

Abstract
Background
The dystrophinopathies include Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), and X‐linked dilated cardiomyopathy (XLDCM). In recent years, co‐ordinated multidisciplinary management for these diseases has improved the quality of care, with early corticosteroid use prolonging independent ambulation, and the routine use of non‐invasive ventilation signficantly increasing survival. The next target to improve outcomes is optimising treatments to delay the onset or slow the progression of cardiac involvement and so prolong survival further.
Objectives
To assess the effects of interventions for preventing or treating cardiac involvement in DMD, BMD, and XLDCM, using measures of change in cardiac function over six months.
Search methods
On 16 October 2017 we searched the Cochrane Neuromuscular Specialised Register, CENTRAL, MEDLINE and Embase, and on 12 December 2017, we searched two clinical trials registries. We also searched conference proceedings and bibliographies.
Selection criteria
We considered only randomised controlled trials (RCTs), quasi‐RCTs and randomised cross‐over trials for inclusion. In the Discussion, we reviewed open studies, longitudinal observational studies and individual case reports but only discussed studies that adequately described the diagnosis, intervention, pretreatment, and post‐treatment states and in which follow‐up lasted for at least six months.
Data collection and analysis
Two authors independently reviewed the titles and abstracts identified from the search and performed data extraction. All three authors assessed risk of bias independently, compared results, and decided which trials met the inclusion criteria. They assessed the certainty of evidence using GRADE criteria.
Main results
We included five studies (N = 205) in the review; four studies included participants with DMD only, and one study included participants with DMD or BMD. All studied different interventions, and meta‐analysis was not possible. We found no studies for XLDCM. None of the trials reported cardiac function as improved or stable cardiac versus deteriorated.
The randomised first part of a two‐part study of perindopril (N = 28) versus placebo (N = 27) in boys with DMD with normal heart function at baseline showed no difference in the number of participants with a left ventricular ejection fraction (LVEF%) of less than 45% after three years of therapy (n = 1 in each group; risk ratio (RR) 1.04, 95% confidence interval (CI) 0.07 to 15.77). This result is uncertain because of study limitations, indirectness and imprecision. In a non‐randomised follow‐up study, after 10 years, more participants who had received placebo from the beginning had reduced LVEF% (less than 45%). Adverse event rates were similar between the placebo and treatment groups (low‐certainty evidence).
A study comparing treatment with lisinopril versus losartan in 23 boys newly diagnosed with Duchenne cardiomyopathy showed that after 12 months, both were equally effective in preserving or improving LVEF% (lisinopril 54.6% (standard deviation (SD) 5.19), losartan 55.2% (SD 7.19); mean difference (MD) −0.60% CI −6.67 to 5.47: N = 16). The certainty of evidence was very low because of very serious imprecision and study limitations (risk of bias). Two participants in the losartan group were withdrawn due to adverse events: one participant developed an allergic reaction, and a second exceeded the safety standard with a fall in ejection fraction greater than 10%. Authors reported no other adverse events related to the medication (N = 22; very low‐certainty evidence).
A study comparing idebenone versus placebo in 21 boys with DMD showed little or no difference in mean change in cardiac function between the two groups from baseline to 12 months; for fractional shortening the mean change was 1.4% (SD 4.1) in the idebenone group and 1.6% (SD 2.6) in the placebo group (MD −0.20%, 95% CI −3.07 to 2.67, N = 21), and for ejection fraction the mean change was −1.9% (SD 9.8) in the idebenone group and 0.4% (SD 5.5) in the placebo group (MD −2.30%, 95% CI −9.18 to 4.58, N = 21). The certainty of evidence was very low because of study limitations and very serious imprecision. Reported adverse events were similar between the treatment and placebo groups (low‐certainty evidence).
A multicentre controlled study added eplerenone or placebo to 42 patients with DMD with early cardiomyopathy but preserved left ventricular function already established on ACEI or ARB therapy. Results showed that eplerenone slowed the rate of decline of magnetic resonance (MR)‐assessed left ventricular circumferential strain at 12 months (eplerenone group median 1.0%, interquartile range (IQR) 0.3 to −2.2; placebo group median 2.2%, IQR 1.3 to −3.1%; P = 0.020). The median decline in LVEF over the same period was also less in the eplerenone group (−1.8%, IQR −2.9 to 6.0) than in the placebo group (−3.7%, IQR −10.8 to 1.0; P = 0.032). We downgraded the certainty of evidence to very low for study limitations and serious imprecision. Serious adverse events were reported in two patients given placebo but none in the treatment group (very low‐certainty evidence).
A randomised placebo‐controlled study of subcutaneous growth hormone in 16 participants with DMD or BMD showed an increase in left ventricular mass after three months' treatment but no significant improvement in cardiac function. The evidence was of very low certainty due to imprecision, indirectness, and study limitations. There were no clinically significant adverse events (very low‐certainty evidence).
Some studies were at risk of bias, and all were small. Therefore, although there is some evidence from non‐randomised data to support the prophylactic use of perindopril for cardioprotection ahead of detectable cardiomyopathy, and for lisinopril or losartan plus eplerenone once cardiomyopathy is detectable, this must be considered of very low certainty. Findings from non‐randomised studies, some of which have been long term, have led to the use of these drugs in daily clinical practice.
Authors' conclusions
Based on the available evidence from RCTs, early treatment with ACE inhibitors or ARBs may be comparably beneficial for people with a dystrophinopathy; however, the certainty of evidence is very low. Very low‐certainty evidence indicates that adding eplerenone might give additional benefit when early cardiomyopathy is detected. No clinically meaningful effect was seen for growth hormone or idebenone, although the certainty of the evidence is also very low.
Plain language summary
Preventing and treating heart complications in Duchenne and Becker muscular dystrophy and X‐linked dilated cardiomyopathy
Review question
What are the effects of treatments used to prevent or treat heart complications in Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), and X‐linked dilated cardiomyopathy (XLDCM)?
Background
The protein dystrophin is essential for muscles to work normally. DMD, BMD and XLDCM are inherited muscle diseases caused by changes in the gene that controls production of dystrophin. People with these conditions develop muscle wasting and weakness. In the heart, a lack of dystrophin causes muscle damage and scarring, which over time causes the heart to fail. Eventually the heart chambers enlarge, which is known as dilated cardiomyopathy. This serious complication can be a cause of death. There are a number of possible treatments for heart problems in these muscle conditions. One option is to reduce the workload of the heart with drugs that lower blood pressure (angiotensin‐converting‐enzyme inhibitors, ACE inhibitors) or slow the heart rate (beta blockers or ivabradine). Another approach is to reduce muscle damage with antioxidants (e.g. idebenone) or medicines that target inflammation (e.g. corticosteroids). Recently, drugs that increase dystrophin have been developed, including ataluren and eteplirsen.
Study characteristics
Cochrane Review authors collected all relevant studies to answer their review question. They searched for trials looking to prevent or treat heart complications in people with DMD, BMD or XLDCM. They limited the review to trials that randomly assign participants to one treatment or another, which usually provide the best evidence. They identified five small trials, with a total of 205 participants.
‐ A three‐year study of perindopril versus placebo (an inactive pill) to prevent heart complications in 57 boys with DMD. The randomised trial was followed by two years of open treatment, then a follow‐up study of 10 years when all children received perindopril.
‐ A one‐year study of lisinopril versus losartan in 23 patients with DMD and newly diagnosed heart complications.
‐ A one‐year study of idebenone versus placebo in 21 boys with DMD, which the manufacturer funded.
‐ A one‐year study of eplerenone versus placebo in 42 patients with DMD who already had heart complications, which the manufacturer partly funded.
‐ A three‐month study of growth hormone versus placebo in 10 patients with DMD or BMD.
Key results and certainty of the evidence
Based on the available evidence from RCTs, early treatment with ACE inhibitors or angiotensin receptor blockers (ARBs) may help people with DMD. In boys with early heart involvement, the effect of ACE inhibitor and ARB may be equivalent; however, the evidence is very uncertain. Findings from non‐randomised studies, some of which have been long term, have led to the use of these drugs in daily clinical practice. Very low‐certainty evidence indicates that adding eplerenone might give additional benefit in DMD when early cardiomyopathy is detected. We did not see a clinically meaningful effect for growth hormone or idebenone in the studies examined, although the certainty of the evidence was also very low. The trials provided only low or very low‐certainty evidence on side effects.
Overall, the numbers of patients in each of these studies was small, and some studies had limitations that might have affected the results, so we are very uncertain about the results.
The evidence is current to October 2017.
Summary of findings
Background
Dystrophinopathies are a group of X‐linked inherited degenerative muscle disorders, including Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD) and X‐linked dilated cardiomyopathy (XLDCM). These three allelic conditions are caused by deletions, duplications or missense mutations in the dystrophin gene at Xp21.2 (Gardner 1995;Koenig 1989; Malhotra 1988; Muntoni 1997). The typical cardiac abnormality found in all dystrophinopathy patients is dilated cardiomyopathy.
DMD is the most severe of these disorders and has an incidence of 1:3500 to 1:6000 live male births (Mendell 2012); muscle biopsy shows dystrophic changes and complete or almost complete absence of the sarcolemmal protein dystrophin. The condition usually presents with muscle weakness by 5 years of age. Without treatment, all affected children lose the ability to walk by their 13th birthday. Once the child is wheelchair dependent, contractures and scoliosis traditionally develop rapidly – often requiring surgery. Premature death from untreated respiratory or cardiac failure occurs on average at 18.5 years (Emery 2003). In recent years, the natural history of the condition has been improved by greater integration of care through multidisciplinary teams. Two developments in particular have led to incremental improvements in survival: routine use of glucocorticoid steroids to improve muscle strength and prolong independent ambulation (Matthews 2016), and routine deployment of non‐invasive nocturnal ventilation using mask bilevel positive airway pressure ventilation (BIPAP) to improve symptoms and delay death from respiratory failure to a mean of 25 years (Eagle 2002). Cardiac involvement culminating in dilated cardiomyopathy with congestive cardiac failure or ventricular arrhythmias remains a key contributor to premature death in DMD. In the absence of cardioactive therapies, the natural history of cardiac involvement has not changed despite the other significant improvements in physical and respiratory management and has become a more common cause of death with 40% to 50% of DMD patients dying as a direct consequence of cardiac involvement (Eagle 2002; Muntoni 2003).
BMD was first described in 1955 (Becker 1955). The condition is less common than DMD, with a reported incidence of between 1:14,000 and 1:18,000 males (Bushby 1991). BMD resembles DMD, but it is milder with a slower progression of muscle weakness because the reading frame of the gene is preserved. This results in the production of a dystrophin molecule which has a lower molecular weight and which is less abundant than normal. There is a broad spectrum of clinical severity in BMD, with onset of symptoms occurring from early childhood to as late as the sixth decade (Emery 1976; Quinlivan 1995). Only 10% of Becker's original series of patients, for example, lost independent ambulation before the age of 40 years, and none lost ambulation before the age of 16 years (Becker 1955). As with DMD, life expectancy can be reduced by respiratory insufficiency and disproportionately by cardiomyopathy.
XLDCM is a rapidly‐progressive cardiomyopathy occurring in teenage boys caused by a deletion in exon 1 of the dystrophin gene. Skeletal muscles are not usually involved (Towbin 1993). Without cardiac transplantation, death occurs within one to two years of the onset of symptoms. In some cases the distinction between XLDCM and a mild variant of BMD can be difficult.
Female carriers of DMD and BMD have been shown to be at increased risk of developing dilated cardiomyopathy (Bushby 1993; Hoogerwaard 1999; Kamamura 1990; Lane 1980; Nolan 2003), although the impact on survival is uncertain (Holloway 2008).
Description of the condition
Dystrophin plays a crucial role in force transduction between cell membranes and the intracellular contractile elements of skeletal and cardiac muscle. When absent or deficient, cell membranes become highly vulnerable to damage, swamping natural repair mechanisms, leading to cell death and tissue fibrosis (Danialou 2001; Menke 1991). Typically the first detectable sign of this process in the heart is found on the electrocardiogram (ECG) with the development of Q waves in the lateral (I and AVL) or inferolateral and apical (II, III, aVF, V5‐V6) leads (Hoogerwaard 1997; Nigro 1990; Nigro 1995), increased voltages in the right precordial leads (V1‐3) (Nikolic 1998), abnormalities in repolarisation (inverted or dysmorphic T waves), and increase in the so‐called 'cardiomyopathic index' (the ratio of QT‐interval (ms)/end‐of‐P wave to QRS onset (ms) (Nigro 1995). These changes can be seen from the age of 6 years in DMD and are almost universal by 12 years (Bies 1992). Although defining the end of dysmorphic T waves may be difficult, some have correlated QT‐prolongation on the surface ECG with increased incidence of sudden death (Nigro 2002). The time‐course and extent of ECG abnormalities are more variable in BMD. In both DMD and BMD, fully evolved ECG changes precede the development of echocardiographically detectable left ventricular dysfunction by many years and thus have no clinical correlation with the degree of cardiomyopathy (Heymsfield 1978)
Although limited in sensitivity and operator dependent, echocardiography is the preferred initial screening method for detecting cardiac involvement in the dystrophinopathies (Nigro 1990). This is because it is readily available, easily repeatable and inexpensive. The first sign of ventricular systolic dysfunction is segmental left ventricular systolic dysfunction, typically found in the postero‐basal segments (Miyoshi 1991; Nigro 1983; Tanaka 1979). Without treatment the extent of abnormality spreads to affect the whole ventricle over time, culminating in chamber dilatation and global systolic dysfunction (Backman 1992; Corrado 2002; De Kermadec 1994; Ferlini 1999; Finsterer 2003; Olfors 1994; Perloff 1984; Takenaka 1993). About 90% of male patients with DMD develop a severe progressive form of cardiac involvement (Heymsfield 1978; Mukoyama 1987), with 20% to 30% having evidence of left ventricular impairment by 10 years of age (Backman 1992; Finsterer 2003). When deploying more sensitive imaging techniques, such as tissue‐Doppler echocardiography (Meune 2004; Mori 2007), 2D‐strain deformation imaging, cardiac magnetic resonance imaging (CMRI), single photon emission tomography (SPECT), positron emission tomography (PET) or 31phosphorous magnetic resonance spectroscopy (31PMRS), abnormalities in left ventricular function are evident in an even larger proportion of patients in their teens (Griffin 2001; Perloff 1984; Quinlivan 1996; Silva 2007; Yamamoto 1988).
In BMD the incidence of cardiac involvement, its age of onset and implications for prognosis are more variable (Angelini 1996; De Visser 1992; Melacini 1996; Steare 1992). Although some 90% of patients with BMD show ECG abnormalities similar to those seen in DMD, only 65% develop left ventricular systolic dysfunction when assessed by echocardiography. However, in some the severity of cardiac involvement may be disproportionate to skeletal muscle weakness and may even be the presenting feature of the condition (Sakata 1990; Steare 1992). In such cases cardiac involvement becomes the determinant of long‐term prognosis (Ishigaki 1997). Best estimates from longitudinal series suggest that cardiac involvement contributes directly to death in up to 50% of male patients with BMD compared with 20% of DMD patients (Angelini 1996; Hoogerwaard 1997; Melacini 1996; Muntoni 2003; Olfors 1994; Steare 1992). However, in recent years with improved care, particularly the use of domiciliary ventilatory support, unpublished estimates of end‐stage dilated cardiomyopathy as a cause of death in DMD are between 40% to 50%.
Some DMD and BMD patients develop a sinus tachycardia unrelated to respiratory failure or other cardiac abnormalities, which is usually attributed to sympathovagal imbalance in cardiac autonomic function (Lanza 2001). Persistent, inappropriate sinus tachycardia may accelerate the development of cardiomyopathy or simply be a sign of subclinical cardiac involvement (Kwon 2012). CMRI can find evidence of left ventricular non‐compaction in a high proportion of DMD patients before any reduction in left ventricular function is identified (Stabile 2013). Atrial natriuretic peptide (ANP) and brain natriouretic peptide (BNP), biomarkers for cardiac impairment, are not sensitive markers for early systolic impairment in DMD; however, once the fractional shortening (FS) is less than 15%, these biomarkers increase and are associated with poor prognosis (Mori 2002).
Complete atrioventricular (AV) block is thought to be uncommon in the dystrophinopathies, but there have been a number of case reports of patients with DMD requiring permanent pacing (Andrikopoulos 2013; Fayssoil 2008; Kono 2015; Kuru 2012). Focal areas of fibrosis in the conducting system have been described in BMD postmortem studies (Donofrio 1989). Abrupt onset of complete heart block without an escape rhythm could account for a proportion of sudden cardiac deaths at more advanced stages of DMD. Prolongation of the QT interval has been noted in a proportion of DMD ECGs and could increase risk of cardiac tachyarrhythmias and sudden death (Nigro 1983). In one BMD patient, complete AV block was reported as the presenting feature, with muscle weakness only developing some years later (Quinlivan 1995). Ventricular tachycardia and fibrillation have been reported in DMD and BMD patients with established cardiomyopathy. However, the extent to which prophylactic use of implantable defibrillators would prolong survival in DMD is unknown.
Histological examination of endocardial biopsies from patients with all types of dystrophinopathy are similar. Typical findings are of hypertrophic cardiomyocytes with increased internal nuclei, endocardial and interstitial fibrosis associated with cytoplasmic lipofuscinosis and focal lymphocytic infiltration, large pleomorphic bizarre nuclei, vacuoles and focal necrosis (Casazza 1988). At postmortem, the pathological features of heart involvement in either DMD or BMD are replacement of cardiac fibres with connective tissue and extensive myocardial fibrosis (Globus 1923; Heymsfield 1978; Olfors 1994).
Description of the intervention
There are a wide range of pharmacological and non‐pharmacological interventions that could potentially preserve or improve cardiac function, alone or in combination, including:
angiotensin converting enzyme (ACE) inhibitors, e.g. ramipril, perindopril, captopril, lisinopril and enalapril;
angiotensin II type I receptor (ATI1) inhibitors (angiotensin receptor blocking agents (ARB)), e.g. losartan, irbesartan, candesartan, and valsartan;
beta‐blockers, e.g. bisoprolol, metoprolol and carvedilol; and sinus node slowing agents, e.g. ivabradine to slow heart rate
diuretics, e.g. aldosterone antagonists such as spironolactone and eplerenone; bendrofluazide; and loop diuretics such as bumetanide and furosemide;
calcium channel blockers, e.g. verapamil, amlodipine, and diltiazem;
magnesium;
phosphodiesterase type 3 (milrinone) and type 5 inhibitors (sildenafil and tadalafil);
positive inotropic agents, e.g. digoxin, bypiridine inhibitors, calcium, catecholamine agonists, and milrinone;
drugs to treat cardiac arrhythmias, e.g. amiodarone, sotalol, and flecainide;
drugs which affect the vascular response to nitric oxide, e.g. sildenafil;
anti‐coagulants e.g. warfarin, coumadin, dabigatran, apixaban, and rivaroxaban
drugs that alter the natural history of the disease (i.e. improve skeletal muscle function or increase dystrophin expression), e.g. glucocorticosteroids, idebenone, coenzyme Q10, ataluren (PTC124), and antisense oligonucleotides for DMD; and
non‐pharmacological interventions, such as single and dual chamber pacemakers, cardiac resynchronisation therapy (CRT) pacemakers, implantable cardioverter defibrillator (ICD or CRT‐D), left ventricular assist devices (LVAD; extravascular counter‐pulsation devices), and cardiac transplantation.
How the intervention might work
We divide interventions into three subsets.
Drugs acting on the cardiovascular system
In the face of damage to the left ventricle, a variety of primitive reflexes activate, and the heart and circulation undergo a process of remodelling, which initially preserves cardiac output and perfusion to vital organs but ultimately causes the heart to progressively decompensate. Several categories of drugs are used routinely in contexts other than DMD/BMD to block these adverse adaptations, thus preventing this downward spiral of ventricular dysfunction.
Blocking the renin‐angiotensin system by ACE inhibitors, ARBs or renin antagonists prevents inappropriate salt and water accumulation by the kidney and the directly toxic effects of excessive angiotensin II, which include vasoconstriction, apoptosis and promotion of cardiac fibrosis (Burnett 2017; Cicoira 2002; Heran 2012; Ponikowski 2016; Zannad 2000).
Blocking the effects of increased circulating endogenous catecholamines and direct neural stimulation by beta‐adrenergic blockers slows the heart rate, reducing myocardial oxygen consumption and peripheral vasoconstriction. These agents also prevent the unhelpful down‐regulation of beta adrenoreceptors in the heart. When doses of beta‐blocking drugs cannot be up‐titrated adequately, the selective sinus node slowing agent ivabradine can be added to improve heart failure by slowing the heart rate further (Abdel‐Salam 2014; Ponikowski 2016; Swedberg 2010).
When there is evidence of fluid retention with overt cardiac failure, loop diuretics promote loss of salt and water by the kidney and so relieve symptoms of congestion and fluid overload. Loop diuretics (e.g. furosemide and bumetanide) are used with ACE inhibitors in this context. Spironolactone and eplerenone are weaker diuretics, which importantly conserve potassium and also have an antifibrotic effect on cardiac muscle (Cicoira 2002; Zannad 2000).
Positive inotropic drugs increase myocardial contractility and can be used to support severely depressed cardiac function. However, their symptomatic benefit is often short‐lived. Type 3 phosphodiesterase inhibitors such as milrinone increase cardiac output at the cost of increased myocardial work, myocardial oxygen consumption and heart rate. Unless used in the context of some reversible cause of cardiac deterioration, positive inotropic agents eventually exacerbate cardiac dysfunction and accelerate its progressive decompensation. However, in end‐stage cardiac failure in DMD/BMD, the prognosis is so poor that these agents may offer short‐term, symptomatic palliative benefits.
Cardiac arrhythmias, such as atrial fibrillation, result in an acute loss of atrial transport to ventricular filling and a sudden increase in ventricular rate. This can precipitate cardiac decompensation acutely with development of heart failure symptoms in patients with reduced left ventricular reserve. Ventricular tachyarrhythmias typically present more dramatically with unheralded acute collapse or virtually instantaneous death in the context of asymptomatic but advanced cardiomyopathy. Anti‐arrhythmic drug therapies other than beta‐blockers have little impact on the occurrence or severity of ventricular tachycardia or ventricular fibrillation in cardiomyopathy of other aetiologies. In other contexts, cardioverter‐defibrillator therapy is the standard recommendation in patients with severe left ventricular dysfunction, but not in those with New York Heart Association (NYHA) functional class IV symptoms, for the primary prevention of sudden cardiac death due to ventricular tachyarrhythmias. An important consideration, given the resting tachycardia in DMD, is that slowing the heart rate in patients with with dilated cardiomyopathy could potentially improve heart function.
Patients with DMD/BMD have dramatically reduced mobility and so are theoretically at risk of developing peripheral venous thrombosis and pulmonary emboli (although there is a surprising dearth of published literature regarding this complication). If small, these can occur silently, but when large they can cause catastrophic haemodynamic collapse and sudden death – indistinguishable clinically from a tachy‐ or bradyarrhythmia. In patients with advanced left ventricular dysfunction, blood clots can form in either the left atrium or left ventricle and result in systemic emboli, most frequently causing stroke. Prophylactic low‐dose or full anticoagulation can prevent venous and arterial thromboembolism, respectively.
Because there is a published review of the effect of calcium antagonists used in DMD to improve skeletal rather than cardiac muscle function, we will not discuss these agents further in this review (Phillips 2008).
Non‐pharmacological treatments for advanced cardiac failure and arrhythmias
Standard dual chamber pacing is indicated in the small subgroup of DMD/BMD patients who develop bradycardia due to sinus or AV‐nodal conduction problems. A more recent pacing indication comes from the realisation that, in hearts with already impaired left ventricular systolic function, the development of left bundle branch block or a non‐specifically widened QRS, causes dyssynchrony of contraction and so a further reduction in left ventricular function. Pacing from two sites on opposite walls of the left ventricle narrows the abnormally widened QRS complex by facilitating faster and more synchronous left ventricular contraction, optimising contraction for the same stage of cardiomyopathy. Recent studies in patients with idiopathic cardiomyopathy show that in appropriately selected patients, CRT significantly improves cardiac function and heart failure symptoms and prolongs life. It also reduces hospitalisations for heart failure (Turley 2008). The role of CRT in DMD/BMD seems limited, however, since most people – even with advanced cardiomyopathy – do not develop QRS‐complex widening. Even in those who do, it remains speculative whether they would respond to CRT. This is because the earliest and most extensively scarred segment of the left ventricle in patients with DMD/BMD is typically epicardial in the postero‐lateral or postero‐basal segments, and the lateral wall is usually the preferred site for left ventricular lead placement to restore synchrony (Bleeker 2006; Hor 2011).
Patients with established cardiomyopathy are at particular risk of developing haemodynamically compromising ventricular tachycardia or ventricular fibrillation, manifesting as sudden cardiac death. By restoring normal rhythm from such unpredictable events, implantable cardioverter‐defibrillators have been shown to significantly reduce the incidence of sudden cardiac death in various subsets of patients with cardiomyopathy (Cevik 2010). All implanted cardioverter‐defibrillators, except those without leads in the heart (i.e. subcutaneous implantable cardioverter‐defibrillators), also contain bradycardia pacing capabilities. The impact of defibrillator therapy on quality of life and overall effect on survival in DMD in particular has yet to be established (Wagner 2007).
When available, cardiac transplantation is an effective treatment for patients with end‐stage cardiomyopathy and short predicted survival, and it could be an option for patients with BMD and XLDCM (Wu 2010). Almost 80% of heart transplant recipients survive for at least one year, and up to 74% survive for five years (Fararolo 2010). However, because of the multisystem nature of DMD and the shortage of suitable donors for all categories of patients who might benefit, cardiac transplantation is rarely considered appropriate in DMD (Papa 2017). The more recent development and increasing availability of a range of battery‐powered, left ventricular mechanical pump support devices (e.g. left ventricular assist device (LVAD), counter‐pulsation devices) offer an alternative which may be more relevant and more widely applicable to DMD patients with heart failure (Abraham 2014; Black 2016; Iodice 2015; Ryan 2014).
Drugs that improve the natural history of the condition
Corticosteroids are known to increase muscle strength in DMD and can prolong ambulation (Matthews 2016), so they have now become part of routine care for DMD. Their precise mechanism of action is not known. It seems likely from non‐randomised retrospective cohort data that corticosteroids also modify the natural history of cardiac involvement in DMD (Barber 2013; Schram 2013; Silversides 2003). One long‐term follow‐up study compared the clinical course of deflazacort‐treated DMD patients with historical untreated DMD patients and demonstrated improved respiratory parameters and echocardiographic measures of left ventricular function in the deflazacort‐treated group (Biggar 2006).
Drugs to reduce oxidative stress (e.g. idebenone and coenzyme Q10) could potentially slow the dystrophic process and have a protective effect.
A range of drugs (e.g. ataluren (Translarna; previously known as PTC 124); antisense oligonucleotides) and cell therapies (stem cells and myoblast transfer and gene therapy) designed to increase dystrophin levels or upregulate utrophin are currently under evaluation. If shown to be clinically effective in improving skeletal muscle function, research would need to independently establish the effect of these potential therapies on the heart. It is already clear, however, that some therapies shown to be of benefit to skeletal muscle in animal models of DMD do not penetrate the heart (Aartsma‐Rus 2013; Wasala 2013). This raises the possibility that some disease modifying approaches to treatment of DMD might even increase the severity of cardiac dystrophinopathy – emphasising the need to include measurement of cardiac function in the overall evaluation of patients.
Why it is important to do this review
Cardiomyopathy is now the most important limiting factor for long‐term survival in BMD and DMD patients. Furthermore, improved management leading to a delay in loss of ambulation could potentially stress an already vulnerable myocardium and thus increase the risk of symptomatic cardiac involvement for this group of patients in the future. The purpose of this review is to systematically review the evidence for early intervention as a means of preventing symptomatic cardiomyopathy, and the best currently available treatments for established cardiac involvement in the dystrophinopathies.
Objectives
To assess the effects of interventions for preventing or treating cardiac involvement in DMD, BMD, and XLDCM, using measures of change in cardiac function over six months.
Methods
Criteria for considering studies for this review
Types of studies
We included double‐ and single‐blind randomised or quasi‐randomised trials and the first arm of cross‐over controlled trials that compared the effects of an intervention versus another intervention, placebo or standard treatment. We did not include longitudinal, observational or open non‐randomised studies in the Results section, but we considered them in the Discussion. (Quasi‐randomised trials use methods of allocation that are not truly random, such as alternation, and allocation by date of birth or case record number.)
Types of participants
All patients, including children and adults of all ages, confirmed to have a dystrophinopathy (DMD, BMD or XLDCM). Diagnosis confirmed by muscle biopsy showing reduced or absent dystrophin staining and/or DNA studies showing a deletion, duplication, nonsense or missense mutation in the dystrophin gene.
Types of interventions
Pharmacological and non‐pharmacological treatments known to have an effect on improving or reversing the physiological effects of dilated cardiomyopathy and pharmacological agents and cell therapies that have an effect on skeletal muscle function (i.e. the natural history of the disease). We planned to analyse data for each type of intervention separately.
Types of outcome measures
Primary outcomes
Dystrophinopathies typically cause profound and progressive physical disability, so even in the context of severe dilated cardiomyopathy, patients usually experience few if any cardiac symptoms. Therefore it is rarely possible to differentiate death in the context of a chest infection with associated respiratory failure from death of primary cardiac aetiology. Indeed it is likely that the occurrence of a lower respiratory infection in a patient with advanced cardiomyopathy can precipitate cardiorespiratory deaths. For this reason we have chosen surrogate measures of cardiac function rather than morbidity and mortality as our primary outcome measure.
We assessed changes in cardiac function following a six‐month period of intervention using 'equivalent techniques' such as echocardiography (ejection fraction (EF), fractional shortening (FS), ventricular dimensions: left ventricular systolic diameter (LVsd), left ventricular diastolic diameter (LVdd), wall motion), tissue Doppler echocardiography, cardiac magnetic resonance imaging (CMRI), and gated radionuclide imaging (ejection fraction). For each, we divided reported outcomes into those measuring benefit (i.e. stable or improved) and those measuring deterioration. In children, in whom echocardiography is usually the preferred intervention, measuring FS and EF have been shown to correlate well with other modalities such as CMRI (Soslow 2016; Spurney 2015).
Secondary outcomes
We planned to assess all secondary outcome measures as either unchanged/improved or worse after a six‐month intervention period.
The size of metabolically abnormal areas of myocardium identified with other forms of cardiac imaging: PET, SPECT and 31PMRS.
Improvements in quality of life measures, such as the Paediatric Quality of Life Inventory (PedsQL) for children (Varni 1999) and, for adults over 16 years of age, the Individualized Neuromuscular Quality of Life Questionnaire (INQol) or Short‐Form 36‐item Health Survey (SF‐36) (Vincent 2007; Ware 2007).
The occurrence of one or more adverse events reported by study investigators.
Search methods for identification of studies
Electronic searches
We searched the following databases on 16 October 2017.
Cochrane Neuromuscular Specialised Register (Appendix 1).
Cochrane Central Register of Controlled Trials (CENTRAL, in the Cochrane Register of Studies; Appendix 2).
MEDLINE (1996 to 16 October 2017; Appendix 3).
Embase (1980 16 October 2017; Appendix 4).
We searched the following trials registries:
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (30 August 2018; Appendix 5)
World Health Organization International Clinical Trials Registry Platform (ICTRP; www.who.int/ictrp/en/) (31 July 2018; Appendix 6).
Searching other resources
We reviewed conference proceedings for non‐published studies identified as published abstracts in our literature search and screened bibliographies of identified manuscripts for studies not identified by the search. We did not perform a separate search for non‐randomised studies but will refer in the Discussion to those non‐randomised studies identified during the search for RCTs.
Data collection and analysis
Selection of studies
All three review authors (RQ, JB, and TB) independently reviewed the titles and abstracts identified from the searches. The authors obtained the full text of all potentially relevant studies for independent assessment. All three authors independently decided which trials met the inclusion criteria. There were no disagreements.
We selected only randomised and quasi‐randomised controlled trials, as well as cross‐over trials, for inclusion. In the Discussion, we reviewed open studies, longitudinal observational studies and individual case reports but only discussed studies in which the diagnosis, intervention, pre‐treatment and post‐treatment states were adequately described and in whom follow‐up for at least six months was available.
The Cochrane Neuromuscular Managing Editor checked results from clinical trials registry searches.
Data extraction and management
Two review authors (TB and RQ) independently extracted data onto pre‐agreed data extraction forms which the third author (JB) then reviewed and approved. There were no disagreements. One author (TB) entered data into the Cochrane statistical software, Review Manager 5 (RevMan 5), and a second author (JB) or a member of the Cochrane Neuromuscular Editorial team (RB) checked data entry (RevMan 2014). We planned to contact trial authors directly in case of any missing data. The Managing Editor entered data into Characteristics of studies awaiting classification and Characteristics of ongoing studies tables.
Assessment of risk of bias in included studies
All three review authors independently assessed studies for risk of bias using pre‐agreed criteria, described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), and we graded each trial as being at high, low or unclear risk of bias for the following domains: sequence generation; allocation concealment; blinding of participants and personnel; blinding of outcome assessors; incomplete outcome data; selective outcome reporting; and other sources of bias.
Measures of treatment effect
Had there been sufficient data, we would have calculated the weighted treatment effect of identified trials using RevMan 2014 to combine risk ratios (RR) with 95% confidence intervals (CIs) and risk differences (RDs) with 95% CIs for dichotomous outcomes, and mean differences (MDs) and 95% CIs for continuous outcomes.
Unit of analysis issues
Because of the progressive nature of dystrophinopathies, a potential source of bias might have occurred if the treatment arm preceded placebo in studies with cross‐over designs. For this reason, we planned to only analyse the first arm of any cross‐over study.
Dealing with missing data
If necessary, we planned to attempt to contact trial authors for missing data, including numbers of dropouts and deaths and whether or not they performed an intention‐to‐treat analysis.
Assessment of heterogeneity
We planned to carefully evaluate all possible causes of heterogeneity and, where appropriate, to report the Chi² and I² statistics. We would have considered Chi² values of P = 0.1 or less to indicate significant heterogeneity.
Assessment of reporting biases
We planned to assess the potential effect of outcome reporting bias by inspecting forest plots and preparing forest plots, if there were sufficient RCTs.
Data synthesis
If we identified two or more studies comparing the same treatments, we planned to use RevMan to pool their results, employing methods appropriate to the type of outcome measures reported. Dichotomous outcomes give proportions for each treatment group and the treatments are usually compared using the ratio of the proportions known as the risk ratio (RR). We planned to combine studies to give an overall RR using fixed‐effect analysis unless there was significant evidence of heterogeneity between studies, in which case a random‐effects analysis would be more appropriate. Counted episodes may be expressed as differences in rates/unit time at risk with standard errors. In that event the simplest analysis would have been to use the generalised inverse variance (GIV) facility in RevMan to obtain and test the pooled difference between treatment effects.
'Summary of findings' tables
We created 'Summary of findings' tables using GRADEpro software (GRADEpro GDT 2015), and presented the following outcomes:
Change in cardiac function after six months;
Size of metabolically abnormal areas of myocardium;
Improvements in quality of life measures; and
Adverse events.
We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the certainty of a body of evidence (studies that contributed data for the prespecified outcomes). We followed methods and recommendations described in Chapters 11 and 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). RQ and JB assessed the certainty of evidence. They downgraded the randomised controlled trial evidence from high to moderate, low or very low certainty depending on the presence of the five GRADE factors. We downgraded once if any single consideration was serious and twice if very serious. We documented decisions to downgrade or upgrade the certainty of evidence using footnotes.
Subgroup analysis and investigation of heterogeneity
We planned to undertake subgroup analysis based on:
diagnosis (DMD, BMD and XLDCM); and
age (adult versus child less than 16 years of age).
Within each group we planned to use the I² statistic for heterogeneity and if its value had been greater than 50% we would have scrutinised the trials and forest plots for differences to explain the heterogeneity. If we found no explanation, we would have repeated the analysis using a random‐effects model.
Sensitivity analysis
We planned to perform a sensitivity analysis to ensure robustness of findings. This could include repeating the analysis but omitting results from studies with cross‐over design, smaller trials, or commercially‐led trials, and those lacking allocation concealment or blinding.
Results
Description of studies
Results of the search
We identified a total of 635 references from searches of the Cochrane Neuromuscular Specialised Register, MEDLINE, Embase, and CENTRAL. After removing duplicate records, we were left with 472 unique records. Following a review of the abstracts, we obtained the full texts of 14 studies, of which only 5 were ultimately suitable for inclusion (see Figure 1 for a flow chart illustrating the study selection process). We described reasons for excluding studies in the Characteristics of excluded studies tables.
1.
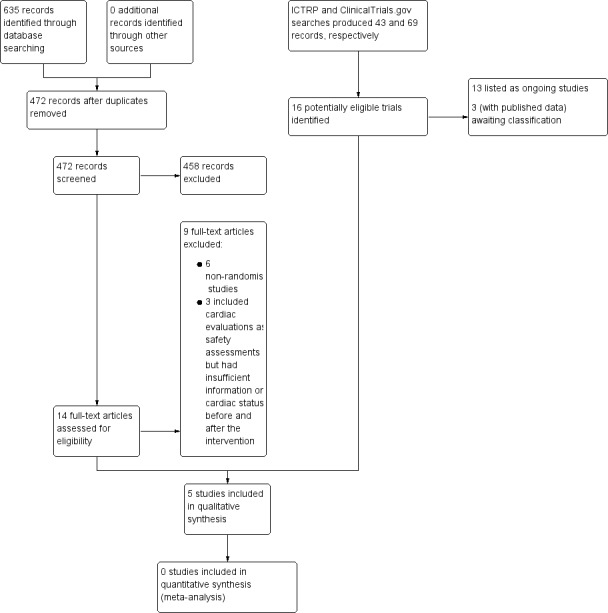
Study flow diagram illustrating the study selection process.
We reviewed and excluded nine other studies of pharmacological agents for cardiomyopathy. Six did not have a randomised controlled design (one was a follow‐up study to one of the RCTs, Duboc 2007, which provided additional outcome information for the original study (Duboc 2005)). We reviewed reports of three RCTs of novel disease‐modifying agents, which included cardiac evaluations as safety assessments. However, ultimately we excluded them because of insufficient information on cardiac status before and after the intervention.
Searches of ICTRP and ClinicalTrials.gov produced 43 and 69 records, respectively, of which we included 13 as Ongoing studies. Three further registrations had published data and we added them to Studies awaiting classification.
Included studies
See Characteristics of included studies.
Prophylactic use of perindopril versus placebo in DMD
Duboc 2005 reported a two‐phase study conducted over five years, comprising an initial 36‐month RCT phase (phase 1) and an open‐label 24‐month continuation phase (phase 2), to assess the effect of prophylactic use of perindopril on the development and progression of left ventricular dysfunction in children with DMD. Participants were recruited from 10 centres in France and had genetically proven DMD, normal cardiac examination and LVEF of more than 55% at baseline as measured by radionuclide ventriculography. Entry criteria required participants to tolerate a 1 mg test dose of perindopril, to have systolic blood pressure of at least 80 mmHg supine or more than 70 mmHg sitting, to be on no other cardioactive drugs, with blood urea nitrogen of more than 7 mmol/L and no contraindications to ACE inhibitor therapy. It was unclear from the initial publication whether participants were taking steroid therapy concurrently or had cardiac devices implanted (Duboc 2005). However, in their subsequent paper reporting long‐term (10‐year) follow‐up (Duboc 2007), the trial authors stated that no other pharmaceutical agent was being administered during the initial randomised phase of the study.
A total of 57 patients aged 9.5 to 13 years were recruited and studied in phase 1 and randomly allocated to receive 2 mg to 4 mg perindopril once a day (active treatment: N = 28; mean age 10.7 years (standard deviation (SD) 1.2); placebo group: N = 29; mean age 10.6 years (SD 1.2)). Baseline characteristics of both groups were similar. Outcome measures included detailed serial clinical and drug tolerance evaluations and routine laboratory blood testing. Resting radionuclide ventriculography was performed at baseline, at 36 months planned study end (phase 1), and at 60 months (phases 1 and 2). Differences between treatment and placebo groups were assessed using Chi² analysis (P < 0.05 for significance). One participant did not complete phase I for reasons unstated. However, as even this patient had LVEF% measured at 36 months, follow‐up in phase I was complete. Mean LVEF at the start of phase I was 65.0% (SD 5.4) in the 57 participants.
During phase 2, the open‐label extension (Duboc 2005), three additional patients withdrew from the study (initial active therapy, n = 1) initial placebo therapy n = 2) for personal reasons, and none had experienced adverse events during phase 1. Furthermore, beta‐blocking drugs were co‐prescribed for supraventricular arrhythmias in nine patients during phase 2 (initial active therapy, n = 4; initial placebo therapy, n = 5). The trial authors do not address the possible confounding effects of these cardioactive drugs but state that none of those on beta‐blockers had LVEF of less than 45% at 60 months.
Lisinopril versus losartan in established cardiomyopathy
Allen 2013 compared the benefits of lisinopril (an ACE inhibitor) 0.07 mg/kg (5 mg/day) with losartan (an ARB) 0.7 mg/kg (25 mg/day) in a randomised, double‐blind, controlled trial of 23 enrolled (22 randomised) DMD patients, newly diagnosed with cardiac dystrophinopathy. After one withdrawal, 12 participants were randomised to lisinopril (median age 12.5 years, range 10 to 21) and 10 to losartan (median age 15.5 years, range 7 to 27 years). Cardiomyopathy was defined on echocardiography by a fall in LVEF of 10% from baseline and subsequently reassessed four‐monthly over 12 months. Median age in the lisinopril group was 12.5 years (range 10 to 21 years) compared to 15.5 years (range 7 to 27 years) in the losartan group. Siblings were randomised to the same treatment arm. Initial doses were doubled if the LVEF decreased by 5% to 10% and participants were withdrawn from further study if the LVEF fell further by more than 10%. Concomitant therapy with corticosteroids, beta‐blockers or both were allowed. Although not stated, the trial authors imply that participants were already taking steroid therapy, but it is unclear whether beta‐blockers could be initiated during the study. Too few participants in the study were taking beta‐blockers (n = 0 in lisinopril group; n = 2 in the losartan group) to allow separate analysis of the effects. Mean ejection fractions were similar at baseline (LVEF lisinopril 47.5% versus losartan 48.4%).
Idebenone versus placebo in subclinical cardiomyopathy
Buyse 2011 conducted a small (N = 21) randomised, double‐blind, placebo‐controlled study of idebenone, an antioxidant, in boys aged 8 to 16 years old with DMD who had subclinical cardiomyopathy, defined by the presence of reduced radial strain measurements in the postero‐lateral segments of the left ventricular wall on echocardiography. Thirteen boys received idebenone 150 mg, and 8 received placebo. The mean age in the idebenone group was 10.8 years (SD 1.9) and in the placebo group 13.4 years (SD 2.1). Exclusion criteria included concomitant use of ACE‐inhibitors or other antioxidants or the presence of an already established cardiomyopathy (fractional shortening of less than 20% or LVEF of less than 40%). The study was partly funded by Santhera Pharmaceuticals, manufacturer of idebenone, and randomisation was 2:1 for idebenone taken three times per day or placebo. The primary outcome was change in measures of peak left ventricular postero‐lateral radial strain between active and placebo treated groups and change within each group from baseline over 12 months. A range of other parameters were also measured, including cardiac biomarkers (troponin‐1 and pro‐BNP) and respiratory and skeletal muscle strength.
Eplerenone versus placebo
In a multicentre, randomised, placebo‐controlled trial, Raman 2014 compared the cardioprotective effect of adding eplerenone (25 mg orally) or placebo to established treatment with an ACE inhibitor or ARB for 12 months in 42 males with DMD. Twenty participants were treated with eplerenone and 22 with placebo, and most participants were already receiving ACE inhibitors (18 in the active eplerenone treatment group; 20 in the placebo group). The median age in years (IQR) in the eplerenone group was 14.5 (12.0 to 18.5) and in the placebo group 15.0 (11.0 to 19.0). Eight participants in the eplerenone group and nine in the placebo group were also taking beta‐blockers, and two were taking regular furosemide. Other concomitant non‐cardiac medications included multivitamins, coenzyme Q10, vitamin D, calcium supplements, proton pump inhibitors, and corticosteroids. Cardiomyopathy was assessed using cardiac magnetic resonance imaging (MRI), which included gadolinium‐based contrast injection. Participants had to have genetically proven DMD or a classical phenotype and be older than 7 years. MRI had to show all of the following features at study entry: myocardial systolic dysfunction, with one or more left ventricular segments showing late gadolinium enhancement but with left ventricular ejection fraction of at least 45%. Exclusion criteria were the presence of an MRI‐incompatible implant, severe claustrophobia, allergy to gadolinium contrast, previous treatment with eplerenone or spironolactone, use of a potassium‐sparing diuretic or other interventional agent within four weeks of the study or five half‐lives of the drug. Eplerenone was administered in a dose of 25 mg on alternate days for the first month then daily if the serum potassium (K+) concentration remained 5.5 mmol/L or below. The primary outcome was change in left ventricular circumferential strain from baseline to 12 months. Secondary outcomes were change in left ventricular circumferential strain from baseline to 6 months and changes in LVEF% and extent of late gadolinium enhancement at 6 and 12 months. Investigators also measured biomarkers: serum creatine kinase‐MB (CK‐MB), troponin‐1 and osteopontin, and adverse events, including admission to hospital for heart failure, cardiac arrhythmia, death and serum K+ of more than 5.5 mmol/L.
Growth hormone versus placebo
One study assessed the effects of growth hormone (GH) therapy on cardiac structure and function in patients with DMD and BMD. Ten consecutive patients with BMD and six with DMD were randomised to receive either recombinant GH (DMD: 0.23 mg/kg/week; BMD: 0.07 mg/kg/week) or placebo for three months (Cittadini 2003). The mean age of the participants was 13 years (SD 2) in those with DMD, and 39 years (SD 3) in those with BMD. The diagnosis was confirmed in all by dystrophin staining of skeletal muscle biopsies. The BMD participants were receiving background therapy including fosinopril 20 mg/day to 30 mg/day (ACE inhibitor), warfarin, magnesium supplements, pidolatum, antioxidants (vitamins E, C, glutathione, ubiquinone), furosemide and deflazacort. One participant in each group was also receiving digoxin and amiodarone. All DMD participants were receiving deflazacort, fosinopril and antioxidants (vitamin E, glutathione and ubiquinone). Cardiac evaluation comprised ECG cardiomyopathic index (QT‐PQ ratio, normal values being 2.2 to 4.6 s), and 24‐hour ECG monitoring and echocardiography (M‐mode, 2D and echo‐Doppler), measures of left ventricular size and function by a sonographer blinded to treatment allocations. Measures of skeletal muscle function included timed function tests (timed Gowers' manoeuvre, time to climb four standard stairs, timed 10‐metre walk, and 'dynamic index'). Pulmonary function measures comprised forced vital capacity (FVC), maximal voluntary ventilation, and maximal expiratory pressure.
Excluded studies
See Characteristics of excluded studies
We excluded five studies that did not have a randomised controlled design (Folkers 1985; Ishikawa 1995; Kajimoto 2006; Matsumura 2010; Rhodes 2008), three safety studies without cardiac outcomes (Mendell 2013; Voit 2014), and one long‐term non‐randomised phase of an included study (Duboc 2007).
Studies awaiting classification
We listed three studies in the Studies awaiting classification section. A trial of oral carvedilol versus ramipril stopped early; the ICTRP record states that no results are available, but this requires confirmation (EUCTR2008‐007236‐18‐IT). We matched two ClinicalTrials.gov records to trial reports (Leung 2014; Salehi 2017). Salehi 2017 studied the effects of coenzyme Q10 in 25 randomised participants, who were said to have genetically confirmed DMD, but as the trial has female participants, we plan to contract the trial authors to confirm eligibility. Leung 2014 was a randomised, placebo‐controlled trial of sildenafil in DMD, which was stopped early for harm (worsening left ventricular end systolic volume on cardiac MRI). We did not initially consider it for inclusion, but as data are available from an interim analysis on 15 participants who completed the six‐month trial, we will re‐assess its eligibility when we update the review.
Ongoing studies
We added 13 trials from searches of clinicaltrials.gov or ICTRP to Characteristics of ongoing studies tables (FOR‐DMD 2012; ISRCTN50395346; NCT00606775; NCT00819845; NCT01126697; NCT01350154; NCT01648634; NCT02354352; NCT02432885; NCT02485938; NCT03340675; NCT03406780; NCT03439670).
Risk of bias in included studies
See Figure 2 for an illustration of the review authors' 'Risk of bias' assessments for all included studies across all domains.
2.
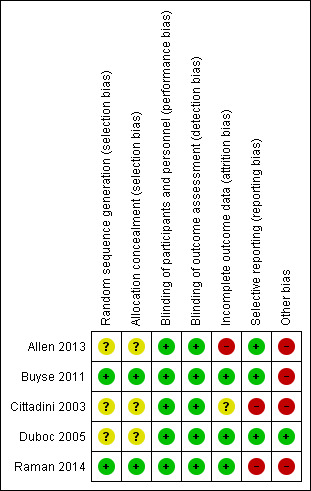
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
The perindopril study did not provide sufficient details on how participants were randomised (Duboc 2005). In Allen 2013, the Nationwide Children's Hospital investigational pharmacy performed the randomisation. Siblings were randomised to the same treatment arm, and we assessed the risk of bias from random sequence generation as unclear. For the idebenone trial (Buyse 2011), randomisation was computer generated by a third party, Averion International, Switzerland. In the eplerenone study (Raman 2014), study participants were randomised using computer‐generated blocks centrally, with only the study statistician and investigational pharmacy aware of the randomisation assignments. No details were provided as to how randomisation was performed in the growth hormone study (Cittadini 2003).
Allocation concealment for Duboc 2005, Allen 2013 and Cittadini 2003 was unclear. A third party (Averion, Switzerland) performed allocation concealment in the idebenone study (Buyse 2011). In Raman 2014 there was good allocation concealment with only the study statistician and institutional pharmacy knowing the randomisation assignments.
Blinding
All five included studies were performed in a double‐blind fashion.
Incomplete outcome data
We considered the following studies as being at low risk of bias for incomplete outcome reporting: Duboc 2005, Buyse 2011 and Raman 2014. Cittadini 2003 did not provide information about compliance or report whether or not there were any dropouts. Six of the 23 participants in Allen 2013 dropped out and we judged the study at high risk of attrition bias.
Selective reporting
Raman 2014 did not provide data to substantiate findings, and Cittadini 2003 presented left ventricular mass index, end‐systolic stress and ejection fraction results graphically. We did not identify any other selective reporting.
Other potential sources of bias
The idebenone trial by Buyse 2011 was in part industry funded, and we assessed the risk of other bias as high. In Cittadini 2003, participants were taking other medications for cardiomyopathy; we considered this to confer a high risk of bias. In Allen 2013 the number of participants receiving corticosteroids was greater in the lisinopril group, and in Raman 2014, although it is not possible to determine whether concomitant therapy confounded the results, we consider the risk high.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5
Summary of findings for the main comparison. Prophylactic perindopril (2 mg to 4 mg daily) versus placebo in DMD.
| Prophylactic perindopril (2 mg to 4 mg daily) versus placebo in DMD | ||||||
| Patient or population: boys with DMD, normal cardiac examination and LVEF > 55% at baseline Setting: 10 clinics in France Intervention: prophylactic perindopril (2 mg to 4 mg daily) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with prophylactic perindopril (2 mg to 4 mg daily) | |||||
| Change in cardiac function: number of patients with EF < 45% Assessed with: radionuclide ventriculography Follow‐up: 36 months | Study population | RR 1.04 (0.07 to 15.77) | 57 (1 RCT) | ⊕⊝⊝⊝ Very lowa | Results of an open‐label extension study are not shown here. See text. | |
| 34 per 1000 | 36 per 1000 (2 to 544) | |||||
| Size of metabolically abnormal areas of myocardium | Not reported | |||||
| Improvements in quality of life measures | Not reported | |||||
| Adverse events Follow‐up: 36 months |
Study population | RR 1.16 (0.78 to 1.72) | 57 (1 RCT) | ⊕⊕⊝⊝ Lowb | — | |
| 586 per 1000 | 680 per 1000 (457 to 1000) | |||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DMD: Duchenne muscular dystrophy; LVEF: left ventricular ejection fraction; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aWe downgraded the certainty of the evidence three times as the method of randomisation was not clear (study limitations), the study was small (imprecision), and the boys in this trial started phase 1 of the trial when their cardiac function was normal (indirectness). The randomised phase of the study lasted three years, not long enough in this phase of the disease for data to determine the effect on decline in cardiac function. bWe downgraded the certainty of the evidence twice as the method of randomisation was not clear (study limitations) and the study was small (imprecision).
Summary of findings 2. Lisinopril (0.7 mg/kg daily) versus losartan (0.7 mg/kg daily) for established cardiomyopathy in DMD.
| Lisinopril (0.7 mg/kg daily) versus losartan (0.7mg/kg daily) for established cardiomyopathy in DMD | ||||
| Patient or population: patients with established cardiomyopathy in DMD Setting: 5 participating centres Intervention: lisinopril (0.7 mg/kg daily) Comparison: losartan (0.7 mg/kg daily) | ||||
| Outcomes | Anticipated absolute effects* (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | |
| Risk with losartan (0.7 mg/kg daily) | Risk with lisinopril (0.7 mg/kg daily) | |||
| Cardiac function: assessed with echocardiography; EF measured by biplane Simpson's rule from the apical 4 chamber view Follow‐up: 12 months (final values) | The mean EF was 55.2% | MD 0.60% lower (6.67 lower to 5.47 higher) | 16 (1 RCT) | ⊕⊝⊝⊝ Very lowa |
| Size of metabolically abnormal areas of myocardium | Not reported | |||
| Improvements in quality of life measures | Not reported | |||
| Adverse events Follow‐up: 12 months | Adverse events are not fully described. There were 2 withdrawals because of adverse events, both in the losartan group (hives and greater than 10% decline in ejection fraction) | 22 (1 RCT) | ⊕⊕⊝⊝ Very lowb | |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DMD: Duchenne muscular dystrophy; EF: ejection fraction; RCT: randomised controlled trial; RR: risk ratio. | ||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||
aWe downgraded the quality of evidence three times: twice for serious imprecision (small study size and CI that included the possibility of clinically important effects in either direction) and once for study limitations (multiple but not controlled concomitant medications, and a large number of dropouts in the lisinopril group). bWe downgraded the quality of evidence three times: once for imprecision (small study size) and twice for study limitations (the report does not provide results from the adverse event questionnaire described in the protocol and methods; and because participants received multiple concomitant medications).
Summary of findings 3. Idebenone (3 daily tablets of 150 mg) versus placebo for subclinical cardiomyopathy in DMD.
| Idebenone (3 daily tablets of 150 mg) versus placebo for subclinical cardiomyopathy in DMD | |||||
| Patient or population: boys (aged 8 to 16 years old) with subclinical cardiomyopathy in DMD Setting: Leuven, Belgium Intervention: idebenone (3 daily tablets of 150 mg) Comparison: placebo | |||||
| Outcomes | Anticipated absolute effects* (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with idebenone (3 daily tablets of 150 mg) | ||||
| Change in cardiac function (change in fractional shortening) assessed with: echocardiography Follow‐up: 12 months | The mean cardiac function (change in fractional shortening) was 1.6% | MD 0.20% lower (3.07 lower to 2.67 higher) | 21 (1 RCT) | ⊕⊕⊝⊝ Very lowa,b | Non‐ significant |
| Change in cardiac function (change in ejection fraction) assessed with: echocardiography Follow‐up: 12 months | The mean cardiac function (change in ejection fraction) was 0.4% | MD 2.3% lower (9.18 lower to 4.58 higher) | 21 (1 RCT) | ⊕⊕⊝⊝ Very lowa,b | Non‐ significant |
| Change in cardiac function (change in peak systolic radial strain in LV lateral wall segments) | The mean cardiac function (change in peak systolic radial strain in LV lateral wall segments) was 7.5% | MD 9.8% higher (1.99 lower to 21.59 higher) | 18 (1 RCT) | ⊕⊕⊝⊝ Very lowa,b | Non‐ significant |
| Change in cardiac function (change in systolic radial strain rate LV inferolateral wall) assessed with: per second | The mean cardiac function (change in systolic radial strain rate LV inferolateral wall) was 0 per second | MD 0.5 per second higher (0.26 lower to 1.26 higher) | 18 (1 RCT) | ⊕⊕⊝⊝ Very lowa,b | Non‐ significant |
| Size of metabolically abnormal areas of myocardium | Not reported | ||||
| Improvements in quality of life measures | Not reported | ||||
| Adverse events Follow‐up: 12 months | 92 adverse events were reported, all rated as mild or moderate, which were equally distributed between the groups. None of these required drug discontinuations or caused participants to drop out from the trial. | 21 (1 RCT) | ⊕⊕⊝⊝ Lowa | — | |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DMD: Duchenne muscular dystrophy; LV: left ventricular; MD: mean difference; RCT: randomised controlled trial; RR: risk ratio. | |||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | |||||
aDowngraded for very serious imprecision (small study size and CI include clinically relevant effects in either direction) and for baseline imbalance (older age in the idebenone group). bThere was also some indirectness as some participants appears to be at a more advanced stage of cardiomyopathy than 'pre‐clinical', with a reduced ejection fraction or fractional shortening, or both. Normally such patients would receive ACE inhibitor therapy but this was an exclusion criterion in the trial.
Summary of findings 4. Eplerenone (25 mg daily) compared to placebo for DMD.
| Eplerenone (25 mg daily) compared to placebo for DMD | |||||
| Patient or population: boys with DMD and left ventricular ejection fraction 45% or more Setting: 3 centres in the USA Intervention: eplerenone (25 mg daily) Comparison: placebo | |||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | |
| Risk with placebo | Risk with eplerenone (25 mg daily) | ||||
| Change in cardiac function: change (decline) from baseline in left ventricular strain assessed with: cardiac magnetic resonance imaging Follow‐up: 12 months | At 12 months, the median decline in left ventricular systolic circumferential strain was less in the eplerenone‐treated group (1.0%, IQR 0.3 to −2.2) than in the placebo group (2.2%, IQR 1.3 to −3.1) (P = 0.020). | — | 42 randomised (1 RCT) | ⊕⊝⊝⊝ Very lowa | |
| Change in cardiac function: change in LVEF (baseline to 6 months) assessed with: cardiac magnetic resonance imaging Follow‐up: 12 months | The median decline of LVEF in the eplerenone group was −1.8% (IQR −2.9 to 6.0) versus −3.7% (IQR −10.8 to 1.0) in the placebo group (P = 0.032) | — | 42 randomised (1 RCT) | ⊕⊝⊝⊝ Very lowa | |
| Size of metabolically abnormal areas of myocardium (baseline to 12 months) | The median change in the eplerenone‐treated group was −1% (IQR −6 to 3) and in the placebo group −3% (IQR −5 to 4), P > 0.999 | — | 42 randomised (1 RCT) | ⊕⊕⊝⊝ Lowb | |
| Improvements in quality of life measures | Not reported | ||||
| Adverse events Follow‐up: 12 months |
Study population | RR 0.37 (0.02 to 8.48) | 42 (1 RCT) | ⊕⊝⊝⊝ Very lowa | |
| 45 per 1000 | 17 per 1000 (1 to 385) | ||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DMD: Duchenne muscular dystrophy; IQR: interquartile range; RCT: randomised controlled trial; RR: risk ratio. | |||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | |||||
aDowngraded three times: once for study limitations (study did not control for concomitant medications, which were numerous) and twice for imprecision; the CI allows for the possibility of a difference in either direction. Additionally LVEF% in some participants would constitute 'definite cardiomyopathy'; however, the criteria for starting ACE inhibitor and beta‐blocking therapy in any of the patients is not stated (yet eplerenone was added to this combination). bDowngraded once for study limitations (study did not control for concomitant medications, which were numerous) and imprecision (N = 39). Additionally, LVEF% in some participants would constitute 'definite cardiomyopathy'; however, the criteria for starting ACE inhibitor and beta‐blocking therapy in any of the patients is not stated (yet eplerenone was added to this combination).
Summary of findings 5. Growth hormone (0.23 mg/kg/week for DMD and 0.07 mg/kg/week in BMD SC injection) versus placebo for DMD and BMD.
| Growth hormone (0.23 mg/kg/week for DMD and 0.07/kg/week in BMD SC injection) versus placebo for DMD and BMD | ||||
| Patient or population: people with DMD or BMD (ages not stated) Setting: Cardiomyology and Myology Centre of Naples Second University Intervention: growth hormone (0.23 mg/kg/week for DMD and 0.07 mg/kg/week in BMD subcutaneous injection) Comparison: placebo | ||||
| Outcomes | Anticipated absolute effects* (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | |
| Risk with placebo | Risk with growth hormone | |||
| Change in cardiac function: ejection fraction assessed with: echocardiography Follow‐up: 3 months | Outcomes were reported at 3 months. No between‐group comparisons of echocardiographic indices for the DMD group (N = 6) were performed due to the small sample size; LV mass index, end‐systolic stress and ejection fraction results were presented graphically The study authors report a non‐significant trend for increase in LV fractional shortening in growth‐hormone‐treated DMD participants. |
16 (1 RCT) | ⊕⊝⊝⊝ Very lowa,b,c | |
| Size of metabolically abnormal areas of myocardium | Not reported | |||
| Improvements in quality of life measures | Not reported | |||
| Adverse events | None reported | 16 (1 RCT) | ⊕⊝⊝⊝ Very lowa,b,c | |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). BMD: Becker muscular dystrophy; CI: confidence interval; DMD: Duchenne muscular dystrophy; LV: left ventricular; RCT: randomised controlled trial; RR: risk ratio. | ||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||
aDowngraded twice for study limitations: inadequate randomisation, confounding from concomitant medications, and selective reporting (numerical results not provided). bDowngraded for imprecision (N = 16). cDowngraded once for indirectness: trial duration 3 months rather than the 6 months specified for this review.
Prophylactic perindopril versus placebo in DMD
One study compared prophylactic perindopril versus placebo in DMD (Duboc 2005).
Primary outcome: change in cardiac function after six months
Duboc 2005 did not provide data on numbers of participants whose cardiac function improved or remained stable versus deteriorated, nor did authors report outcomes for the six‐month time period. The trial authors did report a dichotomous cardiac function outcome: the number of participants with an LVEF of less than 45% at the end of each study phase (36 months and 60 months).
At the end of phase 1 (the randomised phase), LVEF remained normal in most participants, and there was no significant difference in mean LVEF of either group (exact P value not given). Baseline LVEF was 65.0% (SD 5.5) in the treated group (N = 28) and 65.5% (SD 5.5) in the placebo group (N = 29). At the end of 36 months, mean LVEF was 60.7% (SD 7.6) in the treated group versus 64.4% (SD 9.8) in group 2. The difference between groups was not statistically significant (exact P value not given). However, one participant in each group had an LVEF of less than 45% (i.e. established cardiomyopathy) at 36 months (RR 1.04, 95% CI 0.07 to 15.77; very low‐certainty evidence; N = 57; Analysis 1.1).
1.1. Analysis.
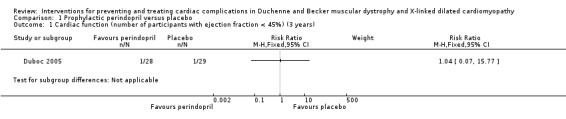
Comparison 1 Prophylactic perindopril versus placebo, Outcome 1 Cardiac function (number of participants with ejection fraction < 45%) (3 years).
We downgraded the certainty of evidence three times, from high to very low, because of serious imprecision (small study size and low event rate), study limitations, and indirectness (a three‐year follow‐up is too short at this stage of DMD to detect effects on cardiac function). See the Discussion for data from the non‐randomised phase 2 of the study and long‐term follow‐up (Duboc 2005; Duboc 2007).
Secondary outcomes
Size of metabolically abnormal areas of myocardium
Not reported.
Improvements in quality of life measures
Not reported.
Adverse events
After 36 months, 19/28 participants in the treatment group had reported at least one adverse event, compared to 17/29 patients in the placebo group (RR 1.16, 95% CI 0.78 to 1.72; N = 57; low‐certainty evidence). The events were similar in nature in each group.
We downgraded the certainty of evidence from high to low because of serious imprecision (small study size) and study limitations.
Lisinopril versus losartan for established cardiomyopathy in DMD
One trial compared lisinopril versus losartan for established cardiomyopathy in DMD (Allen 2013).
Primary outcome: change in cardiac function after six months
In Allen 2013, the authors do not quote the numbers whose LVEF% improved or stabilised versus those in whom it deteriorated, which we specified as our primary outcome; however, the trialists reported the number whose LVEF fell below 45%. The other primary trial outcome was reduction in mean LVEF, which trialists reported after four months, eight months, and one year of therapy.
Mean LVEFs were similar at baseline: 47.5% in the lisinopril group (N = 12) and 48.3% in the losartan group (N = 10) (P = 0.93). At eight months, mean LVEF% was similar in the two groups: 52.9% in the lisinopril group (N = 10) and 53.7% in the losartan group (N = 9). Trialists did not report SDs.
LVEF improved in each group from baseline to 12 months (lisinopril group, P = 0.02 and losartan group, P = 0.03), but there was no important difference in LVEF between the two groups among participants who provided data at 12 months (lisinopril 54.6% (SD 5.19) versus losartan 55.2% (SD 7.19); MD −0.60%, 95% CI −6.67 to 5.47; 16 participants; very low‐certainty evidence). The study was curtailed early because of funding shortfalls, but the trial authors showed clearly where data were missing.
We downgraded the certainty of evidence three times, from high to very low, because of serious imprecision (small study size and CI that included clinically important effects in either direction) and study limitations (multiple but not controlled concomitant medications, and a large number of dropouts in the lisinopril group (due to cessation of funding (n = 5), allergic reaction (n = 1) and poor LVEF at the start of the study or during the study (n = 3)).
Secondary outcomes
Size of metabolically abnormal areas of myocardium
Not reported.
Improvements in quality of life measures
Not reported.
Adverse events
The paper did not report findings from the standardised questionnaire used to collect adverse events. Two participants randomised to the losartan group were removed from the study; one due to an allergic reaction and another who exceeded the safety standard of a greater than 10% decrease in ejection fraction.
We downgraded the certainty of evidence from high to very low because of serious imprecision (small study size) and serious study limitations (selective reporting).
Idebenone versus placebo for subclinical cardiomyopathy in DMD
One trial compared idebenone versus placebo for subclinical cardiomyopathy in DMD (Buyse 2011).
Primary outcome: change in cardiac function after six months
Buyse 2011 reported outcomes at one year but not at six months. The trial authors did not provide data for our primary outcome (number of participants in whom left ventricular function improved or stabilised versus deteriorated). They reported continuous cardiac function outcomes comparing the difference between idebenone treatment and placebo for global ventricular function and left ventricular peak strain measures.
In terms of measures of global ventricular function, the mean change in fractional shortening from baseline to 12 months was 1.4 (SD 4.1) in the idebenone group and 1.6% (SD 2.6) in the placebo group (MD −0.20%, 95% CI −3.07 to 2.67; N = 21). Corresponding changes in ejection fraction were −1.9% (SD 9.8) in the idebenone group and 0.4% (SD 5.5) in the placebo group (MD −2.30%, 95% CI −9.18 to 4.58; N = 19).
We downgraded the certainty of evidence three times for these measures from high to very low because of very serious imprecision (downgraded twice for imprecision because the trial was small and CI included clinically relevant effects in either direction), and once for study limitations (baseline imbalance), and some indirectness (participants appeared to be at a more advanced stage of cardiomyopathy than 'pre‐clinical' but were not receiving ACE inhibitors).
Posterolateral left ventricular peak strain measures were lower at baseline in those randomised to idebenone. This was because those randomised to idebenone (N = 13) were significantly older than those randomised to placebo (N = 8). Idebenone showed an improvement in left ventricular peak systolic radial strain measures from baseline compared to placebo. The mean increase was 17.3% (SD 13.1) in the idebenone group versus 7.5% (SD 12) P = 0.067) in the placebo group (MD 9.80%, 95% CI −1.99 to 21.59; Analysis 3.3; low‐certainty evidence). The change in systolic radial strain rate left ventricular inferolateral wall in the idebenone group was 0.5 s‐1 (SD 0.6; N = 10) and in the placebo group 0.0 s‐1 (SD 0.9; N = 7) (MD 0.50 s‐1, 95% CI −0.26 to 1.26; Analysis 3.4; very low‐certainty evidence).
3.3. Analysis.
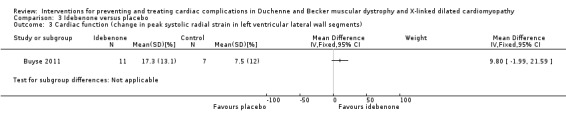
Comparison 3 Idebenone versus placebo, Outcome 3 Cardiac function (change in peak systolic radial strain in left ventricular lateral wall segments).
3.4. Analysis.

Comparison 3 Idebenone versus placebo, Outcome 4 Cardiac function (change in systolic radial strain rate left ventricular inferolateral wall).
We downgraded the certainty of evidence from high to very low because of serious imprecision (the trial was small and CI included clinically relevant effects in either direction), study limitations (baseline imbalance) and indirectness.
Due to the significant age‐related baseline difference between the groups, the authors performed a prespecified secondary analysis to determine percentage change from baseline. This showed a 104.4% change from baseline for idebenone compared to 28.9% for placebo (P = 0.030; SD for changes not given).
See Analysis 3.5; Analysis 3.6; Analysis 3.7; Analysis 3.8 for other cardiac measures from this study.
3.5. Analysis.
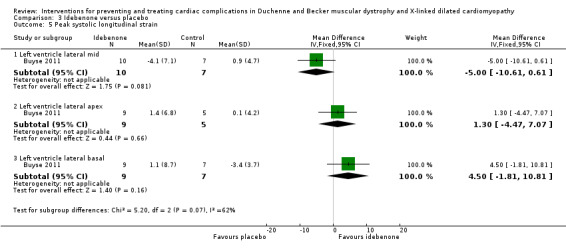
Comparison 3 Idebenone versus placebo, Outcome 5 Peak systolic longitudinal strain.
3.6. Analysis.
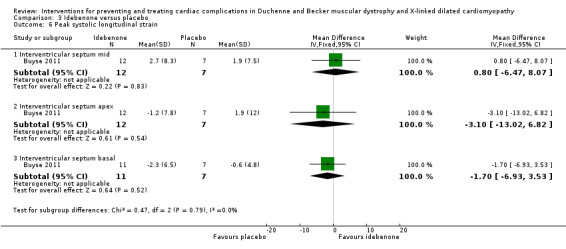
Comparison 3 Idebenone versus placebo, Outcome 6 Peak systolic longitudinal strain.
3.7. Analysis.
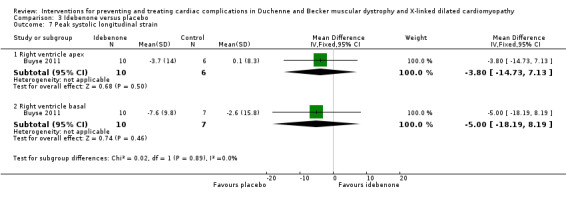
Comparison 3 Idebenone versus placebo, Outcome 7 Peak systolic longitudinal strain.
3.8. Analysis.
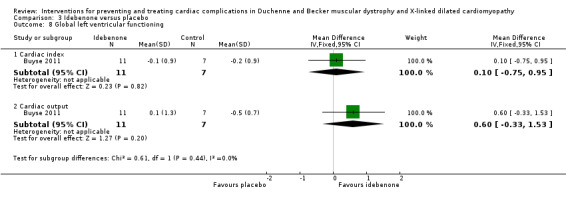
Comparison 3 Idebenone versus placebo, Outcome 8 Global left ventricular functioning.
Secondary outcomes
Size of metabolically abnormal areas of myocardium
Not assessed.
Improvements in quality of life measures
Not assessed.
Adverse events
Trialists noted 92 adverse events, all rated as mild or moderate, which were equally distributed between the groups. None of these resulted in drug discontinuations or dropouts from the trial. The most frequently reported adverse events were gastrointestinal, infections and headache. Two moderately serious adverse events (both traumatic fractures) occurred, one in each group.
We downgraded the certainty of evidence to moderate because of serious imprecision (small study size).
Other outcomes reported in the trial
The study also measured the effect on cardiac biomarkers. Pro‐BNP levels were higher at baseline for those taking idebenone, and there was a non‐significant decrease during treatment. Pro‐BNP levels rose from baseline in the placebo group during the study; however, this was not statistically significant. Cardiac troponin I remained within normal parameters in both groups.
Early respiratory involvement was assessed by measuring peak expiratory flow and static mouth pressures; and restrictive pulmonary changes were measured by spirometry. There was a significant difference in the improvement of peak expiratory flow (PEF) and PEF% predicted (P = 0.039 and P = 0.042, respectively) for idebenone compared to placebo, which tended to have a downward trend. This was despite the older age of the idebenone group.
There were also no significant between‐group differences in upper limb strength.
However, the study is innovative in using highly sensitive measures of early segmental cardiomyopathy as its primary outcome. The study had a number of limitations – older participant age in the idebenone cohort, small sample size, lack of correction in analysis for repeat measures, and use of an idebenone dose not corrected for body mass index. Furthermore, although authors described those recruited as having only preclinical cardiomyopathy, some seemed to be at a more advanced stage on the basis of a reduced ejection fraction (LVEF less than 55%) and/or fractional shortening (FS less than 25%). Such patients would normally be prescribed ACE inhibitor therapy, but this was an exclusion criterion of the study. Overall this was primarily a drug tolerability study, and further therapeutic studies are warranted.
Eplerenone versus placebo for DMD
One trial compared eplerenone versus placebo for DMD (Raman 2014).
Primary outcome: change in cardiac function after six months
Raman 2014 does not state the number of participants whose left ventricular function improved or stabilised versus the number who deteriorated. The study reported results as the difference in changes in LVEF% and circumferential strain between patients receiving eplerenone or placebo from baseline to 6 months, 6 to 12 months, and baseline to 12 months. The study randomised 42 participants and gave the total number of participants completing baseline, 6‐month and 12‐month visits, together with total numbers of analysable examinations, but the number of participants providing data for the outcomes at each time point is not clear.
There was no significant difference in the median decline of LVEF from baseline to six months between the eplerenone group (0%, IQR −3.8 to 4.0) and the placebo group (1.0%, IQR −5.0 to 2.1) (P = 0.474). From 6 months to 12 months, the decline in LVEF was smaller in the eplerenone group (1.6, IQR −0.8 to 2.9) than in the placebo group (−2.8, IQR −5.7 to −1.8) (P = 0.036). This difference was also present in the change from baseline to 12 months, when the median decline of LVEF in the eplerenone group was −1.8% (IQR −2.9 to 6.0) versus −3.7% (IQR −10.8 to 1.0) in the placebo group (P = 0.032).
There were no significant differences in mean decline in left ventricular systolic circumferential strain magnitude between the eplerenone group (0.84% (SD 2.68)) and the placebo group (0.38% (SD 2.56)) from baseline to six months (P = 0.602 or from 6 months to 12 months (P = 0.379). At 12 months, the median decline in left ventricular systolic circumferential strain was less in the eplerenone‐treated group (1.0%, IQR 0.3 to −2.2) than in the placebo group (2.2%, IQR 1.3 to −3.1) (P = 0.020).
The trial authors considered an absolute difference of 1% in strain units at 12 months as clinically significant.
We downgraded the evidence to very low certainty for study limitations (the study did not control for concomitant medications, which were numerous) and twice for imprecision; as the study was small (N = 42 randomised), and measures of variance allow for the possibility of a clinically important difference in either direction. There was also some indirectness. LVEF% in some participants would constitute 'definite cardiomyopathy'.
Secondary outcomes
Size of metabolically abnormal areas of myocardium
The extent of abnormal myocardium, as assessed by extent of late gadolinium enhancement, was reduced over the first six months of the trial by eplerenone therapy (mean change −2% (SD 6) compared to placebo (mean change 4% (SD 6) (MD −6.00%, 95% CI −9.77 to −2.23; low‐certainty evidence)) but not from 6 to 12 months (MD 4.00%, 95% CI 0.23 to 7.77) or from baseline to 12 months (median change in the eplerenone‐treated group −1% (IQR −6 to 3 and in the placebo group −3% (IQR −5 to 4); P > 0.999; low‐certainty evidence).
We downgraded the certainty of evidence twice, from high to low: once for study limitations and once for indirectness. LVEF% in some participants would constitute 'definite cardiomyopathy'; the criteria for starting ACE inhibitor and beta‐blocking therapy in any of the participants was not stated (yet eplerenone was added to this combination).
Improvements in quality of life measures
Not assessed.
Adverse events
In the placebo group, one participant withdrew at three months due to digestive issues and a month after enrolment, one participant died following a fat embolus. Authors did not report any other significant adverse events. Trial authors describe other adverse events as mild. In the placebo group, one participant reported facial flushing after the first two doses and another experienced a panic attack upon commencing the active treatment. We calculated the RR for serious adverse events as RR 0.37, 95% CI 0.02 to 8.48 (very low‐certainty evidence).
We downgraded the certainty of evidence three times, to very low: once for imprecision, as the trial was small (N = 39), once for study limitations, and once for indirectness as the criteria for starting ACE inhibitor and beta‐blocking therapy in any of the participants was not stated (yet eplerenone was added to this combination).
Other outcomes reported in the trial
There were no significant changes in the cardiac biomarkers troponin I, total creatine kinase and creatine kinase MB fraction during the study period.
We have contacted the authors for missing data and are waiting for a response.
Growth hormone versus placebo for DMD and BMD
One trial compared growth hormone (GH) versus placebo for DMD and BMD (Cittadini 2003).
Primary outcome: change in cardiac function after six months
Cittadini 2003 did not provide data on the numbers of participants whose heart function improved or stabilised versus deteriorated, and authors reported outcomes only at three – not at six – months. The study authors reported continuous cardiac function outcomes comparing LVEF and left ventricular mass index in participants receiving GH or placebo for a three‐month time period. There were no between‐group comparisons of echocardiographic indices for the DMD group (N = 6) due to the small sample size; left ventricular mass index, end‐systolic stress and ejection fraction results were presented graphically. Authors reported results in BMD and DMD participants for active versus placebo therapy separately.
We considered the certainty of evidence very low because of study limitations, imprecision (N = 16), and indirectness (three‐month study duration rather than our specified six months).
In participants with BMD (N = 10), left ventricular volumes were larger and LVEF lower compared with DMD participants. In the GH‐treated BMD participants, left ventricular mass increased by approximately 42 g (trend towards increased left ventricular posterior and anterior wall thickness) compared with a slight decrease in these measurements in the placebo group. The authors reported a "concentric remodelling of the left ventricular cavity with a significant increase of the relative wall thickness of 12%" in the GH‐treated BMD group, compared to no change in the placebo‐treated group (no measures of variance reported). End diastolic volumes did not change significantly over time.
In GH‐treated DMD participants, the study authors report a 29% increase in left ventricular mass compared to the placebo‐treated group. The study authors also report a non‐significant trend for increase in left ventricular fractional shortening (LVFS) in GH‐treated DMD participants.
Secondary outcomes
Size of metabolically abnormal areas of myocardium
Not assessed.
Improvements in quality of life measures
Not assessed.
Adverse events
Trial authors reported "no clinical relevant side effect". There were no observed cardiac arrhythmias or haematological adverse effects.
The certainty of this evidence was very low. We downgraded the certainty of evidence three times: twice for study limitations and once for imprecision. Additionally, the trial duration was only three months.
Other outcomes reported in the trial
Authors observed no significant variations in the cardiomyopathic index (which was abnormal at baseline in five BMD and three DMD participants) during the study period.
Treatment and control groups were similar for blood biomarkers at baseline. Seven of the 16 participants showed impairment of GH/IGF1 axis and low circulating IGF1 levels. Plasma IGF‐1 increased by 82% in participants treated with GH but decreased by 9% in those receiving placebo. Thyroid function did not change in either group. Plasma levels of BNP were elevated in all participants compared to controls but decreased by 40% in the treatment group. There were no significant differences in timed function tests or FVC.
Discussion
Summary of main results
This systematic review identified five double‐blind RCTs meeting Cochrane criteria for inclusion (Allen 2013; Buyse 2011; Cittadini 2003; Duboc 2005; Raman 2014). The trials involved a total of 205 participants, and each trial assessed a different intervention. No meta‐analysis was possible because none of the interventions were sufficiently similar for data to be combined.
Two studies, one of GH and one of idebenone, showed no meaningful change in left ventricular function.
A study comparing idebenone with placebo in 21 boys with DMD showed no difference in cardiac function between the two groups after 12 months' treatment. Reported adverse events were similar between the treatment and placebo groups. We rated the certainty of evidence from this study as very low.
The study comparing lisinopril with losartan, which was a short duration study in a small patient cohort, suggested that ACE inhibitors and ARB drugs were equally beneficial in treating the early stages of cardiac dystrophinopathy.
Long‐term follow‐up of perindopril treatment in boys with BMD/DMD found only small changes in cardiac function over long periods of time in most boys, and there were few events, suggesting a need for long‐term studies with more participants.
In boys with DMD and early cardiac involvement who have already been established on ACE inhibitors or ARB drugs, treatment with eplerenone reduced the decline both in measures of left ventricular strain and LVEF% compared with placebo over 12 months.
The results of these studies provide low‐ or very low‐certainty evidence that it may be possible to modify the course of cardiomyopathy in patients with DMD and BMD with early use of ACE inhibitors (Allen 2013; Duboc 2007), ARBs (Allen 2013), and eplerenone (Raman 2014).
Data from open‐label extension studies in a large cohort of DMD patients suggest that corticosteroid treatment – used primarily for muscle strengthening – delays the onset of and slows the course of cardiomyopathy in DMD (Barber 2013; Schram 2013; Silversides 2003). Ongoing trials of newer disease‐modifying agents need to include heart assessments as a key outcome measures.
Evidence from non‐randomised studies
Duboc 2005 reported a two‐phase double‐blind, randomised first phase comparing perindopril 2 mg to 4 mg daily (group 1) and placebo (group 2), lasting three years, followed by a two‐year open‐label second phase at the same dose of perindopril. Duboc 2007 was a follow‐up report after 10 years of treatment.
At the conclusion of phase 2 of this study at 60 months, both groups showed significant decrease in LVEF: from 65.0% (SD 5.5) to 58.6% (SD 8.1) (P = 0.001) in those randomised to active treatment in phase 1; and from 65.4% (SD 5.5) to 56.0% (SD 15.5) (P = 0.006) in those initially randomised to placebo therapy in phase 1. However, the authors stated that there was no statistically significant difference in mean LVEF between the groups (58.6% (SD 8.1) versus 56.0% (SD 15.5)) at 60 months.
Furthermore, only one participant treated with perindopril in phase 1, compared to eight participants treated with placebo in phase 1, had an LVEF of less than 45% (Chi² 5.699, P = 0.02). The mean age of patients with or without depressed LVEF at 60 months was similar, and the benefits of 2 mg versus 4 mg of perindopril were similar. The trialists go on to highlight that three of eight participants from the initial placebo group with LVEF of less than 45% died of congestive heart failure in the year after phase 2 completion, while the one participant from the group initially allocated to perindopril with LVEF of less than 45% remained alive.
The study showed little or no difference between perindopril and placebo groups after three years and, even after 5 years (phases 1 and 2), the mean differences in measures of cardiac function between the groups were not clinically significant. However, more participants from the placebo arm of the RCT had LVEF of less than 45% than in the treatment arm at the end of the open label study, suggesting that a longer time interval may be required to detect changes at follow‐up.
A later publication, reporting 10‐year follow‐up from the original study reported improved survival in the participants in the initial perindopril‐treated arm compared to the initial placebo‐treated arm (26/28 versus 19/29 alive at 10 years; Kaplan‐Meier cumulative survival P = 0.125 and log‐rank Fisher exact test P = 0.02) (Duboc 2007). However, authors did not state the precise mechanism of death, and some participants had also been treated with beta‐blockers (four perindopril‐treated participants; five placebo‐treated participants in phase 1). The study has a number of other weaknesses in that, although it showed that death occurred more often in participants with reduced heart function, it was not powered to assess mortality and, although both active and placebo groups were similar in age, the groups were not standardised by disease severity.
One non‐randomised study compared ACE inhibitors versus ACE inhibitors plus beta‐blockers in patients with DMD‐related cardiomyopathy. Participants were monitored with echo‐measures of LVEF% every 3 to 4 months over 12 months. Prior to ACE inhibitor treatment, 22 participants showed declining LVEF% over time. ACE inhibitor therapy was started and doses modified if further falls in LVEF% were observed. Beta‐blockers were added (N = 24) if resting heart rate on 24‐hour ECG exceeded 100 beats per minute. LVEF% improved compared to baseline in both groups (ACE inhibitors and ACE inhibitors plus beta‐blockers) (P < 0.001). However, the difference between groups was not significant (Viollet 2012). More recently a number of novel disease‐modifying pharmaceutical agents (ataluren, eteplirsen, and drisapersen) have been tested with regard to their effects on skeletal muscle in phase II clinical trials. However, their effects on cardiac function have yet to be reported (Bushby 2014; Mendell 2013, Voit 2014). A meta‐analysis of corticosteroids as a disease‐modifying treatment for DMD showed benefit over six months based upon functional scores, but these trials did not include cardiac outcome measures (Matthews 2016). However, data from long‐term prospective extension studies suggest that corticosteroids do provide some measure of cardiac protection (Burnett 2017; Schram 2013; Silversides 2003). Unfortunately, because corticosteroids are now considered the 'gold standard of care' for patients with DMD, it would no longer be ethical to conduct RCTs to further examine their effects specifically on cardiac function in DMD. One small prospective study assessed cardiac function at baseline and after three months of corticosteroid therapy (prednisolone 5 mg/kg/day on two consecutive days each week) in 25 patients with either BMD, DMD or who were manifesting carriers of DMD (Hussain 2014). LVFS was assessed by echocardiography at baseline and after three months of treatment. LVFS% improved (P = 0.009) and left ventricular mass increased (P = 0.012) in those on prednisolone treatment. Another large, prospective follow‐up study assessed cardiac function by echocardiography in 462 of 797 DMD patients from multiple centres in the USA (MD STARnet), 291 of whom had received corticosteroids and 171 who had never received corticosteroid treatment (Barber 2013). Cardiomyopathy was defined as a fractional shortening of less than 28% or, if fractional shortening measurement was not available, an ejection fraction of less than 55%. Among those who had received corticosteroids, the mean treatment starting age was 7.4 years and the mean treatment duration was 4.1 years. Cardiomyopathy developed in 202 of the 291 corticosteroid‐treated boys, at a mean age of 15.2 (SD 3.4) years, compared to all 171 of those who were untreated, at a mean age of 13.1 (SD 4.8) years. A Kaplan‐Meier curve showed that corticosteroid therapy significantly delayed the cardiomyopathy (P = 0.02, Chi² = 5.27). Furthermore, regression analysis suggested that for every year of corticosteroid treatment, cardiomyopathy was delayed by 4%. Survival was greatest in the group who received carvedilol.
In a non‐randomised study of carvedilol (Matsumara 2010), 41 DMD patients received carvedilol, and 13 did not. All participants were treated with an ACE inhibitor if the ejection fraction was below 50% and were followed up every six months for five years. Symptomatic heart failure occurred in more patients who had not received carvedilol, and more participants in this group died compared with those treated with carvedilol.
A retrospective review of ivabradine treatment in 13 DMD patients with dilated cardiomyopathy (defined as ejection fraction less than 45%) compared with seven untreated patients demonstrated improvement in left ventricular function in the ivabradine‐treated patients (Adorisio 2017).
There have been no RCTs of cardiac transplantation in patients with inherited forms of muscle disease. However, in a retrospective review of 29 transplant centres in the USA covering the period 1990 to 2005 (Wu 2010), outcomes after transplantation were compared between 29 patients with muscular dystrophy (52% had BMD) and 275 age‐ and sex‐ matched 'controls' who underwent transplantation for non‐ischaemic cardiomyopathy without muscle conditions. One‐year survival was 89% and 91% in the muscular dystrophy and the non‐muscle‐affected group, respectively (P > 0.5) and five‐year survival was 83% and 78% (P = 0.5) for muscular dystrophy and non‐muscle‐affected groups. This suggests that a diagnosis of muscular dystrophy should not exclude selected patients from being considered for cardiac transplantation.
At the time of writing this systematic review, several trials are ongoing (see Characteristics of ongoing studies). These include a trial of coenzyme Q10 and lisinopril (NCT01126697), and a randomised, placebo‐controlled trial of ACE inhibitors plus beta‐blockers to prevent the onset or change the course of cardiomyopathy in DMD (ISRCTN50395346). Another ongoing trial is comparing daily corticosteroids (prednisolone or deflazacort) with intermittent prednisolone (10 days on and 10 days off) (FOR‐DMD 2012). The results of these studies are expected, and we plan to include them in the next update of this review.
Overall completeness and applicability of evidence
Currently data are limited to only small studies of a limited range of interventions, but results from larger, ongoing trials are likely to report and add to these data over the next few years. In future updates we will consider including serum biomarkers as a secondary outcome and longer‐term outcome measures.
We found no studies for XLDCM.
Quality of the evidence
The certainty provided by the evidence to date is low or very low due to the paucity of trials and the small numbers of participants studied. Interventions were heterogeneous – none of the interventions were investigated in more than one trial. In addition, data were missing for the eplerenone study, which made further analysis of the results impossible. Most studies had some risk of bias that lowers confidence in our estimates of effect, in some cases substantially. Reporting was not always complete. Several trials did not stratify for age or concomitant medications.
Potential biases in the review process
Studies of disease‐modifying drugs such as eteplirsen (Mendell 2013), which have used cardiac function as a safety measure but have not reported results, could be a potential source of bias for this review as there may be meaningful data not available to the review authors. It is also possible that selective reporting in the literature of only trials with positive results could have potentially biased our review results. Furthermore, the studies that we have reported were of short duration; longer duration non‐randomised studies might potentially show different results.
Agreements and disagreements with other studies or reviews
We did not find data from other studies to support or refute the data presented in this review.
Authors' conclusions
Implications for practice.
Based on the available evidence from RCTs, early treatment with ACE inhibitors or ARBs may be comparably beneficial for people with a dystrophinopathy; however, the certainty of evidence is very low. Findings from non‐randomised studies, some of which have been long term, have led to the use of these drugs in daily clinical practice. Very low‐certainty evidence indicates that adding eplerenone might give additional benefit when early cardiomyopathy is detected. No clinically meaningful effect for growth hormone or idebenone was seen, although the certainty of the evidence was also very low.
Implications for research.
Opportunities to assess the effects of corticosteroids on cardiac involvement in Duchenne muscular dystrophy (DMD) were missed in early randomised controlled trials (RCTs). The opportunity to perform further RCTs to examine the cardiac effects of corticosteroid therapy further has now been lost, as it would be unethical. It is also increasingly difficult to justify placebo‐controlled trials of prophylactic use of ACE inhibitors or ARB therapy in DMD, so we hope that the ongoing study will provide the definitive evidence to guide cardiac management when it reports in 2018. Phase II and phase III trials of novel, disease‐modifying pharmaceutical agents are ongoing. Assessing the effects of these agents on cardiac function from the outset might reduce the need for additional studies specifically to assess cardiac effects later. Future studies could focus on anti‐arrhythmic and heart rate slowing therapies as a potential strategy for preventing further decline in heart function; for example, studies could compare the effect of these agents with beta‐blockers.
A number of pharmacological agents appear to be cardioprotective in DMD and BMD, although the data from RCTs is limited to results from studies of small participant numbers.The plethora of potentially beneficial disease‐modifying therapies and other medications becoming available in the same timeframe on the basis of 'proof of concept' results will make it challenging to recruit sufficient participants from a relatively small pool of eligible patients. Furthermore, even when an individual therapy or medication is shown to be beneficial, there will still be a need to understand how best to combine treatments to optimise patient outcomes without adverse effects or increasing the overall burden of therapy for patients.
We graded the evidence reported as low or very low certainty based upon the studies' short duration, small numbers of participants and missing data. DMD is a rare disorder, and finding sufficient numbers of patients to power a study is not possible without multicentre, multinational collaboration; thus adequately powered RCTs in patients in this population are very expensive, and the costs rise steeply with study duration. In an effort to contain cost and secure funding, therefore, study designs have had to become shorter and use more sensitive surrogate outcomes – as exemplified well by the design and conduct of the eplerenone study (Raman 2014). However, shorter study designs in fewer participants may be insufficient to provide robust clinical evidence to study cardiac involvement in DMD – with its wide inter‐patient variability in which the success of various interventions are probably time dependent. It seems unlikely, however, that the RCTs ideally needed to arrive at the optimum cardiac management in DMD are all affordable. To derive the most from what limited number of studies can be funded, therefore, it is crucial that each addresses an important clinical uncertainty and that the results build on and not duplicate what is already accepted. It is only in this way that cardiac management of patients with DMD can move from theory to evidence‐based care.
What's new
| Date | Event | Description |
|---|---|---|
| 25 February 2020 | Amended | Correction to dates of EMBASE and MEDLINE searches reported in the Methods (correct date October 2017). |
History
Protocol first published: Issue 4, 2011 Review first published: Issue 10, 2018
| Date | Event | Description |
|---|---|---|
| 10 May 2012 | New citation required and minor changes | New author added, one author withdrew. |
| 10 April 2012 | Amended | Change to the protocol title Minor revisions made to the objective, types of interventions, types of studies and outcomes. A statement that we will analyse each type of intervention separately has been included. Embase and CENTRAL search strategies added. |
Acknowledgements
This project was supported by the National Institute for Health Research via Cochrane Infrastructure funding to Cochrane Neuromuscular. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health. Cochrane Neuromuscular is also supported by the MRC Centre for Neuromuscular Disease.
We would like to thank Ruth Brassington, of the Cochrane Neuromuscular disease team, for her help and support in developing this review.
Appendices
Appendix 1. Cochrane Neuromuscular Specialised Register (CRS) search strategy
#1 (duchenne or becker) NEAR5 dystroph* [REFERENCE] [STANDARD] #2 dystrophinopath* or xldcm or "x linked dilated cardiomyopathy" [REFERENCE] [STANDARD] #3 MeSH DESCRIPTOR Dystrophin WITH GE [REFERENCE] [STANDARD] #4 #1 or #2 or #3 [REFERENCE] [STANDARD] #5 cardiomyopathy or cardiomyopathies or "myocardial diseases" or "heart failure" [REFERENCE] [STANDARD] #6 "cardiac protection" or "ventricular dilation" or "heart transplantation" [REFERENCE] [STANDARD] #7 MeSH DESCRIPTOR Pacemaker, Artificial [REFERENCE] [STANDARD] #8 artificial NEAR pacemaker [REFERENCE] [STANDARD] #9 defibrillators or "electric countershock" or "cardiac resynchronisation" [REFERENCE] [STANDARD] #10 "cardiac pacing" NEAR artificial [REFERENCE] [STANDARD] #11 MeSH DESCRIPTOR Angiotensin‐Converting Enzyme Inhibitors Explode All [REFERENCE] [STANDARD] #12 MeSH DESCRIPTOR Calcium Channel Blockers Explode All [REFERENCE] [STANDARD] #13 MeSH DESCRIPTOR Adrenergic beta‐Antagonists Explode All [REFERENCE] [STANDARD] #14 MeSH DESCRIPTOR Cardiotonic Agents Explode All [REFERENCE] [STANDARD] #15 MeSH DESCRIPTOR Diuretics Explode All [REFERENCE] [STANDARD] #16 MeSH DESCRIPTOR Oligonucleotides, Antisense Explode All [REFERENCE] [STANDARD] #17 MeSH DESCRIPTOR Morpholines Explode All [REFERENCE] [STANDARD] #18 (cardiac NEAR1 failure) or (cardiac NEAR1 protection) or (inotropic NEAR1 agent*) [REFERENCE] [STANDARD] #19 (cardiac NEAR1 transplant*) or (cardiac NEAR3 complication*) [REFERENCE] [STANDARD] #20 diuretic* or pacemaker or morpholino [REFERENCE] [STANDARD] #21 #5 or #6 or #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 [REFERENCE] [STANDARD] #22 #4 and #21 [REFERENCE] [STANDARD] #23 (#4 and #21) AND (INREGISTER) [REFERENCE] [STANDARD]
Appendix 2. CENTRAL (CRSO) search strategy
Search run on Mon 16 October 2017 #1 MESH DESCRIPTOR Muscular Dystrophies97 #2 MESH DESCRIPTOR Muscular Dystrophy, Duchenne68 #3 (duchenne NEAR dystrophy):TI,AB,KY300 #4 (becker NEAR dystrophy):TI,AB,KY45 #5 (dystrophinopathy or dystrophinopathies):TI,AB,KY21 #6 MESH DESCRIPTOR Dystrophin18 #7 (xldcm or "x linked dilated cardiomyopathy"):TI,AB,KY0 #8 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7343 #9 MESH DESCRIPTOR Cardiomyopathies EXPLODE ALL TREES1399 #10 MESH DESCRIPTOR Cardiomyopathy, Dilated434 #11 MESH DESCRIPTOR Heart Failure EXPLODE ALL TREES5733 #12 "myocardial diseases"3 #13 ("cardiac protection" or "ventricular dilation"):TI,AB,KY92 #14 MESH DESCRIPTOR Angiotensin‐Converting Enzyme Inhibitors EXPLODE ALL TREES5561 #15 MeSH descriptor Calcium Channel Blockers EXPLODE ALL TREES7997 #16 MESH DESCRIPTOR Adrenergic beta‐Agonists EXPLODE ALL TREES8511 #17 MESH DESCRIPTOR Cardiotonic Agents EXPLODE ALL TREES5163 #18 MESH DESCRIPTOR Diuretics EXPLODE ALL TREES5730 #19 "Heart Transplant*"1059 #20 MESH DESCRIPTOR Pacemaker, Artificial EXPLODE ALL TREES610 #21 defibrillator or "electric countershock"):TI,AB,KY2671 #22 ("cardiac resynchronisaton" or " cardiac pacing"):TI,AB,KY1050 #23 MESH DESCRIPTOR Oligonucleotides, Antisense EXPLODE ALL TREES50 #24 MESH DESCRIPTOR morpholines EXPLODE ALL TREES1900 #25 cardiomyopathy or cardiac NEAR failure or cardiac NEAR protection or inotropic NEAR agent or diuretic7996 #26 (cardiac NEAR therapy or ataluren or ptc124 or antisense NEAR oligonucleotide or morpholino):TI,AB,KY3173 #27 #9 or #10 OR #11 OR #12 OR #13 OR #14 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #25 or #2638015 #28 #8 AND #2764
Appendix 3. MEDLINE OvidSP search strategy
Database: Ovid MEDLINE(R) Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R) <1946 to Present> Ovid MEDLINE(R) Daily Update October 13, 2017 Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 randomized controlled trial.pt. (496904) 2 controlled clinical trial.pt. (99253) 3 randomized.ab. (433409) 4 placebo.ab. (202740) 5 drug therapy.fs. (2114500) 6 randomly.ab. (298737) 7 trial.ab. (457112) 8 groups.ab. (1845391) 9 or/1‐8 (4369043) 10 exp animals/ not humans.sh. (4677556) 11 9 not 10 (3778961) 12 muscular dystrophies/ or muscular dystrophy, duchenne/ (19216) 13 (duchenne adj5 dystroph$).tw. (10171) 14 (becker adj5 dystroph$).tw. (1966) 15 dystrophinopath$.mp. (657) 16 dystrophin/ge (3234) 17 xldcm.tw. (9) 18 x linked dilated cardiomyopathy.tw. (81) 19 or/12‐18 (22625) 20 cardiomyopathies/ or cardiomyopathy, dilated/ (42029) 21 heart failure, congestive/ or myocardial diseases/ (134371) 22 Heart Failure/ (110151) 23 cardiac protection.mp. (734) 24 ventricular dilation.mp. (1667) 25 exp Angiotensin‐Converting Enzyme Inhibitors/ (44109) 26 exp Calcium Channel Blockers/ (83899) 27 exp Adrenergic beta‐Antagonists/ (87190) 28 exp Cardiotonic Agents/ (212589) 29 exp Diuretics/ (81986) 30 Heart Transplantation/ (33069) 31 Pacemaker, Artificial/ (26148) 32 defibrillators/ or defibrillators, implantable/ (17140) 33 Electric Countershock/ (14690) 34 cardiac resynchronisation.mp. (550) 35 Cardiac Pacing, Artificial/ (21708) 36 exp Oligonucleotides, Antisense/ (15589) 37 exp Morpholines/ (24191) 38 (cardiomyopath$ or (cardiac adj1 failure) or (cardiac adj1 protection) or (inotropic adj1 agent$1) or diuretic$1 or (cardiac adj1 transplant$)).tw. (123920) 39 (pacemaker or (resynchronisation adj1 therap$) or ataluren or ptc124 or (antisense adj1 oligonucleotid$) or morpholino).tw. (46306) 40 or/20‐39 (770455) 41 11 and 19 and 40 (367) 42 remove duplicates from 41 (308)
Appendix 4. Embase (OvidSP) search strategy
Database: Embase <1980 to 2017 Week 41> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 crossover‐procedure/ (53437) 2 double‐blind procedure/ (140776) 3 randomized controlled trial/ (471914) 4 single‐blind procedure/ (29732) 5 (random$ or factorial$ or crossover$ or cross over$ or cross‐over$ or placebo$ or (doubl$ adj blind$) or (singl$ adj blind$) or assign$ or allocat$ or volunteer$).tw. (1803990) 6 or/1‐5 (1895784) 7 exp animals/ (23302688) 8 exp humans/ (18903668) 9 7 not (7 and 8) (4399020) 10 6 not 9 (1711898) 11 limit 10 to (conference abstracts or embase) (1434393) 12 muscular dystrophy/ or becker muscular dystrophy/ or duchenne muscular dystrophy/ or dystrophinopathy/ (27272) 13 (xldcm or x linked dilated cardiomyopathy).tw. (85) 14 (duchenne adj5 dystroph$).tw. (11864) 15 (becker adj5 dystroph$).tw. (2220) 16 dystrophinopath$.mp. (927) 17 dystrophin/ (8504) 18 or/12‐17 (30754) 19 cardiomyopathy/ or congestive cardiomyopathy/ (71089) 20 congestive heart failure/ or heart failure/ (257647) 21 myocardial disease/ (5396) 22 heart protection/ (37518) 23 heart dilatation/ (6591) 24 (cardiac protection or ventricular dilation or heart failure or cardiac failure or ventricular dilation or cardiomyopath$).mp. (410670) 25 exp dipeptidyl carboxypeptidase inhibitor/ (158301) 26 exp calcium channel blocking agent/ (203204) 27 exp beta adrenergic receptor blocking agent/ (252354) 28 inotropic agent/ (11044) 29 diuretic agent/ (71583) 30 heart transplantation/ (47005) 31 sinus node/ (7754) 32 defibrillator/ (22735) 33 cardiac resynchronization therapy/ (15287) 34 ataluren/ (504) 35 antisense oligonucleotide/ (16745) 36 (avi or morpholino).mp. (24308) 37 (cardiomyopath$ or (cardiac adj1 failure) or (cardiac adj1 protection) or (inotropic adj1 agent$1) or diuretic$1 or (cardiac adj1 transplant$) or pacemaker or (resynchronisation adj1 therap$) or ataluren or ptc124 or (antisense adj1 oligonucleotid$) or morpholino).tw. (214658) 38 or/19‐37 (1014796) 39 11 and 18 and 38 (192) 40 remove duplicates from 39 (184)
Appendix 5. US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov search strategy
Advanced search
Condition or disease: muscular dystrophy
Other terms: heart
Applied filter: interventional
Appendix 6. World Health Organization International Clinical Trials Registry Platform search strategy
Duchenne AND heart OR Becker AND heart OR dystrophy AND heart
Data and analyses
Comparison 1. Prophylactic perindopril versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cardiac function (number of participants with ejection fraction < 45%) (3 years) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 2. Lisinopril versus losartan.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cardiac function (ejection fraction) (1 year) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
2.1. Analysis.
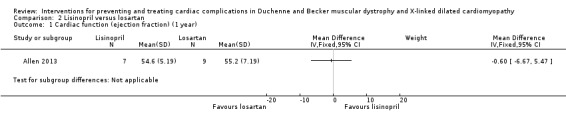
Comparison 2 Lisinopril versus losartan, Outcome 1 Cardiac function (ejection fraction) (1 year).
2.2. Analysis.
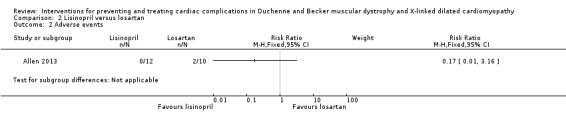
Comparison 2 Lisinopril versus losartan, Outcome 2 Adverse events.
Comparison 3. Idebenone versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cardiac function (change in fractional shortening) (1 year) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2 Cardiac function (change in LVEF) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3 Cardiac function (change in peak systolic radial strain in left ventricular lateral wall segments) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4 Cardiac function (change in systolic radial strain rate left ventricular inferolateral wall) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5 Peak systolic longitudinal strain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Left ventricle lateral mid | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | ‐5.0 [‐10.61, 0.61] |
| 5.2 Left ventricle lateral apex | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 1.30 [‐4.47, 7.07] |
| 5.3 Left ventricle lateral basal | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 4.5 [‐1.81, 10.81] |
| 6 Peak systolic longitudinal strain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 Interventricular septum mid | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | 0.80 [‐6.47, 8.07] |
| 6.2 Interventricular septum apex | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | ‐3.10 [‐13.02, 6.82] |
| 6.3 Interventricular septum basal | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | ‐1.70 [‐6.93, 3.53] |
| 7 Peak systolic longitudinal strain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 Right ventricle apex | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | ‐3.80 [‐14.73, 7.13] |
| 7.2 Right ventricle basal | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | ‐5.0 [‐18.19, 8.19] |
| 8 Global left ventricular functioning | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 Cardiac index | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 0.1 [‐0.75, 0.95] |
| 8.2 Cardiac output | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 0.6 [‐0.33, 1.53] |
3.1. Analysis.
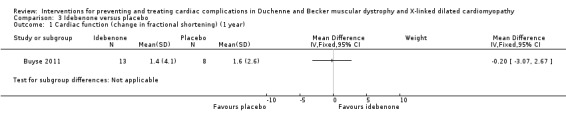
Comparison 3 Idebenone versus placebo, Outcome 1 Cardiac function (change in fractional shortening) (1 year).
3.2. Analysis.
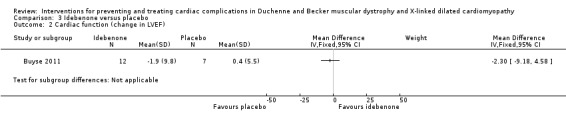
Comparison 3 Idebenone versus placebo, Outcome 2 Cardiac function (change in LVEF).
Comparison 4. Eplerenone versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cardiac function ‐ change (decline) in left ventricular strain (baseline to 6 months) | Other data | No numeric data | ||
| 2 Cardiac function ‐ change (decline) in left ventricular strain (baseline to 12 months) | Other data | No numeric data | ||
| 3 Cardiac function (change in LVEF) (baseline to 6 months) | Other data | No numeric data | ||
| 4 Cardiac function (change in LVEF) from baseline to 12 months | Other data | No numeric data | ||
| 5 Change in size of metabolically abnormal areas of myocardium (baseline to 6 months) | Other data | No numeric data | ||
| 6 Change in size of metabolically abnormal areas of myocardium (baseline to 12 months | Other data | No numeric data | ||
| 7 Adverse events | 1 | 42 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.02, 8.48] |
4.1. Analysis.
Comparison 4 Eplerenone versus placebo, Outcome 1 Cardiac function ‐ change (decline) in left ventricular strain (baseline to 6 months).
| Cardiac function ‐ change (decline) in left ventricular strain (baseline to 6 months) | |||||
|---|---|---|---|---|---|
| Study | Eplerenone (mean) | SD | Placebo (mean) | SD | P value |
| Raman 2014 | 0.84% | 2.68 | 0.38% | 2.56 | 0.602 |
4.2. Analysis.
Comparison 4 Eplerenone versus placebo, Outcome 2 Cardiac function ‐ change (decline) in left ventricular strain (baseline to 12 months).
| Cardiac function ‐ change (decline) in left ventricular strain (baseline to 12 months) | |||||
|---|---|---|---|---|---|
| Study | Eplerenone (median) | IQR | Placebo (median) | IQR | P value |
| Raman 2014 | 1·0% | 0·3 to −2·2 | 2·2% | 1·3 to −3·1 | 0·020 |
4.3. Analysis.
Comparison 4 Eplerenone versus placebo, Outcome 3 Cardiac function (change in LVEF) (baseline to 6 months).
| Cardiac function (change in LVEF) (baseline to 6 months) | |||||
|---|---|---|---|---|---|
| Study | Eplerenone (median) | IQR | Placebo (median) | IQR | P value |
| Raman 2014 | 0% | −3.8 to 4.0 | 1.0% | −5.0 to 2.1 | 0.474 |
4.4. Analysis.
Comparison 4 Eplerenone versus placebo, Outcome 4 Cardiac function (change in LVEF) from baseline to 12 months.
| Cardiac function (change in LVEF) from baseline to 12 months | |||||
|---|---|---|---|---|---|
| Study | Eplerenone (median) | IQR | Placebo (median) | IQR | P value |
| Raman 2014 | −1·8% | −2·9 to 6·0 | −3·7% | −10·8 to 1·0 | 0·032 |
4.5. Analysis.
Comparison 4 Eplerenone versus placebo, Outcome 5 Change in size of metabolically abnormal areas of myocardium (baseline to 6 months).
| Change in size of metabolically abnormal areas of myocardium (baseline to 6 months) | |||||
|---|---|---|---|---|---|
| Study | Eplerenone (mean) | SD | Placebo (mean) | SD | P value |
| Raman 2014 | −2% | 6 | 4% | 6 | 0.034 |
4.6. Analysis.
Comparison 4 Eplerenone versus placebo, Outcome 6 Change in size of metabolically abnormal areas of myocardium (baseline to 12 months.
| Change in size of metabolically abnormal areas of myocardium (baseline to 12 months | |||||
|---|---|---|---|---|---|
| Study | Eplerenone (median) | IQR | Placebo (median) | IQR | P value |
| Raman 2014 | −1% | −6 to 3 | −3% | −5 to 4 | > 0·999 |
4.7. Analysis.
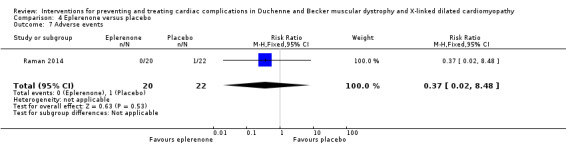
Comparison 4 Eplerenone versus placebo, Outcome 7 Adverse events.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Allen 2013.
| Methods | Randomised, parallel group, double‐blind, multicentre study | |
| Participants | 23 boys with newly diagnosed DMD cardiomyopathy of any age Median age (range) in years: lisinopril 12.5 (10 to 21); losartan 15.5 (7 to 27) Concomitant corticosteroids, beta‐blockers, or both, were allowed Inclusion criteria: Clinical course consistent with DMD, proven mutation of DMD gene or muscle dystrophin levels < 5% on muscle biopsy, Doppler echocardiogram with ejection fraction < 55%, ability to co‐operate with testing Lisinopril ≤ 5 mg, losartan ≤ 25 mg, or enalapril ≤ 5 mg treatment allowed provided 2 weeks washout and LVEF parameters acceptable (see exclusion criteria) Exclusion criteria: Current lisinopril ≤ 5 mg, losartan ≤ 25 mg, or enalapril ≤ 5 mg treatment (no washout) Ejection fraction ≥ 55% or ≤ 40% after at least 2 weeks' washout, of above drugs Skeletal deformities or pulmonary anatomical variants that precluded consistent echocardiography measurements |
|
| Interventions | ACE inhibitor (lisinopril) 0.07 mg/kg (5 mg/day) (N = 12) ARB (losartan) 0.7 mg/kg (25 mg/day) (N = 10) |
|
| Outcomes | Echocardiography at baseline and at 3 subsequent visits at 4‐monthly intervals (4 months, 8 months, 12 months) | |
| Funding sources | Duchenne muscular dystrophy Clinical Research Network grant from the Muscular Dystrophy Association USA | |
| Declarations of interest | No competing interests | |
| Notes | ClinicalTrials.gov NCT01982695 Location: 5 sites in US |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation performed by the Nationwide Children’s Hospital investigational drug pharmacy. Siblings were randomised to the same treatment arm. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The capsules of ACE inhibitor and ARB were identical and the participants were not informed of the treatment they were taking. Siblings were randomised to the same treatment arm to reduce the risk of unblinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study assessors blinded until the termination of the study |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Of 23 participants enrolled, 1 (losartan group) immediately withdrew, 2 were withdrawn due to low ejection fraction and urticaria, respectively, 3 had only 3 echocardiographs performed due to termination of funding |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting for efficacy outcomes. Methods for recording adverse events described but no details reported (other than withdrawals resulting from adverse events) |
| Other bias | High risk | None identified Concomitant medication: 0 in the lisinopril group and 2 in the losartan group receiving beta‐blockers; 8 in the lisinopril group and 2 in the losartan group receiving corticosteroids |
Buyse 2011.
| Methods | Randomised controlled trial | |
| Participants | 21 boys with DMD aged 8 to 16 years with subclinical cardiomyopathy defined by the presence of reduced radial strain measurements in the postero‐lateral segments of the left ventricular wall on echocardiography Mean age: idebenone 10.8 (SD 1.9); placebo 13.4 (SD 2.1) Corticosteroid users: idebenone 5; placebo 8 Other inclusion criteria: If on chronic corticosteroids and/or cardiac medications (beta‐blockers or diuretics, at stable doses for ≥ 6 months and ≥ 3 months, respectively and during the trial Able to perform "reproducible upper limb quantitative muscle testing" Exclusion criteria: Use of ACE inhibitors, coenzyme Q10, idebenone, creatine, glutamine, oxatomide or any herbal medicines within the last 6 months Symptomatic cardiomyopathy or heart failure LVFS (M mode) < 20% and/or ejection fraction < 40%, previous history or presence of ventricular arrhythmias and significant concomitant illness |
|
| Interventions | Idebenone 150 mg (N = 13) or placebo (N = 8) | |
| Outcomes | Primary: change in peak systolic radial strain in left ventricular inferolateral wall | |
| Funding sources | Santhera Pharmaceuticals | |
| Declarations of interest | 1 author an employee and stockholder of funding company which manufactures idebenone; 2 authors "co‐inventors of relevant patent applications" | |
| Notes |
NCT00654784 Enrollment October 2005 to July 2006; follow‐up until August 2007 Location: Leuven, Belgium |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A list of block randomisation numbers and corresponding treatment numbers was computer generated by a third party |
| Allocation concealment (selection bias) | Low risk | Eligible patients were randomised in a double blind fashion and allocated without further stratification in a 2:1 ratio to receive idebenone or matching placebo |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts and specified number of patients with no data available for end of treatment (placebo = 1, idebenone = 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported |
| Other bias | High risk | The idebenone treated group were older than the placebo group. |
Cittadini 2003.
| Methods | Randomised, double‐blind, parallel‐group (pilot study) | |
| Participants | 6 participants with DMD (8 to 19 years) and 10 participants with BMD (24 to 55 years) with documented cardiac involvement Mean age: DMD 13 years (SD 2); BMD 39 years (SD 3) Background therapy unchanged in all participants DMD or BMD diagnosis biopsy‐confirmed; no other inclusion/exclusion criteria specified |
|
| Interventions | Weekly growth hormone 0.23 mg/kg/week (DMD) and 0.07 mg/kg/week (BMD) Placebo Self‐injected, subcutaneously at bedtime for 3 months |
|
| Outcomes | Hormonal measures, ECG (cardiomyopathic index), ECG cardiomyopathic index (QT‐PQ ratio), echocardiography (M‐mode, 2D and echo‐Doppler), measures of left ventricular size and function, timed function tests ((timed Gowers' manoeuvre, time to climb 4 standard stairs, timed 10 metre walk, and 'dynamic index'). Pulmonary function measures comprised forced vital capacity (FVC), maximal voluntary ventilation, and maximal expiratory pressure | |
| Funding sources | Grant from Telethon | |
| Declarations of interest | None given | |
| Notes | Dates of enrollment and follow‐up not reported Location: Italy |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated to be randomised but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Placebo‐controlled |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators performing the echocardiographs were blind to treatment allocations |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The manuscript did not mention if there were any dropouts or withdrawals |
| Selective reporting (reporting bias) | High risk | The report provided no numerical data for left ventricular mass index, end‐systolic stress and ejection fraction outcomes, which were presented graphically |
| Other bias | High risk | The age range of participants was wide and ranged from 8‐19 years for DMD and 24‐55 years for BMD and they were all taking multiple other treatments for cardiomyopathy BMD: 'background' therapy including fosinopril 20 mg/day to 30 mg/day (ACE inhibitor), warfarin, magnesium supplements, pidolatum, antioxidants (vitamins E, C, glutathione, ubiquinone), furosemide, deflazacort. One participant in each group was also receiving digoxin and amiodarone. All DMD participants were receiving deflazacort, fosinopril and antioxidants (vitamin E, glutathione and ubiquinone) |
Duboc 2005.
| Methods | Double‐blind RCT | |
| Participants | 57 children with genetically proven DMD aged 9.5‐13 years with normal cardiac examination, and radionucleotide LVEF > 55% Age range: 9.5 to 13 years Mean age in years: perindopril 10.7 (SD 1.2); placebo 10.6 (SD 1.2) Baseline LVEF%: perindopril 65.0% (SD 5.5); placebo 65.4% (SD 5.5) Other inclusion criteria: Toleration of a 1 mg test dose of perindopril Systolic BP ≥ 80 mmHg supine, > 70 mmHg sitting Exclusion criteria: Treatment with cardioactive drugs Blood urea nitrogen > 7 mmol/L Contraindications to ACE inhibitor therapy |
|
| Interventions | Perindopril 2 mg to 4 mg day for 3 years (N = 28) Placebo (N = 29) |
|
| Outcomes | Reduction in mean LVEF via radionuclide ventriculography, clinical data and tolerance of study drug | |
| Funding sources | Grants from French Association Against Myopathies and from Servier Laboratories | |
| Declarations of interest | None declared. One trial author affiliated to Servier Laboratories | |
| Notes | Dates of enrollment and follow‐up not reported Location: 10 sites in France |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Radionuclide ventriculography was analysed by 2 experts blinded to study data |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Experts blinded to study data |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | One patient did not complete phase 1 |
| Selective reporting (reporting bias) | Low risk | All outcomes reported |
| Other bias | Low risk | None identified |
Raman 2014.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled trial | |
| Participants | 42 boys or young men over 7 years of age with DMD Median age in years: eplerenone 14.5 ( IQR12.0 to 18.5); placebo 15.0 (IQR 11.0 to 19.0) DMD diagnosis by mutation analysis or classic phenotypic features Inclusion criteria: Myocardial damage in one or more left ventricular segments (on late gadolinium enhancement) Preserved left ventricular systolic function (EF ≥ 45%) measured by cine cardiac MRI Current ACE inhibitor or ARB therapy Exclusion criteria: MRI‐incompatible implants Severe claustrophobia Allergy to gadolinium contrast Treatments: eplerenone or spironolactone, potassium‐sparing diuretics, recent experimental treatments (within defined period), CYP3A4 strong inhibitors Scheduled surgery carrying risk of adverse events Baseline serum potassium over 5.5 mmol/L |
|
| Interventions | Eplerenone 25 mg orally alternate days for the 1st month, then daily if the serum potassium (K+) concentration remained ≤ 5.5 mmol/L (N = 20) Placebo (N = 22) |
|
| Outcomes | Primary: change in left ventricular circumferential strain from baseline to 12 months Secondary:
|
|
| Funding sources | BallouSkies, Parent Project for Muscular Dystrophy, US National centre for advancing translational studies and National Institutes of Health. Pfizer supplied active drug and placebo. Funding sources stated to have no involvement in study planning, execution, data analysis or report writing | |
| Declarations of interest | Quote: "The authors were not paid to write this article by a pharmaceutical company or other agency." SVR declared "research support via an institutional agreement from Siemens, one of two manufacturers of MRI equipment used in this study; this company had no active involvement in the study." "Although study drug and matching placebo were obtained from Pfizer Pharmaceuticals, Pfizer had no active involvement in the study" Other authors declared no competing interests. |
|
| Notes | Registered on ClinicalTrials.gov as NCT01521546 Enrolment and follow‐up visits conducted between 3 March 2012 and 1 July 2014 Location: 3 sites in US |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralised computer‐generated randomisation with block sizes of 4 and 6 |
| Allocation concealment (selection bias) | Low risk | Quote: "only the study statistician and the investigational pharmacist had the randomisation assignments" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study personnel and participants were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study personnel and participants were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Complete trial profile |
| Selective reporting (reporting bias) | High risk | All outcomes reported. 12‐month changes from baseline for cardiac measures shown graphically. The report gives the total number of analysable examinations but not numbers for each study visit (baseline, 6 and 12 months); therefore, number of participants providing data for outcomes at each time point is unclear |
| Other bias | High risk | None identified. Participants were receiving many concomitant medications: concomitant ACE inhibitors (18 in eplerenone group; 20 in placebo group). 8 in the eplerenone group and 9 in the placebo group were also taking beta‐blockers and 2 regular furosemide. Other concomitant non‐cardiac medications included: multivitamins, coenzyme Q10, vitamin D, calcium supplements, proton pump inhibitors and corticosteroids. We judged the risk of bias as high as it is not possible to determine whether concomitant therapy confounded the results. |
ACE: angiotensin converting enzyme; ARB: angiotensin receptor blockers; BMD: Becker muscular dystrophy; DMD: Duchenne muscular dystrophy; ECG: echocardiogram; FVC: forced vital capacity; LVEF: left ventricular ejection fraction; LVFS: left ventricular fractional shortening; MRI; magnetic resonance imaging; NCH: Nationwide Children's Hospital.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bushby 2014 | No cardiac outcome measures |
| Duboc 2007 | Non‐randomised study |
| Folkers 1985 | Non‐randomised study |
| Ishikawa 1995 | Non‐randomised study |
| Kajimoto 2006 | Non‐randomised open‐label study |
| Matsumura 2010 | Non‐randomised study |
| Mendell 2013 | No cardiac outcome measures |
| Rhodes 2008 | Non‐randomised open‐label study |
| Voit 2014 | No cardiac outcome measures |
Characteristics of studies awaiting assessment [ordered by study ID]
EUCTR2008‐007236‐18‐IT.
| Methods | Randomised, parallel‐group trial |
| Participants | Adults and children (over 2 years of age) with BMD or DMD and normal baseline cardiac function Inclusion criteria: Immunohystochemical and molecular diagnosis of DMD and BMD No evidence of clinical cardiomyopathy, i.e. no cardiac symptoms, normal ECG, normal 2D‐echocardiography with normal systolic, (left ventricular ejection fraction ≥ 55%, right ventricular ejection fraction ≥ 45% and absence of regional wall motion abnormalities (wall motion score index = 1), and diastolic function Informed consent obtained, able and willing to undergo procedures Exclusion criteria: Cardiological therapy (ACE inhibitors, ARBs or beta‐blockers) Contraindications to carvedilol or ramipril ECG anomalies: in DMD, tall R waves in the right precordial leads with an abnormal RS ratio a deep and narrow Q wave greater than 4 mm over leads I V5 and V6; in BMD, ECG changes suggestive of ischaemic heart disease left bundle‐branch block atrial flutter, fibrillation, ventricular arrhythmias, any degree of atrioventricular block and left ventricular hypertrophy In BMD, hypertension and valvular heart disease (other than trivial) Ventilatory assistance Systolic and/or diastolic dysfunction detected by 2D‐echocardiography Contraindications to cardiac MRI (including any history of claustrophobia) Renal failure, even mild Concomitant steroid therapy allowable |
| Interventions | Carvedilol 6.25 mg, oral Ramipril 2.5 mg, oral |
| Outcomes | Primary: left ventricular ejection fraction, systolic and diastolic left ventricular volumes, late gadolinium enhancement (as a quantitative dimension) and ultrasonic tissue characterisation values |
| Notes | Prematurely ended 20 June 2013. ICTRP record states no results available ‐ to be confirmed before probable exclusion |
Leung 2014.
| Methods | Single‐centre, randomised, double‐blind, placebo‐controlled trial |
| Participants | 20 adults (≥ 15 years) with DMD (defined as absent dystrophin staining on muscle biopsy or a dystrophin mutation predictive of the Duchenne phenotype on genetic testing) and cardiomyopathy Inclusion criteria: Ejection fraction ≤ 45%, concurrent use of an ACE inhibitor or ARB for ≥ 3 months at unchanging dose, unchanged beta‐blocker or corticosteroid dosing for 3 months Exclusion criteria: Contraindications to MRI, implantable cardiac devices, frequent cardiac arrhythmia, hereditary retinal disorders, bleeding disorders, a systolic blood pressure ≤ 85 mmHg or lower, stage 4 or 5 renal failure, active tobacco use, concurrent use of nitrates, alpha‐adrenergic receptor blockers, or phosphodiesterase inhibitors |
| Interventions | Sildenafil (20 mg, 3 times daily) (N = 10) Placebo (N = 10) Treatment duration: 6 months |
| Outcomes | Primary: change in left ventricular end‐systolic volume on cardiac MRI Secondary: cardiac measures (end‐systolic, end‐diastolic, and stroke volumes, left ventricular myocardial mass, and ejection fraction), skeletal muscle function (grip and pinch strength using hand‐held dynamometry), forced vital capacity, quality of life (Short‐Form 36 Health Survey (SF‐36) and Individualized Neuromuscular Quality of Life Questionnaire (INQoL)) and adverse events |
| Notes | ClinicalTrials.gov: NCT01168908 Enrollment stopped early for harm (number experiencing ≥10% increase in LVESV while taking sildenafil) |
Salehi 2017.
| Methods | Randomised, double‐blind, placebo‐controlled trial |
| Participants | Report states: "Children aged 6 to 10 years old were enrolled in a study in which Duchenne muscular dystrophy (DMD) was diagnosed and approved in them by DNA analysis and muscle biopsy (quadriceps or biceps)" Mean age (SD) in years: coenzyme Q10 8.9 (1.7); placebo 8.6 (1.4) Exclusion criteria: Confirmed or suspected heart disease Other concomitant illness Taking herbal medicine, vitamins or enzymes Arrhythmia on ECG 'Inappropriate view' in echocardiography |
| Interventions | Coenzyme Q10 (N = 12) Placebo (N = 13) Treatment duration: 6 months |
| Outcomes | Myocardial performance index |
| Notes | Location: Tehran, Iran Dates: February 2013 to 2015 Iranian Clinical Trials Registry number: IRCT2015070223018N1 The report suggests that the trial included both male and female participants and describes participants as "suspected of DMD". |
ACE inhibitor: angiotensin converting enzyme inhibitor; ARB: angiotensin receptor blocker; BMD: Becker muscular dystrophy; DMD: Duchenne muscular dystrophy; ECG: electrocardiograph; MRI: magnetic resonance imaging
Characteristics of ongoing studies [ordered by study ID]
FOR‐DMD 2012.
| Trial name or title | Finding the optimum regimen for Duchenne muscular dystrophy (FOR‐DMD) |
| Methods | Randomised, parallel‐assignment, quadruple‐blind (participant, care provider, investigator and outcomes assessor), phase 3 trial |
| Participants | Boys with DMD between 4 and 7 years old |
| Interventions | Experimental (3 groups): daily prednisone (0.75 mg/kg/day); intermittent prednisone (0.75 mg/kg/day, 10 days on, 10 days off) daily deflazacort (0.9 mg/kg/day) for 36 to 60 months |
| Outcomes | Cardiac function monitored by trans‐thoracic echocardiogram and 12‐lead ECG was a secondary outcome. Function was categorised as: normal, abnormal but not clinically significant, and abnormal and clinically significant. The earliest definite, echo‐detectable impairment of left ventricular function was defined as ejection fraction < 55% and/or fractional shortening < 28%. Time frame: 1 to 3 months prior to the baseline visit, then every 2 years to the age of 10 years, and annually thereafter or at the onset of cardiac signs and symptoms and the year 3 visit The primary outcome combined FVC, time to stand (log‐transformed) and participant/parent satisfaction with treatment. Other outcomes included timed function tests, range of movement at the ankle, regimen tolerance, adverse events, and quality of life |
| Starting date | January 2013 (estimated primary completion date October 2019) |
| Contact information | Principal Investigator: Robert Griggs, MD, Professor of Neurology, University of Rochester |
| Notes |
ISRCTN50395346.
| Trial name or title | A double‐blind randomised multi‐centre, placebo‐controlled trial of combined angiotensin converting enzyme‐inhibitor and beta‐blocker therapy in preventing the development of cardiomyopathy in genetically characterised males with Duchenne muscular dystrophy without echo‐detectable left ventricular dysfunction |
| Methods | Double‐blind, randomised, multicentre, placebo‐controlled trial |
| Participants | Boys aged 7 to 12 years with genetically confirmed DMD and normal left ventricular function on trans‐thoracic echocardiography |
| Interventions | Perindopril 2 mg/bisoprolol 1.25 mg or placebo for the 1‐month run‐in period
Perindopril 4 mg/bisoprolol 2.5 mg or placebo for the remainder of the trial 2‐year treatment period. Follow‐up period up to 60 months. |
| Outcomes | Primary outcome: change from baseline in left ventricular ejection fraction measured by Simpson's biplane disk method, after a minimum of 2 years' active treatment or placebo Similar comparisons performed for parameters of left ventricular end‐systolic volume and wall motion index Secondary endpoints: death from any cause, development of symptoms and signs of congestive cardiac failure, and sufficient objective deterioration in cardiac function without symptoms to make continued placebo therapy unethical |
| Starting date | September 2007 |
| Contact information | John Bourke, Freeman Hospital, Newcastle upon Tyne, UK |
| Notes |
NCT00606775.
| Trial name or title | The preventive efficacy of carvedilol on cardiac dysfunction in Duchenne muscular dystrophy |
| Methods | Randomised, parallel‐assignment, open‐label, phase 4 trial |
| Participants | Boys and men with DMD, aged 8 to 45 years |
| Interventions | Carvedilol 2.5 mg/day to 5 mg/day No intervention |
| Outcomes | Primary outcome: suppression of minor cardiac damage indicated as elevation of plasma cardiac troponin I (time frame: 2 years) Secondary outcomes: left ventricular function deterioration assessed by echocardiography, in‐hospital mortality for cardiac dysfunction, in‐hospital mortality for any cause, overall mortality (time frame: 5 years) |
| Starting date | December 2007 |
| Contact information | Principal Investigator: Takao Nishizawa, Department of Cardiology, Nagoya University Graduate School of Medicine |
| Notes |
NCT00819845.
| Trial name or title | Ramipril versus carvedilol in Duchenne and Becker patients |
| Methods | Randomised, parallel‐assignment, open‐label, phase 4 trial |
| Participants | Males aged 2 to 45 years with Immunohistochemical and molecular diagnosis of BMD or DMD |
| Interventions | Carvedilol Ramipril |
| Outcomes | Primary outcome: left ventricular ejection fraction, systolic and diastolic left ventricular volumes and late gadolinium enhancement (LGE, as a quantitative measure) detected by MRI and myocardial ultrasound tissue characterisation (UTC) data by echocardiography (time frame: 1 year) Secondary outcome: prevalence of LGE in DMD and BMD, the effects of pharmacological therapy both on LGE evolution and myocardial UTC analysis (time frame: 1 year) |
| Starting date | December 2008 |
| Contact information | Principal Investigator: Vincenzo Giglio, MD, PhD Uildm, Rome |
| Notes |
NCT01126697.
| Trial name or title | Clinical trial of coenzyme Q10 and lisinopril in muscular dystrophies |
| Methods | Randomised, factorial assignment, open‐label, phase 2 or 3 trial |
| Participants | 120 participants aged 8 and above with DMD, BMD, or autosomal recessive limb‐girdle muscular dystrophy (specifically 2C‐2F and 2I) without clinical cardiac symptoms |
| Interventions | Participants randomised to 1 of 4 arms: coenzyme Q10 alone, lisinopril alone, coenzyme Q10 and lisinopril, or no study medication |
| Outcomes | Primary outcome: myocardial performance index (time frame: every 6 months) |
| Starting date | February 2010 |
| Contact information | Cooperative International Neuromuscular Research Group |
| Notes |
NCT01350154.
| Trial name or title | Effect of modulating the nNOS system on cardiac, muscular and cognitive function in Becker muscular dystrophy patients |
| Methods | Randomised, cross‐over assignment, quadruple masking (participant, care provider, investigator and outcomes assessor), phase 2 trial |
| Participants | Males aged 18 years to 80 years with BMD and an established deficiency in muscular content of nNOS protein |
| Interventions | Participants will receive 4 weeks of either sildenafil or placebo with a 2‐week washout period between treatments |
| Outcomes | Primary outcomes were measured as the difference between treatment and placebo groups in the changes between baseline and 4 weeks in: handgrip test with concomitant ultrasound brachial artery flow measurement; resting cardiac end‐diastolic volume measured by MRI; cerebrovascular reactivity to CO2 inhalation and finger stimulation measured by blood oxygen level‐dependent functional MRI (BOLD fMRI); and cognitive function measured by the Cambridge Neuropsychological Test Automated Battery (CANTAB) |
| Starting date | November 2011 |
| Contact information | Neuromuscular Clinic and Research Unit, Department of Neurology, Rigshospitalet, Copenhagen, Denmark |
| Notes |
NCT01648634.
| Trial name or title | Nebivolol for the prevention of left ventricular systolic dysfunction in patients with Duchenne muscular dystrophy (NEBIDYS) |
| Methods | Randomised, parallel‐assignment, double‐blind, phase 3 trial |
| Participants | Boys aged 10 years to 15 years with genetically proven DMD |
| Interventions | Nebivolol Placebo |
| Outcomes | Primary outcome: left ventricular systolic dysfunction Secondary outcomes: right ventricular ejection fraction, N‐terminal pro‐brain natriuretic peptide (NT‐ProBNP), left ventricular dysfunction, hospitalisations, mortality |
| Starting date | February 2012 |
| Contact information | Principal Investigator: Henri‐Marc BECANE, Armand Trousseau Hospital |
| Notes |
NCT02354352.
| Trial name or title | Therapeutic potential for aldosterone inhibition in Duchenne muscular dystrophy |
| Methods | Randomized, single group assignment, quadruple‐blind (participant, care provider, investigator, outcomes assessor) |
| Participants | Boys 7 years and older with DMD with a left ventricular ejection fraction ≥ 45% (+/‐5%) by clinically‐acquired echocardiography, nuclear scan or cardiac MRI done within 2 weeks of enrollment |
| Interventions | Eplerenone (one 50 mg capsule by mouth once daily for 12 months) Spironolactone (one 50 mg capsule by mouth once daily for 12 months) |
| Outcomes | Primary outcome: left ventricular strain at 12 months Secondary outcomes: FVC, muscle injury blood biomarkers |
| Starting date | January 2015 |
| Contact information | Principal investigator: Subha Raman, Ohio State University |
| Notes |
NCT02432885.
| Trial name or title | Myocardial fibrosis progression in Duchenne and Becker muscular dystrophy ‐ ACE inhibitor therapy trial |
| Methods | Randomised, parallel‐assignment, open‐label, phase 3 trial |
| Participants | Male and female participants aged 6 years and older, with biopsy‐proven BMD or DMD |
| Interventions | Enalapril (ACE inhibitor) up to 20 mg twice daily Placebo |
| Outcomes | Primary outcome: quantitative myocardial fibrosis by cardiac MRI in patients with and without ACE inhibitor therapy Secondary outcome: specific genetic mutations as predictors of cardiac involvement Time frame: 2 years |
| Starting date | June 2009 |
| Contact information | Principal Investigator: Carlos E Rochitte, Heart Institute, University of Sao Paulo Medical School |
| Notes |
NCT02485938.
| Trial name or title | HOPE‐Duchenne (Halt cardiomyOPathy progrEssion in Duchenne) (HOPE) |
| Methods | Randomised, parallel‐assignment, open‐label trial |
| Participants | Male participants aged 12 years and over with cardiomyopathy secondary to DMD |
| Interventions | Participants randomised in a 1:1 manner to either intracoronary infusion of CAP‐1002 in 3 coronary arteries supplying the 3 major cardiac territories of the left ventricle of the heart (anterior, lateral, inferior/posterior) or usual care. In the active treatment arm, all 3 major cardiac territories will be treated (infused) during a single procedure in an open‐label fashion. |
| Outcomes | Primary outcome: safety and tolerability composite of CAP‐1002 will be established by summaries of the occurrence of changes in coronary blood flow events, major cardiac events, laboratory assessments, vital signs, physical examination, electrocardiograph, and the occurrence of major adverse events (time frame: 72 hours post infusion) Secondary outcomes: cardiac MRI, functional composite outcome, quality of life composite outcome, biomarkers (time frame: 12 months) |
| Starting date | January 2016 |
| Contact information | Principal Investigator: John L Jefferies, MD, MPH Children's Hospital Medical Center, Cincinnati Study Director: Deborah Ascheim, MD Capricor Inc. |
| Notes |
NCT03340675.
| Trial name or title | Oral ifetroban in subjects with Duchenne muscular dystrophy (DMD) |
| Methods | Randomised, placebo‐controlled, parallel‐assignment, double‐blind, phase 2 trial |
| Participants | Males aged 7 years and older with the diagnosis of DMD (phenotype consistent with DMD and either positive genotype, first degree relative with positive genotype, or confirmatory muscle biopsy) |
| Interventions | Oral ifetroban low dose Oral ifetroban high dose Placebo Administration: once daily for 12 months |
| Outcomes | Primary outcome: incidence of treatment‐emergent adverse events (safety and tolerability) (over 12 months) Secondary outcomes: pharmacokinetics (day 0 and day 7), change (from baseline to 12 months) in left ventricular ejection fraction, change from baseline in pulmonary function, change from baseline in quality of life |
| Starting date | November 2018 |
| Contact information | Sponsors and collaborators: Cumberland Pharmaceuticals, Vanderbilt University Medical Center. |
| Notes |
NCT03406780.
| Trial name or title | A study of CAP‐1002 in ambulatory and non‐ambulatory patients with Duchenne muscular dystrophy (HOPE‐2) |
| Methods | Randomised, placebo‐controlled, parallel‐assignment, quadruple‐blind (participant, care provider, investigator, outcomes assessor) |
| Participants | Male participants, 10 years or older with genetically confirmed DMD, reduced upper arm strength, reduced ability to walk or run (if ambulatory), having received at least 12 months' treatment with corticosteroids at a stable dose for at least 6 months. Exclusion criteria includes ejection fraction < 35% |
| Interventions | CAP‐1002 (cardiosphere‐derived cells (CDCs)) 150 million CDCs via intravenous infusion every 3 months on 4 occasions Placebo intravenous infusions on same schedule |
| Outcomes | Primary outcome: change in the mid‐level (elbow) dimension of the Performance of the Upper Limb (PUL) (time frame: 12 months) Secondary outcomes: change in mid‐level (elbow) dimension of the PUL (time frame: 3, 6, and 9 months), change in regional systolic left ventricular wall thickening as assessed by cardiac MRI (time frame: months 6 and 12) |
| Starting date | April 2018 |
| Contact information | Brian Fedor, Capricor Inc.; HOPE‐2@capricor.com |
| Notes |
NCT03439670.
| Trial name or title | A study to assess the efficacy and safety of vamorolone in boys with Duchenne muscular dystrophy (DMD) |
| Methods | Randomized, parallel group, placebo and active‐controlled, quadruple‐blind (participant, care provider, investigator, outcomes assessor) |
| Participants | Boy aged 4 to 7 years old with confirmed diagnosis of DMD |
| Interventions | Vamorolone, orally at 2.0 mg/kg and 6.0 mg/kg Prednisone 0.75 mg/kg/day Placebo Duration of treatment: 24 weeks |
| Outcomes | Primary outcomes: muscle function; body size as measured by body mass index (time frame 24 weeks) Secondary outcomes: cardiac function (measured by ECG (week 12, week 24, week 40, week 48), 2‐D echocardiogram (week 24, week 48)); treatment‐emergent adverse effects; multiple safety measures; multiple efficacy outcomes |
| Starting date | June 2018 |
| Contact information | Andrea Smith: asmith@trinds.com |
| Notes |
ACE inhibitor: angiotensin converting enzyme inhibitor; BMD: Becker muscular dystrophy; DMD: Duchenne muscular dystrophy; ECG: electrocardiogram; FVC: forced vital capacity; MRI: magnetic resonance imaging
Differences between protocol and review
Where trials reported multiple time points over the 6 months specified we reported the longest time point.
We further clarified our outcomes for inclusion in the 'Summary of findings' table, deciding to report ejection fraction or fractional shortening as measures of cardiac improvement as these are most widely understood and used (Quinlivan 2011 (amended); Quinlivan 2012).
We included a PRISMA flow chart to illustrate the study selection process.
Contributions of authors
All three authors reviewed and agreed on inclusion criteria and studies for inclusion. RQ and JB prepared the manuscript. BT prepared the data extraction forms and completed the tables. All three authors agreed the contents of the manuscript prior to publication.
Sources of support
Internal sources
None, Other.
External sources
None, Other.
-
National Institute of Health Research (NIHR), UK.
Ros Quinlivan is Joint Co‐ordinating Editor of Cochrane Neuromuscular, the position is funded by the National Institute for Health Research via Cochrane Infrastructure funding to Cochrane Neuromuscular
Declarations of interest
John Bourke is a Consultant cardiologist and principal investigator for a multicentre, placebo‐controlled trial for cardiac protection in DMD
Teofila Bueser is a specialist nurse and manages patients with DMD, BMD and X‐linked muscular dystrophy. She has no conflicts of interest.
Dr Quinlivan has received honoraria from PTC bio for teaching on ataluren and Santhera for teaching on idebenone. She is Joint Co‐ordinating Editor of Cochrane Neuromuscular. She was not involved in the editorial process for this review.
Edited (no change to conclusions)
References
References to studies included in this review
Allen 2013 {published data only}
- Allen HD, Flanigan KM, Thrush PT, Viollet‐Callendret L, Dvorchik I, Yin H, et al. A randomized, double‐blind trial of lisinopril and losartan for the treatment of cardiomyopathy in Duchenne muscular dystrophy. PLOS Currents Muscular Dystrophy 2013;5:1‐13. [PMCID: PMC3871420; PUBMED: 24459612] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01982695. Cardiomyopathy in DMD: lisinopril vs. losartan [Compare efficacy of the angiotensin converting enzyme inhibitor (ACEI) lisinopril with angiotensin II receptor antagonist losartan (ARB) for the cardiomyopathy of Duchenne muscular dystrophy]. clinicaltrials.gov/ct2/show/study/NCT01982695 First received: 29 October 2013; results first received 14 August 2015.
Buyse 2011 {published data only}
- Buyse GM, Goemans N, Hauwe M, Thijs D, Groot IJM, Schara U, et al. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: results from a randomized placebo‐controlled trial. Neuromuscular Disorders 2011;21:396‐405. [PUBMED: 21435876] [DOI] [PubMed] [Google Scholar]
Cittadini 2003 {published data only}
- Cittadini A, Ines Comi L, Longobardi S, Rocco Petretta V, Casaburi C, Passamano L, et al. A preliminary randomized study of growth hormone administration in Becker and Duchenne muscular dystrophies. European Heart Journal 2003;24(7):664‐72. [PUBMED: 12657225] [DOI] [PubMed] [Google Scholar]
Duboc 2005 {published data only}
- Duboc D, Meune C, Lerebours G, Devaux J‐Y, Vaksmann G, Bécane H‐M. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. Journal of the American College of Cardiology 2005;45(6):855‐7. [PUBMED: 15766818] [DOI] [PubMed] [Google Scholar]
Raman 2014 {published data only}
- Raman S, Hor K, Mazur W, Halnon N, Kissel J, He X, et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurology 2015;14(2):153‐61. [PUBMED: 25554404] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Bushby 2014 {published data only}
- Bushby K, Finkel R, Wong B, Barohn R, Campbell C, Comi GP, et al. PTC124‐GD‐007‐DMD STUDY GROUP. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle & Nerve 2014;50(4):477‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Duboc 2007 {published data only}
- Duboc D, Meune C, Pierre B, Wahbi K, Eymard B, Toutain T, et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years follow‐up. American Heart Journal 2007;154(3):596‐602. [PUBMED: 17719312] [DOI] [PubMed] [Google Scholar]
Folkers 1985 {published data only}
- Folkers K, Wolaniuk J, Simonsen R, Morishita M, Vadhanavikit S. Biochemical rationale and the cardiac response of patients with muscle disease to therapy with coenzyme Q10. Proceedings of the National Academy of Sciences of the United States of America 1985;82(13):4513‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ishikawa 1995 {published data only}
- Ishikawa Y, Back J, Ishikawa Y, Minami R. A management trial for Duchenne cardiomyopathy. American Journal of Physical Medicine & Rehabilitation 1995;74:345‐50. [PubMed] [Google Scholar]
Kajimoto 2006 {published data only}
- Kajimoto H, Ishigaki K, Okumura K, Tomimatsu H, Nakazawa M, Saito K, et al. Beta‐blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circulation Journal 2006;70(8):991‐4. [DOI] [PubMed] [Google Scholar]
Matsumura 2010 {published data only}
- Matsumura T, Tamura T, Kuru S, Kikuchi Y, Mitsuru K. Carvedilol can prevent cardiac events in Duchenne muscular dystrophy. Internal Medicine 2010;49(14):1357‐63. [DOI] [PubMed] [Google Scholar]
Mendell 2013 {published data only}
- Mendell J, Rodino‐Klapac L, Sahenk Z, Roush K, Bird L, Lowes LP, et al. Eteplirsen Study Group. Eteplirsen for the treatment of DMD. Annals of Neurology 2013;74(5):637‐47. [DOI] [PubMed] [Google Scholar]
Rhodes 2008 {published data only}
- Rhodes J, Margossian R, Darras BT, Colan SD, Jenkins KJ, Geva T, Powell AJ. Safety and efficacy of carvedilol therapy for patients with dilated cardiomyopathy secondary to muscular dystrophy. Pediatric Cardiology 2008;29(2):343‐51. [DOI] [PubMed] [Google Scholar]
Voit 2014 {published data only}
- Voit T, Topaloglu H, Straub V, Muntoni F, Deconinck N, Campion G, et al. Safety and efficacy of Drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised placebo controlled phase II study. Lancet Neurology 2014;13(10):987‐96. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
EUCTR2008‐007236‐18‐IT {published data only}
- EUCTR2008‐007236‐18‐IT. Effects of cardioprotective therapy, carvedilol vs ramipril, in patients affected by Duchenne and Becker muscular dystrophy. Clinical significance and prognostic value of cardiac magnetic resonance study. clinicaltrialsregister.eu/ctr‐search/search?query=eudract_number:2008‐007236‐18 (first received 11 December 2008).
Leung 2014 {published data only}
- Leung DG, Herzka DA, Thompson WR, He B, Bibat G, Tennekoon G, et al. Sildenafil does not improve cardiomyopathy in Duchenne/Becker muscular dystrophy. Annals of Neurology 2014;76(4):541‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01168908. Revatio for heart disease in Duchenne muscular dystrophy and Becker muscular dystrophy (REVERSE‐DBMD). clinicaltrials.gov/show/NCT01168908 (first received 23 July 2010).
Salehi 2017 {published data only}
- IRCT2015070223018N1. Effect of Q10 coenzyme in improving cardiac function [A study on the effect of Q10 coenzyme via tissue doppler method in improving cardiac function in patients with early Duchenne cardiomyopathy aged 6‐10 years]. en.irct.ir/trial/19746 (first received 11 August 2015).
- Salehi F, Zeinaloo A, Riasi HR, Shamloo AS. Effectiveness of Coenzyme Q10 on echocardiographic parameters of patients with Duchenne muscular dystrophy. Electronic Physician 2017;9(3):3896‐3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
FOR‐DMD 2012 {published data only}
- NCT01603407. Finding the optimum regimen for Duchenne muscular dystrophy (FOR‐DMD) [Duchenne muscular dystrophy: double‐blind randomized trial to find optimum steroid regimen]. https://clinicaltrials.gov/ct2/show/NCT01603407 (first received 23 May 2012).
ISRCTN50395346 {published data only}
- ISRCTN50395346. A double‐blind randomised multi‐centre, placebo‐controlled trial of combined angiotensin converting enzyme‐inhibitor and beta‐blocker therapy in preventing the development of cardiomyopathy in genetically characterised males with Duchenne muscular dystrophy without echo‐detectable left ventricular dysfunction. isrctn.com/ISRCTN50395346 (first received 13 August 2007).
NCT00606775 {published data only}
- NCT00606775. The preventive efficacy of carvedilol on cardiac dysfunction in Duchenne muscular dystrophy [Carvedilol for the prevention of minor cardiac damage and cardiac function in Duchenne muscular dystrophy]. clinicaltrials.gov/show/NCT00606775 (first received 5 February 2008).
NCT00819845 {published data only}
- NCT00819845. Ramipril versus carvedilol in Duchenne and Becker patients [Effects of cardioprotective therapy, carvedilol vs ramipril, in patients affected by Duchenne and Becker muscular dystrophy. Clinical significance and prognostic value of cardiac magnetic resonance study]. clinicaltrials.gov/show/NCT00819845 (first received 9 January 2009).
NCT01126697 {published data only}
- NCT01126697. Clinical trial of coenzyme Q10 and lisinopril in muscular dystrophies [PITT0908:clinical trial of coenzyme Q10 and lisinopril in muscular dystrophies]. clinicaltrials.gov/show/NCT01126697 (first received 20 May 2010).
NCT01350154 {published data only}
- NCT01350154. Effect of modulating the nNOS system on cardiac, muscular and cognitive function in Becker muscular dystrophy patients [Does modulation of the nNOS system in patients with muscular dystrophy and defect nNOS signalling affect cardiac, muscular or cognitive function?]. clinicaltrials.gov/show/NCT01350154 (first received 9 May 2011).
NCT01648634 {published data only}
- NCT01648634. Nebivolol for the prevention of left ventricular systolic dysfunction in patients with Duchenne muscular dystrophy (NEBIDYS) [A randomized, double‐blind, placebo‐controlled, multi‐center study to examine the effect of nebivolol, a beta‐blockade drug, for the prevention of ventricular systolic dysfunction in patients with Duchenne muscular dystrophy]. clinicaltrials.gov/show/NCT01648634 (first received 24 July 2012).
NCT02354352 {published data only}
- NCT02354352. Therapeutic potential for aldosterone inhibition in Duchenne muscular dystrophy. https://clinicaltrials.gov/ct2/show/NCT02354352 (first received 21 August 2018).
NCT02432885 {published data only}
- NCT02432885. Myocardial fibrosis progression in Duchenne and Becker muscular dystrophy ‐ ACE inhibitor therapy trial [Myocardial fibrosis progression in Duchenne and Becker muscular dystrophy ‐ angiotensin‐converting‐enzyme (ACE) inhibitor therapy]. clinicaltrials.gov/show/NCT02432885 (first received 4 May 2015).
NCT02485938 {published data only}
- NCT02485938. HOPE‐Duchenne (Halt cardiomyOPathy progrEssion in Duchenne) (HOPE) [A randomized, open‐label study of the safety and efficacy of multi‐vessel intracoronary delivery of allogeneic cardiosphere‐derived cells in patients with cardiomyopathy secondary to Duchenne muscular dystrophy]. clinicaltrials.gov/show/NCT02485938 (first received 30 June 2015).
NCT03340675 {published data only}
- NCT03340675. Oral ifetroban in subjects with Duchenne muscular dystrophy (DMD) [A randomized, double‐blind, placebo‐controlled, multiple dose study to determine the safety, pharmacokinetics and efficacy of oral ifetroban in subjects with Duchenne muscular dystrophy (DMD)]. clinicaltrials.gov/show/NCT03340675 (first received 13 November 2017).
NCT03406780 {published data only}
- NCT03406780. A study of CAP‐1002 in ambulatory and non‐ambulatory patients with Duchenne muscular dystrophy (HOPE‐2) [A phase 2, randomized, double‐blind, placebo‐controlled trial evaluating the safety and efficacy of intravenous delivery of allogeneic cardiosphere‐derived cells in subjects with Duchenne muscular dystrophy]. https://clinicaltrials.gov/ct2/show/NCT03406780 (first received 23 January 2018).
NCT03439670 {published data only}
- NCT03439670. A study to assess the efficacy and safety of vamorolone in boys with Duchenne muscular dystrophy (DMD) [A phase IIb randomized, double‐blind, parallel group, placebo‐ and active‐controlled study with double‐blind extension to assess the efficacy and safety of vamorolone in ambulant boys with Duchenne muscular dystrophy (DMD)]. https://clinicaltrials.gov/ct2/show/NCT03439670 (first received 24 July 2018).
Additional references
Aartsma‐Rus 2013
- Aartsma‐Rus A, Ommen GJ, Kaplan JC. Innovating therapies for muscle diseases. Handbook of Clinical Neurology 2013;113:1497‐501. [DOI] [PubMed] [Google Scholar]
Abdel‐Salam 2014
- Abdel‐Salam Z, Rayan M, Saleh A, Abdel‐Barr MG, Hussain M, Nammas W. I(f) current inhibitor ivabradine in patients with idiopathic dilated cardiomyopathy: impact on the exercise tolerance and quality of life. Cardiology Journal 2014;22(2):227‐32. [DOI] [PubMed] [Google Scholar]
Abraham 2014
- Abraham WT, Aggarwal S, Prabhu SD, Cecere R, Pamboukian SV, Bank AJ, et al. C‐Pulse Trial Group. Ambulatory extra‐aortic counterpulsation in patients with moderate to severe chronic heart failure. JACC: Heart Failure 2014;2(5):526‐33. [DOI] [PubMed] [Google Scholar]
Adorisio 2017
- Adorisio R, Catarutti N, D'Amico A, Chinali M, Baban A, Iorio FS, et al. Heart rate reduction strategy with Ivabradine in reducing acute heart failure in Duchenne dilated cardiomyopathy. European Heart Journal 2017;38(Suppl 1):5275. [Google Scholar]
Andrikopoulos 2013
- Andrikopoulos G, Kourouklis S, Trika C, Tzeis S, Rassias I, Papademetriou C, et al. Cardiac resynchronisation therapy in Becker muscular dystrophy. Hellenic Journal of Cardiology 2013;54(3):227‐9. [PubMed] [Google Scholar]
Angelini 1996
- Angelini C, Fanin M, Freda MP, Martinello F, Miorin M, Melacini P, et al. Prognostic factors in mild dystrophinopathies. Journal of the Neurological Sciences 1996;142(1‐2):70‐8. [DOI] [PubMed] [Google Scholar]
Backman 1992
- Backman E, Nylander E. The heart in Duchenne muscular dystrophy: a non‐invasive study. European Heart Journal 1992;13(9):1239‐44. [DOI] [PubMed] [Google Scholar]
Barber 2013
- Barber B, Andrews J, Lu Z, West N, Meaney J, Price E, et al. Oral corticosteroids and onset of cardiomyopathy in Duchenne muscular dystrophy. Journal of Pediatrics 2013;163(4):1080‐4. [DOI] [PubMed] [Google Scholar]
Becker 1955
- Becker PE, Kiener F. Eine neue X‐chromasomale Muskeldystrophie. Archiv fur Psychiatrie und Nervenkrankheiten 1955;193:427‐48. [DOI] [PubMed] [Google Scholar]
Bies 1992
- Bies RD, Friedman D, Roberts R, Perryman MB, Caskey CT. Expression and localization of dystrophin in human cardiac Purkinje fibres. Circulation 1992;86(1):147‐53. [DOI] [PubMed] [Google Scholar]
Biggar 2006
- Bigggar WD, Harris VA, Eliasoph L, Alman B. Long term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscular Disorders 2006;16(4):249‐55. [DOI] [PubMed] [Google Scholar]
Black 2016
- Black MC, Schumer EM, Rogers M, Trivedi J, Slaughter MS. Sunshine Heart C‐Pulse: device for NYHA III and ambulatory class IV heart failure. Future Cardiolology 2016;12(5):521‐31. [DOI] [PubMed] [Google Scholar]
Bleeker 2006
- Bleeker GB, Kaandorp TA, Lamb HJ, Boersma E, Steendijk P, Roos A, et al. Effect of posterolateral scar tissue on clinical and echocardiographic improvement after cardiac resynchronization therapy. Circulation 2006;113(7):969–76. [DOI] [PubMed] [Google Scholar]
Burnett 2017
- Burnett H, Earley A, Voors AA, Senni M, McMurray JJ, Deschaseaux C, et al. Thirty years of evidence on the efficacy of drug treatments for chronic heart failure with reduced ejection fraction: a network meta‐analysis. Circulation. Heart Failure 2017;10(1):e003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bushby 1991
- Bushby KMD, Thambyayah M, Gardner‐Medwin D. Prevalence and incidence of Becker muscular dystrophy. Lancet 1991;337(8748):1022‐4. [DOI] [PubMed] [Google Scholar]
Bushby 1993
- Bushby K, Goodship JA, Nicholson L, Johnson M, Haggerty ID, Gardner‐Medwin D. Variability in clinical, genetic and protein abnormalities in manifesting carriers of Duchenne and Becker muscular dystrophy. Neuromuscular Disorders 1993;3(1):57‐64. [DOI] [PubMed] [Google Scholar]
Casazza 1988
- Casazza F, Brambilla G, Salvato A, Morandi L, Gronda E, Bonacina E. Cardiac transplantation in Becker muscular dystrophy. Journal of Neurology 1988;235(8):496‐8. [DOI] [PubMed] [Google Scholar]
Cevik 2010
- Cevik C, Nugent K, Perez‐Verdia A, Fish RD. Prophylactic implantation of cardioverter defibrillator in idiopathic non‐ischaemic cardiomyopathy for the primary prevention of death: a narrative review. Clinical Cardiology 2010;33(5):254‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cicoira 2002
- Cicoira M, Zanolla L, Franceschini L, Rossi A, Golia G, Zeni P, et al. Relation of aldosterone 'escape' despite angiotensin‐converting enzyme inhibitor administration to impaired exercise capacity in chronic congestive heart failure secondary to ischaemic or idiopathic dilated cardiomyopathy. American Journal of Cardiology 2002;89(4):403‐7. [DOI] [PubMed] [Google Scholar]
Corrado 2002
- Corrado G, Lissoni A, Beretta S, Terenghi L, Tadeo G, Foglia‐Manzillo G, et al. Prognostic value of echocardiograms, ventricular late potentials, ventricular arrhythmias and left ventricular systolic dysfunction in patients with Duchenne muscular dystrophy. American Journal of Cardiology 2002;89(7):838‐41. [DOI] [PubMed] [Google Scholar]
Danialou 2001
- Danialou G, Comtois AS, Dudley R, Karpati G, Vincent G, Rosiers C, et al. Dystrophin‐deficient cardiomyocytes are abnormally vulnerable to mechanical stress‐induced contractile failure and injury. FASEB Journal 2001;15(9):1655‐7. [DOI] [PubMed] [Google Scholar]
De Kermadec 1994
- Kermadec JM, Bécane HM, Chénard A, Tertrain F, Weiss Y. Prevalence of left ventricular systolic dysfunction in Duchenne muscular dystrophy: an electrocardiographic study. American Heart Journal 1994;127(3):618‐23. [DOI] [PubMed] [Google Scholar]
De Visser 1992
- Visser M, Voogt WG, Rivière GV. The heart in Becker muscular dystrophy, fascioscapulohumeral muscular dystrophy and Bethlem myopathy. Muscle & Nerve 1992;15(5):591‐6. [DOI] [PubMed] [Google Scholar]
Donofrio 1989
- Donofrio PD, Challa VR, Hackshaw BT, Mills SA, Cordell R. Cardiac transplant in a patient with muscular dystrophy and cardiomyopathy. Archives of Neurology 1989;46(6):705‐7. [DOI] [PubMed] [Google Scholar]
Eagle 2002
- Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscular Disorders 2002;12(10):926‐9. [DOI] [PubMed] [Google Scholar]
Emery 1976
- Emery AEH, Skinner R. Clinical studies in benign (Becker type) X‐linked muscular dystrophy. Clinical Genetics 1976;10(4):189‐201. [DOI] [PubMed] [Google Scholar]
Emery 2003
- Emery AEH, Muntoni F. Duchenne Muscular Dystrophy. 3rd Edition. Oxford: Oxford Medical Publications, 2003. [Google Scholar]
Fararolo 2010
- Fararolo R, Peradejord M, Berlotti A, Diez M, Favaloro L, Gomez C, et al. Results of heart transplantation: 16 years experience in a center in Argentina. Transplantation Proceedings 2010;42(1):321‐3. [DOI] [PubMed] [Google Scholar]
Fayssoil 2008
- Fayssoil A, Orlikowski D, Nardi O, Annane D. Complete atrioventricular block in Duchenne muscular dystrophy. Europace 2008;10(11):1351‐2. [DOI] [PubMed] [Google Scholar]
Ferlini 1999
- Ferlini A, Sewry C, Melis MA, Mattedu A, Muntoni F. X‐linked dilated cardiomyopathy and the dystrophin gene. Neuromuscular Disorders 1999;9(5):339‐46. [DOI] [PubMed] [Google Scholar]
Finsterer 2003
- Finserer J, Stöllberger C. The heart in human dystrophinopathies. Cardiology 2003;99(1):1‐19. [DOI] [PubMed] [Google Scholar]
Gardner 1995
- Gardner RJ, Bobrow M, Roberts RG. The identification of point mutations in Duchenne muscular dystrophy patients by using reverse transcription PCR and the protein truncation test. American Journal of Human Genetics 1989;57(2):311‐20. [PMC free article] [PubMed] [Google Scholar]
Globus 1923
- Globus JH. The pathological findings in the heart muscle in progressive muscular dystrophy. Archives of Neurology and Psychiatry 1923;9(1):59‐72. [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime, Inc.). GRADEpro GDT: GRADEpro Guideline Development Tool. Version accessed 20 June 2018. Hamilton (ON): McMaster University (developed by Evidence Prime, Inc.), 2015.
Griffin 2001
- Griffin JL, Williams HJ, Sang E, Clarke K, Rae C, Nicholson JK. Metabolic profiling of genetic disorders: a multi‐tissue (1)H nuclear magnetic resonance spectroscopic and pattern recognition study into dystrophic tissue. Analytical Biochemistry 2001;293(1):16‐21. [DOI] [PubMed] [Google Scholar]
Heran 2012
- Heran BS, Musini VM, Bassett K, Taylor RS, Wright JM. Angiotensin receptor blockers for heart failure. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD003040.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Heymsfield 1978
- Heymsfield SB, McNish T, Perkins JV, Felner JM. Sequence of cardiac changes in Duchenne muscular dystrophy. American Heart Journal 1978;95(3):283‐94. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Holloway 2008
- Holloway SM, Wilcox DE, Wilcox A, Dean JC, Berg JN, Goudie DR, et al. Life expectancy and death from cardiomyopathy amongst carriers of Duchenne and Becker muscular dystrophy in Scotland. Heart 2008;94(5):633‐6. [DOI] [PubMed] [Google Scholar]
Hoogerwaard 1997
- Hoogerwaard EM, Voogt WG, Wilde AA, Wouw PA, Bakker E, Ommen GJ, et al. Evolution of cardiac abnormalities in Becker muscular dystrophy over a 13‐year period. Journal of Neurology 1997;244(10):657‐63. [DOI] [PubMed] [Google Scholar]
Hoogerwaard 1999
- Hoogerwaard EM, Wouw PA, Wilde AA, Bakker E, Ippel PF, Oosterwijk JC, et al. Cardiac involvement in carriers of Duchenne and Becker muscular dystrophy. Neuromuscular Disorders 1999;9(5):347‐51. [DOI] [PubMed] [Google Scholar]
Hor 2011
- Hor KN, Wansapura JP, Al‐Khalidi HR, Gottliebson WM, Taylor MD, Czosek RJ, et al. Presence of mechanical dyssynchrony in Duchenne muscular dystrophy. Journal of Cardiovascular Magnetic Resonance 2011;13(1):12. [DOI: 10.1186/1532-429X-13-12] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hussain 2014
- Hussein G, Mansour L, Ghafor H, Mostafa F, Fawaz L. Short‐term effects of corticosteroid therapy on cardiac and skeletal muscles in muscular dystrophies. Journal of Investigative Medicine 2014;62(6):875‐9. [DOI] [PubMed] [Google Scholar]
Iodice 2015
- Iodice F, Testa G, Averardi M, Brancaccio G, Amodeo A, Cogo P. Implantation of a left ventricular assist device as a destination therapy in Duchenne muscular dystrophy patients with end stage cardiac failure: management and lessons learned. Neuromuscular Disorders 2015;25(1):19‐23. [DOI] [PubMed] [Google Scholar]
Ishigaki 1997
- Ishigaki C, Patria SI, Nishio H, Yoshioka A, Matsuo M. Early cardiac failure in a child with Becker muscular dystrophy is due to an abnormally low amount of dystrophin transcript lacking exon 13. Acta Paediatrica Japonica 1997;39(6):685‐9. [DOI] [PubMed] [Google Scholar]
Kamamura 1990
- Kamakura K, Kawai M, Arahata K, Koizuma H, Watanabe K, Sugita HA. A manifesting carrier of Duchenne muscular dystrophy with severe myocardial symptoms. Journal of Neurology 1990;237(8):483‐5. [DOI] [PubMed] [Google Scholar]
Koenig 1989
- Koenig M, Beggs AH, Moyer M, Scherpf S, Heindrich K, Bettecken T, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Human Genetics 1989;45(4):498‐506. [PMC free article] [PubMed] [Google Scholar]
Kono 2015
- Kono T, Ogimoto A, Nishimura K, Yorozuva T, Okura T, Higaki J. Cardiac resynchronisation therapy in a young patient with Duchenne muscular dystrophy. International Medical Case Reports Journal 2015;21(8):173‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kuru 2012
- Kuru S, Tanahashi T, Matsumoto S, Kitamura T, Konagava M. Complete atrioventricular block in Duchenne muscular dystrophy. Rinsho Shinkeigaku 2012;52(9):684‐7. [DOI] [PubMed] [Google Scholar]
Kwon 2012
- Kwon HW, Kwon BS, Kim GB, Chae JH, Park JD, Bae EJ, et al. The effect of enalapril and carvedilol on left ventricular function in middle childhood and adolescent patients with muscular dystrophy. Korean Circulation Journal 2012;42(3):184‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lane 1980
- Lane RJ, Gardner‐Medwin D, Roses AD. Electrocardiographic abnormalities in carriers of Duchenne muscular dystrophy. Neurology 1980;30(5):497‐506. [DOI] [PubMed] [Google Scholar]
Lanza 2001
- Lanza GA, Dello Russo A, Giglio V, Luca L, Messano L, Santini C, et al. Impairment of cardiac autonomic function in patients with Duchenne muscular dystrophy: relationship to myocardial and respiratory function. American Heart Journal 2001;141(5):808‐12. [DOI] [PubMed] [Google Scholar]
Malhotra 1988
- Malhotra SB, Hart KA, Klamut HJ, Thomas NS, Bodrug SE, Burghes AH, et al. Frame‐shift deletions in patients with Duchenne and Becker muscular dystrophy. Science 1988;242(4879):755‐9. [DOI] [PubMed] [Google Scholar]
Matsumara 2010
- Matsumara T, Tamura T, Kuru S, Kikuchi Y, Kewai M. Carvedilol can prevent cardiac events in Duchenne muscular dystrophy. Internal Medicine 2010;49:1357‐63. [DOI] [PubMed] [Google Scholar]
Matthews 2016
- Matthews E, Brassington R, Kuntzer T, Jichi F, Manzur AY. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database of Systematic Reviews 2016, Issue 5. [DOI: 10.1002/14651858.CD003725.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Melacini 1996
- Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M, et al. Myocardial involvement is very frequent in patients affected with subclinical Becker's muscular dystrophy. Circulation 1996;94(12):3168‐75. [DOI] [PubMed] [Google Scholar]
Mendell 2012
- Mendell JR, Shilling C, Leslie ND, Flanigan KM, al‐Dahhak R, Gastier‐Foster J, et al. Evidence‐based path to newborn screening for Duchenne muscular dystrophy. Annals of Neurology 2012;71(3):304‐13. [DOI] [PubMed] [Google Scholar]
Menke 1991
- Menke A, Jockusch H. Decreased osmotic stability of dystrophin‐less muscle cells from the mdx mouse. Nature 1991;349(6304):69‐71. [DOI] [PubMed] [Google Scholar]
Meune 2004
- Meune C, Pascal O, Bécane HM, Héloire F, Christoforou D, Laforet P, et al. Reliable detection of early myocardial dysfunction by tissue Doppler echocardiography in Becker muscular dystrophy. Heart 2004;90(8):947‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Miyoshi 1991
- Miyoshi K. Echocardiographic evaluation of fibrous replacement in the myocardium of patients with Duchenne muscular dystrophy. British Heart Journal 1991;66(6):452‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mori 2002
- Mori K, Manabe T, Nii M, Hayabuchi Y, Kuroda Y, Tatara K. plasma levels of Natriuretic peptides and echocardiographic parameters in Duchenne's muscular dystrophy. Paediatric Cardiology 2002;23(2):160‐6. [DOI] [PubMed] [Google Scholar]
Mori 2007
- Mori K, Hayabuchi Y, Inoue M, Suzuki M, Sakata M, Nakagawa R, et al. Myocardial strain imaging for early detection of cardiac involvement in patients with Duchenne's progressive muscular dystrophy. Echocardiography 2007;24(6):598‐608. [DOI] [PubMed] [Google Scholar]
Mukoyama 1987
- Mukoyama M, Kondo K, Hizawa K, Nishitani H. Life spans of Duchenne muscular dystrophy patients in the hospital care programme in Japan. Journal of the Neurological Sciences 1987;81(2‐3):155‐8. [DOI] [PubMed] [Google Scholar]
Muntoni 1997
- Muntoni F, Lenarda A, Porcu M, Sinagra G, Mateddu A, Marrosuo G, et al. Dystrophin gene abnormalities in two patients with idiopathic dilated cardiomyopathy. Heart 1997;78(6):608‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Muntoni 2003
- Muntoni F. Cardiomyopathy in muscular dystrophies. Current Opinion in Neurology 2003;16(5):577‐83. [DOI] [PubMed] [Google Scholar]
Nigro 1983
- Nigro G, Comi LI, Limongelli FM, Giugliano MA, Politano L, Petretta V. Prospective study of X‐linked progressive muscular dystrophy in Campania. Muscle & Nerve 1983;6(4):253‐62. [DOI] [PubMed] [Google Scholar]
Nigro 1990
- Nigro G, Comi L, Politano L, Bain RJ. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. International Journal of Cardiology 1990;26(3):271‐7. [DOI] [PubMed] [Google Scholar]
Nigro 1995
- Nigro G, Comi LI, Politano L, Limongelli FM, Nigro V, Rimini ML, et al. Evaluation of the cardiomyopathy in Becker muscular dystrophy. Muscle & Nerve 1995;18(3):283‐91. [DOI] [PubMed] [Google Scholar]
Nigro 2002
- Nigro G, Nigro G, Politano L, Santangelo L, Petretta VR, Passamano L, et al. Is the value of QT dispersion a valid method to foresee the risk of sudden death? A study in Becker patients. Heart 2002;87(2):156‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nikolic 1998
- Nikolíc G. Dominant R wave in lead V1. Heart & Lung 1998;27(5):352‐3. [DOI] [PubMed] [Google Scholar]
Nolan 2003
- Nolan MA, Jones OD, Pedersen RL, Johnston HM. Cardiac assessment in childhood carriers of Duchenne and Becker muscular dystrophies. Neuromuscular Disorders 2003;13(2):129‐32. [DOI] [PubMed] [Google Scholar]
Olfors 1994
- Oldfors A, Eriksson BO, Kyllerman M, Martinsson T, Wahlstrom J. Dilated cardiomyopathy and the dystrophin gene: an illustrated review. British Heart Journal 1994;72(4):344‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Papa 2017
- Papa AA, D'Ambrosio P, Petillo R, Palladino A, Politano L. Heart transplantation in patients with dystrophinopathic cardiomyopathy: review of the literature and personal series. Intractable & Rare Diseases Research 2017;6(2):95‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Perloff 1984
- Perloff JK, Henze E, Schelbert HR. Alterations in regional myocardial metabolism, perfusion, and wall motion in Duchenne muscular dystrophy studied by radionuclide imaging. Circulation 1984;69(1):33‐42. [DOI] [PubMed] [Google Scholar]
Phillips 2008
- Phillips M, Quinlivan R. Calcium antagonists for Duchenne muscular dystrophy. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD004571.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ponikowski 2016
- Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. European Journal of Heart Failure 2016;18(8):891‐975. [DOI] [PubMed] [Google Scholar]
Quinlivan 1995
- Quinlivan R, Ball J, Dunckley M Thomas DJ, Flinter F, Morgan‐Hughes J. Becker muscular dystrophy presenting with complete heart block in the sixth decade. Journal of Neurology 1995;242(6):398‐400. [DOI] [PubMed] [Google Scholar]
Quinlivan 1996
- Quinlivan RM, Lewis P, Marsden P, Dundas R, Robb SA, Baker E. Cardiac function, metabolism and perfusion in Duchenne and Becker muscular dystrophy. Neuromuscular Disorders 1996;6(4):237‐46. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Ryan 2014
- Ryan TD, Jefferies JL, Sawnani H, Wong BL, Gardner A, Corral M, et al. Implantation of the HeartMate II and HeartWare left ventricular assist devices in patients with Duchenne muscular dystrophy: lessons learned from the first applications. ASAIO Journal 2014;60(2):246‐8. [DOI] [PubMed] [Google Scholar]
Sakata 1990
- Sakata C, Sunohara N, Nonaka I, Arahata K, Sugita H. A case of Becker muscular dystrophy presenting cardiac failure as the initial symptom. Rinsho Shinkeigaku 1990;30(2):210‐3. [PubMed] [Google Scholar]
Schram 2013
- Schram G, Fournier A, Leduc H, Dahdah N, Therien J, Vanasse M, et al. All‐cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. Journal of the American College of Cardiology 2013;61(9):948‐54. [DOI] [PubMed] [Google Scholar]
Silva 2007
- Silva MC, Meira ZM, Gurgel Giannetti J, Silva MM, Campos AF, Barbosa Mde M, et al. Myocardial delayed enhancement by magnetic resonance imaging in patients with muscular dystrophy. Journal of the American College of Cardiology 2007;49(18):1874‐9. [DOI] [PubMed] [Google Scholar]
Silversides 2003
- Silversides CK, Webb GD, Harris VA, Biggar DW. Effects of deflazacort on left ventricular function in patients with Duchenne muscular dystrophy. American Journal of Cardiology 2003;91(6):769‐72. [DOI] [PubMed] [Google Scholar]
Soslow 2016
- Soslow J, Xu M, Slaughter J, Stanley M, Crum K, Markham LW, et al. Evaluation of echocardiographic measures of left ventricular function in patients with Duchenne muscular Dystrophy. Journal of the American Society of Echocardiography 2016;29(10):983‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Spurney 2015
- Spurney CF, McCaffrey F, Morgenroth L, Ghelani SJ, Gordish‐Dressman H, Arrieta A, et al. Feasibility and reproducibility of echocardiographic measures in children with muscular dystrophies. Journal of the American Society of Echocardiography 2015;28(8):999‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stabile 2013
- Stabile C, Taylor M, Mazur W, Cripe LH, King E, Pratt J, et al. Left ventricular non‐compaction in Duchenne muscular dystrophy. Journal of Cardiovascular Magnetic Resonance 2013;15(1):67‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Steare 1992
- Steare SE, Dubowitz V, Benatar A. Subclinical cardiomyopathy in Becker muscular dystrophy. British Heart Journal 1992;68(3):304‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Swedberg 2010
- Swedberg K, Komajda M, Böhm M, Borer JS, Ford I, Dubost‐Brama A, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo‐controlled study. Lancet 2010;376(9744):874‐85. [DOI: 10.1016/S0140-6736(10)61198-1] [DOI] [PubMed] [Google Scholar]
Takenaka 1993
- Takenaka A, Yokota M, Iwase M, Miyaguchi K, Hayashi H, Saito H. Discrepancy between systolic and diastolic dysfunction of the left ventricle in patients with Duchenne muscular dystrophy. European Heart Journal 1993;14(5):669‐76. [DOI] [PubMed] [Google Scholar]
Tanaka 1979
- Tanaka H, Nishi S, Katanasako H. Natural course of cardiomyopathy in Duchenne muscular dystrophy. Japanese Circulation Journal 1979;43(11):974‐84. [DOI] [PubMed] [Google Scholar]
Towbin 1993
- Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, Chamberlain JS, et al. X‐linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993;87(6):1854‐65. [DOI] [PubMed] [Google Scholar]
Turley 2008
- Turley A, Raja SG, Salhiyyah K, Nagarajan K. Does cardiac resynchronisation therapy improve survival and quality of life in patients with end‐stage heart failure?. Interactive Cardiovascular and Thoracic Surgery 2008;7(6):141‐6. [DOI] [PubMed] [Google Scholar]
Varni 1999
- Varni JW, Seid M, Rode CA. The PedsQL: measurement model for the pediatric quality of life inventory. Medical Care 1999;37(2):126‐39. [PUBMED: 10024117] [DOI] [PubMed] [Google Scholar]
Vincent 2007
- Vincent KA, Carr AJ, Walburn J, Scott DL, Rose MR. Construction and validation of a quality of life questionnaire for Neuromuscular Disease (INQol). Neurology 2007;68(13):1051‐7. [DOI] [PubMed] [Google Scholar]
Viollet 2012
- Viollet L, Thrush PT, Flanigan KM, Mendell JR, Allen HD. Effects of angiotensin‐converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in Duchenne muscular dystrophy. American Journal of Cardiology 2012;110(1):98‐102. [DOI] [PubMed] [Google Scholar]
Wagner 2007
- Wagner K, Lechtzin N, Judge D. Current treatment of adult Duchenne muscular dystrophy. Biochemica et Biophysica Acta 2007;1772:229‐237. [DOI] [PubMed] [Google Scholar]
Ware 2007
- Ware JE Jr, Kosinski M, Bjorner JB, Turner‐Bowker DM, Gandek B, Maruish ME. User's Manual for the SF‐36v2TM Health Survey. 2nd Edition. Lincoln, RI: Quality Metric Incorporated, 2007. [Google Scholar]
Wasala 2013
- Wasala NB, Bostick B, Yue Y, Duan D. Exclusive skeletal muscle correction does not modulate dystrophic heart disease in the aged mdx model of Duchenne cardiomyopathy. Human Molecular Genetics 2013;22(13):2634‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 2010
- Wu R, Gupta S, Brown R, Yancy C, Wald J, Kaiser P, et al. Clinical outcomes after cardiac transplantation in muscular dystrophy patients. Journal of Heart and Lung Transplantation 2010;29(4):432‐8. [DOI] [PubMed] [Google Scholar]
Yamamoto 1988
- Yamamoto S, Matsushima H, Suzuki A, Sotobata I, Indo T, Matsuoka Y. A comparative study of thallium‐201 single‐photon emission computed tomography and electrocardiography in Duchenne and other types of muscular dystrophy. American Journal of Cardiology 1988;61(10):838‐43. [DOI] [PubMed] [Google Scholar]
Zannad 2000
- Zannad F, Alla F, Dousset B, Perez A, Pitt B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Rales Investigators. Circulation 2000;102(6):2700‐6. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Quinlivan 2011
- Quinlivan R, Chikermane A, Bourke JP. Prevention and treatment for cardiac complications in Duchenne and Becker muscular dystrophy. Cochrane Database of Systematic Reviews 2011, Issue 4. [DOI: 10.1002/14651858.CD009068] [DOI] [PMC free article] [PubMed] [Google Scholar]
Quinlivan 2012
- Quinlivan R, Bourke JP, Bueser T. Prevention and treatment for cardiac complications in Duchenne and Becker muscular dystrophy and X‐linked dilated cardiomyopathy. Cochrane Database of Systematic Reviews 2012, Issue 9. [DOI: 10.1002/14651858.CD009068.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]