

Abstract
Background
Critical illness is associated with uncontrolled inflammation and vascular damage which can result in multiple organ failure and death. Antithrombin III (AT III) is an anticoagulant with anti‐inflammatory properties but the efficacy and any harmful effects of AT III supplementation in critically ill patients are unknown. This review was published in 2008 and updated in 2015.
Objectives
To examine:
1. The effect of AT III on mortality in critically ill participants.
2. The benefits and harms of AT III.
We investigated complications specific and not specific to the trial intervention, bleeding events, the effect on sepsis and disseminated intravascular coagulation (DIC) and the length of stay in the intensive care unit (ICU) and in hospital in general.
Search methods
We searched the following databases from inception to 27 August 2015: Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE (Ovid SP), EMBASE (Ovid SP,), CAB, BIOSIS and CINAHL. We contacted the main authors of trials to ask for any missed, unreported or ongoing trials.
Selection criteria
We included randomized controlled trials (RCTs) irrespective of publication status, date of publication, blinding status, outcomes published, or language. We contacted the investigators and the trial authors in order to retrieve missing data. In this updated review we include trials only published as abstracts.
Data collection and analysis
Our primary outcome measure was mortality. Two authors each independently abstracted data and resolved any disagreements by discussion. We presented pooled estimates of the intervention effects on dichotomous outcomes as risk ratios (RR) with 95% confidence intervals (CI). We performed subgroup analyses to assess risk of bias, the effect of AT III in different populations (sepsis, trauma, obstetrics, and paediatrics), and the effect of AT III in patients with or without the use of concomitant heparin. We assessed the adequacy of the available number of participants and performed trial sequential analysis (TSA) to establish the implications for further research.
Main results
We included 30 RCTs with a total of 3933 participants (3882 in the primary outcome analyses).
Combining all trials, regardless of bias, showed no statistically significant effect of AT III on mortality with a RR of 0.95 (95% CI 0.88 to 1.03), I² statistic = 0%, fixed‐effect model, 29 trials, 3882 participants, moderate quality of evidence). For trials with low risk of bias the RR was 0.96 (95% Cl 0.88 to 1.04, I² statistic = 0%, fixed‐effect model, 9 trials, 2915 participants) and for high risk of bias RR 0.94 (95% Cl 0.77 to 1.14, I² statistic = 0%, fixed‐effect model, 20 trials, 967 participants).
For participants with severe sepsis and DIC the RR for mortality was non‐significant, 0.95 (95% Cl 0.88 to 1.03, I² statistic = 0%, fixed‐effect model, 12 trials, 2858 participants, moderate quality of evidence).
We conducted 14 subgroup and sensitivity analyses with respect to the different domains of risk of bias, but detected no statistically significant benefit in any subgroup analyses.
Our secondary objective was to assess the benefits and harms of AT III. For complications specific to the trial intervention the RR was 1.26 (95% Cl 0.83 to 1.92, I² statistic = 0%, random‐effect model, 3 trials, 2454 participants, very low quality of evidence). For complications not specific to the trial intervention, the RR was 0.71 (95% Cl 0.08 to 6.11, I² statistic = 28%, random‐effects model, 2 trials, 65 participants, very low quality of evidence). For complications other than bleeding, the RR was 0.72 ( 95% Cl 0.42 to 1.25, I² statistic = 0%, fixed‐effect model, 3 trials, 187 participants, very low quality of evidence). Eleven trials investigated bleeding events and we found a statistically significant increase, RR 1.58 (95% CI 1.35 to 1.84, I² statistic = 0%, fixed‐effect model, 11 trials, 3019 participants, moderate quality of evidence) in the AT III group. The amount of red blood cells administered had a mean difference (MD) of 138.49 (95% Cl ‐391.35 to 668.34, I² statistic = 84%, random‐effect model, 4 trials, 137 participants, very low quality of evidence). The effect of AT III in patients with multiple organ failure (MOF) was a MD of ‐1.24 (95% Cl ‐2.18 to ‐0.29, I² statistic = 48%, random‐effects model, 3 trials, 156 participants, very low quality of evidence) and for patients with an Acute Physiology and Chronic Health Evaluation score (APACHE) at II and III the MD was ‐2.18 (95% Cl ‐4.36 to ‐0.00, I² statistic = 0%, fixed‐effect model, 3 trials, 102 participants, very low quality of evidence). The incidence of respiratory failure had a RR of 0.93 (95% Cl 0.76 to 1.14, I² statistic = 32%, random‐effects model, 6 trials, 2591 participants, moderate quality of evidence). AT III had no statistically significant impact on the duration of mechanical ventilation (MD 2.20 days, 95% Cl ‐1.21 to 5.60, I² statistic = 0%, fixed‐effect model, 3 trials, 190 participants, very low quality of evidence); on the length of stay in the ICU (MD 0.24, 95% Cl ‐1.34 to 1.83, I² statistic = 0%, fixed‐effect model, 7 trials, 376 participants, very low quality of evidence) or on the length of stay in hospital in general (MD 1.10, 95% Cl ‐7.16 to 9.36), I² statistic = 74%, 4 trials, 202 participants, very low quality of evidence).
Authors' conclusions
There is insufficient evidence to support AT III substitution in any category of critically ill participants including the subset of patients with sepsis and DIC. We did not find a statistically significant effect of AT III on mortality, but AT III increased the risk of bleeding events. Subgroup analyses performed according to duration of intervention, length of follow‐up, different patient groups, and use of adjuvant heparin did not show differences in the estimates of intervention effects. The majority of included trials were at high risk of bias (GRADE; very low quality of evidence for most of the analyses). Hence a large RCT of AT III is needed, without adjuvant heparin among critically ill patients such as those with severe sepsis and DIC, with prespecified inclusion criteria and good bias protection.
Plain language summary
Antithrombin III for critically ill patients
Background
Antithrombin is a small particle produced by the liver. It deactivates several substances which affect the ability of the blood to form clots. Its activity is increased many‐fold by the drug heparin, which enhances the binding of antithrombin to clotting factors so that the blood does not form clots. Antithrombin also reduces inflammation in the human body. Inflammation is the body's attempt at self protection to remove harmful stimuli and begin healing processes. Inflammation is thus not always a bad process.
This updated review assessed the effects of antithrombin in people recovering from critical illness. Our primary goal was to investigate whether the number of people who died changed by giving antithrombin. We also investigated whether there were more complications among people treated with this drug, the extent of bleeding and the amount of blood given to the critical ill people. Finally we examined the impact of antithrombin on the duration of respiratory therapy, length of stay in the intensive care units and in the hospital in general.
Study characteristics
We included 30 trials with 3933 participants (3882 in our data calculation) in this updated review. We found the overall quality of trials to be poor, with little information on how the experiments were carried out. The results were limited and the included trials were mostly small. In most trials, there was a high risk of misleading information. Thus, the results must be interpreted with caution. The evidence is up to date to 27 August 2015.
Study funding sources
Three of the 30 included trials reported receiving money from drug companies.
Key results
In our review we could not identify a clear advantage of antithrombin for the objectives we examined, overall or among various types of patients or subgroups. In our investigation of bleeding events, however, we found an increased risk of bleeding for patients treated with antithrombin.
Quality of the evidence
Overall, there was a low quality of information from the studies regarding all of the results. We conclude that there is a need for a large‐scale clinical trial with low risk of misleading information to investigate the advantages and harms of this drug among critically ill patients.
Summary of findings
1.
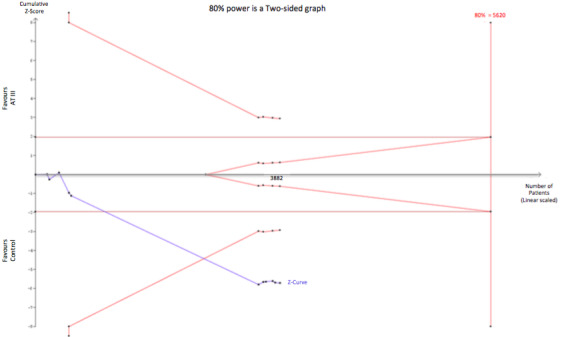
Trial sequential analysis of AT III vs placebo or no intervention on number of bleeding events. The Diversity (D‐square) is 0%, the control event proportion is 13.2% and the required information size is estimated for an anticipated relative risk increase of 20% with a type 1 and 2 error risk of 5% and 20% respectively. The cumulative Z‐curve is breaking through the trial sequential monitoring boundary for harm suggesting, even though the required information size has not been reached, that AT III increases the number of bleeding events. The TSA‐adjusted meta‐analysis yields a RR of 1.85 (95% CI 1.35 to 1.85).
Background
Description of the condition
Despite advances in the medical field, growing numbers of patients are becoming critically ill. Each year, 5,700,000 people in the USA are admitted to intensive care units (ICUs) (Wunsch 2008).
Critical illness is characterized by cellular immune dysfunction, vascular damage and uncontrolled hyperinflammation, even when the cause of illness is not infection. In critical illness, a systemic activation of coagulation may occur which, at its worst, results in a fulminant disseminated intravascular coagulation (DIC). DIC is characterized by simultaneous widespread microvascular thrombosis and profuse bleeding from various sites (Levi 2004). Sepsis resulting from a generalized inflammatory and procoagulant response to an infection is associated with a high risk of mortality. Twenty per cent of patients who develop severe sepsis will die during their hospitalization (Mayr 2014). Septic shock is associated with the highest mortality, approaching 50% (Mayr 2014). This rate increases in the presence of circulatory shock despite aggressive antimicrobial therapy, adequate fluid resuscitation, and optimal care (Periti 2000), and may reach as high as 70% in patients with multiple organ dysfunction (Polderman 2004).
The inflammation associated with critical illness is characterized by an increase in the number and activity of numerous molecules, such as platelet activating factor, von Willebrand factor, and tumour necrosis factor. There is a simultaneous increase in the activity of pro‐inflammatory and pro‐coagulant processes, such as thrombin formation, fibrin deposition at the vascular wall, and the formation of aggregates containing platelets and leukocytes. Leukocyte rolling, adhesion, and transmigration are also important parts of the inflammatory reaction. These processes lead to capillary leakage, severe disturbance of the microcirculation, tissue damage, and eventually multiorgan failure and death (Becker 2000).
Description of the intervention
Any proposed treatment of critical illness should aim to eliminate the underlying disorder or condition and to restore microvascular function, hence reducing organ dysfunction (Levi 2004).
Antithrombin III (AT III) is primarily a potent anticoagulant with independent anti‐inflammatory properties. AT III irreversibly inhibits serine proteases (for example, activated factor X and thrombin) in a one‐to‐one ratio, with the generation of protease‐AT III complexes. Heparin prevents AT III from interacting with the endothelial cell surface by binding to sites on the AT III molecule, competing for the AT III binding site, and reducing AT III ability to interact with its cellular receptor. AT III's anticoagulant effect is thus greatly accelerated (by a factor of 1000) by heparin; heparin reduces AT III's anti‐inflammatory properties, weakens vascular protection, and increases bleeding events (Diaz 2015; Opal 2002; Rublee 2003).
Heparin in patients with sepsis, septic shock, or DIC associated with infection may be associated with decreased mortality (Zarychanski 2015). However, the overall effect is still not clear. Major bleeding events related to heparin administration cannot be excluded (Zarychanski 2015) and safety outcomes have yet to be validated in a multicentre trial setting.
How the intervention might work
The blood concentration of AT III falls by 20% to 40% in septic patients, and these levels correlate with disease severity and clinical outcome (Opal 2002; Wiedermann 2002). This reduction in concentration is due to the combined effect of decreased production of AT III in the liver, inactivation by the enzyme elastase, which is increased during inflammation, and loss of AT III from the circulation into tissues through inflamed and leaking capillary blood vessels. These processes reduce the half‐life of AT III from a mean of 55 hours to 20 hours (Fourrier 2000). The main mechanism of AT III depletion in severe sepsis is linked to consumption of the molecule.
It is this depletion of AT III that has prompted research into the potential benefits of replenishing AT III levels. Investigators have often tried to increase the antithrombin concentration to supranormal values because the activity of pro‐inflammatory and pro‐coagulant molecules are increased in critically ill patients. Thus artificially high levels of AT III may be required to overcome the inhibitory effect of thrombin and other such serine proteases. This is because the normal serum concentration of AT III does not necessarily reflect the amount bound to endothelial receptors and appears insufficient (Fourrier 2000). Finally, by blocking the actions of thrombin, AT III may have anti‐angiogenic and antitumour properties (Larsson 2001).
Why it is important to do this review
Although critically ill patients are a heterogeneous population, they are characterized by having systemic inflammation, no matter what the cause of their illness. This inflammation causes further damage to tissues and organs and can result in multiple organ failure and death. The process of inflammation can be modified by AT III, whether or not clotting is abnormal, and it is possible that AT III can reduce the high death rate or permanent damage experienced by critically ill patients. The benefit of AT III supplementation in critically ill patients is still controversial and its efficacy is still debated (Tagami 2014; Tagami 2015).
Objectives
To examine:
The effect of AT III on mortality in critically ill participants.
The benefits and harms of AT III.
We investigated complications specific and not specific to the trial intervention, bleeding events, the effect on sepsis and DIC and the length of stay in ICU and in hospital in general.
Methods
Criteria for considering studies for this review
Types of studies
We included randomized controlled trials (RCTs) irrespective of publication status, date of publication, blinding status, outcomes published, or language. We contacted the investigators and the authors in order to retrieve relevant data. We included unpublished trials only if trial data and methodological descriptions were either provided in written form or could be retrieved from the trial authors. We excluded cross‐over trials. We include In this updated review trials published as abstracts (Balk 1995; Blauhut 1985; Muntean 1989; Palareti 1995; Schuster 1997).
Types of participants
We included critically ill participants as variously defined by the trial authors. However, we excluded trials of adjuvant AT III administration for the reduction of cardiovascular events in the invasive treatment of acute myocardial infarction.
The terminology for sepsis, as originally proposed by the American College of Chest Physicians and the Society of Critical Care Medicine, is in many ways outdated (Opal 2003). A loose definition of sepsis can easily result in enrolment of a heterogeneous population and hence in exaggerated findings, in either direction, that are difficult to reproduce. However, we accepted the various definitions of sepsis, septic shock, DIC, and other critical illnesses as proposed by the authors; we did not exclude any trial based on their definitions. We chose to accept the term 'standard treatment of sepsis and DIC' as reported by many authors, despite the lack of a generally accepted treatment regimen.
We classified two trials as obstetric trials (Kobayashi 2003; Maki 2000); four trials as paediatric trials (Fulia 2003; Mitchell 2003; Muntean 1989; Schmidt 1998); and a further two trials as trauma trials (Grenander 2001; Waydhas 1998).
Types of interventions
We included AT III versus no intervention or placebo. We included any dose of AT III, any duration of administration, and co‐interventions, but excluded trials that compared different doses of AT III.
Types of outcome measures
Primary outcomes
Overall mortality (longest follow‐up, regardless of the period of follow‐up)
Secondary outcomes
Complications during the inpatient stay specific to the trial intervention, e.g. pneumonia, congestive cardiac failure, respiratory failure, myocardial infarction, renal failure, cerebrovascular accident
Complications during the inpatient stay not specific to the trial intervention, e.g. bleeding, limb venous thrombosis, line sepsis, local haematoma
Complications specific to the trial intervention other than bleeding
Bleeding events
Amount of red blood cells administered
Incidence of surgical intervention
Severity of sepsis (according to different organ dysfunction scores; sepsis versus septic shock if adequately defined by authors)
Incidence of respiratory failure not present at admission (mechanically‐assisted ventilation)
Duration of mechanical ventilation
Length of stay in hospital
Mean length of stay in the intensive care unit (ICU)
Overall mortality among patients with severe sepsis and DIC
We defined bleeding events (4) as intracranial bleeding or bleeding requiring transfusion of at least three units of blood. We counted repeated transfusions in the same participant as a singular event.
Search methods for identification of studies
Electronic searches
In this updated review, we have further extended the original review's search from November 2006 (Afshari 2008). Thus, we searched the Cochrane Central Register of Controlled Trials (CENTRAL; 2015, Issue 8). We updated our search of MEDLINE (Ovid SP, to 27 August 2015), EMBASE (Ovid SP, to 27 August 2015) and CINAHL (to 27 August 2015). The search is now from inception until 27 August 2015.
For specific information regarding our search strategies and results, please see Appendix 1.
Searching other resources
We searched for ongoing clinical trials and unpublished trials on the following Internet sites:
clinicaltrials.gov
We handsearched the reference lists of reviews, randomized and non‐randomized trials, and editorials for additional trials. We contacted the main authors of trials and experts in this field to ask for any missed, unreported, or ongoing studies. We applied no language restrictions to eligible reports. We conducted the latest search on 27 August 2015.
Data collection and analysis
Three review authors (FR, MA and AA) independently screened and classified all citations as potential primary studies, review articles or other. The three review authors also independently examined all potential primary studies and decided on their inclusion in the review. We evaluated all trials for major potential sources of bias (random sequence generation, allocation concealment, blinding, intention‐to‐treat analysis, funding and completeness of follow‐up) (See Figure 2; Figure 3). We assessed each trial quality factor separately and defined the trials as having low risk of bias only if they adequately fulfilled all of the criteria. We independently abstracted and evaluated methodology and outcomes from each trial, in accordance with the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved disagreements by consensus among the review authors.
Selection of studies
We assessed the reports identified from the described searches and excluded obviously irrelevant reports. We screened all articles by title and abstract, and then as full‐text articles for inclusion. We list all excluded studies with reasons for their exclusion in the Characteristics of excluded studies
Two review authors (FR, MA) independently examined the retrieved reports for eligibility. We performed this process without blinding to study authors, institution, journal of publication or results. We resolved disagreements by consensus among the review authors. We provide a detailed description of the search and assessment.
Data extraction and management
We used the above strategy to search for relevant trials. We then screened the titles and abstracts in order to identify studies for eligibility. We independently extracted and collected the data on a standardized paper form. We were not blinded to the study author, source institution, or the publication source of trials. We resolved disagreements by discussion and approached all first authors of the included trials for additional information on risks of bias. For more detailed information please see the section Contributions of authors.
Assessment of risk of bias in included studies
We evaluated the validity and design characteristics of each trial. We evaluated trials for major potential sources of bias (random sequence generation, allocation concealment, blinding, intention‐to‐treat analysis, funding and completeness of follow‐up). We assessed each trial quality factor separately and defined the trials as having a low risk of bias only if they adequately fulfilled all of the criteria.
1. Random sequence generation
Assessment of randomization: sufficiency of the method in producing two comparable groups before intervention.
Grade: ’low risk’: a truly random process (e.g. random computer number generator, coin tossing, throwing dice); ’high risk’: any non‐random process (e.g. date of birth, date of admission by hospital or clinic record number or by availability of the intervention); or ’unclear risk’: insufficient information.
2. Allocation concealment
Allocation method prevented investigators or participants from foreseeing assignment.
Grade: ’low risk’: central allocation or sealed opaque envelopes; ’high risk’: using an open allocation schedule or other unconcealed procedure; or ’unclear risk’: insufficient information.
3. Blinding
Assessment of appropriate blinding of the team of investigators and participants: person responsible for participant care, participants and outcome assessors.
Grade: ’low risk’: blinding considered adequate if participants and personnel were kept unaware of intervention allocations after inclusion of participants into the study, and if the method of blinding involved a placebo indistinguishable from the intervention, as mortality is an objective outcome; ’high risk’: not double‐blinded, categorized as an open‐label study, or without use of a placebo indistinguishable from the intervention; ’unclear risk’: blinding not described.
4. Incomplete outcome data
Completeness of outcome data, including attritions and exclusions.
Grade: ’low risk’: numbers and reasons for dropouts and withdrawals in the intervention groups described, or no dropouts or withdrawals were specified; ’high risk’: no description of dropouts and withdrawals provided; ’unclear risk’: report gave the impression of no dropouts or withdrawals, but this was not specifically stated.
5. Selective reporting
The possibility of selective outcome reporting.
Grade: ’low risk’: reported outcomes prespecified in an available study protocol, or, if this is not available, published report includes all expected outcomes; ’high risk’: not all prespecified outcomes reported, reported using non‐prespecified subscales, reported incompletely or report fails to include a key outcome that would have been expected for such a study; ’unclear risk’: insufficient information.
6. Funding bias
Assessment of any possible funding bias.
Grade: ’low risk’: reported no funding, funding from universities or public institutions; ’high risk’: funding from private investors, pharmaceutical companies or trial investigator employed by the pharmaceutical company; ’unclear risk’: insufficient information.
7. Other bias
Assessment of any possible sources of bias not addressed in domains 1 to 6.
Grade: ’low risk’: report appears to be free of such biases; ’high risk’: at least one important bias is present that is related to study design, early stopping because of some data‐dependent process, extreme baseline imbalance, academic bias, claimed fraudulence or other problems; or ’unclear risk’: insufficient information or evidence that an identified problem will introduce bias.
Measures of treatment effect
We calculated risk ratios (RRs) with 95% confidence intervals (CIs) for dichotomous data (binary outcomes). These included:
Primary outcomes:
Overall mortality (longest follow‐up, regardless of the period of follow‐up)
Secondary outcomes:
Complications during the inpatient stay specific to the trial intervention
Complications during the inpatient stay not specific to the trial intervention
Complications specific to the trial intervention other than bleeding
Bleeding events
Incidence of surgical intervention
Incidence of respiratory failure not present at admission
Overall mortality among patients with severe sepsis and DIC
We used the mean difference (MD) with a 95% confidence interval (CI), if data were continuous and measured in the same way between trials.
Amount of red blood cells administered
Severity of sepsis
Duration of mechanical ventilation
Length of stay in hospital
Mean length of stay in the ICU
Unit of analysis issues
Studies with multiple intervention groups
In accordance with the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), for trials with two or more groups receiving different doses, we combined data for the primary and secondary outcomes. We excluded trials that only compared different doses of AT III and did not have a control group, with or without placebo.
Dealing with missing data
We contacted the authors of trials with missing data in order to retrieve the relevant information. For all included studies we noted levels of attrition and any exclusions. In case of missing data, we chose ’complete‐case analysis’ for our primary outcomes, which excludes from the analysis all participants with the outcome missing.
Selective outcome reporting occurs when nonsignificant results are selectively withheld from publication (Chan 2004), and is defined as the selection, on the basis of the results, of a subset of the original variables recorded for inclusion in publication of trials (Hutton 2000). The most important types of selective outcome reporting are: selective omission of outcomes from reports; selective choice of data for an outcome; selective reporting of different analyses using the same data; selective reporting of subsets of the data and selective underreporting of data (Higgins 2011).
Assessment of heterogeneity
We explored heterogeneity using the I² statistic and Chi² test. An I² statistic above 50% represents substantial heterogeneity (Higgins 2011). In case of I² statistic > 0, we tried to determine the cause of heterogeneity by performing relevant subgroup analyses. We used the Chi² test to provide an indication of heterogeneity between studies, with a P value ≤ 0.1 considered significant.
Assessment of reporting biases
Funding bias is related to the possible publication delay or discouragement of undesired results in trials sponsored by the industry (Higgins 2011).
Data synthesis
Data analysis
We used Review Manager 5 software (RevMan 5.3). We calculated risk ratios (RRs) with 95% confidence intervals (CIs) for dichotomous variables and mean difference (MD) with CI for continuous outcomes. We used the Chi² test to provide an indication of heterogeneity between studies, with a P value < 0.1 considered significant. The degree of heterogeneity observed in the results was quantified using the I² statistic, which can be interpreted as the proportion of the total variation observed between the studies that is attributable to differences between studies rather than to sampling error (Higgins 2002). An I² statistic > 75% is considered as very heterogeneous. We used both a random‐effects model and a fixed‐effect model. If the I² statistic = 0% we only reported the results from the fixed‐effect model, and in the case of I² statistic > 0% we reported only the results from the random‐effects model.
Trial sequential analysis
The risk of type 1 errors in meta‐analyses due to sparse data and repeated significance testing following updates with new trials remains a serious concern (Brok 2009; Thorlund 2009; Wetterslev 2008; Wetterslev 2009). As a result, spurious P values due to systematic errors from trials with high risk of bias, outcome reporting bias, publication bias, early stopping for benefit and small trial bias may result in false conclusions. In a single trial, interim analysis increases the risk of type 1 errors. In order to avoid type 1 errors, group sequential monitoring boundaries (Lan 1983) are used to decide whether a trial could be terminated early because of a sufficiently small P value; thus, the cumulative Z‐curve crosses the monitoring boundary.
Equally, sequential monitoring boundaries can be applied to meta‐analyses and are labelled ’trial sequential monitoring boundaries’ (TSMBs). In ’trial sequential analysis’ (TSA), the addition of each new trial in a cumulative meta‐analysis is viewed as an interim meta‐analysis, which provides useful information on the need for additional trials (Wetterslev 2008).
It is appropriate and wise to adjust new meta‐analyses for multiple testing on accumulating data to control the overall type 1 error risk in cumulative meta‐analysis (Pogue 1997; Pogue 1998; Thorlund 2009; Wetterslev 2008).
When using TSA, if the cumulative Z‐curve crosses the boundary, there is an indication of a sufficient level of evidence having been reached, and as a consequence one may conclude that no further trials may be needed. However, evidence is insufficient to allow a conclusion if the Z‐curve does not cross the boundary or does not surpass the required information size.
In order to construct the TSMBs, one needs a required information size which is calculated as the least number of participants required in a well‐powered single trial with low risk of bias (Brok 2009; Pogue 1998; Wetterslev 2008).
In this updated review, we adjusted the required information size for heterogeneity with the diversity adjustment factor (Wetterslev 2009). We applied TSA, as it prevents an increase in the risk of type 1 errors (20%). If the actual accrued information size was too small, we provided the required information size given the actual diversity (Wetterslev 2009)
Subgroup analysis and investigation of heterogeneity
We conducted the following subgroup analyses:
The effect of AT III in participants given heparin (all types and doses) versus participants not given heparin
Comparing estimates of the pooled intervention effect in trials with low risk of bias to estimates from trials with high risk of bias (i.e. trials having at least one inadequate risk of bias component)
Duration of drug administration (up to one week, more than one week)
Completeness of follow‐up
Comparing the pooled intervention effect in trials with a follow‐up that was longer than the median follow‐up with trials having a follow‐up equal to or shorter than the median follow‐up of trial participants. This was in order to detect a possible dependency of the estimate of intervention effect with length of follow‐up
The effect of AT III in the trauma population
The effect of AT III in obstetrics (eclampsia, pre‐eclampsia)
The effect of AT III in paediatrics (we defined an age below 18 years for our inclusion criteria)
The effect of AT III in sepsis and DIC
If analyses of various subgroups were significant, we performed a test of interaction (Altman 2003). We considered P values < 0.05 as indicating significant interaction between the AT III effect and subgroup category.
We included entire trials for subgroup analysis of trauma, obstetric, and paediatric participants.
Sensitivity analysis
We decided to carry out a sensitivity analysis on the results by applying fixed‐effect and random‐effects models to assess the impact of heterogeneity on our results.
Summary of findings
We used the principles of the GRADE approach to provide an overall assessment of the evidence relating to all of our outcomes. We constructed a 'Summary of findings' table using the GRADEpro software. As outcomes of public interest, we chose to present: mortality, bleeding events, incidence of respiratory failure not present at admission, duration of mechanical ventilation, length of stay in hospital, mean length of stay in ICU and overall mortality among patients with severe sepsis and DIC (see Table 1).
for the main comparison.
| AT III compared to control for critically ill patients | ||||||
|
Setting: Worldwide
Intervention: AT III
Comparison: Control Patient or population: critically ill participants | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with control | Risk with AT III | |||||
| Mortality (subgroup analysis on bias risk) | Study population | RR 0.95 (0.88 to 1.03) | 3882 (29 studies) | ⊕⊕⊕⊝ moderate1,2 | 9 trials had low risk of bias, 20 trials high risk of bias. Trial sequential analysis (TSA) adjusted RR 0.95 (95% CI 0.87 to 1.04). |
|
| 385 per 1000 | 366 per 1000 (339 to 396) | |||||
| Moderate | ||||||
| 310 per 1000 | 295 per 1000 (273 to 320) | |||||
| Bleeding events | Study population | RR 1.58 (1.35 to 1.84) | 3019 (11 studies) | ⊕⊕⊕⊝ moderate3 |
6 trials had low risk of bias, 5 trials high risk of bias. Based on the TSA analysis, there seem to be evidence indicating that AT III increases risk of bleeding (Figure 1). TSA adjusted meta‐analysis yields a RR of 1.85 (95% CI 1.35 to 1.85). | |
| 385 per 1000 | 366 per 1000 (339 to 396) | |||||
| Moderate | ||||||
| 67 per 1000 | 102 per 1000 (87 to 119) | |||||
| Incidence of respiratory failure not present at admission | Study population | RR 0.93 (0.76 to 1.14) | 2591 (6 studies) | ⊕⊕⊕⊝ moderate1,4 | 5 trials had low risk of bias, 1 trial high risk of bias. | |
| 106 per 1000 | 98 per 1000 (80 to 120) | |||||
| Moderate | ||||||
| 317 per 1000 | 294 per 1000 (241 to 361) | |||||
| Duration of mechanical ventilation | The mean duration of mechanical ventilation in the intervention group was 2.2 more (1.21 fewer to 5.6 more) | ‐ | 190 (3 studies) | ⊕⊝⊝⊝ very low5 | 2 trials had low risk of bias, 1 trial high risk of bias | |
| Length of stay in hospital | The mean length of stay in hospital in the intervention group was 1.1 more (7.16 fewer to 9.36 more) | ‐ | 202 (4 studies) | ⊕⊝⊝⊝ very low6 | 2 trials had low risk of bias, 2 trials high risk of bias | |
| Mean length of stay in ICU | The mean length of stay in ICU in the intervention group was 0.24 more (1.34 fewer to 1.83 more) | ‐ | 376 (7 studies) | ⊕⊝⊝⊝ very low1,7 | 3 trials had low risk of bias, 4 trials high risk of bias | |
| Overall mortality among patients with severe sepsis & DIC | Study population |
RR 0.95 (0.88 to 1.03) |
2858 (12 studies) | ⊕⊝⊝⊝ very low1,8 | 4 trials had low risk of bias, 8 trials high risk of bias | |
| 462 per 1000 | 439 per 1000 (407 to 476) | |||||
| Moderate | ||||||
| 392 per 1000 | 372 per 1000 (345 to 404) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1With the exception of Baudo 1998; Fourrier 1993; Fulia 2003; Kobayashi 2003; Maki 2000; Schmidt 1998; Warren 2001; Waydhas 1998 there was a high risk of bias in all trials.
2The entry refers to trials in Analysis 1.1. The outcome was downgraded two levels because of the substantial number of trials with high risk of bias but TSA led to an upgrade of one level to moderate quality of evidence, indicating no benefit for survival. The choice for this TSA upgrade was based on increased precision with continuity correction for zero event trials, adjustment for the risk of random error, a calculation of the required information size leading to rejection of an intervention effect of a RRR of 10% with a power of 80% in 30 randomized trials.
3The entry refers to trials in Analysis 1.14. The outcome was downgraded from high to moderate quality of evidence because majority of trials had high risk of bias.
4The entry refers to trials in Analysis 1.19. The outcome was downgraded from high to moderate quality of evidence since one large trial provided most of the data (Warren 2001).
5The entry refers to trials in Analysis 1.20. The outcome was downgraded from high to very low quality of evidence because of a small number of trials, high risk of bias, a small numbers of participants and imprecision of results with a wide confidence interval.
6The entry refers to trials in Analysis 1.21. The outcome was downgraded from high to very low quality of evidence because of a small number of trials, high risk of bias, a small numbers of participants and imprecision of results with a wide confidence interval.
7The entry refers to trials in Analysis 1.22. The outcome was downgraded from high to very low quality of evidence because of a small number of trials, most of them with high risk of bias.
8The entry refers to trials in Analysis 1.10. The outcome was downgraded from high to very low quality of evidence because of numerous trials with high risk of bias. The vast majority of trials were small and poorly described.
Results
Description of studies
See: Characteristics of included studies; Characteristics of excluded studies; Characteristics of studies awaiting classification; Characteristics of ongoing studies
Results of the search
Through electronic searches and from reading the references of potentially relevant articles, we identified 11287 publications on AT III (see Figure 2). After reading the abstracts, we could directly exclude 11,173 publications. We retrieved 65 relevant publications for further assessment. From these 65 publications, we included 30 (Albert 1992; Balk 1995; Baudo 1992; Baudo 1998; Blauhut 1985; Diaz‐Cremades 1994; Eisele 1998; Fourrier 1993; Fulia 2003; Gando 2013; Grenander 2001; Haire 1998; Harper 1991; Inthorn 1997; Kobayashi 2003; Langely 1993; Lavrentieva 2008; Maki 2000; Mitchell 2003; Muntean 1989; Neporada 2008A; Nishiyama 2011; Palareti 1995; Schmidt 1998; Schorr 2000; Schuster 1997; Smith‐Erichsen 1996; Vorobyeva 2007; Warren 2001; Waydhas 1998), which randomized a total of 3933 participants. One trial (Blauhut 1985) was only published as an abstract and the data were so inadequate that they could not be used for further processing. Our analyses include a total of 3882 participants. The sample size varied from 16 to 2314 participants. We excluded 24 publications for the reasons detailed in the Characteristics of excluded studies. We found one ongoing trial (D'angelo 2005) but no data were provided for this trial (see Characteristics of ongoing studies).
2.

Flow
One trial was published in Russian only (Vorobyeva 2007), and one excluded trial was in German (Angstwurm 2009, a secondary publication of Warren 2001). We had these studies translated into English. Five trials had multiple full‐text publications or published post hoc subgroup analyses (Inthorn 1997; Maki 2000; Mitchell 2003; Neporada 2008A; Warren 2001).
In this updated review, we also included trials that were only published as abstracts in order to avoid publication bias (Balk 1995; Blauhut 1985; Muntean 1989; Palareti 1995; Schuster 1997).
Types of participants
We classified two trials as obstetric studies (Kobayashi 2003; Maki 2000); four trials as paediatric trials (Fulia 2003; Mitchell 2003; Muntean 1989; Schmidt 1998); and a further two trials as trauma studies (Grenander 2001; Waydhas 1998). The remaining trials consisted of mixed populations of critically ill participants, mainly with sepsis.
Types of interventions
The duration of the intervention varied from less than 24 hours to four weeks. Three trials had a median duration of AT III intervention that was longer than one week (Inthorn 1997; Mitchell 2003; Smith‐Erichsen 1996). Follow‐up ranged from seven to 90 days.
Included studies
The 30 included trials were all published between 1985 and 2013. Five trials of AT III were multicentre trials (Baudo 1998; Eisele 1998; Gando 2013; Mitchell 2003; Warren 2001). Five trials of AT III were multinational trials including Germany, Belgium, the Netherlands, Canada and the USA (Baudo 1992; Eisele 1998; Gando 2013; Mitchell 2003; Warren 2001). One trial was carried out in 19 countries (Warren 2001). Two trials did not state the location (Blauhut 1985; Palareti 1995).
The 30 included trials involved a total of 3933 participants. The details of the included studies are provided in the table Characteristics of included studies.
Fourteen trials recruited more male than female participants (Baudo 1998; Diaz‐Cremades 1994; Eisele 1998; Fourrier 1993; Fulia 2003; Gando 2013; Grenander 2001; Inthorn 1997; Lavrentieva 2008; Mitchell 2003; Nishiyama 2011; Smith‐Erichsen 1996; Warren 2001; Waydhas 1998). One trial recruited only men (Baudo 1992), two trials recruited only women (Kobayashi 2003; Maki 2000), six did not report the gender of the participants (Blauhut 1985; Harper 1991; Neporada 2008A; Palareti 1995; Schuster 1997; Vorobyeva 2007) and two studies had more female than male participants (Balk 1995; Haire 1998). The age of the participants included extends from the premature infant to the elderly intensive‐care participant. It therefore makes little sense to calculate the average age of the participants included. One trial however, excluded participants older than 75 (Neporada 2008A).
Eighteen trials used an initial loading dose either based on weight (u/kg) or as a fixed dose (Albert 1992; Baudo 1992; Diaz‐Cremades 1994; Eisele 1998; Fourrier 1993; Fulia 2003; Grenander 2001; Haire 1998; Harper 1991; Inthorn 1997; Langely 1993; Palareti 1995; Schmidt 1998; Schorr 2000; Schuster 1997; Smith‐Erichsen 1996; Vorobyeva 2007; Warren 2001). All trials except two (Balk 1995; Blauhut 1985) stated the use of a maintenance dose. Nine trials used albumin as the control intervention (Baudo 1998; Diaz‐Cremades 1994; Fourrier 1993; Haire 1998; Maki 2000; Schmidt 1998; Schuster 1997; Warren 2001; Waydhas 1998); two trials used fresh frozen plasma as the control intervention (Neporada 2008A; Vorobyeva 2007); three trials only stated the use of an unknown placebo (Balk 1995; Eisele 1998; Kobayashi 2003). Nine trials used no placebo (Albert 1992; Baudo 1992; Grenander 2001; Harper 1991; Inthorn 1997; Langely 1993; Mitchell 2003; Schorr 2000; Smith‐Erichsen 1996). Four trials did not state which control they applied (Baudo 1998; Gando 2013; Lavrentieva 2008; Palareti 1995).
Excluded studies
We excluded 24 randomized trials (Aibiki 2006; Dietrich 2013; Doi 2012; Hoffmann 2010; Ilias 2000; Jochum 1995; Kanbak 2011; Kim 2013; Korninger 1987; Leitner 2006; Maki 1987; Mayumi 2011; Neporada 2008B; Nishiyama 2006; Paparella 2014; Paternoster 2000; Paternoster 2004; Ranucci 2013; Sawamura 2009; Scherer 1997; Shimada 1994; Terao 1989; Valsecchi 2008; Vinazzer 1995), for the reasons detailed in the Characteristics of excluded studies.
Ongoing studies
One trial is stated to be ongoing (D'angelo 2005); see the Characteristics of ongoing studies
Awaiting classification
No studies are awaiting classification.
Risk of bias in included studies
All of the included studies were randomized controlled trials. However, the risk of bias of the included studies was high (Figure 3). The trials often failed to report trial methodology in sufficient detail. See Characteristics of included studies.
3.
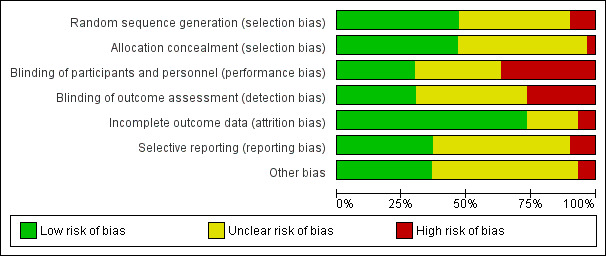
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included trials.
Allocation
Generation of allocation sequence was adequately reported in 15 trials (Albert 1992; Baudo 1998; Fourrier 1993; Fulia 2003; Gando 2013; Grenander 2001; Haire 1998; Kobayashi 2003; Lavrentieva 2008; Maki 2000; Mitchell 2003; Neporada 2008A; Schmidt 1998; Warren 2001; Waydhas 1998) (Figure 4).
4.
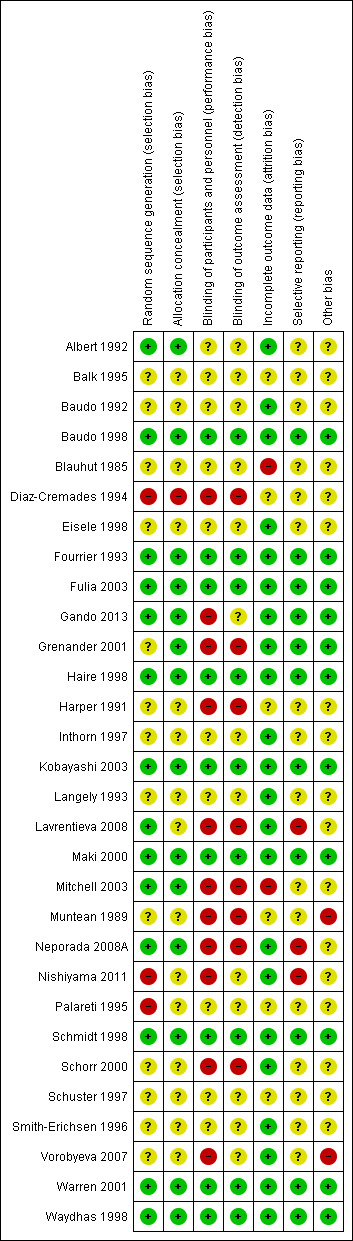
Risk of bias graph summarized
Allocation concealment was adequately reported in 14 trials (Albert 1992; Baudo 1998; Fourrier 1993; Fulia 2003; Gando 2013; Grenander 2001; Haire 1998; Kobayashi 2003; Maki 2000; Mitchell 2003; Neporada 2008A; Schmidt 1998; Warren 2001; Waydhas 1998) (Figure 4).
Blinding
Nine trials provided sufficient data to be categorized as double‐blinded (Baudo 1998; Fourrier 1993; Fulia 2003; Haire 1998; Kobayashi 2003; Maki 2000; Schmidt 1998; Warren 2001; Waydhas 1998). The remaining 21 trials were either open‐label or did not provide sufficient data on how the double‐blinding was achieved (Figure 4).
Incomplete outcome data
Two trials did not provide sufficient data (high risk) on follow‐up (Blauhut 1985; Mitchell 2003). Six trials did not provide any data on follow‐up (unclear risk) (Balk 1995; Diaz‐Cremades 1994; Harper 1991; Muntean 1989; Palareti 1995; Schuster 1997). Twenty‐two trials had adequate follow‐up (low risk) (Albert 1992; Baudo 1992; Baudo 1998; Eisele 1998; Fourrier 1993; Fulia 2003; Gando 2013; Grenander 2001; Haire 1998; Inthorn 1997; Kobayashi 2003; Langely 1993; Lavrentieva 2008; Maki 2000; Neporada 2008A; Nishiyama 2011; Schmidt 1998; Schorr 2000; Smith‐Erichsen 1996; Vorobyeva 2007; Warren 2001; Waydhas 1998).
Selective reporting
Ten trials provided adequate information to be classified as low‐risk trials (Baudo 1998; Fourrier 1993; Fulia 2003; Gando 2013; Grenander 2001; Kobayashi 2003; Maki 2000; Schmidt 1998; Warren 2001; Waydhas 1998). This was often due to supplementary information provided based on online registration, protocol availability or authors providing supplementary information while responding to our questions.
Eighteen trials did not provide sufficient data on selective reporting (unclear risk) (Albert 1992; Balk 1995; Baudo 1992; Blauhut 1985; Diaz‐Cremades 1994; Eisele 1998; Grenander 2001; Haire 1998; Harper 1991; Inthorn 1997; Langely 1993; Mitchell 2003; Muntean 1989; Palareti 1995; Schorr 2000; Schuster 1997; Smith‐Erichsen 1996; Vorobyeva 2007).
Three trials had substantial methodological shortcomings across multiple domains of bias (Lavrentieva 2008; Neporada 2008A; Nishiyama 2011).
Other potential sources of bias
Fourteen trials performed analysis according to the intention‐to‐treat (ITT) method or provided sufficient data for us to perform ITT analyses (Baudo 1992; Eisele 1998; Fourrier 1993; Fulia 2003; Haire 1998; Harper 1991; Inthorn 1997; Kobayashi 2003; Langely 1993; Maki 2000; Schmidt 1998; Schorr 2000; Warren 2001; Waydhas 1998).
Eight trials reported sample size calculations (Baudo 1998; Fourrier 1993; Haire 1998; Inthorn 1997; Kobayashi 2003; Mitchell 2003; Schmidt 1998; Warren 2001).
Three trials (Grenander 2001; Langely 1993; Warren 2001) reported receiving pharmaceutical company funding.
Effects of interventions
See: Table 1
Primary outcomes
1. Overall mortality (longest follow‐up, regardless of the period of follow‐up)
Combining all trials showed no statistically significant effect of AT III on mortality, with a risk ratio (RR) of 0.95 (95% CI 0.88 to 1.03, I² statistic = 0%, P value = 0.91), based on data from 3882 participants in 29 trials. Results were analysed using a fixed‐effect model because heterogeneity was low. We downgraded the outcome from high to moderate quality of evidence because of 20 trials with high risk of bias. However, trial sequential analysis (TSA) led us to upgrade the overall assessment. Equally, for trials with only low risk of bias (Baudo 1998; Fourrier 1993; Fulia 2003; Haire 1998; Kobayashi 2003; Maki 2000; Schmidt 1998; Warren 2001; Waydhas 1998) we found no statistically significant effect; RR 0.96 (95% 0.88 to 1.04, I² statistic = 0%, fixed‐effect model, 9 trials, 2915 participants). Trials with only high risk of bias (Albert 1992; Balk 1995; Baudo 1992; Diaz‐Cremades 1994; Eisele 1998; Gando 2013; Grenander 2001; Harper 1991; Inthorn 1997; Langely 1993; Lavrentieva 2008; Mitchell 2003; Muntean 1989; Neporada 2008A; Nishiyama 2011; Palareti 1995; Schorr 2000; Schuster 1997; Smith‐Erichsen 1996; Vorobyeva 2007) had a non‐significant RR of 0.94 (95% CI 0.77 to 1.14, I² statistic = 0%, fixed‐effect model, 20 trials, 967 participants) (Analysis 1.1).
1.1. Analysis.
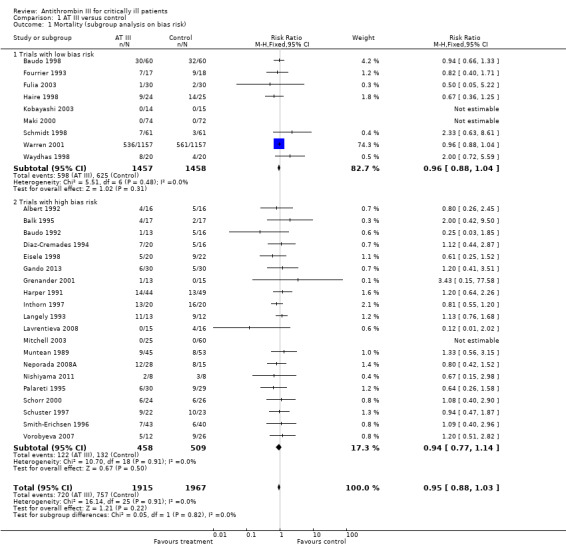
Comparison 1 AT III versus control, Outcome 1 Mortality (subgroup analysis on bias risk).
We conducted three subgroup analyses concerning the use of heparin (Analysis 1.7; Analysis 1.8; Analysis 1.9), but detected no statistically significant effects between the groups.
1.7. Analysis.
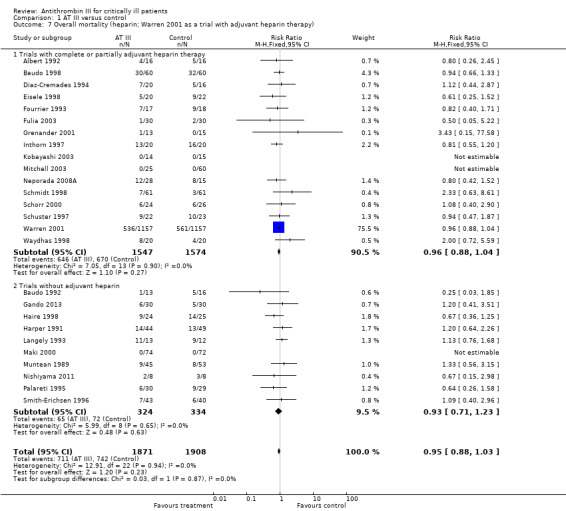
Comparison 1 AT III versus control, Outcome 7 Overall mortality (heparin; Warren 2001 as a trial with adjuvant heparin therapy).
1.8. Analysis.
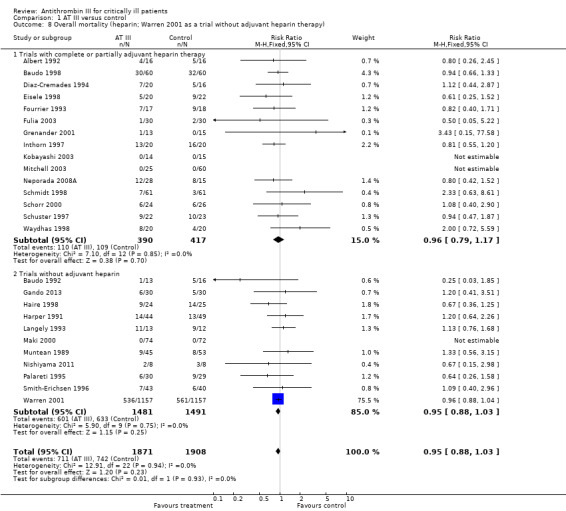
Comparison 1 AT III versus control, Outcome 8 Overall mortality (heparin; Warren 2001 as a trial without adjuvant heparin therapy).
1.9. Analysis.
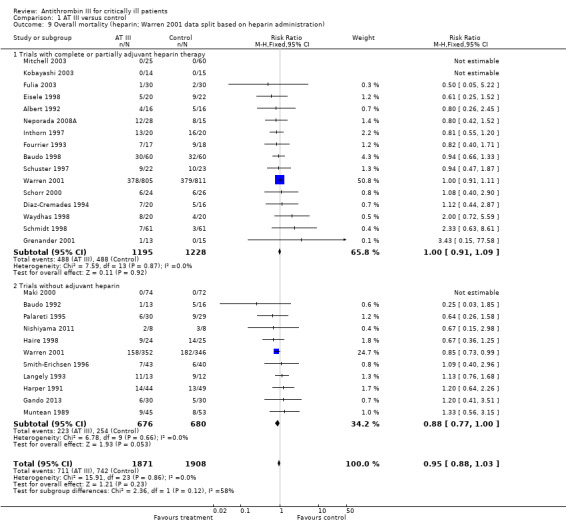
Comparison 1 AT III versus control, Outcome 9 Overall mortality (heparin; Warren 2001 data split based on heparin administration).
We carried out 15 subgroup and sensitivity analyses in regard to our primary outcome, and found no statistically significant effect in any of them (Analysis 1.1; Analysis 1.2; Analysis 1.3; Analysis 1.4; Analysis 1.5; Analysis 1.6; Analysis 1.7; Analysis 1.8; Analysis 1.9; Analysis 1.10; Table 2).
1.2. Analysis.
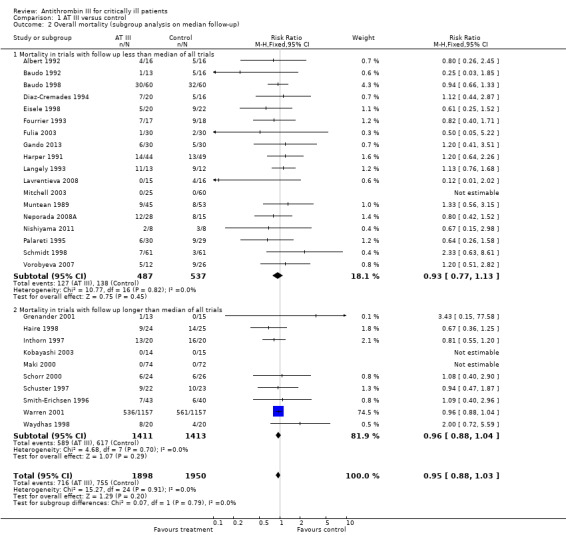
Comparison 1 AT III versus control, Outcome 2 Overall mortality (subgroup analysis on median follow‐up).
1.3. Analysis.
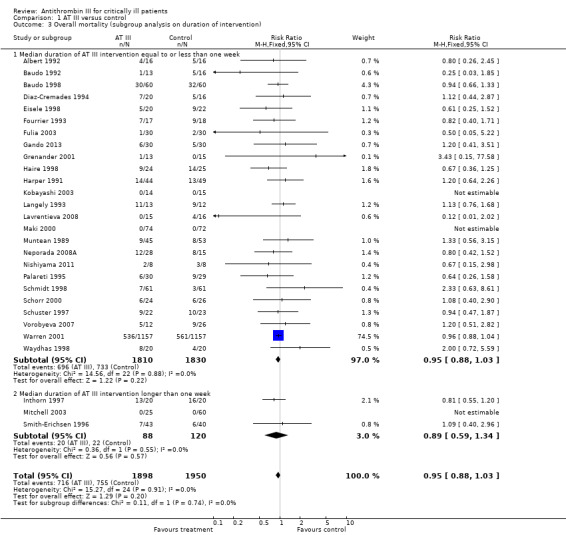
Comparison 1 AT III versus control, Outcome 3 Overall mortality (subgroup analysis on duration of intervention).
1.4. Analysis.
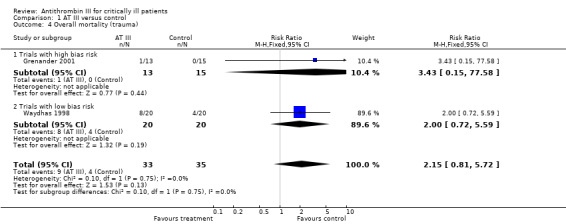
Comparison 1 AT III versus control, Outcome 4 Overall mortality (trauma).
1.5. Analysis.
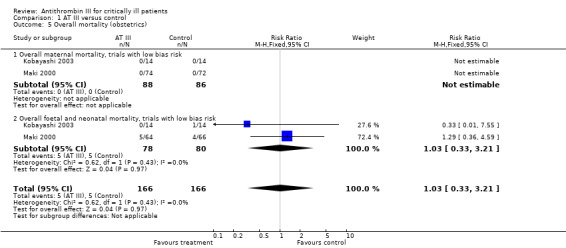
Comparison 1 AT III versus control, Outcome 5 Overall mortality (obstetrics).
1.6. Analysis.
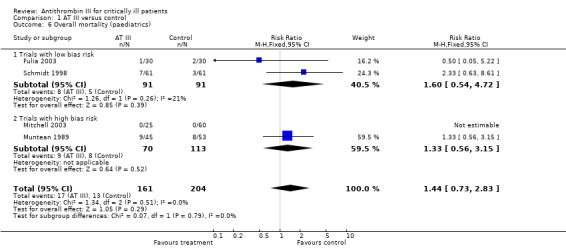
Comparison 1 AT III versus control, Outcome 6 Overall mortality (paediatrics).
1.10. Analysis.
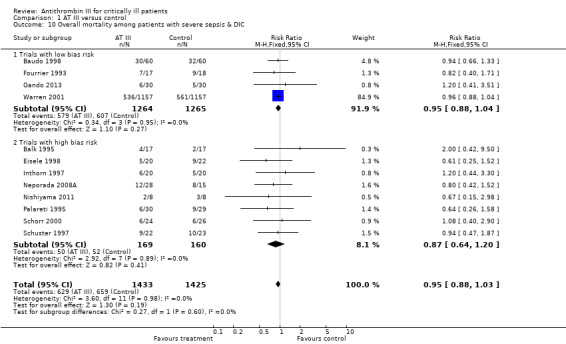
Comparison 1 AT III versus control, Outcome 10 Overall mortality among patients with severe sepsis & DIC.
1. Subgroup analysis (Overall mortality).
| AT III n/N |
Control n/N |
Low risk of bias trials RR (95% CI) |
High risk of bias trials RR (95% CI) |
Overall RR (95% CI) | Heterogeneity | |
| Overall mortality (subgroup analysis on random sequence generation)1 | 720/1915 | 757/1967 | 0.95 (0.88 to 1.03) | 0.98 (0.79 to 1.21) | 0.95 (0.88 to 1.03) | I² = 0% |
| Overall mortality (subgroup analysis on allocation concealment)2 | 720/1915 | 757/1967 | 0.96 (0.88 to 1.04) | 0.93 (0.75 to 1.15) | 0.95 (0.88 to 1.03) | I² = 0% |
| Overall mortality (subgroup analysis on blinding)3 | 720/1915 | 757/1967 | 0.96 (0.88 to 1.04) | 0.94 (0.77 to 1.14) | 0.95 (0.88 to 1.03) | I² = 0% |
| Overall mortality (subgroup analysis on completeness of follow‐up)4 | 720/1915 | 757/1967 | 0.95 (0.88 to 1.02) | 1.08 (0.77 to 1.51) | 0.95 (0.88 to 1.03) | I² = 0% |
| Overall mortality (subgroup analysis on ITT)5 | 705/1856 | 736/1895 | 0.98 (0.79 to 1.22) | 0.95 (0.88 to 1.03) | 0.95 (0.88 to 1.03) | I² = 0% |
1Low risk of bias trials (adequate random sequence generation): Albert 1992; Baudo 1998; Fourrier 1993; Fulia 2003; Gando 2013; Haire 1998; Kobayashi 2003; Lavrentieva 2008; Maki 2000; Mitchell 2003; Neporada 2008A; Schmidt 1998; Warren 2001; Waydhas 1998. High risk of bias trials (inadequate or unclear random sequence generation): Balk 1995; Baudo 1992; Diaz‐Cremades 1994; Eisele 1998; Grenander 2001; Harper 1991; Inthorn 1997; Langely 1993; Muntean 1989; Nishiyama 2011; Palareti 1995; Schorr 2000; Schuster 1997; Smith‐Erichsen 1996; Vorobyeva 2007.
2Low risk of bias trials (adequate allocation concealment): Albert 1992; Baudo 1998; Fourrier 1993; Fulia 2003; Gando 2013; Grenander 2001; Haire 1998; Kobayashi 2003; Maki 2000; Mitchell 2003; Neporada 2008A; Schmidt 1998; Warren 2001; Waydhas 1998. High risk of bias trials (inadequate or unclear allocation concealment): Balk 1995; Baudo 1992; Diaz‐Cremades 1994; Eisele 1998; Harper 1991; Inthorn 1997; Langely 1993; Lavrentieva 2008; Muntean 1989; Nishiyama 2011; Palareti 1995; Schorr 2000; Schuster 1997; Smith‐Erichsen 1996; Vorobyeva 2007
3Low risk of bias trials (blinded): Baudo 1998; Fourrier 1993; Fulia 2003; Haire 1998; Kobayashi 2003; Maki 2000; Schmidt 1998; Warren 2001; Waydhas 1998. High risk of bias trials (not blinded or unclear blinding): Albert 1992; Balk 1995; Baudo 1992; Diaz‐Cremades 1994; Eisele 1998; Gando 2013; Grenander 2001; Harper 1991; Inthorn 1997; Langely 1993; Lavrentieva 2008; Mitchell 2003; Muntean 1989; Neporada 2008A; Nishiyama 2011; Palareti 1995; Schorr 2000; Schuster 1997; Smith‐Erichsen 1996; Vorobyeva 2007
4Low risk of bias trials (complete follow‐up): Balk 1995; Diaz‐Cremades 1994; Harper 1991; Mitchell 2003; Muntean 1989; Palareti 1995; Schuster 1997. High risk of bias trials (absence of complete follow‐up): Albert 1992; Baudo 1992; Baudo 1998; Eisele 1998; Fourrier 1993; Fulia 2003; Gando 2013; Grenander 2001; Haire 1998; Inthorn 1997; Kobayashi 2003; Langely 1993; Maki 2000; Neporada 2008A; Nishiyama 2011; Schmidt 1998; Schorr 2000; Smith‐Erichsen 1996; Vorobyeva 2007; Warren 2001; Waydhas 1998.
5Low risk of bias trials (ITT): Baudo 1992; Eisele 1998; Fourrier 1993; Fulia 2003; Gando 2013; Haire 1998; Harper 1991; Inthorn 1997; Langely 1993; Maki 2000; Schmidt 1998; Schorr 2000; Smith‐Erichsen 1996; Warren 2001; Waydhas 1998. High risk of bias trials (no ITT): Albert 1992; Baudo 1998; Diaz‐Cremades 1994; Grenander 2001; Kobayashi 2003; Lavrentieva 2008; Mitchell 2003; Muntean 1989; Neporada 2008A; Nishiyama 2011; Schuster 1997.
Secondary outcomes
Summarized in Table 3
2. Secondary outcomes; refers to 'Data and analyses' 1.11; 1.12; 1.13; 1.14; 1.16; 1.19.
| Secondary outcomes | AT III n/N |
Control n/N |
Low risk of bias trials RR (95% CI) |
High risk of bias trials RR (95% CI) |
Overall RR (95% CI) |
| Analysis 1.11: Intracranial bleeding | 36/1226 | 26/1228 | 1.62 (0.96 to 2.73) | 0.92 (0.51 to 1.66) | 1.26 (0.83 to 1.92) |
| Analysis 1.12: Renal failure | 2/33 | 3/32 | 3.00 (0.13 to 69.52) | 0.31 (0.04 to 2.57) | 0.71 (0.08 to 6.11) |
| Analysis 1.13: Complications other than bleeding | 14/75 | 33/112 | 0.75 (0.18 to 3.07) | 0.72 (0.40 to1.30) | 0.72 (0.42 to 1.25) |
| Analysis 1.14: Bleeding events | 317/1492 | 201/1527 | 1.58 (1.35 to 1.85) | 1.57 (0.71 to 3.49) | 1.58 (1.35 to 1.84) |
| Analysis 1.16: Incidence of surgical intervention | 31/51 | 30/52 | 1.04 (0.85 to 1.27) | NA | 1.04 (0.85 to 1.27) |
| Analysis 1.19: Respiratory failure not present at admission | 115/1293 | 137/1298 | 0.97 (0.77 to 1.22) | 0.73 (0.45 to 1.18) | 0.93 (0.76 to 1.14) |
1. Complications during the inpatient stay specific to the trial intervention
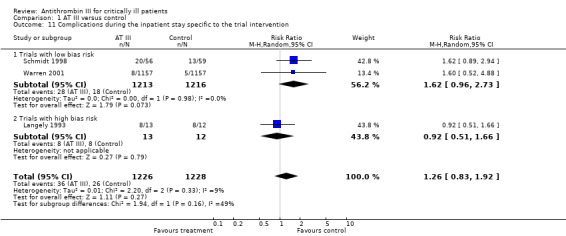
Two trials with low risk of bias (Schmidt 1998; Warren 2001) and one trial with high risk of bias (Langely 1993) demonstrated a statistically significant increase in complications specific to the trial intervention: RR 1.26 (95% CI 0.83 to 1.92, I² statistic = 9%, P value = 0.33), based on data from 2454 participants in the three trials. We analysed results using a random‐effects model. We downgraded the outcome from high to very low quality because of the small number of trials (Analysis 1.11)
1.11. Analysis.
Comparison 1 AT III versus control, Outcome 11 Complications during the inpatient stay specific to the trial intervention.
2. Complications during the inpatient stay not specific to the trial intervention
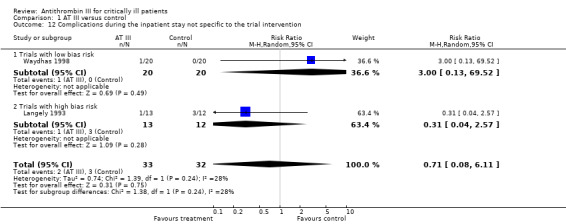
Two trials (Langely 1993; Waydhas 1998) did not reach statistical significance assessing complications not specific to the trial intervention: RR 0.71 (95% CI 0.08 to 6.11, I² statistic = 28%, P value = 0.24), based on data from 65 participants. We analysed results using a random‐effects model. We downgraded the outcome from high to very low quality because of the small number of trials (Analysis 1.12)
1.12. Analysis.
Comparison 1 AT III versus control, Outcome 12 Complications during the inpatient stay not specific to the trial intervention.
3. Complications specific to the trial intervention other than bleeding
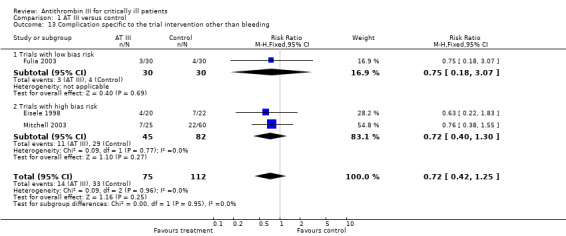
Three trials, one with low risk of bias (Fulia 2003) and two with high risk of bias (Eisele 1998; Mitchell 2003), examined complications specific to the trial intervention other than bleeding: RR 0.72 (95% CI 0.42 to 1.25, I² statistic = 0%, P value = 0.95), based on data from 187 participants in the three trials. We analysed results using a fixed‐effect model. We downgraded the outcome from high to very low quality because of the small number of trials (Analysis 1.13)
1.13. Analysis.
Comparison 1 AT III versus control, Outcome 13 Complication specific to the trial intervention other than bleeding.
4. Bleeding events
Six trials with low risk of bias (Baudo 1998; Fulia 2003; Kobayashi 2003; Maki 2000; Schmidt 1998; Warren 2001) and five with high risk of bias (Gando 2013; Grenander 2001; Langely 1993; Mitchell 2003; Neporada 2008A) demonstrated a statistically significant increase in bleeding events in the intervention group compared to the control group, with a RR of 1.58 (95% CI 1.35 to 1.84, I² statistic = 0%, P value = 0.57), based on data from 3019 participants in the 11 trials. We analysed results using a fixed‐effect model. We downgraded the outcome from high to moderate quality because of the proportion of trials with high risk of bias (Analysis 1.14)
1.14. Analysis.
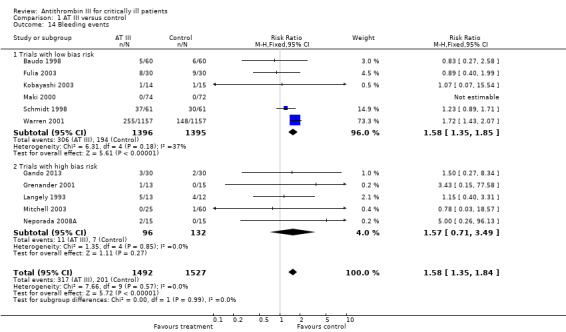
Comparison 1 AT III versus control, Outcome 14 Bleeding events.
5. Amount of red blood cells administered
Four trials referred to the amount of red blood cells administered; one with low risk of bias (Fourrier 1993) and three with high risk of bias (Baudo 1992; Blauhut 1985; Inthorn 1997) with a mean difference (MD) of 138.49 (95% CI ‐391.35 to 668.34, I² statistic = 88%, P value = 0.0001), based on data from 137 participants. We analysed results using a random‐effects model. We downgraded the outcome from high to very low quality because of the small number of trials, three of them with high risk of bias (Analysis 1.15)
1.15. Analysis.
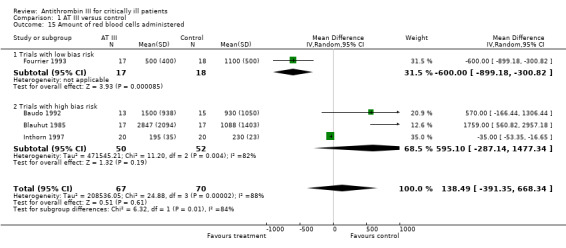
Comparison 1 AT III versus control, Outcome 15 Amount of red blood cells administered.
6. Incidence of surgical intervention
Three trials referred to the incidence of surgical intervention, all with low risk of bias (Fourrier 1993; Kobayashi 2003; Waydhas 1998) with a RR of 1.04 (95% CI 0.85 to 1.27, I² statistic = 0%, P value = 0.61), based on data from 103 participants. We analysed results using a fixed‐effect model. We downgraded the outcome from high to very low quality because of the small number of trials with few participants (Analysis 1.16)
1.16. Analysis.
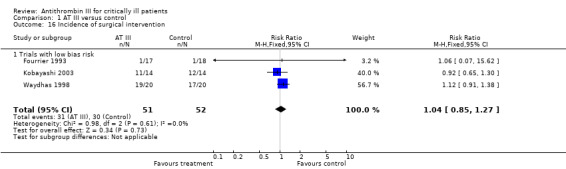
Comparison 1 AT III versus control, Outcome 16 Incidence of surgical intervention.
7. Severity of sepsis
Only Analysis 1.17, of the three different analyses (Analysis 1.17; Analysis 1.18; Table 4) reached statistical significance, with a MD of ‐1.24 (95% CI ‐2.18 to ‐0.29, I² statistic = 48%, P value = 0.015, random‐effects model, 3 trials, 156 participants) (Baudo 1998; Eisele 1998; Inthorn 1997) when examining the effect of AT III on various illness scores. Six trials provided data (Baudo 1998; Diaz‐Cremades 1994; Eisele 1998; Haire 1998; Inthorn 1997; Schorr 2000). However, the trials that did provide data adequate for meta‐analysis were quite heterogenous in their application of various scores and their choice of time points.
1.17. Analysis.
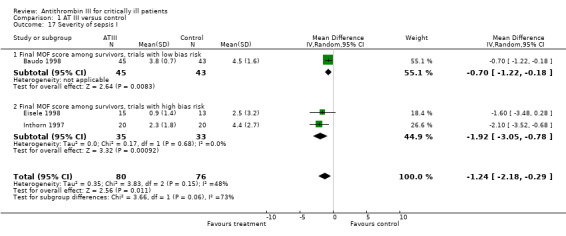
Comparison 1 AT III versus control, Outcome 17 Severity of sepsis I.
1.18. Analysis.
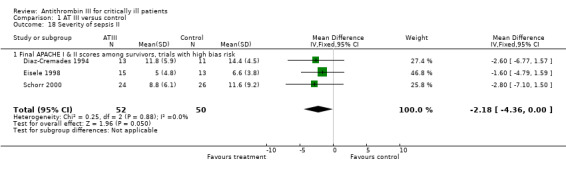
Comparison 1 AT III versus control, Outcome 18 Severity of sepsis II.
3. Severity of sepsis III.
| Trial | AT III | Control | Mean Difference | ||||
| Mean | SD | Total | Mean | SD | Total | IV, Fixed, 95% CI | |
| Eisele 1998 | 0.3 | 0.5 | 15 | 0.5 | 0.7 | 13 | ‐0.20 (‐0.66 to 0.26) |
Subgroup analysis where only one trial had the relevant endpoints.
8. Incidence of respiratory failure not present at admission
Six trials examined the effect of AT III on the incidence of respiratory failure (not present at admission) (Eisele 1998; Fourrier 1993; Kobayashi 2003; Maki 2000; Warren 2001; Waydhas 1998). There was no statistically significant difference, with a RR of 0.93 (95% CI 0.76 to 1.14, I² statistic = 32%, P value = 0.22), based on data from 2591 participants in six trials. We analysed results using a random‐effects model. We downgraded the outcome from high to moderate quality because one trial contributed with the majority of patients (Warren 2001. It is considered to skew the finding; however, it is rated low risk of bias, and contributes a weighting of only 30.7%. A sensitivity analysis removing it from the plot eliminates all heterogeneity in that subgroup.
9. Duration of mechanical ventilation
Three trials examined the effect of the trial intervention on duration of mechanical ventilation (Grenander 2001; Schmidt 1998; Waydhas 1998). There was no statistically significant difference, with a MD of 2.20 (95% CI ‐1.21 to 5.60, I² statistic = 0%, P value = 0.89), based on data from 190 participants. We analysed results using a fixed‐effect model. We downgraded the outcome from high to very low quality of evidence because of the small number of trials, few participants and imprecision of results with a wide confidence interval. The mean duration of mechanical ventilation in the intervention group was 2.2 days more (1.21 fewer to 5.6 more). (Analysis 1.20).
1.20. Analysis.
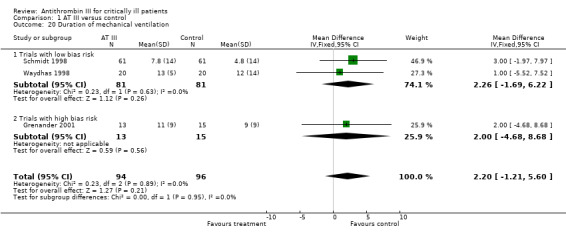
Comparison 1 AT III versus control, Outcome 20 Duration of mechanical ventilation.
10. Length of stay in hospital
Four trials examined the intervention effect on the length of stay in hospital (Haire 1998; Neporada 2008A; Smith‐Erichsen 1996; Waydhas 1998) with a MD of 1.10 (95% CI ‐7.16 to 9.36, I² statistic = 74%, P value = 0.009), based on data from 202 participants. We analysed results using a random‐effects model. We downgraded the outcome from high to very low quality of evidence because of the small number of trials, few participants and imprecision of results with a wide confidence interval. The mean length of stay in hospital in the intervention group was 1.1 days more (7.16 fewer to 9.36 more). (Analysis 1.21)
1.21. Analysis.
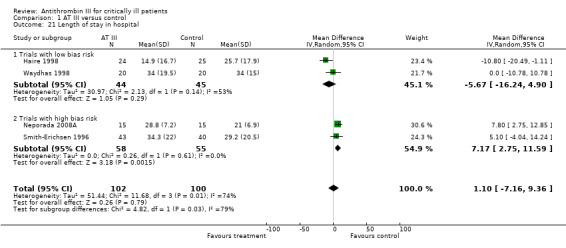
Comparison 1 AT III versus control, Outcome 21 Length of stay in hospital.
11. Mean length of stay in the ICU
Three trials with low risk of bias (Baudo 1998; Fourrier 1993; Waydhas 1998) and four trials with high risk of bias (Albert 1992; Diaz‐Cremades 1994; Neporada 2008A; Smith‐Erichsen 1996) examined the intervention effect on length of stay in the ICU. There was insufficient evidence to support any beneficial effect of the intervention, with a MD of 0.24 (95% CI ‐1.34 to 1.83, I² statistic = 0%, P value = 0.70), based on data from 376 participants. We analysed results using a fixed‐effect model. We downgraded the outcome from high to very low quality of evidence because of the small number of trials, most of them with high risk of bias. The mean length of stay in ICU in the intervention group was 0.24 days more (1.34 fewer to 1.83 more). (Analysis 1.22).
1.22. Analysis.
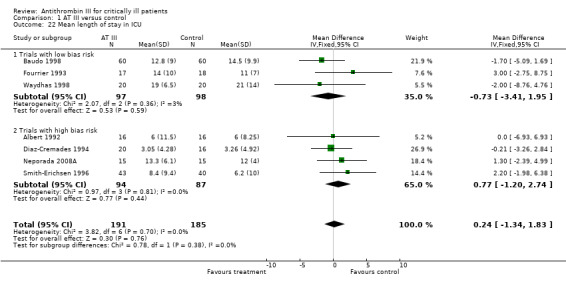
Comparison 1 AT III versus control, Outcome 22 Mean length of stay in ICU.
12. Overall mortality among participants with severe sepsis and DIC
Twelve trials examined the intervention effect on mortality among participants with severe sepsis and disseminated intravascular coagulation (DIC) (Balk 1995; Baudo 1998; Eisele 1998; Fourrier 1993; Gando 2013; Inthorn 1997; Neporada 2008A; Nishiyama 2011; Palareti 1995; Schorr 2000; Schuster 1997; Warren 2001). The trials demonstrated a statistically significant decrease in mortality in favour of the trial intervention: RR 0.95 (95% CI 0.88 to 1.03, I² statistic = 0%, P value = 0.98), based on data from 2858 participants. We analysed results using a fixed‐effect model. We downgraded the outcome from high to very low quality of evidence, because of numerous trials with high risk of bias. The vast majority of trials were small and poorly described.
Subjective overall quality‐of‐life assessment
Only one trial examined the intervention's effect on quality of life (Rublee 2003: based on data from Warren 2001). There was an objective assessment of physical performance and dependency, and a subjective overall quality‐of‐life assessment analysis. Neither assessment supported intervention with AT III, with a MD of ‐2.00, (95% CI ‐4.49 to 0.49, fixed‐effect model, 897 participants) and a MD of ‐2.00, (95% CI ‐5.01 to 1.01, fixed‐effect model, 897 participants) respectively, both rated at very low quality (Table 5; Table 6).
4. Subjective overall quality of life assessment.
| Trial | At III | Control | Mean Difference | ||||
| Mean | SD | Total | Mean | SD | Total | IV, Fixed, 95% CI | |
| Warren 2001 | 47 | 23 | 460 | 49 | 23 | 437 | ‐2.00 (‐5.01 to 1.01) |
Subgroup analysis where only one trial had the relevant endpoints.
5. Objective assessment of physical performance and dependency (Karnofsky).
| Trial | AT III | Control | Mean Difference | ||||
| Mean | SD | Total | Mean | SD | Total | IV, Fixed, 95% CI | |
| Warren 2001 | 53 | 19 | 460 | 55 | 19 | 437 | ‐2.00 (‐4.49 to 0.49) |
Subgroup analysis where only one trial had the relevant endpoints.
Subgroup and sensitivity analyses
The heparin issue
A detrimental interaction between AT III and heparin was suspected before the Warren 2001 trial, and we predefined use of AT III with and without heparin in the protocol for secondary analyses. However, the participants were not stratified according to heparin administration and the protocol allowed concomitant use of heparin by indication, after randomization to AT III or placebo. Even if the baseline comparison of participants allocated to AT III and placebo, in the subgroup without heparin, showed similar characteristics, the randomization is violated in the subgroup analysis.
Pooling all trials with and without concomitant use of heparin, with the Warren 2001 trial as either a trial with concomitant use of heparin or as a trial without use of heparin, does not provide evidence of a statistically significant intervention effect of AT III (Analysis 1.7; Analysis 1.8). Even when splitting the Warren 2001 trial into two 'separate trials', with and without concomitant use of heparin, and pooling these results with the other trials, we found no statistically significant intervention effect of AT III in the subgroup of trials without adjuvant heparin administration (RR 0.95, 95% CI 0.88 to 1.03; I² statistic = 0%, fixed‐effects model, Analysis 1.9). However, splitting the Warren 2001 trial violates the randomization procedure.
Trial sequential analysis
We conducted trial sequential analysis (TSA) of AT III versus control on longest follow‐up mortality (Analysis 1.1; Figure 5). The TSA‐adjusted confidence interval for the meta‐analysis of the primary outcome with continuity correction for zero events trials (0.001 event in each arm) in a fixed‐effect model results in a RR of 0.95 (95% CI 0.88 to 1.03; I² statistic = 0%, Diversity D² = 0%). The point estimate of the potential intervention effect as suggested by the low risk of bias trials in the meta‐analysis of the effect of AT III on mortality is a relative risk reduction (RRR) of 5% and the low‐bias heterogeneity‐adjusted information size (LBHIS) calculated based on this intervention effect (with 80% power and alpha 0.05, assuming a double‐sided type I risk of 5% and a type II risk of 20%) is 23,634 participants (Figure 6). With an accrued information size of 3882 participants and no boundaries crossed so far, only 16.43% of the required information size is actually available at this stage to reject or accept a 4% RRR for overall mortality.
5.
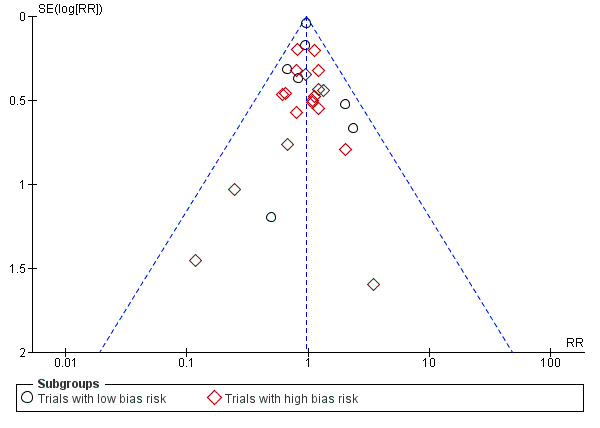
Funnel plot, overall mortality regardless of follow‐up and bias Analysis 1.1
6.
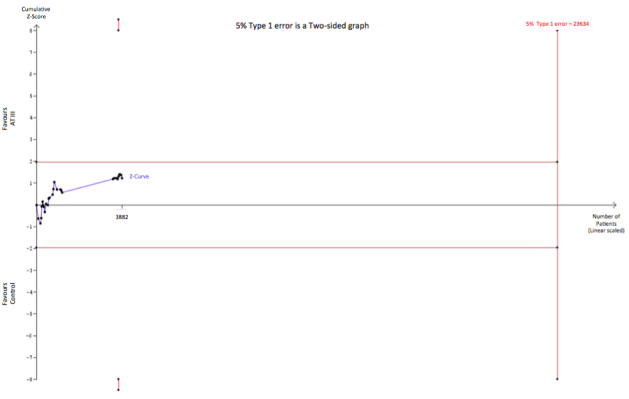
Trial sequential analysis of all trials with low risk of bias of the effect of AT III on mortality. Cumulative Z‐curve in blue does not cross the trial sequential monitoring boundary (full red line with open diamonds) constructed for a low‐bias heterogeneity‐adjusted information size of 23,634 participants corresponding to a RRR of 5% with an α = 0.05 and a power of 80% (β = 0.20). Only 16.43 % of the required information size has been reached so far.
However, solid evidence may be obtained with fewer participants if eventually the cumulative meta‐analysis Z‐curve crosses the trial sequential monitoring boundary constructed for a required information size of 23,634 randomized participants.
On the other hand, to demonstrate or reject an a priori anticipated intervention effect of a RRR of 10%, 5037 should be randomized. In this analysis, the cumulative Z‐curve breaks through the boundary for futility (non‐superiority) (Figure 7). As 3882 participants are included in the present meta‐analyses on mortality without the meta‐analysis becoming statistically significant and since the futility boundary is crossed, an intervention effect of 10% RRR or more on mortality is unlikely.
7.
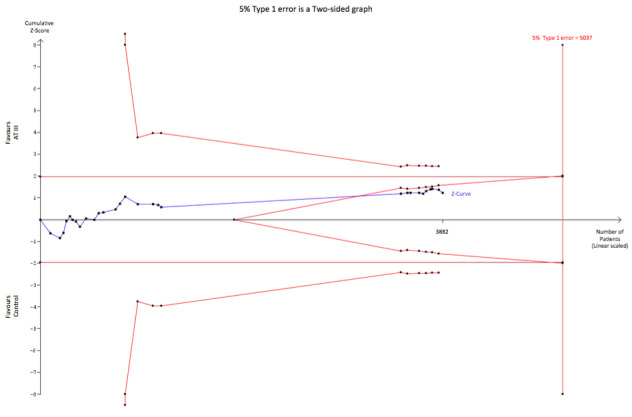
Trial sequential analysis (TSA) of all trials of the effect of AT III on mortality. Cumulative Z‐curve in blue does not cross the boundary constructed for an information size of 5037 in the meta‐analysis (full red line with open diamonds) with a RRR of 10% (α = 0.05) and a power of 80% (β = 0.20). However, the cumulative Z‐curve breaks through the boundary for futility (non‐superiority). The analysis therefore led to rejection of an intervention effect of a RRR of 10% with a power of 80% in 30 randomized trials with a total number of accrued participants of 3882.
When carrying out the same TSA analyses as above for trials of sepsis and DIC only (Analysis 1.10) with an anticipated RRR of 10%, the required information size is 3794 participants without the meta‐analysis becoming statistically significant, and with the boundary for futility being crossed, thus indicating that a RRR of 10% is to be rejected. (Figure 8)
8.
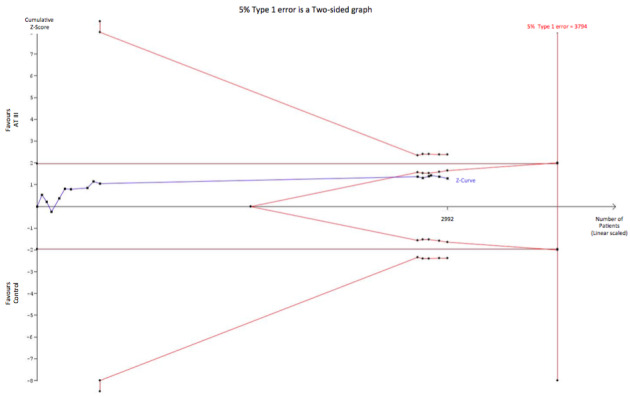
Trial sequential analysis (TSA) of all trials of sepsis and DIC examining the effect of AT III on mortality. Cumulative Z‐curve in blue does not cross the boundary constructed for an information size of 3794 in the meta‐analysis (full red line with open diamonds) with a RRR of 10% (α = 0.05) and a power of 80% (β = 0.20). However, the cumulative Z‐curve breaks through the boundary for futility (non‐superiority). The analysis therefore led to rejection of an intervention effect of a RRR of 10% with a power of 80% in 30 randomized trials with a total number of accrued participants of 2992.
TSA analysis based on a potential RRR of 5% as indicated by the meta‐analysis for studies on sepsis and DIC (Analysis 1.10) yields a LBHIS of 21,657 participants and with an accrued information size of 2992 participants and no boundaries being crossed so far, only 13.82% of the required information size is actually available (Figure 9).
9.
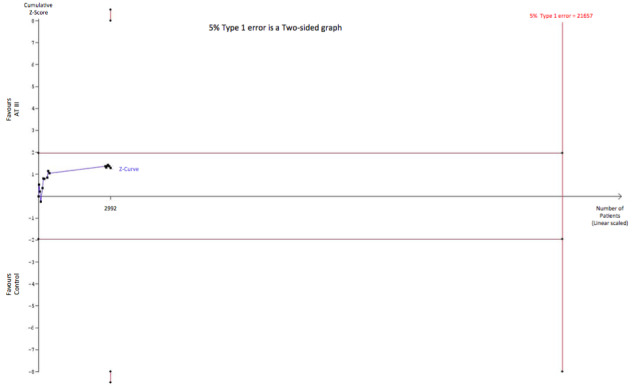
Trial sequential analysis of all trials of Sepsis and DIC with low risk of bias examining the effect of AT III on mortality. Cumulative Z‐curve in blue does not cross the trial sequential monitoring boundary (full red line with open diamonds) constructed for a low bias heterogeneity adjusted information size of 21,657 participants corresponding to a RRR of 5% with an α = 0.05 and a power of 80% (β = 0.20). Only 13.82 % of the required information size has been reached so far.
Discussion
Summary of main results
In this systematic review of 30 trials with 3933 participants we found no significant beneficial effect of AT III on mortality (Analysis 1.1).
The analyses on mortality showed no heterogeneity and were robust when performing different subgroup analyses. Conversely, AT III increased the risk of bleeding (Analysis 1.14) and it appeared to improve only one of the reported severity of sepsis scores (multiple organ failure syndrome, MOFS) with limited data included in the analysis (inadequate power and precision) and remains a surrogate outcome (Analysis 1.17). None of the other secondary outcomes reached statistical significance.
Neither the meta‐analysis nor the subgroup analyses demonstrated a statistically significant effect of AT III on mortality. However, this is not evidence of the absence of a beneficial effect, but the data suggest that a potentially beneficial effect of AT III must be at best modest compared to what had been expected. Trial sequential analysis added valuable information to the level of evidence and as such we are able to reject an intervention effect of 10% RRR or more for all trials, as well as trials including participants with sepsis and DIC. Additionally, based on the existing level of evidence from trials with low risk of bias, only 16.43% of the required information size is available to reject or accept a 5% RRR for overall mortality.
Subgroup analysis of duration of intervention and length of follow‐up
Based on follow‐up less than or longer than the median of all trials, we undertook a subgroup analysis to examine the intervention effect on mortality. However, there was no statistically significant association between follow‐up and mortality (see Analysis 1.2). The median follow‐up time was 32 days.
We also examined the intervention effect based on the median duration of intervention being less than or longer than one week (Analysis 1.3). Only three trials with a total of 208 participants had a median duration of intervention longer than one week (Inthorn 1997; Mitchell 2003; Smith‐Erichsen 1996). The current evidence does not support a longer duration of intervention.
Subgroup analyses on paediatric, obstetric and trauma populations
Based on the existing data, we have to conclude that there is insufficient data to help us support or refute the use of AT III intervention among trauma, obstetric, or paediatric populations.
Subgroup analyses regarding septic populations
Very few trials met our requirements in terms of trial intervention effect on various illness scores. We accepted the various definitions provided by the authors and undertook four different meta‐analyses. The participant numbers in these analyses ranged from 28 to 156, and only one meta‐analysis reached statistical significance (Analysis 1.17). The meta‐analyses examining the overall mortality in the septic population, based on 2918 participants, also failed to demonstrate a statistically significant reduction of mortality (Analysis 1.10).
The heparin issue
We examined a potential detrimental interaction of AT III with heparin by carrying out three separate analyses pooling mortality data from trials with concomitant heparin use against those without (Analysis 1.7; Analysis 1.8; Analysis 1.9), while examining the impact of data from Warren 2001. The latter trial was either defined as a trial with or without heparin use (Analysis 1.7; Analysis 1.8) and finally we chose to split data from Warren 2001 in order to examine the hypothesis. As such, this is to be considered a post hoc analysis violating the randomization procedure (Analysis 1.9). However, none of these analyses demonstrated any statistically significant interaction effects.
Overall completeness and applicability of evidence
The authors of this updated review are confident that our search strategy obtained all available studies. We have contacted several authors. We identified two additional trials by Neporada et al. One was included (Neporada 2008A) and one excluded Neporada 2008B.
Quality of the evidence
The randomized controlled trial (RCT) is considered the most rigorous method of determining whether a cause‐effect relationship exists between an intervention and outcome. The strength of the RCT lies in the process of randomization.
We rank the quality of findings from moderate to very low quality of evidence across the different outcomes. The main limiting factors were high risk of bias and small and poorly described trials.
Nine trials were reported as being at completely low risk of bias (Baudo 1998; Fourrier 1993; Fulia 2003; Haire 1998; Kobayashi 2003; Maki 2000; Schmidt 1998; Warren 2001; Waydhas 1998) (Figure 4; Figure 3). 16 of the of 30 included trials were at high risk or unclear risk of bias in random sequence generation (selection bias) (Balk 1995; Baudo 1992; Blauhut 1985; Diaz‐Cremades 1994; Eisele 1998; Grenander 2001; Harper 1991; Inthorn 1997; Langely 1993; Muntean 1989; Nishiyama 2011; Palareti 1995; Schorr 2000; Schuster 1997; Smith‐Erichsen 1996; Vorobyeva 2007). Only one trial was registered on an available trial database (Gando 2013) Three trials had received funding from the pharmaceutical industry (Grenander 2001; Langely 1993; Warren 2001).
The five trials (Balk 1995; Blauhut 1985; Muntean 1989; Palareti 1995; Schuster 1997) only published as abstracts lack a great amount of valuable information with regard to methodology and outcomes, and we consequently rate them at high risk of bias.
Application of the GRADE approach enables us to incorporate risk of bias, directness of evidence, heterogeneity, precision of effect estimate, and risk of publication bias. Based on these criteria, the quality of evidence in this review was very low and there was a high risk of bias.
Potential biases in the review process
Our systematic review has several potential limitations. As for all systematic reviews, our findings and interpretations are limited by the quality and quantity of the available evidence on the effects of AT III on mortality. We assessed the risk of bias of the included trials by using the published data, which ultimately may not reflect the truth. We tried to contacted all authors but only a few responded and provided further information. Three trials with 260 participants reported zero mortality in both trial groups (Kobayashi 2003; Maki 2000; Mitchell 2003).
We include five trials submitted only as abstracts in this updated review (Balk 1995; Blauhut 1985; Muntean 1989; Palareti 1995; Schuster 1997). These abstracts lack important information. Nevertheless, a sensitivity analysis on the mortality data of only these trials yielded a RR of 0.91 (95% CI 0.55 to 1.52, I² statistic = 0%, fixed‐effect model). The result of the sensitivity analysis is highly comparable with and supportive of the overall mortality (Analysis 1.1).
Due to a lack of convincing evidence in favour of AT III in settings without heparin, we chose to implement trial sequential analysis results, since the hypothesis of a beneficial effect of AT III in critically ill people still generates much attention.
Although there was minimal heterogeneity among trial results on mortality, we are aware that we pooled very heterogeneous trials in terms of participants, settings, and treatment regimens. However, all the included conditions cause low levels of AT III, can result in DIC, and have similar inflammatory pathways. We therefore think that there is a biologically plausible reason to perform an inclusive meta‐analysis, which also considerably increases the generalizability and usefulness of the review. Furthermore, a broad meta‐analysis increases power, reduces the risk of erroneous conclusions, and facilitates exploratory analyses which can generate hypotheses for future research (for example, adjuvant heparin) (Gotzsche 2000).
We have adhered to Cochrane methodology and applied additional statistical methods, such as TSA, to strengthen our conclusions and reduce the risk of random error.
Agreements and disagreements with other studies or reviews
The overall results of this updated systematic review are in accordance with the two previously published papers on the same topic, and only highlight the previously stated findings through inclusion of several additional studies. (Afshari 2007; Afshari 2008)
Authors' conclusions
Implications for practice.
There is insufficient evidence to support AT III substitution in any category of critically ill people. We did not find a statistically significant effect of AT III on mortality, but AT III increased the risk of bleeding events. Subgroup analyses performed according to duration of intervention, length of follow‐up, different patient groups, and use of adjuvant heparin did not show differences in the estimates of intervention effects. Serious methodological shortcomings of the included studies are, however, likely to have influenced the overall intervention effect (Table 1).
Trial sequential analysis showed that there is sufficient evidence to reject a beneficial effect of more than 10% RRR (4% absolute risk reduction) on overall mortality and for trials including participants with sepsis and DIC. There also remains the possibility that the use of AT III may be harmful.
The GRADE approach only reaffirmed our interpretation of the level of evidence, and we are confident that at this stage the quality of evidence in regard to our primary outcomes is moderate, despite the fact that many of the trials have high risk of bias.
Implications for research.
To our knowledge only one trial (D'angelo 2005) is ongoing. The participants are women with pre‐eclampsia occurring before the 30th week of gestation. We have found no trials registered dealing with the effect of AT III in septic patients.
There is a need for a large‐scale randomized controlled trial with low risk of bias to evaluate the effectiveness of AT III without heparin, before this intervention can be used routinely in critically ill patients. We recognize the heterogeneity in the patient population in the included trials and, as a consequence of the high mortality rate in the septic population, we believe that a new trial should address the effect of AT III in septic patients.
What's new
| Date | Event | Description |
|---|---|---|
| 13 December 2018 | Amended | Editorial team changed to Cochrane Emergency and Critical Care |
History
Protocol first published: Issue 3, 2005 Review first published: Issue 3, 2008
| Date | Event | Description |
|---|---|---|
| 4 January 2016 | New citation required but conclusions have not changed | A new lead author (Allingstrup M) has updated this review in collaboration with the original authors. In this updated review we chose to include trials which had only been published as abstracts (Balk 1995; Blauhut 1985; Muntean 1989; Palareti 1995; Schuster 1997). These trials report mortality data from 236 participants. The trial abstracts do not provide us with much data to examine. However, we provide the limited mortality data that we were able to retrieve from these trials. We classify all of these trials as being at high risk of bias. The conclusions have not changed in this updated review. |
| 4 January 2016 | New search has been performed | We searched the databases until 27 August 2015. We included 10 new antithrombin III (AT III) trials in this updated review. This review now has 30 included studies in total (3933 participants). We have updated the Methods section, included full 'Risk of bias' tables and 'Summary of findings' tables. We applied trial sequential analysis (TSA). |
| 11 February 2013 | Amended | Contact details updated; order of tables corrected |
| 12 October 2010 | Amended | Contact details updated. |
| 11 May 2010 | Amended | Contact details updated. |
| 7 August 2008 | Amended | Minor edits to text |
| 12 May 2008 | Amended | converted to new Review format |
Acknowledgements
We first thank Dr John Carlisle, Prof Marcus Müllner, Dr Francois Fourrier, Dr Geoffrey Playford, Dr Christian Josef Wiedermann, Kathie Godfrey, and Nete Villebro for their help and editorial advice during the preparation of the protocol (Afshari 2007) for the original review (Afshari 2008).
We also thank Dr Nadezda Vorobyeva, Dr Elena Neporada, Dr Domenico Paparella, Dr Athina Lavrentieva, and Dr Armando D’Angelo for providing further information about their trials.
We would like to thank Dr Mikhail Zemtsovski for his help with translation of two manuscripts from Russian, and librarian Tove Svendsen (Medical Research Library, Copenhagen University Hospital, Rigshospitalet) for help with updating our literature search.
From the Cochrane Group we would like to thank Nicola Petrucci (Content Editor) and Jing Xie (Sophia) (Statistical Editor), Karen Hovhannisyan (Trials Search Co‐ordinator) for his assistance in providing our different search strategies, and Jane Cracknell (Managing Editor, Cochrane Anaesthesia Critical and Emergency Care Group (ACE)) for her valuable assistance during the entire process.
Appendices
Appendix 1. Search strategies
| Database | Search terms |
| The Cochrane Central Register of Controlled Trials (CENTRAL) | #1 MeSH descriptor: (Antithrombin III) explode all trees #2 (antithrombin III or AT III):ti,ab #3 #1 or #2 |
| CINAHL (EBSCO host) | S1 TI ( antithrombin*) OR AB ( antithrombin*) S2 (random* or ((controlled or clinical) N3 trial*) or placebo* or multicenter or prospective*) or ((blind* or mask*) and (single or double or triple or treble)) S3 S1 and S2 |
| MEDLINE (Ovid SP) | 1. Antithrombins/ or AT?III.mp. or antithrombin*.mp. 2. ((randomised controlled trial or controlled clinical trial).pt. or randomized.ab. or placebo.ab. or clinical trials as topic.sh. or randomly.ab. or trial.ti.) not (animals not (humans and animals)).sh. 3. 1 and 2 () (())) |
| EMBASE (Ovid SP) | 1. antithrombin/ or exp antithrombin III/ or (AT?III or antithrombin*).ti,ab. 2. (randomized‐controlled‐trial/ or randomization/ or controlled‐trial/ or multicenter‐trial/ or phase‐3‐clinical‐trial/ or phase‐4‐clinical‐trial/ or double‐blind‐procedure/ or single‐blind‐procedure/ or (random* or cross?over* or multicenter* or factorial* or placebo* or volunteer*).mp. or ((singl* or doubl* or trebl* or tripl*) adj3 (blind* or mask*)).ti,ab. or (latin adj square).mp.) not (animals not (humans and animals)).sh. 3. 1 and 2 |
| Science Citation Index EXPANDED | #1 TS=(antithrombin* or AT?III) #2 TS=(random* or ((controlled or clinical) SAME trial*) or placebo* or multicenter or prospective) or TS=((blind* or mask*) SAME (single or double or triple or treble)) #3 #1 and #2 |
| Latin American Caribbean Health Sciences Literature (LILACS | "antithrombin$" or "AT III" |
Data and analyses
Comparison 1. AT III versus control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality (subgroup analysis on bias risk) | 29 | 3882 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.88, 1.03] |
| 1.1 Trials with low bias risk | 9 | 2915 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.88, 1.04] |
| 1.2 Trials with high bias risk | 20 | 967 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.77, 1.14] |
| 2 Overall mortality (subgroup analysis on median follow‐up) | 28 | 3848 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.88, 1.03] |
| 2.1 Mortality in trials with follow up less than median of all trials | 18 | 1024 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.77, 1.13] |
| 2.2 Mortality in trials with follow up longer than median of all trials | 10 | 2824 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.88, 1.04] |
| 3 Overall mortality (subgroup analysis on duration of intervention) | 28 | 3848 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.88, 1.03] |
| 3.1 Median duration of AT III intervention equal to or less than one week | 25 | 3640 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.88, 1.03] |
| 3.2 Median duration of AT III intervention longer than one week | 3 | 208 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.59, 1.34] |
| 4 Overall mortality (trauma) | 2 | 68 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.15 [0.81, 5.72] |
| 4.1 Trials with high bias risk | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.43 [0.15, 77.58] |
| 4.2 Trials with low bias risk | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.72, 5.59] |
| 5 Overall mortality (obstetrics) | 2 | 332 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.33, 3.21] |
| 5.1 Overall maternal mortality, trials with low bias risk | 2 | 174 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 Overall foetal and neonatal mortality, trials with low bias risk | 2 | 158 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.33, 3.21] |
| 6 Overall mortality (paediatrics) | 4 | 365 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.73, 2.83] |
| 6.1 Trials with low bias risk | 2 | 182 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.6 [0.54, 4.72] |
| 6.2 Trials with high bias risk | 2 | 183 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.56, 3.15] |
| 7 Overall mortality (heparin; Warren 2001 as a trial with adjuvant heparin therapy) | 26 | 3779 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.88, 1.03] |
| 7.1 Trials with complete or partially adjuvant heparin therapy | 16 | 3121 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.88, 1.04] |
| 7.2 Trials without adjuvant heparin | 10 | 658 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.71, 1.23] |
| 8 Overall mortality (heparin; Warren 2001 as a trial without adjuvant heparin therapy) | 26 | 3779 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.88, 1.03] |
| 8.1 Trials with complete or partially adjuvant heparin therapy | 15 | 807 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.79, 1.17] |
| 8.2 Trials without adjuvant heparin | 11 | 2972 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.88, 1.03] |
| 9 Overall mortality (heparin; Warren 2001 data split based on heparin administration) | 26 | 3779 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.88, 1.03] |
| 9.1 Trials with complete or partially adjuvant heparin therapy | 16 | 2423 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.91, 1.09] |
| 9.2 Trials without adjuvant heparin | 11 | 1356 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.77, 1.00] |
| 10 Overall mortality among patients with severe sepsis & DIC | 12 | 2858 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.88, 1.03] |
| 10.1 Trials with low bias risk | 4 | 2529 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.88, 1.04] |
| 10.2 Trials with high bias risk | 8 | 329 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.64, 1.20] |
| 11 Complications during the inpatient stay specific to the trial intervention | 3 | 2454 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.83, 1.92] |
| 11.1 Trials with low bias risk | 2 | 2429 | Risk Ratio (M‐H, Random, 95% CI) | 1.62 [0.96, 2.73] |
| 11.2 Trials with high bias risk | 1 | 25 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.51, 1.66] |
| 12 Complications during the inpatient stay not specific to the trial intervention | 2 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.08, 6.11] |
| 12.1 Trials with low bias risk | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 69.52] |
| 12.2 Trials with high bias risk | 1 | 25 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.04, 2.57] |
| 13 Complication specific to the trial intervention other than bleeding | 3 | 187 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.42, 1.25] |
| 13.1 Trials with low bias risk | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.18, 3.07] |
| 13.2 Trials with high bias risk | 2 | 127 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.40, 1.30] |
| 14 Bleeding events | 11 | 3019 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [1.35, 1.84] |
| 14.1 Trials with low bias risk | 6 | 2791 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [1.35, 1.85] |
| 14.2 Trials with high bias risk | 5 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.57 [0.71, 3.49] |
| 15 Amount of red blood cells administered | 4 | 137 | Mean Difference (IV, Random, 95% CI) | 138.49 [‐391.35, 668.34] |
| 15.1 Trials with low bias risk | 1 | 35 | Mean Difference (IV, Random, 95% CI) | ‐600.0 [‐899.18, ‐300.82] |
| 15.2 Trials with high bias risk | 3 | 102 | Mean Difference (IV, Random, 95% CI) | 595.10 [‐287.14, 1477.34] |
| 16 Incidence of surgical intervention | 3 | 103 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.85, 1.27] |
| 16.1 Trials with low bias risk | 3 | 103 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.85, 1.27] |
| 17 Severity of sepsis I | 3 | 156 | Mean Difference (IV, Random, 95% CI) | ‐1.24 [‐2.18, ‐0.29] |
| 17.1 Final MOF score among survivors, trials with low bias risk | 1 | 88 | Mean Difference (IV, Random, 95% CI) | ‐0.70 [‐1.22, ‐0.18] |
| 17.2 Final MOF score among survivors, trials with high bias risk | 2 | 68 | Mean Difference (IV, Random, 95% CI) | ‐1.92 [‐3.05, ‐0.78] |
| 18 Severity of sepsis II | 3 | 102 | Mean Difference (IV, Fixed, 95% CI) | ‐2.18 [‐4.36, ‐0.00] |
| 18.1 Final APACHE I & II scores among survivors, trials with high bias risk | 3 | 102 | Mean Difference (IV, Fixed, 95% CI) | ‐2.18 [‐4.36, ‐0.00] |
| 19 Incidence of respiratory failure not present at admission | 6 | 2591 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.76, 1.14] |
| 19.1 Trials with low bias risk | 5 | 2564 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.77, 1.22] |
| 19.2 Trials with high bias risk | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.45, 1.18] |
| 20 Duration of mechanical ventilation | 3 | 190 | Mean Difference (IV, Fixed, 95% CI) | 2.20 [‐1.21, 5.60] |
| 20.1 Trials with low bias risk | 2 | 162 | Mean Difference (IV, Fixed, 95% CI) | 2.26 [‐1.69, 6.22] |
| 20.2 Trials with high bias risk | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | 2.0 [‐4.68, 8.68] |
| 21 Length of stay in hospital | 4 | 202 | Mean Difference (IV, Random, 95% CI) | 1.10 [‐7.16, 9.36] |
| 21.1 Trials with low bias risk | 2 | 89 | Mean Difference (IV, Random, 95% CI) | ‐5.67 [‐16.24, 4.90] |
| 21.2 Trials with high bias risk | 2 | 113 | Mean Difference (IV, Random, 95% CI) | 7.17 [2.75, 11.59] |
| 22 Mean length of stay in ICU | 7 | 376 | Mean Difference (IV, Fixed, 95% CI) | 0.24 [‐1.34, 1.83] |
| 22.1 Trials with low bias risk | 3 | 195 | Mean Difference (IV, Fixed, 95% CI) | ‐0.73 [‐3.41, 1.95] |
| 22.2 Trials with high bias risk | 4 | 181 | Mean Difference (IV, Fixed, 95% CI) | 0.77 [‐1.20, 2.74] |
1.19. Analysis.
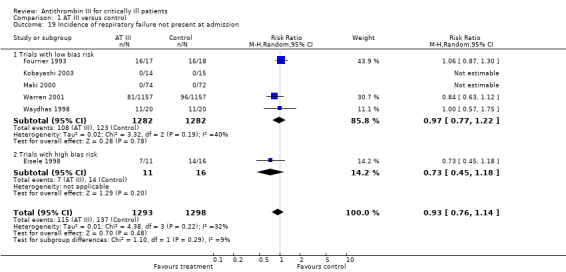
Comparison 1 AT III versus control, Outcome 19 Incidence of respiratory failure not present at admission.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Albert 1992.
| Methods | 2‐group parallel randomized clinical trial ITT: no No sample size calculation was reported | |
| Participants | 32 patients in the ICU with plasma AT III levels less than 70%. All participants included in the trial within 2 days after admission to the ICU AT III group: 16 participants, 8 men, median age: 63.5 years. Median APACHE II score on inclusion: 14, TISS : 34. Reason for admission: surgery 7 patients, trauma 4 patients, infection 3 patients, and other causes: 2 patients Control group: 16 participants, 9 men, median age: 66 years. Median APACHE II score on inclusion: 15, TISS : 39. Reason for admission: surgery 5 patients, trauma 4 patients, infection 5 patients, and other causes: 2 patients Exclusion criteria: under 18 years old, liver disease, HIV infection, severe brain damage, patients admitted for postoperative pain relief only, anorexia nervosa, and pregnancy |
|
| Interventions | AT III: concentrate by intravenous infusion for 20 ‐ 30 minutes twice daily as long as AT III < 90%, in a dose intended to raise the level above 100% until the patients were discharged from the ICU. First dose: 2000 ‐ 4000 IU, total amount: 3500 ‐ 17000 IU Control: no placebo Unfractionated heparin 2500 or 5000 IU x 2 or x 3 daily was given subcutaneously to all patients for thromboprophylaxis. If continuous arteriovenous haemofiltration for acute anuria, then 20000 ‐ 40000 IU heparin per 24 hours was administered as continuous infusion. Blood samples from each patient were taken daily for 7 days or until discharge. Standard intensive care treatments were administered to all patients in both groups. |
|
| Outcomes | Primary: mortality Secondary: days in ICU, bleeding and transfusion, AT III, CRP, tissue plasminogen activator inhibitor, partial thromboplastin time, prothrombin complex (prothrombin time), fibrinogen degradation products, lactoferrin/serum, lactoferrin/plasma, lysozyme, side effects, APACHE II score and TISS (during the first 24 hours) |
|
| Notes | Country: Sweden
Letter sent to authors in November 2005. Reply received in December 2005
2 participants randomized but not evaluated: 1 transferred to another hospital, the other had received AT III although randomized to the non‐AT III group. There were 3 cases of cross‐over: these participants received AT III despite being allocated to the control group. These participants were evaluated up to the day when they received AT III. No side effects were reported. Funding bias: No funding by industry |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random computer number generator |
| Allocation concealment (selection bias) | Low risk | Sealed opaque envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unclear |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals were specified. |
| Selective reporting (reporting bias) | Unclear risk | Unclear |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Balk 1995.
| Methods | 2‐group parallel randomized clinical trial | |
| Participants | Adults patients with SEPSIS, cared for in a tertiary care centre, Chicago, IL, USA 34 participants, 20 women, 14 men. 65% hypotensive at the time of enrolment Median APACHE score for the intervention group was 21.5 (range 2 ‐ 29) and for the control group 17 (range 6 ‐ 31). The AT III % activity at entry was 75.8 + 18.0% for the intervention group and 78.1 + 16.8% for control group. The article does not state how many participants there are in each group. In this review we assume that there are 17 in each group. |
|
| Interventions | AT III (Kypernin) and placebo. No details stated Use of heparin not stated |
|
| Outcomes | Follow for 28 days. Mortality and organ failure was measured. | |
| Notes | Country: Germany Trial only published as an abstract. No data on follow‐up, no data on treatment duration Funding bias: Not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Double‐blinded but no additional information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | Unclear risk | Not stated |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Baudo 1992.
| Methods | 2‐group randomized clinical trial ITT: yes No sample size calculation was reported | |
| Participants | 29 patients with post‐necrotic cirrhosis undergoing liver transplantation were randomized to receive AT III (13 participants) or to control group (no placebo, 16 participants) ‐ before the induction of anaesthesia and during surgery. AT III group: 13 men, 4 patients with cryptogenetic cirrhosis, 4 with post‐necrotic cirrhosis, 5 with alcoholic cirrhosis, average age 44 ± 9 years, duration of surgery (mins): 625 ± 68 Control group: 16 men, 5 patients with cryptogenetic cirrhosis, 7 with post necrotic cirrhosis, 4 with alcoholic cirrhosis, average age 42 ± 9 years, duration of surgery (mins): 642 ± 113 |
|
| Interventions | AT III: administered by bolus infusion before the induction of anaesthesia in order to obtain a pre‐operative activity of 100%. Thereafter 1000 units per hour were given by continuous infusion until the end of surgery. No systemic heparinization Control: no placebo |
|
| Outcomes | Mortality. Blood loss, transfusion requirements, activation of coagulation and fibrinolysis (thrombin‐antithrombin complexes), activated partial thromboplastin time, prothrombin time, thrombin time, reptilase time, expressed as ratio, fibrinogen, plasminogen, alfa2‐antiplasmin, AT III, platelet count, thrombin‐antithrombin complexes, total fibrinogen, fibrinogen and fibrin degradation products | |
| Notes | Country: Italy
Letter sent to authors in January 2006, no reply received Funding bias: Unclear. Not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals were specified. |
| Selective reporting (reporting bias) | Unclear risk | Not stated |
| Other bias | Unclear risk | Not stated |
Baudo 1998.
| Methods | 2‐group parallel randomized clinical trial ITT: no Sample size calculation reported | |
| Participants | Multicentre trial, 120 patients admitted to the ICU with an AT III concentration of < 70% AT III group: 60 participants, 42 men, age distribution: 58.6 ± 13.8 years, MOFS 5.6 ± 2.5, SAPS: 15.6 ± 4.4, 49% sepsis, 33% septic shock, 51% respiratory support, 53% haemodynamic support, haemorrhages: 5%, surgery 47%, surgery with sepsis 43%, AT III: 52.8 ± 15.5%. Control group: 60 participants, 41 men, age distribution 62±12.2 years, MOFS: 4.8 ± 2.3, SAPS: 16.5 ± 5.5, 51% sepsis, 23% septic shock, 48% respiratory support, 42% haemodynamic support, haemorrhages: 6%, surgery 45%, surgery with sepsis 41%, AT III: 52.9 ± 14.5% Inclusion criteria: 1) respiratory and/or haemodynamic support 2) sepsis: systemic response to infection manifested by 2 or more of a) tp > 38.5º C, b) heart rate > 90/min, c) respiratory rate > 20/min, d) leukocytosis > 15 x 10⁹/l or leukopenia < 4 x 10⁹/l, e) septic shock with sepsis‐induced hypotension requiring vasoactive drugs for > 24 hrs persisting despite adequate fluid replacement along with presence of hypoperfusion abnormalities that may include but are not limited to lactic acidosis, oliguria, or an acute alteration of mental status, f) post‐surgical complications requiring respiratory and/or haemodynamic support, g) respiratory support with assisted or controlled ventilation > 24 hrs, h) haemodynamic support inotropic (dopamine/dobutamine ≥ 5 mg/kg/min) and/or vasoactive (epinephrine or norepinephrine) 3) 18 ‐ 75 years 4) AT III activity < 70% Exclusion criteria: 1) Multiple trauma, 2) liver cirrhosis/acute liver failure, 3) Cancer in a terminal phase, 4) Immunodeficiency, 5) leukaemia, 6) pregnancy, 7) heparin therapy (except prophylaxis heparin, 5000 units subcut) |
|
| Interventions | AT III: total dose 24,000 units, fixed dose of 4000 AT III and 2000 IU every 12 hours, 5 days, infusion by pump. Control: human albumin, 50 g/l, 4 g albumin bolus in 30 mins, 1 bottle containing 2 g albumin every 12 hrs for 5 days, pump‐driven Standard intensive care treatment, FFP infused when bleeding and/or prothrombin time ratio > 2, platelet concentration infused when ≤ 50 (1 unit/10 kg weight) |
|
| Outcomes | Primary: mortality, survival for 30 days, MOFS for 7 days, FFP and PC requirements Secondary: days in ICU, bleeding and transfusion, MOFS, septic shock survival, AT III plasma concentration |
|
| Notes | Country: Italy
Letter sent to authors in December 2005, no reply received.
Balanced randomization within each centre, post hoc analysis of septic shock subgroup. The distribution of participants with septic shock on haemodynamic support were unbalanced in the 2 groups at admission. SAPS and MOFS recorded for each participant at admission 4 participants received therapy for < 24 hrs; 1 participant in the placebo group was transferred to another hospital after the bolus infusion and was considered not evaluable; 3 participants with septic shock (2 AT III and 1 placebo) died within 24 hrs. A total of 119 were included in the analysis. Funding bias: ATIII concentrate and placebo (albumin solution 50 g/t) were supplied by Immuno (Wien). No additional information |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization was balanced within each centre by coded envelopes centrally provided and centrally generated by computer. |
| Allocation concealment (selection bias) | Low risk | Randomization was balanced within each centre by coded envelopes centrally provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The attending physicians, evaluating the clinical course, were blind to treatment allocation and to sequential measurements of ATIII levels. Bottles, syringes, and infusion sets were black and identical. The analysis was carried out before the codes were broken. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals were specified. |
| Selective reporting (reporting bias) | Low risk | Published paper appears to include all outcomes expected. |
| Other bias | Low risk | Appears free of such biases |
Blauhut 1985.
| Methods | 3‐group parallel randomized clinical trial ITT: no No sample size calculation was reported | |
| Participants | 51 patients with DIC and septic shock Inclusion criteria: AT III activity < 70% and at least 3 of the following: platelet count < 100, thrombin time > 24 sec, thrombin coagulase time > 22 sec, fibrinogen level < 150 mg/dl, ethanol gelatin test positive |
|
| Interventions | Control group 1: Heparin: iv heparin 3000 IU followed by continuous infusion of 250 IU/h Intervention group 2: AT III substitution with the aim to keep ATIII activity constantly around 100% Intervention group 3: Same AT III substitution as group 2 with heparin 1000 IU iv and continuous infusion of 100 IU/h |
|
| Outcomes | Blood loss, concentration of ATIII, duration of symptoms of DIC, platelet count, esterase inhibitor level, blood loss | |
| Notes | Country: Austria
No primary endpoint, unable to retrieve additional data from authors despite several attempts to contact them. 12 participants died during the trial but no data on distribution between the groups except no statistically significant difference. Funding bias: No information |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Unable to retrieve relevant data on mortality |
| Selective reporting (reporting bias) | Unclear risk | No information or protocol available |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Diaz‐Cremades 1994.
| Methods | 2‐group parallel randomized clinical trial ITT: no No sample size calculation was reported | |
| Participants | 195 critically ill patients admitted to ICU, without previous history of hepatic disease, 36 patients were chosen with AT III levels below 70% as inclusion criteria. None of the patients had manifest DIC. AT III: 20 participants (10 with sepsis, 6 with trauma, 1 with sepsis + trauma, 3 with shock), 12 men, mean age 46.25 ± 11.79 years Control group: 16 participants (8 with sepsis, 4 with trauma, 2 with sepsis + trauma and 2 with shock), 10 men, mean age 56.37 ± 25.78 years Inclusion or exclusion criteria were not provided. |
|
| Interventions | AT III group: initial dose 60 U/kg of body weight, then 10 U/kg every 6 hrs, without dose adjustment depending on the AT III levels. The mean AT III dose received was 11.17 ± 5.98 U. Treatment interrupted when the participants were discharged from ICU, or if they achieved AT III levels equal or superior to 90% during at least 48 hrs. Mean length of treatment: 5.8 ± 4.2 days Control: human albumin 0.6% Standard intensive care treatment in both groups. All participants received 7500 IU of prophylactic subcutaneous calcium heparin every 12 hrs. |
|
| Outcomes | Primary: mortality Secondary: 1) platelet count, 2) side effects, 3) prothrombin time, 4) activated thromboplastin time, 5) fibrinogen, 6) fibrinogen degradation products, 7) AT III, 8) protein C, 9) APACHE II score |
|
| Notes | Country: Spain
Letter sent to authors in December 2005, no reply received. No adverse effects reported. The only detectable alterations in the haemostasis were a mild thrombocytopenia and low levels of AT III and protein C. Manifest DIC was not reported in any participant. Funding bias: AT III concentrate infusion provided by Kybernin‐P, Institute Behring, Barcelona. No additional information |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | No information available |
| Allocation concealment (selection bias) | High risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open, unblinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | Unclear risk | Not stated |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Eisele 1998.
| Methods | Randomized clinical trial ITT: yes No sample size calculation was reported | |
| Participants | Multicentre, multinational trial of patients admitted to ICU AT III group: 20 participants, 11 men, mean age: 57 ± 16 yrs, weight (kg): 71 ± 14, height (cm): 171 ± 9 Underlying disease: peritonitis 2, mediastinitis 1, neoplasia (non‐haematologic) 4, others: 12. Severity of sepsis at trial entry (mean ± SD): APACHE II: 13.5 ± 5.1, MOF: 6.1 ± 2.1, OFS:1.2 ± 0.9, AT III activity: 45.7 ± 14.4% Coagulation failure: 3, acute hepatic dysfunction: 5, acute renal dysfunction: 8, acute respiratory dysfunction: 8, acute neurological dysfunction: 6. Control group: 22 patients, 10 men, mean age: 58 ± 14 yrs, weight (kg): 73 ± 15, height (cm): 169 ± 8 Underlying disease: peritonitis 3, mediastinitis 1, adult respiratory distress syndrome 1, acute pancreatitis 3, neoplasia (non‐haematologic) 3, other: 11. Severity of sepsis at trial enter (mean ± SD): APACHE II: 15.5 ± 5.7, MOF: 6.7 ± 2.0, OFS: 0.9 ± 0.8, AT III activity: 49.0 ± 19.1%. Coagulation failure: 2, acute hepatic dysfunction: 8, acute renal dysfunction: 8, acute respiratory dysfunction: 6, acute neurological dysfunction: 10 Inclusion criteria: < 80 and > 18 years old; fulfilled the following criteria within 6‐hr window prior to initiation of treatment: 1) clinical evidence of sepsis with a suspected source of infection; 2) temperature rectally > 38.5º C or < 35.5º C; 3) leukocyte count > 10000/mm³ or < 3500. Furthermore 3 of the following had to be fulfilled: a) heart rate > 100/min; b) breath rate > 24/min or mechanical ventilation because of septic indication; c) hypotension, sys BP < 90 mm Hg when no vasoactive agent or fluid replacement; d) platelet count < 100,000/mm³; e) lactate > 1.6 mmol/l; f) urine output < 20 ml/h. Exclusion criteria: a) heparin (except sc low dose); b) malignancies (incurable with metastases); c) haematologic neoplasia (except complete remission); d) chronic treatment with high‐dose immunosuppressive drugs; e) high‐dose NSAID within previous 2 days; f) known bleeding disorder; g) ongoing massive surgical bleeding; h) multiple organ failure, at least 4 organs involved existing for over 24 hrs; i) acute myocardial Infarction; j) chronic compensated organ dysfunction (chronic liver disease, dialysis, NYHA 3 ‐ 4); k) complicated pre‐existing diabetes mellitus; l) severe obstructive pulmonary disease; m) burn; n) AIDS; o) transplant; p) severe cranial trauma and/or pathologic changes on CT and GCS < 6; q) pregnancy; r) treatment with plasma expanders (Haes); s) prior enrolment in the trial or another clinical trial within the last 30 days |
|
| Interventions | Experimental: AT III iv loading dose: 3000 units AT III, maintenance 1500 IU every 12 hours, 5 days treatment, cumulative dose: 18,000 IU Control: placebo equivalent amounts, unknown agent Standard intensive care treatment in both groups |
|
| Outcomes | Primary: 30‐day all‐cause mortality Secondary: survival days, days in ICU, safety (bleeding and transfusion), MOFS, APACHE II, OSFS, AT III activity, AT III plasma concentration, resolution of acute pre‐existing organ dysfunction (neurological, respiratory, renal, hepatic and coagulation) and incidence of organ dysfunction |
|
| Notes | Country: Germany, Belgium, the Netherlands
A letter sent to authors in December 2005, no answer received APACHE II, MOF and OSF scores were calculated on days 1, 2, 3, 7, 10, 20 and 30. No adverse events reported Funding bias: AT III by Kybernin R Centeon Pharma GmbH, Marburg, Germany. No additional information provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No indications of incomplete data |
| Selective reporting (reporting bias) | Unclear risk | Unable to assess |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Fourrier 1993.
| Methods | 2‐group parallel randomized clinical trial ITT: yes Sample size was calculated to detect a 50% reduction in mortality in ICU | |
| Participants | Critically ill patients with septic shock and DIC and MOF with at least 1 organ failure added to circulatory and haematologic failures AT III group: 17 participants, 10 men, mean age (SD): 52 ± 22 yrs. Underlying disease: alcoholism 5; chronic cardiac failure 8; COPD 2. No underlying disease 3; recent surgery 6. SAPS: 20 ± 7. Organ failures: 2.8 ± 1.0, renal failure: 57%, respiratory failure: 93% Control group: 18 patients, 10 men, mean age (SD): 52 ± 18 yrs. Underlying disease: alcoholism 3; chronic cardiac failure 9; COPD 3. No underlying disease 4; recent surgery 6. SAPS: 19 ± 6. Organ failures: 2.8 ± 0.9, renal failure: 56%, respiratory failure: 89% Inclusion criteria: a) mandatory criteria for septic shock: 1) bacteraemia or evidence of septic focus; 2) fever > 38.5º C; 3) leukocyte count > 15 x 10⁹/L or < 4 x 10⁹; 4) hypotension, sys BP < 90 mmHg despite adequate vascular filling; 5) requirement of vasoactive agent (dopamine > 15 µg/kg/min, dobutamine, epinephrine or norepinephrine) during more than 12 hrs b) Mandatory DIC criteria: 1) platelet counts < 100x10⁹/L or a decrease of > 100 x 10⁹ over the last 24 hrs; 2) decrease in factor V level < 70%; 3) presence of soluble complexes of fibrin monomers Exclusion criteria: a) < 16 years old; b) suspected pregnancy; c) sepsis or DIC within 8 days after delivery; d) chronically immunocompromised; e) bone marrow aplasia or acute leukaemia; f) chronic liver failure with cirrhosis, recent hepatic surgery or transplant; g) multiple trauma or uncontrolled haemorrhage that required massive blood transfusion or undergone plasma exchange (< 5 days) |
|
| Interventions | Experimental: 90 ‐ 120 IU/kg, loading dose over 3 hrs (3 ml/kg) + 3 ml/kg over 21 hrs, then 90 ‐ 120 IU/kg/day for 3 days by continuous infusion. The aim: supranormal AT III levels. Average of 6000 IU/day Control: human albumin 0.6%, equivalent amount in ml Standard intensive care treatment to all participants. FFP, PC, and fibrinogen concentrates restricted to haemorrhages and severe decreases in prothrombin time to < 30%, platelet count to < 50 x 10⁹/L (1/10 kg of body weight), and fibrinogen levels to < 1 g/L. LMWH 0.5 mg/kg of body weight when used in lines and filters of haemodialysis or haemofiltration |
|
| Outcomes | Primary: 28‐day mortality. Secondary: recovery of DIC, side effects, length of stay, transfusion and vascular requirements, bleeding, numbers and types of organ failure, organ function scores, protein C and S activity, fibrinogen and AT III activity, fibrin degradation products, factor V and VII, cardiac index, systemic vascular resistances, blood lactate, total bilirubin, blood creatinine, omega score, surgery during treatment |
|
| Notes | Country: France
Letter sent to authors in November 2005, authors replied in December 2005. Heparin therapy was not used as control to avoid haemorrhagic hazards and discrepancies related to heparin‐induced AT III consumption. Since FFP contains significant amount of AT III, infusion was restricted. No side effects reported. Funding bias: Supported by a grant from the French National Institute of Health and Medical Research: INSERM |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central computer‐generated |
| Allocation concealment (selection bias) | Low risk | Central computer‐generated with central allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The treatment‐allocation code, randomization, preparation of AT III, and placebo vials were designed and conducted by a co‐ordinator blinded to clinical data and patients’ inclusion. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Clinical analysis was done by physicians blinded to the treatment allocation and results of AT III measurements. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No indications of incomplete data |
| Selective reporting (reporting bias) | Low risk | All outcomes assessed |
| Other bias | Low risk | Appears free of such biases |
Fulia 2003.
| Methods | 2‐group parallel randomized clinical trial ITT: yes No sample size calculation was reported | |
| Participants | 60 infants born before 30 weeks of gestation randomized to a loading dose of AT III or placebo AT III: 30 infants, 16 male, gestational age (weeks): 28.5 ± 1.5, birth weight (g): 1060 ± 218, vaginal delivery: 12, elective caesarean: 18, antenatal steroids: 16, APGAR score at 1 min: 5.83 ± 0.75, APGAR score at 5 min: 7 ± 0.64, cord ABG pH: 7.28 ± 0.07 Control: 30 infants, 17 male, gestational age (weeks): 28.5 ± 1.3, birth weight (g): 1054 ± 233, vaginal delivery: 11, elective caesarean: 19; antenatal steroids: 17, APGAR score at 1 min: 5.63 ± 0.82, APGAR score at 5 min: 6.88 ± 0.76, umbilical cord arterial blood gas pH: 7.31 ± 0.08 Inclusion criteria: gestational age < 30 weeks, postnatal age < 12 hrs, AT III activity < 40% (normal value 20 ‐ 72%) Exclusion criteria: sepsis, congenital malformations, cerebral haemorrhage, bleeding disorders, thrombocytopenia (platelet count of 50 x 10⁹ or less) |
|
| Interventions | AT III group: loading dose of 2 ml/kg equivalent to 100 U/kg of AT III followed by 1 ml/kg equivalent to 50 U/kg every 8 hrs for 48 hrs Control: 5% glucose in equivalent amounts (ml) Arterial catheters were perfused with 1 ml/h of 5% glucose containing I U/ml of unfractionated heparin. No other anticoagulants were administered. All newborns received the same exogenous surfactant |
|
| Outcomes | Primary: death and intraventricular haemorrhage (grade 0 ‐ IV) Secondary: partial thromboplastin time, quick prothrombin time, platelet count, surfactant need, pneumothorax, pulmonary haemorrhage, patent ductus arteriosus inotropes, bronchopulmonary dysplasia, level of AT III, degree of IVH measured by ultrasonography. |
|
| Notes | Country: Italy
Letter sent to authors in January 2006, no answer received Funding bias: Not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central number generation (computer) |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals were specified. |
| Selective reporting (reporting bias) | Low risk | All expected outcomes appear included. |
| Other bias | Low risk | Appears free of such biases |
Gando 2013.
| Methods | 2‐group parallel randomized clinical trial ITT: Yes |
|
| Participants | 60 DIC patients with sepsis and antithrombin levels of 50 to 80% Location: 13 critical care centres of tertiary care hospitals from April 2008 to February 2012 Inclusion criteria: Patients with a diagnosis of DIC (JAAM DIC score ≥ 4) with sepsis and levels of antithrombin ranging from 50 ‐ 80% were eligible in the trial Exclusion criteria: Age > 15 years. A history of haematopoietic malignancy. A history of liver cirrhosis classified as Child‐Pugh grade C. Receiving concomitant treatment with chemotherapies or irradiation. A history of known clotting disorders or receiving anticoagulant therapy. In an early phase of trauma or burn injuries. A life expectancy of less than 28 days Sex: 25 women and 35 men Age: Case: 73 ± 15, control 67 ± 17 yrs |
|
| Interventions | Timing of intervention: Immediately after the participants met the inclusion criteria, they were randomly assigned to either a group receiving antithrombin at a dose of 30 IU/kg (given over 60 minutes) per day for 3 days, or to the control group with no intervention The trial reported only whether the participants received blood products or not. The quantity received was not stated No heparin used under the trial |
|
| Outcomes | Length of follow‐up: 28 days
Primary outcome: recovery from DIC on day 3 Secondary outcome: 28‐day all‐cause mortality |
|
| Notes | Country: Japan Unable to contact the authors for additional data despite attempts in 2015 Funding bias: Funded and supported by the Japanese Association for Acute Medicine (JAAM). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Web‐based randomization was generated by the University Hospital Medical Information Network centre. |
| Allocation concealment (selection bias) | Low risk | Web‐based allocation ratio of 1:1 for the control and antithrombin groups was generated by the University Hospital Medical Information Network centre. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. Neither the physicians nor the patients were blinded to the treatment assignment. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals were specified. |
| Selective reporting (reporting bias) | Low risk | Trial registration available on UMIN Clinical Trials Registry. Protocol not published |
| Other bias | Low risk | Appears free of such biases |
Grenander 2001.
| Methods | 2‐group parallel prospective randomized trial ITT: no No sample size calculation was provided | |
| Participants | 28 patients with purely traumatic brain injury and no other major trauma AT III group: 13 participants, 10 men, mean age: 48 yrs. Mean APACHE II score: 14.2, mean GCS: 7.7, median: 7.0; 50% had positive blood alcohol detection Control group: 15 participants, 10 men, mean age: 44 years. Mean APACHE II score: 14.8, mean GCS: 7.5, median: 7.0; 50% had positive blood alcohol detection Inclusion criteria: 1) 14 ‐ 70 yrs old; 2) brain injury was CT‐verified and assessed by GCS; 3) only GCS between 4 ‐ 12 included; 4) inclusion within 12 hrs from the time of injury Exclusion criteria: 1) ongoing dialysis‐dependent renal disorder; 2) bleeding or coagulation disorder such as haemophilia or von Willebrand's disease; 3) anticoagulant therapy |
|
| Interventions | AT III: 60 IU/kg body weight initially. 8 and 16 hrs later an additional 20 IU/kg body weight was given ‐ a total of 100 IU/kg body weight during 24 hrs. The doses were adjusted to the nearest 500 IU. Duration: 24 hrs. Average total dose of AT given was 8269 ± 1562 IU Control: no placebo Both groups received additional treatment according to local standards for brain injury including surgery, ventricular drain, or intraparenchymatous intracranial pressure (ICP) device, moderate hyperventilation (pCO₂ 4 ‐ 4.5 kPa), sedation, drainage of cerebrospinal fluid, low‐dose barbiturates, mannitol infusion, inotropic drugs, clonidine and dihydroergotamine. Treatment goals were ICP < 20 mm Hg and CPP > 60 mm Hg. LMWH 2500 IU sc only to a random group of participants in few events (1 in AT group and 3 in control group). 3 participants in each group received FFP for volume replacement. |
|
| Outcomes | Primary outcome: time of coagulopathy in participants with brain contusion and with or without intracranial haemorrhage, coagulation parameters improvement: soluble fibrin (SF), AT III‐activity, D‐dimer, thrombin‐antithrombin complex (TAT), platelet count, prothrombin complex and fibrinogen, mortality at 3 months Secondary: duration of mechanical ventilation, APACHE II score (slightly modified in order to estimate the severity of illness), improved outcome assessed by GCS (once daily and at 3 months), brain injury progress monitored by CT, bleeding events, days with mechanical ventilation, need for intensive care, and evaluation of severe disability |
|
| Notes | Country: Sweden
Letter sent to authors in December 2005, no answer received. 2 withdrawals (late presentation of exclusion criteria and a revoked consent), 1 late exclusion resulting from the revelation of improper dosage of antithrombin because of incorrect information regarding body weight 1 case of cross‐over. 1 participant in the control group was given AT III by the attending physician as part of the treatment for a septic reaction. Partially missing data on this participant Funding bias: This trial was supported by Pharmacia & Upjohn, Stockholm, Sweden. They provided antithrombin concentrate and funded the trial. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Unclear |
| Allocation concealment (selection bias) | Low risk | Sealed randomization envelopes were opened in numerical order |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not blinded. Open |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label. No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | Low risk | Published report appears to repot all expected outcomes. |
| Other bias | Low risk | Appears free of such biases |
Haire 1998.
| Methods | ||
| Participants | 49 people undergoing haematopoietic stem cell transplantation AT III group: 24 participants, 10 men. Mean age (SD): 46.1 ± 10.3 yrs. Underlying disease: acute myeloid leukaemia 5, breast carcinoma 8, chronic myeloid leukaemia 1, multiple myeloma 3, lymphoma 7 Control group: 25 participants, 11 men. Mean age (SD): 46.0 ± 10.5 yrs. Underlying disease: acute lymphocytic leukaemia 1, acute myeloid leukaemia 4, breast carcinoma 4, chronic lymphocytic leukaemia 2, chronic myeloid leukaemia 4, Hodgkin's disease 1, multiple myeloma 3, lymphoma 6 Inclusion criteria: malignant disease admitted for HSCT, not committed to other confounding clinical trials, English their first language (to allow reliable use of the mini mental status examination) |
|
| Interventions | AT III: 70 U/kg within 24 hrs of organ dysfunction detection, followed by 50 U/kg 8,16, 48 and 72 hrs later. Total dose of AT III: 270 U/kg. Estimated to provide an increment of about 250% activity above pretreatment levels after the 3rd dose. Mean total dose: 20,520 U/participant based on the mean weight of the randomized participants (76 kg) Control: 5% human albumin, volume equal to the calculated volume of AT III concentrate |
|
| Outcomes | Primary: mortality, severity‐of‐illness score, length of hospital stay Secondary: daily screening for pulmonary, hepatic or CNS dysfunction, MODS. Correlation between AT III, number of organ failure and MODS. Number of participants progressing from single through multiple organ dysfunction |
|
| Notes | Country: USA
Letter sent to authors in December 2005, no answer received.
One participant was randomized and received the 1st dose of the trial drug based on an erroneous laboratory report. This participant was not included in the final analysis.
Organ dysfunction defined as: 1) CNS ‐ a drop in the score of standardized mini mental status examination of at least 4 points from the pre‐chemotherapy score; 2) pulmonary ‐ finger oximetry reading of SpO₂ < 90% on 2 occasions on the same day separated by at least 2 hours; 3) peptic ‐ a combination of bilirubin > 2.0 mg%, a weight gain of > 5% over pre‐chemotherapy weight, and abdominal pain of possible hepatic origin
Participants with single‐organ dysfunction with a concomitant AT III activity of < 84% of normal were defined as having MODS Funding bias: Trial drugs were provided by Baxter Healthcare, Glendale, CA, USA. No additional information is provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated list of random numbers |
| Allocation concealment (selection bias) | Low risk | Sealed opaque envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | An unblinded research pharmacist reconstituted the requisite volume of trial drug and provided it to the ward personnel in unmarked containers. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | Low risk | Unable to assess adequately but published report does not appear to exclude outcomes |
| Other bias | Low risk | Appears to be free of other biases |
Harper 1991.
| Methods | 2‐group parallel randomized controlled trial ITT: yes No sample size calculation was provided | |
| Participants | Patients with AT III activity of < 70% admitted to the ICU. Patients with an initial AT activity > 70% monitored daily and were randomized into the trial if their AT activity at any time during their stay fell below 70%. AT III group: 44 participants, median age: 49 yrs (range 19 ‐ 78). Admission AT III activity: 51% (range 12 ‐ 68); APACHE II score median: 16 (range 6 ‐ 33) Control group: 49 participants, median age: 49 yrs (range 16 ‐ 77). Admission AT III activity: 50% (range 12 ‐ 64); APACHE II score median: 14 (range 4 ‐ 26) Exclusion criteria: acute or chronic renal failure prior to admission to the ICU. If readmitted to ICU, the participants were allocated to the same randomization as on their previous admission. Participants were excluded if they stayed less than 4 days on ICU. 10 liver transplant recipients and 15 participants with septicaemia, major trauma, or following major surgery in each group |
|
| Interventions | Treatment: AT III, aiming at 120%. Loading dose was calculated from the initial AT III activity and the participant's weight ((120‐AT III activity (%)) x weight (kg)). Maintenance doses were given iv over 10 mins. Treatment was continued until the participant was discharged from the ICU, twice daily. Control: no placebo Standard intensive care treatment to all participants |
|
| Outcomes | Primary: mortality, coagulation parameters (AT III activity, AT‐antigen concentration, fibrinogen concentration, platelet count). Renal function impaired if creatinine clearance < 20 ml/min at any time during their stay on the ICU Secondary: days in ICU, side effects, APACHE II |
|
| Notes | Country: UK
Letter sent to authors in November 2005, no answer received. 81 participants had an AT III activity below 70% on admission to the ICU and in a further 12 participants the plasma AT III fell below 70% whilst on the ICU. Thus a total of 93 patients were randomized: 44 to the treatment arm and 49 in the no‐treatment arm. 9 participants in the AT III arm did not receive AT III replacement; 3 died before treatment began; and 6 were transferred from the ICU within a few hours of randomization. A total of 35 participants received AT III replacement. Of these 25 remained on the ICU for at least 4 days. 32 participants in the control arm remained on the ICU for at least 4 days. 25 of these were selected by diagnosis to act as controls. The authors provided mortality data on all participants randomized, and we have chosen to include these in our analysis. Funding bias: Unable to assess |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unable to assess |
| Selective reporting (reporting bias) | Unclear risk | Unable to assess |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Inthorn 1997.
| Methods | 2‐group parallel randomized clinical trial ITT: yes Sample size calculation reported | |
| Participants | 40 surgical patients with severe sepsis, post‐traumatic or postoperative admitted to surgical ICU AT III group: 20 participants. Demographic characteristics only provided for the 14 participants included: 13/14 were men. Source of sepsis: peritonitis (10), pneumonia (2), catheter‐related infection (1), late abscess (1). Age: 62.3 ± 3.8 y, MOF score: 4.1 ± 0.5 Control group: 20 participants. Demographic characteristics only provided for the 15 participants included: 11/15 were men. Source of sepsis: peritonitis (12), pneumonia (2), Mediastinitis (1). Age: 61.5 ± 3.6 yrs, MOF score: 3.9 ± 0.4 Inclusion criteria: a) 1 or more blood cultures positive for gram‐positive or ‐negative bacteria or evidence of septic focus; b) temperature above 38.5º C; c) leukocyte count > 15 g/L or < 5% or > 20% of immature (band) forms; d) thrombocyte count < 100 g/L or a decrease of > 20% during the last 24 hours; e) at least 1 organ dysfunction Exclusion criteria: 1) < 18 yrs old; 2) received AT within 21 days prior to the trial |
|
| Interventions | AT III: continuous AT III infusion over 14 days in order to obtain plasma AT III > 120%. Average of 6000 IU AT III was administered on 1st day and 4000 IU on the subsequent days. Control: no placebo treatment All participants received standard relevant intensive care treatment that was comparable between groups and continuous low‐dose heparinization (4 IU/kg bodyweight/h). FFP was only given to participants with severe haemorrhage and markedly impaired plasmatic coagulation (contains AT III); prothrombin complex given only during excessive clinical bleeding; platelets given only when < 50 g/L; packed red blood given when < 10 g/dL |
|
| Outcomes | Primary: 14‐ and 90‐day mortality; mortality between days 7 ‐ 14, hospital discharge Secondary: transfusions, MOFS, frequency of surgical interventions, frequency of DIC, AT III plasma activity, severity of organ dysfunction, effect of AT III on clotting system and MOF, haemodynamic and respiratory profile |
|
| Notes | Country: Germany
Letter sent to authors in November 2005, no answer received.
The results of this trial were also published in 2 additional articles (See Included studies, Inthorn 1997). Comparable participants in both groups and no significant baseline differences in measured outcomes. Peritonitis most frequent source of sepsis (70%). Due to the presumed mechanisms of AT III action, only long‐term supplementation was assumed to affect parameters of organ function. Thus, 11 participants were prospectively excluded from the final evaluation since they died during the 14‐day period. However their data were included in an intention‐to‐treat analysis. No side effects were reported. Funding bias: Unable to assess |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | Unclear risk | Unable to assess |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Kobayashi 2003.
| Methods | 2‐group parallel randomized clinical trial ITT: yes Sample size calculation reported | |
| Participants | 29 severe pre‐eclamptic women (24 to 36 weeks of gestation, gestosis index (GI ≥ 6). Severe pre‐eclampsia was defined by the presence of hypertension plus proteinuria. AT III: 14 women; age: 30 ± 6 yrs; pregnancy weight (kg): 51 ± 7 SD; systolic BP (mm Hg): 163 ± 21, diastolic BP (mmHg): 104 ± 15; gestosis index: 7.2 ± 1.6; gestational age (weeks): 32.3 ± 3.1; biophysical profile score: 7.0 ± 2.0; primigravida (%): 71.4 Control: 15 women; age: 30 ± 4 y; pregnancy weight (kg): 53 ± 9; systolic BP (mm Hg): 165 ± 12, diastolic BP (mmHg): 100 ± 14; gestosis index: 6.6 ± 1.8; gestational age (weeks): 29.8 ± 3.7; biophysical profile score: 7.4 ± 1.9; primigravida (%): 60.0. Inclusion criteria: systolic BP ≥ 160 mm Hg and/or diastolic BP ≥ 110 mm Hg on 2 occasions 6 hours or more apart; and/or proteinuria ≥ 2 g/L in a 24‐hr urine collection; and a GI ≥ 6 points on 2 occasions at least 6 hours apart at bed rest Gestosis index score was calculated as: a) oedema after bed rest: 0 (none), 1 (tibia), 2 (generalized); b) proteinuria (g protein/L) 0 (< 0.5), 1 (≥ 0.5 ‐ 1.99), 2 (≥ 2.0 ‐ 4.99), 3 (≥ 5.0); c) systolic BP (mm Hg) 0 (< 140), 1 (140 ‐ 159), 2 (160 ‐ 179), 3 (> 180); d) diastolic (BP mm Hg) 0 (< 90), 1 (90 ‐ 99), 2 (100 ‐ 109), 3 (≥ 110). Exclusion criteria: patients with chronic hypertension, renal disease, diabetes mellitus, systemic lupus erythematosus, multiple pregnancies, AT deficiency and other severe medical conditions |
|
| Interventions | AT III: 1500 U/day once daily for 7 consecutive days Control group: unknown placebo 5000 U/day of unfractionated heparin was given simultaneously in both groups. Heparin was infused by 24‐hr intravenous drip with saline. Concomitant therapy with other anticoagulants, antiplatelet agents, and blood preparations except albumin was not permitted during the trial. Only hydralazine hydrochloride at a 30 mg/day was allowed in both groups. |
|
| Outcomes | Neonatal and foetal outcomes: mortality (neonatal and foetal), bleeding disorder, weeks of gestation at delivery, birth weight (g), APGAR score at 1 minute, neonatal distress (%), response on the biophysical profile score Maternal outcomes: gestosis index improvement, improvement of coagulation index, adverse events, blood loss |
|
| Notes | Country: Japan
Letter sent to authors in December 2005, reply received in January 2006.
No adverse event related to AT III was reported. The criteria used to initiate a preterm delivery were based on sustained severe hypertension (≥ 180/110 mm Hg), thrombocytopenia (≤ 100 x (10⁹/L), markedly worsened liver or renal function (serum aspartate aminotransferase or serum alanine amino transferase ≥ 100 IU/L, serum creatinine ≥ 15 mg/L, blood urea nitrogen ≥ 200 mg/L), severe maternal symptoms (eclampsia, HELLP syndrome, placental abruption, pulmonary oedema, etc) or impaired foetal status evidenced by intrauterine growth retardation (≤ ‐2 standard deviations) and oligohydramnios (amniotic pocket ≥ 1 cm), or persistently severe abnormal foetal heart rates or worsened biophysical profile test results (based on foetal breathing movements, foetal tone, reactive foetal heart rate, gross body movements, qualitative amniotic fluid volume). Funding bias: Appears to be based on institutional funding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A central 24‐hrs registration service |
| Allocation concealment (selection bias) | Low risk | A randomization envelope in order to allocate the drug treatment group |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | Low risk | Published report include all expected outcomes. |
| Other bias | Low risk | Appears free of such biases |
Langely 1993.
| Methods | 2‐group parallel randomized clinical trial ITT: yes Sample size calculation was not reported | |
| Participants | 25 patients in grade III or IV coma were selected on the basis of evidence of sepsis, intravascular coagulation, and a high risk of developing multiorgan failure. AT III: 13 participants, 7 men, 12 with severe hepatic dysfunction on entry of which 9 also had further complications of fulminant hepatic failure. Underlying aetiology: paracetamol overdose (9), hepatotoxicity to phenytoin (1), hepatitis B (1), non‐A non‐B hepatitis (2). Time from the onset of illness to entry into the trial: median 3 (2 ‐ 28) hrs Control: 12 participants, 4 men, all with severe hepatic dysfunction on entry of which 6 also had further complications of FHF. Underlying aetiology: paracetamol overdose (8), hepatitis B (2), non‐A non‐B hepatitis (1), halothane hepatitis (1). Time from the onset of illness to entry into the trial: median 3 (2 ‐ 37) hrs No statistically significant differences between the 2 groups on admission with respect to age, sex, prothrombin ratio, and initial AT III level Inclusion criteria: 1) severe hepatic dysfunction, prothrombin ratio expressed as an international normalized ratio (INR) > 6.7 for paracetamol overdose or INR > 3.3 for other aetiologies; 2) evidence of sepsis with temperature higher than 39º C, white cell count > 15 x 10⁹/L, or microbiologically proven sepsis; 3) presence of disseminated intravascular coagulation with spontaneous bleeding and a platelet count of less than 50 x 10⁹/L; 4) multiorgan failure with systolic blood pressure < 80 mm Hg, requiring inotropic support to maintain blood pressure and blood pH ≤ 7.3 Exclusion criteria: not provided |
|
| Interventions | AT III group: 13 participants received 3000 units of AT III on entry into the trial followed by a further 1000 units every 6 hours unless AT III levels were within the normal range. Total amount of AT III given to the participants during the course of their illness ranged from 3000 units to 23,000 U (mean 7000 ± 5000 U). Control: no placebo All participants received standard supportive therapy for FHF which included haemodynamic monitoring, haemodialysis or haemofiltration for renal failure, and continuous intracranial monitoring with treatment for raised intracranial pressure where necessary. Mechanical ventilation was used to treat respiratory dysfunction or pulmonary complications. |
|
| Outcomes | Primary: mortality, plasma levels of AT III Secondary: renal failure, cerebral oedema, incidence of infection, liver transplantation, coma grade, effect of AT III on bleeding and coagulation parameters, transfusion requirements |
|
| Notes | Country: United Kingdom
Letter sent to authors in December 2005, no reply received. Funding bias: This trial was supported by Behringwerke AG including provision of the antithrombin III concentrate, and assay kits for antithrombin III, thrombin‐antithrombin Ill complex and prothrombin fragment (Fl +2) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | Unclear risk | Unable to assess |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Lavrentieva 2008.
| Methods | 2‐group parallel prospective randomized clinical trial. ITT: Not stated | |
| Participants | 31 patients with severe burn injury Location: Burn Unit in Papanicolau General Hospital, Thessaloniki, Greece from April 2004 to December 2005 Inclusion criteria: Patients with severe burn injury admitted to the Burn Unit Exclusion criteria: Patients with known haematological disease, hepatic and renal failure, malignancies and associated trauma Sex: 7 women 24 men Age: Case: 37.1 ± 13.6, control 44.1 ± 21.5 yrs |
|
| Interventions | Timing of intervention: AT administration was started from the 1st post‐burn day and continued for the next 3 consecutive days at a dose of 64.9 ± 11.4 U/kg/day. The AT dose was titrated depending on the desired target value of plasma AT activity > 150% Use of heparin not stated. |
|
| Outcomes | Length of follow‐up: 28 days
Primary outcome: Diagnosis af DIC Secondary outcome: 28‐day mortality |
|
| Notes | Country: Greece
We contacted the author 23rd April 2015 and he replied on the same day with detailed answers. Funding bias: The authors received no funding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly‐generated zeros and ones according to SPSS 14 statistical software |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding of participants and personnel |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No blinding of outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | High risk | Protocol or trial registration not available. The trial was not registered online according to the author. |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Maki 2000.
| Methods | 2‐group parallel randomized clinical trial ITT: yes No sample size calculation was reported | |
| Participants | Severe pre‐eclamptic women (24 to 35 weeks of gestation) AT III: 74 women, age: 30 ± 5 yrs; pregnancy weight (kg): 66 ± 12; systolic BP (mm Hg): 170 ± 16, diastolic BP (mmHg): 103 ± 12; gestosis index: 7.7 ± 1.4; gestational age (weeks): 31.8 ± 3.2; biophysical profile score: 8.6 ± 2.0; primigravida (%): 34.8 Control: 72 women, age: 30 ± 5 yrs; pregnancy weight (kg): 65 ±12; systolic BP (mm Hg): 169 ± 17, diastolic BP (mmHg): 105 ± 12; gestosis index: 7.7 ± 1.6; gestational age (weeks): 31.7 ± 2.7; biophysical profile score: 8.7 ± 1.7; primigravida (%): 37.3 Inclusion criteria: systolic BP ≥ 160 mmHg and/or diastolic BP ≥ 110 mmHg on 2 occasions 6 hrs or more apart; and/or proteinuria ≥ 2 g/L of protein in a 24‐hr urine collection; GI ≥ 6 points on 2 occasions at least 6 hrs apart despite bedrest. GI is calculated based on oedema, proteinuria, systolic BP and diastolic BP. Exclusion criteria: chronic hypertension, multiple pregnancy, renal disease, diabetes mellitus, systemic lupus erythematosus, other severe medical conditions and people with AT deficiency. Similar baseline characteristics, proportion of severe early‐onset pre‐eclampsia was equally distributed within the trial groups. For definitions of GI and bio‐profile scoring technique, please see Kobayashi 2003 characteristics. |
|
| Interventions | Treatment: 3000 IU intravenous AT III once daily for 7 consecutive days Control: 582 mg albumin intravenously, once daily for 7 consecutive days as placebo Concomitant therapy with other anticoagulants, antiplatelet agents and blood preparations except albumin was not permitted during the trial. Other relevant treatment such as antihypertensive agents, magnesium sulfate allowed in both groups. No heparin was used. |
|
| Outcomes | Primary endpoints: improvement of GI, improvement in foetal findings (biophysical profile score, and the estimated foetal weight gain), duration of pregnancy, birth weight, gestational age at delivery, improvement of coagulation parameters (TAT‐complexes, plasmin‐plasmin inhibitor complexes, PC, D‐dimer, AT activity, AT antigen), stillbirth or neonatal death ascribed to pre‐eclampsia, maternal death from any cause, or death of neonate at any time attributed to pre‐eclampsia or associated with intra‐uterine growth retardation. Secondary: adverse events related to AT Stillbirth included all intrauterine deaths at or after 24 weeks, and neonatal death included all deaths after birth up to the age of 28 days. |
|
| Notes | Country: Japan
Letter sent to authors in December 2005, reply received in January 2006.
Intervention had to be stopped in 23 participants in the AT III group and 29 participants in the control group prematurely due to worsening of maternal and foetal findings. Funding bias: Appears to be based on institutional funding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | 24 hrs registration service (randomization co‐ordinating centre) |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | Low risk | Published report includes all expected outcomes. |
| Other bias | Low risk | Appears free of such biases |
Mitchell 2003.
| Methods | 2‐group parallel randomized clinical trial ITT: no Sample size calculation reported | |
| Participants | Paediatric acute lymphoblastic leukaemia (ALL) patients AT III: 25 participants, 15 boys, median age 3.8 (1.6 ‐ 17.2) yrs, weight 18.3 (9.7 ‐ 76.7) kg Control: 60 participants, 37 boys, median age 5.9 (1.9 ‐ 16.7) yrs, weight 20.6 (10.8 ‐ 83.4) kg Inclusion criteria: age > 6 months and < 18 years, newly diagnosed with ALL at the beginning of the induction of chemotherapy, a functioning central venous line placed within 2 weeks of initiating induction chemotherapy and obtaining informed consent Exclusion criteria: previous treatment with L‐asparaginase, a known hypersensitivity to any of the ingredients in antithrombin concentrate, medical conditions that could have interfered with participation or assessment of the trial drug, received other investigational drugs within 30 days of enrolment, or required treatment with therapeutic anticoagulation All participants received unfractionated heparin for prophylaxis of central venous line blockage either by continuous infusion (1 ‐ 3 units/mL) or intermittent flushes (50 ‐ 100 units/mL up to 4 times per day). The decision to use additional anticoagulants was left to the patient's physician. |
|
| Interventions | AT III: infusions once weekly for 4 weeks (days 1, 8, 15, 22) to increase plasma concentrations of AT to approximately 3.0 units/mL but no more than 4.0 units/mL Control: no placebo |
|
| Outcomes | Primary: prevalence of thrombotic events Secondary: bleeding events, plasma markers, efficacy and safety |
|
| Notes | Country: Canada and USA
Letter sent to authors in December 2005, no answer received.
No data were provided on mortality. Based on the detailed follow‐up, we concluded that there was no mortality in either group. The trial was not powered to prove efficacy or safety of AT III supplementation but rather to look for trends. 24 participants excluded from analysis (12 in each group). The reasons for exclusion in the AT III group were: 3 participants due to withdrawal of consent, 2 participants categorized as noncompliant due to an underdosing with AT concentrate, 7 participants due to the absence of an exit venogram. In the control group: 3 participants due to withdrawal of consent, 1 participant had an adverse event, and 8 participants were excluded due to the absence of an exit venogram. Funding bias: Unclear |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated |
| Allocation concealment (selection bias) | Low risk | Sealed opaque envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Incomplete follow‐up |
| Selective reporting (reporting bias) | Unclear risk | Unable to assess |
| Other bias | Unclear risk | Unabel to assess adequately |
Muntean 1989.
| Methods | 2‐group parallel open randomized clinical trial. ITT: Not stated | |
| Participants | 98 preterm neonates Inclusion criteria: premature infants Exclusion criteria: Not stated |
|
| Interventions | Intervention group: n = 45. Single bolus AT III immediately after birth. Birth weight under 1500 g was given 100 U, over 1500 g 200 U Control group: n = 53, standard treatment. No details |
|
| Outcomes | Length of follow‐up: Not stated Outcomes: Participants received artificial ventilation. Duration of artificial ventilation. Intraventricular haemorrhage and mortality | |
| Notes | Location: Department of Gynaecology and Obstetrics, University of Graz, Austria
Trial only published as an abstract. Letter sent to authors, no reply received. Funding bias: Not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unable to assess |
| Selective reporting (reporting bias) | Unclear risk | Unable to assess |
| Other bias | High risk | Only published as abstract |
Neporada 2008A.
| Methods | 3‐group parallel randomized clinical trial. ITT: Not stated | |
| Participants | 43 patients with DIC and AT activity ≤ 70% Inclusion criteria: 43 consecutive patients were enrolled with DIC diagnostic criteria by the JAAM and AT activity ≤ 70% admitted to the ICU Exclusion criteria: age < 16 and > 75 yrs; body weight < 50 or > 100 kg, malignant neoplasms, bleeding, haemostatic therapy, platelet count over 50 10⁹/L Interventions group: 15 participants were randomized into AT III group, mean age 42 (95% CI 32 to 52), the proportion of men was 53%. Sepsis was present in 9 participants (60%). Control group: 15 participants were randomized to FFP group. Mean age 56 (95% CI 48 to 64), the proportion of men 60%. 8 participants had sepsis. |
|
| Interventions | Intervention group 1: 15 participants received AT III 500 ‐ 1000 IU/day Control group: 15 participants received FFP (10 ‐ 17 ml/kg) Intervention group 2 (initiated at a later stage): n = 13, same intervention as group 1 and control group combined. If AT activity persists < 70% therapy protocol continued during 4 days. Concomitant therapy with nadroparin (95 IU/kg/day) was used in all groups. |
|
| Outcomes | Length of follow‐up: 30‐days Outcomes: Respiratory function, organ failure assessment, DIC JAAM score, bleeding complications, allergy and all‐cause 30‐day mortality | |
| Notes | Location: ICU, City Hospital #1, Arkhangelsk, Russia
Article sent by e‐mail. Published in Russian magazine in 2008. The author, Elena Neporada, contributed with further details about the trial. Funding bias: No funding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomized |
| Allocation concealment (selection bias) | Low risk | State envelope method. No further details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | High risk | Protocol or trial registration not available. The trial was not registered online according to the author. |
| Other bias | Unclear risk | Baseline and design questions remain unclear. |
Nishiyama 2011.
| Methods | 2‐group parallel prospective randomized clinical trial. ITT: Not stated | |
| Participants | 16 patients Inclusion criteria: 16 adults were enrolled, diagnosed as having DIC with infection assessed with an acute DIC score 4 or higher at admission to the ICU. Exclusion criteria: Haematologic diseases, liver cirrhosis classified as Child‐Pugh grade C, burn, or obstetric diseases or patients who had received anticancer agents, radiation, or anticoagulant were excluded from the trial Sex: 5 women and 11 men Age: range 30 ‐ 85 yrs |
|
| Interventions | Intervention group: received antithrombin (Mitsubishi Tanabe Pharm, Co, Ltd, Osaka, Japan) as a 1500 unit infusion for 30 min/d for 5 days. Control group: received gabexate mesilate (Ono Pharm, Co, Ltd, Osaka, Japan) as a 2000 mg infusion for 24 hrs/d for 5 days. |
|
| Outcomes | Length of follow‐up: 28 days Outcomes: White blood cell counts, C‐reactive protein, platelet counts, antithrombin, fibrin and fibrinogen degradation product, D‐dimer, fibrinogen, thrombin antithrombin complex, plasmin plasminogen complex, prothrombin time, activated partial thrombin time, as ordinally measurable parameters of inflammation, coagulation, and fibrinolysis, were monitored on the day of ICU admission and on days 1, 3, 5, and 7 thereafter. Mortality over 28 days was compared between the 2 groups. | |
| Notes | Country: Japan
Attempts to reach the author failed. No reply. Funding bias: None stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Participants were stratified randomly into 2 groups using an envelope method. No further details |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Single‐blinded trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | High risk | Protocol or trial registration not available. Protocol not registered on www.clinicaltrials.gov |
| Other bias | Unclear risk | Unable to assess adequately |
Palareti 1995.
| Methods | 2‐group parallel randomized clinical trial | |
| Participants | 119 participants who met the inclusion criteria: 18 ‐ 75 yrs, AT III < 70%, Sepsis and/or post‐surgical complications requiring haemodynamic or respiratory support. 60 participants in the intervention group, 59 in the control group | |
| Interventions | AT III: loading dose 4000 U followed by 2000 U/12hrs by continuous dose over 5 days Control: Placebo (no details described). |
|
| Outcomes | AT III activity, 7(6)‐day mortality, D‐dimer, plasinogen, PAP‐complex, α2‐antiplasmin | |
| Notes | Trial only published as abstract Available data are based on only the 1st included 59 participants (30 in the intervention group, 29 in the placebo group) on day 6. Use of heparin was an exclusion criterion, so it is assumed that no heparin was given during the trial. Funding bias: Not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomized, but no further details |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | Unclear risk | Not stated |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Schmidt 1998.
| Methods | 2‐group parallel randomized clinical trial ITT: yes Sample size calculation reported | |
| Participants | Premature infants with respiratory distress syndrome (RDS) in the neonatal ICU, 942 screened with birth weights 750 ‐ 1900 g AT III: 61 participants. 33 male, gestational age (weeks) 28.3 ± 2.03, birth weight: 1.198 ± 300.9 g, venous cord blood pH: 7.31 ± 0.10, APGAR score at 1 min: 5.4 ± 1.95, APGAR score at 5 min: 7.6 ± 1.24, AT III, U/mL: 0.33 ± 0.08, maternal age: 28.1 ± 6.68, maternal steroids: 40 (65.6%) Control: 61 participants. 31 male, gestational age (weeks) 28.8 ± 2.25, birth weight: 1.201 ± 314.4 g, venous cord blood pH: 7.28 ± 0.15, APGAR score at 1 min: 5.3 ± 2.22, APGAR score at 5 min: 7.7 ± 1.16, AT III, U/mL: 0.32 ± 0.08, maternal age: 28.9 ± 5.16, maternal steroids: 35 (57.4%). Inclusion criteria: 224 met inclusion criteria; (1) age between 2 ‐ 12 hrs; 2) endotracheal intubation and positive pressure ventilation for RDS; 3) indwelling arterial catheter; 4) ratio of arterial to alveolar oxygen pressure PO₂ < 0.3 after the 1st dose of exogenous surfactant) Exclusion criteria: 1) congenital infection; 2) congenital malformation(s); 3) hydrops; 4) pulmonary hypoplasia; 5) clinically apparent bleeding disorder; 6) thrombocytopenia (platelets ≤ 50 x 10⁹/L); 7) moribund |
|
| Interventions | AT III: loading dose 100 U/kg followed by 50 U/kg every 6 hrs for 48 hrs Control: human albumin 1%, equivalent dose All participants received the same standard intensive care treatment and the same exogenous surfactant. Arterial catheters were perfused with 1 ml/h of 5% dextrose containing 1 IU/ml of unfractionated heparin. No additional administration of anticoagulants |
|
| Outcomes | Primary: mortality. Secondary: days in ICU, plasma AT activity, TAT‐complex (thrombin‐AT), prothrombin fragment (F1 + 2), ratio of arterial to alveolar oxygen pressure (a/A) PO₂ and ventilator efficiency index (VEI), thrombin formation, improved gas exchange, duration of mechanical ventilation, duration of supplemental oxygen therapy, safety of AT therapy evaluated primarily by comparing the incidence of severe (Grade 3) intraventricular haemorrhage and of periventricular echodensities in the 2 groups, bleeding events |
|
| Notes | Country: Canada
Letter sent to authors in November 2005, no answer received.
AT levels in healthy preterm infants are about 40% of normal adult values, and even lower levels have been reported in sick premature infants with RDS. The therapy was initiated on average 7.2 hrs after delivery in each group.
Having observed for some time an apparent imbalance in deaths between the treatment groups, the external safety and efficacy monitoring committee decided to break the code and make its recommendation because 7 deaths had occurred in participants receiving AT and 2 with placebo. Funding bias: Supported by Physicians’ Services Incorporated Foundation, Toronto, and Behringwerke AG, Germany. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer programme developed by Hoechst AG |
| Allocation concealment (selection bias) | Low risk | Adequate strata and central allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Adequate blinding on all levels |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | Low risk | Appears to report on all outcomes |
| Other bias | Low risk | Appears free of such biases |
Schorr 2000.
| Methods | 2‐group parallel randomized clinical trial ITT: yes Sample size calculation was not reported | |
| Participants | Patients with secondary peritonitis, surgical population in ICU AT III: 24 participants, 12 men, median age 60 yrs (33 ‐ 86), APACHE II 14.4 (6 ‐ 30), MOF 4.6 (0 ‐ 10), SOFA 7.7 (0 ‐ 18), MPI 26.7 (12 ‐ 38), malignant diseases (%) 33. Control: 26 participants, 12 men, median age: 62 yrs (28 ‐ 85), APACHE II 14.6 (4 ‐ 28), MOF 4.9 (0 ‐ 11), SOFA 8.7 (0 ‐ 16), MPI 27.8 (16 ‐ 38), malignant diseases (%) 35. Inclusion criteria: 1) > 18 years; 2) diffuse secondary peritonitis affecting 2+ quadrants after perforation of an intraperitoneal organ or failed anastomosis; 3) focus of infection had to be surgically eliminated Exclusion criteria: 1) acute pancreatitis; 2) peritonitis due to peritoneal dialysis; 3) primary peritonitis; 4) ascites due to cirrhosis of the liver or due to malignant underlying disease; 5) pregnancy; 6) incapacitated persons; 7) prisoners |
|
| Interventions | AT III: continuous IV AT III and 2 intraperitoneal installations of fresh frozen serum. The aim was to achieve AT III level of at least 140% of the normal plasma value for 4 days. A calculated bolus was given in 1 hr followed by a continuous infusion of AT III (200 IU‐800 IU per hr) depending on the 6‐hourly measurements of plasma AT III. 300 ml of blood group‐compatible FFS was applied intraperitoneally after the operation and 6 hours postoperatively through especially installed drains. The first FFS was supplemented with 1500 IU AT III to equalize the lack of AT III in FFS. Mean AT III administered was 26196 (± 299 SEM) IU. Control: no placebo All participants: routine intensive care treatment. If no risk of postoperative bleeding, heparin iv 200 ‐ 400 IU/h 6 hrs after operation until discharge from ICU |
|
| Outcomes | Primary: 90‐day mortality. Secondary: number of days at ICU, mechanical ventilation, organ function scores, side effects, organ failure and function, bleeding and transfusions, AT III activity, prothrombin concentrations in exudate, TAT‐complex in exudate, opsonic capacity and opsonin concentration in exudate |
|
| Notes | Country: Germany
Letter sent to authors in November 2005, no reply received. Funding bias: This trial was supported by the Bundesministerium für Forschung und Technologie (National research grant). ATIII was a gift from Pharmacia, Stockholm. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | Unclear risk | Unable to assess |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Schuster 1997.
| Methods | 2‐group parallel randomized clinical trial ITT: Unclear Sample size calculation: not reported | |
| Participants | 45 patients with sepsis without DIC | |
| Interventions | Intervention group: allocated to AT III supplementation (loading dose of 3000 IU followed by 500 IU every 4 hrs for 7 days, total dose 17,000 IU) Control: placebo (albumin) Both groups received iv heparin (6 IU/kg/h) |
|
| Outcomes | Secondary outcomes (not reported) number of days at ICU, mechanical ventilation, organ function scores, side effects, organ failure and function, bleeding and transfusions, AT III activity | |
| Notes | Country: Germany
Only presented as abstract. No additional data are available. Several previous attempts to reach the authors or find a manuscript failed to provide more information. Funding bias: Not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | Unclear risk | Not stated |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Smith‐Erichsen 1996.
| Methods | 2‐group parallel randomized clinical trial ITT: no No sample size calculation was reported | |
| Participants | Critically ill and trauma patients in ICU
83 participants allocated to intervention and control groups. Within each of these groups, the participants were then allocated to 2 groups (group 1 and group 2) according to the type of surgery or trauma. Group 1: multiple trauma, defined as injury of at least 2 body regions, but without abdominal injury Group 2: abdominal operations and multiple trauma with abdominal injury: 2.1) total gastrectomy, oesophageal resection, rectal resection/amputation in participants over 70 years old; 2.2) pancreatic resection; 2.3) liver resection; 2.4) secondary gastro‐intestinal interventions due to postoperative complications; 2.5) multiple trauma with abdominal injury. The participants were randomized in blocks with 4 participants in each block. AT III group: 43 participants, 30 men, age: 60.1 ± 16.3 yrs. 5 allocated to group 1 (trauma). Number of participants in group 2 as defined above; Group 2.1 17; Group 2.2 9; Group 2.3 5; Group 2.4 6; Group 2.5 1 Control group: 40 participants, 25 men, age: 59.0 ± 16.2 yrs. 5 allocated to group 1 (trauma). Number of participants in group 2 as defined above; Group 2.1 17; Group 2.2 8; Group 2.3 4; Group 2.4 5; Group 2.5 1 Inclusion criteria: AT III plasma level less than 80% measured the day after trauma or major abdominal surgery Exclusion criteria: trauma patients with head injury and patients below the age of 18 years |
|
| Interventions | AT III: the aim was to maintain plasma AT III activity in the range of 100% ± 10%. No data were provided on average or total dose. Based on daily samples, the substitution dose was calculated. A maintenance dose of 500 IU was given at midnight and 10 hours the following day. Treatment was continued until the plasma AT III activity was maintained within the normal range for 3 consecutive days without AT III substitution but was discontinued after 14 days, irrespective of the plasma activity. Control: no placebo All participants were treated according to the standard management routines of each hospital. 500 ml of dextran‐70 was given every 2nd day during the 1st week as antithrombotic prophylaxis. No participant received heparin or LMWH during the observations period. |
|
| Outcomes | Primary: plasma protease changes, mortality, days in ICU, days in hospital Secondary: AT III level, prothrombin, prekallikrein, functional kallikrein inhibition, plasma kallikrein‐like activity, plasminogen, functional antiplasmin activity and plasmin‐like activity, proenzyme functional inhibition index and adverse effects | |
| Notes | Country: Norway
Letter sent to authors in December 2006, no answer received. Funding bias: Unclear |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | Unclear risk | Unclear |
| Other bias | Unclear risk | Unable to assess, insufficient information |
Vorobyeva 2007.
| Methods | Prospective randomized clinical trial ITT: Not stated | |
| Participants | 38 patients with DIC. 26 participants in the control group, 12 participants in the intervention group. Age 16 ‐ 69 years. Gender is not described. | |
| Interventions | Intervention group (AT III): (100% minus measured AT III%) x kg bodyweight, 1000 IU/h, max 1500 IU/day Control group (FFP): 10 ‐ 17 ml/kg, max 1000 ml/day The duration of treatment not described. |
|
| Outcomes | Mortality. Other outcomes are not described in the paper. | |
| Notes | Country: Russia
We tried to contact the author, Nadejda Vorobeva on April 28th 2015, but received no reply. Funding bias: Supported by grants from the Arkhangelsk region Government |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | Unclear risk | Unable to assess |
| Other bias | High risk | Country: Russia Abstracts and article were written in Russian. Poorly‐described section of participants, inclusion criteria, outcome and intervention |
Warren 2001.
| Methods | 2‐group parallel randomized multicentre clinical trial ITT: yes Sample size calculation reported | |
| Participants | Critically ill population of ICU patients with severe sepsis and septic shock AT III: 1157 participants, men 62%; age mean (SD) 57 (17) years; SAPS II (SD) 49 (17); body weight mean (SD) 77 (19) kg. Underlying problem or site of infection: 1) respiratory system 35%; 2) intra‐abdominal infection 27%; 3) genitourinary system 6%; 4) injury 7%; 5) other 24%. Surgical status: A) no 54%, yes 46%. Circulatory shock: 49%; blood culture results: a) gram‐negative 15%, b) gram‐positive 15%, c) other/mixed 4%, d) not done/not verified 66%. Control: 1157 participants, men 61%; age mean (SD) 58 (17) years; SAPS II (SD) 49 (16); body weight mean (SD) 77 (20) kg. Underlying problem or site of infection: 1) respiratory system 34%; 2) intra‐abdominal infection 28%; 3) genitourinary system 8%; 4) injury 6%; 5) other 24%. Surgical status: A) no 53%, yes 47%. Circulatory shock: 47%; blood culture results: a) gram‐negative 16%, b) gram‐positive 17%, c) other/mixed 2%, d) not done/not verified 64%. Inclusion criteria: 18 years or above, and met following criteria within 6 hours: 1) clinical evidence of sepsis with a suspected source of infection; 2) body temperature (rectal or core) > 38.5º C or < 35.5º C; 3) leukocyte count > 10,000/µL or < 3500/µL. Additionally, 3 of the following 6 signs had to be met within the same 6‐hour period: a) heart rate > 100/min; b) tachypnoea > 24/min or mechanical ventilation because of septic indication; c) hypotension, systolic BP < 90 mmHg despite sufficient fluid replacement or the need for vasoactive agents to maintain systolic BP of 90 mmHg or greater; d) platelet count < 100,000/µL; e) elevated lactate above upper limits of normal range or metabolic acidosis (pH < 7.3 or BE≤ ‐10 mmol/L) not secondary to respiratory alkalosis; f) oliguria (< 20 ml/h despite sufficient fluid replacement). Exclusion criteria: a) advanced directive to withhold life‐sustaining treatment (except cardiopulmonary resuscitation); b) condition other than sepsis anticipated to be fatal within 28 days; c) pregnancy or breast feeding; d) history of hypersensitivity to trial medication; e) treatment with other investigational drugs within the last 30 days; f) treatment with an AT III concentrate within the last 48 hours; g) treatment with heparin (except sc low dose or iv line flushing) or coumarin derivatives; h) NSAID treatment within previous 2 days; i) known bleeding disorder or ongoing massive surgical bleeding; j) platelet count < 30 x 1000/µL; k) immunocompromised status; l) acute myocardial infarction (within previous 7 days); m) 3rd‐degree burns ≥ 20% of total body area; n) incurable malignancy with metastases and life expectancy of < 3 months; o) haematologic neoplasia during cytostatic treatment; p) bone marrow aplasia; q) pre‐existing dialysis‐dependent renal failure; r) end‐stage liver disease; s) transplantation (postoperative state); t) history of stroke within the last year; u) severe cranial or spinal trauma within the last year; v) planned cranial or spinal surgery (except nontraumatic lumbar puncture) within the next 48 hours |
|
| Interventions | AT III: loading dose of 6000 IU given over 30 minutes, followed by a continuous iv infusion of 6000 IU per day for 4 days, total of 30000 IU Control: equivalent volume of placebo solution (1% of human albumin) Heparin was permitted to be used in prophylactic doses as an adjunct to standard therapy. Administration of heparin was left to the attending physicians. Not all participants received heparin (698 participants received no heparin while receiving AT). Heparin dosage: unfractionated or LMWH for venous thrombosis prophylaxis (≤ 1000 IU subcutaneous per day) and heparin flushes for vascular catheter patency (iv of ≤ 2I U/kg body weight/h) |
|
| Outcomes | Primary: all‐cause mortality at 28 days (subgroup 28‐ and 90‐day survival for participants not receiving heparin). Secondary: mortality at 56 and 90 days, survival time, length of ICU stay, occurrence of new organ dysfunction within 7 days (according to logistic organ dysfunction score), severity of sepsis (SAPSII), circulatory shock index (i.e. ratio of heart rate (beats/minute) and systolic BP (mmHg) exceeded the value of 1.5); surgical interventions and bleeding events for 28 days, major bleeding if intracranial or required transfusion of > 3 units of blood, other serious adverse events, AT III plasma concentration, activated partial thromboplastin time and prothrombin time |
|
| Notes | Country: KyberSept Trial trial Group, 211 contributing centres in 19 countries including USA, Germany Letter sent to authors November 2005, reply by authors in December 2005 and January 2006. Funding bias: Aventis Behring sponsored the trial and the manuscript. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Phone to a randomization centre, available on a 24‐hr basis. Randomization plans had been prepared in advance with a block size of 4 participants and investigators were told the medication package number to be used. |
| Allocation concealment (selection bias) | Low risk | Packages for individual participants consisted of vials and labels identical in appearance for antithrombin III and placebo. Central allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reason for dropouts specified |
| Selective reporting (reporting bias) | Low risk | Reported outcomes prespecified in an available study protocol |
| Other bias | Low risk | Appears free of such biases |
Waydhas 1998.
| Methods | 2‐group parallel randomized clinical trial ITT: yes Sample size calculation not reported | |
| Participants | Trauma patients in ICU. AT III: 20 participants, 13 men, age 32 yrs (26 ‐ 42), systolic blood pressure (SBP) on the scene 120 mmHg (90 ‐ 130), 2 participants with SBP < 80 mmHg, GCS 13 (6 ‐ 15), use of catecholamines on the scene in 2 cases, intubation on the scene 15 (75%), fluids on the scene 1.5 L (1.0 ‐ 3.0), time from accident until admission 53 mins (33 ‐ 75), lactate 3.2 mmol/L (2.7 ‐ 8.0), haemoglobin 8.9 g/100 mL (7.0 ‐ 12‐0), PH 7.30 (7.27 ‐ 7.36), base deficit 6.2 (8.3 ‐ 3.7) Control: 20 participants, 13 men, age 35 yrs (27 ‐ 47), SBP on the scene 100 mmHg (93 ‐ 120), 2 patients on the scene with SBP < 80 mmHg, GCS 13 (4 ‐ 15), use of catecholamines on the scene in 2 cases, intubation on the scene 10 (50%), fluids on the scene 2.0 L (1.0 ‐ 3.0), time from accident until admission 58 mins (30 ‐ 105), lactate 3.7 mmol/L (2.1 ‐ 6.0), haemoglobin 9.5 g/100 mL (8.0 ‐ 11‐0), PH 7.29 (7.10 ‐ 7.32), base deficit 7.6 (13.1 ‐ 4.2) Inclusion criteria: 1) ISS of 29 or greater (because an ISS of 29 or greater was associated with an organ failure rate of > 60%); 2) admission within 6 hrs after injury; 3) 18 ‐ 70 yrs; 4) blunt trauma; 5) if the head region contributed to calculation of the ISS, the sum of squared abbreviated ISS of the 2 other regions that were included in the calculation had to be equal to or greater than 20 points in order to rule out predominant head injury and ensure severe multiple injuries. Exclusion criteria: a) resuscitation with epinephrine; b) if closed chest compression performed; c) severe brain injury with uncertain outcome (both pupils unresponsive to light for more than 30 minutes, midline shift > 1 cm, or extensive intracerebral haemorrhage on the initial computed tomographic scan) The only post hoc exclusion criterion was survival of < 24 hrs after trauma. |
|
| Interventions | AT III: a total of 20,000 IU (16,125 ‐ 22,875), vials containing 500 IU diluted in 8 mL of water. Infusion rate of 96mL/h via a pump. AT III levels assessed every 6 hrs during the first 48 hrs. Additional AT III or placebo was substituted to keep the AT III concentration at 140% of normal. In addition, on the next 2 days the test substance was administered once daily in the morning. Control: placebo, 20% human albumin in corresponding doses and volume For prophylaxis of DVT and pulmonary embolism: standard sodium heparin iv at a rate of 400 IU/h, started within the first 24 hrs after trauma unless contraindications (active bleeding, intracranial haemorrhage, or others). Participants with traumatic intracranial bleeding were started on heparin on day 5, when the acute phase of brain swelling had subsided and there were no signs of ongoing haemorrhage. |
|
| Outcomes | Primary: incidence and severity of multiple organ dysfunction, mortality, incidence of respiratory failure, severity of organ failure, duration of mechanical ventilation and length of stay in the ICU and hospital Secondary: plasma concentration of indicators of DIC and systemic inflammatory response (prothrombin, prothrombin fragment F1 + F2, TAT‐complex lower, partial thromboplastin time, prothrombin time, PC, plasminogen activator inhibitor I, soluble TNF receptor II, IL1 receptor antagonist, IL6, IL8, neutrophil elastase), transfusions, respiratory dysfunction, ARDS, renal and liver dysfunction, incidence of DIC |
|
| Notes | Country: Germany
Letters sent to authors in November and December 2005, authors replied both times. Funding bias: This trial was supported by a grant from the Sonderforschungsbereich SFB 207 (project G5), the University of Munich, and the Bundesministerium fuer Bildung und Forschung (FKZ: 01 KE 8912). Antithrombin III (Atenativ) was supplied by Pharmacia & Upjohn (Erlangen, Germany). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer number generator |
| Allocation concealment (selection bias) | Low risk | Closed envelopes that had been prepared in blocks of 10, each block containing 5 decisions for placebo and 5 for the test substance |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The envelopes were opened by a person not involved in the care of the patient or the collection of the data. This person also prepared the test infusion. The attending team and the trial group were thus blinded to the type of substance given to the patient. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts or withdrawals were specified. |
| Selective reporting (reporting bias) | Low risk | Appears to include all expected outcomes |
| Other bias | Low risk | Appears free of such biases |
CABG = coronary artery bypass graft; ALL = acute lymphatic leukaemia; APACHE = acute physiology and chronic health evaluation; APGAR = appearance, pulse, grimace, activity, and respiration; APTT = activated partial thromboplastin time; ARDS = acute respiratory distress syndrome; BP = blood pressure; CI = confidence interval; CNS = central nervous system; COPD = chronic obstructive pulmonary disease; CPP = cerebral perfusion pressure; CRP = C‐reactive protein; DIC = disseminated intravascular coagulation; DVT = deep vein thrombosis; FFP = fresh frozen plasm; GCS = Glasgow coma scale; hr = hour; HELLP = haemolysis, elevated liver enzymes, and low platelet count; HIV = human immunodeficiency virus; HSCT = haematopoietic stem cell transplantation; ICP = intracranial pressure; INR = international normalized ratio; ISS = injury severity score; ITT = intention‐to‐treat; IU = international unit; JAAM = Japanese association for acute medicine; LMWH = low molecular weight heparin; MODS = multiorgan dysfunction syndrome ; MOFS = multiorgan failure score; MPI = Mannheimer peritonitis index; NSAID = nonsteroidal anti‐inflammatory drug; NYHA = New York Heart Association functional classification; OFS = organ failure score; OSFS = organ system failure scoring; RDS = respiratory distress syndrome; SAPS = simplified acute physiology score; SBP = systolic blood pressure; SD = standard deviation; sys = systolic; TAT = thrombin‐antithrombin complex; TISS = therapeutic intervention scoring system; Tp = temperature; yr = year
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aibiki 2006 | This randomized controlled trial compared 2 active interventions of AT‐III, 500 U/8 hrs and 1500 U/24 hrs. Reason for exclusion: No placebo group, no control group |
| Dietrich 2013 | 41 participants scheduled for primary CABG. Reason for exclusion: elective participants with heart disease, meets our exclusion criteria: AT III administration for the reduction of cardiovascular events. No primary endpoints. |
| Doi 2012 | Trial only published as abstract. Contains no relevant endpoints. |
| Hoffmann 2010 | Trial only published as abstract. Trial on elective participants. Contains no relevant endpoints. |
| Ilias 2000 | An open randomized clinical trial, evaluating the safety, pharmacokinetics and practicability of 2 different regimens of AT III treatment (intermittent bolus infusions vs continuous infusion), both aiming at a total dose of 30,000 IU AT III with concomitant heparin during a 4‐day period. Reason for exclusion: 2 different regimens of AT III treatment |
| Jochum 1995 | This review includes 1 unpublished, not double‐blinded trial of AT III supplementation in a septic population aiming at AT III activity > 120%. AT III group (n = 20) received 2000 ‐ 8000 U/d AT III over 21 days and heparin (4 U/kg body weight/hour). Control group (n = 20) received no placebo. Outcome measures were various organ dysfunction analyses, inflammatory and coagulatory parameters and mortality (control: 80%; AT III: 65%). The results were not statistically significant. Study excluded due to lack of outcomes of relevance and since we were unable to contact the authors in order to retrieve additional information |
| Kanbak 2011 | Randomized trial made in CABG participants. Reason for exclusion: Meets our exclusion criteria: AT III administration for the reduction of cardiovascular events. No primary endpoints. |
| Kim 2013 | The purpose of this trial was to compare the pharmacokinetic and pharmacodynamic characteristics of 2 AT III formulations. Reason for exclusion: Trial on healthy Korean volunteers |
| Korninger 1987 | A randomized trial of AT III in participants undergoing peritoneo‐venous shunt operation because of intractable ascites. 10 participants with alcoholic liver cirrhosis were randomized according to the stage of liver disease and pre‐operative AT III levels. AT III was infused in 5 participants, twice daily for 4 days, at a dose of 20 U/kg body weight starting 12 hours prior to operation. 5 participants were allocated to placebo. Coagulatory parameters were examined. Reason for exclusion: only published as abstract, no data on mortality |
| Leitner 2006 | This randomized, double‐blinded, placebo‐controlled trial examined 30 healthy male volunteers. The active treatment groups received infusions of AT III to achieve levels of 200% and 500% before infusion of 2 ng/kg endotoxin. Reason for exclusion: experimental trial among healthy volunteers |
| Maki 1987 | This randomized controlled trial compared 2 active interventions, in participants with obstetric complications. Reason for exclusion: 2 different active interventions |
| Mayumi 2011 | Trial only published as an abstract. We assume that the abstract is a subgroup analysis of an earlier trial. We cannot contact the author as it is not possible to find contact information. |
| Neporada 2008B | Data based on the same trial as Neporada 2008A |
| Nishiyama 2006 | This randomized, unblinded trial investigated the effects of AT III on coagulation, fibrinolysis, production of cytokines and adhesion molecules in abdominal aortic aneurysm repair surgery. 16 participants for Y‐shaped graft replacement of abdominal aortic aneurysm were divided into an AT III group and a control group. In the AT III group, 3000 U AT III was infused over 30 min before heparin administration and 24 hrs later. Reason for exclusion: no data on mortality, nor any data relevant for our review. We emailed the author on 17th April 2015, but received no reply. |
| Paparella 2014 | Randomized trial made in CABG participants. Reason for exclusion: Meets our exclusion criteria: AT III administration for the reduction of cardiovascular events. No primary endpoints. |
| Paternoster 2000 | This prospective trial evaluated the modifications of clotting and clinical parameters before and immediately after delivery amongst a pre‐eclamptic population either treated with AT III (n = 18) or acting as control (n = 21). Reason for exclusion: only published as a brief communication, no data on mortality, randomization or blinding |
| Paternoster 2004 | In this trial, 23 pre‐eclamptic women were randomly subdivided into 2 groups: 1st group (n = 10) were treated with 3000 units AT III once daily for 5 days or until delivery, while the 2nd group (n = 13) were treated with doses of AT III sufficient to maintain at least 80% of the activity. The endpoints were the prolongation of pregnancy, assessment of the maternal bleeding and haemostasis, and inflammatory markers. Reason for exclusion: comparison of 2 different regimens of AT III |
| Ranucci 2013 | 200 participants given preoperative AT supplementation in elective participants during cardiac surgery with CPB. Reason for exclusion: elective participants with heart disease |
| Sawamura 2009 | 23 DIC participants were treated with either high‐dose (60 IU/kg/day) or low‐dose (30 IU/kg/day) antithrombin concentrates for 3 days. The trial tested the hypotheses that AT III (antithrombin) improves DIC when applied to DIC participants. Reason for exclusion: No placebo group, no control group |
| Scherer 1997 | A randomized prospective trial examining the effects of AT III substitution in order to achieve supranormal values in participants with liver cirrhosis scheduled for liver transplantation. 19 participants were given AT III aiming at either 100% (n = 10) or 175% (n = 9) AT III activity. Control group (n = 5) received saline 0.9%. The endpoints were various coagulatory variables measured prior to AT III infusion and 60 min thereafter. Reason for exclusion: no data on mortality, 3 groups |
| Shimada 1994 | A randomized prospective trial of AT III efficacy in hepatic resection among 24 participants with hepatocellular carcinoma. 13 participants were given 1500 IU AT III immediately before operation, during hepatectomy, and immediately after operation. Control group (n = 11) received no placebo. Coagulant and fibrinolytic profiles were determined. Reason for exclusion: no data on mortality, different population than defined in our review |
| Terao 1989 | A randomized trial of 40 participants with pre‐eclampsia comparing AT III intervention with no treatment on coagulatory findings, gestosis index, abortion times, and incidence of newborn asphyxia. Reason for exclusion: no data on mortality |
| Valsecchi 2008 | Only published as an abstract. Trial was terminated by the sponsor with insufficient information. |
| Vinazzer 1995 | This review mentioned one unpublished trial of participants with DIC due to sepsis or septic shock in which 170 participants were evenly allocated to 2 groups (heparin versus AT III). This uncontrolled open clinical trial showed positive outcome of AT III substitution. Study excluded due to lack of outcomes of relevance and since we were unable to contact the authors in order to retrieve additional information |
ALL = acute lymphatic leukaemia; AT III = Antithrombin III ; CABG = coronary artery bypass graft; CPB = cardiopulmonary bypass; DIC = disseminated intravascular coagulation; d = day: H =; Hours; Kg = Kilogram; min= minute(s); U= units; U/d = units/day
Characteristics of ongoing studies [ordered by study ID]
D'angelo 2005.
| Trial name or title | EPAS |
| Methods | Double‐blind RCT |
| Participants | Women with pre‐eclampsia occurring before the 30th week of gestation |
| Interventions | 7‐day course of antithrombin 3000 IU/day. No heparin prophylaxis will be permitted during the time of AT III administration. |
| Outcomes | Primary outcome: reduction of the combined endpoint of foetoneonatal mortality and severe neonatal morbidity. Secondary outcomes: reduction in maternal complications and stay in neonatal ICU and the evaluation of the changes in coagulation and inflammation parameters. |
| Starting date | December 2004 |
| Contact information | D'angelo A (1),Valsecchi L (2) 1): Coagulation Service and Thrombosis Research Unit 2): Department of Obstetrics and Gynecology, Irccs H S. Raffaele Milano, Italy |
| Notes | D'angelo A,Valsecchi L; A double‐blind trial of high‐dose antithrombin supplementation in early pre‐eclampsia. Thrombosis Research. 2005; 115 Suppl 1:117 Prof. Dr. Armando D'Angelo was contacted 9th April 2015 and answer the same day. The trial results will be published in the near future. |
AT III = Antithrombin III; EPAS = Early pre‐eclampsia antithrombin trial; IU = international unit
Differences between protocol and review
We decided after publication of our protocol (Afshari 2007) for this review to also include randomized trials examining the effect of AT III on malignant diseases and cirrhosis. The decision was based on the desire to increase precision and power without confounding our data. The generalizability and usefulness of meta‐analyses are increased considerably if the individual trials cover different patient populations, settings, and treatment regimen. Furthermore, a broad (inclusive) meta‐analysis increases power, reduces the risk of erroneous conclusions, and facilitates exploratory analyses, which can generate hypotheses for future research. Usually it is also recommended to pool a broad range of studies (Gotzsche 2000).
We decided to employ trial sequential analyses (TSA) as a way of estimating the level of evidence of the experimental intervention. In our protocol we defined the secondary outcome 'Bleeding events within 14 days'. However, we changed this to 'Bleeding events', since we found it more relevant to look at the overall bleeding without applying any time constraint. Additionally, we found very few trials reporting bleeding events within the first 14 days.
We defined bleeding events as noted by the authors (requiring transfusions) and related to the intervention.
In this updated review we decided to expand our 'Risk of bias' table. We therefore added the following domains: blinding of participants and personnel (performance bias), blinding of outcome assessment (detection bias), incomplete outcome data (attrition bias), selective reporting (reporting bias), and other bias. We also added two 'Risk of bias' figures Figure 4; Figure 3).
We have removed five subgroup analyses from this updated review, since we found them redundant. We present the results in Table 2.
We also applied the principles of the GRADE approach to provide an overall assessment of the evidence relating to all of our outcomes. We constructed a 'Table 1' table using the GRADEpro software.
In this updated review, we also included trials that were only published as abstracts, in order to avoid publication bias.
Four subgroup analyses contained only one individual study. These are listed in Table 4; Table 5; Table 6; Table 8.
6. Severity of illness score.
| Trial | AT III | Control | Mean Difference | ||||
| Mean | SD | Total | Mean | SD | Total | IV, Fixed, 95% CI | |
| Haire 1998 | 15.7 | 19.2 | 24 | 25.7 | 17.9 | 25 | ‐10.00 (‐20.40 to 0.40) |
Subgroup analysis where only one trial had the relevant endpoints.
Contributions of authors
Updated review
Mikkel Allingstrup (MA), Jørn Wetterslev (JW), Frederikke B Ravn (FR), Ann Merete Møller (AM), Arash Afshari (AA). MA, FR and AA were involved in literature searching, quality assessment and data abstraction of trials, and writing the review. JW was involved in statistics and TSA analysis. AM was involved in proofreading the manuscript
Original published review
All three authors (Arash Afshari, Jørn Wetterslev and Ann Merete Møller) were involved in protocol development, literature searching, quality assessment and data abstraction of trials, and writing the review.
Sources of support
Internal sources
Cochrane Anaesthesia Critical and Emergency Care Group (ACE), Denmark.
External sources
No sources of support supplied
Declarations of interest
Mikkel Allingstrup declares that there are no conflicts of interest Jørn Wetterslev declares that he is a member of the task force on trial sequential analysis (TSA) at Copenhagen Trial Unit developing and programming TSA (please see www.ctu.dk/tsa) Frederikke B Ravn declares that there are no conflicts of interest Ann Merete Møller declares that there are no conflicts of interest Arash Afshari declares that there are no conflicts of interest
Edited (no change to conclusions)
References
References to studies included in this review
Albert 1992 {published data only}
- Albert J, Blomqvist H, Gardlund B, Jakobsson J, Svensson J, Blomback M. Effect of antithrombin concentrate on haemostatic variables in critically ill patients. Acta Anaesthesiologica Scandinavia 1992;36(8):745‐52. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Balk 1995 {published data only}
- Balk RA, Bedrosian C, McCormick L, Baughman J, Eisele B, Keinecke HO, et al. Prospective double blind placebo‐controlled trial of ATIII substitution in sepsis. Intensive Care Medicine 1995;21 Suppl 1:17. [Google Scholar]
Baudo 1992 {published data only}
- Baudo F, DeGasperi A, Cataldo F, Caimi TM, Cattaneo D, Redaelli R, et al. Antithrombin III supplementation during orthotopic liver transplantation in cirrhotic patients: a randomized trial. Thrombosis Research 1992;68(4‐5):409‐16. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Baudo 1998 {published data only}
- Baudo F, Caimi TM, Cataldo F, Ravizza A, Arlati S, Casella G, et al. Antithrombin III (ATIII) replacement therapy in patients with severe sepsis and/or postsurgical complications: a controlled double‐blind, randomized, multicenter study. Intensive Care Medicine 1998;24(4):336‐42. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Blauhut 1985 {published data only}
- Blauhut B, Kramar H, Vinazzer H, Bergmann H. Substitution of antithrombin III in shock and DIC: a randomized study. Thrombosis Research 1985;39(1):81‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Diaz‐Cremades 1994 {published data only}
- Diaz‐Cremades JM, Lorenzo R, Sánchez M, Moreno MJ, Alsar MJ, Bosch JM, et al. Use of antithrombin III in critical patients. Intensive Care Medicine 1994;20(8):577‐80. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Eisele 1998 {published data only}
- Eisele B, Lamy M, Thijs LG, Keinecke HO, Schuster HP, Matthias FR, et al. Antithrombin III in patients with severe sepsis. A randomized, placebo‐controlled, double‐blind multicenter trial plus a meta‐analysis on all randomized, placebo‐controlled, double‐blind trials with antithrombin III in severe sepsis.. Intensive Care Medicine 1998;24(7):663‐72. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fourrier 1993 {published data only}
- Fourrier F, Chopin C, Huart JJ, Runge I, Caron C, Goudemand J. Double‐blind, placebo‐controlled trial of antithrombin III concentrates in septic shock with disseminated intravascular coagulation. Chest 1993;104(3):882‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fulia 2003 {published data only}
- Fulia F, Cordaro S, Meo P, Gitto P, Gitto E, Trimarchi G, et al. Can the administration of antithrombin III decrease the risk of cerebral hemorrhage in premature infants?. Biology of the Neonate 2003;83(1):1‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Gando 2013 {published data only}
- Gando S, Saitoh D, Ishikura H, Ueyama M, Otomo Y, Oda S, et al. Japanese Association for Acute Medicine Disseminated Intravascular Coagulation (JAAM DIC) Study Group for the JAAM DIC Antithrombin Trial (JAAMDICAT). A randomized, controlled, multicenter trial of the effects of antithrombin on disseminated intravascular coagulation in patients with sepsis. Critical Care 2013;17(6):297. [PUBMED: 24342495] [DOI] [PMC free article] [PubMed] [Google Scholar]
- UMIN‐CTR Clinical Trial. Trial registration. upload.umin.ac.jp/cgi‐open‐bin/ctr/ctr.cgi?function=brows&action=brows&type=summary&recptno=R000001064&language=E (accessed 21 April 2015). [https://]
Grenander 2001 {published data only}
- Grenander A, Bredbacka S, Rydvall A, Aroch R, Edner G, Koskinen LO, et al. Antithrombin treatment in patients with traumatic brain Injury; a pilot study. Journal of Neurosurgical Anesthesiology 2001;13(1):49‐56. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Haire 1998 {published data only}
- Haire WD, Ruby EI, Stephens LC, Reed E, Tarantolo SR, Pavletic ZS, et al. A prospective randomized double‐blind trial of antithrombin III concentrate in the treatment of multiple‐organ dysfunction syndrome during hematopoietic stem cell transplantation. Biology of Blood and Marrow Transplantation 1998;4(3):142‐50. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Harper 1991 {published data only}
- Harper PL, Williamson L, Park G, Smith JK, Carrell RW. A pilot study of antithrombin replacement in intensive care management: the effects on mortality, coagulation and renal function. Transfusion Medicine 1991;1(2):121‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Inthorn 1997 {published data only}
- Hoffmann JN, Mühlbayer D, Jochum M, Inthorn D. Effect of long‐term and high‐dose antithrombin supplementation on coagulation and fibrinolysis in patients with severe sepsis. Critical Care Medicine 2004;32(9):1851‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Inthorn D, Hoffmann JN, Hartl WH, Mühlbayer D, Jochum M. Antithrombin III supplementation in severe sepsis: beneficial effects on organ dysfunction. Shock 1997;8(5):328‐34. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Inthorn D, Hoffmann JN, Hartl WH, Mühlbayer D, Jochum M. Effect of antithrombin III supplementation on inflammatory response in patients with severe sepsis. Shock 1998;10(2):90‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kobayashi 2003 {published data only}
- Kobayashi T, Terao T, Ikenoue T, Sameshima H, Nakabayashi M, Kajiwara Y, et al. BI 51 017 Study Group. Treatment of severe preeclampsia with antithrombin concentrate: results of a prospective feasibility study. Seminars in Thrombosis and Hemostasis 2003;29(6):645‐52. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Langely 1993 {published data only}
- Langley PG, Hughes RD, Forbes A, Keays R, Williams R. Controlled trial of antithrombin III supplementation in fulminant hepatic failure. Journal of Hepatology 1993;17(3):326‐31. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lavrentieva 2008 {published data only}
- Lavrentieva A, Kontakiotis T, Bitzani M, Parlapani A, Thomareis O, Scourtis H, et al. The efficacy of antithrombin administration in the acute phase of burn injury.. Thrombosis and Haemostasis 2008;100(2):286–90. [PUBMED: 18690349] [PubMed] [Google Scholar]
Maki 2000 {published data only}
- Maki M, Kobayashi T, Terao T, Ikenoue T, Satoh K, Nakabayashi M, et al. BI51.017 Study Group. Antithrombin therapy for severe preeclampsia: results of a double‐blind, randomized, placebo‐controlled trial. Thrombosis and Haemostasis 2000;84(4):583‐90. [MEDLINE: ] [PubMed] [Google Scholar]
- Sameshima H, Kodama Y, Ikenoue T, Kajiwara Y. Antithrombin improves fetal condition in women with severe pre‐eclampsia before 32 weeks of gestation;a randomized, double‐blind, placebo‐controlled trial. Journal of Obstetrics and Gynaecology Research 2008;34(1):34–9. [PUBMED: 18226126] [DOI] [PubMed] [Google Scholar]
Mitchell 2003 {published data only}
- Mitchell L, Andrew M, Hanna K, Abshire T, Halton J, Wu J, et al. Trend to efficacy and safety using antithrombin concentrate in prevention of thrombosis in children receiving l‐asparaginase for acute lymphoblastic leukemia. Results of the PAARKA study. Thrombosis and Haemostasis 2003;90(2):235‐44. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Mitchell LG, Andrew M, Hanna K, Abshire T, Halton J, Anderson R, et al. A prospective cohort study determining the prevalence of thrombotic events in children with acute lymphoblastic leukemia and a central venous line who are treated with L‐asparaginase ‐ Results of the prophylactic antithrombin replacement in kids with acute lymphoblastic leukemia treated with asparaginase (PARKAA) study. Cancer 2003;97(2):508‐16. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Muntean 1989 {published data only}
- Muntean W, Rossegger H. Antithrombin III concentrate in preterm infants with IRDS: an open, controlled, randomized clinical trial (abstract). Thrombosis and Haemostasis 1989;62:288. [Google Scholar]
Neporada 2008A {published and unpublished data}
- Neporada E, Vorobyeva N, Nedashkovsky E. Antithrombin III deficiency correction in patients with disseminated intravascular coagulation. Obshaya Reanimatologia 2008;5:49‐54. [Google Scholar]
Nishiyama 2011 {published data only}
- Nishiyama T, Kohno Y, Koishi K. Effects of antithrombin and gabexate mesilate on disseminated intravascular coagulation: a preliminary study.. American Journal of Emergency Medicine 2012;30(7):1219–23. [PUBMED: 22204993] [DOI] [PubMed] [Google Scholar]
Palareti 1995 {published data only}
- Palareti G, Legnani C, Coccheri S, Ridolfi L, Baudo F, Caimi TM, et al. Laboratory effects of antithrombin‐III (ATIII) replacement in patients with sepsis and or postsurgical complications requiring hemodynamic and or respiratory support ‐ a controlled, double‐blind, randomized multicenter study. Thrombosis and Haemostasis 1995;73(6):1251. [Google Scholar]
Schmidt 1998 {published data only}
- Schmidt B, Gillie P, Mitchell L, Andrew M, Caco C, Roberts R. A placebo‐controlled randomized trial of antithrombin therapy in neonatal respiratory distress syndrome. American Journal of Respiratory and Critical Care Medicine 1998;158(2):470‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schorr 2000 {published data only}
- Schorr M, Siebeck M, Zugel N, Welcker K, Gippner‐Steppert C, Czwienzek E, et al. Antithrombin III and local serum application: adjuvant therapy in peritonitis. European Journal of Clinical Investigation 2000;30(4):359‐66. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schuster 1997 {published data only}
- Schuster HP, Eisele B, Keinecke HO, Heinrichs H, Mescheder A, Knaub S. S‐AT III study: antithrombin III in patients with sepsis. Intensive Care Medicine 1997;23 Suppl 1:76. [Google Scholar]
Smith‐Erichsen 1996 {published data only}
- Smith‐Erichsen N, Aasen AO, Kongsgaard UE, Bakka A, Bergen A, Bjerkelund CE, et al. The effect of antithrombin III substitution therapy on components of the plasma protease systems in surgical patients. Clinical Intensive Care 1996;7:291‐6. [Google Scholar]
Vorobyeva 2007 {published data only}
- Vorob'eva N, Neporada E, Turundaevskaia O, Mel'nikova G. Place of antithrombin III concentrate in the intensive care of disseminated intravascular coagulation. Anesteziologiia i Reanimatologiia 2007;2:42‐4. [PUBMED: 17563999] [PubMed] [Google Scholar]
Warren 2001 {published data only}
- Angstwurm M, Hoffmann J, Ostermann H, Frey L, Spannagl M. Severe sepsis and disseminated intravascular coagulation. Supplementation with antithrombin [Schwere sepsisund disseminierte intravasale erinnung. Substitution von antithrombin]. Anaesthesist 2009;58(2):171–9. [PUBMED: 19189066] [DOI] [PubMed] [Google Scholar]
- Eid A, Wiedermann CJ, Kinasewitz GT. Early administration of high‐dose antithrombin in severe sepsis: single center results from the KyberSept‐trial. Anesthesia and Analgesia 2008;107(5):1633‐8. [PUBMED: 18931224] [DOI] [PubMed] [Google Scholar]
- Gonano C, Sitzwohl C, Meitner E, Weinstabl C, Kettner SC. Four‐day antithrombin therapy does not seem to attenuate hypercoagulability in patients suffering from sepsis. Critical Care 2006;10(6):160. [PUBMED: 17107615] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann JN, Wiedermann CJ, Juers M, Ostermann H, Kienast J, Briegel J, et al. Benefit/risk profile of high‐dose antithrombin in patients with severe sepsis treated with and without concomitant heparin. Thrombosis and Haemostasis 2006;95(5):850‐6. [MEDLINE: ] [PubMed] [Google Scholar]
- Kienast J, Juers M, Wiedermann CJ, Hoffmann JN, Ostermann H, Strauss R, et al. Treatment effects of high‐dose antithrombin without concomitant heparin in patients with severe sepsis with or without disseminated intravascular coagulation. Journal of Thrombosis and Haemostasis 2006;4(1):90‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Rublee D, Opal SM, Schramm W, Keinecke HO, Knaub S. Quality of life effects of antithrombin III in sepsis survivors: results from the KyberSept trial [ISRCTN22931023]. Critical Care 2002;6(4):349‐56. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeuwen HJ, Heezius EC, Dallinga GM, Strijp JA, Verhoef J, Kessel KP. Lipoprotein metabolism in patients with severe sepsis. Critical Care Medicine 2003;31(5):1359‐66. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, et al. Caring for the critically ill patient. High‐dose antithrombin III in severe sepsis, a randomized controlled trial. JAMA 2001;286(15):1869‐78. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Wiedermann CJ, Hoffmann JN, Juers M, Ostermann H, Kienast J, Briegel J, et al. High‐dose antithrombin III in the treatment of severe sepsis in patients with a high risk of death: efficacy and safety. Critical Care Medicine 2006;34(2):285‐92. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Waydhas 1998 {published data only}
- Waydhas C, Nast‐Kolb D, Gippner‐Steppert C, Trupka A, Pfundstein C, Schweiberer L, et al. High‐dose antithrombin III treatment of severely injured patients: results of a prospective study. Journal of Trauma, Injury, Infection and Critical Care 1998;45(5):931‐40. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Aibiki 2006 {published data only}
- Aibiki M, Fukuoka N, Nishiyama T, Maekawa S, Shirakawa Y. Differences in antithrombin III activities by administration method in critical patients with disseminated intravascular coagulation: a pharmacokinetic study. Shock 2007;28(2):141‐7. [PUBMED: 17515857] [DOI] [PubMed] [Google Scholar]
Dietrich 2013 {published data only}
- Dietrich W, Busley R, Spannagl M, Braun S, Schuster T, Lison S. The influence of antithrombin substitution on heparin sensitivity and activation of hemostasis during coronary artery bypass graft surgery: a dose‐finding study. Anesthesia and Analgesia 2013;116(6):1223‐30. [PUBMED: 23408673] [DOI] [PubMed] [Google Scholar]
Doi 2012 {published data only}
- Doi T, Kozawa O, Enomoto Y, Ogura S. Analysis of the anti‐inflammatory effects of antithrombin III on human platelets. 7th Congress of the International Federation of Shock Societies ‐ 35th Annual Conference on Shock Miami Beach, FL United States 2012;1:198. [Google Scholar]
Hoffmann 2010 {published data only}
- Hoffmann JN, Fertmann JM, Illner W, Arbogast H, Land H, Jauch K. High‐dose antithrombin can not improve kidney function despite improved graft perfusion in human kidney transplantation‐final results of a prospective, controlled trial. 23rd International Congress of the Transplantation Society, TTS 2010 Vancouver, BC Canada. 2010; Vol. 1:1882.
Ilias 2000 {published data only}
- Ilias W, List W, Decruyenaere J, Lignian H, Knaub S, Schindel F, et al. Antithrombin III in patients with severe sepsis: a pharmacokinetic study. Intensive Care Medicine 2000;26(6):704‐15. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Jochum 1995 {published and unpublished data}
- Jochum M. Influence of high‐dose antithrombin concentrate therapy on the release of cellular proteinases, cytokines, and soluble adhesion molecules in acute inflammation. Seminars in Hematology 1995;32 Suppl 2(4):19‐32. [MEDLINE: ] [PubMed] [Google Scholar]
Kanbak 2011 {published data only}
- Kanbak M, Oc B, Salman MA, Ocal T, Oc M. Peroperative effects of fresh frozen plasma and antithrombin III on heparin sensitivity andcoagulation during nitroglycerine infusion in coronary artery bypass surgery. Blood Coagulation and Fibrinolysis 2011;22(7):593‐9. [PUBMED: 21799398] [DOI] [PubMed] [Google Scholar]
Kim 2013 {published data only}
- Kim KA, Lim YY, Kim SH, Park JY. Bioequivalence of two intravenous formulations of antithrombin III: a two‐way crossover study in healthy Korean subjects. Clinical Therapeutics 2013;35(11):1752‐61. [PUBMED: 24094463] [DOI] [PubMed] [Google Scholar]
Korninger 1987 {published data only}
- Korninger C, Klepteko W, Miholic J, Schwarz C, Lechner K. Randomized trial of antithrombin‐III versus placebo in patients undergoing peritoneo‐venous shunt operation. Thrombosis and Haemostasis 1987;58(1):426. [Google Scholar]
Leitner 2006 {published data only}
- Leitner JM, Firbas C, Mayr FB, Reiter RA, Steinlechner B, Jilma B. Recombinant human antithrombin inhibits thrombin formation and interleukin 6 release in human endotoxemia. Clinical Pharmacology and Therapeutics 2006;79(1):23‐34. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Maki 1987 {published data only}
- Maki M, Terao T, Ikenoue T, Takemura T, Sekiba K, Shirakawa K, et al. Clinical evaluation of antithrombin III concentrate (BI 6.013) for disseminated intravascular coagulation in obstetrics. Well‐controlled multicenter trial. Gynecology and Obstetric Investigation 1987;23(4):230‐40. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Mayumi 2011 {published data only}
- Mayumi T, Suzuki S, Yamamoto T, Ichikawa T, Onodera M, Tsuzuki M, et al. Levels of antithrombin activity after antithrombin administration indicate prognosis of septic DIC patients. Shock Society 2011; Vol. 1:www.shocksociety.org/assets/docs/shock%202011%20oral%20abstracts.pdf.
Neporada 2008B {unpublished data only}
- Neporada E, Turundayevskaya O, Vorobyeva N. Antithrombin III concentrate in comparison with FFP in patients with disseminated intravascular coagulation [КОНЦЕНТРАТ АНТИТРОМБИНА III В СРАВНЕНИИ СО СВЕЖЕЗАМОРОЖЕННОЙ ПЛАЗМОЙ ПРИ СИНДРОМЕ ДИССЕМИНИРОВАННОГО ВНУТРИСОСУДИСТОГО СВЕРТЫВАНИЯ]. Human Ecology 2008;7:47‐51. [Google Scholar]
Nishiyama 2006 {published data only}
- Nishiyama T. Antithrombin can modulate coagulation, cytokine production, and expression of adhesion molecules in abdominal aortic aneurysm repair surgery. Anesthesia and Analgesia 2006;102(4):1007‐11. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Paparella 2014 {published data only}
- NCT01201070. Trial registration. clinicaltrials.gov/ct2/show/NCT01201070 (accessed 21 April 2015).
- Paparella D, Rotunno C, Palo M, Finamore S, Guida P, Rubino G, et al. Antithrombin administration in patients with low antithrombin values after cardiac surgery: a randomized controlled trial. Annals of Thoracic Surgery 2014;97(4):1207‐13. [PUBMED: 24507941] [DOI] [PubMed] [Google Scholar]
Paternoster 2000 {published data only}
- Paternoster DM, Fusco D, Tambuscio B. Treatment with antithrombin III (AT III) concentrate in pre‐eclampsia. International Journal of Gynaecology and Obstetrics 2000;71(2):175‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Paternoster 2004 {published data only}
- Paternoster DM, Fantinato S, Manganelli F. Efficacy of AT in pre‐eclampsia: a case‐control prospective trial. Thrombosis and Haemostasis 2004;91(2):283‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ranucci 2013 {published data only}
- Ranucci M, Baryshnikova E, Crapelli GB, Woodward MK, Paez A, Pelissero G. Preoperative antithrombin supplementation in cardiac surgery: a randomized controlled trial. Journal of Thoracic and Cardiovascular Surgery 2013;145(5):1393‐9. [PUBMED: 23102903] [DOI] [PubMed] [Google Scholar]
Sawamura 2009 {published data only}
- Sawamura A, Gando S, Hayakawa M, Hoshino H, Kubota N, Sugano M. Effects of antithrombin III in patients with disseminated intravascular coagulation diagnosed by newly developed diagnostic criteria for critical illness. Clinical and Applied Thrombosis/Hemostasis 2009;15(5):561‐6. [PUBMED: 18840625] [DOI] [PubMed] [Google Scholar]
Scherer 1997 {published data only}
- Scherer R, Kabatnik M, Erhard J, Peters J. The influence of antithrombin III (AT III) substitution to supranormal activities on systemic procoagulant turnover in patients with end‐stage chronic liver disease. Intensive Care Medicine 1997;23(11):1150‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Shimada 1994 {published data only}
- Shimada M, Matsumata T, Kamakura T, Hayashi H, Urata K, Sugimachi K. Modulation of coagulation and fibrinolysis in hepatic resection: a randomized prospective control study using antithrombin III concentrates. Thrombosis Research 1994;74(2):105‐14. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Terao 1989 {published data only}
- Terao T, Kobayashi T, Imai N, Oda H, Karasawa T. Pathological state of the coagulatory and fibrinolytic system in preeclampsia and the possibility of its treatment. Asia‐Oceania Journal of Obstetrics and Gynaecology 1988;15(1):25‐32. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Valsecchi 2008 {published data only}
- Valsecchi L, D’Angelo A. High‐dose antithrombin supplementation in early pre‐eclampsia: a double‐blind, placebo controlled study (on behalf of the ATIII‐EPAS study group). Journal of Thrombosis and Haemostasis 2009;7:297‐8. [Google Scholar]
Vinazzer 1995 {published data only}
- Vinazzer HA. Antithrombin III in shock and disseminate intravascular coagulation. Clinical and Applied Thrombosis/Hemostasis 1995;1(1):62‐5. [Google Scholar]
References to ongoing studies
D'angelo 2005 {published data only}
- D'angelo A, Valsecchi L. A double‐blind trial of high‐dose antithrombin supplementation in early pre‐eclampsia. Thrombosis Research 2005;115(Suppl 1):117. [DOI] [PubMed] [Google Scholar]
Additional references
Altman 2003
- Altman DG, Bland JM. Interaction revisited: the difference between two estimates. BMJ 2003;326(7382):219. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Becker 2000
- Becker BF, Heindl B, Kupatt C, Zahler S. Endothelial function and hemostasis. Zeitschrift fur Kardiologie 2000;89(3):160‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta‐analyses may be inconclusive‐‐Trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta‐analyses. International Journal of Epidemiology 2009;38(1):287‐98. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Chan 2004
- Chan AW, Hróbjartsson A, Haahr MT, Gøtzsche PC, Altman DG. Empirical evidence for selective reporting of outcomes in randomized trials: comparison of protocols to published articles. JAMA 2004;291(20):2457‐65. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Diaz 2015
- Diaz R, Moffett BS, Karabinas S, Guffrey D, Mahoney DH Jr, Yee DL. Antithrombin concentrate use in children receiving unfractionated heparin for acute thrombosis. Journal of Pediatrics 2015;167(3):645‐9. [DOI: 10.1016/j.jpeds.2015.06.006; PUBMED: 26148660] [DOI] [PubMed] [Google Scholar]
Fourrier 2000
- Fourrier F, Jourdain M, Tournoys A. Clinical trial results with antithrombin III in sepsis. Critical Care Medicine 2000;28(9 Suppl):S38‐43. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Gotzsche 2000
- Gotzsche PC. Why we need a broad perspective on meta‐analysis. It may be crucially important for patients. BMJ 2000;321(7261):585‐6. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JP, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;15(11):1539‐58. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. [DOI: 10.1002/9780470712184] [DOI]
Hutton 2000
- Hutton JL, Williamson PR. Bias in meta‐analysis due to outcome variable selection within studies. Journal of the Royal Statistical Society Series 2000;49(3):359‐70. [DOI: 10.1111/1467-9876.00197] [DOI] [Google Scholar]
Lan 1983
- Lan KKG, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika 1983;70(3):659‐63. [Google Scholar]
Larsson 2001
- Larsson H, Åkerud P, Nordling K, Raub‐Segall E, Claesson‐Welsh L, Bjork I. A novel anti‐angiogenic form of antithrombin with retained proteinase binding ability and heparin affinity. Journal of Biological Chemistry 2001;276(15):11996‐2002. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Levi 2004
- Levi M, Jonge E, Poll T. New treatment strategies for disseminated intravascular coagulation based on current understanding of the pathophysiology. Annals of Medicine 2004;36(1):41‐9. [DOI] [PubMed] [Google Scholar]
Mayr 2014
- Mayr F, Yende S, Angus D. Epidemiology of severe sepsis. Virulence 2014;5(1):4‐11. [PUBMED: 24335434] [DOI] [PMC free article] [PubMed] [Google Scholar]
Opal 2002
- Opal SM, Kessler CM, Roemisch J, Knaub S. Antithrombin, heparin, and heparan sulfate. Critical Care Medicine 2002;30(5 Suppl):S325‐31. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Opal 2003
- Opal SM. Clinical trial design and outcomes in patients with severe sepsis. Shock 2003;20(4):295‐302. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Periti 2000
- Periti P. Current treatment of sepsis and endotoxaemia. Expert opinion of Pharmacotherapy 2000;1(6):1203‐17. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pogue 1997
- Pogue JM, Yusuf S. Cumulating evidence from randomized trials: Utilizing sequential monitoring boundaries for cumulative meta‐analysis. Controlled Clinical Trials 1997;18(6):580‐93. [DOI] [PubMed] [Google Scholar]
Pogue 1998
- Pogue J, Yusuf S. Overcoming the limitations of current meta‐analysis of randomised controlled trials. Lancet 1998;351(9095):47‐52. [DOI] [PubMed] [Google Scholar]
Polderman 2004
- Polderman KH, Girbes ARJ. Drug intervention trials in sepsis: divergent results. Lancet 2004;363(9422):1721‐3. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
RevMan 5.3 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rublee 2003
- Rublee D, Opal SM, Schramm W, Keinecke HO, Knaub S. Quality of life effects of antithrombin III in sepsis survivors: results from the KyberSept trial. Critical Care 2002;Aug; 6(4):349‐56. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tagami 2014
- Tagami T, Matsui H, Horiguchi H, Fushimi K, Yasunaga H. Antithrombin and mortality in severe pneumonia patients with sepsis‐associated disseminated intravascular coagulation: an observational nationwide study. Journal of Thrombosis and Haemostasis September 2014;12(9):1470–9. [PUBMED: 24943516] [DOI] [PubMed] [Google Scholar]
Tagami 2015
- Tagami T, Matsui H, Fushimi K, Yasunaga H. Supplemental dose of antithrombin use in disseminated intravascular coagulation patientsafter abdominal sepsis. Thrombosis and Haemostasis 2015;114(3):537‐45. [PUBMED: 25948492] [DOI] [PubMed] [Google Scholar]
Thorlund 2009
- Thorlund K, Devereaux PJ, Wetterslev J, Guyatt G, Ioannidis JP, Thabane L, et al. Can trial sequential monitoring boundaries reduce spurious inferences from meta‐analyses?. International Journal of Epidemiology 2009;38(1):276‐86. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61(1):64‐75. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wetterslev 2009
- Wetterslev J, Thorlund K, Brok J, Gluud C. Estimating required information size by quantifying diversity in random‐effects model meta‐analyses. BMC Medical Research Methodology 2009;30(9):86. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiedermann 2002
- Wiedermann CJ, Römisch J. The anti‐inflammatory actions of antithrombin ‐ a review. Acta Medica Austriaca 2002;29(3):89‐92. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wunsch 2008
- Wunsch H, Angus D, Harrison D, Collange O, Fowler R, Hoste E, et al. Variation in critical care services across North America and Western Europe. Critical Care Medicine 2008;36(10):2787‐93. [PUBMED: 18766102] [DOI] [PubMed] [Google Scholar]
Zarychanski 2015
- Zarychanski R, Abou‐Setta AM, Kanji S, Turgeon AF, Kumar A, Houston DS, et al. The efficacy and safety of heparin in patients with sepsis: a systematic review and metaanalysis. Critical Care Medicine 2015;43(3):511–8. [PUBMED: 25493972] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Afshari 2007
- Afshari A, Wetterslev J, Brok J, Møller A. Antithrombin III in critically ill patients: systematic review with meta‐analysis and trial sequential analysis. BMJ 2007;335(7632):1248‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Afshari 2008
- Afshari A, Wetterslev J, Brok J, Møller A. Antithrombin III for critically ill patients. Cochrane Database of Systematic Reviews 2008, Issue 3. [DOI: 10.1002/14651858.CD005370.pub2] [DOI] [PubMed] [Google Scholar]