

Abstract
Background
Dyspepsia is a common condition associated with gastrointestinal (GI) disease. Prokinetics are the treatment of choice for functional dyspepsia (FD). However, the role of prokinetics in FD treatment is still controversial.
Objectives
We conducted a systematic review and meta‐analysis of randomised control trials (RCTs) examining the efficacy of prokinetics in the treatment of FD. The primary outcome was overall absence of or improvement of symptoms and symptom scores at the end of treatment. We also evaluated quality of life (QoL) and adverse events as secondary outcomes.
Search methods
We performed a systematic search of MEDLINE, Embase, the Cochrane Library, and CINAHL, from 1946 until September 2017. RevMan 5.3 was used to calculate pooled risk ratios (RR) of symptoms persisting or without improved QoL or adverse events, mean difference (MD) or standardised mean difference (SMD) of post‐treatment symptoms scores, changes of symptom scores, and QoL, when appropriate with 95% confidence intervals (CI), using a random‐effects model. Quality of evidence was evaluated using GRADE methodology.
Selection criteria
We included studies that were parallel group RCTs comparing one prokinetic with either placebo or another prokinetic of the same or different class for the treatment of FD. Studies involved adults who presented with dyspepsia symptoms and who had negative or insignificant findings on endoscopy as well as no other organic and metabolic disorders. Studies only including participants with primarily reflux or heartburn symptoms were excluded.
Data collection and analysis
Two review authors independently assessed study eligibility, study quality and performed data extraction.
Main results
From an initial 1388 citations, we identified 43 studies in 40 papers. Of those, 29 studies with 10,044 participants compared six prokinetics with placebo for the outcome of absence of symptoms or symptom improvement. There was a statistically significant effect of prokinetic treatment in reducing global symptoms of FD (RR of remaining dyspeptic = 0.81, 95% CI 0.74 to 0.89; number needed to treat for an additional beneficial outcome (NNTB) =7, very low‐quality evidence) with considerable heterogeneity; I2 = 91% (P < 0.00001). After removing cisapride from the analysis, the effect of prokinetics in global symptom improvement still persisted, compared to placebo (RR 0.87, 95% CI 0.80 to 0.94), but was still based on very low‐quality evidence. The result showed persistence of significant improvement in subgroups of studies at unclear or at low risk of bias (RR 0.86, 95% CI 0.80‐0.92), and in subgroups by molecules of cisapride (RR 0.71, 95% CI 0.54 to 0.93; NNTB = 4), acotiamide (RR 0.94, 95% CI 0.91 to 0.98; NNTB = 20) and tegaserod(RR 0.89, 95% CI 0.82 to 0.96; NNTB = 14).
Ten studies compared different types of prokinetics with each other and the most commonly used comparator was domperidone, 10 mg three times a day (eight of the 10 studies). There was a significantly better post‐treatment symptom score in other prokinetics, compared to domperidone (SMD ‐0.19, 95% CI ‐0.35 to ‐0.03, very low‐quality evidence), but no difference in reducing global symptom (RR 0.94, 95% CI 0.83 to 1.07), and mean difference symptom scores (SMD ‐0.13, 95% CI ‐0.31 to 0.05). We found five studies that assessed quality of life, but there was no benefit in improving quality of life with prokinetic treatment (SMD 0.11, 95% CI ‐0.10 to 0.33; participants = 1774). The adverse events in individual prokinetics was not different from placebo (RR 1.09, 95% CI 0.95 to 1.25; participants = 3811; studies = 17). However, when we looked at the adverse effects by each prokinetic, there were overall greater adverse effects in the active treatment group with cisapride (RR 1.31, 95% CI 1.03 to 1.65; P = 0.03). The most common side effects were diarrhoea, abdominal discomfort and nausea. The funnel plot was asymmetric (Egger's test, P = 0.02) implying reporting bias or other small‐study effects may be, in part, driving the benefit of prokinetics compared to placebo in this meta‐analysis. The GRADE assessment of the quality of the evidence in each outcome are mostly low or very low due to concerns around risk of bias in study design, unexplained heterogeneity and possible publication bias.
Authors' conclusions
Due to low, or very low, quality of evidence, we are unable to say whether prokinetics are effective for the treatment of functional dyspepsia . We are uncertain which of the individual prokinetic drugs is the most effective as well as whether prokinetics can improve quality of life. Apart from cisapride, prokinetics are well‐tolerated. Good quality RCTs are needed to verify the efficacy of prokinetics.
Plain language summary
Medications which promote stomach movement to relieve upper abdominal discomfort that does not have a specific cause
Background
Functional dyspepsia occurs in people with upper abdominal discomfort that does not have an obvious, specific cause. Prokinetics may help people with functional dyspepsia, by promoting stomach movement.
Study characteristics
We included 43 studies that compared prokinetics with either placebo (powder that has the appearance similar to drug) or another prokinetic for treatment of functional dyspepsia. The studies were limited to those which assessed only adults who presented with upper abdominal discomfort but who did not have a specific cause after investigation.
Key results
We are uncertain whether prokinetics reduce dyspeptic symptoms, compared to no prokinetic treatment. We are also uncertain which prokinetics had the most efficacy in reducing dyspeptic symptoms, improving post‐treatment symptom scores, or improving the mean difference of symptom score. We are uncertain whether prokinetic treatment can improve quality of life. We are uncertain whether prokinetics (except cisapride) differ from no prokinetic in producing unpleasant symptoms. The most common unpleasant symptoms from prokinetics were diarrhoea, abdominal discomfort and nausea.
Quality of the evidence
The quality of the evidence was graded as low or very low. We need more research to prove the benefit of prokinetics for treatment in people with functional dyspepsia.
Summary of findings
Background
Description of the condition
Dyspepsia is a common condition in gastrointestinal disease with a global prevalence of at least 20% (Ford 2015; Tack 2006). It is defined by epigastric pain or discomfort as described in the Rome criteria definition, which has had four iterations (Stanghellini 2016; Tack 2006; Talley 1991; Tally 1999). Nevertheless, 72% to 82% of people presenting with dyspepsia have no evidence of structural disease on endoscopic findings (Ford 2010) that is likely to explain the symptom, called functional dyspepsia (FD) (Stanghellini 2016; Tack 2006; Talley 1991; Tally 1999).
The pathophysiology of FD is likely multifactorial and not fully understood (Stanghellini 2016). However, several factors have been identified as relevant, including abnormality of gastroduodenal motor (delayed gastric emptying or impaired gastric accommodation) and sensory (gastric and duodenal hypersensitivity) mechanisms (Stanghellini 2016; Vanheel 2013).
A prokinetic is one of the rescue medications for FD, which according to Lacy and colleagues, aims to improve gastric emptying (Lacy 2012). Additionally, prokinetic provided a significant benefit over placebo with a relative risk reduction of 33% and number needed to treat for an additional beneficial outcome (NNTB) of six; however, this evidence had the major concern of publication bias (Stanghellini 2016). Moreover, Moayyedi and colleagues reported the significant effect of prokinetic treatment in reducing overall symptoms of FD with a relative risk of remaining dyspeptic in the prokinetic group of 0.92 (95% confidence interval (CI) 0.88 to 0.97) with a NNTB of 12.5 (95% CI 8 to 25) (Moayyedi 2017).
Currently, a prokinetic is recommended as first‐line treatment in people with postprandial distress syndrome (PDS) subtype (Stanghellini 2016). On the other hand, it is suggested as the third‐line treatment by recent guidelines on dyspepsia from the American College of Gastroenterology (ACG) and the Canadian Association of Gastroenterology (CAG), regardless of FD subtypes (Moayyedi 2017).
Description of the intervention
The intervention addressed in this review is the use of prokinetic agents to treat FD. Prokinetics are agents that accelerate gastric emptying and intestinal transit time.
How the intervention might work
FD is likely to be a multifactorial disease (Tack 2011). A significant proportion of people with FD have delayed gastric emptying and this is present much more commonly than people without symptoms (Tack 2004). Delayed gastric emptying may explain some patients' symptoms of postprandial fullness, nausea and epigastric pain and improving gastric emptying with a prokinetic drug may improve these symptoms.
Why it is important to do this review
FD is a clinical entity of significant disease and economic burden on both patients and the healthcare system (Lacy 2013; Moayyedi 2002; van Zanten 2011). Its pathophysiology remains elusive and as such, so has its appropriate management. However, a subtype of this population (e.g. PDS) may experience symptoms secondary to dysmotility, which drives the use of prokinetics as a potential therapeutic intervention (Tally 2005). A Cochrane systematic review on the pharmacological Interventions for FD has evaluated the effectiveness of six classes of drugs for the treatment of FD (Moayyedi 2011). Prokinetics were found to be and efficacious drug class with relative risk reduction 33%; 95% CI 18% to 45% (Moayyedi 2011). However, several new prokinetics have since been developed and, added to this; cisapride, the most heavily studied drug of this class, is no longer available in many markets thus necessitating a more up‐to‐date review.
Objectives
By meta‐analysis and systematic review of randomised controlled trials (RCTs), to evaluate the role of prokinetics in the treatment of functional dyspepsia (FD) as reflected by improvement of either individual or global (overall) dyspepsia symptom scores and quality of life scores. The primary comparisons were as follows.
Are prokinetic drugs in general better than placebo?
Which of the individual prokinetic drugs is the most effective?
Methods
Criteria for considering studies for this review
Types of studies
Any parallel group randomised controlled trials (RCTs) comparing one prokinetic with either placebo or another prokinetic of the same or different class for the treatment of functional dyspepsia (FD) were included. Cross‐over trials were eligible for inclusion, but only the first period of the trial prior to cross‐over would have been included..
Types of participants
Adults with dyspepsia, as defined by either Rome Criteria I to IV (Stanghellini 2016; Tack 2006; Talley 1991; Tally 1999), or non‐Rome criteria but using the criteria compatible with the Rome criteria. Specifically, we included studies on adults presenting with dyspepsia symptoms who have had negative or insignificant findings on their endoscopy as well as no other organic (pancreatico‐biliary disease, oesophagitis, peptic ulcer disease and neoplastic disease) and drug‐induced (non‐steroidal anti‐inflammatory drugs) and metabolic disorders. Studies only including participants with primarily reflux or heartburn symptoms were excluded.
Types of interventions
Only studies that considered the use of prokinetics for the treatment of FD were considered. Prokinetics included: erythromycin, metoclopramide, domperidone, cisapride, mosapride, itopride, ABT‐229, alosetron, tegaserod, mosapride, and acotiamide, as well as any other prokinetics identified through a literature review. Only studies that provided treatment duration of at least seven days were eligible for inclusion.
Types of outcome measures
Primary outcomes
Global (overall) symptoms of dyspepsia, reported as binary outcome (yes or no) or symptom scores. We used the most stringent definition of not symptom‐free or no overall symptom improvement by the patient at the end of treatment. If that was not available, we used overall symptom assessment as assessed by the doctor/researcher. If global symptoms were not reported, we used epigastric pain/discomfort improvement as the outcome measure, but these studies were removed in the sensitivity analysis.
Secondary outcomes
Quality of life (QoL),reported as a binary outcome (improved or not improved) or symptom scores changed.
Adverse events, reported as binary outcome (yes or no)
Search methods for identification of studies
Electronic searches
In an effort to identify RCTs comparing a prokinetic either with placebo or with another prokinetic of the same or different class, we searched the individual names of prokinetics that were available, have been, or were under investigation.
We searched the following databases:
Cochrane Central Register of Controlled Trials (CENTRAL) and Cochrane Database of Systematic Reviews (CDSR) in the Cochrane Library (OvidSP)(Issue 9, 2017) (2005 to 13 September 2017) (Appendix 2);
MEDLINE via OvidSP (1946 to 14 September 2017 ) (Appendix 3);
Embase via OvidSP (1974 to 14 September 2017) (Appendix 4);
CINAHL(1981 to 14 September 2017) (Appendix 5).
Searching other resources
We searched all reference lists of the articles retrieved. Additionally, we contacted experts within the field of FD as well as pharmaceutical companies regarding ongoing clinical studies and relevant unpublished data.
Data collection and analysis
Two review authors (RP and YY) evaluated each retrieved RCT for its eligibility, risk of bias and results.
Selection of studies
Two review authors (RP and YY) independently reviewed studies retrieved by the search strategies and excluded studies based on titles, abstracts, or both. Both review authors independently reviewed selected studies for complete analysis.
Data extraction and management
A data collection form specifically designed for this review was used for data collection. One review author extracted data and entered it into RevMan. The other review author served to ensure the accuracy of this process.
The data collected included the following.
Participant characteristics: demographics, recruitment source, diagnostic criteria used by study authors, symptoms at the study's start, most prevalent type of dyspepsia.
Details of interventions: name of medication, dose, schedule.
Dyspeptic symptoms before and after the intervention: number of people with dyspepsia symptom, global Dyspepsia Symptom Scores, quality of life, adverse events.
Data were managed and analysed according to an intention‐to‐treat analysis.
Assessment of risk of bias in included studies
All studies were assessed using Cochrane's 'Risk of bias' tool, which evaluates the following domains: random sequence generation (selection bias), allocation concealment (selection bias), blinding of participants and personnel (performance bias), blinding of outcome assessment (detection bias), incomplete outcome data addressed at short and long term (attrition bias), selective reporting (reporting bias) and other bias. Each domain was described according to what happened in the individual study, followed by a judgment as to the risk of bias relating to that domain. 'Other bias' refers to any other study‐specific characteristic that confers a risk of bias on the results (for example, early stopping, baseline imbalance, blocked randomisation in unblinded studies, and differential diagnostic activity).
Measures of treatment effect
For the binary outcomes, we presented the results as risk ratio (RR) with 95% confidence intervals (CIs). For the continuous outcomes, we presented the results as mean difference (MD) with 95% CIs. If all studies did not use the same scales, the results were presented as a standardised mean difference (SMD) with 95% CIs.
Unit of analysis issues
Only a simple parallel group design for clinical studies such that the number of observations matches the number of individuals randomised. Had cross‐over studies been identified, only the results from the first phase of the study prior to cross‐over would have been included.
Dealing with missing data
Any data that were missing were noted on the data collection form and taken into consideration when evaluating the overall quality of the study. We also attempted to contact the study authors.
Assessment of heterogeneity
We assessed heterogeneity with the Chi² test (P < 0.10 = significant heterogeneity) and I² statistic (> 50% = substantial heterogeneity) using a random‐effects model along with visual inspection of the forest plots. Possible sources for heterogeneity were evaluated by subgroup analyses according to the following criteria.
Outcome of not symptom‐free
Subtypes of functional dyspepsia (postprandial distress syndrome (PDS) and epigastric pain syndrome (EPS))
Length of follow‐up
Use of validated dyspepsia questionnaires
Studies assessed as hIgh risk of bias versus low or unclear risk of bias
Prokinetic subtype and dose
Type of publication
Assessment of reporting biases
In order to assess the presence of small‐study effects in the meta‐analysis, a funnel plot was used. We assessed publication bias by examining the relationship between the treatment effects and the standard error of the estimate using a funnel plot and Egger's test (Egger 1997).
Data synthesis
Global symptoms of dyspepsia were categorised as not symptom‐free or no overall symptom improvement (if "not symptom‐free" was unavailable, which included unchanged or worsened symptoms). The relative risk reduction (RR) and 95% CIs were recorded, and number needed to treat for an additional beneficial outcome (NNTB) (if a significant difference was seen) were all calculated. We recorded the mean and standard deviation (SD) of global symptom score at pre‐ and post‐ treatment as well as mean and SD of change scores from baseline in each group, if available. We calculated the mean and SD of change scores from baseline if only pre‐ and post‐treatment scores were reported, using the methods proposed in the Cochrane Handbook for Systematic Reviews of Interventions(Higgins 2011b). An analysis based on changes from baseline was preferred as it was more efficient and powerful than a comparison of final values. For studies that did not report change scores from baseline, or for scores which were not calculable, we used the final values as the difference in mean final values are on average be the same as the difference in mean change scores in RCTs. Mean difference (MD) and 95% CIs were calculated as the summary statistic for symptom scores, for studies that used the same scales. The SMD and 95% CIs were calculated between two groups if different scales were used in the primary studies. However, final value and change scores were not combined together as SMD (Deeks 2011), and they were reported separately.
For QoL, we calculated the RR and 95%CI of people without QoL improvement, MD or SMD for post‐treatment QoL scores and difference in mean change of QoL scores, when appropriate.
For adverse events, we calculated the RR, with 95% CIs and the number need to treat for an additional harmful outcome (NNTH) if a significant difference was seen. MD and SMD and 95% CIs were used to report changes of quality scores, for similar or different QOL scales, respectively. A meta‐analysis of all data was conducted, if possible. Mantel‐Haenszel (M‐H) methods (random‐effects model) was used to synthesise data in the meta‐analysis (Mantel 1959).
'Summary of findings' tables
We created 'Summary of findings' tables for the following comparisons:
prokinetics versus placebo,
one prokinetic versus domperidone.
We used the following primary and secondary outcomes:
global symptom and symptom score of dyspepsia,
quality of life,
adverse events.
We used the GRADE considerations (study design, study limitations (risk of bias), inconsistency of effect, imprecision, indirectness of evidence, and publication bias) to assess the quality of a body of evidence as it related to the studies which contribute data to the meta‐analyses for the pre‐specified outcomes (GRADEpro GDT). We used methods and recommendations described in Section 8.5 and Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a) and used the GRADEpro software (GRADEpro GDT). We justified all decisions to down grade the quality of studies using footnotes and made comments to aid a reader's understanding of the review where necessary. If meta‐analysis was not possible, we presented the results in a narrative format. We considered whether there was any additional outcome information that was not able to be incorporated into meta‐analyses and noted this in the comments, and stated if it supported or contradicted the information from the meta‐analyses.
Subgroup analysis and investigation of heterogeneity
Subgroups that we planed to analyse included the predominant type of dyspepsia: postprandial distress syndrome (PDS), epigastric pain syndrome (EPS) and mixed type. If heterogeneity was substantial (I2 statistic > 50%, or P value for Chi2 test < 0.10), we explored whether it was explained by methodological or clinical heterogeneity, or both, among the studies. Issues that may explain observed heterogeneity include the following.
Subtypes of functional dyspepsia (PDS, EPS or mixed participants in a study).
Length of follow‐up (> one month versus < one month).
Use of validated versus non‐validated symptoms assessment tools (e.g. dyspepsia questionnaires or symptoms scale).
Studies assessed as hIgh risk of bias versus low or unclear risk of bias.
Prokinetic subtype, recommended versus below recommended versus above recommended dose as per manufacturer.
Type of publication (full paper versus conference abstract).
For not symptom‐free or no symptom improvement, we performed subgroup analysis for not symptom‐free versus studies only reported no symptom improvement.
For mean of change symptoms scores from baseline, we also performed a subgroup analysis for reported MD and calculated the MD (we calculated the MD symptom scores when the primary studies only reported pre‐treatment and post‐treatment scores).
Sensitivity analysis
We conducted sensitivity analysis depending on study characteristics identified during the review process. Studies using individual symptom improvement as the outcome were excluded in the sensitivity analysis. Studies with significant clinical heterogeneity were excluded from the sensitivity analysis. Pre‐specified sensitivity analyses included: fixed‐effect model analysis, outcomes expressed as odds ratios versus relative risks.
Results
Description of studies
We found 43 randomised controlled trials (RCTs) that were eligible for inclusion (Abid 2017; Al‐Quorain 1995;Amarapurkar 2004; Champion 1997; Chen 2004; Choi 2015; de Groot 1997; De Nutte 1989; Du 2014; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Holtmann 2006; Jian 1989; Kellow 1995; Kusunoki 2012; Li 2005; Lin 2009; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Mo 2003; Nakamura 2017; Rösch 1987; Shen 2014; Singh 2015; Sun 2003; Tack 2009; Tack 2011; Talley 2000; Talley 2008a; Talley 2008b; Talley 2008c; Teixeira 2000; Vakil 2008a; Vakil 2008b; Wang 1995; Wong 2014; Yeoh 1997; Zhou 2000; Zhu 2005). Three articles reported the results of two RCTs each (Matsueda 2010a; Matsueda 2010b; Talley 2008b; Talley 2008c; Vakil 2008a; Vakil 2008b. In order to analyse each study individually, the citations are duplicated, while their particular study characteristics detailed in the Characteristics of included studies tables reflect the results of the individual studies.
Of those, 33 studies evaluated the efficacy of a prokinetic and placebo (Abid 2017; Al‐Quorain 1995; Champion 1997; de Groot 1997; De Nutte 1989; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Holtmann 2006; Jian 1989; Kellow 1995; Kusunoki 2012; Lin 2009; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Nakamura 2017; Rösch 1987; Shen 2014; Tack 2009; Tack 2011; Talley 2000; Talley 2008a; Talley 2008b; Talley 2008c; Teixeira 2000; Vakil 2008a; Vakil 2008b; Wang 1995; Wong 2014; Yeoh 1997) whereas 10 studies compared two types of prokinetics (Amarapurkar 2004; Chen 2004; Choi 2015; Du 2014; Li 2005; Mo 2003; Singh 2015; Sun 2003; Zhou 2000; Zhu 2005).
Amongst 33 RCTs comparing a prokinetic and placebo, 18 studies reported only a dichotomous outcome (not symptom‐free or no symptom improvement) (Al‐Quorain 1995; Champion 1997; de Groot 1997; De Nutte 1989; Francois 1987; Hallerback 2002; Hansen 1998; Kusunoki 2012; Lin 2009; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Rösch 1987; Tack 2011; Talley 2008a; Teixeira 2000; Wang 1995; Wong 2014), four studies reported only a continuous outcome (symptom scores) (Abid 2017; Jian 1989; Nakamura 2017; Tack 2009), and 11 studies reported both outcomes (Holtmann 2002; Holtmann 2006; Kellow 1995; Ma 2012; Shen 2014; Talley 2000; Talley 2008b; Talley 2008c; Vakil 2008a; Vakil 2008b; Yeoh 1997).
Amongst 10 RCTs comparing two types of prokinetics, four studies reported only a dichotomous outcome (not symptom‐free or no symptom improvement)(Mo 2003; Sun 2003; Zhou 2000; Zhu 2005), one study reported only a continuous outcome (symptom scores) (Singh 2015),and five studies reported both outcomes (Amarapurkar 2004; Chen 2004; Choi 2015; Du 2014; Li 2005).
See the details in Characteristics of included studies and Characteristics of excluded studies.
Results of the search
Iniitially 1336 citations were retrieved and reviewed based on title/abstract for eligibility: 308 from MEDLINE, 845 from Embase, 164 from the Cochrane Library and 19 from CINAHL. An additional 52 citations were identified from other sources by review authors. After removing duplicate citations, 1038 records were screened. Then 966 citations were excluded after screening of title/abstract eligibility, thus 72 studies were further assessed in the full text, and 43 studies met the inclusion criteria and were included in the meta‐analyses.(Figure 1)
1.
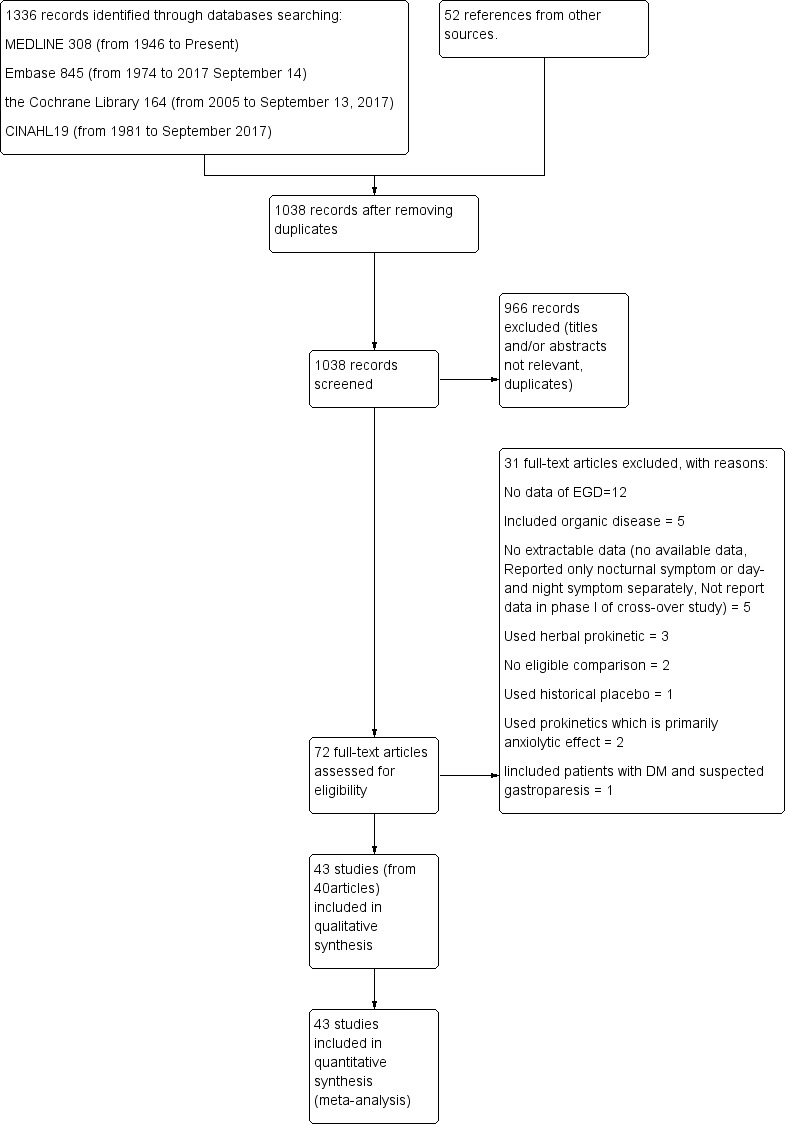
Study flow diagram.
Included studies
All 72 studies were reviewed in detail for eligibility and finally 43 studies in 40 papers (nine in Chinese (Chen 2004; Li 2005; Lin 2009; Mo 2003; Shen 2014; Sun 2003; Wang 1995; Zhou 2000; Zhu 2005) and one in Portuguese (Teixeira 2000)) with 18,227 participants were included: 40 full‐text studies (Abid 2017; Al‐Quorain 1995; Amarapurkar 2004; Champion 1997; Chen 2004; Choi 2015; de Groot 1997; De Nutte 1989; Du 2014; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Holtmann 2006; Jian 1989; Kellow 1995; Kusunoki 2012; Li 2005; Lin 2009; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Mo 2003; Nakamura 2017; Rösch 1987; Shen 2014; Singh 2015; Sun 2003; Tack 2009; Talley 2000; Talley 2008b; Talley 2008c; Teixeira 2000; Vakil 2008a; Vakil 2008b; Wang 1995; Yeoh 1997; Zhou 2000; Zhu 2005) and three conference abstracts (Tack 2011; Talley 2008a; Wong 2014).
Excluded studies
Of those, 31 studies were excluded after being reviewed for eligibility; 12 from the original review as they did not exclude organic disease using esophago‐gastro‐duodenoscopy (EGD) (Agorastos 1991; Bekhti 1979; Chey1982; Chung1993; Creytens 1984; Davis1988; Deruyttere 1987; Haarmann 1979; Hannon 1987; Hausken 1992; Kearney 2000; Van de Mierop 1979), five which included organic disease (De Loose 1979; Milo 1984; Testoni 1990; Van Ganse 1978; Van Outryve M), and 15 which failed eligibility upon closer review such as use of a herbal prokinetic (Kim 2010; Liu 2013; Shim 2015), reported only nocturnal symptom or day and night symptoms separately (Chen 2010; Wood 1993; Yan 2012), no eligible comparison (Manayagi 2014; Yamawaki 2016), used prokinetic which is primarily anxiolytic effect (Miwa 2009; Tack 2012), included participants with diabetes mellitus (DM) and suspected gastroparesis (Tack 2016), did not report data in phase I of a cross‐over study (Goethals 1987), used historical placebo (Kas'ianenko 2014), and no available data (Talley 2001).
Risk of bias in included studies
Risk of bias of the included studies is summarised in Characteristics of included studies. The results are shown in the 'Risk of bias' graph (Figure 2) and the 'Risk of bias' summary table (Figure 3).
2.
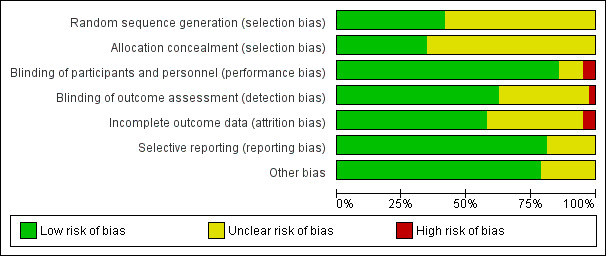
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
For random sequence generation, we considered 18 studies to be at low risk of bias as they reported adequate sequence generation of randomisation (Amarapurkar 2004; Chen 2004; Choi 2015; Hansen 1998; Holtmann 2006, Li 2005; Lin 2009; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Nakamura 2017; Singh 2015; Talley 2008b; Talley 2008c; Vakil 2008a; Vakil 2008b; Zhou 2000). We considered 25 studies to be at unclear risk of bias as no specific information was provided regarding to randomisation process (Abid 2017; Al‐Quorain 1995; Champion 1997; de Groot 1997; De Nutte 1989; Du 2014; Francois 1987; Hallerback 2002; Holtmann 2002; Jian 1989; Kellow 1995; Kusunoki 2012; Mo 2003; Rösch 1987; Shen 2014; Sun 2003; Tack 2009; Tack 2011; Talley 2000; Talley 2008a; Teixeira 2000; Wang 1995; Wong 2014; Yeoh 1997; Zhu 2005). No study with high risk of bias in this domain was identified.(Figure 3)
For allocation concealment, 15 from 43 studies explicitly mentioned concealed allocation of participants to study groups and were rated as low risk of bias(Chen 2004; Choi 2015; Holtmann 2006; Lin 2009; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Nakamura 2017; Talley 2008a; Talley 2008b; Talley 2008c; Vakil 2008a; Vakil 2008b; Zhou 2000). The remaining 28 studies had uncertain concealment (Abid 2017; Al‐Quorain 1995; Amarapurkar 2004; Champion 1997; de Groot 1997; De Nutte 1989; Du 2014; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Jian 1989; Kellow 1995; Kusunoki 2012; Li 2005; Mo 2003; Rösch 1987; Shen 2014; Singh 2015; Sun 2003; Tack 2009; Tack 2011; Talley 2000; Teixeira 2000; Wang 1995; Wong 2014; Yeoh 1997; Zhu 2005).
Blinding
Only one study was of open‐labelled design and considered to have a high risk of performance and detection bias (Wang 1995).
There were 37 studies that indicated they were double‐blinded, thus they were at low risk of bias for participant and personnel blinding (Al‐Quorain 1995; Amarapurkar 2004; Champion 1997; Chen 2004; Choi 2015; de Groot 1997; De Nutte 1989; Du 2014; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Holtmann 2006; Jian 1989; Kellow 1995; Kusunoki 2012; Li 2005; Lin 2009; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Mo 2003; Nakamura 2017; Rösch 1987; Sun 2003; Tack 2009; Talley 2000; Talley 2008a; Talley 2008b; Talley 2008c; Teixeira 2000; Vakil 2008a; Vakil 2008b; Wong 2014; Yeoh 1997; Zhou 2000; Zhu 2005). Four studies did not mention participant or personnel blinding in randomisation process and were considered as unclear risk of bias (Abid 2017; Shen 2014; Singh 2015; Tack 2011). One study was unable to blind the physician because the placebo was sham acupuncture (Ma 2012).
Apart from Wang 1995, which was at high risk of bias for assessor blinding as it was open‐labelled study, there were 27 studies blinding assessor and rated as low risk of bias (Al‐Quorain 1995; Amarapurkar 2004; Champion 1997; Chen 2004; Choi 2015; Du 2014; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Holtmann 2006; Jian 1989; Kellow 1995; Kusunoki 2012; Lin 2009; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Tack 2009; Talley 2000; Talley 2008b; Talley 2008c; Teixeira 2000; Vakil 2008a; Vakil 2008b; Wong 2014) and 15 studies were unclear in assessor blinding(Abid 2017; de Groot 1997; De Nutte 1989; Li 2005; Mo 2003; Nakamura 2017; Rösch 1987; Shen 2014; Singh 2015; Sun 2003; Tack 2011; Talley 2008a; Yeoh 1997; Zhou 2000; Zhu 2005).
Incomplete outcome data
We considered 25 studies to be low risk of bias for incomplete outcome data as they had less than 20% dropout and no other concern in this domain (Abid 2017; Al‐Quorain 1995; Amarapurkar 2004; Choi 2015; de Groot 1997; De Nutte 1989; Du 2014; Francois 1987; Hansen 1998; Holtmann 2002; Holtmann 2006; Jian 1989; Kellow 1995; Kusunoki 2012; Li 2005; Ma 2012; Matsueda 2012; Mo 2003; Rösch 1987; Shen 2014; Vakil 2008a; Vakil 2008b; Wong 2014; Zhou 2000; Zhu 2005). Although a small number of dropouts, we considered 16 studies to be unclear risk of bias due to unbalance rate of loss follow‐up in both groups or no reason of loss follow‐up provided (Chen 2004; Hallerback 2002; Lin 2009; Matsueda 2010a; Matsueda 2010b; Nakamura 2017; Singh 2015; Sun 2003; Tack 2009; Tack 2011; Talley 2000; Talley 2008a; Talley 2008b; Talley 2008c; Teixeira 2000; Wang 1995).Two studies were at high risk of bias for incomplete data; Champion 1997 with 22% who did not complete the treatment and the reason unknown for loss of follow‐up in each group, and Yeoh 1997 with 27% accounted for incomplete outcome data at the end of treatment.
Selective reporting
Most (35 studies) reported all pre‐specified outcomes and were considered as low risk of bias for selective reporting (Abid 2017; Al‐Quorain 1995; Amarapurkar 2004; Chen 2004; Choi 2015; De Nutte 1989; Du 2014; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Holtmann 2006; Jian 1989; Kellow 1995; Kusunoki 2012; Li 2005; Lin 2009; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Nakamura 2017; Rösch 1987; Shen 2014; Singh 2015; Tack 2009; Talley 2000; Talley 2008b; Talley 2008c; Teixeira 2000; Vakil 2008a; Vakil 2008b; Wang 1995; Yeoh 1997; Zhou 2000). Eight studies were at unclear reporting bias; three (Mo 2003; Sun 2003; Zhu 2005) reported only individual symptoms (instead of global symptoms), three (Tack 2011; Talley 2008a; Wong 2014) were conference abstracts and two (Champion 1997; de Groot 1997) combined excellent and good global response rate as an outcome.
Other potential sources of bias
We considered 34 studies to be low risk of bias as no other risk found (Abid 2017; Al‐Quorain 1995; Champion 1997; Chen 2004; Choi 2015; de Groot 1997; De Nutte 1989; Du 2014; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Holtmann 2006; Jian 1989; Kellow 1995; Li 2005; Lin 2009; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Mo 2003; Nakamura 2017; Rösch 1987; Shen 2014; Singh 2015; Sun 2003; Talley 2008b; Talley 2008c; Teixeira 2000; Vakil 2008a; Vakil 2008b; Yeoh 1997; Zhu 2005). Nine studies had unclear other risk of bias. Of those, three (Tack 2011; Talley 2008a; Wong 2014) were conference abstracts, three (Amarapurkar 2004; Tack 2009; Wang 1995) had significant imbalance of baseline characteristic in each group, one (Kusunoki 2012) used range in the follow‐up period, one (Talley 2000) reported a difference baseline characteristic data in table and figure, one (Zhou 2000) used inconsistent terms in the report. In the main text in Chinese, "lack of appetite" was used, but in their English table, "anorexia" was used.
Effects of interventions
Summary of findings for the main comparison. Prokinetic compared to placebo for functional dyspepsia.
| Prokinetic compared to placebo for functional dyspepsia | ||||||
| Patient or population: functional dyspepsia Setting: out‐patient clinic Intervention: prokinetic Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Prokinetic | |||||
|
Not symptom‐free or no symptom improvement Follow‐up: 2 to 12 weeks |
Study population | RR 0.81 (0.74 to 0.89) | 10,044 (29 RCTs) | ⊕⊝⊝⊝ VERY LOW1 2 3 4 | ||
| 74 per 100 | 60 per 100 (55 to 66) | |||||
|
Post‐treatment symptoms scores Follow‐up: 2 to 6 weeks |
The mean post‐treatment symptoms scores was 2.3 to 5.6 (different scales were used) | SMD 0.36 lower (0.65 lower to 0.07 lower) | ‐ | 2914 (6 RCTs) | ⊕⊕⊝⊝ LOW5 6 7 | Higher scores means worse symptoms |
|
Mean difference symptoms scores Follow‐up: 2 to 12 weeks |
The mean difference symptoms scores was ‐10 to 3.43 (different scales were used) | SMD 0.65 lower (1.5 lower to 0.2 higher) | ‐ | 1822 (11 RCTs) | ⊕⊝⊝⊝ VERY LOW1 3 5 8 | Positive scores means worse symptoms |
|
Change of QoL scores Follow‐up: 3 to 12 weeks |
The mean change of QoL scores was 2.8 to 13.2 (different scales were used) | SMD 0.11 higher (0.1 lower to 0.33 higher) | ‐ | 1774 (5 RCTs) | ⊕⊝⊝⊝ VERY LOW5 6 9 | Higher scores means better quality of life |
|
Adverse events Follow‐up: 2 to 8 weeks |
Study population | RR 1.09 (0.95 to 1.25) | 3811 (17 RCTs) | ⊕⊝⊝⊝ VERY LOW1 2 4 5 8 | ||
| 31 per 100 | 34 per 100 (29 to 39) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded one level due to study limitations: most information (>60%) were obtained from studies with unclear risk of bias for random sequence generation and/or allocation, one or more study were considered to be high risk of bias.
2 One study was open‐labelled design.
3 Downgraded one level due to serious inconsistency: significant heterogeneity without plausible explanations.
4 Downgraded one level due to other considerable in publication bias: the funnel plot was asymmetrical, probably from small‐study effect.
5 One study was considered to be high risk of bias.
6 Downgraded one level due to serious inconsistency: significant heterogeneity with some possible explanations.
7 Downgraded one level due to imprecision (95% CI of pooled data was very close to no effect).
8 Downgraded one level due to imprecision (95% CI of pooled data included no effect).
9 Downgraded two levels due to imprecision (95% CI of pooled data included no effect and small number of included studies).
Summary of findings 2. Other prokinetics compared to domperidone 10mg three times a day for functional dyspepsia.
| Other prokinetics compared to domperidone 10mg three times a day for functional dyspepsia | ||||||
| Patient or population: functional dyspepsia Setting: out‐patient clinic Intervention: other prokinetics Comparison: domperidone 10mg three times per day | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with domperidone 10mg three times a day | Risk with Other prokinetics | |||||
|
Not symptom‐free or no symptoms improvement (itopride 50 three times per day, cinitapride 1 mg three times per day, mosapride 5 mg three times per day versus domperidone 10 mg three times per day) Follow‐up: 2 to 4 weeks |
Study population | RR 0.94 (0.83 to 1.07) | 1527 (7 RCTs) | ⊕⊝⊝⊝ VERY LOW1 2 | ||
| 42 per 100 | 39 per 100 (35 to 45) | |||||
|
Post‐treatment scores Follow‐up: 2 to 4 weeks |
The mean post‐treatment scores was 1.0 to 5.4 (different scales were used) | SMD 0.19 lower (0.35 lower to 0.03 lower) | ‐ | 617 (3 RCTs) | ⊕⊝⊝⊝ VERY LOW1 3 | Higher scores means worse symptoms |
|
Mean difference symptoms scores Follow‐up: 2 to 4 weeks |
The mean difference symptoms scores was ‐0.35 to ‐13 (different scales were used) | SMD 0.13 lower (0.31 lower to 0.05 higher) | ‐ | 839 (4 RCTs) | ⊕⊝⊝⊝ VERY LOW1 2 | Positive scores means worse symptoms |
|
Adverse events Follow‐up: 2 to 4 weeks |
Study population | RR 0.69 (0.50 to 0.97) | 1557 (7 RCTs) | ⊕⊝⊝⊝ VERY LOW1 4 5 | ||
| 10 per 100 | 7 per 100 (5 to 9) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded one level due to study limitation (most information (> 60%) were obtained from studies with unclear risk of bias for random sequence generation and/or allocation).
2 Downgraded two levels due to imprecision (95% CI of pooled data included no effect and small number of included studies).
3 Downgraded two levels due to imprecision (95% CI of pooled data was very close to no effect and small number of included studies).
4 Downgraded one level due to imprecision (there were fewer than 300 events and wide 95% CI).
5 Downgraded one level due to imprecision (95% CI of pooled data was very close to no effect)
All analyses used a random‐effects model unless otherwise stated.
1 Prokinetic versus placebo
Thirty‐three studies evaluated the efficacy of a prokinetic versus placebo (Abid 2017; Al‐Quorain 1995; Champion 1997; de Groot 1997; De Nutte 1989; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Holtmann 2006; Jian 1989; Kellow 1995; Kusunoki 2012; Lin 2009; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Nakamura 2017; Rösch 1987; Shen 2014; Tack 2009; Tack 2011; Talley 2000; Talley 2008a; Talley 2008b; Talley 2008c; Teixeira 2000; Vakil 2008a; Vakil 2008b; Wang 1995; Wong 2014; Yeoh 1997). Of these, 18 studies reported only dichotomous outcome (not symptom‐free or no symptom improvement) (Al‐Quorain 1995; Champion 1997; de Groot 1997; De Nutte 1989; Francois 1987; Hallerback 2002; Hansen 1998; Kusunoki 2012; Lin 2009; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Rösch 1987; Tack 2011; Talley 2008a; Teixeira 2000; Wang 1995; Wong 2014), four studies reported only continuous outcome (symptom scores) (Abid 2017; Jian 1989; Nakamura 2017; Tack 2009), and 11 studies reported both outcomes (Holtmann 2002; Holtmann 2006; Kellow 1995; Ma 2012; Shen 2014; Talley 2000; Talley 2008b; Talley 2008c; Vakil 2008a; Vakil 2008b; Yeoh 1997).
1.1 Not symptom‐free or no symptom improvement
Twenty‐nine studies that comprised a total of 10,044 participants (5949 participants in prokinetic group and 4095 controls in placebo group) reported the number of participants without resolution of symptom or no symptom improvement at the end of the study period (Al‐Quorain 1995; Champion 1997; de Groot 1997; De Nutte 1989; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Holtmann 2006; Kellow 1995; Kusunoki 2012; Lin 2009; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Rösch 1987; Shen 2014; Tack 2011; Talley 2000; Talley 2008a; Talley 2008b; Talley 2008c; Teixeira 2000; Vakil 2008a; Vakil 2008b; Wang 1995; Wong 2014; Yeoh 1997). The average percentage of not symptom‐free or no symptom improvement was 59.5% in prokinetic group, compared to 73.9% in placebo group. There was a statistically significant effect of prokinetic treatment in reducing global symptoms of functional dyspepsia (FD) (risk ratio (RR) of remaining dyspeptic symptom 0.81, 95% confidence interval (CI) 0.74 to 0.89; P < 0.00001) with considerable heterogeneity; I2 = 91% (P < 0.00001) (Analysis 1.1). NNTB = 7 (95% CI 5 to 12).The funnel plot was asymmetric (Figure 4); significant publication bias or small‐study effect is suggested (Egger's test, P = 0.02). Various subgroup analyses were done to explore the factors influencing heterogeneity.
1.1. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 1 Not symptom‐free or no symptom improvement.
4.
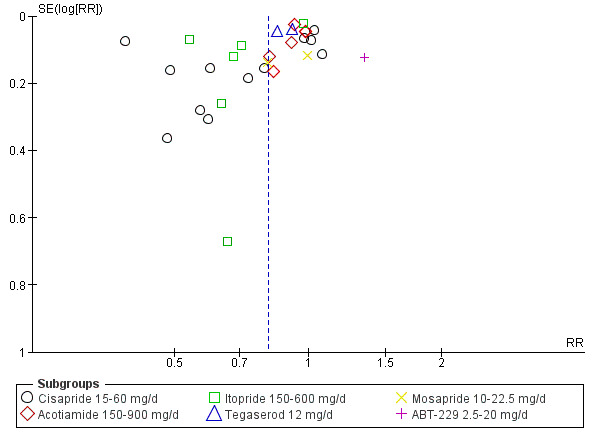
Funnel plot of comparison: 1 Prokinetic versus placebo, outcome: 1.1 Not symptom‐free or no symptom improvement.
Not symptom‐free or no symptom improvement according to individual prokinetic
Cisapride ๅ1 versus placebo
Twelve studies used cisapride as the active comparator (N = 1647) (Al‐Quorain 1995; Champion 1997; de Groot 1997; De Nutte 1989; Francois 1987; Hansen 1998; Holtmann 2002; Kellow 1995; Rösch 1987; Teixeira 2000; Wang 1995; Yeoh 1997). There was significant heterogeneity amongst the studies (I2 = 95%, P < 0.00001). In this subgroup of studies, there was a statistically significant effect of cisapride in reducing global symptoms of FD (RR 0.71, 95% CI 0.54 to 0.93; P = 0.01); NNTB = 4 (95% CI 3 to 17). In sensitivity analyses, the results remained robust with odds ratio (OR) or a fixed‐effect model or removal of three studies that were considered to be at high risk of bias (Champion 1997; Wang 1995; Yeoh 1997).
Acotiamide versus placebo
Six studies (Kusunoki 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Tack 2011; Talley 2008a) compared acotiamide with placebo (N = 2429) and showed statistically significant effect of acotiamide in global symptom improvement without heterogeneity amongst studies (RR 0.94, 95% CI 0.91 to 0.98 ; test of heterogeneity I2 = 0%, P = 0.49). NNTB 20 (95% CI 13 to 60). In sensitivity analyses, the results remained robust with OR or a fixed‐effect model.
Itopride versus placebo
Six studies (Holtmann 2006; Ma 2012; Shen 2014; Talley 2008b; Talley 2008c; Wong 2014) compared itopride with placebo and did not show a statistically significant difference (RR 0.70, 95% CI 0.47 to 1.03; P = 0.07; participants = 2066). There was significant heterogeneity amongst the studies (I2 = 97%, P < 0.00001). In sensitivity analysis, the results remained non‐significant after removing one study that was considered to be at high risk of bias (Ma 2012), but became statistically significant with OR (OR 0.39, 95% CI 0.22 to 0.71; P = 0.002) and a fixed‐effect model (RR 0.76, 95% CI 0.72 to 0.80; P < 0.00001) favouring itopride treatment.
Tegaserod versus placebo
Two studies (Vakil 2008a; Vakil 2008b) assessed the efficacy of tegaserod versus placebo in 2667 participants.There was insignificant moderate heterogeneity in these studies ( I2 = 41%; P = 0.19). They showed an evidence of better global symptom improvement in tegaserod compared to placebo, (RR 0.89, 95% CI 0.82 to 0.96; P = 0.003; NNTB 14, 95% CI 8 to 38). In sensitivity analyses, the results remained robust with OR or a fixed‐effect model.
Mosapride versus placebo
Two studies (Hallerback 2002; Lin 2009) used mosapride as an active comparator (N = 626) and did not demonstrate significant difference between mosapride and placebo in reducing global symptom (RR 0.91, 95% CI 0.73 to 1.13; I2 = 35%, P = 0.22). In sensitivity analyses, the results remained non‐significant with OR or a fixed‐effect model.
ABT‐229 versus placebo
One study (Talley 2000) compared ABT‐229 and placebo in 609 participants. The efficacy of ABT‐229 was significantly worse than placebo for global symptom improvement (RR 1.33, 95% CI 1.05 to 1.70; P = 0.02). In sensitivity analyses, the results remained the same with OR or a fixed‐effect model.
There was a significant heterogeneity amongst individual prokinetic (test for subgroup differences, I2 = 69.8%, P = 0.005). Recently, cisapride is not commercially available in many countries. When we removed cisapride from the analysis, the effect of a prokinetic in global symptom improvement remained robust in 8397 participants (RR 0.87, 95% CI 0.80 to 0.94; P = 0.0004). Furthermore, the significant heterogeneity amongst the rest of studies (I2 = 86%; P < 0.00001) and between individual prokinetic (I2 = 68.2%; P = 0.01) persisted.
1.2 Not symptom‐free or no symptom improvement according to each outcome
There was a statistically significant effect of prokinetic treatment in reducing global symptoms of FD (RR of remaining dyspeptic symptom = 0.81, 95% CI 0.74 to 0.89; participants = 10,044; studies = 29; I2 = 91%).
Sixteen of 29 studies reported incomplete symptom resolution in 4356 participants (Al‐Quorain 1995; De Nutte 1989; Francois 1987; Hansen 1998; Holtmann 2002; Kellow 1995; Lin 2009; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Rösch 1987; Shen 2014; Talley 2008b; Talley 2008c; Wang 1995; Yeoh 1997). Prokinetics showed the better efficacy in global symptom‐free, compared to placebo (RR 0.78, 95% CI 0.68 to 0.89; P = 0.0003) with considerable heterogeneity (I2 = 95%; P < 0.00001) (Analysis 1.2). In sensitivity analyses, the results remained robust with OR or a fixed‐effect model, or removal of two studies that were considered to be at high risk of bias (Wang 1995; Yeoh 1997).
1.2. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 2 Not symptom‐free or no symptom improvement, subgroup by definition.
The other 13 studies (Champion 1997; de Groot 1997; Hallerback 2002; Holtmann 2006; Kusunoki 2012; Ma 2012; Tack 2011; Talley 2000; Talley 2008a; Teixeira 2000; Vakil 2008a; Vakil 2008b; Wong 2014) in 5688 participants demonstrated the better global symptom improvement of prokinetic when compared to placebo (RR 0.86, 95% CI 0.78 to 0.94; P = 0008) with substantial heterogeneity (I2 = 59%: P = 0.004). (Analysis 1.2). In sensitivity analyses, the results remained robust with OR or a fixed‐effect model or removal of two studies that were considered to be at high risk of bias (Champion 1997; Ma 2012).
No statistical significant difference was seen in a test for subgroup differences (I2 = 26%, P = 0.24).
1.3 Not symptom‐free or no symptom improvement according to functional dyspepsia subtype
There was a statistically significant effect of prokinetic treatment in reducing global symptoms of FD (RR of remaining dyspeptic symptom = 0.81, 95% CI 0.74 to 0.89; participants = 10,044; studies = 29; I2 = 91%).
Nine studies included 5068 participants with postprandial distress syndrome (PDS) subtype (2828 prokinetics users and 2240 controls) and showed significant difference in global symptom‐free or improvement in prokinetic group (RR 0.78, 95% CI 0.65 to 0.92; P = 0.004) with considerable heterogeneity (I2 = 94%; P < 0.00001) (Analysis 1.3) (Lin 2009; Matsueda 2010b; Matsueda 2012; Shen 2014; Tack 2011; Talley 2008b; Talley 2008c; Wang 1995; Wong 2014). In sensitivity analyses, the results remained robust with OR or a fixed‐effect model, or removal of one study that was considered to be at high risk of bias (Wang 1995).
1.3. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 3 Not symptom‐free or no symptom improvement, subgroup by FD subtype.
There were 19 studies with 4944 participants that evaluated symptom improvement in both PDS and epigastric pain syndrome (EPS) subtypes (Al‐Quorain 1995; Champion 1997; de Groot 1997; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Holtmann 2006; Kellow 1995; Kusunoki 2012; Ma 2012; Matsueda 2010a; Rösch 1987; Talley 2000; Talley 2008a; Talley 2008b; Talley 2008c; Teixeira 2000; Yeoh 1997). This still showed the efficacy of prokinetic in reducing global dyspeptic symptom (RR 0.83, 95% CI 0.75 to 0.93; P = 0.001) with considerable heterogeneity (I2 = 89%: P < 0.00001) (Analysis 1.3). In sensitivity analyses, the results remained robust with OR or a fixed‐effect model or removal of three studies that were considered to be at high risk of bias (Champion 1997; Ma 2012; Yeoh 1997).
One small study (De Nutte 1989) evaluated participants with only EPS subtype showed the efficacy of prokinetic in global symptom improvement (RR 0.48, 95% CI 0.24 to 0.98; participants = 32). The results remained robust with OR in sensitivity analysis.
No statistical significant difference is seen in test for subgroup differences (I2 = 22.5%, P = 0.28).
1.4 Not symptom‐free or no symptom improvement according to type of publication
There was a statistically significant effect of prokinetic treatment in reducing global symptoms of FD (RR of remaining dyspeptic symptom = 0.81, 95% CI 0.74 to 0.89; participants = 10,044; studies = 29; I2 = 91%)
Most studies (26/29) were published as full‐text articles and showed the efficacy of prokinetics in global dyspeptic symptom‐free or improvement with considerable heterogeneity (RR 0.81, 95% CI 0.74 to 0.89; participants = 9309; I2 = 92%, P < 0.0001) (Al‐Quorain 1995; Champion 1997; de Groot 1997; De Nutte 1989; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Holtmann 2006; Kellow 1995; Kusunoki 2012; Lin 2009; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Rösch 1987; Shen 2014; Talley 2000; Talley 2008b; Talley 2008c; Teixeira 2000; Vakil 2008a; Vakil 2008b; Wang 1995; Yeoh 1997) (Analysis 1.4). In sensitivity analysis, the results remained significance after removing four studies that were considered to be at high risk of bias (Champion 1997; Ma 2012; Wang 1995; Yeoh 1997) as well as with OR or a fixed‐effect model.
1.4. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 4 Not symptom‐free or no symptom improvement, subgroup by publication type.
Three studies were published as conference abstracts only (Tack 2011; Talley 2008a; Wong 2014). This demonstrated no significant difference between prokinetics and placebo in global symptom‐free or improvement without heterogeneity (RR 0.88, 95% CI 0.77 to 1.00; participants = 735; I2 = 0%) (Analysis 1.4). In sensitivity analysis, the results became statistically significant with a fixed‐effect model (RR 0.87, 95% CI 0.76 to 0.99; P = 0.04) favouring prokinetic treatment, but remained non‐significance with OR.
No statistical significant difference was seen between the two subgroups (I2 = 14.4%, P = 0.28).
1.5 Not symptom‐free or no symptom improvement according to validity of assessment tool
There was a statistically significant effect of prokinetic treatment in reducing global symptoms of FD (RR of remaining dyspeptic symptom = 0.81, 95% CI 0.74 to 0.89; participants = 10,044; studies = 29; I2 = 91%).
Only one small study in 30 individuals (Wong 2014) used valid assessment tool (Leeds Dyspepsia questionnaire; LDQ) and reported no statistically significant difference between prokinetics and placebo (RR 0.66, 95% CI 0.18 to 2.44).
Twenty‐eight studies used non‐valid assessment tool and showed the efficacy of prokinetics in global symptom‐free or improvement (RR 0.81, 95% CI 0.74 to 0.89; participants = 10,044; I2 = 91%, P < 0.00001).(Al‐Quorain 1995; Champion 1997; de Groot 1997; De Nutte 1989; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Holtmann 2006; Kellow 1995; Kusunoki 2012; Lin 2009; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Rösch 1987; Shen 2014; Tack 2011; Talley 2000; Talley 2008a; Talley 2008b; Talley 2008c; Teixeira 2000; Vakil 2008a; Vakil 2008b; Wang 1995; Yeoh 1997) (Analysis 1.5)
1.5. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 5 Not symptom‐free or no symptom improvement, subgroup by assessment tool.
No statistical significant difference is seen in test for subgroup differences (I2 = 0%, P = 0.75).
1.6 Not symptom‐free or no symptom improvement according to duration of follow‐up (< 1 month versus 1 month or more)
There was a statistically significant effect of prokinetic treatment in reducing global symptoms of FD (RR of remaining dyspeptic symptom = 0.81, 95% CI 0.74 to 0.89; participants = 10,044; studies = 29; I2 = 91%).
Six studies (473 participants) were conducted for less than one month's duration of treatment and follow‐up(Francois 1987; Hansen 1998; Kusunoki 2012; Lin 2009; Shen 2014; Teixeira 2000). There was no significant difference between prokinetic and placebo (RR 0.77, 95% CI 0.59 to 1.01; P = 0.06). There was significant heterogeneity amongst studies (I2 = 77%, P = 0.0007) (Analysis 1.6). In sensitivity analyses, the results became statistically significant, favouring prokinetics with OR (OR 0.48, 95% CI 0.27 to 0.86; P = 0.01) or a fixed‐effect model (RR 0.87, 95% CI 0.79 to 0.96; P = 0.005).
1.6. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 6 Not symptom‐free or no symptom improvement, subgroup by follow‐up period.
In contrast, there was evidence of better efficacy in prokinetic treatment, compared to placebo when the duration of treatment and follow‐up was at least one month (N = 9571) (RR 0.81, 95% CI 0.74 to 0.90; I2 = 92%, P < 0.00001) (Al‐Quorain 1995; Champion 1997; de Groot 1997; De Nutte 1989; Hallerback 2002; Holtmann 2002; Holtmann 2006; Kellow 1995; Ma 2012; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Rösch 1987; Tack 2011; Talley 2000; Talley 2008a; Talley 2008b; Talley 2008c; Vakil 2008a; Vakil 2008b; Wang 1995; Wong 2014; Yeoh 1997) (Analysis 1.6). In sensitivity analysis, the results remained significant with OR or a fixed‐effect model or after removing four studies that were considered to be at high risk of bias (Champion 1997; Ma 2012; Wang 1995; Yeoh 1997).
No statistically significant difference was seen in a test for subgroup differences (I2 = 0%, P = 0.73).
1.7 Not symptom‐free or no symptom improvement according to risk of bias
There was a statistically significant effect of prokinetic treatment in reducing global symptoms of FD (RR of remaining dyspeptic symptom = 0.81, 95% CI 0.74 to 0.89; participants = 10,044; studies = 29; I2 = 91%).
Four studies with 1049 participants were assessed to be at high risk of bias and showed insignificant difference between prokinetics and placebo (RR 0.67, 95% CI 0.39 to 1.15) with significant heterogeneity between studies (I2 = 97%, P < 0.00001) ( Analysis 1.7) (Champion 1997; Ma 2012; Wang 1995; Yeoh 1997). In a sensitivity analysis, the results remained non‐significant with OR but became significant with a fixed‐effect model (RR 0.56, 95% CI 0.51 to 0.62).
1.7. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 7 Not symptom‐free or no symptom improvement, subgroup by risk of bias.
On the other hand, 21 studies with unclear risk of bias (N = 4883) (Al‐Quorain 1995; de Groot 1997; De Nutte 1989; Francois 1987; Hallerback 2002; Hansen 1998; Holtmann 2002; Kellow 1995; Kusunoki 2012; Lin 2009; Matsueda 2010a; Matsueda 2010b; Rösch 1987; Shen 2014; Tack 2011; Talley 2000; Talley 2008a; Talley 2008b; Talley 2008c; Teixeira 2000; Wong 2014) and four studies with low risk of bias (N = 4112) (Holtmann 2006; Matsueda 2012; Vakil 2008a; Vakil 2008b) showed significant efficacy of a prokinetic in global symptom improvement with RR 0.84, 95% CI 0.76 to 0.93; I2 = 87%, P < 0.0001; and RR 0.87, 95% CI 0.80 to 0.95; test for heterogeneity, I2 = 76%, P < 0.0001, respectively (Analysis 1.7). In sensitivity analysis, the results remained robust with OR or a fixed‐effect model in both groups.
No significant difference was seen between subgroups (I2 = 0%, P = 0.59).
1.8 Post‐treatment symptom scores
Six studies (N = 2914) reported post‐treatment symptom scores in 1459 participants taking prokinetic and 1455 participants taking placebo (Kellow 1995; Nakamura 2017; Shen 2014; Vakil 2008a; Vakil 2008b; Yeoh 1997). Overall, prokinetics showed statistically significant lower global symptom scores after treatment, compared to placebo (SMD ‐0.36, 95% CI ‐0.65 to ‐0.07; P = 0.02) There was significant heterogeneity amongst studies (I2 = 89%; P < 0.00001) (Analysis 1.8). In a sensitivity analysis, the results remained robust with a fixed‐effect model or when one study was removed, which was classified as high risk of bias (Yeoh 1997). Various subgroup analyses were done to explore the factors influencing heterogeneity.
1.8. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 8 Post‐treatment symptom scores (different scales used).
Post‐treatment symptom scores according to individual prokinetic
Six studies evaluated four different prokinetics versus placebo: tegaserod (Vakil 2008a; Vakil 2008b; N = 2656).
Tegaserod versus placebo: (SMD ‐0.13, 95% CI ‐0.24 to ‐0.02; participants = 2656; studies = 2; I2 = 50%)(Vakil 2008a; Vakil 2008b).
Cisapride versus placebo: (SMD ‐0.06, 95% CI ‐0.40 to 0.28; participants = 132; studies = 2; I2 = 0%)(Kellow 1995; Yeoh 1997.
Itopride versus placebo: (SMD ‐1.88, 95% CI ‐2.41 to ‐1.35; participants = 80; studies = 1; I2 = 0%)(Shen 2014).
Acotiamide versus placebo: (SMD ‐0.30, 95% CI ‐0.88 to 0.28; participants = 46; studies = 1; I2 = 0%)(Nakamura 2017).
Acotiamide measured post‐treatment symptoms scores. Only tegaserod and itopride showed significant lower post‐treatment symptom scores, compared to placebo (Analysis 1.8). In a sensitivity analysis, the results remained robust with a fixed‐effect model or when we removed one study which was classified as high risk of bias (Yeoh 1997). Signficant difference is seen amongst individual prokinetics (I2 = 92.7%, P < 0.00001).
1.9 Post‐treatment symptom scores according to functional dyspepsia subtype
Overall, prokinetics showed statistically significant lower global symptom scores after treatment, compared to placebo. (SMD ‐0.36, 95% CI ‐0.65 to ‐0.07; participants = 2914; studies = 6; I2 = 89%).
Four studies (N = 2782) assessed participants presenting with PDS subtype and showed significant efficacy of prokinetic in better post‐treatment symptom score (SMD ‐0.50, 95% CI ‐0.87 to ‐0.13; P = 0.008) with considerable heterogeneity (I2 = 93%; P < 0.00001) (Nakamura 2017; Shen 2014; Vakil 2008a; Vakil 2008b). In contrast, there was no evidence of post‐treatment symptom score difference in people with mixed subtypes of FD treated by prokinetic and placebo (SMD ‐0.06, 95% CI ‐0.40 to 0.28; N = 132; I2 = 0%, P = 0.45) (Analysis 1.9) (Kellow 1995; Yeoh 1997). In a sensitivity analysis, the results remained robust with fixed‐effect model or when removed Yeoh 1997 which was classified as high risk of bias. No FD patient with only EPS subtype was included in this outcome measurement. Signficant difference is seen between two subgroups (I2 = 65.6%, P = 0.09).
1.9. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 9 Post‐treatment symptom scores (different scales used), subgroup by FD subtype.
1.10 Post‐treatment symptom scores according to validity of assessment tool
Overall, prokinetics showed statistically significant lower global symptom scores after treatment, compared to placebo. (SMD ‐0.36, 95% CI ‐0.65 to ‐0.07; participants = 2914; studies = 6; I2 = 89%).
One study evaluated post‐treatment symptom scores in 46 participants by using a validated assessment tool, the validated gastrointestinal symptom rating scale (GSRS) in the Japanese edition, and found no significant difference between prokinetic and placebo (SMD ‐0.30, 95% CI ‐0.88 to 0.28; N = 46)(Nakamura 2017) . The other five studies used a non‐validated assessment tool and showed significantly lower post‐treatment symptom scores in prokinetic treatment.(SMD ‐0.37, 95% CI ‐0.69 to ‐0.05; N = 2868), with significant heterogeneity between studies (I2 = 91%, P < 0.00001)(Kellow 1995; Shen 2014; Vakil 2008a; Vakil 2008b; Yeoh 1997). (Analysis 1.10). In a sensitivity analysis, the results remained robust with a fixed‐effect model or when one study which was classified as high risk of bias was removed (Yeoh 1997) . No significant difference is seen between the two subgroups (I2 = 0%, P = 0.84).
1.10. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 10 Post‐treatment symptom scores (different scales used), subgroup by assessment tool.
1.11 Post‐treatment symptom scores according to duration of follow‐up (less than one month versus greater than one month)
Overall, prokinetics showed statistically significant lower global symptom scores after treatment, compared to placebo. (SMD ‐0.36, 95% CI ‐0.65 to ‐0.07; participants = 2914; studies = 6; I2 = 89%) (Analysis 1.11).
1.11. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 11 Post‐treatment symptom scores (different scales used), subgroup by follow‐up period.
Two studies (Nakamura 2017; Shen 2014) treated people with FD for less than one month and followed up the symptom at the end of treatment. They found no significant difference in post‐treatment symptom scores between prokinetic and placebo (SMD ‐1.09, 95% CI ‐2.64 to 0.45; N = 126), with significant heterogeneity between studies (I2 = 94%, P < 0.00001). On the other hand, four studies using at least one month of treatment and follow‐up in 2788 participants showed significantly lower post‐treatment symptom scores in prokinetic without heterogeneity (SMD ‐0.13, 95% CI ‐0.20 to ‐0.05; I2 = 0%, P = 0.44) (Analysis 1.11) (Kellow 1995; Vakil 2008a; Vakil 2008b; Yeoh 1997). In a sensitivity analysis, the results remained robust when we removed one study which was classified as high risk of bias (Yeoh 1997), but became favourable towards a prokinetic (SMD ‐1.16, 95% CI ‐1.56 to ‐0.77) with a fixed‐effect model in participants with less than one month of treatment duration and follow‐up period. No significant difference was seen between the two subgroups (I2 = 33.5%, P = 0.22).
1.12 Post‐treatment symptom scores according to risk of bias
Overall, prokinetics showed statistically significant lower global symptom scores after treatment, compared to placebo. (SMD ‐0.36, 95% CI ‐0.65 to ‐0.07; participants = 2914; studies = 6; I2 = 89%).
One small study with high risk of bias (Yeoh 1997; N = 76) and three studies with unclear risk of bias (Kellow 1995; Nakamura 2017; Shen 2014; N = 182) showed no evidence of using prokinetic for improving post‐treatment symptom scores (SMD ‐0.17, 95% CI ‐0.62 to 0.28 and SMD ‐0.70; 95% CI ‐1.91 to 0.51, respectively). Significant heterogeneity was seen between studies with and unclear risk of bias (I2 = 93%, P < 0.00001). Two studies with low risk of bias (Vakil 2008a; Vakil 2008b; N = 2656) demonstrated the efficacy of prokinetic in post‐treatment symptom scores (SMD ‐0.13, 95% CI ‐0.24 to ‐0.02; P = 0.02), with substantial heterogeneity (I2 = 50%; P =0.16) (Analysis 1.12). No significant difference was seen between subgroups (I2 = 0%; P = 0.65). In a sensitivity analysis, the results remained robust with a fixed‐effect model.
1.12. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 12 Post‐treatment symptom scores (different scales used), subgroup by risk of bias.
1.13 Mean difference symptom scores (post‐treatment minus pre‐treatment)
Eleven studies (N = 1622) evaluating four different prokinetics reported pre‐ and post‐treatment symptom scores or the change in symptom scores (prokinetic versus placebo) (Abid 2017; Holtmann 2002; Holtmann 2006; Jian 1989; Kellow 1995; Ma 2012; Nakamura 2017; Shen 2014; Tack 2009; Talley 2000; Yeoh 1997). There was no difference in mean difference symptom scores (SMD ‐0.65, 95% CI ‐1.50 to 0.20; I2 = 98%, P < 0.00001) (Analysis 1.13). In a sensitivity analysis, the results remained robust when we removed two studies and which were classified as high risk of bias, but became favourable towards prokinetics with a fixed‐effect model (SMD ‐0.73, 95% CI ‐0.84 to ‐0.62) (Ma 2012; Yeoh 1997).
1.13. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 13 Mean difference symptom scores (post‐treatment ‐ pre‐treatment, different scales used), subgroup by prokinetic.
Mean difference symptom scores according to individual prokinetic
All four individual prokinetics measuring the change in symptom scores failed to show the efficacy of prokinetic in symptom score reduction.
Itopride versus placebo: (SMD ‐1.53, 95% CI ‐3.42 to 0.37; participants = 860; studies = 4) (Abid 2017; Holtmann 2006; Ma 2012; Shen 2014).
Cisapride versus placebo: (SMD ‐0.28, 95% CI ‐0.71 to 0.16; participants = 280; studies = 4) (Holtmann 2002; Jian 1989; Kellow 1995; Yeoh 1997).
Acotiamide versus placebo: (SMD ‐0.06, 95% CI ‐0.47 to 0.35; participants = 108; studies = 2) (Nakamura 2017; Tack 2009).
ABT‐229 versus placebo: (SMD 0.14, 95% CI ‐0.07 to 0.34; participants = 574; studies = 1) (Talley 2000).
A test for subgroup differences showed moderate heterogeneity ( I2 = 48.0%, P = 0.12). When we removed two studies which were classified as high risk of bias from itopride (Ma 2012) and cisapride (Yeoh 1997), respectively, the results remained robust. In contrast, the results became favourable towards itopride (SMD ‐1.75, 95% CI ‐1.93 to ‐1.57) and cisapride (SMD ‐0.35, 95% CI ‐0.58 to ‐0.11) when a fixed‐effect model was applied in a sensitivity analysis.
1.14 Mean difference symptom scores according to functional dyspepsia subtype
There was no difference in mean difference symptom scores between prokinetic and placebo. (SMD ‐0.65, 95% CI ‐1.50 to 0.20; participants = 1822; studies = 11; I2 = 98%).
There was little to no difference in the efficacy of prokinetics in reducing symptom scores assessed in participants with only the PDS subtype of FD (SMD ‐0.68, 95% CI ‐1.65 to 0.29; participants = 154; studies = 3)(Jian 1989; Nakamura 2017; Shen 2014). There was little or no difference in the studies evaluating people with mixed subtypes (SMD ‐0.64, 95% CI ‐1.70 to 0.42; participants = 1668; studies = 8)(Abid 2017; Holtmann 2002; Holtmann 2006; Kellow 1995; Ma 2012; Tack 2009; Talley 2000; Yeoh 1997). There was no EPS subtype studies in this outcome.Analysis 1.17. No significant difference was seen between subgroups (I2 = 0%, P = 0.88). In a sensitivity analysis, the results remained robust when we removed two studies which were classified as high risk of bias (Ma 2012; Yeoh 1997), but became favourable towards prokinetics in the PDS subtype (SMD ‐0.81, 95%CI ‐1.15 to ‐0.47) and mixed subtype (SMD ‐0.72, 95%CI ‐0.84 to ‐0.60) with a fixed‐effect model.
1.17. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 17 Mean difference symptom scores (post‐treatment ‐ pre‐treatment, different scales used), subgroup by follow‐up period.
1.15 Mean difference symptom scores according to method of calculating mean difference
There was no difference in mean difference symptom scores between prokinetic and placebo. (SMD ‐0.65, 95% CI ‐1.50 to 0.20; participants = 1822; studies = 11; I2 = 98%).
There were seven studies (N = 1564) reporting the mean difference between pre‐ and post‐treatment symptom scores (Abid 2017; Holtmann 2002; Holtmann 2006; Jian 1989; Ma 2012; Tack 2009; Talley 2000). There was no difference in reducing symptom scores with prokinetic treatment when compared to placebo (SMD ‐0.78, 95% CI ‐1.98 to 0.42). In a sensitivity analysis, the results remained robust when we removed one study which was at high risk of bias (Ma 2012), but became favourable towards prokinetics (SMD ‐0.80, 95%CI ‐0.93 to ‐0.68) with a fixed‐effect model. Another four studies (N = 258) reported only pre‐ and post symptom scores, and for which we calculated the mean difference and SD (Kellow 1995; Nakamura 2017; Shen 2014; Yeoh 1997). The pooled data did not show efficacy of prokinetics over placebo (SMD ‐0.42, 95% CI ‐1.20 to 0.36; I2= 99%, P < 0.00001). No difference is seen between subgroups (I2 = 0%, P = 0.62). In a sensitivity analysis, the results remained robust when we removed a study which was classified as a high risk of bias (Yeoh 1997) but became favourable toward prokinetics (SMD ‐0.44, 95%CI ‐0.70 to ‐0.19) with a fixed‐effect model.
1.16 Mean difference symptom scores according to validity of assessment tool
There was no difference in mean difference symptom scores between prokinetic and placebo. (SMD ‐0.65, 95% CI ‐1.50 to 0.20; participants = 1822; studies = 11; I2 = 98%).
Neither four studies (N = 720) (Abid 2017; Holtmann 2002; Holtmann 2006; Nakamura 2017) using a validated assessment tool, nor seven studies (N = 1102) (Jian 1989; Kellow 1995; Ma 2012; Shen 2014; Tack 2009; Talley 2000; Yeoh 1997) using a non‐validated assessment tool showed efficacy of prokinetics in reducing symptom score after treatment (SMD ‐1.24, 95% CI ‐3.25 to 0.78; and SMD ‐0.30, 95% CI ‐0.72 to 0.11, respectively,). Signficant heterogeneity was seen in both subgroups (I2 = 99%, P < 0.0001 and I2 = 87%, P < 0.0001, respectively). No significant difference was seen between subgroups (I2=0%, P = 0.37). Analysis 1.16. In a sensitivity analysis, the results remained robust when we removed two studies which were considered to be at high risk of bias, but became favourable towards prokinetics in both validated (SMD ‐2.05, 95%CI ‐2.26 to ‐1.84) and non‐validated assessment tool (SMD ‐0.18, 95%CI ‐0.32 to ‐0.05) with a fixed‐effect model (Ma 2012; Yeoh 1997).
1.16. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 16 Mean difference symptom scores (post‐treatment ‐ pre‐treatment, different scales used), subgroup by assessment tool.
1.17 Mean difference symptom scores according to duration of follow‐up (< 1 month versus 1 month or more)
There was no difference in mean difference symptom scores between prokinetic and placebo. (SMD ‐0.65, 95% CI ‐1.50 to 0.20; participants = 1822; studies = 11; I2 = 98%).
Three studies were conducted during less than one month in duration of treatment and follow‐up (Nakamura 2017; Shen 2014; Tack 2009) (N = 188).There was no difference in reducing symptom scores between prokinetic and placebo in pooled data from these three studies (SMD ‐0.56, 95% CI ‐1.59 to 0.46). There was no difference in reducing symptom scores between prokinetic and placebo in pooled data from the remaining eight studies (N = 1634) which treated people with FD and assessed symptom scores at least one month after treatment (SMD ‐0.68, 95% CI ‐1.75 to 0.38)(Abid 2017; Holtmann 2002; Holtmann 2006; Jian 1989; Kellow 1995; Ma 2012; Talley 2000; Yeoh 1997). Signficant heterogeneity was seen in both subgroups (I2 = 90%, P < 0.0001 and I2 = 99%, P < 0.0001, respectively). No significant difference is seen between subgroups (I2 = 0%, P = 0.88) Analysis 1.17. In a sensitivity analysis, the results remained robust when we removed two studies which were classified as high risk of bias, but became favourable towards prokinetics in both less than one month's treatment (SMD ‐0.66, 95%CI ‐0.97 to ‐0.34) and one month or more in duration of treatment and follow‐up (SMD ‐0.74, 95%CI ‐0.86 to ‐0.62) with a fixed‐effect model (Ma 2012; Yeoh 1997).
1.18 Mean difference symptom scores according to risk of bias
There was no difference in mean difference symptom scores between prokinetic and placebo. (SMD ‐0.65, 95% CI ‐1.50 to 0.20; participants = 1822; studies = 11; I2 = 98%).
Two studies (N = 302) with high risk of bias (Ma 2012; Yeoh 1997), and one study (N = 523) with low risk of bias (Holtmann 2006) showed an efficacy of prokinetic in reducing symptom scores (SMD ‐0.40, 95% CI ‐0.63 to ‐0.17; and SMD ‐3.80, 95% CI ‐4.10 to ‐3.50, respectively). There was no heterogeneity between high risk of bias studies (I2 = 0%). In a sensitivity analysis, the results remained robust with a fixed‐effect model. In contrast, eight studies with unclear risk of bias (Abid 2017; Holtmann 2002; Jian 1989; Kellow 1995; Nakamura 2017; Shen 2014; Tack 2009; Talley 2000) demonstrated no difference in symptom scores reduction between prokinetics and placebo (SMD 0.32;95% CI ‐0.76 to 0.11;I2= 86%, P < 0.001) Analysis 1.18. In a sensitivity analysis, the results became favourable towards prokinetics (SMD ‐0.17, 95%CI ‐0.32 to ‐0.03) with a fixed‐effect model. Significant subgroup difference was seen (I2 = 99.4%, P < 0.001).
1.18. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 18 Mean difference symptom scores (post‐treatment ‐ pre‐treatment, different scales used), subgroup by risk of bias.
Quality of life (QoL)
Most studies (four of six) that reported data related to QoL reported overall QoL (Holtmann 2006; Ma 2012; Tack 2009; Wong 2014).Two studies did not report overall QoL, thus the most generalised sub‐domain was chosen; daily activity score from short form Nepean Dyspepsia Index (NDI) (Matsueda 2012) and physical functioning score from SF‐8 (Nakamura 2017).
1.19 Improved QoL
One small study (Wong 2014) (N = 30) reported data for the number of participants with improved QoL, it did not show the benefit of itopride over placebo in QoL improvement (RR 1.17, 95% CI 0.54 to 2.54) (Analysis 1.19).
1.19. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 19 Improved QoL.
1.20 Post QoL scores
One small study (Nakamura 2017) (N = 46) did not show the benefit of acotiamide over placebo in physical functioning sub‐domain of post‐treatment QoL scores (SMD 0.24, 95% CI ‐0.34 to 0.82; participants = 46; studies = 1; I2 = 0%) (Analysis 1.20).
1.20. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 20 Post QoL scores.
1.21 Mean difference QoL scores (post‐treatment ‐ pre‐treatment)
The pooled data from five studies (N = 1774)failed to show the difference in QoL score change when using prokinetics versus placebo (SMD 0.11, 95% CI ‐0.10 to 0.33; I2 = 32%, P = 0.23)(Analysis 1.21)(Holtmann 2006; Ma 2012; Matsueda 2012; Nakamura 2017; Tack 2009). Of those, three studies (N = 1000) (Matsueda 2012; Nakamura 2017; Tack 2009), showed no difference between acotiamide and placebo in the change of QoL scores (SMD ‐0.16, 95% CI ‐0.79 to 0.47; I2 = 84%, P = 0.002), whereas two studies (N = 774) (Holtmann 2006; Ma 2012), demonstrated a change of QoL score after itopride treatment, compared to placebo without heterogeneity (SMD 0.24, 95% CI 0.08 to 0.39; I2 = 0%, P = 0.80). No significant difference was seen between subgroups (I2 = 32%, P = 0.23). In a sensitivity analysis, the overall result remained non‐significant when we removed the studies that did not report overall QoL (Matsueda 2012; Nakamura 2017).
1.21. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 21 Change of QoL scores (post‐treatment ‐ pre‐treatment, different scales).
1.22 Adverse events
From pooled data of four different prokinetics in 17 studies, the adverse events were found to be 29.3% with prokinetic treatment and 30.8% with placebo. There was no association between prokinetic and any adverse events (RR 1.09, 95% CI 0.95 to 1.25; participants = 3811; studies = 17; I2 = 18%, P = 0.25) (Abid 2017; Al‐Quorain 1995; Champion 1997; De Nutte 1989; Hansen 1998; Holtmann 2002; Holtmann 2006; Jian 1989; Kellow 1995; Li 2005; Matsueda 2010a; Matsueda 2010b; Matsueda 2012; Rösch 1987; Wang 1995; Wong 2014; Yeoh 1997)(Analysis 1.22). The following adverse events of individual prokinetic were analysed.
1.22. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 22 Adverse events.
Cisapride versus placebo: Ten studies (N = 1482) were pooled (Al‐Quorain 1995; Champion 1997; De Nutte 1989; Hansen 1998; Holtmann 2002; Jian 1989; Kellow 1995; Rösch 1987; Wang 1995; Yeoh 1997). This prokinetic was the sole medication that was significantly associated with the occurrence of adverse effect (RR 1.31, 95% CI 1.03 to 1.65; P = 0.03) No heterogeneity was detected amongst the studies ( I2 = 0%; P = 0.58).
Acotiamide versus placebo: Three studies (N = 1660) showed no significantly adverse event in acotiamide treatment, compared to placebo (RR 0.98, 95% CI 0.83 to 1.16) without significant heterogeneity (I2 = 18%, P = 0.29)(Matsueda 2010a; Matsueda 2010b; Matsueda 2012).
Itopride versus placebo: Three studies (N = 609) did not show adverse events from itopride (RR 1.02, 95% CI 0.80 to 1.31)(Abid 2017; Holtmann 2006; Wong 2014). A test for heterogeneity was not possible, because only one study reported any events.
Mosapride versus placebo: One small study (N = 60) did not demonstrate any adverse events from mosapride (RR 1.50, 95% CI 0.27 to 8.34)(Li 2005).
No significant difference was seen between subgroups (I2 = 25.9%, P = 0.26). In a sensitivity analysis, the results remained robust when calculating an OR or applying a fixed‐ effect model.
The funnel plot displayed asymmetry (Figure 5) (Egger's test, P = 0.005).
5.
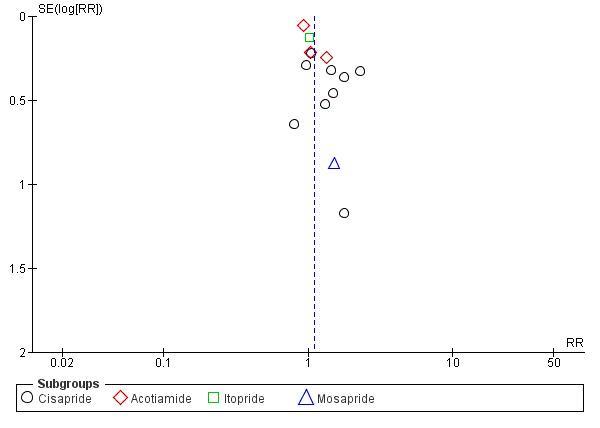
Funnel plot of comparison: 1 Prokinetic versus placebo, outcome: 1.22 Adverse events.
2 Prokinetic versus prokinetic
Six prokinetics contributed data for six comparisons between prokinetics, Nine of 10 included studies reported data for symptom‐free or no symptom improvement (Analysis 2.1).
2.1. Analysis.
Comparison 2 Prokinetic versus prokinetic, Outcome 1 Not symptom‐free or no symptom improvement.
2.1 Not symptom‐free or no symptom improvement
Itopride 50 mg three times a day versus domperidone 10 mg three times a day
All five studies (N = 932) were from China and used non‐validated assessment tools. All studies were at an unclear risk of bias. They failed to demonstrate the difference in symptom improvement when comparing two drugs (RR 1.00, 95% CI 0.89 to 1.13; I2 = 0%, P = 0.56) (Analysis 3.1) (Li 2005; Mo 2003; Sun 2003; Zhou 2000; Zhu 2005).There was no difference in the efficacy of the two drugs in reducing symptoms by subgroup analysis of not symptom‐free (RR 0.95, 95% CI 0.82 to 1.12)(Li 2005; Zhou 2000; Zhu 2005) or no symptom improvement (RR 1.09, 95% CI 0.89 to 1.33)(Mo 2003; Sun 2003) , with no significant difference between subgroups (I2 = 0%, P = 0.56). Three studies did not report global symptoms, thus the individual symptom most experienced by the participants was extracted; epigastric discomfort (Mo 2003), post‐prandial fullness (Sun 2003), and early satiety (Zhu 2005). For the two studies that reported global symptoms, the result remained robust (RR 0.92, 95% CI 0.73 to 1.17; N = 417; I2 = 43%, P = 0.18), which was not significantly different than the three studies that reported individual symptoms only (RR 1.08, 95%CI 0.90 to 1.29; N = 515), I2=5.4%, P = 0.30 (Analysis 3.2).
3.1. Analysis.
Comparison 3 Itopride 50 mg three times a day versus domperidone 10 mg three times a day, Outcome 1 Not symptom‐free or no symptom improvement.
3.2. Analysis.
Comparison 3 Itopride 50 mg three times a day versus domperidone 10 mg three times a day, Outcome 2 Not symptom‐free or no symptom improvement, subgroup by symptom type.
One study (Li 2005) (N = 200) reported post‐treatment symptom score as well as mean difference symptom score and showed no difference between two drugs (MD ‐0.20, 95% CI ‐0.68 to 0.28; Analysis 3.3 and MD 0.00, 95% CI ‐1.11 to 1.11; Analysis 3.4).
3.3. Analysis.
Comparison 3 Itopride 50 mg three times a day versus domperidone 10 mg three times a day, Outcome 3 Post treatment scores.
3.4. Analysis.
Comparison 3 Itopride 50 mg three times a day versus domperidone 10 mg three times a day, Outcome 4 Mean difference symptom scores (post‐treatment ‐ pre‐treatment).
Five studies (N = 952) assessed adverse events. Pooled data did not show a significant difference in adverse events between itopride and domperidone (RR 0.88, 95% CI 0.48 to 1.61; P=0.68,I2 = 0%, P = 0.81) (Analysis 3.5) (Li 2005; Mo 2003; Sun 2003; Zhou 2000; Zhu 2005).
3.5. Analysis.
Comparison 3 Itopride 50 mg three times a day versus domperidone 10 mg three times a day, Outcome 5 Adverse events.
DA‐9701 30 mg three times a day versus itopride 50 mg three times a day
Only one study from Korea for this comparator was found (Choi 2015). This study was assessed to be at a low risk of bias. It used a non‐validated assessment tool and did not show significant difference in not symptom‐free (RR 0.99, 95% CI 0.96 to 1.01; N = 464) (Analysis 4.1), post‐treatment epigastric pain scores (MD 0.09, 95% CI ‐0.08 to 0.26; N = 455) (Analysis 4.2), mean difference epigastric pain scores (MD ‐0.05, 95% CI ‐0.28 to 0.18; N = 455) (Analysis 4.3), post‐treatment QoL score assessed by daily activity scores of NDI (MD 0.29, 95% CI ‐2.94 to 3.52; N = 455) (Analysis 4.4), and change of QoL assessed by daily activity scores of NDI (MD ‐0.56, 95% CI ‐3.78 to 2.66; N = 455) (Analysis 4.5), as well as adverse events (RR 1.12, 95% CI 0.61 to 2.06; N = 464) (Analysis 4.6) between these two prokinetics.
4.1. Analysis.
Comparison 4 DA‐9701 30 mg three times a day versus itopride 50 mg three times a day, Outcome 1 Not symptom‐free.
4.2. Analysis.
Comparison 4 DA‐9701 30 mg three times a day versus itopride 50 mg three times a day, Outcome 2 Post‐treatment scores.
4.3. Analysis.
Comparison 4 DA‐9701 30 mg three times a day versus itopride 50 mg three times a day, Outcome 3 Mean difference symptom scores.
4.4. Analysis.
Comparison 4 DA‐9701 30 mg three times a day versus itopride 50 mg three times a day, Outcome 4 Post‐treatment NDI QoL score (interference with daily activities).
4.5. Analysis.
Comparison 4 DA‐9701 30 mg three times a day versus itopride 50 mg three times a day, Outcome 5 Change of NDI QoL score (interference with daily activities).
4.6. Analysis.
Comparison 4 DA‐9701 30 mg three times a day versus itopride 50 mg three times a day, Outcome 6 Adverse events.
Cinitapride 1 mg three times a day versus domperidone 10 mg three times a day
Only one study from China compared cinitapride 1 mg three times a day with domperidone 10 mg three times a day by using validated 7‐point Likert score (Du 2014). This study was assessed to be at an unclear risk of bias. It demonstrated a significantly better post‐treatment symptom scores compared to domperidone (MD ‐1.10, 95% CI ‐2.03 to ‐0.17; N = 344) (Analysis 5.2), but no difference in global symptom improvement (RR 0.80, 95% CI 0.51 to 1.27; N = 383) (Analysis 5.1), calculated mean difference symptom score (MD ‐0.50, 95% CI ‐2.03 to 1.03; N = 344) (Analysis 5.3), and adverse events were significantly lower in cinitapride (RR 0.60, 95% CI 0.37 to 0.97; N = 383) (Analysis 5.4), between these two prokinetics.
5.2. Analysis.
Comparison 5 Cinitapride 1 mg three times a day versus domperidone 10 mg three times a day, Outcome 2 Post‐treatment scores.
5.1. Analysis.
Comparison 5 Cinitapride 1 mg three times a day versus domperidone 10 mg three times a day, Outcome 1 No symptom improvement.
5.3. Analysis.
Comparison 5 Cinitapride 1 mg three times a day versus domperidone 10 mg three times a day, Outcome 3 Mean difference symptom scores (post‐treatment ‐ pre‐treatment).
5.4. Analysis.
Comparison 5 Cinitapride 1 mg three times a day versus domperidone 10 mg three times a day, Outcome 4 Adverse events.
Mosapride 5 mg three times a day versus domperidone 10 mg three times a day
One Chinese‐language study compared mosapride 5 mg three times a day with domperidone 10 mg three times a day (Chen 2004). This study was considered to be at an unclear risk of bias and used an un‐validated assessment tool. It showed better efficacy of mosapride in complete upper abdominal distension resolution (RR 0.72, 95% CI 0.54 to 0.97; N = 212) (Analysis 6.1), and reported mean difference overall symptom scores (MD ‐1.40, 95% CI ‐2.36 to ‐0.44; N = 222) (Analysis 6.2), compared to domperidone. The adverse events were not different between the two prokinetics (RR 0.68, 95% CI 0.33 to 1.42; N = 222) (Analysis 6.3).
6.1. Analysis.
Comparison 6 Mosapride 5 mg three times a day versus domperidone 10 mg three times a day, Outcome 1 Not symptom‐free.
6.2. Analysis.
Comparison 6 Mosapride 5 mg three times a day versus domperidone 10 mg three times a day, Outcome 2 Mean difference symptom scores.
6.3. Analysis.
Comparison 6 Mosapride 5 mg three times a day versus domperidone 10 mg three times a day, Outcome 3 Adverse events.
Itopride 50 mg three times a day versus mosapride 5 mg three times a day
One small study (N = 60) from India demonstrated significantly better efficacy of itopride 50 mg three times a day than mosapride 5 mg three times a day in complete global symptom resolution (RR 0.62, 95% CI 0.39 to 0.99) (Analysis 7.1), and post‐treatment epigastric pain scores (MD ‐0.36, 95% CI ‐0.62 to ‐0.10) (Analysis 7.2), but not in calculated mean difference epigastric pain scores (MD 0.17, 95% CI ‐0.31 to 0.65) (Analysis 7.3), compared to mosapride (Amarapurkar 2004). All assessment tools were un‐validated. The adverse events were not different between the two prokinetics (RR 0.09, 95% CI 0.01 to 1.57) (Analysis 7.4).This study was assessed to be at an unclear risk of bias.
7.1. Analysis.
Comparison 7 Itopride 50 mg three times a day versus mosapride 5 mg three times a day, Outcome 1 Not symptom‐free.
7.2. Analysis.
Comparison 7 Itopride 50 mg three times a day versus mosapride 5 mg three times a day, Outcome 2 Post‐treatment scores.
7.3. Analysis.
Comparison 7 Itopride 50 mg three times a day versus mosapride 5 mg three times a day, Outcome 3 Mean difference symptom scores (post‐treatment ‐ pre‐treatment).
7.4. Analysis.
Comparison 7 Itopride 50 mg three times a day versus mosapride 5 mg three times a day, Outcome 4 Adverse events.
Metoclopramide 10 mg three times a day versus domperidone 10 mg three times a day
Another small study (N = 73) from India applied the validated Short‐Form Leeds Dyspepsia Questionnaire (SF‐LDQ) for evaluating dyspeptic symptom but failed to show difference in post‐treatment (MD ‐0.14, 95% CI ‐0.58 to 0.30) (Analysis 8.1) (Singh 2015). The study calculated mean difference symptom score (MD ‐0.02, 95% CI ‐0.61 to 0.57) (Analysis 8.2) between metoclopramide 10 mg three times a day and domperidone 10 mg three times a day. This study was assessed to be at unclear risk of bias .
8.1. Analysis.
Comparison 8 Metoclopramide 10 mg three times a day versus domperidone 10 mg three times a day, Outcome 1 Post‐treatment scores.
8.2. Analysis.
Comparison 8 Metoclopramide 10 mg three times a day versus domperidone 10 mg three times a day, Outcome 2 Mean difference symptom scores.
Other prokinetics versus domperidone 10 mg three times a day
The most commonly used comparator was domperidone 10 mg three times a day, which was reported in eight of the 10 studies. We combined data for other prokinetics with domperidone 10 mg three times a day.
Not symptom‐free or no symptom improvement
Seven pooled studies (N = 1527) failed to demonstrate the difference between three different prokinetics (itopride 50 mg three times a day, cinitapride 1 mg three times a day and mosapride 5 mg three times a day) and domperidone (RR 0.94, 95% CI 0.83 to 1.07; P = 0.35) with low heterogeneity (I2 = 22%; P = 0.27)(Chen 2004; Du 2014; Li 2005; Mo 2003; Sun 2003; Zhou 2000; Zhu 2005) . There was no statistically significant difference amongst three type of prokinetics (I2 = 55.2%; P = 0.11) Analysis 9.1. In a sensitivity analysis, the results remained robust with OR or a fixed‐effect model.
9.1. Analysis.
Comparison 9 Other prokinetics versus domperidone 10 mg three times a day, Outcome 1 Not symptom‐free or no symptom improvement.
Post‐treatment symptom score
Three prokinetics (cinitapride 1 mg three times a day, itopride 50 mg three times a day and metoclopramide 10 mg three times a day) from three studies showed better post‐treatment overall symptom score, compared to domperidone without heterogeneity (SMD ‐0.19, 95% CI ‐0.35 to ‐0.03; N = 617; I2 = 0%, P = 0.74) (Analysis 9.2) (Du 2014; Li 2005; Singh 2015). Although SMD was used in the pooled analysis, significant difference was still only seen between cinitapride and domperidone. In a sensitivity analysis, the results remained robust with a fixed‐effect model.
9.2. Analysis.
Comparison 9 Other prokinetics versus domperidone 10 mg three times a day, Outcome 2 Post‐treatment scores.
Mean difference symptom score
Four prokinetics (cinitapride 1 mg three times a day, mosapride 5 mg three times a day, itopride 50 mg three times a day and metoclopramide 10 mg three times a day) from four studies in 839 participants failed to show the efficacy of other prokinetics over domperidone in reducing mean symptom scores (SMD ‐0.13, 95% CI ‐0.31 to 0.05; test for subgroup differences, I2 = 38%, P = 0.19) (Analysis 9.3) (Chen 2004; Du 2014; Li 2005; Singh 2015). Although SMD is used in the pooled analysis, a significant difference was still only seen between mosapride and domperidone. A sensitivity analysis was done with a fixed‐effect model and after removing two studies which used a calculated mean difference, the result remained robust (Du 2014; Singh 2015).
9.3. Analysis.
Comparison 9 Other prokinetics versus domperidone 10 mg three times a day, Outcome 3 Mean difference symptom scores.
Adverse events
Pooled data from seven studies; five of itopride (Li 2005; Mo 2003; Sun 2003; Zhou 2000; Zhu 2005), one of cinitapride (Du 2014) and one of mosapride (Chen 2004), demonstrated the overall adverse events from these three prokinetics were lesser than domperidone, without heterogeneity (RR 0.69, 95% CI 0.50 to 0.97; participants = 1557; , I2 = 0%, P = 0.86) (Analysis 9.4). Of those, significant difference was only seen between cinitapride 1 mg three times a day versus domperidone 10 mg three times a day (RR 0.60, 95% CI 0.37 to 0.97). No significant difference was seen between subgroups (I2 = 0%, P = 0.62). In a sensitivity analysis, the results remained robust when calculating an OR or using a fixed‐effect model.
9.4. Analysis.
Comparison 9 Other prokinetics versus domperidone 10 mg three times a day, Outcome 4 Adverse events.
Discussion
Summary of main results
We included 43 randomised controlled trials (RCTs) in this meta‐analysis. Of those, 33 studies evaluated the efficacy of prokinetic and placebo, whereas 10 studies compared two types of prokinetics. Of the 33 RCTs comparing prokinetic and placebo, 18 studies reported only a dichotomous outcome (not symptom‐free or no symptom improvement),four studies reported only a continuous outcome (symptom scores), and 11 studies reported both outcomes.
Amongst the 10 RCTs comparing two types of prokinetics, four studies reported only dichotomous outcome (not symptom‐free or no symptom improvement), one study reported only a continuous outcome (symptom scores), and five studies reported both outcomes.
The primary outcome of this review was "not symptom‐free" or "no symptom improvement". We used the most stringent definition of not symptom‐free or no overall symptom improvement at the end of treatment. We included both outcomes of "not symptom‐free" and "no symptom improvement" since not all studies evaluated "not symptom‐free", which is the ultimate endpoint of treatment in dyspepsia.
Prokinetic versus placebo
This review found that a prokinetic can improve dyspeptic symptoms (RR 0.81, 95% CI 0.74 to 0.89; number needed to treat for an additional beneficial outcome (NNTB) 7;95% CI 5 to 12), either in studies that reported "symptom‐free" or only reported symptom improvement. Of those, cisapride, acotiamide and tegaserod were efficacious in reducing dyspeptic symptoms with an NNTB of 4, 20 and 14, respectively. In contrast, the efficacy of ABT‐229 was significantly worse than placebo for global symptom improvement. Itopride and mosapride were not different from placebo, when assessing symptom‐free or symptom improvement. This outcome changed to favouring itopride in sensitivity analysis using an OR and a fixed‐effect model, whereas the results from other prokinetics were robust.
In subgroup analysis, studies published solely as a conference abstract, duration of treatment/follow‐up less than a month, used a validated assessment tool, or that were at a high risk of bias resulted in an insignificant difference in reducing dyspeptic symptoms between prokinetics and placebo, although the number of participants was small. A significant difference between prokinetic versus placebo was seen in other subgroups and in all dyspepsia subtypes.
Significant heterogeneity was seen between studies. There was substantial heterogeneity amongst individual prokinetics, but no statistically significant subgroup differences related to the type of reported outcomes (not symptom‐free or no symptom improvement), dyspepsia subtypes ( postprandial distress syndrome (PDS), epigastric pain syndrome (EPS) or mixed), type of publication (full‐text article or abstract), type of assessment tools (validated or non‐validated tool), duration of treatment and follow‐up (less than one month or greater than one month), and risk of bias (high, unclear or low risk).
Cisparide is not available in most countries, but the effect of a prokinetic in global symptom improvement remained robust when cisapride was excluded from the analysis. The significant heterogeneity amongst the rest of studies and between individual prokinetics persisted even when cisapride was excluded. Choice of prokinetic drugs is still limited to acotiamide and tegaserod with a high NNTB. No studies comparing metoclopramide or domperidone versus placebo were identified.
Most studies in this analysis did not define random sequence generation or allocation method; four studies were considered at high risk of bias.Thus, they were considered as serious in study limitations for quality of study assessment. Significant publication bias or small‐study effect is suggested (Egger's test, P = 0.02). Therefore, the results from this analysis should be interpreted with caution as the GRADE assessment suggested very low‐quality evidence.
Prokinetic versus prokinetic
There were six comparisons between prokinetics. However, five comparisons contained only one study each; DA‐9701 30 mg three times a day versus itopride 50 mg three times a day, cinitapride 1 mg three times a day versus domperidone 10 mg three times a day, mosapride 5 mg three times a day versus domperidone 10 mg three times a day, itopride 50 mg three times a day versus mosapride 5 mg three times a day, metoclopramide 10 mg three times a day versus domperidone 10 mg three times a day. Of them, mosapride 5 mg three times a day versus domperidone 10 mg three times a day, and itopride 50 mg three times a day versus mosapride 5 mg three times a day showed marginally significant differences between the two prokinetics. In addition, five studies compared itopride 50 mg three times a day versus domperidone 10 mg three times a day, with a total sample size of only 932; no significant difference was seen between the two prokinetics.
The most common comparator was domperidone 10 mg three times a day, thus we analysed other prokinetics versus domperidone. When seven RCTs (N = 1527) were pooled, the efficacy of other prokinetics (itopride, cinitapride, mosapride) was comparable to domperidone in symptom‐free or symptom improvement. However, when we investigated individual prokinetics, the significant difference was seen only between mosapride and domperidone (1 study, N = 212). There was no consistent evidence to support the efficacy of any individual prokinetic over another one in terms of number of people with symptom‐free or improvement.
Post‐treatment symptom score
In our opinion, not‐symptom‐free or no symptom improvement is more reliable to evaluate the efficacy of any treatment, compared to rating symptom score. However, we included post‐treatment symptom score and mean difference symptom score as secondary outcomes in this review in order to assess the efficacy of prokinetic over placebo/another prokinetic in all aspects of possible outcome measurements.
Prokinetic versus placebo
This review demonstrated the benefit of prokinetics in post‐treatment symptom score (SMD ‐0.36, 95% CI ‐0.65 to ‐0.07), six studies, N = 2914. This finding remained robust in all sensitivity analyses. However, significant heterogeneity was seen between studies. In subgroup analyses, only tegaserod and itopride, studies that only included participants with PDS subtype or assessed symptom by non‐validated tool, or had duration of treatment/follow‐up at least one month, or with low risk of bias showed significant efficacy of prokinetic in functional dyspepsia (FD) treatment. However, due to the observational nature of subgroup analyses and the small sample sizes of individual studies, the results of the subgroup analysis should be interpreted with caution.
Prokinetic versus prokinetic
Only five studies were included in this analysis. No difference was seen between itopride 50 mg three times a day or metoclopramide 10 mg three times a day, versus domperidone 10 mg three times a day; or DA‐9701 30 mg three times a day versus itopride 50 mg three times a day. Significant difference was seen for cinitapride 1 mg three times a day versus domperidone 10 mg three times a day, and itopride 50 mg three times a day versus mosapride 5 mg three times a day.
When data were pooled for other prokinetics versus domperidone 10 mg three times a day in three studies, significant difference was seen between other prokinetics versus domperidone in post‐treatment symptom scores (SMD ‐0.19, 95%CI ‐0.35 to ‐0.03) (N = 617), the only significant difference is seen between cinitapride versus domperidone. The lack of heterogeneity in this analysis should be also interpreted with caution due to the few included studies (Loannidis 2007).
Mean difference symptom score
Prokinetic versus placebo
Eleven studies (N = 1822) reported data for this outcome. We found no significant difference in mean difference symptom score between any prokinetic (itopride, cisapride, acotiamide, ABT‐229) and placebo. However, significant heterogeneity was seen between studies. This result changed to favouring prokinetics in a sensitivity analysis with a fixed‐effect model, and in subgroup analysis when limited to the subgroup of studies with a high risk and low risk of bias. The change of result in a fixed‐effect model demonstrated the results were not robust. The results remained insignificant in subgroup analysis related to dyspepsia subtypes (PDS, EPS or mixed), method of calculating mean difference (reporting or calculating), type of assessment tools (validated or non‐validated tool), and duration of treatment and follow‐up (less than one month or one month or more). Except for subgroups of risk of bias, there were no significant seen in subgroups .
Prokinetic versus prokinetic
This analysis showed no difference between three prokinetics (cinitapride 1 mg three times a day, itopride 50 mg three times a day and metoclopramide 10 mg three times a day) and domperidone 10 mg three times a day, DA‐9701 30mg three times a day and itopride 50 mg three times a day as well as itopride 50 mg three times a day and mosapride 5 mg three times a day in reducing symptom score after treatment. A significant favourable result was seen only when comparing mosapride 5 mg three times a day compared with domperidone 10 mg three times a day. When data were pooled for four prokinetics versus domperidone 10 mg three times a day (one study for each prokinetic), no significant difference was seen between other prokinetics and domperidone. The lack of heterogeneity in this analysis should be interpreted with caution due to the few included studies (Loannidis 2007).
Qaulity of life
Quality of life (QiL) is an another important outcome in functional gastrointestinal measurement. However, only five RCTs evaluated this outcome.
Prokinetic versus placebo
We found no difference in number of people with improved QoL, post‐treatment QoL score and changed in QoL between prokinetic and placebo;however, only one, one and five studies provided data for these outcomes, respectively. In addition, only itopride and acotiamide were assessed for QoL. In change of QoL scores from baseline, itopride significantly improved QoL scores after treatment, whereas acotiamide could not. There was no significant heterogeneity between subgroups, thus the difference in subgroup analysis should be interpreted with caution.
Prokinetic versus prokinetic
Only one study compared post‐treatment Nepean Dyspepsia Index (NDI) QoL score (interference with daily activities) and in change of NDI QoL score between DA‐9701 3 mg three times a day and itopride 50 mg three times a day, no significant difference was seen. There was lack of evidence to assess this outcome between other prokinetics.
Adverse events
Adverse event is an important outcome when assessing the efficacy of any medication. However, only some studies reported this outcome.
Prokinetic versus placebo
Overall, we found no difference in adverse events when four prokinetics were pooled (cisapride, acotiamide, itopride and mosapride) and compared with placebo at the end of treatment (RR 1.09, 95% CI 0.95 to 1.25) (N = 3811). This finding was robust in all sensitivity analyses. However, cisapride was the only prokinetic that had significant adverse events than placebo in subgroup analysis. Although, there was no heterogeneity amongst studies and between subgroups, the funnel plot displayed asymmetry which means possible publication bias or small‐sample effect.
Prokinetic versus prokinetic
This analysis demonstrated greater adverse events in domperidone when compared to other prokinetics (data pooled for itopride, cinitapride and mosapride) (RR 0.69, 95%CI 0.50 to 0.97; 7 studies, N = 1557). This finding remained robust in all sensitivity analyses. However, only cinitapride, which had the largest sample size (N = 383), showed significantly more people with adverse events of domperidone in subgroup analysis. No significant difference was seen between itopride 50 mg three times a day versus domperidone 10 mg three times a day, mosapride 5 mg three times a day versus domperidone 10 mg three times a day, DA‐9701 30 mg three times a day versus itopride 50 mg three times a day, and Itopride 50 mg three times a day versus mosapride 5 mg three times a day. Therefore, the result of adverse events should be carefully interpreted.
Overall completeness and applicability of evidence
This systematic review and meta‐analysis was designed to include RCTs comparing a prokinetic with placebo or two types of prokinetics, regardless of publication language and publication status. We planned to evaluate global dyspeptic symptom as we believed it is the best representative of efficacy in dyspepsia treatment. When global symptoms were not reported, epigastric pain/discomfort symptom were extracted. We found few studies assessing individual symptom (Chen 2004; Choi 2015; Mo 2003; Sun 2003; Zhu 2005). Nevertheless, we carried out sensitivity analysis removing the studies that reported only individual symptoms, calculating an odds ratio for a dichotomous outcome and using a fixed‐effect model in all outcomes, to confirm the robustness of the results. We also performed subgroup analyses to explore the sources of heterogeneity of each outcome. Although we failed to obtain unpublished data from one study (Talley 2001), we believe this review is comprehensive, and the results reflect the best available evidence for demonstrating the efficacy of prokinetics in functional dyspepsia (FD) treatment.
Quality of the evidence
Overall, for prokinetics versus placebo, the quality of evidence for the outcome of not symptom‐free or no symptom improvement was very low because of study limitations (unclear risk of bias for random sequence generation and/or allocation, one study was an open‐labelled design, and another could not blind the physician as the placebo group was sham acupuncture), significant heterogeneity and publication bias or small‐study effect (Table 1).
The quality of evidence was low for the outcome of post‐treatment symptom scores due to unclear risk of bias for random sequence generation and/or allocation in half of the studies. It was downgraded one level due to imprecision (the 95% confidence interval (CI) of pooled data was very close to no effect). One study was considered to be at high risk of bias; significant heterogeneity with some possible explanations. Two items were considered together and the quality was downgraded one level. The number of studies was less than 10, so we did not create a funnel plot and this quality assessment should be interpreted with caution.
For mean difference symptom scores outcome, we judged the quality of evidence as very low because of study limitations (unclear risk of bias for random sequence generation and/or allocation, two studies were considered at high risk of bias), significant heterogeneity without plausible explanations and imprecision (95% CI of pooled data included no effect).
We assessed the quality of evidence in quality of life as very low, due to inconsistency of effect and impression. One of the five studies was considered to be at high risk of bias.
We assessed the quality of evidence in adverse events outcome as very low, due to study limitations (most studies had an unclear risk of bias for random sequence generation and/or allocation, three studies were considered at high risk of bias), imprecision and publication bias or small‐study effect.
For comparisons between prokinetics, the quality of evidence was only assessed for other prokinetics versus domperidone (Table 2). For outcomes of "not symptom‐free" or "no symptom improvement", mean difference scores and adverse events, we judged the quality of evidence to be very low due to study limitations and impression. For post‐treatment scores, we judged the quality of evidence to be very low due to study limitations and imprecision.
Potential biases in the review process
We did not identify any potential bias in the review process.
Agreements and disagreements with other studies or reviews
A previous systematic review and meta‐analysis evaluated a dichotomous outcome (not symptom‐free or no symptom improvement), and suggested the efficacy of a prokinetic in non‐ulcer dyspepsia (which means functional dyspepsia in this review); however, it concluded that the effect of prokinetic therapy is difficult to interpret due to publication bias or other small‐study effect (Moayyedi 2011). With newer studies and newer prokinetics added to this review, the results supported the data from the previous publication. Furthermore, this review showed the persistent benefit of a prokinetic when cisapride is removed from the analyses, as it is not currently available in many regions. Additionally, this review assessed the studies comparing two types of prokinetics as well as the continuous outcomes (post‐treatment symptom score and mean difference of symptom score), which were not reported in the previous review.
Authors' conclusions
Implications for practice.
Prokinetics showed benefit only on being symptom‐free or symptom improvement and post‐treatment symptom scores, but not mean difference symptom scores or quality of life, with low to very low quality of evidence. Thus, the usefulness of a prokinetic in functional dyspepsia treatment is still questionable. Moreover, there was insufficient evidence to determine which prokinetic is most effective for functional dyspepsia treatment. Additionally, prokinetics cannot change the quality of life. Apart from cisapride, prokinetics were well‐tolerated in short‐term treatment. Therefore, if a prokinetic is available, the physician can use it in short‐term duration aiming for symptom improvement if the patient has not responded to other effective therapies.
Implications for research.
Additional randomised controlled trials (RCTs) of people with functional dyspepsia, of good methodology, and large sample size is still warranted in order to clarify the efficacy of prokinetics, especially newer prokinetics.
History
Protocol first published: Issue 11, 2011 Review first published: Issue 10, 2018
| Date | Event | Description |
|---|---|---|
| 10 October 2017 | New citation required and major changes | New author team formed and protocol updated to include new interventions |
Notes
A new review team was formed and the following protocol sections updated.
Background: recent citations to support this section were included.
-
Methods
Types of participants: ROME criteria were expanded to 1 to 4 (from 1 to 3) since the new Rome IV criteria was published in 2016 after Rome III in 2006 (Stanghellini 2016).
Types of outcome measures: we revised the primary outcome from "proportion of patients with any improvement of symptoms" to "global symptoms of dyspepsia" (using the most stringent definition of not symptom‐free or not overall symptom improvement given by the patient at the end of treatment), because overall symptom improvement is a more reliable measure than one or more symptoms when assessing the treatment efficacy; using the unfavourable outcome (not symptom‐free or not improved) makes the risk ratio (RR) easier to be interpreted by clinicians.
Search methods: we updated the search strategies to include the most recent filters and capture new drugs.
-
Data collection and analysis
We removed individual symptom scores from the outcome because overall symptom improvement is a more reliable measure than improvement of a single symptom.
We also assessed continuous outcome (global symptom scores) to make this review comprehensive.
Assessment of risk of bias in included studies: we included the most recent version of the 'Risk of bias' domains to be assessed.
Subgroup analysis and investigation of heterogeneity: we included a subgroup to stratify studies according to their 'Risk of bias' assessment.
Sensitivity analysis: we excluded studies with significant clinical heterogeneity in sensitivity analysis, and excluded studies that only assessed individual symptoms without data for overall symptom improvement in the sensitivity analysis.
This new review partially updates a portion of the published review Moayyedi 2011.
Acknowledgements
We would like to acknowledge the assistance of the Canadian Cochrane Centre.
Appendices
Appendix 1. Glossary of medical terms
Dyspepsia: upper abdominal discomfort.
Endoscopy: inserting the camera into gastrointestinal tract.
Reflux: acid in the stomach moves up to oesophagus.
Placebo: powder that has the appearance similar to drug.
Efficacy: advantage, benefit.
Heterogeneity: diversity.
Postprandial: after eating.
Dysmotility: abnormal movement of gastrointestinal tract.
Pancreatico‐biliary: pancreas and bile duct.
Oesophagitis: inflammation of oesophagus.
Neoplastic: malignancy.
Epigastric; over the stomach.
Dichotomous; two ways eg. yes/no.
Global symptom of FD: overall dyspeptic symptom.
Appendix 2. CENTRAL search strategy (Ovid)
exp Dyspepsia/
(dyspep* or "NUD" or "FD").tw,kw.
(indigestion or indigestive).tw.
or/1‐3
(prokinetic* or gastroprokinetic* or gastrokinetic* or gastro‐kinetic*).tw,kw.
(antiemetic* or anti‐emetic).tw,kw.
exp Benzamides/
(Benzoic Acid Amide or Amides or Phenyl Carboxyamide or Benzamide* or Benzoylamide or benzoates).tw,kw.
(Phenylcarboxyamide or Phenylcarboxamide or Benzenecarboxamide or Amid kyseliny benzoove).tw,kw.
exp Domperidone/
(domperidon* or domidon or Domperi or Domstal or evoxin or gastrocure or motilium or motilium).tw,kw.
(motis or nauzelin or Motinorm Costi or Nomit or Brulium or Molax).tw,kw.
exp Antiemetics/
exp Metoclopramide/
(Metoclopramide or cerucal or clopra or gastrese or gastrobid or gastroflux or gastromax or maxolon).tw,kw.
(metaclopramide or metozolv or metramid or migravess or mygdalon or octamide or parmid).tw,kw.
(primperan or reglan or reliveran or rimetin or Degan or Maxeran or Pylomid or Pramin).tw,kw.
exp Cisapride/
(Cisapride or alimix or Prepulsid or Propulsid).tw,kw.
exp Cholinesterase Inhibitors/
(Itopride or ganaton).tw,kw.
Mosapride.tw,kw.
exp Erythromycin/
(erythromycin or aknemycin or emcin or emgel or emycin or eryderm or erygel or erymax).tw,kw.
(erymin or eryped or gallimycin or ilosene or ilosone or ilotycin or lauromicina or maracyn).tw,kw.
(monomycin or ornacyn or retcin or rommix or romycin or roymicin or staticin or stiemycin or theramycin or tiloryth or wyamycin).tw,kw.
(Motilin adj3 (receptor* or agonist*)).tw,kw.
((5HT3 or 5HT 3 or 5‐HT3 or 5‐HT 3) adj3 antagonist*).tw,kw.
((5HT or 5‐HT or 5‐hydroxytryptamine*) adj3 (agonist* or antagonist*)).tw,kw.
((5‐HT1A or 5HT1A or 5‐HT 1A or 5HT 1A) adj3 agonist*).tw,kw.
exp Serotonin Antagonists/
exp Serotonin 5‐HT3 Receptor Antagonists/
exp Serotonin 5‐HT4 Receptor Agonists/
exp Serotonin 5‐HT1 Receptor Agonists/
(serotonin adj3 receptor adj3 (agonist* or antagonist* or block*)).tw,kw.
(tegaserod or Zelnorm or Zelmac).tw,kw.
ABT‐229.tw,kw.
(Tandospirone or Sediel or metanopirone or buspirone).tw,kw.
(alosetron or Lotronex).tw,kw.
(Acotiamide or YM‐443 or Z‐338D).tw,kw.
(acetylcholinesterase inhibitor* or cholinesterase Inhibitor* or anti‐cholinesterase* or anticholinesterase*).tw,kw.
((5HT‐4 or 5HT4 or 5‐HT 4 or 5‐HT4) adj3 agonist*).tw,kw.
or/5‐42
4 and 43
Appendix 3. MEDLINE search strategy
exp Dyspepsia/
(dyspep* or "NUD" or "FD").tw,kw.
(indigestion or indigestive).tw.
or/1‐3
(prokinetic* or gastroprokinetic* or gastrokinetic* or gastro‐kinetic*).tw,kw.
(antiemetic* or anti‐emetic).tw,kw.
exp Benzamides/
(Benzoic Acid Amide or Amides or Phenyl Carboxyamide or Benzamide* or Benzoylamide or benzoates).tw,kw.
(Phenylcarboxyamide or Phenylcarboxamide or Benzenecarboxamide or Amid kyseliny benzoove).tw,kw.
exp Domperidone/
(domperidon* or domidon or Domperi or Domstal or evoxin or gastrocure or motilium or motilium).tw,kw.
(motis or nauzelin or Motinorm Costi or Nomit or Brulium or Molax).tw,kw.
exp Antiemetics/
exp Metoclopramide/
(Metoclopramide or cerucal or clopra or gastrese or gastrobid or gastroflux or gastromax or maxolon).tw,kw.
(metaclopramide or metozolv or metramid or migravess or mygdalon or octamide or parmid).tw,kw.
(primperan or reglan or reliveran or rimetin or Degan or Maxeran or Pylomid or Pramin).tw,kw.
exp Cisapride/
(Cisapride or alimix or Prepulsid or Propulsid).tw,kw.
exp Cholinesterase Inhibitors/
(Itopride or ganaton).tw,kw.
Mosapride.tw,kw.
exp Erythromycin/
(erythromycin or aknemycin or emcin or emgel or emycin or eryderm or erygel or erymax).tw,kw.
(erymin or eryped or gallimycin or ilosene or ilosone or ilotycin or lauromicina or maracyn).tw,kw.
(monomycin or ornacyn or retcin or rommix or romycin or roymicin or staticin or stiemycin or theramycin or tiloryth or wyamycin).tw,kw.
(Motilin adj3 (receptor* or agonist*)).tw,kw.
((5HT3 or 5HT 3 or 5‐HT3 or 5‐HT 3) adj3 antagonist*).tw,kw.
((5HT or 5‐HT or 5‐hydroxytryptamine*) adj3 (agonist* or antagonist*)).tw,kw.
((5‐HT1A or 5HT1A or 5‐HT 1A or 5HT 1A) adj3 agonist*).tw,kw.
exp Serotonin Antagonists/
exp Serotonin 5‐HT3 Receptor Antagonists/
exp Serotonin 5‐HT4 Receptor Agonists/
exp Serotonin 5‐HT1 Receptor Agonists/
(serotonin adj3 receptor adj3 (agonist* or antagonist* or block*)).tw,kw.
(tegaserod or Zelnorm or Zelmac).tw,kw.
ABT‐229.tw,kw.
(Tandospirone or Sediel or metanopirone or buspirone).tw,kw.
(alosetron or Lotronex).tw,kw.
(Acotiamide or YM‐443 or Z‐338D).tw,kw.
(acetylcholinesterase inhibitor* or cholinesterase Inhibitor* or anti‐cholinesterase* or anticholinesterase*).tw,kw.
((5HT‐4 or 5HT4 or 5‐HT 4 or 5‐HT4) adj3 agonist*).tw,kw.
or/5‐42
4 and 43
randomized controlled trial.pt.
controlled clinical trial.pt.
random*.mp.
placebo.ab.
drug therapy.fs.
trial.ab.
groups.ab.
or/45‐51
exp animals/ not humans.sh.
52 not 53
44 and 54
Appendix 4. Embase search strategy
exp dyspepsia/
(dyspep* or "NUD" or "FD").tw,kw.
(indigestion or indigestive).tw.
or/1‐3
(prokinetic* or gastroprokinetic* or gastrokinetic* or gastro‐kinetic*).tw,kw.
(antiemetic* or anti‐emetic).tw,kw.
exp benzamide derivative/
(Benzoic Acid Amide or Amides or Phenyl Carboxyamide or Benzamide* or Benzoylamide or benzoates).tw,kw.
(Phenylcarboxyamide or Phenylcarboxamide or Benzenecarboxamide or Amid kyseliny benzoove).tw,kw.
exp domperidone/
(domperidon* or domidon or Domperi or Domstal or evoxin or gastrocure or motilium or motilium).tw,kw.
(motis or nauzelin or Motinorm Costi or Nomit or Brulium or Molax).tw,kw.
exp antiemetic agent/
exp metoclopramide/
(Metoclopramide or cerucal or clopra or gastrese or gastrobid or gastroflux or gastromax or maxolon).tw,kw.
(metaclopramide or metozolv or metramid or migravess or mygdalon or octamide or parmid).tw,kw.
(primperan or reglan or reliveran or rimetin or Degan or Maxeran or Pylomid or Pramin).tw,kw.
exp cisapride/
(Cisapride or alimix or Prepulsid or Propulsid).tw,kw.
exp cholinesterase inhibitor/
(Itopride or ganaton).tw,kw.
exp mosapride/
Mosapride.tw,kw.
exp erythromycin/
(erythromycin or aknemycin or emcin or emgel or emycin or eryderm or erygel or erymax).tw,kw.
(erymin or eryped or gallimycin or ilosene or ilosone or ilotycin or lauromicina or maracyn).tw,kw.
(monomycin or ornacyn or retcin or rommix or romycin or roymicin or staticin or stiemycin or theramycin or tiloryth or wyamycin).tw,kw.
exp motilin receptor agonist/
(Motilin adj3 (receptor* or agonist*)).tw,kw.
((5HT3 or 5HT 3 or 5‐HT3 or 5‐HT 3) adj3 antagonist*).tw,kw.
((5HT or 5‐HT or 5‐hydroxytryptamine*) adj3 (agonist* or antagonist*)).tw,kw.
((5‐HT1A or 5HT1A or 5‐HT 1A or 5HT 1A) adj3 agonist*).tw,kw.
exp serotonin antagonist/
exp serotonin 3 antagonist/
exp serotonin 4 agonist/
exp serotonin 1 agonist/
(serotonin adj3 receptor adj3 (agonist* or antagonist* or block*)).tw,kw.
exp tegaserod/
(tegaserod or Zelnorm or Zelmac).tw,kw.
ABT‐229.tw,kw.
exp tandospirone/
(Tandospirone or Sediel or metanopirone or buspirone).tw,kw.
exp alosetron/
(alosetron or Lotronex).tw,kw.
exp acotiamide/
(Acotiamide or YM‐443 or Z‐338D).tw,kw.
(acetylcholinesterase inhibitor* or cholinesterase Inhibitor* or anti‐cholinesterase* or anticholinesterase*).tw,kw.
((5HT‐4 or 5HT4 or 5‐HT 4 or 5‐HT4) adj3 agonist*).tw,kw.
or/5‐48
4 and 49
random*.mp.
placebo:.mp.
clinical trial:.mp.
double‐blind:.mp. or blind:.tw.
or/51‐54
exp animal/ not human/
55 not 56
50 and 57
remove duplicates from 58
Appendix 5. CINAHL Search strategy
(MH "Dyspepsia") or TX (dyspep* or "NUD" or "FD")
TX prokinetic* or gastroprokinetic* or gastrokinetic* or gastro‐kinetic*
TX antiemetic* or anti‐emetic
TX Benzoic Acid Amide or Amides or Phenyl Carboxyamide or Benzamide* or Benzoylamide or benzoates
TX Phenylcarboxyamide or Phenylcarboxamide or Benzenecarboxamide or Amid kyseliny benzoove
TX domperidon* or domidon or Domperi or Domstal or evoxin or gastrocure or motilium or motilium
TX motis or nauzelin or Motinorm Costi or Nomit or Brulium or Molax
TX Metoclopramide or cerucal or clopra or gastrese or gastrobid or gastroflux or gastromax or maxolon
TX metaclopramide or metozolv or metramid or migravess or mygdalon or octamide or parmid
TX primperan or reglan or reliveran or rimetin or Degan or Maxeran or Pylomid or Pramin
TX Cisapride or alimix or Prepulsid or Propulsid
TX Itopride or ganaton
TX Mosapride
TX erythromycin or aknemycin or emcin or emgel or emycin or eryderm or erygel or erymax
TX erymin or eryped or gallimycin or ilosene or ilosone or ilotycin or lauromicina or maracyn
TX monomycin or ornacyn or retcin or rommix or romycin or roymicin or staticin or stiemycin or theramycin or tiloryth or wyamycin
TX Motilin and (receptor* or agonist*)
TX (5HT3 or 5HT 3 or 5‐HT3 or 5‐HT 3) and antagonist*
TX (5HT or 5‐HT or 5‐hydroxytryptamine*) and (agonist* or antagonist*)
TX (5‐HT1A or 5HT1A or 5‐HT 1A or 5HT 1A) and agonist*
TX serotonin and receptor and (agonist* or antagonist* or block*)
TX tegaserod or Zelnorm or Zelmac
TX ABT‐229
TX Tandospirone or Sediel or metanopirone or buspirone
TX alosetron or Lotronex
TX Acotiamide or YM‐443 or Z‐338D
TX acetylcholinesterase inhibitor* or cholinesterase Inhibitor* or anti‐cholinesterase* or anticholinesterase*
TX (5HT‐4 or 5HT4 or 5‐HT 4 or 5‐HT4) and agonist*
(MH "Serotonin Agonists+")
S2 or S3 or S4 or S5 or S6 or S7 or S8 or S9 or S10 or S11 or S12 or S13 or S14 or S15 or S16 or S17 or S18 or S19 or S20 or S21 or S22 or S23 or S24 or S25 or S26 or S27 or S28 or S29
S1 and S31
Data and analyses
Comparison 1. Prokinetic versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Not symptom‐free or no symptom improvement | 29 | 10044 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 1.1 Cisapride 15‐60 mg/d | 12 | 1647 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.54, 0.93] |
| 1.2 Acotiamide 150‐900 mg/d | 6 | 2429 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.91, 0.98] |
| 1.3 Itopride 150‐600 mg/d | 6 | 2066 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.47, 1.03] |
| 1.4 Tegaserod 12 mg/d | 2 | 2667 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.82, 0.96] |
| 1.5 Mosapride 10‐22.5 mg/d | 2 | 626 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.73, 1.13] |
| 1.6 ABT‐229 2.5‐20 mg/d | 1 | 609 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [1.05, 1.70] |
| 2 Not symptom‐free or no symptom improvement, subgroup by definition | 29 | 10044 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 2.1 Not symptom‐free | 16 | 4356 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.68, 0.89] |
| 2.2 No symptom improvement | 13 | 5688 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.78, 0.94] |
| 3 Not symptom‐free or no symptom improvement, subgroup by FD subtype | 29 | 10044 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 3.1 EPS | 1 | 32 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.24, 0.98] |
| 3.2 PDS | 9 | 5068 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.65, 0.92] |
| 3.3 EPS and PDS | 19 | 4944 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.75, 0.93] |
| 4 Not symptom‐free or no symptom improvement, subgroup by publication type | 29 | 10044 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 4.1 Full paper | 26 | 9309 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 4.2 Conference abstract | 3 | 735 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.77, 1.00] |
| 5 Not symptom‐free or no symptom improvement, subgroup by assessment tool | 29 | 10044 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 5.1 Validated tool | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.18, 2.44] |
| 5.2 Non‐validated tool | 28 | 10014 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 6 Not symptom‐free or no symptom improvement, subgroup by follow‐up period | 29 | 10044 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 6.1 Less than one month | 6 | 473 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.59, 1.01] |
| 6.2 Greater than or equal to one month | 23 | 9571 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.90] |
| 7 Not symptom‐free or no symptom improvement, subgroup by risk of bias | 29 | 10044 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 7.1 High risk of bias | 4 | 1049 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.39, 1.15] |
| 7.2 Unclear risk of bias | 21 | 4883 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.76, 0.93] |
| 7.3 Low risk of bias | 4 | 4112 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.80, 0.95] |
| 8 Post‐treatment symptom scores (different scales used) | 6 | 2914 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.65, ‐0.07] |
| 8.1 Tegaserod | 2 | 2656 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.24, ‐0.02] |
| 8.2 Cisapride | 2 | 132 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.40, 0.28] |
| 8.3 Itorpide | 1 | 80 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.88 [‐2.41, ‐1.35] |
| 8.4 Acotiamide | 1 | 46 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.88, 0.28] |
| 9 Post‐treatment symptom scores (different scales used), subgroup by FD subtype | 6 | 2914 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.65, ‐0.07] |
| 9.1 PDS | 4 | 2782 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐0.87, ‐0.13] |
| 9.2 EPS and PDS | 2 | 132 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.40, 0.28] |
| 10 Post‐treatment symptom scores (different scales used), subgroup by assessment tool | 6 | 2914 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.65, ‐0.07] |
| 10.1 Validated tool | 1 | 46 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.88, 0.28] |
| 10.2 Non‐validated tool | 5 | 2868 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.37 [‐0.69, ‐0.05] |
| 11 Post‐treatment symptom scores (different scales used), subgroup by follow‐up period | 6 | 2914 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.65, ‐0.07] |
| 11.1 Less than one month | 2 | 126 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.09 [‐2.64, 0.45] |
| 11.2 Greater than or equal to one month | 4 | 2788 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.20, ‐0.05] |
| 12 Post‐treatment symptom scores (different scales used), subgroup by risk of bias | 6 | 2914 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.65, ‐0.07] |
| 12.1 High risk of bias | 1 | 76 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.62, 0.28] |
| 12.2 Unclear risk of bias | 3 | 182 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.70 [‐1.91, 0.51] |
| 12.3 Low risk of bias | 2 | 2656 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.24, ‐0.02] |
| 13 Mean difference symptom scores (post‐treatment ‐ pre‐treatment, different scales used), subgroup by prokinetic | 11 | 1822 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.65 [‐1.50, 0.20] |
| 13.1 Itorpide | 4 | 860 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.53 [‐3.42, 0.37] |
| 13.2 Cisapride | 4 | 280 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.71, 0.16] |
| 13.3 Acotiamide | 2 | 108 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.47, 0.35] |
| 13.4 ABT‐229 | 1 | 574 | Std. Mean Difference (IV, Random, 95% CI) | 0.14 [‐0.07, 0.34] |
| 14 Mean difference symptom scores (post‐treatment ‐ pre‐treatment, different scales used), subgroup by FD subtype | 11 | 1822 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.65 [‐1.50, 0.20] |
| 14.1 PDS | 3 | 154 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.68 [‐1.65, 0.29] |
| 14.2 EPS and PDS | 8 | 1668 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.64 [‐1.70, 0.42] |
| 15 Mean difference symptom scores (post‐treatment ‐ pre‐treatment, different scales used), subgroup by method of calculating MD | 11 | 1822 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.65 [‐1.50, 0.20] |
| 15.1 Reported mean difference | 7 | 1564 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.78 [‐1.98, 0.42] |
| 15.2 Calculated mean difference | 4 | 258 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.42 [‐1.20, 0.36] |
| 16 Mean difference symptom scores (post‐treatment ‐ pre‐treatment, different scales used), subgroup by assessment tool | 11 | 1822 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.65 [‐1.50, 0.20] |
| 16.1 Validated tool | 4 | 720 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.24 [‐3.25, 0.78] |
| 16.2 Non‐validated tool | 7 | 1102 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.72, 0.11] |
| 17 Mean difference symptom scores (post‐treatment ‐ pre‐treatment, different scales used), subgroup by follow‐up period | 11 | 1822 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.65 [‐1.50, 0.20] |
| 17.1 Less than one month | 3 | 188 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.56 [‐1.59, 0.46] |
| 17.2 Greater than or equal to one month | 8 | 1634 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.68 [‐1.75, 0.38] |
| 18 Mean difference symptom scores (post‐treatment ‐ pre‐treatment, different scales used), subgroup by risk of bias | 11 | 1822 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.65 [‐1.50, 0.20] |
| 18.1 High risk of bias | 2 | 302 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐0.63, ‐0.17] |
| 18.2 Unclear risk of bias | 8 | 997 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.32 [‐0.76, 0.11] |
| 18.3 Low risk of bias | 1 | 523 | Std. Mean Difference (IV, Random, 95% CI) | ‐3.80 [‐4.10, ‐3.50] |
| 19 Improved QoL | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 19.1 Itopride | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.54, 2.54] |
| 20 Post QoL scores | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 20.1 Acotiamide | 1 | 46 | Std. Mean Difference (IV, Random, 95% CI) | 0.24 [‐0.34, 0.82] |
| 21 Change of QoL scores (post‐treatment ‐ pre‐treatment, different scales) | 5 | 1774 | Std. Mean Difference (IV, Random, 95% CI) | 0.11 [‐0.10, 0.33] |
| 21.1 Acotiamide | 3 | 1000 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.16 [‐0.79, 0.47] |
| 21.2 Itopride | 2 | 774 | Std. Mean Difference (IV, Random, 95% CI) | 0.24 [0.08, 0.39] |
| 22 Adverse events | 17 | 3811 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.95, 1.25] |
| 22.1 Cisapride | 10 | 1482 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [1.03, 1.65] |
| 22.2 Acotiamide | 3 | 1660 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.83, 1.16] |
| 22.3 Itopride | 3 | 609 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.80, 1.31] |
| 22.4 Mosapride | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.5 [0.27, 8.34] |
1.14. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 14 Mean difference symptom scores (post‐treatment ‐ pre‐treatment, different scales used), subgroup by FD subtype.
1.15. Analysis.
Comparison 1 Prokinetic versus placebo, Outcome 15 Mean difference symptom scores (post‐treatment ‐ pre‐treatment, different scales used), subgroup by method of calculating MD.
Comparison 2. Prokinetic versus prokinetic.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Not symptom‐free or no symptom improvement | 9 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Itopride 50 mg three times a day versus domperidone 10 mg three time a day | 5 | 932 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.89, 1.13] |
| 1.2 DA‐9701 30 mg three times a day versus itopride 50 mg three times a day | 1 | 464 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.96, 1.01] |
| 1.3 Cinitapride 1 mg three times a day versus domperidone 10 mg three times a day | 1 | 383 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.51, 1.27] |
| 1.4 Mosapride 5 mg three times a day versus domperidone 10 mg three times a day | 1 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.54, 0.97] |
| 1.5 Itopride 50 mg three times a day versus mosapride 5 mg three times a day | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.39, 0.99] |
Comparison 3. Itopride 50 mg three times a day versus domperidone 10 mg three times a day.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Not symptom‐free or no symptom improvement | 5 | 932 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.89, 1.13] |
| 1.1 Not symptom‐free | 3 | 621 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.82, 1.12] |
| 1.2 No symptom improvement | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.89, 1.33] |
| 2 Not symptom‐free or no symptom improvement, subgroup by symptom type | 5 | 932 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.89, 1.13] |
| 2.1 Assessed by global symptoms | 2 | 417 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.73, 1.17] |
| 2.2 Assessed by individual symptoms | 3 | 515 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.90, 1.29] |
| 3 Post treatment scores | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4 Mean difference symptom scores (post‐treatment ‐ pre‐treatment) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5 Adverse events | 5 | 952 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.48, 1.61] |
Comparison 4. DA‐9701 30 mg three times a day versus itopride 50 mg three times a day.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Not symptom‐free | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2 Post‐treatment scores | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3 Mean difference symptom scores | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4 Post‐treatment NDI QoL score (interference with daily activities) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5 Change of NDI QoL score (interference with daily activities) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6 Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only |
Comparison 5. Cinitapride 1 mg three times a day versus domperidone 10 mg three times a day.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 No symptom improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2 Post‐treatment scores | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3 Mean difference symptom scores (post‐treatment ‐ pre‐treatment) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4 Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only |
Comparison 6. Mosapride 5 mg three times a day versus domperidone 10 mg three times a day.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Not symptom‐free | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2 Mean difference symptom scores | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3 Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only |
Comparison 7. Itopride 50 mg three times a day versus mosapride 5 mg three times a day.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Not symptom‐free | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2 Post‐treatment scores | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3 Mean difference symptom scores (post‐treatment ‐ pre‐treatment) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4 Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only |
Comparison 8. Metoclopramide 10 mg three times a day versus domperidone 10 mg three times a day.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Post‐treatment scores | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2 Mean difference symptom scores | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only |
Comparison 9. Other prokinetics versus domperidone 10 mg three times a day.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Not symptom‐free or no symptom improvement | 7 | 1527 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.83, 1.07] |
| 1.1 Itopride 50 mg three times a day | 5 | 932 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.89, 1.13] |
| 1.2 Cinitapride 1 mg three times a day | 1 | 383 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.51, 1.27] |
| 1.3 Mosapride 5 mg three times a day | 1 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.54, 0.97] |
| 2 Post‐treatment scores | 3 | 617 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.19 [‐0.35, ‐0.03] |
| 2.1 Cinitapride 1 mg three times a day | 1 | 344 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.25 [‐0.46, ‐0.04] |
| 2.2 Itopride 50 mg three times a day | 1 | 200 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.39, 0.16] |
| 2.3 Metoclopramide 10 mg three times a day | 1 | 73 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.61, 0.31] |
| 3 Mean difference symptom scores | 4 | 839 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.31, 0.05] |
| 3.1 Cinitapride 1 mg three times a day | 1 | 344 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.28, 0.14] |
| 3.2 Mosapride 5 mg three times a day | 1 | 222 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.38 [‐0.65, ‐0.11] |
| 3.3 Itopride 50 mg three times a day | 1 | 200 | Std. Mean Difference (IV, Random, 95% CI) | 0.0 [‐0.28, 0.28] |
| 3.4 Metoclopramide 10 mg three times a day | 1 | 73 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.02 [‐0.47, 0.44] |
| 4 Adverse events | 7 | 1557 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.50, 0.97] |
| 4.1 Itopride 50 mg three times a day | 5 | 952 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.48, 1.61] |
| 4.2 Cinitapride 1 mg three times a day | 1 | 383 | Risk Ratio (M‐H, Random, 95% CI) | 0.60 [0.37, 0.97] |
| 4.3 Mosapride 5 mg three times a day | 1 | 222 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.33, 1.42] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Abid 2017.
| Methods | Randomised, placebo‐controlled trial. Single‐centre. Criteria for FD: Rome III but unclear most prevalent type of FD. | |
| Participants | N = 31 Female: 32% Mean age: 33 years for overall Country of study: Pakistan |
|
| Interventions | Intervention: Itopride 150 mg/day Comparator: placebo Rescue medication: antacid as required Duration: 4 weeks | |
| Outcomes | Validated 7point global overall symptom scale by patient | |
| Notes | Only symptom score, no dichotomous outcome | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly allocated |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropout |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Al‐Quorain 1995.
| Methods | Randomised. double‐blind. placebo‐controlled trial. Single‐centre. Criteria for FD: upper gastrointestinal symptoms indicative of FD but unclear most prevalent type of FD. Follow‐up: 4 weeks. | |
| Participants | N = 98 Female: 49% Mean age:32.5 ±8.5 years for prokinetic group and 33.7 ± 6.8 years for placebo group Country of study: Saudi Arabia |
|
| Interventions | Intervention: cisapride 5 mg orally three times a day. Comparator: placebo. Rescue medication: none Duration 4 weeks |
|
| Outcomes | Non‐validated global assessment in four categories assessed by participants | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blinded study, likely outcome assessors (participants) were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 10% loss follow‐up |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcome |
| Other bias | Low risk | No other risk found |
Amarapurkar 2004.
| Methods | Randomised to two drugs. Double‐blind. Single‐centre. Criteria for FD: equivalent symptom to Rome but unclear most prevalent type of FD. | |
| Participants | N = 60 Female: 50% Mean age: 45.2 ± 13.1 years for prokinetic group and 39.8 ± 10.8 years for placebo group Country of study: India |
|
| Interventions | Intervention: itopride hydrochloride 50 mg orally three times a day Comparator: mosapride citrate 5 mg orally three times a day Rescue medication: none Duration 2 weeks |
|
| Outcomes | Severity of functional dyspepsia symptoms: 0 to 3 (0 = none, 1 = mild, 2 = moderate, 3 = severe), non‐validated global evaluation of efficacy rated by participants and physicians separately: 4‐point scale (excellent, good, fair, poor) | |
| Notes | Unclear other risk: Duration of symptoms and number of males significantly higher in the mosapride group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Predetermined randomization table" |
| Allocation concealment (selection bias) | Unclear risk | No information is provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by participants who were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropout |
| Selective reporting (reporting bias) | Low risk | Reported pre‐defined outcome |
| Other bias | Unclear risk | Duration of symptom and number of males were significantly higher in mosapride group |
Champion 1997.
| Methods | Randomised, double‐blind, placebo‐controlled trial. 6 centres. Criteria for FD: equivalent symptom to Rome but unclear most prevalent type of FD. | |
| Participants | N = 123 Female 69% Mean age: 41 years Country of study: Canada |
|
| Interventions | Intervention: cisapride 10 mg orally three times a day or cisapride 20 mg orally three times a day Comparator: placebo Rescue medication: aluminium hydroxide Duration 6 weeks |
|
| Outcomes | Investigator assessed 10 symptoms for severity using 4‐point scale and frequency using 5‐point scale. Combined scores 0 to 12. Participants rated 8 symptoms using 4‐point scale. Overall 5‐point scale by participants and physicians. "Symptoms clusters"= severity* frequency | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly and equally assigned |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 27/123 (22%) did not complete the treatment. It is not clear if lost to follow up reasons balanced between groups |
| Selective reporting (reporting bias) | Unclear risk | Combined excellent and good global response rated |
| Other bias | Low risk | No other risk found |
Chen 2004.
| Methods | Randomised to two drugs. Double‐blind. multi‐centre (6). Criteria for FD: equivalent symptom to Rome and included both types of FD. | |
| Participants | N = 231 Female 53.2% Mean age: 44 years for mosapride group and 43 years for domperidone group Country of study: China |
|
| Interventions | Intervention: mosapride 5 mg orally three times a day Comparator: domperidone 10 mg orally three times a day Rescue medication: not mentioned Duration 4 weeks |
|
| Outcomes | Symptom scores 4 grades: 0 to3. Cure = symptoms disappeared, significant improvement = symptoms improved two grades but not yet symptoms free, improvement = symptoms improved one grade but not yet symptom‐free, failure= symptoms worse or no change. For overall improvement scores = sum of six individual symptoms scores. Unlcear who was the assessor. | |
| Notes | Article In Chinese. Reported individual symptom‐free (we use abdominal distension free) and overall mean symptom score difference (continuous outcome) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐randomised, in blocks of 4. Participants were assigned to treatment group according to the randomised number in sequence |
| Allocation concealment (selection bias) | Low risk | Double‐blinded,To maintain blinding, both medications were identical in appearance. On the package only the randomised number is shown |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded, likely outcomes were assessed by participants who were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The blinding list was opened after all participants completed the F/U |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Outcomes were analysed in PP sample size. Details for excluded reasons were provided. Seven participants were lost to follow‐up, it is not clear whether more participants in control group were lost to follow‐up due to side effects related to study medication. Two participants (1 in each group )were excluded from the analysis because the participants changed the treatment medication. Authors reported individual symptom resolution or improvement stratified according to symptom subtype. Symptoms improvement only reported for individual symptoms, since not all participants had all symptoms, thus we could not use ITT sample size to consider those withdrawn. |
| Selective reporting (reporting bias) | Low risk | Reported all predefined‐outcomes |
| Other bias | Low risk | No other risk found |
Choi 2015.
| Methods | Randomised to two drugs. Double‐blind. 18 centres. Criteria for FD: modified Rome II with both types of FD. | |
| Participants | N = 464 Female 69.6% Mean age: 41.3 ±13.4 years for DA‐9701 group and 40.3 ± 14 years for itopride group Country of study: Korea |
|
| Interventions | Intervention: DA‐9701 (motilitone) 30 mg three times a day Comparator: itopride 50 mg three times a day Rescue medication: not mentioned Duration: 4 weeks |
|
| Outcomes | The change from baseline in composite score of the 8 dyspeptic symptoms (1 to 100 for each) and the overall treatment effect. Responder was defined as ≥ 5 of the 7‐point Likert scale, a scale of and 0 (not at all) to 4 (extremely bothersome).‐complete relief to no response or aggravation. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | By a computerised random number table.The random number table was created by a block randomisation method |
| Allocation concealment (selection bias) | Low risk | Quote: "conceal allocation", "by key code securely stored" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded, same shaped counterpart placebo was used |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 9% participants discontinued intervention, balanced between two groups |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcome |
| Other bias | Low risk | No other risk found |
de Groot 1997.
| Methods | Randomised. double‐blind. placebo‐controlled trial. Single‐centre. Criteria for FD: equivalent symptom to Rome with both type of FD | |
| Participants | N = 121 Female: 54% Mean age: 40.9 years for the prokinetic group and 43.9 years for the placebo group Country : the Netherlands |
|
| Interventions | Intervention: cisapride 10 mg three times a day Comparator: placebo No rescue medication Duration of treatment: 4 weeks |
|
| Outcomes | Physician's assessment of overall result as on 4‐point scales (excellent, good, moderate, poor), data reported as excellent or good | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomized" |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Assessed by doctor but we do not know if he was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 7% not included in the analyses |
| Selective reporting (reporting bias) | Unclear risk | Combined excellent and good global response |
| Other bias | Low risk | No other risk found |
De Nutte 1989.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Single‐centre. Criteria for FD: equivalent symptom with only epigastric pain (EPS) | |
| Participants | N = 32 Female 37.5% Mean age: 41 ± 23 years. SD for overall Country of study: Belgium |
|
| Interventions | Intervention: cisapride 5 mg orally three times a day. Comparator: placebo. Rescue medication: antacid Duration 4 weeks |
|
| Outcomes | Symptoms scores 0 to 3. Global scores: excellent = complete relief of symptoms, good =improvement with occasional symptoms, fair =slight general improvement, poor = persisted | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomly assigned" |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Assessed by doctor but we do not know if he was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropout |
| Selective reporting (reporting bias) | Low risk | Reported pre‐defined outcome |
| Other bias | Low risk | No other risk found |
Du 2014.
| Methods | Randomised to two drugs. Double‐blind. 13 centres. Criteria for FD: Rome III with only postprandial distress syndrome (PDS) | |
| Participants | N = 383 Female 59.7% Mean age: 43.7 ±11.9 years for intervention group and 41.3 ± 12 years for comparator group Country of study: China |
|
| Interventions | Intervention: cinitapride hydrogen tartrate 1 mg and domperidone analogue three times a day Comparator: domperidone 10 mg and cinitapride analogue three times a day Rescue medication: none Duration 4 weeks |
|
| Outcomes | Global improvement: primary outcome = total score decreasing > 50%, secondary outcome = 7‐point Likert scale ranging from strongly improved, to strongly deteriorated. also assessed overall severity of postprandial fullness, early satiation, and bloating gastric emptying by participants | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly allocated |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | One patient was not included in the full analysis set; 11% from intervention group versus 9% from comparator group were excluded in the per protocol analysis, dropouts = 10.5% from intervention group versus 8.4% from comparator group. Lost to follow‐up was balanced between groups. |
| Selective reporting (reporting bias) | Low risk | Reported all predefined‐outcomes |
| Other bias | Low risk | No other risk found |
Francois 1987.
| Methods | Randomised. double blind, placebo‐controlled trial. Single‐centre. Criteria for FD: equivalent symptom with both type of FD | |
| Participants | N = 34 Female: 71% Age: 21 to 70 years Country: Belgium |
|
| Interventions | Intervention: cisapride 5 mg to 10mg orally three times a day Comparator: placebo Rescue medication: antacid and/or benzodiazepine tranquillizers Duration of treatment: 3 weeks |
|
| Outcomes | Scale (excellent, good, fair and poor) better, as good as, or worse), not validate. Assessed by participants | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly order |
| Allocation concealment (selection bias) | Unclear risk | No info |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 5.6 % (2/36) withdrew after randomisation, not clear from which group |
| Selective reporting (reporting bias) | Low risk | Reported pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Hallerback 2002.
| Methods | Randomised, double blind, placebo‐controlled trial. Multi‐centre (79). Criteria for FD: equivalent symptom with both types of FD | |
| Participants | N = 566 Female: 64% Mean age: not provided Countries: Denmark, Germany, France, Sweden and the UK |
|
| Interventions | Intervention: mosapride 5 mg twice a day, 10 mg twice a day, 7.5 mg three times a day Comparator: placebo Duration of treatment: 6 weeks Rescue medication: not mentioned |
|
| Outcomes | Global symptom severity assessed by participants (7‐point Likert scale), proportion of participants improved (investigator asked) | |
| Notes | We used authors' ITT sample in the analysis, because the number of randomised participants minus those who found ineligible after randomisation does not match authors' ITT | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomized" |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind, double‐dummy, treatment code was breaking after symptoms assessment |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 606 were randomised but 17 were found ineligible, only 566 (93%) were included in the author's ITT analysis. It is not clear from table 2 what patients were included in the ITT analysis, except those who did not fulfilled the eligibility after randomisation. Of those, 16% did not complete the study. It is not clear if the reasons of missing data were balanced between groups. |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Hansen 1998.
| Methods | Randomised, double blind, placebo‐controlled trial. Single‐centre. Criteria for FD: equivalent symptom with mixed ulcer‐like, reflux‐like, dysmotility‐like | |
| Participants | N = 219 Female: 68% Mean age: 43 ±15 years for prokinetic group and 42± 14 years for placebo group Country: Denmark. |
|
| Interventions | Intervention: cisapride 10 mg orally three times a day Comparator: placebo Rescue medication: not mentioned Duration of treatment: 2 weeks |
|
| Outcomes | Symptomatic response, global resolved, improved, unchanged or worse; also individual symptoms on 0‐3 Likert scale by participants | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized according to a computer generated randomization code." |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Doubl‐ blinded, double dummy |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind study, outcomes were assessed by participants who were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 15% dropout in the whole study (three arms) were withdrawn from the study, authors reported "reasons were equally distributed among the three groups" |
| Selective reporting (reporting bias) | Low risk | Reported pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Holtmann 2002.
| Methods | Randomised. double blind, placebo‐controlled trial. Multi‐centres (16) from private practice. Criteria for FD: equivalent symptoms with both type of FD | |
| Participants | N = 120 Female: 48.3% Mean age: 50 ±14.3 years for prokinetic group and 51.8± 13 years for placebo group Country: Germany |
|
| Interventions | Intervention: cisapride 10 mg three times a day Comparator: placebo Rescue medication: not mentioned Duration of treatment: 8 weeks |
|
| Outcomes | O’Brien global measure of the participants’ rating of 10 upper gastrointestinal symptoms:0 to 3 scores, VAS scale for the intensity of discomfort by participants | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded double‐dummy |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 9% dropout (8% in cisapride versus 10% placebo), reasons balanced between groups |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Holtmann 2006.
| Methods | Randomised. double‐blind, placebo‐controlled trial. Multi‐centres (79). Criteria for FD: Rome II with both type of FD. | |
| Participants | N = 548 Female: 63.5% Mean age: 47.8 ±16.1 years for prokinetic group and 49.3± 15.5 years for placebo group Country: Germany |
|
| Interventions | Intervention: itopride 50 mg three times a day, 100 mg three times a day, and 200 mg three times a day Comparator: placebo Rescue treatment: not mentioned Duration of treatment: 8 weeks |
|
| Outcomes | Valided Leeds Dyspepsia Questionnaire (LDQ), 6 grades administered by an investigator, summary scores ranged 0 to 40. Participants’ global assessments of efficacy were evaluated at eight weeks with the use of a global scale with the following five grades: symptom‐free, markedly improved, moderately improved, not changed, and deteriorated by participants | |
| Notes | 554 participants were randomised, however, data reported for 548 who received at least one medication as our ITT sample in the meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computor0generated randomisation |
| Allocation concealment (selection bias) | Low risk | The study medication was packed identically for four groups and was identified by a randomised number |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded double‐dummy |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 9% cisapride users versus 13% placebo users did not completed study medications. Reasons were balanced between groups |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcome |
| Other bias | Low risk | No other risk found |
Jian 1989.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Single‐centre. Criteria of FD: equivalent symptom with PDS | |
| Participants | N = 28 Female: 64% Mean age: 41 ±17 years for prokinetic group and 36± 17 years for placebo group Country: France |
|
| Interventions | Intervention: cisapride 10 mg orally three times a day Comparator: placebo Rescue medication: not allowed Duration of treatment: 6 weeks |
|
| Outcomes | Symptoms were evaluated by a diary GDS sum 14 and a VAS by participants | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomized" |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants had completed outcomes |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Kellow 1995.
| Methods | Randomised. double‐blind, placebo‐controlled trial. Single‐centre. Criteria for FD: equivalent symptom with mixed ulcer‐ like and dysmotility‐like | |
| Participants | N = 61 Female: 70% Mean age: 50 ±18 years for prokinetic group and 46 ± 15 years for placebo group Country : Australia |
|
| Interventions | Intervention: cisapride 10 mg orally three times a day Comparator: placebo Rescue medication: antacid Duration of treatment: 4 weeks |
|
| Outcomes | Total score of symptoms based on scale 0 to 3 assessed by doctor Global assessment: marked improvement‐complete or near complete resolution of symptoms: moderate improvement‐partial remission of symptoms; minimal improvement‐slight improvement of symptoms: unchanged‐no change in symptoms; deteriorated‐symptoms worsened as compared to before treatment assessed by participants |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomized" |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by both investigators and participants who were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 8% participants withdrawn (6.5% from prokinetic versus 10% from placebo), balanced between two groups |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Kusunoki 2012.
| Methods | Randomised, double blind, placebo‐controlled trial. Single‐centre. Criteria for FD: Rome II with both type of FD | |
| Participants | N = 42 Female: 65% Mean age: 40.3 ±13.2 years for the prokinetic group and 40.6± 18 years for the placebo group Country: Japan |
|
| Interventions | Intervention: acotiamide 100 mg orally three times a day Comparator: placebo Rescue medication:not mentioned Duration of treatment: 14 to 18 days |
|
| Outcomes | 7‐point Likert scale, ‘markedly improved in comparison with the baseline period.’ The worst condition was ‘markedly aggravated in comparison with the baseline period by participants | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only one from placebo group versus zero from prokinetic group lost to follow‐up, but the authors excluded 5 participants (12%) from the analysis (three from placebo versus two from prokinetic) due to protocol violation or noncompliance etc |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Unclear risk | The treatment duration was 14 to 18 days and both groups were in the same duration, likely participants were not measured on the same duration |
Li 2005.
| Methods | Randomised to two drugs. Double‐blind.Not clear number of centres but the authors are from 4 different hospitals. Criteria for FD: equivalent symptom to Rome and included both types of FD. | |
| Participants | N = 209 Female 53% Mean age: 38 years (overall) Country of study: China |
|
| Interventions | Intervention: itopride 50 mg orally three times a day Comparator: domperidone 10 mg three times a day Rescue medication: not allowed Duration 4 weeks |
|
| Outcomes | Symptoms scores 0 to 3, reported overall scores and individual symptoms scores in table 4. Global assessment: (pre‐treatment score minus post‐treatment score)/pre‐treatment score *100%. cure = Symptom‐free, significant improvement = scores >= 80%, improvement = scores < 80% but > 50%, not efficacy = =<50%, worse = scores < 0. Total improvement rate= cure+ significant improvement. Reported individual symptoms and global symptoms. Assessed by physician. | |
| Notes | Article In Chinese | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random digits tables, by statistician |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Drugs are provided by the pharmaceutical company, identical appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Accessed by doctors, not clear if outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Four from itopride group versus five from domperidone group lost to follow up. The authors excluded participants who had loss of follow‐up in the analysis, (we used the ITT sample in the meta‐analysis) |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Lin 2009.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Single‐centre Criteria for FD: Rome III and included only PDS core > 2 | |
| Participants | N = 60 Female 67% Mean age: 41 years for the prokinetic group and 40 years for the placebo group Country of study: China |
|
| Interventions | Intervention: mosapride dispersible table 5 mg three times a day plus hydrotalcite 1000mg three times a day Comparator: hydrotalcite 1000 mg plus placebo three times a day Rescue medication: not allowed Duration 2 weeks |
|
| Outcomes | PDS symptoms scores from 0 to 3, for 5 symptoms. 0 = no symptoms, 3 = severe symptoms, have severe impact at work and life need to be controlled by medications. global Symptoms improvement (efficacy rate). table 1, Point 0 = no symptoms. Symptoms improvement = scores reduced >=2, failure = scores reduced < 2 points or symptoms worse. Unclear assessor. | |
| Notes | Ariticle In Chinese | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random digits table |
| Allocation concealment (selection bias) | Low risk | First assigned medications the sequence numbers according to the random table, then the continuous participants received the medications in sequent |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The codes were blocked after the study was finished |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Two participants who were lost to follow‐up and two participants who were excluded (received antibiotics for other reasons) were not included in the report, it is not clear which groups these participants were assigned |
| Selective reporting (reporting bias) | Low risk | Reported pre‐specified outcomes |
| Other bias | Low risk | No other risk found |
Ma 2012.
| Methods | Randomised, double‐blind, placebo‐ (sham acupuncture) controlled trial. 8 Multi‐centre. Criteria for FD: Rome III with both type of FD | |
| Participants | N = 239 Female: 70% Mean age: 36.2± 13.9 years for the prokinetic group and 36.8 ±13.1 years for the control group Country: China |
|
| Interventions | Intervention: itopride 50 mg three times a day Comparator: sham acupuncture Rescue medication:not allowed Duration of treatment: 4 weeks but follow‐up at 12 weeks |
|
| Outcomes | Symptom Index of Dyspepsia scale (0 to 4). The improvement of at least two scores or no occurrence of any symptom included in the Symptom Index of Dyspepsia scale was regarded as the positive response. All assessed by participants | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation process was pre‐programmed and carried out by central computer. |
| Allocation concealment (selection bias) | Low risk | The allocation sequence was generated by a permuted‐block randomisation.Investigators received a confirmation email at the same time, containing a random number, the group assignment code and the patient’s basic information. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Although participants were blinded for which acupoints, participants and doctors could not be blinded for treatment interventions (sham acupuncture versus drugs) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded Quote: "the outcome assessors and statistical analysis were unaware of the intervention assignments throughout the trial" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 5.4% (8 from prokinetic group versus 5 from control group) could not complete 4‐week treatment (including dropout and violations), balanced between two groups |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Matsueda 2010a.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Multi‐centre (33). Criteria for FD: Rome II with both type of FD | |
| Participants | N = 323 Female: 56% Mean age: range 37.3 to 38.6 years Country: Japan |
|
| Interventions | Intervention: acotiamide 100 mg or 300 mg three times a day Comparator: placebo Rescue medication: not allowed Duration of treatment: 4 weeks |
|
| Outcomes | Daily basis, 9 symptoms, on 0 to 3 severity scale, on weekly basis, global assessment of overall treatment efficacy (OTE) on 7‐point Likert scales, elimination rate of postprandial fullness by participants | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed through a computer‐generated program |
| Allocation concealment (selection bias) | Low risk | Eligible participants were assigned a randomisation number according to a predetermined list at each site. These numbers were allocated in sequential order and registered in the patient enrolment list, and ensured appropriate concealed allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded, the study code and randomisation were examined at the end of the study to ensure that the study blind had been maintained |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Only 5% were excluded or discontinued but did not provide the reasons of exclusion, it is not clear if balanced between two groups |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Matsueda 2010b.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Multi‐centre (46). Criteria for FD: Rome II with PDS | |
| Participants | N = 462 Female: 65.4% Mean age: range 38.0 to 40.6 years Country: Japan |
|
| Interventions | Intervention: acotiamide 50 mg, 100 mg or 300 mg three times a day Comparator: placebo Rescue medication: not allowed Duration of treatment: 4 weeks |
|
| Outcomes | Daily basis, 9 symptoms, on 0 to 3 severity scale, on weekly basis, global assessment of overall treatment efficacy (OTE) on 7‐point Likert scales, elimination rate of postprandial fullness by participants | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed through a computer‐generated program. |
| Allocation concealment (selection bias) | Low risk | Eligible participants were assigned a randomization number according to a predetermined list at each site. These numbers were allocated in sequential order and registered in the patient enrolment list, and ensured appropriate concealed allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Only 5% were excluded or discontinued but did not provide the reasons of exclusion, it is not clear if balanced between two groups |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Matsueda 2012.
| Methods | Randomised, double‐blind, placebo‐controlled trials. Multi‐centre (67). Criteria for FD: Rome III with only PDS. | |
| Participants | N = 897 Female: 59.3% Mean age: 37.6±10.7 years for the prokinetic group and 37.1 ±9.9 years for the placebo group Country: Japan |
|
| Interventions | Intervention: acotiamide 100 mg three times a day Comparator: placebo Rescue medication: not mention Duration of treatment: 4 weeks |
|
| Outcomes | Daily basis, 9 symptoms, on 0 to 3 severity scale, on weekly basis, subjects global assessment of overall treatment efficacy (OTE) on 7‐point Likert scales, elimination rate of postprandial fullness by participants | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed through a computer‐generated program |
| Allocation concealment (selection bias) | Low risk | Eligible participants were assigned a randomisation number according to a predetermined list at each centre.These numbers were allocated in sequential order and registered in the patient enrolment list and the allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded. Emergency envelopes containing the randomisation code were provided to the investigators and were examined at the end of the study to ensure that the study blinding had been maintained. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only 5% dropout, with balanced reasons |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Mo 2003.
| Methods | Randomised to two drugs. Double‐blind. Multi‐centre (4). Criteria for FD: Rome II and included mainly PDS (99%) | |
| Participants | N = 80 Female 64% Mean age: 48,79 years for the itopride group and 47.39 years for the domperidone group Country of study: China |
|
| Interventions | Intervention: itopride hydrochloride 50 mg three times a day Comparator: domperidone 10 mg three times a day Rescue medication: not clear Duration 2 weeks |
|
| Outcomes | Data reported for individual symptoms in table 1 for upper abdominal uncomfortable, postprandial fullness, early satiety, decreased appetite, nausea, vomiting, acid reflux and regurgitation , improvement (significantly improved+ improved), we can consider "significantly improved"= symptom‐free. Unclear assessor. | |
| Notes | Article In Chinese. Use epigastric discomfort improvement because no global symptom was reported post treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly divided |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is not clear if outcomes were assessed by doctors or participants |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropout |
| Selective reporting (reporting bias) | Unclear risk | Reported planned outcomes. However, data were not reported for all participants as "global improvement", but reported for individual participants who had the symptoms at baseline. Therefore, we could not use randomised sample in our analysis, we picked the most reported symptoms (epigastric discomfort and early satiety N = 79). That is, we are not able to know which participants had symptoms free for all symptoms. Did not provide data as per ITT or PP sample (i.e. symptom response or resolution for each group of participants). For example, only 1 patient had belching in itopride group at baseline, then this symptom outcome after 2 weeks was reported as improved in 1/1 patient. |
| Other bias | Low risk | No other risk found |
Nakamura 2017.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Single‐centre. Criteria for FD: Rome III with only PDS. | |
| Participants | N = 50 Female: 74% Mean age: 56.3 ± 16.9 years Country: Japan |
|
| Interventions | Intervention: acotiamide 100 mg three times a day Comparator: placebo Rescue medication: not mentioned Duration of treatment: 2 weeks |
|
| Outcomes | Gastrointestinal symptom rating scale (GSRS)‐ 15 questions, 5 lower domains, (reflux, abdominal pain, dyspepsia, diarrhoea, constipation), 7‐point Likert scale by unclear assessor | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Drawing lots from a sealed envelop that contained pre‐assigned random group |
| Allocation concealment (selection bias) | Low risk | The code was concealed until the end of the study |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Do not know if assessors were doctors or participants and if outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Although only 8% (4/50) filed to complete. 12% (3) in active treatment stopped treatment versus 4% (1) in placebo 8% dropout |
| Selective reporting (reporting bias) | Low risk | Reported all prespecified outcomes |
| Other bias | Low risk | No other risk found |
Rösch 1987.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Single‐centre. Criteria for FD: equivalent symptom with both types of FD | |
| Participants | N = 114 Female: not reported Mean age: not reported Country: Germany |
|
| Interventions | Intervention: cisapride 10 mg orally three times a day Comparator: placebo Rescue medication: not mentioned Duration of treatment: 4 weeks |
|
| Outcomes | Physicians assessed: frequency 0 to 7, severity 0 to 3, participants assessed: VAS 0‐100 | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | quote: "Randomized" |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Global outcomes assessed by both doctors and participants but do not know if outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 5% from prokinetic group versus 4% from placebo group drop‐out |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Shen 2014.
| Methods | Randomised to two drugs. (aimed to assess the add‐on effect of itopride to azintamide.Single‐centre. Criteria for FD: Rome III and assessed only abdominal distension.(PDS) | |
| Participants | N = 80 Female 41% Mean age: 57.42 years for the intervention group and 64.17 years for the control group (P > 0.05) Country of study: China |
|
| Interventions | Intervention: itopride one tablet three times a day + azintamide three times a day (no dose provided) Comparator: azintamide three times a day (no dose provided) Rescue medication: azintamide Duration of treatment: 2 weeks |
|
| Outcomes | Physicians assessed abdominal distension, duration 0 to 3 scores, severity 0 to 3 | |
| Notes | Mean age was significant different between two groups Article In Chinese |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Singh 2015.
| Methods | Randomised to two drugs. Signle‐centre. Criteria for FD: equivalent symptom with both types of FD | |
| Participants | N = 120 Female: 68% Mean age: 47.1 years for the metoclopramide group and 35.8 years for the domperidone group Country: India |
|
| Interventions | Intervention: metoclopramide 10 mg three times a day Comparator: domperidone 10 mg three times a day Rescue medication: not allow Duration of treatment: 4 weeks |
|
| Outcomes | Short‐Form Leeds Dyspepsia Questionnaire (SF‐LDQ) , 5 questions, 5 symptoms, 5‐point scale but unclear assessor | |
| Notes | One of the arm was "Levosulpride". The authors considered as one of the prokinetics. We decided not to consider it as a prokinetic because of its antidepressant effect, so this arm is not included in the meta‐analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly with randomised block design |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 8.8% dropout but imbalance between groups and no detail of dropout participants |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Sun 2003.
| Methods | Randomised to two drugs. Double‐blinded. Multi‐centre (4). Criteria for FD: equivalent symptom to Rome and mainly included PDS (99%) | |
| Participants | N = 240 % Female: not reported Mean age: not reported (range: 18 to 70) Country of study: China |
|
| Interventions | Intervention: itopride hydrochloride 50 mg three times a day Comparator: domperidone 10 mg three times a day Rescue medication: not allowed Duration of treatment: 2 weeks |
|
| Outcomes | Symptoms scores 0 to 3. For each participants calculated Imprevement rate = ((pre total scores ‐ post total scores)/ pre total scores) *100%". global assessment: cure= symptoms free, significant effective = symptoms improved significantly, scores reduced >=2 scores, but not yet symptoms free, effective=symptoms improved, but scores reduced < 2 scores, but >=1. Failure= symptoms no change or worse. Total effective rate= (number of cure + number of significant effective)/ (cure+ significant effective+ effective+ failure cases) *100%. reported effective rates of abolishing postprandial full ness, early satiety and epigastric discomfort. ‐ not for all symptoms. Unclear assessor. | |
| Notes | Article In Chinese Use postprandial fullness symptom improvement due to no global symptom reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded, double‐dummy |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Do not know if assessors were doctors or participants and if outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 4% (N = 10) lost to follow‐up, 5 in each group, Since outcome reported for 115 (itopride) versus 117 (domperidone), we do not know if all lost to follow‐up participants also had postprandial symptoms so we could not add these participants in the ITT analysis forms, effective rate was reported for three major symptoms, therefore we were not able to use the ITT sample to calculate the overall global improvement rate |
| Selective reporting (reporting bias) | Unclear risk | Since not all participants had all symptoms, effective rate was reported for three major symptoms, therefore we were not able to use the ITT sample to calculate the overall global improvement rate |
| Other bias | Low risk | No other risk found |
Tack 2009.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Multi‐centre (8). Criteria for FD: Rome II with both type of FD | |
| Participants | N = 71 Female: 54% Mean age: 40 years for the prokinetic group and 49.1 years for the placebo group Countries: Belgium, the Netherlands, UK, Norway, Poland |
|
| Interventions | Intervention: acotiamide 50 mg, 100 mg and 300 mg three times a day Comparator: placebo Rescue medication: not mentioned Duration of treatment: 3 weeks |
|
| Outcomes | Participants assessed the severity (0 = absent; 1 = mild; 2 = moderate; 3 = severe) and the frequency (0 = absent; 1 = rarely; 2 = occasionally; 3 = often; 4 = whole day) of nine dyspeptic symptoms (upper abdominal pain, upper abdominal discomfort, postprandial fullness, upper abdominal bloating, early satiety, nausea, vomiting, excessive belching and heartburn). An overall symptom score was calculated as the sum of the mean weekly individual symptom scores. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 13% were excluded from the efficacy analysis but unbalance in each group. |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Unclear risk | Imbalance in age and gender, although did not reach statistical significance |
Tack 2011.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Multi‐centre. Criteria for FD: Rome II with PDS. | |
| Participants | N = 289 Female: no data Mean age: no data Countries: US, Japan and Europe |
|
| Interventions | Intervention: acotiamide 100 mg and 300 mg three times a day Comparator: placebo Rescue medication: not mentioned Duration of treatment: 12 weeks |
|
| Outcomes | Global Subject Outcome Assessment (GSOA), at Week 12. The secondary endpoints were weekly GSOA, 50% response rate of GSOA and the individual symptom score (5 Likert scale) of 5 dyspepsia symptoms (PF, ES, upper abdominal bloating, nausea and epigastric pain (EP)) by unclear assessor | |
| Notes | Conference abstract | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Ramdomised |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Reported number of participants were treated, randomized sample is not provided |
| Selective reporting (reporting bias) | Unclear risk | No information |
| Other bias | Unclear risk | Conference abstract, no other information |
Talley 2000.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Multi‐centre (at least 2). Criteria for FD: equivalent symptom. with both types of FD | |
| Participants | N = 609 Female: 69% Mean age: 46.3 years for the prokinetic group and 46.1 years for the placebo group Countries: USA and Europe |
|
| Interventions | Intervention: ABT‐229 1.25 mg, 2.5 mg, 5 mg and 10 mg twice a day Comparator: placebo Rescue medication: not mentioned Duration 4 weeks |
|
| Outcomes | Participant questionnaire, VAS 0‐100 used to assess severity, frequency and impact measured by 5‐graded Likert scale, duration one a 7‐graded Likert scale. Patient diary, severity of postprandial fullness, bloating, epigastric discomfort and postprandial nausea recorded on 7‐point Likert scale. Global evaluation at week 4 for excellent (complete or near complete resolution of symptoms), good (distinct improvement), moderate (some improvement), or poor (no change or deterioration). Assesed by participants. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomized" |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐bledind. Placebo was identical to active therapy. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 8% (47/609) participants prematurely discontinued, the reasons were balanced between groups. The authors only reported outcome for 589 (96%) participants who had baseline data and at least one follow up assessment no more than 5 days and considered these participants as ITT population, which were 99%, 98%, 98% and 96% in active treatment but 95% in placebo group. |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Unclear risk | Difference in number baseline scores in table 1 and figure 2 |
Talley 2008a.
| Methods | Randomised, double‐blinded, placebo‐controlled trial. Not clear number of centres. Criteria for FD: Rome II with both types of FD | |
| Participants | N = 416 Female: not reported Mean age: not reported Country: Not clear but international multi‐centre |
|
| Interventions | Intervention: acotiamide 300 mg, 600 mg and 900 mg three times a day Comparator: placebo Rescue medication: not mentioned Duration of treatment: 12 weeks |
|
| Outcomes | Primary endpoints were overall (adequate) relief of stomach symptoms in past 7 days (ORS, %) and overall treatment evaluation past 7 days (OTE: 9 graded). ORS responder as yes ≥ 50% weeks. We use ORS for outcome. Unclear assessor | |
| Notes | Conference abstract. Treatment duration was 12 weeks, the primary outcome OTE reported for four weeks | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised |
| Allocation concealment (selection bias) | Low risk | Concealed allocation was strictly maintained |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Do not know if assessors were doctors or participants and if outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information. Did not report randomized number and dropout numbers |
| Selective reporting (reporting bias) | Unclear risk | No information. Data for QoL and AEs only mentioned not significant difference |
| Other bias | Unclear risk | Conference abstract. No sufficient information. Treatment duration was 12 weeks, the primary outcome OTE reported for 4 weeks |
Talley 2008b.
| Methods | Randomised, double‐blind. placebo‐controlled trial. Multi‐centre (170). Criteria for FD: Rome II with both types of FD | |
| Participants | N = 525 Female: 64.7% Mean age: 42.9 +/‐12.9 years for the prokinetic group and 43.3 +/‐12.9 years for the placebo group Countries: Germany, France, the Netherlands, Belgium, Poland, UK , USA, and Canada |
|
| Interventions | Intervention: itopride 100 mg three times a day Comparator: placebo Rescue medication: not mention Duration of treatment: 8 weeks |
|
| Outcomes | (1) global patient assessment (GPA) of efficacy; ‐ symptom‐free, markedly improved, slightly improved, unchanged, worse’’. Symptom‐free or markedly improved was defined as a responder.and (2) Leeds Dyspepsia Questionnaire (LDQ).‐LDQ questions 1 and 8,measuring pain in the upper abdomen and feeling of fullness, respectively, were the primary end point questions by participants. We used the information from LDQ. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | All participants had a four‐digit number assigned. The randomisation code was generated by Quintiles Inc. using a computer‐generated program. |
| Allocation concealment (selection bias) | Low risk | At the baseline visit, eligible participants were assigned a randomisation number according to the pre‐determined list at each site. These numbers were allocated in sequential order and registered in the patient enrolment list. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded, participants took identical active or placebo medication, and participants and investigators were blinded at all sites. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded.Emergency envelopes were provided to the investigators with the study code and randomisation, and these were examined at the end of the study to ensure the study blind being maintained |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 525 participants were included in ITT analysis. However, 14% (80) from intervention group versus 12% (69) from placebo group did not complete the study (both studies combined).(in two studies, 1170 were randomised, data only reported for 1150 without further information for each group in each study) (see table 1 and table 3). |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk seen |
Talley 2008c.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Multi‐centre (not clear in numbers). Criteria for FD: Rome II with both types of FD | |
| Participants | N = 645 Female: 67.2% Mean age: 42.6 +/‐12.8 years for the prokinetic group and 43.0+/‐ 12.5 years for the placebo group Countries: USA, Canada, Poland, Germany |
|
| Interventions | Intervention: itopride 100 mg three times a day Comparator: placebo Rescue medication: not mention Duration of treatment: 8 weeks |
|
| Outcomes | (1) global patient assessment (GPA) of efficacy; ‐ symptom‐free, markedly improved, slightly improved, unchanged, worse’’. Symptom‐free or markedly improved was defined as a responder.and (2) Leeds Dyspepsia Questionnaire (LDQ). LDQ questions 1 and 8, measuring pain in the upper abdomen and feeling of fullness, respectively, were the primary endpoint questions by participants. We used the information from LDQ. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | All participants had a four‐digit number assigned. The randomisation code was generated by Quintiles Inc. using a computer‐generated program. |
| Allocation concealment (selection bias) | Low risk | At the baseline visit, eligible participants were assigned a randomisation number according to the pre‐determined list at each site. These numbers were allocated in sequential order and registered in the patient enrolment list. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded, participants took identical active or placebo medication, and participants and investigators were blinded at all sites. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded. Emergency envelopes were provided to the investigators with the study code and randomisation, and these were examined at the end of the study to ensure the study blind being maintained |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 626 / 645 (97%) participants included in ITT, but no detail informations given for each group (in two studies, 1170 were randomised, data only reported for 1150 without further information for each group in each study) (see table 1 and table 3), even the authors claimed to use ITT. 14% (80) from prokinetic group versus 12% (69) from placebo group did not complete the study (both studies combined). |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Teixeira 2000.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Single‐centre. Criteria for FD: Rome II but unclear most prevalent type of FD | |
| Participants | N = 38 Female 71% Mean age: range 18 to 74 years (median 39) Country of study: Portugual |
|
| Interventions | Intervention: cisapride 10 mg orally three times a day Comparator: placebo Rescue medication: not mention Duration 15 days |
|
| Outcomes | Participants' symptoms assessed though a questionnaire of symptoms point scale. Participants were asked about intensity, frequency, duration and factors that triggered or relief symptoms, as well as the daily activities related to symptoms | |
| Notes | Article In Protuguese | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No mention of dropout rate |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcomes |
| Other bias | Low risk | No other risk found |
Vakil 2008a.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Mulit‐centres (675). Criteria for FD: Rome II but subgroup analysis based on Rome III and include only dysmotility‐like symptom (PDS) | |
| Participants | N = 1360 Female: 100% Mean age: 47.3+/‐13.2 years for the prokinetic group and 44.2+/‐14.5 years for the placebo group Countries: USA, UK, Canada, and South Africa |
|
| Interventions | Intervention: tegaserod 6 mg twice a day Comparator: placebo Rescue medication: not reported Duration 6 weeks |
|
| Outcomes | Daily assess 7‐point scale, weekly assessed global assessment of change question, 7‐point Likert scale. Outcomes: (a) percentage of days with satisfactory relief of dyspepsia symptoms, and (b) composite average daily severity score (CADSS) for the three cardinal dyspepsia symptoms (post‐prandial fullness, early satiety, and bloating). had not been validated‐calculated by averaging the responses to the daily questions regarding individual dyspepsia symptom severity. Assessed by participants. | |
| Notes | Study of women only | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation (1:1 allocation ratio) was performed using a computer‐generated sequence in each treatment centre using permutated blocks of size 4 |
| Allocation concealment (selection bias) | Low risk | The randomisation scheme was reviewed by a biostatistics quality assurance group, locked on their approval, and concealed from participants and study personnel at both the site and the sponsor offices until after both studies were completed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blind |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 13% from prokinetic group versus 10% from placebo group did not completed the study, reasons were balanced between groups |
| Selective reporting (reporting bias) | Low risk | Reported symptom data. However, no QoL raw data were reported except Quote: " the greatest improvement during tegaserod versus placebo therapy was observed in the eating/drinking domain for this subgroup of participants (0.77 point, P = 0.0005)" (combined two studies) |
| Other bias | Low risk | No other risk found |
Vakil 2008b.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Mulit‐centre (675). Criteria for FD: Rome II but subgroup analysis based on Rome III and include only dysmotility‐like symptom. | |
| Participants | N = 1307 Female 100% Mean age:43.4+/‐13.7 years for the prokinetic group and 43.6+/‐13.2 years for the placebo group Countries: USA, UK, Canada, and South Africa |
|
| Interventions | Intervention: tegaserod 6 mg twice a day Comparator: placebo Rescue medication: not reported Duration 6 weeks |
|
| Outcomes | Daily assess 7‐point scale, weekly assessed global assessment of change question, 7‐point Likert scale. Outcomes: (a) percentage of days with satisfactory relief of dyspepsia symptoms, and (b) composite average daily severity score (CADSS) for the three cardinal dyspepsia symptoms (post‐prandial fullness, early satiety, and bloating). had not been validated‐calculated by averaging the responses to the daily questions regarding individual dyspepsia symptom severity. Assessed by participants. | |
| Notes | Study only in women | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation (1:1 allocation ratio) was performed using a computer‐generated sequence in each treatment centre using permutated blocks of size 4 |
| Allocation concealment (selection bias) | Low risk | The randomisation scheme was reviewed by a biostatistics quality assurance group, locked on their approval, and concealed from participants and study personnel at both the site and the sponsor offices until after both studies were completed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blind |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 17% from prokinetic group versus 11% from placebo group did not completed the study, reasons were balanced between groups, reasons were balanced between groups |
| Selective reporting (reporting bias) | Low risk | Reported symptom data. However, no QoL raw data were reported exceptQuote: "the greatest improvement during tegaserod versus placebo therapy was observed in the eating/drinking domain for this subgroup of participants (0.77 point, P = 0.0005)" (combined two studies) |
| Other bias | Low risk | No other risk found |
Wang 1995.
| Methods | Randomised, placebo‐controlled, open‐labelled trial. Multi‐centres (16). Criteria for FD: equivalent symptoms and mixed type of FD but PDS predominant (70%) | |
| Participants | N = 609 Female: 54% Mean age: 43 years for prokinetic group and 41 years for placebo group Country: China |
|
| Interventions | Intervention: cisapride 5 mg three times a day Comparator: placebo Rescue medication: not allowed Duration of treatment: 4 weeks |
|
| Outcomes | Symptoms scores 0 to 3 scale, 0 = no symptoms, 3 = severe symptoms, could not have routine work. Symptoms improvement assessment: effective = improved one score, significant effective = improved two scores or symptoms disappeared, failure = worse or no change. Assessed by participants. | |
| Notes | Article In Chinese Open‐label study PDS predominant |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label study. No party was blinded, medication and placebo were different in appearance |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label study. No party was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 3% (N = 19) were excluded from the analyses because they could not finish the treatment. However, detailed reasons were provided for all participants but not by treatment group, except all four discontinuation due to AEs were from cisapride; therefore, we were not able to use ITT sample (609) in the analysis |
| Selective reporting (reporting bias) | Low risk | Reported pre‐specified outcomes |
| Other bias | Unclear risk | More participants in cisapride had longer diseases duration (> 2 years, P < 0.05). |
Wong 2014.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Not clear number of centres but all authors are from one centre. Criteria for FD: equivalent symptom but include PDS and PDS overlap symptom. | |
| Participants | N = 30 Female: not reported Mean age: not reported Country: Malaysia |
|
| Interventions | Intervention: itopride 100 mg three times a day Comparator: placebo Rescue medication: not reported Duration 8 weeks |
|
| Outcomes | Leeds Dyspepsia questionnaire (LDQ), Functional Dyspepsia Questionnaire (FDQ) to assess symptoms improvement. Assessed by participants. | |
| Notes | Conference abstract | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Radomized |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessed by patient who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No information |
| Other bias | Unclear risk | Conference abstract, no other information is provided |
Yeoh 1997.
| Methods | Randomised, double‐blind,placebo‐controlled trial. Single‐centre, Criteria for FD: equivalent symptom and included both types of FD | |
| Participants | N = 104 Female: 53% Mean age:43.5+/‐2.2 years for the gastritis group and 35.6+/‐1.5 years for the non‐gastritis group Country: Singapore |
|
| Interventions | Intervention: cisapride 10 mg orally three times a day Comparator: placebo Rescue medication: antacid (open‐labelled) Duration of treatment: 4 weeks |
|
| Outcomes | Symptoms of epigastric pain, bloating, nausea, belching, early satiety and heartburn were graded on a 4‐point scale were assessed by participants A global response by physicians‐(i) poor, no change or deterioration of symptoms; (ii) fair, clear but limited improvement; (iii) good, considerable overall improvement; and (iv) excellent, complete or almost complete disappearance of symptoms |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomly allocated" |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Scores were assessed by participants, global response was assessed by physicians. It is not clear if outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 27% (28/104) participants did not complete the study |
| Selective reporting (reporting bias) | Low risk | Reproted all pre‐defined outcomes |
| Other bias | Low risk | No other risk of bias found |
Zhou 2000.
| Methods | Randomised to two drugs. Double‐blinded. Multi‐centre (5). Criteria for FD: equivalent symptoms but unclear predominant symptom. | |
| Participants | N = 208 Female: 64% Mean age: 42.8 years for itopride group and 42.6 years for domperidone group Country: China |
|
| Interventions | Intervention: itopride hydrochloride 50 mg + placebo domperidone three times a day Comparator: domperidone 10 mg + placebo itopride three times a day Rescue medication: not mentioned Duration of treatment: 2 weeks |
|
| Outcomes | Global, A = complete resolution = the primary symptom disappeared, B = Significantly effective = the primary symptom improved at least two grades; C = improved: primary symptom improved one grade, D = treatment failure, no improvement or worsening of primary symptom. Response rate = (A+B) / (A+B+C+D). We used (A+B+C) / total as improvement rate, to be consistent with other studies. Unclear assessor. | |
| Notes | Article In Chinese The authors used Qupte: “lack of appetite” in Chinese in the main text, however, they translated it as “anorexia” in the tables in English. We believe “lack of appetite” is a more appropriate translation for the term. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | All medications were packed in bottles separately, randomised by the pharmaceutical company stratified for each hospital, the codes were mixed, coded for treatment or control, the physicians distributed the medication according to the sequence of the code” |
| Allocation concealment (selection bias) | Low risk | All medications were packed in bottles separately, randomized by the pharmaceutical company stratified for each hospital, the codes were mixed, coded for treatment or control, the physicians distributed the medication according to the sequence of the code” |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is not clear if doctors or participants assessed the outcomes and if outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Global response rate reported in PP sample, missing outcome data for 3% participants. Reported the detail of participants who were lost to follow‐up or withdrew from treatment, They were balanced between the two groups. Symptom outcomes were reported based on PP sample. However, since authors reported baseline characteristics in a figure instead of providing the raw data, it is difficult to retrieve the patient numbers with symptoms at baseline. |
| Selective reporting (reporting bias) | Low risk | Reported all pre‐defined outcome |
| Other bias | Unclear risk | The authors used "lack of appetite" in Chinese in the main text but they translated it as "anorexia" in the table in English. We believed that "lack of appetite" is a more appropriate translation for the term. |
Zhu 2005.
| Methods | Randomised to two drugs. Double‐blinded. Multi‐centre. Criteria for FD: equivalent symptoms and all participants must have epigastric distention or early satiety (severity score > / = 2) (PDS) | |
| Participants | N = 236 % Female: not reported Mean age: not reported Country: China |
|
| Interventions | Intervention: itopride hydrochloride 50 mg + placebo domperidone three times a day Comparator: domperidone 10 mg + placebo itopride three times a day Rescue medication: not mentioned Duration of treatment: 4 weeks |
|
| Outcomes | Reported global symptoms improvement rates were not proportion of participants, but proportion of symptoms, symptom‐free reported for two major symptoms, but not all participants had this symptom so we can not use ITT sample in the outcome calculation. Unclear assessor. | |
| Notes | Article in Chinese Use early satiety symptom‐free due to no global symptom reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly allocated |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is not clear if doctors or participants assessed the outcomes and if outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants had outcomes reported |
| Selective reporting (reporting bias) | Unclear risk | Reported global symptoms improvement rates were not proportion of participants, but proportion of symptoms, symptom‐free reported for two major symptoms, but not all participants had this symptom so we can't use ITT sample in the outcome calculation |
| Other bias | Low risk | No other risk found |
AE= adverse event; EPS= epigastric pain syndrome; FD = functional dyspepsia; GDS = global dyspepsia symptoms; ITT = intention to treat; ORS = overall resolution of symptoms; OTE = overall treatment efficacy; PDS= post‐prandial distress syndrome; PP = per‐protocol; QoL = quality of life; SD ‐ standard deviation; VAS = visual analogue scale;
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Agorastos 1991 | Less than 90% underwent EGD |
| Bekhti 1979 | Participants did not undergo EGD |
| Chen 2010 | Only nocturnal symptom |
| Chey1982 | EGD and/or x‐ray used to exclude organic disease and do not know the number of participants who underwent EGD |
| Chung1993 | EGD and/or x‐ray used to exclude organic disease and do not know the number of participants who underwent EGD |
| Creytens 1984 | EGD and/or x‐ray used to exclude organic disease and do not know the number of participants who underwent EGD |
| Davis1988 | Participants did not undergo EGD |
| De Loose 1979 | Included participants with biliary disease and did not mention EGD |
| Deruyttere 1987 | EGD or x‐ray used to exclude organic disease and do not know the number of participants underwent EGD |
| Goethals 1987 | No data in phase I of cross‐over study |
| Haarmann 1979 | Participants did not undergo EGD |
| Hannon 1987 | EGD and/or x‐ray using to exclude organic disease and do not know the number of participants who underwent EGD |
| Hausken 1992 | Participants were not required to have negative endoscopy |
| Kas'ianenko 2014 | Historical placebo |
| Kearney 2000 | Participants did not undergo EGD (uninvestigated dyspepsia) |
| Kim 2010 | Herbal prokinetics |
| Liu 2013 | Herbal prokinetics |
| Manayagi 2014 | Only one arm of study, with no control group, just add‐on PPI to prokinetic in one arm |
| Milo 1984 | Included organic causes |
| Miwa 2009 | Used tandospirone citrate, which is primarily anxiolytic drug |
| Shim 2015 | Herbal prokinetics |
| Tack 2012 | Using buspirone which is primarily anxiolytic drug |
| Tack 2016 | 25% of participants had diabetes mellitus and all cases are suspected gastroparesis |
| Talley 2001 | No data available |
| Testoni 1990 | Included organic causes |
| Van de Mierop 1979 | Participants did not undergo EGD |
| Van Ganse 1978 | Included organic diseases |
| Van Outryve M | Included organic diseases |
| Wood 1993 | Reported day‐ and night‐time symptoms separately |
| Yamawaki 2016 | Add‐on effect of prokinetic to PPI (prokinetic versus PPI versus PPI only), not eligible comparison |
| Yan 2012 | Reported only nocturnal symptom |
EGD = esophago‐gastro‐duodenoscopy; PPI = proton pump inhibitor
Differences between protocol and review
We removed "complete resolution of global symptoms of dyspepsia" from secondary outcome because it should be considered as a subgroup analysis.
We removed "tandospirone citrate" from types of intervention because it is primarily an anxiolytic drug.
We changed "one prokinetic versus another prokinetic" to "one prokinetic versus domperidone" in the 'Summary of findings' table because domperidone is the most commonly used comparator in prokinetic versus prokinetic.
Contributions of authors
RP: drafted the revised protocol, performed the literature search for the systematic review and meta‐analysis, extracted data, evaluated study quality and wrote the manuscript. YY: revised the protocol, performed the literature search for the systematic review and meta‐analysis, extracted data, evaluated study quality and edited the manuscript. NB: drafted the first version of the protocol RK: drafted the first version of the protocol GL: supervising author and edited the manuscript PM: supervising author and edited the manuscript
Sources of support
Internal sources
McMaster University, Canada.
External sources
No sources of support supplied
Declarations of interest
RP: none known. YY: none known. NB: has received speaker honoraria, consulting and reimbursement for expenses from AbbVie. RK: has received fees for consulting from Takeda, AbbVie, Jansen, Shire, Pfizer and Robarts Clinical Trials. GL: none known. PM: attends advisory board meetings of Allergan, and has received grants from Allergan and Takeda to support a research network evaluating irritable bowel syndrome and inflammatory bowel disease across Canada.
New
References
References to studies included in this review
Abid 2017 {published data only}
- Abid S, Jafri W, Zaman MU, Bilal R, Awan S, Abbas A. Itopride for gastric volume, gastric emptying and drinking capacity in functional dyspepsia. World Journal of Gastrointestinal Pharmacology and Therapeutics 2017;8(1):74‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Al‐Quorain 1995 {published data only}
- Al‐Quorain A, Larbi EB, Al‐Shedoki F. A double‐blind, randomized, placebo‐controlled trial of cisapride in Saudi Arabs with functional dyspepsia. Scandavian Journal of Gastroenterology 1995;30:531‐4. [DOI] [PubMed] [Google Scholar]
Amarapurkar 2004 {published data only}
- Amarapurkar DN, Rane P. Randomised, double‐blind, comparative study to evaluate the efficacy and safety of ganaton (itopride hydrochloride) and mosapride citrate in the management of functional dyspepsia. Journal of the Indian Medical Association 2004;102(12):735‐7. [PubMed] [Google Scholar]
Champion 1997 {published data only}
- Champion MC, MacCannell KL, Thomson AB, Tanton R, Eberhard S, Sullivan SN, et al. A double‐blind randomized study of cisapride in the treatment of nonulcer dyspepsia. Canadian Journal of Gastroenterology 1997;11(2):127‐34. [DOI] [PubMed] [Google Scholar]
Chen 2004 {published data only}
- Chen SY, Wang JY, Zhu CW, Yuan YZ, Zou B, Xia L, et al. A randomized controlled multi‐center clinical trial on mosapride in the treatment of functional dyspepsia. Zhonghua Liu Xing Bing Xue za Zhi 2004;25(2):165‐8. [PubMed] [Google Scholar]
Choi 2015 {published data only}
- Choi MG, Rhee PL, Park H, Lee OY, Lee KJ, Choi SC, et al. Randomized, controlled, multi‐center trial: comparing the safety and efficacy of DA‐9701 and itopride hydrochloride in patients with functional dyspepsia. Journal of Neurogastroenterology and Motility 2015;21(3):414‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
de Groot 1997 {published data only}
- Groot GH, Both PS. Cisapride in functional dyspepsia in general practice. A placebo‐controlled, randomized, double‐blind study. Alimentary Pharmacology & Therapeutics 1997;11(1):193‐9. [DOI] [PubMed] [Google Scholar]
De Nutte 1989 {published data only}
- Nutte N, Ganse W, Witterhulghe M, Defrance P. Relief of epigastric pain in nonulcer dyspepsia: controlled trial of the promotility drug cisapride. Clinical Therapeutics 1989;11(1):62‐8. [PubMed] [Google Scholar]
Du 2014 {published data only}
- Du Y, Su T, Song X, Gao J, Zou D, Zuo C, et al. Efficacy and safety of cinitapride in the treatment of mild to moderate postprandial distress syndrome‐predominant functional dyspepsia. Journal of Clinical Gastroenterology 2014;48(4):328‐35. [DOI] [PubMed] [Google Scholar]
Francois 1987 {published data only}
- Francois I, Nutte N. Nonulcer dyspepsia: effect of the gastrointestinal prokinetic drug cisapride. Current Therapeutic Research 1987;41(6):891‐8. [Google Scholar]
Hallerback 2002 {published data only}
- Hallerback BI, Bommelaer G, Bredberg E, Campbell M, HellblomM, Lauritsen K, et al. Dose finding study of mosapride in functional dyspepsia: a placebo‐controlled, randomized study. Alimentary Pharmacology & Therapeutics 2002;16:959‐67. [DOI] [PubMed] [Google Scholar]
Hansen 1998 {published data only}
- Hansen JM, Bytzer P, Schaffalitzky de Muckadell OB. Placebo‐controlled trial of cisapride and nizatidine in unselected patients with functional dyspepsia. American Journal of Gastroenterology 1998;93(3):368‐74. [DOI] [PubMed] [Google Scholar]
Holtmann 2002 {published data only}
- Holtmann G, Gschossmann J, Mayr P, Talley NJ. A randomized placebo‐controlled trial of simethicone and cisapride for the treatment of patients with functional dyspepsia. Alimentary Pharmacology & Therapeutics 2002;16:1641‐8. [DOI] [PubMed] [Google Scholar]
Holtmann 2006 {published data only}
- Holtmann G, Talley NJ, Liebregts T, Adam B, Parow C. A placebo‐controlled trial of itopride in functional dyspepsia. New England Journal of Medicine 2006;354(8):832‐40. [DOI] [PubMed] [Google Scholar]
Jian 1989 {published data only}
- Jian R, Ducrot F, Ruskone A, Chaussade S, Rambaud JC, Modiglian R, et al. Symptomatic, radionuclide and therapeutic assessment of chronic idiopathic dyspepsia. A double‐blind placebo‐controlled evaluation of cisapride. Digestive Diseases and Sciences 1989;34(5):657‐64. [DOI] [PubMed] [Google Scholar]
Kellow 1995 {published data only}
- Kellow JE, Cowan H, Shuer B, Riley JW, Lunzer MR, Eckstein RP, et al. Efficacy of cisapride therapy in functional dyspepsia. Alimentary Pharmacology & Therapeutics 1995;9:153‐60. [DOI] [PubMed] [Google Scholar]
Kusunoki 2012 {published data only}
- Kusunoki H, Haruma K, Manabe N, Imamura H, Kamada T, Shiotani A, et al. Therapeutic efficacy of acotiamide in patients with functional dyspepsia based on enhanced postprandial gastric accommodation and emptying: randomized controlled study evaluation by real‐time ultrasonography. Neurogastroenterology and Motility 2012;24(6):540–5. [DOI] [PubMed] [Google Scholar]
Li 2005 {published data only}
- Li YH, Gong PL, Hou XH, Chen J, Liu NZ, Tian DA, et al. Itopride in treatment of 104 patients with functional dyspepsia: a randomized, double blind controlled clinical trial. Zhongguo Xinyao Yu Linchuang Zazhi 2005;7:524‐8. [Google Scholar]
Lin 2009 {published data only}
- Lin J, Ren M, Peng X, Xiao Y, Wang S, Yang L, et al. Short‐term efficacy of mosapride dispersible tablet on postprandial distress syndrome [莫沙必利分散片治疗餐后不适综合征的近期疗效观察]. 胃肠病学 (Chinese Journal of Gastrenterology) 2009;14(8):488–90. [Google Scholar]
Ma 2012 {published data only}
- Ma TT, Yu SY, Li Y, Liang FR, Tian XP, Zheng H, et al. Randomised clinical trial: an assessment of acupuncture on specific meridian or specific acupoint vs. sham acupuncture for treating functional dyspepsia. Alimentary Pharmacology & Therapeutics 2012;35:552–61. [DOI] [PubMed] [Google Scholar]
Matsueda 2010a {published data only}
- Matsueda K, Hongo M, Sasaki D, Kusano M, Harasawa S, Arakawa T, et al. Therapeutic efficacy of novel agent (Z‐338) in functional dyspepsia (FD). Gastroenterology 2005;128 (supplement1):A467. [Google Scholar]
- Matsueda K, Hongo M, Tack J, Aoki H, Saito Y, Kato H. Clinical trial: dose‐dependent therapeutic efficacy of acotiamide hydrochloride (Z‐338) in patients with functional dyspepsia ‐ 100mg tid is an optimal dosage. Neurogastroenterology and Motility 2010;22:618‐e173. [DOI] [PubMed] [Google Scholar]
Matsueda 2010b {published data only}
- Matsueda K, Hongo M, Tack J, Aoki H, Saito Y, Kato H. Clinical trial: dose‐dependent therapeutic efficacy of acotiamide hydrochloride (Z‐338) in patients with functional dyspepsia ‐ 100mg tid is an optimal dosage. Neurogastroenterology and Motility 2010;22:618‐e173. [DOI] [PubMed] [Google Scholar]
Matsueda 2012 {published data only}
- Matsueda K, Hongo M, Tack J, Saito Y, Kato H. A placebo‐controlled trial of acotiamide for meal‐related symptoms of functional dyspepsia. Gut 2012;61(6):821–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mo 2003 {published data only}
- Mo JZ, Li DG, Jiang JH, Jiang YB, Wang XP, Gong ZH, et al. A multi‐center clinical trial of itopride hydrochloride in the treatment of functional dyspepsia [盐酸伊托必利治疗功能性消化不良的多中心临床研究]. 中国新药杂志 Zhongguo Xinyao Zazhi (Chinese Journal of New Drugs) 2003;12(6):467‐9. [Google Scholar]
Nakamura 2017 {published data only}
- Nakamura K, Tomita T, Oshima T, Asano H, Yamasaki T, Okugawa T, et al. A double‐blind placebo controlled study of acotiamide hydrochloride for efficacy on gastrointestinal motility of patients with functional dyspepsia. Journal of Gastroenterology 2017;52(5):602‐10. [DOI] [PubMed] [Google Scholar]
Rösch 1987 {published data only}
- Rösch W. Cisapride in non‐ulcer dyspepsia. Results of a placebo‐controlled trial. Scandinavian Journal of Gastroenterology 1987;22(2):161‐4. [DOI] [PubMed] [Google Scholar]
Shen 2014 {published data only}
- Shen Y, Fang N, Yang C‐H. Azintamide combined with itopride hydrochloride for treatment of functional dyspepsia [联合治疗功能性消化不良患者的临床疗效]. Shi Jie Wei Chang Bing Xue Za Zhi She [World Chinese Journal of Digestology] 2014;22(20):2993‐6. [Google Scholar]
Singh 2015 {published data only}
- Singh H, Bala R, Kaur K. Efficacy and tolerability of levosulipride, domperidone and metoclopramide in patients with non‐ulcer functional dyspepsia: a comparative analysis. Journal of Clinical and Diagnostic Research: JCDR 2015;9(4):FC09‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sun 2003 {published data only}
- Sun J, Zhang CL, Chu Y, Yuan YZ, Li ZS, Liu XG, et al. A multi‐center, double‐blind, randomized and controlled trial of itopride hydrochloride in treatment of functional dyspepsia [伊托必利治疗功能性消化不良多中心、随机、双盲、双模拟临床研究]. 上海医学 [Shanghai Medical Journal] 2003;26(4):227‐9. [Google Scholar]
Tack 2009 {published data only}
- Tack J, Masclee A, Heading R, Berstad A, Piessevaux H, Popiela T, et al. A dose‐ranging, placebo‐controlled, pilot trial of Acotiamide in patients with functional dyspepsia. Neurogastroenterology and Motility 2009;21:272‐80. [DOI] [PubMed] [Google Scholar]
Tack 2011 {published data only}
- Tack JF, Stanghellini V, Holtmann G, Varannes SB, Vandenberghe A, Morooka K, et al. Efficacy and safety study of acotiamide (Z‐338) in European patients with functional dyspepsia. Gastroenterology 2011;140:S805. [Google Scholar]
Talley 2000 {published data only}
- Talley NJ, Verlinden M, Snape W, Beker JA, Ducrotte P, Dettmer A, et al. Failure of a motilin receptor agonist (ABT‐229) to relieve the symptoms of functional dyspepsia in patients with an without, delayed gastric emptying: a randomized double‐blind placebo‐controlled trial. Alimentary Pharmacology & Therapeutics 2000;14(12):1653‐61. [DOI] [PubMed] [Google Scholar]
Talley 2008a {published data only}
- Talley NJ, Tack JF, Kowalski DL, Borton MA, Barve A. A novel acetylcholine esterase inhibitor acotiamide hydrochloride (YM443) in functional dyspepsia: efficacy in a randomized, double‐blind, placebo controlled dose ranging trial. Gastroenterology 2008;134(4 Suppl 1):A‐157. [Google Scholar]
Talley 2008b {published data only}
- Talley NJ, Tack J, Ptak T, Gupta R, Giguère M. Itopride in functional dyspepsia: results of two phase III multicentre, randomised, double‐blind, placebo‐controlled trials. Gut 2008;57(6):740‐6. [DOI] [PubMed] [Google Scholar]
Talley 2008c {published data only}
- Talley NJ, Tack J, Ptak T, Gupta R, Giguère M. Itopride in functional dyspepsia: results of two phase III multicentre, randomised, double‐blind, placebo‐controlled trials. Gut 2008;57(6):740‐6. [DOI] [PubMed] [Google Scholar]
Teixeira 2000 {published data only}
- Teixeira CR, Abdalla Kurban AC, Denicol IP, Vieira Coelho NH, Spinato Torresini RJ, Peter RB, et al. Randomized, double‐blind study of functional dyspepsia with cisapride, trimebutine and placebo ‐ relationship with variation of the gastric emptying time. Revista Brasileira de Medicina 2000;57:729‐35. [Google Scholar]
Vakil 2008a {published data only}
- Vakil N, Laine L, Talley NJ, Zakko SF, Tack J, Chey WD, et al. Tegaserod treatment for dysmotility‐like functional dyspepsia: results of two randomized, controlled trials. American Journal of Gastroenterology 2008;103(8):1906–19. [DOI] [PubMed] [Google Scholar]
Vakil 2008b {published data only}
- Vakil N, Laine L, Talley NJ, Zakko SF, Tack J, Chey WD, et al. Tegaserod treatment for dysmotility‐like functional dyspepsia: results of two randomized, controlled trials. American Journal of Gastroenterology 2008;103(8):1906–19. [DOI] [PubMed] [Google Scholar]
Wang 1995 {published data only}
- Wang B, Liang X, Jia B, Jia B. A controlled multi‐centre clinical trial on cisapride in treatment of functional dyspepsia [西沙必利治疗功能性消化不良的多中心临床疗效观察]. Zhonghua Nei Ke za Zhi [Chinese Journal of Internal Medicine] 1995;34(3):180‐4. [PubMed] [Google Scholar]
Wong 2014 {published data only}
- Wong Z, Nadirah Daud U, Naidu J, Soon Ngiu C, Affendi Raja Ali R, Palaniappan S, et al. Randomised, double‐blind placebo controlled trial assessing the efficacy of itopride in postprandial distress syndrome (PDS): a pilot study: P‐215. Journal of Gastroenterology and Hepatology 2014;29:124‐5. [Google Scholar]
Yeoh 1997 {published data only}
- Yeoh KG, Kang JY, Tay HH, Gwee KA, Tan CC, Wee A, et al. Effect of cisapride on functional dyspepsia in patients with an without histological gastritis: A double‐blind placebo‐controlled trial. Journal of Gastroenterology and Hepatology 1997;12(1):13‐8. [DOI] [PubMed] [Google Scholar]
Zhou 2000 {published data only}
- Yuan YZ, Zhou LY, Li BQ, Lin SR, Wang AY, Dong XY, et al. A multicenter clinical trial on itopride hydrochloride for treatment of functional dyspepsia [(盐酸伊托必利治疗功能性消化不良的多中心临床研究]. 中国临床药理学杂志 Zhongguo Linchuang Yaolixue Zazhi [Chinese Journal of Clinical Pharmacology] 2000;16(6):403‐7. [Google Scholar]
Zhu 2005 {published data only}
- Zhu CQ, Mao YM, Zeng MD, Dong SX, Xu GM, Wang GS, et al. A clinical study of hydrochloride itopride in the treatment of functional dyspepsia. Zhongguo Yaoke Daxue Xuebao [Journal of China Pharmaceutical University] 2005;6:580‐3. [Google Scholar]
References to studies excluded from this review
Agorastos 1991 {published data only}
- Agorastos I, Akriviadis E, Goulis G. Effect of cisapride in nonulcer dyspepsia: A placebo‐controlled trial. Current Therapeutic Research, Clinical and Experimental 1991;49(5):870‐7. [Google Scholar]
Bekhti 1979 {published data only}
- Bekhti A, Rutgeerts L. Domperidone in the treatment of functional dyspepsia in patients with delayed gastric emptying. Postgraduate Medical Journal 1979;55(Suppl 1):30‐2. [PubMed] [Google Scholar]
Chen 2010 {published data only}
- Chen S, Ji J, Xu P, Cao Z, Mo J, Fang J, et al. Effect of domperidone therapy on nocturnal dyspeptic symptoms of functional dyspepsia patients. World Journal of Gastroenterology 2010;16(5):613‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chey1982 {published data only}
- Chey WY, You CH, Ange DA. Open and double blind clinical trials of domperidone in patients with unexplained nausea, vomiting, abdominal bloating and early satiety. Gastroenterology 1982;82(5):1033. [Google Scholar]
Chung1993 {published data only}
- Chung JM. Cisapride in chronic dyspepsia: results of a double‐blind, placebo‐controlled trial. Scandinavian Journal of Gastroenterology 1993;28(9):11‐4. [DOI] [PubMed] [Google Scholar]
Creytens 1984 {published data only}
- Creytens G. Effect of the non‐antidopaminergic drug cisapride on postprandial nausea. Current Therapeutic Research, Clinical and Experimental 1984;36(5II):1063‐70. [Google Scholar]
Davis1988 {published data only}
- Davis RH, Clench MH, Mathias JR. Effects of domperidone in patients with chronic unexplained upper gastrointestinal symptoms: a double‐blind,placebo‐controlled study. Digestive Diseases and Sciences 1988;33(12):1501‐11. [DOI] [PubMed] [Google Scholar]
De Loose 1979 {published data only}
- Loose F. Domperidone in chronic dyspepsia: a pilot open study and a multicentre general practice crossover comparison with metoclopramide and placebo. Pharmatherapeutica 1979;2:140‐6. [Google Scholar]
Deruyttere 1987 {published data only}
- Deruyttere M, Lepoutre L, Heyen H, Samain H, Pennoit H. Cisapride in the management of chronic functional dyspepsia: a multicenter, double‐blind, placebo‐controlled study. Clinical Therapeutics 1987;10(1):44‐51. [PubMed] [Google Scholar]
Goethals 1987 {published data only}
- Goethals C, Mierop L. Cisapride in the treatment of chronic functional dyspepsia. Current Therapeutic Research, Clinical and Experimental 1987;42(2):261‐7. [Google Scholar]
Haarmann 1979 {published data only}
- Haarmann K, Lebkuchner F, Widmann A, Kief W, Esslinger M. A double‐blind study of domperidone in the symptomatic treatment of chronic post‐prandial upper gastrointestinal distress. Postgraduate Medical Journal 1979;55(Suppl 1):24‐7. [PubMed] [Google Scholar]
Hannon 1987 {published data only}
- Hannon R. Efficacy of cisapride in patients with nonulcer dyspepsia. Current Therapeutic Research 1987;42(5):814‐22. [Google Scholar]
Hausken 1992 {published data only}
- Hausken T, Berstad A. Cisapride treatment of patients with non‐ulcer dyspepsia and erosive prepyloric changes. A double‐blind, placebo‐controlled trial. Scandinavian Journal of Gastroenterology 1992;27(3):213‐7. [DOI] [PubMed] [Google Scholar]
Kas'ianenko 2014 {published data only}
- Kas'ianenko VI, Denisov NL, Vasil'ev IV. Use of itopride in the symptoms of functional dyspepsia in Russia: results of a phase IV prospective open‐label multicenter clinical trial. Terapevticheskii Arkhiv 2014;86(8):35‐41. [PubMed] [Google Scholar]
Kearney 2000 {published data only}
- Kearney DJ, Avins AL, McQuaid KR. Treatment of uninvestigated dyspepsia with cisapride for patients with negative Helicobacter pylori serologies. American Journal of Gastroenterology 2000;95(9):2212‐17. [DOI] [PubMed] [Google Scholar]
Kim 2010 {published data only}
- Kim YM, Park YC, Jo JH, Kang WC, Son MW, Hong KE. Effect of herb medicine treatment for functional dyspepsia: a randomized placebo‐controlled and compared standard treatment trial. Journal of Korean Medicine 2010;31(1):1‐13. [Google Scholar]
Liu 2013 {published data only}
- Liu B, Piao X, Guo L. Effect of herbal formula xiao pi‐II on functional dyspepsia. Journal of Traditional Chinese Medicine / Chung i Tsa Chih Ying Wen Pan 2013;33(3):298‐302. [DOI] [PubMed] [Google Scholar]
Manayagi 2014 {published data only}
- Mayanagi S, Kishino M, Kitagawa Y, Sunamura M. Efficacy of acotiamide in combination with esomeprazole for functional dyspepsia refractory to proton‐pump inhibitor monotherapy. Tohoku Journal of Experimental Medicine 2014;234(4):237‐40. [DOI] [PubMed] [Google Scholar]
Milo 1984 {published data only}
- Milo R. Non‐cholinergic, non‐antidopaminergic treatment of chronic digestive symptoms suggestive of a motility disorder: a two‐step pilot evaluation of cisapride. Current Therapeutic Research, Clinical and Experimental 1984;36:1053‐62. [Google Scholar]
Miwa 2009 {published data only}
- Miwa H, Nagahara A, Tominaga K, Yokoyama T, Sawada Y, Inoue K, et al. Efficacy of the 5‐HT1A agonist tandospirone citrate in improving symptoms of patients with functional dyspepsia: a randomized controlled trial. American Journal of Gastroenterology 2009;104:2779‐87. [DOI] [PubMed] [Google Scholar]
Shim 2015 {published data only}
- Shim YK, Lee JY, Kim NY, Park YH, Yoon H, Shin CM, et al. Efficacy and safety of new prokinetic agent benachio Q solution in patients with postprandial distress syndrome subtype in functional dyspepsia: a single‐center, randomized, double‐blind, placebo‐controlled pilot study. Taehan Sohwagi Hakhoe Chi [Korean Journal of Gastroenterology] 2015;66(1):17‐26. [DOI] [PubMed] [Google Scholar]
Tack 2012 {published data only}
- Tack J, Janssen P, Masaoka T, Farré R, Oudenhove L. Efficacy of buspirone, a fundus‐relaxing drug, in patients with functional dyspepsia. Clinical Gastroenterology & Hepatology 2012;10(11):1239‐45. [DOI] [PubMed] [Google Scholar]
Tack 2016 {published data only}
- Tack J, Rotondo A, Meulemans A, Thielemans L, Cools M. Randomized clinical trial: A controlled pilot trial of the 5‐HT4 receptor agonist revexepride in patients with symptoms suggestive of gastroparesis. Neurogastroenterology and Motility 2016;28(4):487‐97. [DOI] [PubMed] [Google Scholar]
Talley 2001 {published data only}
- Talley NJ, Zanten SV, Saez LR, Dukes G, PerschyT, Heath M, et al. A dose‐ranging, placebo‐controlled, randomized trial of alosetron in patients with functional dyspepsia. Alimentary Pharmacology & Therapeutics 2001;15:525‐37. [DOI] [PubMed] [Google Scholar]
Testoni 1990 {published data only}
- Testoni PA, Masci E, Bagnolo F, Passaretti S, Pellegrini A, Ronchi G, et al. Effect of long‐term oral therapy with cisapride in reflux antral gastritis: results of a double‐blind placebo‐controlled trial. Current Therapeutic Research 1990;47(1):156‐65. [Google Scholar]
Van de Mierop 1979 {published data only}
- Mierop L, Rutgeerts L, Langenbergh B, Staessen A. Oral domperidone in chronic postprandial dyspepsia. Digestion 1979;19(4):244‐50. [DOI] [PubMed] [Google Scholar]
Van Ganse 1978 {published data only}
- Ganse W, Damme L, Mierop L, Deruyttere M, Lauwers W, Coegegrachts J. Chronic dyspepsia: double‐blind treatment with domperidone (R 33812) or a placebo. A multicentre therapeutic evaluation. Current Therapeutic Research, Clinical and Experimental 1978;23(11):695‐701. [Google Scholar]
Van Outryve M {published data only}
- Outryve M, Lauwers W, Verbeke S. Domperidone for the symptomatic treatment of chronic post‐prandial nausea and vomiting.. Postgraduate Medical Journal 1979;55(Suppl 1):33‐5. [PubMed] [Google Scholar]
Wood 1993 {published data only}
- Wood SF, Penney SC, Cochran KM. Cisapride in functional dyspepsia: a double‐blind, placebo‐controlled randomized trial in general practice patientsFunctional Dyspepsia: A Double‐Blind, Placebo‐Controlled Randomized Trial in General Practice Patients. Scandinavian Journal of Gastroenterology. Supplement 1993;28(195):5‐10. [DOI] [PubMed] [Google Scholar]
Yamawaki 2016 {published data only}
- Yamawaki H, Futagami S, Kawagoe T, Maruki Y, Hashimoto S, Nagoya H, et al. Improvement of meal‐related symptoms and epigastric pain in patients with functional dyspepsia treated with acotiamide was associated with acylated ghrelin levels in Japan. Neurogastroenterology and Motility 2016;28(7):1037‐47. [DOI] [PubMed] [Google Scholar]
Yan 2012 {published data only}
- Yan X, Feng C, Liu Z, Ji J, Chen S. A randomized, double‐blinded, placebo‐controlled, parallel study on efficacy of mosapride on nocturnal dyspeptic symptoms. Chinese Journal of Gastroenterology 2012;17(3):164‐7. [Google Scholar]
Additional references
Deeks 2011
- Deeks JJ, Higgins JPT, Deeks JJ, Altman DG, on behalf of the Cochrane Statistical Methods Group, editors. Chapter 9.4: Analysing data and undertaking meta‐analyses. In: Higgins JP, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ford 2010
- Ford AC, Marwaha A, Lim A, Moayyedi P. What is the prevalence of clinically significant endoscopic findings in subjects with dyspepsia? Systematic review and meta‐analysis. Clinical Gastroenterology and Hepatology 2010;8:830‐7. [DOI] [PubMed] [Google Scholar]
Ford 2015
- Ford AC, Marwaha A, Sood R, Moayyedi P. Global prevalence of, and risk factors for, uninvestigated dyspepsia: a meta‐analysis. Gut 2015;64(7):1049–57. [DOI] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- GRADE Working Group, McMaster University. GRADE Working Group, McMaster University. GRADEpro GDT. Version (accessed prior to April 22, 2017). Hamilton (ON): GRADE Working Group, McMaster University, 2015.
Higgins 2011a
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011].The Cochrane Collaboration, 2011. .Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins JPT, Deeks JJ, Altman DG, on behalf of the Cochrane Statistical Methods Group, editors. Chapter 16.1.3.2: Imputing standard deviations for changes from baseline. In: Higgins JP, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Lacy 2012
- Lacy BE, Talley NJ, Locke GR 3rd, Bouras EP, DiBaise JK, El‐Serag HB, et al. Review article: current treatment options and management of functional dyspepsia. Alimentary Pharmacology & Therapeutics 2012;36(1):3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lacy 2013
- Lacy BE, Weiser KT, Kennedy AT, Crowell MD, Talley NJ. Functional dyspepsia: the economic impact to patients. Alimentary Pharmacology & Therapeutics 2013;38:170‐7. [DOI] [PubMed] [Google Scholar]
Loannidis 2007
- Loannidis JP, Patsopoulos NA, Evangelou E. Uncertainty in heterogeneity estimates in meta‐analyses. BMJ 2007;335(7626):914‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mantel 1959
- Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. Journal of the National Cancer Institute 1959;22(4):719‐48. [PubMed] [Google Scholar]
Moayyedi 2002
- Moayyedi P, Mason J. Clinical and economic consequences of dyspepsia in the community. Gut 2002;50(Suppl 4):10‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moayyedi 2011
- Moayyedi P, Soo S, Deeks JJ, Delaney B, Innes M, Forman D. Pharmacological interventions for non‐ulcer dyspepsia. Cochrane Database of Systematic Reviews 2011, Issue 12. [DOI: 10.1002/14651858.CD001960.pub4] [DOI] [Google Scholar]
Moayyedi 2017
- Moayyedi PM, Lacy BE, Andrews CN, Enns RA, Howden CW, Vakil N. Corrigendum: ACG and CAG clinical guideline: management of dyspepsia. American Journal of Gastroenterology 2017;112:1484. [DOI: 10.1038/ajg.2017.238] [DOI] [PubMed] [Google Scholar]
Stanghellini 2016
- Stanghellini V, Chan FK, Hasler WL, Malagelada JR, Suzuki H, Tack J, et al. Gastroduodenal disorders. Gastroenterology 2016;150:1380‐92. [DOI] [PubMed] [Google Scholar]
Tack 2004
- Tack J, Bisschops R, Sarnelli G. Pathophysiology and treatment of functional dyspepsia. Gastroenterology 2004;127:1239‐55. [DOI] [PubMed] [Google Scholar]
Tack 2006
- Tack J, Talley MJ, Camilleri M, Holtmann G, Hu P, Malagelada JR, et al. Functional gastroduodenal disorders. Gastroenterology 2006;130:1466‐79. [DOI] [PubMed] [Google Scholar]
Talley 1991
- Talley NJ, Colin‐Jones DG, Koch KL, Koch M, Nyren O, Stanghellini V. Functional dyspepsia: a classification with guidelines for diagnosis and management. Gastroenterology International 1991;4:145‐60. [Google Scholar]
Tally 1999
- Talley NJ, Stanghellini V, Heading RC, Koch KL, Malagelada JR, Tytgat GN. Functional gastroduodenal disorders. Gut 1999;45 (Suppl 2):II37‐42. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tally 2005
- Talley NJ, Vakil NB, Moayyedi P. American gastroenterological association technical review on the evaluation of dyspepsia. Gastroenterology 2005;129:1756‐80. [DOI] [PubMed] [Google Scholar]
van Zanten 2011
- Zanten SV, Wahlqvist P, Talley NJ, Halling K, Vakil N, Lauritsen K, et al. Randomised clinical trial: the burden of illness of uninvestigated dyspepsia before and after treatment with esomeprazole—results from the STARS II study. Alimentary Pharmacology & Therapeutics 2011;34(7):714‐23. [DOI] [PubMed] [Google Scholar]
Vanheel 2013
- Vanheel H, Farré R. Changes in gastrointestinal tract function and structure in functional dyspepsia. Nature Reviews. Gastroenterology & Hepatology 2013;10:142–9. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Bollegala 2011
- Bollegala NP, Khanna R, Moayyedi P, Leontiadis GI. Prokinetics for functional dyspepsia. Cochrane Database of Systematic Reviews 2011, Issue 11. [DOI: 10.1002/14651858.CD009431] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pittayanon 2017
- Pittayanon R, Yuan Y, Bollegala NP, Khanna R, Leontiadis GI, Moayyedi P. Prokinetics for functional dyspepsia. Cochrane Database of Systematic Reviews 2017, Issue 11. [DOI: 10.1002/14651858.CD009431.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]