of heterogeneity
Due to the strong clinical belief that some antiepileptic drugs are more effective in some seizure types than others, we stratified all analyses by seizure type (focal onset versus generalised onset), according to the classification of main seizure type at baseline. We classified focal seizures (simple or complex) and focal secondarily generalised seizures as focal epilepsy.
We classified primarily generalised seizures as generalised epilepsy. We conducted a Chi² test of interaction between treatment and seizure type. If we found significant statistical heterogeneity to be present, we performed meta‐analysis with a random‐effects model in addition to a fixed‐effect model, presenting the results of both models and performing sensitivity analyses to investigate differences in study characteristics.
Sensitivity analysis
The two trials which provided IPD were double‐blind. After completion of the maintenance period, some participants continued to be followed up taking 'open‐label' (unblinded) treatment. The primary analyses included data from this open‐label period. We repeated the analysis, including only data from the double‐blind period of 392 days (eight‐week titration period plus the 48‐week maintenance period).
Misclassification of seizure type is a recognised problem in epilepsy, whereby some people with generalised seizures have been mistakenly classed as having focal onset seizures, and vice versa. There is clinical evidence that individuals with generalised onset seizures are unlikely to have an 'age of onset' greater than 25 to 30 years (Malafosse 1994). Such misclassification impacted upon the results of three reviews in our series of pair‐wise reviews for monotherapy in epilepsy comparing carbamazepine to phenobarbitone, phenytoin and sodium valproate, in which around 30% to 50% of participants analysed may have had their seizure type misclassified as generalised onset (Marson 2000; Nevitt 2018b; Nevitt 2017b). Given the potential biases introduced into those reviews, we examined the distribution of age at onset for individuals with generalised seizures in the trials included in this review, to assess the potential impact of misclassification of seizure type on the outcomes.
One trial was a paediatric trial so no individuals over the age of 30 were recruited (Guerreiro 1997). In Bill 1997, 104 individuals were classified as having generalised onset seizures and 30 of these individuals were over the age of 30 at entry into the trial. Therefore, up to 29% of individuals classified as having generalised onset seizures may have had their seizure type misclassified. Such a misclassification could bias our results against finding an interaction between treatment and seizure types (focal onset versus generalised onset). We undertook the following two analyses to investigate misclassification.
We reclassified all individuals with generalised seizures and age at onset greater than 30 into an 'uncertain seizure type' group.
We reclassified individuals with generalised seizures and age at onset greater than 30 as having focal onset seizures.
Summary of findings and quality of the evidence (GRADE)
For the 2013 update, in a post‐hoc change from protocol, we added two 'Summary of findings' tables to the review (outcomes in the tables were decided before the update started based on clinical relevance).
Table 1 reports the primary outcome of 'time to treatment failure (for any reason related to treatment)' and 'time to treatment failure due to adverse events' in the subgroups of participants with focal onset seizures, generalised onset seizures and overall adjusted by epilepsy type.
Summary of findings for the main comparison. Summary of findings: oxcarbazepine versus phenytoin (primary outcomes).
| Oxcarbazepine compared with phenytoin for epilepsy | ||||||
|
Patient or population: adults and children with newly diagnosed epilepsy Settings: outpatients Intervention: oxcarbazepine Comparison: phenytoin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenytoin | Oxcarbazepine | |||||
|
Time to treatment failure (any reason related to treatment) All participants Range of follow‐up: 1 to 779 days |
The 20th percentile** of time to treatment failure was 263 days in the phenytoin group | The 20th percentile** of time to treatment failure was 342 days (79 days longer) in the oxcarbazepine group |
HR 0.78 (0.53 to 1.14)a |
476 (2 trials) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical advantage for oxcarbazepine |
|
Time to treatment failure (any reason related to treatment) Subgroup: focal onset seizures Range of follow‐up: 1 to 532 days |
The 20th percentile** of time to treatment failure was 230 days in the phenytoin group | The 20th percentile** of time to treatment failure was 414 days (184 days longer) in the oxcarbazepine group |
HR 0.69 (0.43 to 1.09) |
333 (2 trials) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical advantage for oxcarbazepine |
|
Time to treatment failure (any reason related to treatment) Subgroup: generalised onset seizures Range of follow‐up: 1 to 779 days |
The 20th percentile** of time to treatment failure was 306 days in the phenytoin group | The 20th percentile** of time to treatment failure was 268 days (38 days shorter) in the oxcarbazepine group |
HR 1.03 (0.51 to 2.08) |
143 (2 trials) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical advantage for oxcarbazepine |
|
Time to treatment failure due to adverse events All participants Range of follow‐up: 1 to 779 days |
32 out of 240 (13%) withdrew due to adverse events in the phenytoin groupc | 7 out of 240 (3%) withdrew due to adverse events in the oxcarbazepine groupc |
HR 0.22 (0.10 to 0.51) |
480 (2 trials) | ⊕⊕⊕⊕ high | HR < 1 indicates a clinical advantage for oxcarbazepine |
|
Time to treatment failure due to lack of efficacy All participants Range of follow‐up: 1 to 779 days |
4 out of 240 (2%) withdrew due to lack of efficacy in the phenytoin groupc | 5 out of 240 (2%) withdrew due to lack of efficacy in the phenytoin groupc |
HR 1.17 (0.31 to 4.35) |
480 (2 trials) | ⊕⊕⊕⊝ moderated | HR < 1 indicates a clinical advantage for oxcarbazepine |
| * Illustrative risks in the oxcarbazepine and phenytoin groups are calculated at the median time to treatment failure (i.e. the time to 50% of participants failing or withdrawing from allocated treatment) within each group across all trials. The relative effect (pooled hazard ratio) shows the comparison of 'time to treatment failure' between the treatment groups. ** The 20th percentile of time to treatment failure (i.e. the time to 20% of participants failing or withdrawing from allocated treatment) is presented for the subgroup with generalised seizures as less than 50% of participants failed/withdrew from treatment in both groups, therefore the median time could not be calculated. Abbreviations: 95% CI: 95% confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
a. Pooled hazard ratio for all participants adjusted for epilepsy type.
b. Downgraded once due to risk of bias: up to 29% of adult participants classified as experiencing generalised onset seizures (in Bill 1997) may have had their epilepsy type wrongly classified and sensitivity analyses show that misclassification may have had an impact on the conclusions drawn for individuals with generalised seizures and whether an interaction between treatment effect and epilepsy type exists.
c. Medians or 20th percentile of time to treatment failure could not be calculated due to small numbers of participants withdrawing due to adverse events or lack of efficacy in one or both of the treatment groups. We were unable to perform subgroup analysis for 'time to treatment failure due to adverse events' and 'time to treatment failure due to lack of efficacy' due to small numbers of participants with each epilepsy type failing treatment for these reasons.
d. Downgraded once due to imprecision: there are wide confidence intervals around the pooled HR so it is unclear whether there is an advantage to either drug, or no difference between drugs.
Table 2 reports the secondary outcomes of 'time to first seizure' and 'time to 12‐month remission' in the subgroups of participants with focal onset seizures, generalised onset seizures and overall, adjusted by epilepsy type.
Summary of findings 2. Summary of findings: oxcarbazepine versus phenytoin (secondary outcomes).
| Oxcarbazepine compared with phenytoin for epilepsy | ||||||
|
Patient or population: adults and children with newly diagnosed epilepsy Settings: outpatients Intervention: oxcarbazepine Comparison: phenytoin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenytoin | Oxcarbazepine | |||||
|
Time to first seizure (post‐randomisation) All participants Range of follow‐up: 1 to 779 days |
The median time to first seizure was 230 days in the phenytoin group | The median time to first seizure was 252 days (22 days longer) in the oxcarbazepine group |
HR 0.97 (0.75 to 1.26)a |
468 (2 trials) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical advantage for oxcarbazepine |
|
Time to first seizure (post‐randomisation) Subgroup: focal onset seizures Range of follow‐up: 1 to 498 days |
The median time to treatment first seizure was 224 days in the phenytoin group | The median time to first seizure was 233 days (9 days longer) in the oxcarbazepine group |
HR 0.92 (0.68 to 1.25) |
326 (2 trials) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical advantage for oxcarbazepine |
|
Time to first seizure (post‐randomisation) Subgroup: generalised onset seizures Range of follow‐up: 1 to 779 days |
The 25th percentile** of time to first seizure was 78 days in the phenytoin group | The 25th percentile** of time to first seizure was 28 days (50 days shorter) in the oxcarbazepine group |
HR 1.11 (0.66 to 1.86) |
142 (2 trials) | ⊕⊕⊝⊝ lowb,c | HR < 1 indicates a clinical advantage for oxcarbazepine |
|
Time to achieve 12‐month remission (seizure‐free period) All participants Range of follow‐up: 1 to 532 days |
The median time to 12‐month remission was 401 days in the phenytoin group | The median time to 12‐month remission was 365 days (36 days shorter) in the oxcarbazepine group |
HR 1.04 (0.77 to 1.41)a |
468 (2 trials) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical advantage for phenytoin |
|
Time to achieve 12‐month remission (seizure‐free period) Subgroup: focal onset seizures Range of follow‐up: 1 to 532 days |
The median time to 12‐month remission was 401 days in the phenytoin group | The median time to 12‐month remission was 390 days (11 days shorter) in the oxcarbazepine group |
HR 1.09 (0.75 to 1.57) |
326 (2 trials) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical advantage for phenytoin |
|
Time to achieve 12‐month remission (seizure‐free period) Subgroup: generalised onset seizures Range of follow‐up: 1 to 532 days |
The median time to 12‐month remission was 365 days in the phenytoin group | The median time to 12‐month remission was 365 days (0 days shorter or longer) in the oxcarbazepine group |
HR 0.94 (0.55 to 1.62) |
142 (2 trials) | ⊕⊕⊝⊝ lowb,c | HR < 1 indicates a clinical advantage for phenytoin |
| * Illustrative risks in the oxcarbazepine and phenytoin groups are calculated at the median time to first seizure or time to 12‐month remission (i.e. the time to 50% of participants experiencing a first seizure or 12‐months of remission) within each group across all trials. The relative effect (pooled hazard ratio) shows the comparison of 'time to first seizure' or 'time to 12‐month remission' between the treatment groups. ** The 25th percentile of time to first seizure (i.e. the time to 25% of participants experiencing a first seizure) is presented for the subgroup with generalised seizures as less than 50% of participants experienced a seizure recurrence in both groups, therefore the median time could not be calculated. Abbreviations: 95% CI: 95% confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
a. Pooled hazard ratio for all participants adjusted for epilepsy type.
b. Downgraded once due to risk of bias: as participants who failed treatment or withdrew from the treatment were no longer followed up in the study, remission and seizure outcomes had to be censored at time of treatment failure, therefore remission and seizure outcomes could not be analysed with an intention‐to‐treat approach.
c. Downgraded once due to risk of bias: up to 29% of adult participants classified as experiencing generalised onset seizures (in Bill 1997) may have had their epilepsy type wrongly classified, and sensitivity analyses show that misclassification may have had an impact on the conclusions drawn for individuals with generalised seizures and whether an interaction between treatment effect and epilepsy type exists.
We determined the quality of the evidence using the GRADE approach, where we downgraded our assessment in the presence of high risk of bias in at least one trial, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results and high probability of publication bias. We downgraded evidence by one level if the limitation was considered serious and by two levels if considered very serious, as judged by the review authors.
Results
Description of studies
Results of the search
We identified 125 records from the databases and search strategies outlined in Electronic searches. We found no further records through searching other resources. We removed 36 duplicate records and screened 89 records (title and abstract) for inclusion in the review. We excluded 80 records based on title and abstract and assessed eight full‐text articles and records for inclusion in the review. One study was excluded from the review and three studies (reported in seven full‐text articles) were included in the review.
We identified no new studies in this update of the review. See Figure 1 for details of eligibility screening.
1.

PRISMA Study flow diagram.
Included studies
We included a total of three randomised controlled trials in which participants were randomised to oxcarbazepine or phenytoin; see Characteristics of included studies for further details.
Individual participant data (IPD) were available for a total of 480 participants from two studies (Bill 1997; Guerreiro 1997), and computerised data were provided directly for both trials. Data were available for the following percentages of participants, on the following characteristics: randomised drug (100%); time between first seizure and randomisation (100%); number of seizures prior to randomisation (100%); sex (100%); age (99.8%); seizure types (99.2%); electroencephalographic (EEG) results (98.1%); and computerised tomography (CT) scan results (79.2%). Neurological examination findings were not available for either trial. See the Characteristics of included studies and Table 4 for further details.
2. Demographic characteristics of trial participants (trials providing individual participant data).
| Bill 1997 | Guerreiro 1997 | |||||
| OXC | PHT | Missing | OXC | PHT | Missing | |
| Focal seizures: n(%) | 84 (59%) | 98 (68%) | 1 | 73 (77%) | 78 (82%) | 3 |
| Male gender: n (%) | 82 (57%) | 92 (64%) | 0 | 46 (47%) | 50 (52%) | 0 |
| Age at entry (years): mean (SD), range | 27.1 (11.3), 16 to 63 |
26.5 (10.2), 15 to 91 |
1 | 10.2 (3.1), 5 to 17 |
10.9 (3.1), 6 to 17 |
0 |
| Aged > 30 and generalised seizures: n (%) | 14 | 16 | 1 | NA | NA | NA |
| Epilepsy duration (years): mean (SD), range | 1.8 (3.7), 0 to 22 | 1.7 (3.7), 0 to 25 | 0 | 0.6 (0.9), 0 to 5 | 0.7 (1.8), 0 to 14 | 0 |
| Number of seizures in prior 6 months: median (range) | 3 (0 to 252) | 3 (0 to 157) | 0 | 2 (0 to 70) | 2 (0 to 108) | 0 |
| EEG normal: n (%) | 82 (60%) | 70 (49%) | 9 | 49 (51%) | 52 (54%) | 0 |
| CT scan normal: n (%) | 31 (27%) | 38 (30%) | 45 | 6 (8%) | 6 (9%) | 55 |
CT= computerised tomography; EEG = electroencephalographic; n = number of participants; NA = not available; OXC=oxcarbazepine; PHT= Phenytoin; SD = standard deviation
Proportions (%) are calculated based on non‐missing data
The two trials were similar in design and recruited participants with newly diagnosed and previously untreated epilepsy, however one trial recruited adults only (Bill 1997), and one trial recruited children and adolescents only (Guerreiro 1997), which is a potential source of heterogeneity. Both trials recruited participants with focal onset seizures (simple/complex focal or secondary generalised tonic‐clonic) and participants with generalised tonic‐clonic seizures without focal onset. In the trial including adults only 61% of participants were male (57% males in the oxcarbazepine group and 64% in the phenytoin group) (Bill 1997). In the trial including children and adolescents (Guerreiro 1997), 50% of participants were male (47% males in the oxcarbazepine group and 52% in the phenytoin group). To be included in the two trials (Bill 1997; Guerreiro 1997), participants had to have a minimum of two seizures, separated by at least 48 hours, in the six months before entering the study. In both trials the baseline assessment included a medical and seizure history, physical examination, laboratory evaluations, ECG, EEG and an optional cranial computed tomography (CT) scan to rule out any progressive neurological disorder such as a brain tumour. In Bill 1997 and Guerreiro 1997, seizures were classified according to the 1981 International Classification of seizure types (Commission 1981). The study by Guerreiro and colleagues also used the 1989 classification of epilepsies and epileptic syndromes (Commission 1989).
During the eight‐week titration period, treatment was started with daily doses of:
300 mg oxcarbazepine or 100 mg phenytoin and then increased bi‐weekly (every two weeks) based on clinical response (for adults) (Bill 1997);
150 mg oxcarbazepine or 50 mg phenytoin and then increased gradually based on clinical response (for children and adolescents) (Guerreiro 1997).
No fixed titration schedule was used except that after eight weeks participants were to be on a tid (three times per day) regimen of oxcarbazepine or phenytoin with daily doses of 450 to 2400 mg and 150 to 800 mg, respectively. The daily dose range and tid regimen were to be continued during the subsequent 48‐week maintenance period. However, adjustment of the daily dose according to clinical response was possible during this period. The median daily dose actually taken (with lower and upper quartiles) for oxcarbazepine was 900 mg (900 mg; 1200 mg) in Bill 1997, and 600 mg (450 mg; 900 mg) in Guerreiro 1997. The median daily dose (with lower and upper quartiles) for phenytoin was 300 mg (300 mg; 300 mg) in Bill 1997, and 200 mg (150 mg; 300 mg) in Guerreiro 1997.
Individual participant data were not available for the remaining trial (Aikia 1992), a single‐centre trial conducted in Finland which recruited 37 adult participants with newly onset seizures (19 to oxcarbazepine and 18 to phenytoin) and reported the characteristics and results for the 29 participants completing the study. The majority of participants had focal onset seizures (72%), 38% of participants were male and the mean age of included participants was approximately 33 years. The outcomes evaluated were neuropsychological assessment and cognitive functioning in three major areas (verbal learning and memory, sustained attention, simple psychomotor speed), and they were measured at baseline, six months' and 12 months' follow‐up. MANOVA (repeated measures analysis of variance) for these assessments showed no statistically significant interaction between treatment group and time.
Excluded studies
We excluded one study from the review as it was not fully randomised (Sabers 1995, see Characteristics of excluded studies).
Risk of bias in included studies
For further details, see Characteristics of included studies, Figure 2 and Figure 3.
2.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
We judged two studies to be at low risk of selection bias (Bill 1997; Guerreiro 1997). Randomisation numbers were sequentially assigned across centres within each country. A computer‐generated randomisation scheme was used to provide balanced blocks of participant numbers for each of the two treatment groups within each centre. A block size of six was used (Pohlmann 2005 [pers comm]).
The trial reports for two studies did not provide details on allocation concealment (Bill 1997; Guerreiro 1997), but the trial statistician explained in personal correspondence that allocation concealment was achieved as follows: sequentially numbered packages were prepared which were identical and contained identical tablets; and recruiting clinicians were asked to allocate each participant the package with the lowest number available at the centre (Pohlmann 2005 [pers comm]). We deemed these trials to be at low risk of selection bias.
Participants were "randomly allocated" to treatment in Aikia 1992, however the trial report did not provide methods used to generate a random sequence or to conceal allocation, therefore we judged the study to be at unclear risk of selection bias.
Blinding
We judged all three studies to be at low risk of performance bias and detection bias.Two studies were double‐blinded by using divisible tablets with identical appearance (Bill 1997; Guerreiro 1997). The third study was also double‐blinded, however the method of achieving the double‐blind was not provided in the trial report (Aikia 1992).
Incomplete outcome data
We judged all three studies to be at high risk of attrition bias.
Analyses were not performed using an intention‐to‐treat approach in Aikia 1992: 29 participants who completed 12 months of follow‐up were included in analyses; and eight participants who experienced inadequate seizure control, adverse events or were non‐compliant were withdrawn from the study and excluded from analysis.
For the studies for which IPD were provided (Bill 1997; Guerreiro 1997), analyses for time to six‐month remission, time to 12‐month remission and time to first seizure could not be performed using an intention‐to‐treat approach as participants who failed treatment in both studies were not followed up after the time of treatment failure.
In Bill 1997 and Guerreiro 1997, the numbers for premature discontinuation in the titration period differed from the numbers reported in the publications (49 participants — 25 on oxcarbazepine and 24 on phenytoin — in Bill 1997; and 31 participants — 15 on oxcarbazepine and 16 on phenytoin — in Guerreiro 1997). We corresponded with the trial statistician about these differences and they proposed a possible explanation: it is likely that the raw premature discontinuation data (0 or 1) as collected in the clinical record file (CRF) were provided for this Cochrane Review, but for the time to premature discontinuation analyses in the publication, a derived premature discontinuation variable based on the "time under assessment" was created (Pohlmann 2005 [pers comm]). If certain participants had empty records in the maintenance period, the created variable will indicate a premature discontinuation at the end of the titration period, although in the CRF they were coded as discontinuing during the maintenance period. If this was the case, it is possible that we find fewer participants who discontinued during the titration period, compared to the publication. (Note that the trial statistician who proposed this explanation was not the original trial statistician and could only explain how they handled data at the time of the trial (in the 1990s). The data used for the publication were not accessible at the time of our query (Pohlmann 2005 [pers comm]).
Selective reporting
We judged all three studies to be at low risk of reporting bias. Protocols were not available for the three included studies (Aikia 1992; Bill 1997; Guerreiro 1997), however unpublished IPD were provided in order to calculate outcomes used in this review for two studies (Bill 1997; Guerreiro 1997), and neuropsychological and cognitive outcomes were well reported in Aikia 1992.
Other potential sources of bias
We detected no other sources of bias in any of the three studies.
Effects of interventions
Details regarding the number of participants contributing to each analysis are given in Table 5. One participant in Bill 1997, and three in Guerreiro 1997, had missing data for the main seizure type at baseline and therefore their epilepsy type could not be derived. We excluded these four participants from subgroup analyses according to epilepsy types.
3. Number of participants contributing to analysis.
| Trial | Epilepsy type | Number randomised | Time to treatment failurea | Time to first seizure | Time to 12 month remission | Time to 6 month remission | |||||
| OXC | PHT | OXC | PHT | OXC | PHT | OXC | PHT | OXC | PHT | ||
|
Bill 1997 |
Focal | 84 | 98 | 84 | 98 | 81 | 97 | 81 | 97 | 81 | 97 |
| Generalised | 58 | 46 | 59 | 46 | 58 | 46 | 58 | 46 | 58 | 46 | |
| Total classified | 142 | 144 | 142 | 144 | 138 | 143 | 138 | 143 | 138 | 143 | |
| Unclassified/missing | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | |
| TOTAL | 143 | 144 | 143 | 144 | 139 | 143 | 139 | 143 | 139 | 143 | |
|
Guerreiro 1997 |
Focal | 73 | 78 | 73 | 78 | 72 | 76 | 72 | 76 | 72 | 76 |
| Generalised | 22 | 17 | 22 | 17 | 22 | 17 | 22 | 17 | 22 | 17 | |
| Total classified | 95 | 95 | 95 | 95 | 94 | 93 | 94 | 93 | 94 | 93 | |
| Unclassified/missing | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | |
| TOTAL | 97 | 96 | 97 | 96 | 96 | 94 | 96 | 94 | 96 | 94 | |
| TOTAL | 237 | 239 | 237 | 239 | 235 | 237 | 235 | 237 | 235 | 237 | |
OXC: oxcarbazepine, PHT: phenytoin
a. All participants were included in analyses of time to treatment failure for any reason related to the treatment, time to treatment failure due to adverse events and time to treatment failure due to lack of efficacy.
See Table 1 for a summary of the results for the primary outcome 'time to treatment failure (for any reason related to treatment)' and 'time to treatment failure due to adverse events' (stratified by epilepsy type), and Table 2 for a summary of results for the secondary outcomes 'time to first seizure' and 'time to 12‐month remission'. Survival curve plots are shown in Figure 4; Figure 5; Figure 6; Figure 7; Figure 8; Figure 9; Figure 10; Figure 11; Figure 12; and Figure 13. All survival curve plots were produced in Stata software version 14 (Stata 2015) using data from all trials providing IPD combined.
4.

Time to treatment failure (for any reason related to treatment)
PHT= phenytoin; OXC=oxcarbazepine
5.

Time to treatment failure due to adverse events
PHT= phenytoin; OXC=oxcarbazepine. Y‐axis of the figure scaled down to a maximum of 0.3 as a small number of individuals failed treatment due to adverse events
6.
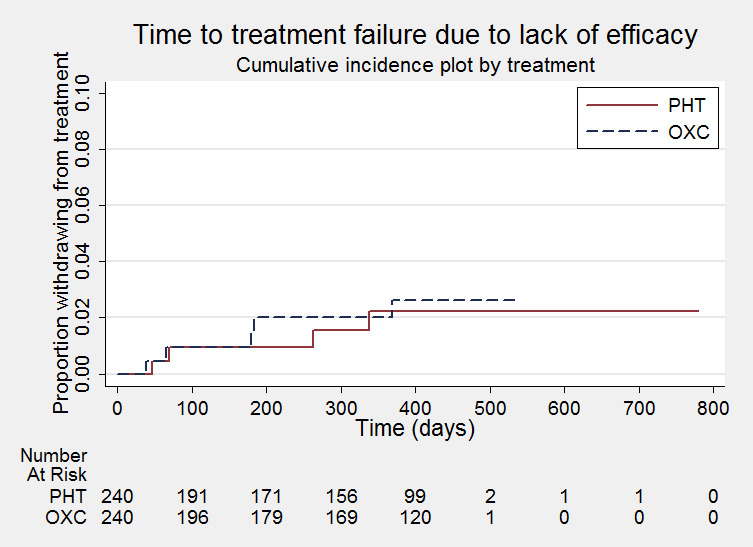
Time to treatment failure due to lack of efficacy
PHT= phenytoin; OXC=oxcarbazepine. Y‐axis of the figure scaled down to a maximum of 0.1 as a small number of individuals failed treatment due to lack of efficacy and censoring marks removed to allow steps (events) on the curve to be seen.
7.
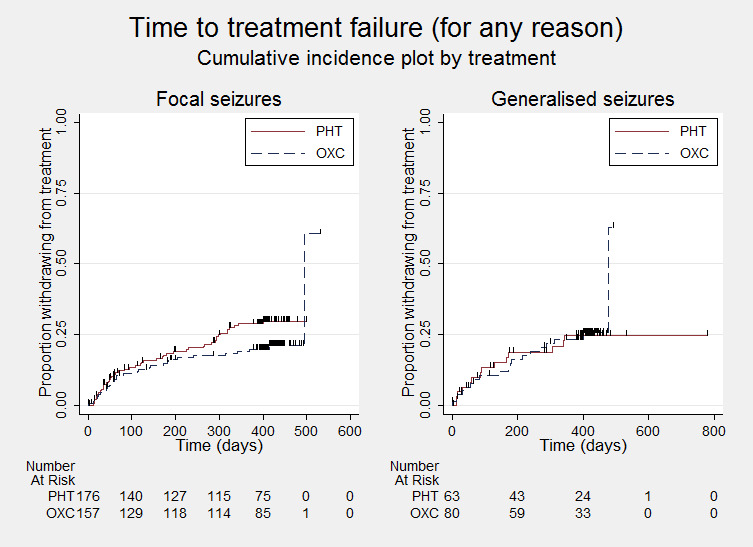
Time to treatment failure (for any reason related to treatment) ‐ by epilepsy type
PHT = phenytoin; OXC = oxcarbazepine
8.
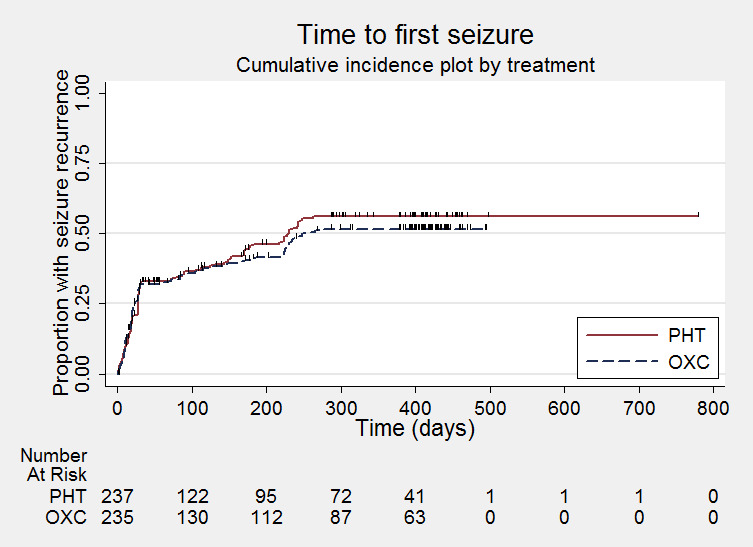
Time to first seizure post‐randomisation
PHT = phenytoin; OXC = oxcarbazepine
9.

Time to first seizure post‐randomisation ‐ by epilepsy type
PHT = phenytoin; OXC = oxcarbazepine
10.

Time to 12‐month remission
PHT = phenytoin; OXC = oxcarbazepine
11.
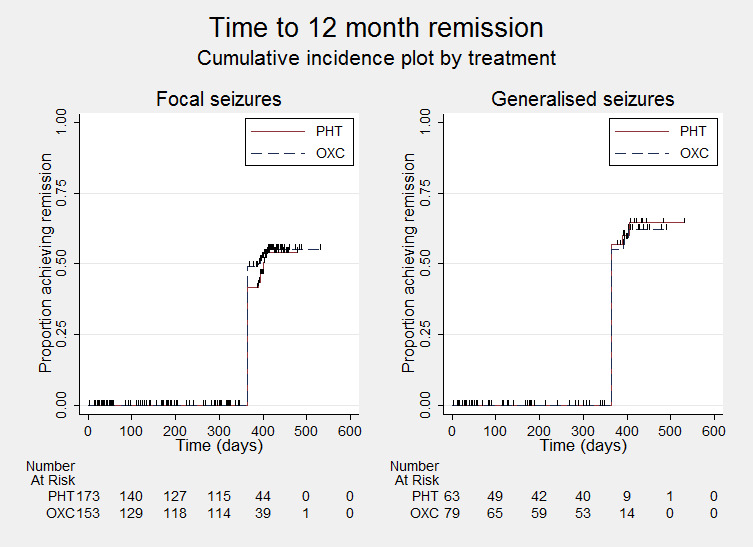
Time to 12‐month remission ‐ by epilepsy type
PHT = phenytoin; OXC = oxcarbazepine
12.

Time to six‐month remission
PHT = phenytoin; OXC = oxcarbazepine
13.

Time to six‐month remission ‐ by epilepsy type
PHT= phenytoin; OXC=oxcarbazepine
We note that participants with event times of zero (i.e. those who experienced treatment failure or experienced seizure recurrence on the day of randomisation) are not included in the 'Numbers at risk' on the graphs and that data are not stratified by trial within these survival curve plots. All figures are intended to provide a visual representation of outcomes, extent of follow‐up and visual differences between seizure types. These graphs are not intended to show statistical significance and numerical values may vary compared to the text due to differences in methodology.
We calculated all hazard ratios (HRs) presented below by generic inverse variance fixed‐effect meta‐analysis unless otherwise stated. All analyses met the assumption of proportional hazards (the addition of a time‐varying covariate into the model was non‐significant).
Primary outcome
Time to treatment failure (retention time)
For this outcome, a HR of less than one indicates a clinical advantage for oxcarbazepine.
Times to treatment failure and reasons for treatment failure were available for all 480 participants from the two trials providing IPD (100% of IPD available). See Table 3 for reasons for premature discontinuation of treatment (treatment failure) by treatment and how we classified these reasons in analysis.
In Bill 1997, of the 287 participants who were randomised (143 to oxcarbazepine and 144 to phenytoin), 117 participants discontinued prematurely from the trial (40.4%); 56 (39.2%) in the oxcarbazepine group and 61 (42.2%) in the phenytoin group. Of these participants, 28 (12 on oxcarbazepine and 16 on phenytoin) discontinued during the eight‐week titration period. An additional 89 participants (44 on oxcarbazepine and 45 on phenytoin) discontinued during the 48‐week maintenance period.
In Guerreiro 1997, of the 193 participants who were randomised (97 to oxcarbazepine and 96 to phenytoin), 58 participants discontinued prematurely from the trial (30.1%); 24 (24.7%) in the oxcarbazepine group and 34 (35.4%) in the phenytoin group. Of these participants, 14 (6 on oxcarbazepine and 8 on phenytoin) discontinued during the eight‐week titration period. An additional 44 participants (18 on oxcarbazepine and 26 on phenytoin) discontinued during the 48‐week maintenance period.
Therefore in total, 175 participants prematurely withdrew from treatment (36.5% of 480 participants): 80 out of 240 participants randomised to oxcarbazepine (33.3%) and 95 out of 240 participants randomised to phenytoin (39.6%).
We deemed 113 participants (64.6% of 175 treatment failures) to have withdrawn for reasons related to the study drug: 50 (62.5%) on oxcarbazepine and 63 (66.3%) on phenytoin, and we classed these reasons as 'events' in analysis. The most common treatment‐related reasons for treatment failure were:
non‐compliance with treatment (or protocol violation, or both), which accounted for 65 withdrawals (37.1% of total treatment failures), 38 (47.5% of total treatment failures) on oxcarbazepine and 27 (28.4% of total treatment failures) on phenytoin; and
adverse events: 39 withdrawals (22.3% of total treatment failures), 7 (8.8% of total treatment failures) on oxcarbazepine and 32 (33.7% of total treatment failures) on phenytoin.
Only nine participants (1.8%) across the two trials withdrew due lack of efficacy (i.e. continued seizures).
We classed the other 62 reasons (30 on oxcarbazepine and 32 on phenytoin), which were mostly losses to follow‐up (53 participants lost to follow‐up, 11% of other withdrawals), to be not related to the treatment and we censored these participants in the analysis, in addition to the 305 participants (160 on oxcarbazepine and 145 on phenytoin) who completed the trial without withdrawing or failing treatment.
Time to treatment failure for any treatment related reason
The overall pooled HR for this outcome (for 480 participants providing IPD from two trials) was 0.77 (95% confidence interval (CI) 0.53 to 1.12, P=0.17, high‐quality evidence; Analysis 1.1), indicating a potential advantage with oxcarbazepine (i.e. treatment failure may occur later with oxcarbazepine than phenytoin) which is not statistically significant. No important heterogeneity was present between trials (I² = 4%).
1.1. Analysis.
Comparison 1 Oxcarbazepine versus phenytoin, Outcome 1 Time to treatment failure (any reason related to the treatment).
Time to treatment failure due to adverse events
Considering time to treatment failure due to adverse events (all other reasons for treatment failure or treatment withdrawal censored in analysis), the overall pooled HR (for 480 participants providing IPD from two trials) was 0.22 (95% CI 0.10 to 0.51, P = 0.0004, high‐quality evidence, Analysis 1.2), indicating a statistically significant advantage with oxcarbazepine (i.e. treatment failure due to adverse events occurs significantly later with oxcarbazepine than phenytoin). No heterogeneity was present between trials (I²= 0%).
1.2. Analysis.
Comparison 1 Oxcarbazepine versus phenytoin, Outcome 2 Time to treatment failure due to adverse events.
Time to treatment failure due to lack of efficacy
Considering time to treatment failure due to lack of efficacy (all other reasons for treatment failure or treatment withdrawal censored in analysis), the overall pooled HR (for 480 participants providing IPD from two trials) was 1.17 (95% CI 0.31 to 4.35, P = 0.82, moderate‐quality evidence, Analysis 1.3). As the pooled HR is quite imprecise, it is unclear whether there is an advantage to either drug, or no difference between drugs. No heterogeneity was present between trials (I² = 0%) and we note that the confidence intervals of this pooled HR are very wide because of the small number of participants across the two trials failing treatment due to lack of efficacy, and therefore a small number of events within the analysis (see Table 3).
1.3. Analysis.
Comparison 1 Oxcarbazepine versus phenytoin, Outcome 3 Time to treatment failure due to lack of efficacy.
Subgroup analyses: seizure type (focal versus generalised onset)
For four participants, information on the type of seizures was not available (one participant in Bill 1997, and three in Guerreiro 1997); we could not classify their epilepsy type and so we did not include these participants in the subgroup analyses.
Considering time to treatment failure for any reason related to the treatment, the overall pooled HR (adjusted by epilepsy type for 476 participants from two trials) was 0.78 (95% CI 0.53 to 1.14, P = 0.20, moderate‐quality evidence, see Analysis 1.4). This result is similar to the unadjusted pooled HR (Analysis 1.1) and conclusions remain unchanged following the exclusion of four individuals with missing epilepsy type. Again, no important heterogeneity was present between trials (I² = 10%).
1.4. Analysis.
Comparison 1 Oxcarbazepine versus phenytoin, Outcome 4 Time to treatment failure (any reason related to the treatment) ‐ by epilepsy type.
For individuals with focal onset seizures (333 participants from two trials), the pooled HR was 0.69 (95% CI 0.43 to 1.09, P = 0.11, moderate‐quality evidence, Analysis 1.4), indicating a potential advantage with oxcarbazepine which is not statistically significant. A moderate amount of heterogeneity was present between the two studies for individuals with focal onset seizures (I² = 35%) and when we repeated the analysis using a random‐effects model, the numerical results were relatively similar and the results unchanged (pooled HR 0.67, 95% CI 0.37 to 1.20; P = 0.17). For individuals with generalised onset seizures (143 participants from two trials), the pooled HR was 1.03 (95% CI 0.51 to 2.08, P = 0.93, low‐quality evidence, Analysis 1.4). There are wide confidence intervals around the pooled HR so it is unclear whether there is an advantage to either drug, or no difference between drugs. No heterogeneity was present between trials (I² = 0%).
There was no statistically significant evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (test of subgroup differences: P = 0.34; I² statistic for variability due to subgroup differences = 0%, Analysis 1.4).
We were unable to perform subgroup analysis for 'time to treatment failure due to adverse events' and 'time to treatment failure due to lack of efficacy' due to small numbers of participants with each epilepsy type failing treatment for these reasons (see Table 3).
Sensitivity analysis: only data from the double‐blind period of 392 days
Five participants had a recorded time of treatment failure greater than 392 days (four in Bill 1997, and one in Guerreiro 1997); two with generalised onset seizures and three with focal onset seizures. When values greater than 392 days were censored in the analyses (adjusted for epilepsy type), the numerical results were very similar to the uncensored analyses and conclusions were unchanged (see Table 6).
4. Results of sensitivity analyses.
| Analysisa |
Time to treatment failure (for any reason related to treatment) |
Time to first seizure | Time to 12‐month remission | Time to 6‐month remission | |
| Original analysis (adjusted for epilepsy type) |
Participants | Overall:476 (Foc: 333; Gen: 143) |
Overall: 468 (Foc: 326; Gen: 142) |
Overall: 468 (Foc: 326; Gen: 142) |
Overall: 468 (Foc: 326; Gen: 142) |
|
Pooled HR (95% CI), P value, I² (%) |
Foc: 0.69 (0.43 to 1.09) P=0.11, I² = 35% Gen: 1.03 (0.51 to 2.08) P=0.93, I² = 0% Overall: 0.78 (0.53 to 1.14) P=0.20, I² = 10% |
Foc: 0.92 (0.68 to 1.25), P=0.61, I² = 0% Gen: 1.11 (0.66 to 1.86), P=0.69, I² = 0% Overall: 0.97 (0.75 to 1.26), P=0.81, I² = 0% |
Foc: 1.09 (0.75 to 1.57), P=0.66, I² = 0% Gen: 0.94 (0.55 to 1.62), P=0.83, I² = 0% Overall: 1.04 (0.77 to 1.41), P=0.81, I² = 0% |
Foc: 1.17 (0.87 to 1.58), P=0.29, I² = 0% Gen: 0.83 (0.53 to 1.31), P=0.43, I² = 0% Overall: 1.06 (0.82 to 1.36), P=0.65, I² = 0% |
|
| Test of subgroup differences | P = 0.34, I² = 0% | P = 0.55, I² =0% | P = 0.67, I² = 0% | P = 0.21, I² = 35.8% | |
| Sensitivity analysis ‐ events in the double‐blind period only (events censored at 392 days ‐ 56 weeks)b |
Participants | Overall:476 (Foc: 333; Gen: 143) |
Unchanged from original analysis (no first seizure events in the open label phase) |
Overall: 468 (Foc: 326; Gen: 142) |
Unchanged from original analysis (no six‐month remission events in the open label phase) |
|
Pooled HR (95% CI), P value, I² (%) |
Foc: 0.68 (0.43 to 1.09) P=0.11, I² = 34% Gen: 0.92 (0.45 to 1.88) P=0.82 I² = 0% Overall: 0.75 (0.51 to 1.10) P=0.14, I² = 0% |
Foc: 1.19 (0.81 to 1.76), P = 0.37, I² = 0% Gen: 0.92 (0.53 to 1.61), P = 0.91, I² = 0% Overall: 1.10 (0.80 to 1.51), P = 0.57, I² = 0% |
|||
| Test of subgroup differences | P = 0.49, I² = 0% | P = 0.46, I² = 0% | |||
| Sensitivity analysis classifying generalised onset seizures and age at onset > 30 classified as focal onset seizures |
Participants | Overall:476 (Foc: 363; Gen: 113) |
Overall: 468 (Foc: 355; Gen: 113) |
Overall: 468 (Foc: 355; Gen: 113) |
Overall: 468 (Foc: 355; Gen: 113) |
|
Pooled HR (95% CI), P value, I² (%) |
Foc: 0.80 (0.52 to 1.24) P=0.32, I² = 66% Gen: 0.67 (0.31 to 1.46) P=0.32, I² = 58% Overall: 0.77 (0.52 to 1.13) P=0.18, I² = 45% |
Foc: 0.87 (0.65 to 1.17), P = 0.36, I² = 0% Gen: 1.44 (0.79 to 2.62), P = 0.23, I² = 0% Overall: 0.97 (0.73 to 1.28), P = 0.82, I² = 7% |
Foc: 1.12 (0.79 to 1.59), P = 0.53, I² = 0% Gen: 0.75 (0.40 to 1.41), P = 0.37, I² = 0% Overall: 1.02 (0.75 to 1.38), P = 0.91, I² = 0% |
Foc: 1.19 (0.90 to 1.58), P = 0.23, I² = 0% Gen: 0.72 (0.43 to 1.23), P = 0.23, I² = 0% Overall: 1.06 (0.82 to 1.37), P = 0.66, I² = 5% |
|
| Test of subgroup differences | P = 0.70, I² = 0% | P = 0.14, I² = 54% | P = 0.28, I² = 14.9% | P = 0.10, I² = 62.4% | |
| Sensitivity analysis classifying generalised onset seizures and age at onset > 30 classified as uncertain epilepsy type |
Participants | Overall:476 (Foc: 333; Gen: 113; Unc: 30) |
Overall: 468 (Foc: 326; Gen: 113; Unc: 29) |
Overall: 468 (Foc: 326; Gen: 113; Unc: 29) |
Overall: 468 (Foc: 326; Gen: 113; Unc: 29) |
|
Pooled HR (95% CI), P value, I² (%) |
Foc: 0.69 (0.43 to 1.09) P=0.11, I² = 35% Gen: 0.67 (0.31 to 1.46) P=0.32, I² = 58% Unc: 6.23 [0.73, 53.44] P=0.09, I² = NA% Overall: 0.73 [0.50, 1.08] P=0.12, I² = 49% |
Foc: 0.92 (0.68 to 1.25), P=0.61, I² = 0% Gen: 1.44 (0.79 to 2.62), P = 0.23, I² = 0% Unc: 0.38 (0.10 to 1.44), P=0.16, I² =NA Overall: 0.97 (0.75 to 1.27), P=0.85, I² =18% |
Foc: 1.09 (0.75 to 1.57), P=0.66, I² = 0% Gen: 0.75 (0.40 to 1.41), P = 0.37, I² = 0% Unc: 2.05 (0.68 to 6.17), P = 0.20, I² = NA Overall: 1.05 (0.77 to 1.42), P = 0.77, I² = 0% |
Foc: 1.17 (0.87 to 1.58), P=0.29, I² = 0% Gen: 0.72 (0.43 to 1.23), P = 0.23, I² = 0% Unc: 1.51 (0.59 to 3.86), P=0.40, I² = NA Overall: 1.07 (0.83 to 1.38), P = 0.48, I² = 0% |
|
| Test of subgroup differences | P = 0.14, I² = 49.2% | P = 0.16, I² = 45.5% | P = 0.77, I² = 21.4% | P = 0.22, I² = 33.2% | |
CI: confidence interval; Foc: focal onset seizures; Gen=generalised onset seizures; HR: hazard ratio; NA: Not applicable, Unc= uncertain seizure type.
a. For time to treatment failure and time to first seizure, HR < 1 indicates a clinical advantage for oxcarbazepine and for time to 12‐month and 6‐month remission, HR < 1 indicates a clinical advantage for phenytoin. All results presented are calculated from fixed‐effect meta‐analysis.
b. Five participants with time to treatment failure greater than 392 days (within open‐label treatment phase); two with generalised epilepsy and three with focal epilepsy. Fifteen participants with time to 12‐month remission greater than 392 days (within open‐label treatment phase); three with generalised epilepsy and 12 with focal epilepsy. No participants with six‐month remission achieved or first seizure recorded in the open‐label treatment phase.
Sensitivity analysis: reclassification of epilepsy type
In the sensitivity analyses to investigate misclassification of epilepsy type, we reclassified the 30 participants aged 30 or older with new onset generalised seizures from one study (Bill 1997), to focal onset seizures or an uncertain seizure type. Overall the numerical results (adjusted by epilepsy type) were quite similar, but substantially more heterogeneity was present between the studies in both analyses compared to the original analysis (Analysis 1.4) and some numerical results within the subgroups by epilepsy type are quite different (see Table 6).
Following reclassification, for the smaller subgroup of individuals with generalised onset seizures and age of onset of 30 years or less (113 participants from two trials), the pooled HR was 0.67 (95% CI 0.31 to 1.46; P = 0.32; I²= 58%, see Table 6, Analysis 1.5 and Analysis 1.6), indicating a potential advantage with oxcarbazepine which is not statistically significant. When we repeated the analysis using a random‐effects model, the pooled HR was 0.85 (95% CI 0.21 to 3.46; P = 0.82), indicating no clear advantage to either drug (Analysis 1.5).
1.5. Analysis.
Comparison 1 Oxcarbazepine versus phenytoin, Outcome 5 Time to treatment failure (any reason related to treatment) ‐ sensitivity analysis: epilepsy type reclassified to focal onset for generalised onset and age > 30 years.
1.6. Analysis.
Comparison 1 Oxcarbazepine versus phenytoin, Outcome 6 Time to treatment failure (any reason related to treatment) ‐ sensitivity analysis: epilepsy type reclassified to uncertain seizure type for generalised onset and age > 30 years.
For the larger subgroup of individuals with focal onset seizures (including the 30 participants aged 30 or older with new onset generalised seizures, a total of 363 participants from two trials), the pooled HR was 0.80 (0.52 to 1.24; P = 0.32; I²= 66%, see Table 6 and Analysis 1.5), indicating a potential advantage with oxcarbazepine which is not statistically significant. When we repeated the analysis using a random‐effects model, the pooled HR was 0.74 (95% CI 0.34 to 1.61; P = 0.44), so conclusions were unchanged.
For the 30 participants aged 30 or older with new onset generalised seizures, the HR is quite imprecise so it is unclear whether there is an advantage to either drug, or no difference between drugs (6.23, 95% CI 0.73 to 53.44, P = 0.09; see Table 6 and Analysis 1.6). As in the original analysis, there was also no statistically significant evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect in either of the sensitivity analyses (see Table 6, Analysis 1.5 and Analysis 1.6 for P values of test for subgroup differences). However, in the sensitivity analysis reclassifying to uncertain seizure type, the I² statistic for variability due to subgroup differences increased from 0% to I² = 49.2%, suggesting some potential differences in treatment effect by epilepsy type following reclassification of epilepsy type.
We were unable to perform sensitivity analyses for 'time to treatment failure due to adverse events' and 'time to treatment failure due to lack of efficacy' due to small numbers of participants with each epilepsy type failing treatment for this reason (see Table 3).
Secondary outcomes
Time to first seizure post‐randomisation
For this outcome, a HR of less than one indicates a clinical advantage for oxcarbazepine.
Data for 472 participants (98.3% of those providing IPD) from two trials were available for the analysis of this outcome. This outcome could not be calculated for eight participants (five from Bill 1997, and three from Guerreiro 1997), due to missing data (no data for mean frequency of seizures in the maintenance period as well as the titration period; number of days on trial medication ranged between one and 36 days for these eight participants). Analyses could not be performed with an intention‐to‐treat approach as participants who withdrew from both studies were not followed up after time of treatment failure.
The overall pooled HR (for 472 participants providing IPD from two trials) was 0.93 (95% CI 0.72 to 1.21, P = 0.60, moderate‐quality evidence, Analysis 1.7), indicating no clear advantage of either drug. No heterogeneity was present between trials (I² = 0%).
1.7. Analysis.
Comparison 1 Oxcarbazepine versus phenytoin, Outcome 7 Time to first seizure.
Subgroup analyses: seizure type (focal versus generalised onset)
For four participants, information on the type of seizures was not available (one participant in Bill 1997, and three in Guerreiro 1997); we could not classify their epilepsy type and so we did not include these participants in the subgroup analyses.
The overall pooled HR (adjusted by epilepsy type for 468 participants from two trials) was 0.97 (95% CI 0.75 to 1.26, P = 0.81, moderate‐quality evidence, see Analysis 1.8). This result is similar to the unadjusted pooled HR (Analysis 1.7) and conclusions remain unchanged following the exclusion of four individuals with missing epilepsy type. No heterogeneity was present between trials (I² = 0%).
1.8. Analysis.
Comparison 1 Oxcarbazepine versus phenytoin, Outcome 8 Time to first seizure ‐ by epilepsy type.
For individuals with focal onset seizures (326 participants from two trials), the pooled HR was 0.92 (95% CI 0.68 to 1.25, P = 0.61, moderate‐quality evidence, Analysis 1.8), indicating no clear advantage of either drug. No heterogeneity was present between trials (I² = 0%). For individuals with generalised onset seizures (142 participants from two trials), the pooled HR was 1.11 (95% CI 0.66 to 1.86, P = 0.69, low‐quality evidence, Analysis 1.8), indicating no clear advantage for either drug. No heterogeneity was present between trials (I² = 0%).
There was no statistically significant evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (test of subgroup differences: P = 0.55, I² statistic for variability due to subgroup differences = 0% (Analysis 1.8).
Sensitivity analysis: only data from the double‐blind period of 392 days
No participants had a time of first seizure greater than 392 days into the follow‐up time, therefore results were identical for the analysis of the full follow‐up and for the double‐blind period only (see Table 6).
Sensitivity analysis: reclassification of epilepsy type
In the sensitivity analyses to investigate misclassification of epilepsy type, we reclassified the 29 participants aged 30 or older with new onset generalised seizures from one study (Bill 1997), to focal onset seizures or an uncertain seizure type. The numerical results overall and for individuals with focal onset seizures were similar to the original analyses and conclusions were unchanged.
Following reclassification, for the smaller group of individuals with generalised onset seizures (113 participants from two trials), the pooled HR was 1.44, 95% CI 0.79 to 2.62, P = 0.23, I² = 0%, suggesting a potential advantage with phenytoin (i.e. first seizure recurrence may occur later on phenytoin than oxcarbazepine), but this potential advantage is quite imprecise so an advantage with oxcarbazepine, or no difference between drugs, cannot be ruled out. Also for the 29 participants aged 30 or older with new onset generalised seizures, the HR is quite imprecise so it is unclear whether there is an advantage to either drug, or no difference between drugs (0.38, 95% CI 0.10 to 1.44, P = 0.16) (see Table 6).
Within the original analysis, there was no evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (see Analysis 1.8). However, within the two sensitivity analyses, while there was no statistically significant evidence of an interaction, the I² statistics for variability due to subgroup differences were relatively high (I² = 54% and 45.5% respectively), suggesting some potential differences in treatment effect by epilepsy type following reclassification of epilepsy type (see Table 6).
Time to achieve 12‐month remission
For this outcome, a HR of less than one indicates a clinical advantage for phenytoin.
Data for 472 participants (98.3% of those providing IPD) from two trials were available for the analysis of this outcome. We could not calculate this outcome for eight participants (five from Bill 1997, and three from Guerreiro 1997) due to missing data (no data for mean frequency of seizures in the maintenance period as well as the titration period; number of days on trial medication ranged between one and 36 days for these eight participants). Analyses could not be performed with an intention‐to‐treat approach as participants who withdrew from both studies were not followed up after time of treatment failure.
The overall pooled HR (for 472 participants providing IPD from two trials) was 1.09 (95% CI 0.80 to 1.47, P = 0.58, moderate‐quality evidence, Analysis 1.9), indicating no clear advantage of either drug. No heterogeneity was present between trials (I² = 0%).
1.9. Analysis.
Comparison 1 Oxcarbazepine versus phenytoin, Outcome 9 Time to achieve 12‐month remission.
Subgroup analyses: seizure type (focal versus generalised onset)
For four participants, information on the type of seizures was not available (one participant in Bill 1997, and three in Guerreiro 1997); we therefore could not classify their epilepsy type and so we did not include these participants in the subgroup analyses.
The overall pooled HR (adjusted by epilepsy type for 468 participants from two trials) was 1.04 (95% CI 0.77 to 1.41, P = 0.81, moderate‐quality evidence, see Analysis 1.10). This result is similar to the unadjusted pooled HR (Analysis 1.9) and conclusions remain unchanged following the exclusion of four individuals with missing epilepsy type. No heterogeneity was present between trials (I² = 0%).
1.10. Analysis.
Comparison 1 Oxcarbazepine versus phenytoin, Outcome 10 Time to achieve 12‐month remission ‐ by epilepsy type.
For individuals with focal onset seizures (326 participants from two trials), the pooled HR was 1.09 (95% CI 0.75 to 1.57, P = 0.66, moderate‐quality evidence, Analysis 1.10), indicating no clear advantage of either drug. No heterogeneity was present between trials (I² = 0%). For individuals with generalised onset seizures (142 participants from two trials), the pooled HR was 0.94 (95% CI 0.55 to 1.62, P = 0.83, low‐quality evidence, Analysis 1.10), indicating no clear advantage of either drug. No heterogeneity was present between trials (I² = 0%).
There was no statistically significant evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (test of subgroup differences: P = 0.67, I² statistic for variability due to subgroup differences = 0% (Analysis 1.10).
Sensitivity analysis: only data from the double‐blind period of 392 days
Fifteen participants had a time to 12‐month remission greater than 392 days (nine participants in Bill 1997, and six in Guerreiro 1997); three with generalised onset seizures and 12 with focal onset seizures. When we censored values greater than 392 days in analyses (adjusted for epilepsy type), numerical results overall and for individuals with generalised onset seizures were very similar to the original analyses and conclusions were unchanged. For individuals with focal onset seizures there was a slight, but not statistically significant, potential advantage with oxcarbazepine (i.e. 12‐month remission may occur earlier on oxcarbazepine than phenytoin), compared to the original analysis which showed no clear advantage to either drug (see Table 6).
Sensitivity analysis: reclassification of epilepsy type
In the sensitivity analyses to investigate misclassification of epilepsy type, we reclassified the 29 participants aged 30 or older with new onset generalised seizures from one study (Bill 1997), to focal onset seizures or an uncertain seizure type. The numerical results overall and for individuals with focal onset seizures were similar to the original analyses and conclusions were unchanged.
Following reclassification, for the smaller group of individuals with generalised onset seizures (113 participants from two trials), the pooled HR was 0.75, 95% CI 0.40 to 1.41, P = 0.37, I² = 0%, suggesting a potential advantage with phenytoin (i.e. 12‐month remission may occur earlier with phenytoin than oxcarbazepine). For the 29 participants aged 30 or older with new onset generalised seizures, the HR is quite imprecise so it is unclear whether there is an advantage to either drug, or no difference between drugs (2.05, 95% CI 0.68 to 6.17, P = 0.20) (see Table 6). As in the original analysis, there is also no statistically significant evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (see Table 6).
Time to achieve six‐month remission
For this outcome, a HR less than one indicates a clinical advantage for phenytoin.
Data for 472 participants (98.3% of those providing IPD) from two trials were available for the analysis of this outcome. We could not calculate this outcome for eight participants (five from Bill 1997, and three from Guerreiro 1997) due to missing data (no data for mean frequency of seizures in the maintenance period as well as the titration period; number of days on trial medication ranged between one and 36 days for these eight participants). Analyses could not be performed with an intention‐to‐treat approach as participants who withdrew from both studies were not followed up after time of treatment failure.
The overall pooled HR (for 472 participants providing IPD from two trials) was 1.12 (95% CI 0.87 to 1.43, P = 0.38, moderate‐quality evidence, Analysis 1.12), indicating no clear advantage to either drug. No heterogeneity was present between trials (I² = 0%).
1.12. Analysis.
Comparison 1 Oxcarbazepine versus phenytoin, Outcome 12 Time to achieve 6‐month remission ‐ by epilepsy type.
Subgroup analyses: seizure type (focal versus generalised onset)
For four participants, information on the type of seizures was not available (one participant in Bill 1997, and three in Guerreiro 1997); therefore we could not classify their epilepsy type and so we did not include these participants in the subgroup analyses.
The overall pooled HR (adjusted by epilepsy type for 468 participants from two trials) was 1.06 (95% CI 0.82 to 1.36, P = 0.65, moderate‐quality evidence, see Analysis 1.12). This result is similar to the unadjusted pooled HR (Analysis 1.11) and conclusions remain unchanged following the exclusion of four individuals with missing epilepsy type. No heterogeneity was present between trials (I² = 0%).
1.11. Analysis.
Comparison 1 Oxcarbazepine versus phenytoin, Outcome 11 Time to achieve 6‐month remission.
For individuals with focal onset seizures (326 participants from two trials), the pooled HR was 1.17 (95% CI 0.87 to 1.58, P = 0.29, moderate‐quality evidence, Analysis 1.12), suggesting a slight potential advantage with oxcarbazepine (i.e. that six‐month remission may occur earlier on oxcarbazepine than phenytoin) which is not statistically significant. No heterogeneity was present between trials (I² = 0%). For individuals with generalised onset seizures (142 participants from two trials), the pooled HR was 0.83 (95% CI 0.53 to 1.31, P = 0.43, low‐quality evidence, Analysis 1.12), suggesting a slight potential advantage with phenytoin. No heterogeneity was present between trials (I²= 0%).
There was no statistically significant evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (test of subgroup differences: P = 0.21, I² statistic for variability due to subgroup differences = 35.8%, Analysis 1.12).
Sensitivity analysis: only data from the double‐blind period of 392 days
No participants achieved six‐month remission after 392 days into the follow‐up time, therefore results were identical for the analysis of the full follow‐up and for the double‐blind period only (see Table 6).
Sensitivity analysis: reclassification of epilepsy type
In the sensitivity analyses to investigate misclassification of epilepsy type, we reclassified the 29 participants aged 30 or older with new onset generalised seizures from one study (Bill 1997), to focal onset seizures or an uncertain seizure type. Numerical results overall and for individuals with focal onset seizures were similar to the original analyses and conclusions were unchanged.
Following reclassification, the smaller group of individuals with generalised onset seizures (113 participants from two trials), the pooled HR was 0.72, 95% CI 0.43 to 1.23, P = 0.23, I² = 0%, suggesting a potential advantage with phenytoin (i.e. six‐month remission may occur earlier with phenytoin than oxcarbazepine). For the 29 participants aged 30 or older with new onset generalised seizures, the HR is quite imprecise so it is unclear whether there is an advantage to either drug, or no difference between drugs (1.51, 95% CI 0.59 to 3.86, P = 0.40) (see Table 6).
Within the original analysis, there was no statistically significant evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (see Analysis 1.12) and within the sensitivity analysis of reclassification to uncertain seizure type (test of subgroup differences: P = 0.22, I² statistic for variability due to subgroup differences = 33.2%, see Table 6). However, within the sensitivity analysis of reclassification to focal onset seizures, while there was no statistically significant evidence of an interaction (P = 0.10), the I² statistics for variability due to subgroup differences were relatively high in the sensitivity analyses (I² = 62.4%), suggesting some potential differences in treatment effect by epilepsy type following reclassification of epilepsy type (see Table 6).
Incidence of adverse events
See Table 7 for details of all adverse event data provided in the studies included in this review. We note that adverse event information was not included in IPD requests conducted for the original version of this review, therefore we extracted information on adverse events from the published reports of two studies (Bill 1997; Guerreiro 1997). No adverse event information was provided in Aikia 1992.
5. Adverse events reported.
| Adverse event | Bill 1997 | Guerreiro 1997 | Total | ||||
| OXC | PHT | OXC | PHT | OXC | PHT | Total | |
| Somnolence | 41 | 41 | 24 | 28 | 65 | 69 | 134 |
| Headache | 20 | 27 | 13 | 14 | 33 | 41 | 74 |
| Dizziness | 18 | 22 | 9 | 21 | 27 | 43 | 70 |
| Gum Hyperplasia | 2 | 18 | 2 | 24 | 4 | 42 | 46 |
| Nausea | 13 | 16 | 5 | 7 | 18 | 23 | 41 |
| Rash | 12 | 16 | 4 | 5 | 16 | 21 | 37 |
| Nervousness | 2 | 9 | 2 | 11 | 4 | 20 | 24 |
| Apathy | 0 | 0 | 11 | 10 | 11 | 10 | 21 |
| Tremor | 4 | 10 | 0 | 0 | 4 | 10 | 14 |
| Ataxia | 0 | 0 | 0 | 13 | 0 | 13 | 13 |
| Diplopia | 1 | 11 | 0 | 0 | 1 | 11 | 12 |
| Acne | 9 | 3 | 0 | 0 | 9 | 3 | 12 |
| Nystagmus | 3 | 8 | 0 | 0 | 3 | 8 | 11 |
| Abnormal thinking | 0 | 0 | 5 | 6 | 5 | 6 | 11 |
| Abdominal pain | 0 | 0 | 5 | 4 | 5 | 4 | 9 |
| Hypertricosis | 0 | 0 | 0 | 8 | 0 | 8 | 8 |
| Vomiting | 0 | 0 | 0 | 5 | 0 | 5 | 5 |
Overall, most adverse events were reported in more participants taking phenytoin than those taking oxcarbazepine (although we did not compare the rates of adverse events by drug statistically). The most common adverse events reported in more than 10% of participants on either drug were as follows.
Somnolence: reported by 134 participants on both drugs (28% of total participants), with similar rates across the two drugs.
Headache: reported by 74 participants on both drugs (15% of total participants), with similar rates across the two drugs.
Dizziness: reported by 70 participants on both drugs (14.5% of total participants), reported by slightly more participants on phenytoin (18%) than oxcarbazepine (11%).
Gum hyperplasia: reported by 46 participants on both drugs (9.5% of total participants), reported by substantially more participants on phenytoin (18%) than oxcarbazepine (2%).
Additional specific adverse events reported in less than 5% of total participants (which were reported by slightly more participants on phenytoin than oxcarbazepine) were: tremor, ataxia, diplopia, hypertrichosis and vomiting. Slightly more participants reported acne on oxcarbazepine than phenytoin.
Discussion
Summary of main results
In this review we included individual participant data (IPD) from 480 out of 517 participants (93%), from two out of three trials in which participants were randomised to either oxcarbazepine or phenytoin.
Analyses of all participants (without adjustment for epilepsy type) indicated that treatment failure due to adverse events occurs significantly later with oxcarbazepine than phenytoin (pooled hazard ratio (HR) 0.22, 95% confidence interval (CI) 0.10 to 0.51, P = 0.0004, high‐quality evidence). The analyses also showed no clear difference between the drugs in terms of time to treatment failure due to lack of efficacy (pooled HR 1.17, 95% CI 0.31 to 4.35, P = 0.82, moderate‐quality evidence), although relatively few participants withdrew from the two trials due to lack of efficacy.
Moderate‐quality evidence from analyses with and without adjustment for epilepsy type, as well as sensitivity analysis, suggested a potential clinical advantage with oxcarbazepine in terms of time to treatment failure for any reason related to treatment, though this effect was not statistically significant (pooled HR 0.78 (95% CI 0.53 to 1.14, P = 0.20). This non‐significant clinical advantage was also consistently observed for individuals with focal onset seizures (pooled HR 0.69, 95% CI 0.43 to 1.09, P = 0.11, moderate‐quality evidence), but results were more imprecise and less consistent following sensitivity analysis for individuals with generalised onset seizures (pooled HR 1.03 (95% CI 0.51 to 2.08, P = 0.93, low‐quality evidence). Substantial heterogeneity was introduced into analysis following reclassification of individuals aged 30 or older with new onset generalised seizures, and therefore it is difficult to make a conclusion regarding the comparative time to treatment failure of the two treatments due to variability and potential confounding from seizure misclassification.
The results of this review provide moderate‐ to low‐quality evidence for secondary efficacy outcomes (time to first seizure post‐randomisation and time to six‐ and 12‐month remission from seizures). No statistically significant differences were found overall or by epilepsy‐type subgroups, and no clear differences between drugs were observed overall. There are consistent trends in all of the analyses (including sensitivity analyses) indicating a potential advantage with oxcarbazepine for participants with focal onset seizures (i.e. first seizure recurrence may occur later and seizure remission may occur earlier on oxcarbazepine than phenytoin), but results are less consistent and more imprecise for individuals with generalised onset seizures, and the results for this epilepsy‐type subgroup may have been confounded by seizure misclassification.
In most cases, adverse events were reported in more participants on phenytoin than participants on oxcarbazepine (although we did not compare the rates of adverse events by drug statistically). The most common adverse events reported in more than 10% of participants on either drug were somnolence (28% of total participants, with similar rates across the two drugs), headache (15% of total participants, with similar rates across the two drugs), dizziness (14.5% of total participants, reported by slightly more participants on phenytoin (18%) than oxcarbazepine (11%)) and gum hyperplasia (reported by substantially more participants on phenytoin (18%) than oxcarbazepine (2%)).
Overall completeness and applicability of evidence
We have gratefully received IPD for 480 out of 517 participants (93%) from two trials (Bill 1997; Guerreiro 1997), but we were not able to make contact with the authors of a third eligible trial of 37 participants (Aikia 1992), and no relevant data were published within the trial report to include within the analyses of this review.
Both trials included in IPD meta‐analysis used adequate methods of randomisation and adequate methods of allocation concealment, both were double‐blinded and attrition rates (loss to follow‐up and exclusions) were similar in the oxcarbazepine and phenytoin groups, with slightly more participants failing or withdrawing from treatment in the phenytoin group compared to the oxcarbazepine group in both trials (see Table 3). The main difference between the trials is that one recruited adults whilst the other recruited children, which is a potential source of heterogeneity. The majority of participants (333, 70%) had focal onset seizures while 143 (30%) were classified as having generalised onset seizures. The follow‐up period in both trials was less than two years, which is relatively short given that epilepsy is a chronic condition often requiring many years of treatment. Other reviews within this series of pairwise IPD monotherapy reviews have included studies with over 10 years of follow‐up (Marson 2000; Nevitt 2017b; Nevitt 2018a; Nevitt 2018b; Nolan 2013c; Nevitt 2018c; Nolan 2016b).
A major methodological issue in both of the trials is that participants were no longer followed up after the allocated treatment was withdrawn, and hence had to be censored at the time of treatment failure for the analyses of seizure and remission outcomes. This failure to follow participants up after the failure or withdrawal of allocated treatment violates the principle of intention‐to‐treat and may bias the seizure and remission analyses, as treatment may have been withdrawn for differing reasons which may have lead to informative censoring. For these reasons the analyses of seizure and remission outcomes require cautious interpretation, although no statistically significant differences between treatments or between epilepsy types were found in any case.
Quality of the evidence
The two trials with IPD available for this review were generally of good quality (Bill 1997; Guerreiro 1997), however, both trials were at high risk of attrition bias because individuals who failed treatment were not followed up in terms of seizures and remission outcomes (see Incomplete outcome data (attrition bias) and Overall completeness and applicability of evidence). Furthermore, for Bill 1997, there was evidence that up to 29% of adult participants classified as experiencing generalised onset seizures may have had their epilepsy type wrongly classified; and sensitivity analyses show that misclassification may have had an impact on the conclusions drawn for individuals with generalised seizures and whether an interaction between treatment effect and epilepsy type exists.
The trial for which IPD was not provided was generally unclear regarding methodology employed for randomisation, allocation concealment and blinding and was also of high risk of attrition bias (Aikia 1992).
Overall, due to the documented methodological issues that may have introduced heterogeneity, biases and imprecision into our meta‐analyses, we rated the evidence provided in this review as moderate to low quality according to GRADE criteria (See Table 1 and Table 2). The limited evidence in this review is insufficient for this review alone to inform clinical decisions to use one drug over the other.
Potential biases in the review process
We were able to include IPD for 480 out of 517 participants (93%) from two out of three trials included in this review, and conducted all analyses as IPD analyses. Such an approach has many advantages, such as enabling the standardisation of definitions of outcomes across trials. Attrition and reporting biases can also be reduced as additional analyses can be performed and additional outcomes calculated from unpublished data. For the outcomes we used in this review that are of a time‐to‐event nature, an IPD approach is considered to be the 'gold standard' approach to analysis (Parmar 1998).
However, despite the advantages of this approach, for reasons out of our control we were not able to obtain IPD for 37 participants from one eligible trial and no aggregate data were available for our outcomes of interest in study publications; therefore, we had to exclude 7% of eligible participants from our analyses, which may have introduced bias to the review. However, given the relatively small amount of data excluded, and that no differences were found between treatments in terms of the outcomes measures in Aikia 1992, we do not believe that our conclusions would have changed had the IPD for this trial been available.
Finally, we made some assumptions in the statistical methodology used in this review. We received information regarding the mean number of seizures per week in the titration (eight weeks) and maintenance phases (48 weeks) for both trials but we did not receive precise dates of seizures. Using these data, we were able to interpolate the dates of seizures assuming a uniform distribution so that we could calculate the outcomes time to first seizure and time to six‐ and 12‐month remission. We are aware that an individual's seizure patterns may be non‐linear; therefore, we recommend caution when interpreting the numerical results of the seizure‐related outcomes.
Agreements and disagreements with other studies or reviews
To our knowledge, together with previous versions of this review, this is the only systematic review and meta‐analysis that compares oxcarbazepine and phenytoin monotherapy for focal onset seizures and generalised onset tonic‐clonic seizures. A network meta‐analysis has been published (Nevitt 2017a), which compares all direct and indirect evidence on oxcarbazepine, phenytoin, and other standard and new antiepileptic drugs licensed for monotherapy. The results of this review generally agree with the results of the network meta‐analysis; results of the network meta‐analysis showed no statistically significant differences between oxcarbazepine and phenytoin for any of the outcomes considered (treatment failure, seizure recurrence and seizure remission).
Authors' conclusions
Implications for practice.
Current UK guidelines recommend carbamazepine or lamotrigine as first‐line treatment for adults and children with new onset focal seizures and sodium valproate for adults and children with new onset generalised seizures (NICE 2012).
Evidence provided by this review for individuals with generalised onset seizures is of moderate to low quality and does not inform current treatment policy. For all individuals regardless of epilepsy type, high‐quality evidence provided by this review indicates that treatment failure due to adverse events occurs significantly later on oxcarbazepine than phenytoin. For individuals with focal onset seizures, moderate‐quality evidence provided by this review suggests that oxcarbazepine may be superior to phenytoin in terms of treatment failure for any reason, seizure recurrence and seizure remission. Therefore, where first‐line recommended treatments are not suitable for an individual and where an alternative treatment option is required, oxcarbazepine may be a preferable alternative treatment than phenytoin, particularly for individuals with focal onset seizures.
Implications for research.
This review highlights the need for comparative antiepileptic drug monotherapy trials that measure longer‐term outcomes, as well as the need to continue to follow participants up after randomised treatment has been withdrawn in order to comply with the principle of intention‐to‐treat and to avoid the problems of informative censoring. It is essential that the designs of future antiepileptic drug monotherapy trials aim to recruit individuals with specific epilepsy syndromes, and that future trials are powered to detect a difference between particular antiepileptic drugs. An approach likely to reflect and inform clinical practice, and be statistically powerful, would be to recruit heterogeneous populations for whom epilepsy syndromes have been adequately defined, with testing for interaction between treatment and epilepsy syndrome. In view of potential problems of misclassification, syndromes need to be well defined, with use of adequate checking mechanisms to ensure that classifications are accurate, and a system to recognise uncertainty surrounding epilepsy syndromes in individuals within trials.
The choice of outcomes at the design stage of a trial and the presentation of the results of outcomes, particularly of a time‐to‐event nature, require very careful consideration. While the majority of trials of a monotherapy design record an outcome measuring efficacy (seizure control) and an outcome measuring tolerability (adverse events), there is little uniformity between the definition of the outcomes and the reporting of the summary statistics related to the outcomes (Nolan 2013b), which precludes the use of an aggregate data approach to meta‐analysis in reviews of monotherapy. Where trial authors cannot or will not make individual participant data available for analysis, we are left with no choice but to exclude a proportion of relevant evidence from the review, which may impact upon the interpretation of the results of the review and the applicability of the evidence and conclusions. The International League Against Epilepsy recommends that trials of a monotherapy design should adopt a primary effectiveness outcome of time to treatment failure (i.e. retention time) and should be of at least 48 weeks' duration to allow for assessment of longer‐term outcomes, such as remission (ILAE 1998; ILAE 2006). If trials followed these recommendations, an aggregate data approach to meta‐analysis may be feasible, which would reduce the resources and time required from an individual participant data approach.
What's new
| Date | Event | Description |
|---|---|---|
| 20 August 2018 | New search has been performed | Searches updated 20 August 2018; we included no new studies and the conclusions are unchanged. |
| 1 August 2018 | New citation required but conclusions have not changed | The term "partial" has been replaced by "focal", in accordance with the most recent classification of epilepsies of the International League Against Epilepsy (Scheffer 2017). The lead author, previously Sarah Nolan, is now Sarah Nevitt. |
History
Protocol first published: Issue 2, 2002 Review first published: Issue 2, 2006
| Date | Event | Description |
|---|---|---|
| 10 February 2015 | New search has been performed | Searches updated 3 February 2013; no new studies identified. Conclusions unchanged |
| 11 December 2014 | Amended | Title changed to specify that the review uses individual participant data |
| 12 March 2013 | New search has been performed | Searches updated 22 January 2013; one new trial included (Aikia 1992). Conclusions remain unchanged. |
| 22 January 2013 | New citation required but conclusions have not changed | Analyses and text updated. 'Risk of bias' assessments and 'Summary of findings' table added. |
| 7 September 2010 | Amended | Contact author's details updated. |
| 7 August 2009 | Amended | Copy edits made at editorial base. |
| 24 October 2008 | Amended | Search strategy amended to comply with RevMan 5. |
| 12 August 2008 | New search has been performed | Searches were re‐run on 4 April 2008; no new studies were found. |
| 12 August 2008 | Amended | Converted to new review format. |
Notes
Sarah J Nolan (lead author of the 2013 update) is now Sarah J Nevitt.
Acknowledgements
We are grateful to Martie Muller for her contributions to the original review, to Jennifer Pulman for providing support and advice on 'Risk of bias' and quality assessments for the 2013 review update, to Paula Williamson for contributions to the original review and to the Cochrane Epilepsy Information Specialists, Alison Beamond and Graham Chan, for performing all the electronic searches.
We are greatly indebted to the original trial investigators for providing individual participant data for this review and to Harald Pohlmann who very kindly responded to our requests for additional information and data.
Appendices
Appendix 1. CRS Web search strategy
1. MeSH DESCRIPTOR Phenytoin AND CENTRAL:TARGET
2. (Difenilhidantoin* or Dihydantoin or Dilantin or Diphenylan or Diphenylhydantoin* or Diphenylhydatanoin* or Dwufenylohydantoin* or Epanutin or Eptoin or Fenitoin* or Fenytoin* or Phenytek or Phenytoin*):AB,KW,MC,MH,TI AND CENTRAL:TARGET
3. #1 OR #2 AND CENTRAL:TARGET
4. (oxcarbazepi* or carbox or OCBZ or oxcarbamaz* or trilept*):AB,KW,MC,MH,TI AND CENTRAL:TARGET
5. #3 AND #4 AND >22/01/2013:CRSCREATED AND CENTRAL:TARGET
6. MESH DESCRIPTOR Epilepsy EXPLODE ALL AND CENTRAL:TARGET
7. MESH DESCRIPTOR Seizures EXPLODE ALL AND CENTRAL:TARGET
8. (epilep* OR seizure* OR convuls*):AB,KW,MC,MH,TI AND CENTRAL:TARGET
9. #6 OR #7 OR #8 AND CENTRAL:TARGET
10. #5 AND #9
Appendix 2. MEDLINE search strategy
The following search is based on the Cochrane Highly Sensitive Search Strategy for identifying randomised trials in MEDLINE (Lefebvre 2011).
1. (randomized controlled trial or controlled clinical trial or pragmatic clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
2. clinical trials as topic.sh.
3. trial.ti.
4. 1 or 2 or 3
5. exp animals/ not humans.sh.
6. 4 not 5
7. exp Epilepsy/
8. exp Seizures/
9. (epilep$ or seizure$ or convuls$).tw.
10. 7 or 8 or 9
11. exp Phenytoin/
12. (Difenilhidantoin$ or Dihydantoin or Dilantin or Diphenylan or Diphenylhydantoin$ or Diphenylhydatanoin$ or Dwufenylohydantoin$ or Epanutin or Eptoin or Fenitoin$ or Fenytoin$ or Phenytek or Phenytoin$).tw.
13. (oxcarbazepi$ or carbox or OCBZ or oxcarbamaz$ or trilept$).tw.
14. (11 or 12) and 13
15. 6 and 10 and 14
16. limit 15 to ed=20130122‐20180820
17. 15 not (1$ or 2$).ed.
18. 17 and (2013$ or 2014$ or 2015$ or 2016$ or 2017$ or 2018$).dt.
19. 16 or 18
Appendix 3. ClinicalTrials.gov search strategy
Interventional Studies | Epilepsy | Oxcarbazepine AND phenytoin
Appendix 4. ICTRP search strategy
Condition: epilepsy
Intervention: oxcarbazepine AND phenytoin
Recruitment status: all
Phases: 2, 3, 4
Data and analyses
Comparison 1. Oxcarbazepine versus phenytoin.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Time to treatment failure (any reason related to the treatment) | 2 | 480 | Hazard Ratio (Fixed, 95% CI) | 0.77 [0.53, 1.12] |
| 2 Time to treatment failure due to adverse events | 2 | 480 | Hazard Ratio (Fixed, 95% CI) | 0.22 [0.10, 0.51] |
| 3 Time to treatment failure due to lack of efficacy | 2 | 480 | Hazard Ratio (Fixed, 95% CI) | 1.17 [0.31, 4.35] |
| 4 Time to treatment failure (any reason related to the treatment) ‐ by epilepsy type | 2 | 476 | Hazard Ratio (Fixed, 95% CI) | 0.78 [0.53, 1.14] |
| 4.1 Focal onset seizures | 2 | 333 | Hazard Ratio (Fixed, 95% CI) | 0.69 [0.43, 1.09] |
| 4.2 Generalised onset seizures | 2 | 143 | Hazard Ratio (Fixed, 95% CI) | 1.03 [0.51, 2.08] |
| 5 Time to treatment failure (any reason related to treatment) ‐ sensitivity analysis: epilepsy type reclassified to focal onset for generalised onset and age > 30 years | 2 | 476 | Hazard Ratio (Fixed, 95% CI) | 0.77 [0.52, 1.13] |
| 5.1 Focal onset seizures | 2 | 363 | Hazard Ratio (Fixed, 95% CI) | 0.80 [0.52, 1.24] |
| 5.2 Generalised onset seizures | 2 | 113 | Hazard Ratio (Fixed, 95% CI) | 0.67 [0.31, 1.46] |
| 6 Time to treatment failure (any reason related to treatment) ‐ sensitivity analysis: epilepsy type reclassified to uncertain seizure type for generalised onset and age > 30 years | 2 | 476 | Hazard Ratio (Fixed, 95% CI) | 0.73 [0.50, 1.08] |
| 6.1 Focal onset seizures | 2 | 333 | Hazard Ratio (Fixed, 95% CI) | 0.69 [0.43, 1.09] |
| 6.2 Generalised onset seizures | 2 | 113 | Hazard Ratio (Fixed, 95% CI) | 0.67 [0.31, 1.46] |
| 6.3 Uncertain seizure type | 1 | 30 | Hazard Ratio (Fixed, 95% CI) | 6.23 [0.73, 53.44] |
| 7 Time to first seizure | 2 | 472 | Hazard Ratio (Fixed, 95% CI) | 0.93 [0.72, 1.21] |
| 8 Time to first seizure ‐ by epilepsy type | 2 | 468 | Hazard Ratio (Fixed, 95% CI) | 0.97 [0.75, 1.26] |
| 8.1 Focal onset seizures | 2 | 326 | Hazard Ratio (Fixed, 95% CI) | 0.92 [0.68, 1.25] |
| 8.2 Generalised onset seizures | 2 | 142 | Hazard Ratio (Fixed, 95% CI) | 1.11 [0.66, 1.86] |
| 9 Time to achieve 12‐month remission | 2 | 472 | Hazard Ratio (Fixed, 95% CI) | 1.09 [0.80, 1.47] |
| 10 Time to achieve 12‐month remission ‐ by epilepsy type | 2 | 468 | Hazard Ratio (Fixed, 95% CI) | 1.04 [0.77, 1.41] |
| 10.1 Focal onset seizures | 2 | 326 | Hazard Ratio (Fixed, 95% CI) | 1.09 [0.75, 1.57] |
| 10.2 Generalised onset seizures | 2 | 142 | Hazard Ratio (Fixed, 95% CI) | 0.94 [0.55, 1.62] |
| 11 Time to achieve 6‐month remission | 2 | 472 | Hazard Ratio (Fixed, 95% CI) | 1.12 [0.87, 1.43] |
| 12 Time to achieve 6‐month remission ‐ by epilepsy type | 2 | 468 | Hazard Ratio (Fixed, 95% CI) | 1.06 [0.82, 1.36] |
| 12.1 Focal onset seizures | 2 | 326 | Hazard Ratio (Fixed, 95% CI) | 1.17 [0.87, 1.58] |
| 12.2 Generalised onset seizures | 2 | 142 | Hazard Ratio (Fixed, 95% CI) | 0.83 [0.53, 1.31] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Aikia 1992.
| Methods | Randomised, double‐blinded, parallel‐group study | |
| Participants | Adult participants with newly diagnosed epilepsy and "normal intellectual capacity" with a minimum of 2 seizures in the last 2 years or 1 seizure and an epileptiform EEG Number randomised: total = 37, OXC = 19, PHT = 18 Mean age of 29 included participants (SD): OXC = 33.6 (14) years, PHT = 32.7 (12.5) years 11 out of 29 included participants male (38%), 21 out of 29 included participants with focal epilepsy (72%); see Notes |
|
| Interventions | Monotherapy with oxcarbazepine or phenytoin 4‐ to 8‐week titration period until serum concentrations reached 30 μmol/litre to 120 μmol/litre for OXC and 40 μmol/litre to 80 μmol/litre for PHT, followed by a maintenance phase of 12 months |
|
| Outcomes | Neuropsychological assessment and cognitive functioning in 3 major areas at baseline, 6 months and 12 months follow‐up:
|
|
| Notes | Participants experiencing inadequate seizure control, adverse events or those who were non‐compliant were withdrawn from the study and excluded from analysis (5 from OXC group and 3 from PHT group). Results are presented only for 29 participants (OXC = 14 and PHT = 15) completing the study. Outcomes for this review were not reported; IPD were not available (we could not make contact with original trial authors to request IPD). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were "randomly assigned" to treatment; no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "The study followed a double blind design"; no further information provided regarding how the double‐blind was achieved |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Intention‐to‐treat approach not taken: results reported only for 29 participants (OXC = 14 and PHT = 15) who completed 12‐month follow‐up. Eight participants experiencing inadequate seizure control, adverse events or those who were non‐compliant (OXC = 5 and PHT = 3) were excluded from analysis and results. |
| Selective reporting (reporting bias) | Low risk | No protocol available and outcomes chosen for this review not reported Neuropsychological and cognitive outcomes well reported and treatment withdrawal rates reported |
| Other bias | Low risk | No other sources of bias detected |
Bill 1997.
| Methods | Multicentre, double‐blind, parallel‐group trial conducted in centres in Argentina, Brazil, Mexico, South Africa Written informed consent obtained from participants or parents/guardians Approved by local ethics committees Conducted 1991 to first quarter of 1995 | |
| Participants | Participants aged between 16 and 65 years with newly diagnosed epilepsy with focal seizures or generalised tonic‐clonic seizures 1 participant above the upper age limit (aged 91) included in efficacy and tolerability analyses A minimum of 2 seizures, separated by at least 48 hours, within 6 months preceding trial entry No previous AED, except emergency treatment of seizures for a maximum of 3 weeks prior to trial entry Number randomised: total = 287, OXC = 143, PHT = 144 174 male (61%); 182 focal epilepsy (63%) |
|
| Interventions | Monotherapy with oxcarbazepine or phenytoin. Eight‐week titration period started with 300 mg OXC or 100 mg PHT, increased bi‐weekly, based on clinical response After 8 weeks participants were to be on a tid regimen with daily doses of 450 mg to 2400 mg OXC or 150 mg to 800 mg PHT Continued during 48‐week maintenance with adjustment according to clinical response A third long‐term, open‐label extension phase followed the maintenance period. Double‐blind results only are reported. |
|
| Outcomes | Efficacy: proportion of seizure‐free participants who had at least 1 seizure assessment during the maintenance period Tolerability: comparison of participants who prematurely discontinued because of adverse experiences Clinical utility: comparing premature discontinuation | |
| Notes | IPD provided for all outcomes of the review | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Treatment groups randomised in 1:1 ratio across centres via computer‐generated randomisation numbers over balanced blocks of size 6 |
| Allocation concealment (selection bias) | Low risk | Allocation concealment was achieved with sequentially numbered packages which were identical and contained identical tablets (information provided by trial statistician) |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Trial conducted in 2 phases: 56‐week, double‐blind phase followed by long‐term, open‐label extension Double‐blind phase results reported only Blind achieved with divisible OXC and PHT tablets identical in appearance |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rates reported in both treatment phases, participants withdrawing from treatment were no longer followed up so seizure outcomes had to be censored at time of treatment failure and therefore analyses for remission and seizure outcomes could not adopt an ITT approach |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | No other sources of bias detected |
Guerreiro 1997.
| Methods | Multicentre, double‐blind, parallel‐group trial conducted in centres in Argentina and Brazil Written informed consent obtained from participants or parents/guardians Approved by local ethics committees Conducted 1991 to first quarter of 1995 | |
| Participants | Participants aged 5 to 18 years with newly diagnosed epilepsy with focal seizures or generalised tonic clonic seizures A minimum of 2 seizures, separated by at least 48 hours, within 6 months preceding trial entry No previous AED, except emergency treatment of seizures for a maximum of 3 weeks prior to trial entry Number randomised: total = 193, OXC = 97, PHT = 96 96 male (50%); 151 focal epilepsy (78%) |
|
| Interventions | Monotherapy with oxcarbazepine or phenytoin. Eight‐week titration period started with 150 mg OXC or 50 mg PHT, increased bi‐weekly, based on clinical response After 8 weeks participants were to be on a tid regimen with daily doses of 450 mg to 2400 mg OXC or 150 mg to 800 mg PHT Continued during 48‐week maintenance with adjustment according to clinical response A third long‐term, open‐label extension phase followed the maintenance period. Double‐blind results only are reported |
|
| Outcomes | Efficacy: proportion of seizure‐free participants who had at least 1 seizure assessment during the maintenance period Tolerability: comparison of participants who prematurely discontinued because of adverse experiences Clinical utility: comparing the rate of premature discontinuation | |
| Notes | IPD provided for all outcomes of the review | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Treatment groups randomised in 1:1 ratio across centres via computer‐generated randomisation numbers over balanced blocks of size 6 |
| Allocation concealment (selection bias) | Low risk | Allocation concealment was achieved with sequentially numbered packages which were identical and contained identical tablets (information provided by trial statistician) |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Trial conducted in 2 phases: 56‐week, double‐blind phase followed by long‐term, open‐label extension. Double‐blind phase results reported only Blind achieved with divisible OXC and PHT tablets identical in appearance |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rates reported in both treatment phases, participants withdrawing from treatment were no longer followed up so seizure outcomes had to be censored at time of treatment failure and therefore analyses for remission and seizure outcomes could not adopt an ITT approach |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | No other sources of bias detected |
1. Abbreviations: AED: antiepileptic drug; EEG: electroencephalogram; IPD: individual participant data; ITT: intention‐to‐treat; OXC: oxcarbazepine; PHT: phenytoin; tid: three times per day
2. For studies where IPD were provided for all randomised participants (Bill 1997; Guerreiro 1997), attrition and reporting bias are reduced as attrition rates and unpublished outcome data are requested.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Sabers 1995 | Not fully randomised: "The treatment was chosen at random unless the individual diagnoses required a specific drug". |
IPD: individual participant data
Differences between protocol and review
In the 2013 update, we added sensitivity analyses for misclassification of epilepsy type following the discovery of this potential classification bias in other reviews in the series of Cochrane IPD reviews investigating pair‐wise monotherapy comparisons. We added 'Summary of findings' tables and added text in the Methods section for 'Summary of findings' tables.
In December 2014 we changed the title to specify that the review uses individual participant data.
In the 2018 update, we added the outcome 'incidence of adverse events' and removed the outcome 'quality of Life', for consistency with the other reviews in the series of Cochrane IPD reviews investigating pair‐wise monotherapy comparisons.
In the 2018 update, we redefined 'time to withdrawal of allocated treatment' as 'time to treatment failure', due to feedback received from the Cochrane Editorial Unit regarding potential confusion regarding 'withdrawal' as a positive or negative outcome of antiepileptic monotherapy. The definitions of reasons for treatment failure/withdrawal for some individuals were reclassified as events or censored observations in line with the definitions of a treatment related treatment failure used across the series of Cochrane IPD reviews investigating pair‐wise monotherapy comparisons.
We added analyses of 'time to treatment failure' (due to lack of efficacy and due to adverse events) following feedback on published antiepileptic drug monotherapy reviews that these sub‐outcomes would be useful for clinical practice.
We replaced the term 'partial' with 'focal', in accordance with the most recent classification of epilepsies of the International League Against Epilepsy (Scheffer 2017).
Contributions of authors
SJ Nevitt assessed studies for inclusion in the review update, obtained individual participant data from trial investigators for the review update, assessed risk of bias in all included studies, performed analyses in Stata version 14, added survival plots and a 'Summary of findings' table, and updated the text of the review.
M Muller was the lead investigator on the original review and was involved in developing the original protocol, assessing eligibility of trials for inclusion in the review and obtaining, validating and checking individual participant data and assessing risk of bias in all included studies.
C Tudur Smith provided statistical supervision and was involved with data analysis in the original review.
AG Marson was involved in obtaining individual participant data from original trial investigators and provided guidance with the clinical interpretation of results
Sources of support
Internal sources
University of Liverpool, UK.
South African Cochrane Centre, Medical Research Council, South Africa.
Biostatistics Unit, Medical Research Council, South Africa.
Institute for Maritime Technology, Simon's Town, South Africa.
External sources
Effective Health Care Alliance, UK.
National Health Service, Research and Development, UK.
Department for International Development, UK.
-
National Institute of Health Research (NIHR), UK.
This review presents independent research commissioned by the National Institute for Health Research (NIHR). The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Declarations of interest
SJN: none known. CTS: none known. AGM: a consortium of pharmaceutical companies (GSK, EISAI, UCB Pharma) funded the National Audit of Seizure Management in Hospitals (NASH) through grants paid to the University of Liverpool. Professor Tony Marson is part funded by the National Institute for Health Research Collaboration for Leadership in Applied Health Research and Care North West Coast (NIHR CLAHRC NWC).
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Aikia 1992 {published data only}
- Aikia M, Kalviainen R, Sivenius J, Halonen T, Riekkinen PJ. Cognitive effects of oxcarbazepine and phenytoin monotherapy in newly diagnosed epilepsy: one year follow‐up. Epilepsy Research 1992;11(3):199‐203. [DOI] [PubMed] [Google Scholar]
Bill 1997 {published data only}
- Bill PA, Vigonius U, Pohlmann H, Guerreiro CAM, Kochen S, Saffer D, et al. A double‐blind controlled clinical trial of oxcarbazepine versus phenytoin in adults with previously untreated epilepsy. Epilepsy Research 1997;27(3):195‐204. [DOI] [PubMed] [Google Scholar]
- D'Souza J, Miller M, Sturm YE. Oxcarbazepine is at least as effective and well‐tolerated as phenytoin with a superior safety profile over long‐term treatment in patients with partial seizures. Epilepsia 2003;44(Suppl 9):265. [Google Scholar]
- Schmidt D. How reliable is early treatment response in predicting long‐term seizure outcome?. Epilepsy & Behavior 2007;10(4):588‐94. [DOI] [PubMed] [Google Scholar]
Guerreiro 1997 {published data only}
- D'Souza J, Miller M, Sturm YE. Oxcarbazepine is at least as effective and well‐tolerated as phenytoin with a superior safety profile over long‐term treatment in patients with partial seizures. Epilepsia 2003;44(Suppl 9):265. [Google Scholar]
- Guerreiro M. Better seizure control and tolerability over the long term with oxcarbazepine (Trileptal (R)) monotherapy compared with phenytoin in newly diagnosed children and adolescents with partial and generalised tonic‐clonic seizures. Epilepsia 2003;44(Suppl 8):148‐9. [Google Scholar]
- Guerreiro MM, Vigonius U, Pohlmann H, Manreza ML, Fejerman N, Antoniuk SA, et al. A double‐blind controlled clinical trial of oxcarbazepine versus phenytoin in children and adolescents with epilepsy. Epilepsy Research 1997;27(2):205‐13. [DOI] [PubMed] [Google Scholar]
- Schmidt D. How reliable is early treatment response in predicting long‐term seizure outcome?. Epilepsy & Behavior 2007;10(4):588‐94. [DOI] [PubMed] [Google Scholar]
- Sturm YE, Miller M. Long‐term oxcarbazepine therapy in children is at least as effective as and better tolerated than phenytoin. Epilepsia 2003;44(Suppl 9):272. [Google Scholar]
References to studies excluded from this review
Sabers 1995 {published data only}
- Sabers A, Maller A, Dam M, Smed A, Arlien‐Saborg P, Buchman J, et al. Cognitive function and anticonvulsant therapy: effect of monotherapy in epilepsy. Acta Neurologica Scandinavica 1995;92(1):19‐27. [DOI] [PubMed] [Google Scholar]
Additional references
Annegers 1999
- Annegers JF, Dubinsky S, Coan SP, Newmark ME, Roht L. The incidence of epilepsy and unprovoked seizures in multiethnic, urban health maintenance organizations. Epilepsia 1999;40(4):502‐6. [DOI] [PubMed] [Google Scholar]
Bang 2003
- Bang L, Goa K. Oxcarbazepine: a review of its use in children with epilepsy. Paediatric Drugs 2003;5(8):557‐73. [DOI] [PubMed] [Google Scholar]
Beydoun 2000
- Beydoun A, Sachdeo RC, Rosenfeld WE, Krauss GL, Sessler N, Mesenbrink P, et al. Oxcarbazepine monotherapy for partial‐onset seizures: a multicenter, double‐blind, clinical trial. Neurology 2000;54(12):2245‐51. [DOI] [PubMed] [Google Scholar]
Carl 1992
- Carl GF, Smith ML. Phenytoin‐folate interactions: differing effects of the sodium salt and the free acid of phenytoin. Epilepsia 1992;33(2):372‐5. [DOI] [PubMed] [Google Scholar]
Christe 1997
- Christe W, Krämer G, Vigonius U, Pohlmann H, Steinhoff BJ, Brodie MJ, et al. A double‐blind controlled clinical trial: oxcarbazepine versus sodium valproate in adults with newly diagnosed epilepsy. Epilepsy Research 1997;26(3):451‐60. [DOI] [PubMed] [Google Scholar]
Cockerell 1995
- Cockerell OC, Johnson AL, Sander JW, Hart YM, Shorvon SD. Remission of epilepsy: results from the National General Practice Study of Epilepsy. Lancet 1995;346(8968):140‐4. [DOI] [PubMed] [Google Scholar]
Commission 1981
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 1981;22(4):489‐501. [DOI] [PubMed] [Google Scholar]
Commission 1989
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30(4):389‐99. [DOI] [PubMed] [Google Scholar]
Dam 1989
- Dam M, Ekberg R, Loyning Y, Waltimo O, Jakobsen K. A double blind study comparing oxcarbazepine and carbamazepine in patients with newly diagnosed, previously untreated epilepsy. Epilepsy Research 1989;3(1):70‐6. [DOI] [PubMed] [Google Scholar]
Faigle 1990
- Faigle JW, Menge GP. Metabolic characteristics of oxcarbazepine (Trileptal) and their beneficial implications for enzyme induction and drug interactions. Behaviour Neurology 1990;3(1):21‐30. [DOI] [PubMed] [Google Scholar]
Gladstone 1992
- Gladstone DJ, Bologa M, Maguire C, Pastuszak A, Koren G. Course of pregnancy and fetal outcome following maternal exposure to carbamazepine and phenytoin: a prospective study. Reproductive Toxicology 1992;6(3):257‐61. [DOI] [PubMed] [Google Scholar]
Grant 1992
- Grant SM, Faulds D. Oxcarbazepine. A review of its pharmacology and therapeutic potential in epilepsy, trigeminal neuralgia and affective disorders. Drugs 1992;43:873‐88. [DOI] [PubMed] [Google Scholar]
Hauser 1993
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota 1935 ‐ 1984. Epilepsia 1993;34:453‐68. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hirtz 2007
- Hirtz D, Thurman DJ, Gwinn‐Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. How common are the "common" neurologic disorders?. Neurology 2007;68:326‐37. [DOI] [PubMed] [Google Scholar]
ILAE 1998
- ILAE Commission on Antiepileptic Drugs. Considerations on designing clinical trials to evaluate the place of new antiepileptic drugs in the treatment of newly diagnosed and chronic patients with epilepsy. Epilepsia 1998;39(7):799‐803. [DOI] [PubMed] [Google Scholar]
ILAE 2006
- Glauser T, Ben‐Menachem E, Bourgeois B, Cnaan A, Chadwick D, Guerreiro C, et al. ILAE treatment guidelines: evidence based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 2006;47(7):1094‐120. [DOI] [PubMed] [Google Scholar]
Juul‐Jenson 1983
- Juul‐Jenson P, Foldspang A. Natural history of epileptic seizures. Epilepsia 1983;24:297‐312. [DOI] [PubMed] [Google Scholar]
Kwan 2000
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. New England Journal of Medicine 2000;342:314‐9. [DOI] [PubMed] [Google Scholar]
Kwan 2003
- Kwan P, Brodie MJ. Clinical trials of antiepileptic medications in newly diagnosed patients with epilepsy. Neurology 2003;60(11 Suppl 4):S2‐12. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
MacDonald 2000
- MacDonald BK, Johnson AL, Goodridge DM, Cockerell OC, Sander JWA, Shorvon SD. Factors predicting prognosis of epilepsy after presentation with seizures. Annals of Neurology 2000;48:833‐41. [PubMed] [Google Scholar]
Malafosse 1994
- Malfosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R. Idiopathic Generalised Epilepsies: Clinical, Experimental and Genetic. Eastleigh: John Libbey and Company, 1994. [Google Scholar]
Marson 2000
- Marson AG, Williamson PR, Hutton JL, Clough HE, Chadwick DW. Carbamazepine versus valproate monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2000, Issue 3. [DOI: 10.1002/14651858.CD001030] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mattson 1985
- Mattson RH, Cramer JA, Collins JF, Smith DB, Delgado‐Escueta AV, Browne TR, et al. Comparison of carbamazepine, phenobarbital, phenytoin, and primidone in partial and secondarily generalized tonic‐clonic seizures. New England Journal of Medicine 1985;313(3):145‐51. [DOI] [PubMed] [Google Scholar]
Meador 2008
- Meador K, Reynolds MW, Crean S, Fahrbach K, Probst C. Pregnancy outcomes in women with epilepsy: a systematic review and meta‐analysis of published pregnancy registries and cohorts. Epilepsy Research 2008;81(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Murray 1994
- Murray CJL, Lopez AD. World Health Organization. Global comparative assessments in the health sector: disease burden, expenditures and intervention packages. apps.who.int/iris/handle/10665/41177 (accessed 01/08/2018).
Nevitt 2017a
- Nevitt SJ, Sudell M, Weston J, Tudur Smith C, Marson AG. Antiepileptic drug monotherapy for epilepsy: a network meta‐analysis of individual participant data. Cochrane Database of Systematic Reviews 2017, Issue 12. [DOI: 10.1002/14651858.CD011412.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nevitt 2017b
- Nevitt SJ, Marson AG, Weston J, Tudur‐Smith C. Carbamazepine versus phenytoin monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.CD001911.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nevitt 2018a
- Nevitt SJ, Tudur Smith C, Weston J, Marson AG. Lamotrigine versus carbamazepine monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2018, Issue 6. [DOI: 10.1002/14651858.CD001031.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nevitt 2018b
- Nevitt SJ, Marson AG, Weston J, Tudur Smith C. Sodium valproate versus phenytoin monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2018, Issue 8. [DOI: 10.1002/14651858.CD001769.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nevitt 2018c
- Nevitt SJ, Marson AG, Weston J, Tudur Smith C. Carbamazepine versus phenobarbitone monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2018, Issue 10. [DOI: 10.1002/14651858.CD001904.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ngugi 2010
- Ngugi AK, Bottomley C, Kleinschmidt I, Sander JW, Newton CR. Estimation of the burden of active and life‐time epilepsy: a meta‐analytic approach. Epilepsia 2010;51:883‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
NICE 2012
- National Institute for Health and Care Excellence. The epilepsies: the diagnosis and management of the epilepsies in adults and children in primary and secondary care. www.nice.org.uk/guidance/cg137 (accessed 01/08/2018).
Nolan 2013b
- Nolan SJ, Sutton L, Marson A, Tudur Smith C. Consistency of outcome and statistical reporting of time‐to‐event data: the impact on Cochrane Reviews and meta‐analyses in epilepsy. 21st Cochrane Colloquium: Better Knowledge for Better Health, Quebec City. 2013:114‐5.
Nolan 2013c
- Nolan SJ, Tudur Smith C, Pulman J, Marson AG. Phenobarbitone versus phenytoin monotherapy for partial onset seizures and generalised onset tonic‐clonic seizures. Cochrane Database of Systematic Reviews 2013, Issue 1. [DOI: 10.1002/14651858.CD002217.pub2] [DOI] [PubMed] [Google Scholar]
Nolan 2016b
- Nolan SJ, Sudell M, Tudur Smith C, Marson A. Topiramate versus carbamazepine monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2016, Issue 12. [DOI: 10.1002/14651858.CD012065.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nulman 1997
- Nulman I, Scolnik D, Chitayat D, Farkas LD, Koren G. Findings in children exposed in utero to phenytoin and carbamazepine monotherapy: independent effects of epilepsy and medications. American Journal of Medical Genetics 1997;68(1):18‐24. [PubMed] [Google Scholar]
Olafsson 2005
- Olafsson E, Ludvigsson P, Gudmundsson G, Hesdorfer D, Kjartansson O, Hauser WA. Incidence of unprovoked seizures and epilepsy in Iceland and assessment of the epilepsy syndrome classification: a prospective study. Lancet Neurology 2005;4:627‐34. [DOI] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [DOI] [PubMed] [Google Scholar]
Pohlmann 2005 [pers comm]
- Pohlmann H. Personal communication 23 September 2005.
Sander 1996
- Sander JW, Shorvon SD. Epidemiology of the epilepsies. Journal of Neurology, Neurosurgery, and Psychiatry 1996;61(5):433‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sander 2004
- Sander JW. The use of anti‐epileptic drugs ‐ principles and practice. Epilepsia 2004;45(6):28‐34. [DOI] [PubMed] [Google Scholar]
Schachter 1999
- Schachter SC, Vazquez B, Fisher RS, Laxer KD, Montouris GD, Combs‐Cantrell DT, et al. Oxcarbazepine: double‐blind, randomized, placebo‐control, monotherapy trial for partial seizures. Neurology 1999;52(4):732‐7. [DOI] [PubMed] [Google Scholar]
Scheffer 2017
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Scheinfeld 2003
- Scheinfeld N. Phenytoin in cutaneous medicine: its uses, mechanisms and side effects. Dermatology Online Journal 2003;9(3):6. [PubMed] [Google Scholar]
Stata 2015 [Computer program]
- StataCorp. Stata Statistical Software: Release 14. College Station, TX: StataCorp LP, 2015.
Tennis 1997
- Tennis P, Stern RS. Risk of serious cutaneous disorders after initiation of use of phenytoin, carbamazepine, or sodium valproate: a record linkage study. Neurology 1997;49(2):542‐6. [DOI] [PubMed] [Google Scholar]
Tudur Smith 2007
- Tudur Smith C, Marson AG, Chadwick DW, Williamson PR. Multiple treatment comparisons in epilepsy monotherapy trials. Trials 2007;5(8):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wallace 1997
- Wallace H, Shorvon SD, Hopkins A, O'Donoghue M. Guidelines for the clinical management of adults with poorly controlled epilepsy. National Society of Epilepsy guidelines. London: Royal College of Physicians, 1997. [Google Scholar]
Wellington 2001
- Wellington K, Goa KL. Oxcarbazepine: an update of its efficacy in the management of epilepsy. CNS Drugs 2001;15(2):137‐63. [DOI] [PubMed] [Google Scholar]
Wilder 1995
- Wilder BJ. Phenytoin: clinical use. Antiepileptic Drugs. 4th Edition. New York: Raven Press, 1995:339‐44. [Google Scholar]
Williamson 2000
- Williamson PR, Marson AG, Tudur C, Hutton JL, Chadwick DW. Individual patient data meta‐analysis of randomized anti‐epileptic drug monotherapy trials. Journal of Evaluation in Clinical Practice 2000;6(2):205‐14. [DOI] [PubMed] [Google Scholar]
Williamson 2002
- Williamson PR, Tudur Smith C, Hutton JL, Marson AG. Aggregate data meta‐analysis with time‐to‐event outcomes. Statistics in Medicine 2002;21(22):3337‐51. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Muller 2006
- Muller M, Marson AG, Williamson PR. Oxcarbazepine versus phenytoin monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2006, Issue 2. [DOI: 10.1002/14651858.CD003615.pub2] [DOI] [PubMed] [Google Scholar]
Nolan 2013a
- Nolan SJ, Muller M, Tudur Smith C, Marson AG. Oxcarbazepine versus phenytoin monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2013, Issue 5. [DOI: 10.1002/14651858.CD003615.pub3] [DOI] [PubMed] [Google Scholar]