

Abstract
This is a protocol for a Cochrane Review (Intervention). The objectives are as follows:
We aim to review and synthesise evidence from randomised controlled trials of hormonal and non‐hormonal treatments in women with catamenial epilepsy. The primary objectives are to evaluate the efficacy and safety of hormonal and non‐hormonal treatments for catamenial seizures.
Background
Description of the condition
Studies have shown that in developed countries, prevalence rates for active epilepsy are between 4 and 10 per 1000 (Sander 1996). In a systematic review of incidence studies, the median annual incidence of epilepsy was 50.7 per 100,000 for males and 46.2 per 100,000 for females (Kotsopoulos 2002). Globally, 50% of women and girls with epilepsy are in the reproductive age range of 15 to 49 years.
Catamenial epilepsy describes a worsening of seizures in relation to the menstrual cycle (peri‐menstrual seizures); it may affect around 40% of women with epilepsy (Herzog 1997). Studies examining day‐to‐day comparisons of seizures throughout the menstrual cycle have consistently shown a greater likelihood of seizures on day one (the start of menstruation), with the lowest risk of seizures on day 20 (the mid‐luteal phase) (Laidlaw 1956; Rosciszewska 1980; Ansell 1986; Tauboll 1991; Herzog 1997). The menstrual cycle is characterised by two phases: the follicular phase (day one to day 13), which comprises menstruation (day one to five) followed by ovulation (day 14), and the luteal phase (day 15 to 28). There are two major hormonal changes: a preovulatory surge in oestradiol (day 10 to 15), and a premenstrual drop in progesterone levels (day 25 to 28). In one study of 184 women with focal epilepsy, there was statistically significant evidence for greater seizure occurrences around the time of these two critical hormonal changes, compared with the mid‐follicular and mid‐luteal phases. These time periods were categorised as catamenial type 1 (C1) pattern (day ‐3 (25) to day 3) and catamenial type 2 (C2) pattern (day 10 to 15). A third pattern — catamenial type 3 (C3) — was noted in patients experiencing anovulatory cycles (where no ovulation occurs during the cycle), whereby a lack of progesterone secretion during the luteal phase predisposed to a higher mid‐luteal ratio of oestradiol to progesterone, which placed the patient at risk of seizures throughout the luteal phase (Herzog 1997). The hormonal changes and catamenial seizure patterns during a menstrual cycle are summarised in Figure 1.
Figure 1.
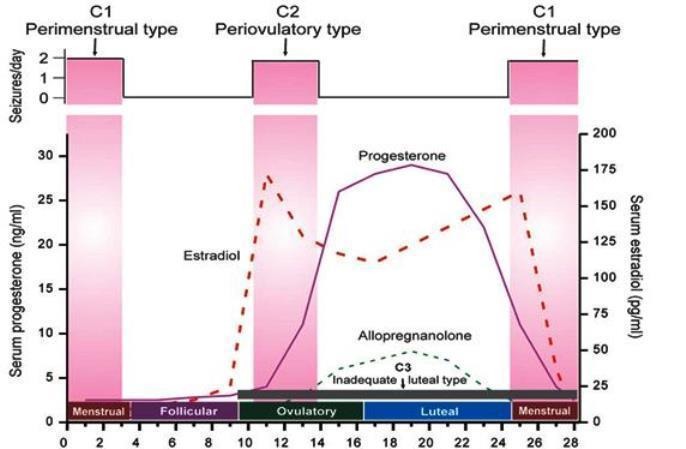
Figure 1: Hormonal changes and catamenial seizures patterns during the menstrual cycle
Approximately 10% of menstrual cycles in healthy women are anovulatory, whereas 35% are anovulatory in women with temporal lobe epilepsy (Herzog 2001). In a study conducted in 1997, around 42% of women demonstrated at least one of the three patterns of catamenial epilepsy. Around 36% had C1 pattern, 29% had C2 pattern, and 42% had C3 pattern (Herzog 1997). Other studies have reported higher prevalence rates (between 63% and 78%), however they compared seizures in just peri‐menstrual phases versus other phases of the cycle (Laidlaw 1956; Rosciszewska 1980; Ansell 1986; Tauboll 1991). When a similar comparison was made in the 1997 study, a prevalence rate of 71% was found (Herzog 1997). Reported clinical risk factors for catamenial epilepsy are: younger age, temporal lobe seizures and a left‐sided epileptogenic foci, which implies that cyclical seizure rhythms are affected by the neuroanatomic substrate of the seizure focus (Quigg 2009).
Description of the intervention
In patients with catamenial seizures, non‐hormonal and hormonal treatments may be considered in addition to regular medication. Non‐hormonal treatments include pulsed clobazam and acetazolamide. Hormonal treatments include natural progesterone supplements, synthetic oral or intramuscular progesterones, allopregnanolone, and gonadotropin‐releasing hormone (GnRH) analogues (triptorelin and goserelin).
For women with catamenial epilepsy who have regular menstrual cycles, intermittent treatment approaches are considered. These interventions target vulnerable days of the menstrual cycle peri‐menstrually (C1 pattern), at ovulation (C2 pattern), and during the luteal phase (C3 pattern). The NIH progesterone trial (Herzog 2012) assigned 462 women with drug‐resistant seizures to either oral progesterone or placebo taken during days 14 to 28, and observed changes in seizure frequency (a reduction of more than 50%) between the three‐month baseline and three‐month treatment period. The study found comparable outcomes for progesterone and placebo overall. However, a secondary analysis identified that the women most likely to respond were those with a C1 pattern seizure type (secondary generalised seizures and focal seizures with altered awareness) and a threefold higher peri‐menstrual seizure frequency. The study demonstrated a favourable short‐term safety profile (Herzog 2012). However, the study did not show a clear effect in women with C2 or C3 pattern, which may reflect differences in underlying pathophysiology. Other intermittent cyclic treatments include benzodiazepines, acetazolamide, or increasing the dose of an anti‐seizure drug already in use.
For women with irregular menstrual cycles, or in those for whom the intermittent cyclic treatments are not effective, the option of pharmacologically stopping the menstrual cycle altogether may be considered, either by using synthetic hormones such as medroxyprogesterone (Depo‐Provera), GnRH analogues (triptorelin and goserelin), or sustained oral contraceptives.
How the intervention might work
Preclinical studies have demonstrated that withdrawal of progesterone or its reduced metabolite allopregnanolone, as occurs premenstrually, can cause insensitivity to the inhibitory neurotransmitter gamma‐Aminobutyric acid (GABA) and also to benzodiazepines that act to enhance GABA transmission (Gangisetty 2010). This is thought to occur by the alteration in the subunit composition of the GABA‐A receptor (Maguire 2005). In animal models, progesterone has been found to reduce neuronal firing and decrease spontaneous and induced epileptiform discharges (Reddy 2004). Progesterone has demonstrated effects on reducing the number of excitatory synapses and the number of oestrogen receptors (McEwan 2001). Other experimental studies support the role of allopregnanolone (a metabolite of progesterone) as conferring seizure protection, with the role of progesterone largely unexplained (Kokate 1999).
The mechanism by which oestradiol causes seizures is uncertain (Osbourne 2009). It may regulate the limbic system; there is evidence of oestradiol synthesising enzymes present within the hippocampus of the temporal lobe. It has also been hypothesised that oestradiol increases excitation by enhancing glutamate transmission and associated receptors (Woolley 1994; Smejkalova 2010). Several studies of chronic oestrogen administration in females, however, show either anticonvulsant effects or no effect of oestrogen on seizures. Studies have also demonstrated that, in low doses, oestradiol can produce neuro‐protective effects (Velísková 2000; Kalkbrenner 2003). Modulation of enzymes involved in glutamate breakdown to GABA have been proposed as neuroprotective mechanisms (Joh 2006; Ledoux 2009).
A detailed understanding of the patterns and pathophysiology is paramount for the development of rational approaches for preventing and treating catamenial epilepsy.
Why it is important to do this review
Catamenial epilepsy and seizure exacerbation is common in women with epilepsy, and may have a significant negative impact on quality of life. Patients may not be receiving appropriate treatment for their catamenial seizures because of uncertainty regarding which treatment works best and when in the menstrual cycle treatment should be taken, as well as the possible impact on fertility, the menstrual cycle, bone health and cardiovascular health. This review aims to address these issues and inform clinical practice and future research.
Objectives
We aim to review and synthesise evidence from randomised controlled trials of hormonal and non‐hormonal treatments in women with catamenial epilepsy. The primary objectives are to evaluate the efficacy and safety of hormonal and non‐hormonal treatments for catamenial seizures.
Methods
Criteria for considering studies for this review
Types of studies
We will include randomised and quasi‐randomised controlled studies of blinded or open‐label design. We will only include studies which randomise participants individually (i.e. cluster‐randomised trials will not be included). We will include trials with a cross‐over design if each treatment period was at least 12 weeks in length and the trial had a suitable washout period.
Types of participants
We will include women of childbearing age who have experienced a catamenial pattern of seizures in at least two baseline cycles, defined as one or more of the following.
C1 pattern: a greater average daily seizure frequency during the peri‐menstrual phase (days ‐3 to +3) compared with the mid‐follicular phase (days 4 to 9) and mid‐luteal phase (days ‐12 to 14) in normal ovulatory cycles.
C2 pattern: a greater average daily seizure frequency during the peri‐ovulatory phase (days 10‐ to ‐13) compared to the mid‐follicular phase (days 4 to 9) and mid‐luteal phase (days ‐12 to 14) in normal ovulatory cycles.
C3 pattern: a greater average daily seizure frequency during the luteal phase (days 15 to 28) compared to the follicular phase (days 1 to 14) in anovulatory cycles.
Types of interventions
We will include the following intervention and control groups.
Intervention group: women who received a hormonal or non‐hormonal drug intervention, in addition to an existing antiepileptic drug regimen for a minimum treatment duration of 12 weeks.
Control group(s): women who received a placebo, comparative drug intervention, or no treatment, in addition to an existing antiepileptic drug regimen for a minimum treatment duration of 12 weeks.
Types of outcome measures
Primary outcomes
Seizure freedom, defined as the proportion of women who become seizure‐free over the treatment period.
Responder rate, defined as the proportion of women with a 50% reduction in seizure frequency compared to baseline.
Change in seizure frequency, defined as the absolute and percentage change in seizure frequency compared to baseline.
Secondary outcomes
-
Withdrawals, defined as the number of withdrawals from allocated treatment or from the trial.
Withdrawals for any reason
Withdrawals due to lack of efficacy
Withdrawals due to adverse events
-
Adverse events: of interest (outlined below), including serious adverse events; and other events reported in the trials regardless of relationship to treatment.
Seizure exacerbation
Cardiac events
Thromboembolic events
Osteoporosis and bone health
Mood disorders
Sedation
Menstrual cycle disorders
Fertility issues
-
Quality of life, according to validated general scales such as the 36‐Item Short Form Health Survey (SF‐36), EuroQol 5‐Dimensions (EQ‐5D) or epilepsy‐specific scales such as the Quality Of Life In Epilepsy ‐31 (QOLIE‐31).
Total quality‐of‐life score
Domain‐specific scores of quality‐of‐life scales
In the first instance, we intend to report change from baseline in quality of life. If change‐from‐baseline scores are not available, we will report the final scores.
Search methods for identification of studies
Electronic searches
We will search the following databases, with no language restrictions. We will seek translation of reports published in any languages other than English.
Cochrane Epilepsy Group Specialized Register
Cochrane Central Register of Controlled Trials (CENTRAL), via the Cochrane Register of Studies Online (CRSO)
MEDLINE (Ovid) 1946 to present date
ClinicalTrials.gov
WHO International Clinical Trials Registry Platform (ICTRP)
The proposed search strategy for MEDLINE is set out in Appendix 1. This strategy will be modified for use with the other databases.
Searching other resources
We will review the reference lists of retrieved trials to check for additional reports of relevant studies.
Data collection and analysis
Selection of studies
The two review authors (MM and SJN) will independently assess trials for inclusion using the Cochrane Covidence software. Firstly we will screen the titles and abstracts of the records, and exclude any that are clearly irrelevant. Subsequently, full‐text articles will be screened for inclusion and any found not to be eligible will be excluded, and the reason for exclusion will be recorded. Any disagreements between the authors regarding eligibility of trials will be resolved by discussion. The screening process will be displayed in a PRISMA study flow diagram (Moher 2009).
Data extraction and management
We will extract the following information for each trial, using a data extraction form.
Methodology/trial design
Method of randomisation and concealment
Method of blinding
Trial inclusion and exclusion criteria
Number of people excluded from analyses
Duration of trial periods, e.g. baseline, treatment, and follow‐up periods, and total trial duration
Trial intervention treatment: type of drug and dose
Trial control treatment: type of control (including type of drug and dose if applicable)
Source of funding of the trial and author disclosures
Participant demographics
Total number of women randomised to each group
Age (overall and by treatment group)
Epilepsy/seizure type
Epilepsy duration and aetiology
Existing anti‐epileptic drug regimen (including dose, overall and by treatment group)
Baseline seizure frequency (overall and by treatment group)
Proportion with C1, C2 and C3 catamenial pattern of seizures
Results
Number of women included in analysis of each outcome by treatment group
Outcome summary data for each intervention (see Types of outcome measures)
The two review authors (MM and SJN) will independently extract data for each trial and compare extractions. We will pilot the content of the form on an eligible trial and add to the content if required. Any discrepancies in data extracted by the two review authors will be resolved by discussion.
If any of the above information appears to have been recorded but not published within the trial reports, or if information is unclear, we will attempt to contact original trial authors for clarification.
Assessment of risk of bias in included studies
The two review authors (MM and SJN) will independently assess the risk of bias for each trial using the Cochrane 'Risk of bias' tool, as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We will rate each of the following six domains as low, unclear or high risk of bias: method of generating random sequence, allocation concealment, blinding methods, incomplete outcome data, selective outcome reporting and other sources of bias. Any discrepancies in the 'Risk of bias' judgements of the two review authors will be resolved by discussion.
Measures of treatment effect
We will report dichotomous data (seizure freedom, responder rate, withdrawals and adverse events) as risk ratios (RRs) with 95% confidence intervals (CIs). If a large number of different adverse events are reported across the studies (for example, more than five different adverse events), 99% CIs will be reported for this outcome to account for multiplicity of statistical testing.
We will report change in seizure frequency as the mean difference (MD) in the change from baseline, with 95% CIs. We will also report quality of life as MD, with 95% CIs, where the same scales are used across studies. If different quality‐of‐life scales are reported across studies, we will consider the similarity of the domains and questions of the scales. If we deem the different scales to be sufficiently similar, we will report pooled quality‐of‐life scores as the standardised mean difference (SMD), with 95% CIs. If the different scales are not deemed sufficiently similar to combine, we will report each scale in separate analyses (where data allow), or in a narrative review.
Unit of analysis issues
We will only include studies which randomise participants individually (i.e. cluster‐randomised trials will not be included). We will include cross‐over trials if each treatment period was a least 12 weeks in length and the trial had a suitable washout period.
If cross‐over studies are included, we will use methods recommended by Elbourne for pooling cross‐over data, which take account of the correlation between measurements taken from the same group of participants via paired analyses (Elbourne 2002). If limited data are available, we will only be able to either use the first‐period data only, or to treat the cross‐over studies as if they are parallel studies, which is a conservative approach and will not take account of within‐patient correlation.
If trials with more than two treatment arms are identified (e.g. drug A, drug B, and placebo), we will construct separate head‐to‐head comparisons to consider the different pairs of interventions and controls.
Dealing with missing data
We will record the attrition rates reported in each trial and, if appropriate, we will attempt to contact the original trial authors if the extent of missing data is unclear. In order to allow an intention‐to‐treat analysis within this review, we will extract and report data by randomised treatment groups where possible, irrespective of compliance with allocated treatment, exclusion from analysis or loss to follow‐up.
In the event of substantial amounts of missing outcome data, we will consider the potential bias that may have been introduced when interpreting the results, particularly if missing data are deemed to not be missing at random.
If appropriate for primary outcomes of seizure freedom and responder rate, we will consider sensitivity analyses such as best‐case scenario and worst‐case scenario analyses (in the best‐case scenario, individuals in the treatment group are assumed to have a good outcome and those in the control group are assumed to have a bad outcome; in the worst‐case scenario the opposite is assumed).
Assessment of heterogeneity
We will assess clinical heterogeneity by reviewing the differences across trials in design, characteristics of recruited participants and interventions. We will also estimate heterogeneity statistically using a Chi2 test for heterogeneity (with a conservative judgement of P value less than 0.1 suggesting heterogeneity), and the I2 statistic. We will interpret the I2 statistic as follows (Deeks 2011):
0% to 40% might not be important;
30% to 60% may represent moderate heterogeneity;
50% to 90% may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity.
Assessment of reporting biases
If a sufficient number of trials (10 or more) are included in any comparison, we will investigate publication bias by using a funnel plot and examining any asymmetry.
To assess selective reporting bias, we will compare the measurements and outcomes planned by the original investigators during the trial with those reported within the published paper, by checking the trial protocols (when available) against the information in the final publication. Where published protocols are not available and trial authors do not provide an unpublished protocol on request, we will compare the 'Methods' and the 'Results' sections of the published papers. We will also use our knowledge of the clinical area to identify where trial investigators have not reported commonly used outcome measures.
Data synthesis
If we deem that trials are sufficiently homogenous in design, participant characteristics and interventions, we will perform meta‐analysis using Mantel‐Haenzel methodology for dichotomous data and inverse variance methodology for continuous data (see Measures of treatment effect). We intend to use a fixed‐effect meta‐analysis model in the first instance. If substantial or considerable heterogeneity is found (i.e. an I2 value of more than 50%), we will repeat the meta‐analysis with a random‐effects model and compare the results of both models.
If we deem that the designs, participant characteristics and interventions are too heterogeneous to combine data, we will report results in a narrative review. Where appropriate, we will present outcome data in tables or enter trial‐specific data into forest plots for visual purposes, without pooling any outcome data.
Subgroup analysis and investigation of heterogeneity
We will assess clinical and statistical heterogeneity using the methods outlined in Assessment of heterogeneity.
If appropriate, and if data allow, we plan to conduct the following subgroup analyses for all outcomes.
Type of epilepsy (focal versus generalised onset, and temporal versus extratemporal onset)
Catamenial pattern (C1, C2, and C3)
Age groups, as defined by the trials (e.g. puberty, sexual maturity, peri‐menopausal)
Sensitivity analysis
As outlined in Dealing with missing data, if a substantial amount of outcome data is missing, we will consider sensitivity analyses such as best‐case scenario and worst‐case scenario analyses.
We will also consider, if appropriate, performing a sensitivity analysis excluding studies with a high risk of bias across any of the domains outlined in Assessment of risk of bias in included studies.
Summary of findings and quality of the evidence
We will generate a 'Summary of findings' table for each comparison of the review, including all outcomes (Schünemann 2011): seizure freedom, responder rate, change in seizure frequency, withdrawals, adverse events, and quality of life.
For clarity and brevity in the tables, we will report a general statement about the summary of findings for secondary outcomes (withdrawals, adverse events, quality of life), based on different reasons for withdrawal, different adverse events and different quality‐of‐life scales.
We will determine the quality of the evidence using the GRADE approach (GRADE 2004), and downgrade evidence in the presence of a high risk of bias in at least one study, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results, and high probability of publication bias. We will downgrade evidence by one level if we consider the limitation to be serious, and by two levels if very serious.
Acknowledgements
We are grateful to Graham Chan for providing an example search strategy, and to the Cochrane Epilepsy Group for editorial support.
This protocol was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Epilepsy Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. MEDLINE search strategy
This strategy is based on the Cochrane Highly Sensitive Search Strategy for identifying randomized trials (Lefebvre 2011).
1. catamenial epilep$.tw.
2. catamenial seizure$.tw.
3. perimenstrual seizure$.tw.
4. perimenstrual epilep$.tw.
5. (menstrua$ adj4 epilep$).tw.
6. (menstrua$ adj4 seizure$).tw.
7. 1 or 2 or 3 or 4 or 5 or 6
8. (randomized controlled trial or controlled clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
9. clinical trials as topic.sh.
10. trial.ti.
11. 8 or 9 or 10
12. exp animals/ not humans.sh.
13. 11 not 12
14. 7 and 13
15. remove duplicates from 14
Contributions of authors
MM and SJN wrote the protocol. Both review authors approved the final version of the protocol.
Sources of support
Internal sources
No sources of support supplied
External sources
National Institute for Health Research (NIHR), UK.
Declarations of interest
MM has no conflicts of interest.
SJN has no conflicts of interest.
New
References
Additional references
- Ansell B, Clarke E. Ovarian hormones, anticonvulsant drugs and seizures during the menstrual cycle in women with epilepsy. Journal of Neurology, Neurosurgery, and Psychiatry 1986;49:47‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veritas Health Innovation. Covidence systematic review software. Melbourne, Australia: Veritas Health Innovation.
- Deeks JJ, Higgins JPT, Altman DG (Editors), Cochrane Statistical Methods Group. Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
- Gangisetty O, Reddy DS. Neurosteroid withdrawal regulates GABA‐A receptor α4‐subunit expression and seizure susceptibility by activation of progesterone receptor‐independent early growth response factor‐3 pathway. Neuroscience 2010;170(3):865‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRADE Working Group. Grading quality of evidence and strength of recommendations. British Medical Journal 2004;328:1490‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog AG, Klein P, Ransil BJ. Three patterns of catamenial epilepsy. Epilepsia 1997;38 (10):1082‐8. [DOI] [PubMed] [Google Scholar]
- Herzog AG, Friedman MN. Menstrual cycle interval and ovulation in women with localization‐related epilepsy. Neurology 2001;57(11):2133–5. [DOI] [PubMed] [Google Scholar]
- Herzog AG, Fowler KM, Smithson SD, Kalayjian LA, Heck CN, Sperling MR, et al. Progesterone Trial Study Group. Progesterone vs placebo therapy for women with epilepsy: a randomized clinical trial. Neurology 2012;78(24):1959‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Joh HD, Searles RV, Selmanoff M, Alkayed NJ, Koehler RC, Hurn PD, et al. Estradiol alters only GAD67 mRNA levels in ischemic rat brain with no consequent effects on GABA. Journal of Cerebral Blood Flow & Metabolism 2006;26(4):518–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkbrenner KA, Standley CA. Estrogen modulation of NMDA‐induced seizures in ovariectomized and non‐ovariectomized rats. Brain Research 2003;964(2):244‐9. [DOI] [PubMed] [Google Scholar]
- Kokate TG, Juhng KN, Kirkby RD, Llamas J, Yamaguchi S, Rogawski MA. Convulsant actions of the neurosteroid pregnenolone sulfate in mice. Brain Research 1999;831(1–2):119‐24. [DOI] [PubMed] [Google Scholar]
- Kotsopoulos IA, Van MT, Kessels FG, Krom MC, Knottnerus JA. Systematic review and meta analysis of incidence studies of epilepsy and unprovoked seizures. Epilepsia 2002;43:1402‐9. [DOI] [PubMed] [Google Scholar]
- Laidlaw J. Catamenial epilepsy. Lancet 1956;271:1235‐7. [DOI] [PubMed] [Google Scholar]
- Ledoux VA, Smejkalova T, May RM, Cooke BM, Woolley CS. Estradiol facilitates the release of neuropeptide Y to suppress hippocampus‐ dependent seizures. Journal of Neuroscience 2009;29(5):1457–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Maguire JL, Stell BM, Rafizadeh M, Mody I. Ovarian cycle‐linked changes in GABA(A) receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nature Neuroscience 2005;8(6):797‐804. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Invited review: estrogens effects on the brain: multiple sites and molecular mechanisms. Journal of Applied Physiology 2001;91(6):2785–801. [DOI] [PubMed] [Google Scholar]
- Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. British Medical Journal 2009;339:b2535. [PMC free article] [PubMed] [Google Scholar]
- Osborne DM, Frye CA. Estrogen increases latencies to seizures and levels of 5alpha‐pregnan‐3alpha‐ol‐20‐one in hippocampus of wild‐type, but not 5alpha‐reductase knockout, mice. Epilepsy Behavior 2009;16(3):411‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigg M, Smithson SD, Fowler KM, Sursal T, Herzog AG, for NIH Progesterone Trial Study Group. Laterality and location influence catamenial seizure expression in women with partial epilepsy. Neurology 2009;73(3):223‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Castaneda DC, O’Malley BW, Rogawski MA. Anticonvulsant activity of progesterone and neurosteroids in progesterone receptor knockout mice. Journal of Pharmacology and Experimental Therapeutics 2004;10(1):230‐39. [DOI] [PubMed] [Google Scholar]
- Rosciszewska D. Analysis of seizure dispersion during menstrual cycle in women with epilepsy. Monographs in Neural Sciences 1980;5:280‐4. [PubMed] [Google Scholar]
- Sander JW, Shorvon SD. Epidemiology of the epilepsies. Journal of Neurology, Neurosurgery and Psychiatry 1996;61:433‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and ‘Summary of findings' tables. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Smejkalova T, Woolley CS. Estradiol acutely potentiates hippocampal excitatory synaptic transmission through a presynaptic mechanism. Journal of Neuroscience 2010;30(48):16137‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubøll E, Lundervold A, Gjerstad L. Temporal distribution of seizures in epilepsy. Epilepsy Research 1991;8(2):153‐65. [DOI] [PubMed] [Google Scholar]
- Velísková J, Velísek L, Galanopoulou AS, Sperber EF. Neuroprotective effects of estrogens on hippocampal cells in adult female rats after status epilepticus. Epilepsia 2000;41(suppl 6):S30‐5. [DOI] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Estradiol regulates hippocampal dendritic spine density via an N‐methyl‐D‐aspartate receptor‐dependent mechanism. Journal of Neuroscience 1994;14(12):7680–7. [DOI] [PMC free article] [PubMed] [Google Scholar]