

Abstract
Purpose of Review
This review summarizes research on the physiological changes that occur with aging and the resulting effects on fracture healing.
Recent Findings
Aging affects the inflammatory response during fracture healing through senescence of the immune response and increased systemic pro-inflammatory status. Important cells of the inflammatory response, macrophages, T cells, mesenchymal stem cells, have demonstrated intrinsic age-related changes that could impact fracture healing. Additionally, vascularization and angiogenesis are impaired in fracture healing of the elderly. Finally, osteochondral cells and their progenitors demonstrate decreased activity and quantity within the callus.
Summary
Age-related changes affect many of the biologic processes involved in fracture healing. However, the contributions of such changes do not fully explain the poorer healing outcomes and increased morbidity reported in elderly patients. Future research should address this gap in understanding in order to provide improved and more directed treatment options for the elderly population.
Keywords: Fracture healing, Elderly, Senescence, Inflammatory response, Inflamm-aging
Introduction
The elderly population in the USA has been steadily increasing, and those aged 65 years old and older are expected to comprise 17% of the population by 2030 [1, 2]. This growing population presents their own unique health needs, and in order to meet these needs a better understanding of the physiologic changes that occur with aging is necessary. The skeletal system exhibits physiologic changes that occur with increasing age. Conditions such as osteoporosis and osteoarthritis increase with age. Additionally, many reports demonstrate a higher rate of bone fracture, and these are associated with increased morbidity and mortality [3–5]. A decline in healing potential is observed in the elderly, and this may result in increased rates of delayed healing or nonunions [6]. Delayed healing, and the resulting incapacitation, can have more severe and systemic consequences in the elderly, which poses unique challenges for the treating clinician [3, 7]. While increased age has been generally associated with a wide range of physiological changes, the mechanisms that result in decreased capacity for fracture healing are not fully understood.
An understanding of the age-related effects to fracture healing is complicated by a lack of a complete understanding of fracture healing in healthy and in young individuals. However, by analyzing the individual facets of fracture healing that we do understand we can compare differences in fracture healing between the young and old humans and animals. This review will highlight the phases of fracture healing and the respective physiologic changes that occur with age. Cellular, molecular, and genetic differences between young and old humans and animal models will be characterized to illustrate the current understanding of the effect of age on bone fracture healing. Figure 1 summarizes the effect of age on critical cells that contribute to fracture healing.
Fig. 1.
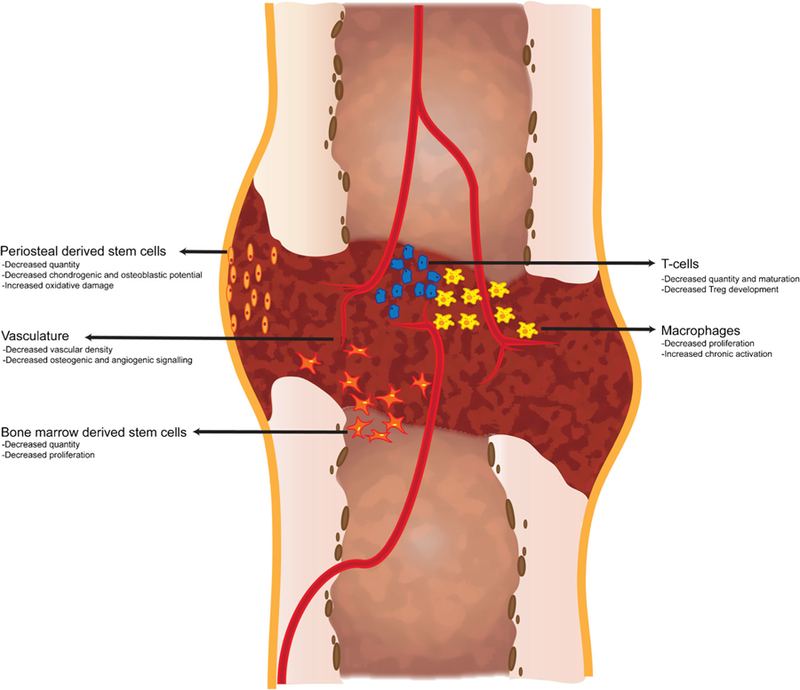
The effect of age on the cellular contribution to fracture healing. Stem cells and immune cells involved in fracture healing demonstrate age-related changes that may negatively affect fracture healing. Osteochondral stem cells arise from the periosteum and bone marrow and demonstrate decreased quantity, increased oxidative damage, and decreased osteoblastic and chondrogenic differentiation potential with age. T-cells contribute to fracture healing and production and maturation of T cells is negatively affected by age-related changes to the bone marrow hematopoietic compartment and to the thymus. Macrophages are important regulators of inflammation during fracture healing. Aged macrophages demonstrate decreased proliferation and increased activation that may contribute to the poorer healing outcomes associated with aged macrophages compared to young. Finally, adequate vascularization is required for successful fracture healing. Aged animals demonstrate decreased vascular density within the callus which is associated with decreased levels of key angiogenic factors required for healing
Physiology of Bone Fracture Healing
Fracture healing proceeds through multiple phases characterized by anabolic and catabolic processes [8]. The early healing stage is characterized by a robust inflammatory response that is responsible for debriding the fracture site and contributing to the signaling milieu that will propagate the successive stages of healing, including recruitment and differentiation of skeletal tissue progenitor cells [9]. The anabolic phase follows the initial inflammatory response. Progenitor cells give rise to a soft callus with a central cartilaginous region and new bone formation at the periphery [9, 10]. The soft callus is characterized by avascular cartilage tissue that induces vascularization [11]. A hard callus begins to develop through endochondral ossification with increased mineralization and replacement of chondrocytes with osteoblast, in part, through transdifferentiation [12, 13]. Finally, remodeling of the callus occurs through catabolic processes. The callus is reduced in size and osteoblastic and osteoclastic processes alternate to reestablish the normal hematopoietic and trabecular structure, restoring bone to its pre-injured state [14].
Histological and molecular changes within the callus have been described during the stages of fracture healing as mentioned above and provide healing comparisons between old and young animals. Numerous studies have reported delayed fracture healing in elderly animals and have shown decreased cartilage and bone formation, delayed cartilage resorption, and slower mineralization within the callus [15–17]. Delayed bone healing may be associated with age-related changes in the osteochondral stem cells. In general, there is an age-related decrease of stem cell quantity compounded by a decrease in proliferation and differentiation potential as demonstrated in humans and animal models [18–20]. Lopas et al. demonstrated a decrease in osteochondral stem cell proliferation associated with a significant decrease in bone and cartilage content within the facture callus of aged mice compared to young [15].
Chondrocytes and osteoblasts arise from stem cells predominantly located in the periosteum during fracture healing [9, 21, 22]. Senescence and greater oxidative damage was associated with periosteal-derived progenitor cells from old humans compared to young [23]. Additionally, a decreased number of periosteal cells were able to be derived from the periosteum of old humans compared to young [21]. The chondrogenic potential of stem cells in the periosteum is decreased in elderly mice compared to young mice, and chondrogenic differentiation from periosteal cells is delayed in old versus young mice [22, 24]. Older animals have delayed expression of type 2 collagen (ColII) and delayed cartilage matrix deposition at early time points of fracture healing [24]. Similarly, osteoblast differentiation and osteocalcin expression is delayed from periosteal cells at the fracture site in elderly mice compared to young mice [24].
Stem cells contributing to fracture healing may also arise from other tissue sources that could be negatively affected by age-related changes. Cells located in the muscle appear to contribute to bone fracture healing. While muscle stem cells, known as satellite cells, may contribute only a small number of cells that comprise the skeletal tissues, they appear to regulate fracture healing possibly through signaling interactions [25]. With aging, satellite cell quantity and function decline which may negatively affect their ability to support fracture healing [26]. However, the role of other cell types that reside in the muscle is not known and worthy of investigation. The bone marrow is also a potential source of osteochondral stem cells [27, 28]. Similar to satellite cells, aging results in decreased function and quantity of bone marrow stem cells that could have a negative effect on fracture healing [29, 30].
Age also affects later stages of healing during endochondral ossification. The characteristic histological findings that describe endochondral ossification, hypertrophic chondrocytes, expression of type 10 collagen (ColX), and vascular invasion were all delayed in elderly mice compared to young mice [24]. Completion of endochondral ossification, characterized by complete conversion of cartilage to bone within the callus, was also delayed in elderly mice versus young mice [24].
The age-related alterations to the cellular processes that are evident during facture healing are accompanied by changes in the regulation of critical genes involved in bone fracture healing. By using the whole genome expression analysis of fracture calluses in rats, significant differential regulation of 144 genes was found in young compared to elderly rats [31]. Functional analysis of these genes suggested they were largely involved in cell migration [31]. More specifically, bone morphogenic protein (BMP-2) and Indian Hedgehog (IHH) expression in the fracture callus of elderly rats were decreased compared to young rats [32]. Other studies have shown comparable levels of gene expression in the early time points of fracture healing; however, in elderly rats, expression of IHH, BMP, and TGF-β decreased at the same rate as the young rats despite requiring almost twice as long for complete healing to be detected radiographically [33]. Thus, molecular changes occurring in cells comprising the fracture callus are evident in animals of advanced age, and these changes may contribute to the alterations in healing that are observed in older animals. However, systemic changes as a result of the normal process of aging also occur and could contribute to the delays in healing.
Inflammation and Fracture Healing
The initial phase of fracture healing is characterized by a robust inflammatory response. Secretion of pro-inflammatory cytokines at this time is necessary to initiate the healing process and achieve adequate healing [34]. Temporal control of the inflammatory response is crucial for proper bone fracture healing. After the initial pro-inflammatory phase, inflammation must be resolved in order to allow the anabolic processes to begin and continue the subsequent healing phases. However, changes in the inflammatory system occur with age, and chronic inflammation and/or a decreased ability to resolve inflammatory processes could negatively affect bone fracture healing.
Inflamm-Aging and Immunosenescence
An excessive or prolonged inflammatory phase can have detrimental effects on fracture healing [35, 36]. In animal models, prolonged inflammation results in delayed chondrogenesis and smaller callus size [37]. Recently, it was shown that elevated systemic levels of a pro-inflammatory cytokine, TNFα, in induced diabetic mice negatively affected angiogenesis during fracture healing [38]. In humans, an elevated inflammatory status is related to certain systemic conditions, including diabetes, smoking, and increased age. These conditions are all associated with poorer fracture healing outcomes, but the mechanisms remain incompletely understood. The term “inflamm-aging” has been used to describe a chronic increased pro-inflammatory status in the elderly [39]. Elderly people are found to have higher levels of circulating proinflammatory cytokines, even in healthy individuals. It appears that the increased pro-inflammatory status in the elderly predisposes them to the range of systemic diseases including osteoporosis, Alzheimer’s disease, Type II diabetes, atherosclerosis, and Parkinson’s disease [40–42]. Currently, it is unclear what drives this increased inflammation. Inflamm-aging has been suggested to be the result of a defect in the proper resolution of the normal inflammatory response, or the result of an unknown chronic mechanism that signals and prolongs the inflammatory response [43•, 44]. As the inflammatory response is a critical step in proper fracture healing, any disruption of the inflammatory response could negatively affect fracture healing.
Inflamm-aging may also be a result of age-related changes to the immune response. Aging of the adaptive immune response has been described as immunosenescence [45]. Immunosenescence describes a loss of immune function that is associated with a predisposition to infection and disease in the elderly [45]. Increased age is associated with changes in T and B cell production and maturation. T cell production and maturation is negatively affected by age-related changes to the bone marrow hematopoietic compartment and to the thymus [46, 47]. With increasing age, the source of T cell progenitors, the hematopoietic compartment, decreases in size and is associated with a decrease in T cell progenitor quantity and proliferation potential [47]. The T cell progenitors migrate to the thymus for further differentiation and maturation. A decrease in thymus function and T cell output is seen with increasing age as involution and atrophic changes are present [46]. The lack of a sufficient quantity of T cells produced by the thymus negatively affects the ability to mount an effective immune response. Additionally, the lack of T cells limits the availability of regulatory T cells that are present to resolve the mounted immune and inflammatory response [46, 48]. Immune cell trafficking has also been shown to be of importance in fracture healing studies showing impaired fracture healing in CCR2 deficient mice [49]. CCR2 is a chemokine receptor for the ligands Ccl2, Ccl7, and Ccl12 that is expressed on monocytes, myeloid-derived cells, a subset of T cells, and mesenchymal stem cells. Recent studies in a muscle regeneration model have suggested that CCR2 deficiency in young mice results in an inflamm-aging environment similar to changes seen with aging [50]. In this way, it has been proposed that the elevated pro-inflammatory status of inflamm-aging may be a response to age-related defects of the immune response. Figure 2 demonstrates a conceptual model of the effects of inflammatory response dysregulation on the subsequent stages of fracture healing.
Fig. 2.
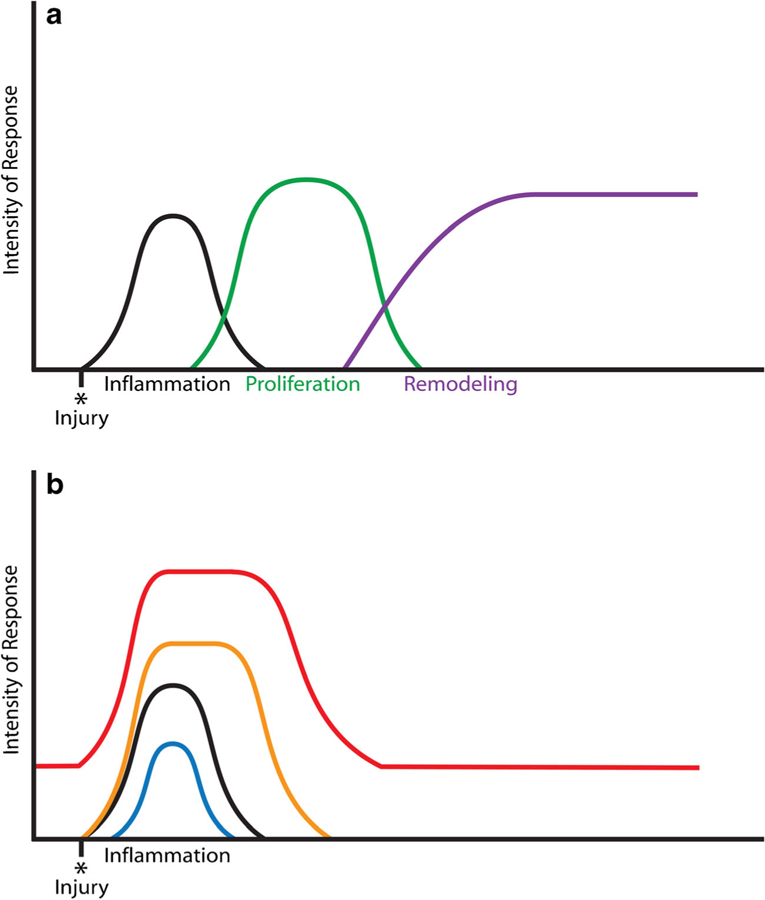
The effects of inflammatory response perturbation on the stages of fracture healing. a Fracture healing follows three general stages of inflammation, proliferation, and remodeling. The initial inflammatory response is tightly regulated and crucial in initiating the subsequent stages of healing. b Systemic conditions, including increased age, have an effect on inflammation and may result in differential inflammatory responses during fracture healing. Senescence of the inflammatory response results in a decreased and limited inflammatory response (blue curve) that may result in inadequate activation of the proceeding healing stages. An exaggerated and sustained response (orange and red curves) can result from inadequate resolution of the response and may negatively affect the proceeding stages. An increased basal level of inflammation (red curve) is proposed to occur with inflamm-aging and would have possible negative effects throughout the healing process
The age-related effects on inflammation during fracture healing can be investigated independently of other age related changes to the organism by using a chimeric animal model. Xing et al. used such a chimeric model of lethal irradiation followed by bone marrow transplant [51]. By irradiating aged mice and transplanting the bone marrow of young mice to those animals, investigators found that the osteochondral stem cells were derived from the aged host and the inflammatory cells were derived from the young donor in a fracture model. In this manner, the older mice receiving young bone marrow had larger calluses and more bone in the early healing time points, more rapid callus remodeling at later stages of healing, and a rejuvenation of the inflammatory response compared to older mice receiving age-matched bone marrow transplants [51]. A similar study by Baht et al. utilized a parabosis and a bone marrow transplant model of old and young mice in a fracture healing study [52•]. Fractures in old mice healed significantly better with shared circulation from a young mouse or with bone marrow transplants from young mice compared to aged-matched old mouse donors. No cells from the young donor circulation or bone marrow differentiated into skeletal cells in the callus suggesting a component of the circulation or bone marrow could revert osteochondral stem cells of old mice to a more youthful phenotype. They further found that β-catenin was differentially expressed in old and young mice during fracture healing. Shared circulation with young mice resulted in decreased levels of β-catenin and was associated with improved fracture healing. Decreasing β-catenin activity in old mice resulted in similar improvements in healing as sharing circulation with young mice. The two studies described above suggest a crucial component regulating fracture healing of the hematopoietic environment that is affected by age-related changes.
Cellular Regulation of Inflammation
An understanding of the intrinsic age-related changes to cells involved in the inflammatory response may explain, in part, the poorer healing potential in the elderly. Cells of the innate and adaptive immune system local to the fracture site assist in regulating the inflammatory response. Macrophages are powerful regulators of inflammation. In the early inflammatory stage of healing, macrophages are classically activated and have an M1 phenotype [53, 54]. M1 macrophages are proinflammatory and release cytokines IL-1, IL-6, TNF-α, and iNOS to elicit and propagate the inflammatory response [53, 54]. As inflammation is down-regulated in the later phases of fracture healing, the macrophages acquire an alternatively activated M2 state. M2 macrophages express anti-inflammatory cytokines, such as IL-10 and promote healing through secretion of growth factors TGFβ, VEGF, and PDGF [53, 55]. Temporal control of the polarization of M1 and M2 macrophages is important to regulate inflammation during the healing process.
Intrinsic age-related changes to macrophages may perturb the inflammatory response in the elderly and may have negative consequences for fracture healing. Aged macrophages were found to be less responsive to granulocyte macrophage colony-stimulating factor (GMCSF) that resulted in decreased proliferation compared to young macrophages [56]. Additionally, elevated serum levels of chitotriosidase, a marker for chronically activated macrophages, was found in elderly humans compared to young controls [57]. The negative effects of age-related changes to macrophages are further supported in animal models of healing. Cutaneous wounds heal slower in aged mice and can be rescued with transplants of macrophages from young mice [58•]. Further, aged macrophages appear to be detrimental to healing. In blocking macrophage recruitment to the fracture site, elderly mice appear to exhibit better fracture healing compared to elderly mice that had normal macrophage activity [59]. Conversely, in the same experiment, blocking macrophage recruitment to the fracture site in young mice had a negative effect on fracture healing [59].
Macrophages are known to contribute to bone healing. A tissue resident population of macrophages, termed “osteomacs,” has recently been described [60•]. Osteomacs regulate osteoblast function and promote fracture healing; blocking their activity has deleterious effects on bone healing [60•, 61]. While it appears clear that circulating macrophages that are recruited to the site of bone injury have age-related changes that hinder fracture healing, whether age-related changes occur in osteomacs is not known.
Cells of the adaptive immune system also contribute to fracture healing [62]. T and B cells are present within the early callus during the inflammatory phase [63]. Regulatory B cells negatively regulating inflammation through expression of IL-10 and downregulating pro-inflammatory cytokine expression in Tcells within the callus [64]. B cells from patients with poor fracture healing outcomes had decreased expression of IL-10 compared to patients that healed normally [64]. As healing continues, cartilage formation within the callus is associated with a local increase in Treg cells [65]. At later time points of healing, T and B cells are readily present at sites of mineralization and in direct contact with osteoblasts and osteoclasts [63]. Models of fracture healing using mice with genetic modifications have further elucidated the role of T and B cells. Deficient T and B cell function in Rag1−/− mice demonstrated impaired fracture healing which was associated with decreased IL-17F expression [66]. IL-17F was shown to promote osteoblast maturation in vivo [66]. Additionally, dysregulated T cell recruitment and activation within the bone in a mouse model of lupus was associated with increased bone turnover and decreased bone fraction within the fracture callus [65]. As discussed above, T and B cell quantity and function are negatively affected by age. Due to the involvement of T and B cells in fracture healing, such age-related changes could have a negative effect on fracture healing.
Mesenchymal stem cells are also involved in the inflammatory response and act as powerful immunomodulators at the site of injury to control excessive inflammation and promote repair [67–69]. Suppression of inflammation mediated by MSCs occurs, in part, through signaling and interactions with local inflammatory cells. MSCs have been shown to interact with T cells, B cells, NK cells, and dendritic cells through signaling that limits proliferation of inflammatory cells or promotes secretion of anti-inflammatory molecules [70]. Additionally, research has suggested that MSC and macrophages interact to promote alternatively activated macrophages in the downregulation of inflammation and promotion of healing [71]. The immunomodulatory properties of MSCs have been utilized clinically and have been reported beneficial in the treatment of graft versus host disease, Crohn’s disease, renal failure, and heart failure [72–75].
Intrinsic age-related changes to MSCs could explain the impaired healing in older humans and animals as a result of dysregulated inflammatory response. The quantity of MSCs isolated from bone marrow is decreased with age [30]. Additionally, there are increased markers for oxidative damage of MSCs from elderly human samples [58•]. These agerelated changes to MSCs corroborate findings in experimental wound healing studies that show a benefit of therapeutic administration of MSCs from young animals but no benefit in healing with aged MSCs [58•].
Vasculature and Fracture Healing
Successful bone fracture healing requires adequate vascularization of the tissue [76]. The contribution of the vasculature to fracture healing includes providing blood supply for delivery of nutrients and cells, providing the endothelial cells that express angiogenic and osteogenic signaling molecules locally, and providing the source of oxygen to the healing callus [9, 77, 78]. However, the complete contribution of the vasculature to fracture healing is not fully understood.
With increasing age, perturbations in bone fracture healing are associated with age-related dysfunction to the bone vascular system and its ability to regenerate in healing. Generally, the vascular perfusion of the skeleton decreases with age [79]. Elderly rats display significantly higher ossifications and decreased patency of bone marrow blood vessels compared to younger rats [80]. The decrease in vascularization at the time of fracture may delay angiogenesis during the fracture healing. The fracture callus at early healing time points in young mice have a higher surface density of blood vessels compared to the elderly mice [77]. The increase in vascular density was associated with an early detection of vascular endothelial growth factor (VEGF) and hypoxia inducible factor 1α (HIF-1α) in young mice but not elderly [77]. Additionally, increased Mmp9 and Mmp13 transcripts were detected throughout early and late stage healing in young compared to old mice [ 77 ]. Differential expression of angiogenic factors, including VEGF, HIF-1α, and Mmps, has been well demonstrated in young animals versus old during bone fracture healing [81–83].
Conclusions
Increasing age has been shown to negatively affect the cellular and molecular processes throughout the different stages of bone fracture healing. Inflammatory regulation, cellular differentiation, and signaling cascades are all affected, in part, by age-related changes. Our current understanding of these age related changes explains, only partially, the decreased healing potential and increased complications observed during fracture healing in elderly patients. A more complete understanding is necessary to allow for therapeutics to target the specific age-related deficiencies and provide better care for the increasing geriatric population.
Acknowledgments
This work was funded by NIH/NIA R01AG046282.
Theordore Miclau reports grants and personal fees from the Foundation for Orthopedic Trauma, grants from National Institutes of Health and Baxter, and personal fees from Depuy-Synthes, Acelity, Surrozen, and Arquos outside the submitted work.
Footnotes
Conflict of Interest Daniel Clark, Mary Nakamura, and Ralph Marcucio declare no conflict of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
- 1.Iorio R, Robb WJ, Healy WL, Berry DJ, Hozack WJ, Kyle RF, et al. Orthopaedic surgeon workforce and volume assessment for total hip and knee replacement in the United States: preparing for an epidemic. J Bone Joint Surg Am 2008;90:1598–605. [DOI] [PubMed] [Google Scholar]
- 2.UScensus. U.S. Department of Commerce, Economics and Statistics Administration, U.S. Census Bureau, Washington: 2015. http://www.census.gov/content/dam/Census/library/2015 Accessed 29 July 2015. [Google Scholar]
- 3.Rose S, Maffulli N. Hip fractures: an epidemiological review. Bull Hosp Jt Dis 1999;58:197–201. [PubMed] [Google Scholar]
- 4.Green E, Lubahn JD, Evans J. Risk factors, treatment, and outcomes associated with nonunion of the midshaft humerus fracture. J Surg Orthop Adv 2005;14:64–72. [PubMed] [Google Scholar]
- 5.Cauley JA, Thompson DE, Ensrud KC, Scott J, Black D. Risk of mortality following clinical fractures. Osteoporos Int 2000;11: 556–61. [DOI] [PubMed] [Google Scholar]
- 6.Nieminen S, Nurmi M, Satokari K. Healing of femoral neck fractures; influence of fracture reduction and age. Ann Chir Gynaecol 1981;70:26–31. [PubMed] [Google Scholar]
- 7.Geerts WH, Heit JA, Clagett GP, Pineo GF, Colwell CW, Anderson FA, et al. Prevention of venous thromboembolism. Chest 2001;119:132S–75S. [DOI] [PubMed] [Google Scholar]
- 8.Little DG, Ramachandran M, Schindeler A. The anabolic and catabolic responses in bone repair. J Bone Joint Surg Br 2007;89:425–33. [DOI] [PubMed] [Google Scholar]
- 9.Hankenson KD, Zmmerman G, Marcucio R. Biological perspectives of delayed fracture healing. Injury 2014;45:S8–S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phillips AM. Overview of the fracture healing cascade. Injury 2005;36:55–7. [DOI] [PubMed] [Google Scholar]
- 11.Kurdy NM, Weiss JB, Bate A. Endothelial stimulating angiogenic factor in early fracture healing. Injury 1996;27:143–5. [DOI] [PubMed] [Google Scholar]
- 12.Hu DP, Ferro F, Yang F, Taylor AJ, Chang W, Miclau T, et al. Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development 2017;15:221–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bahney CS, Hu DP, Taylor AJ, Ferro F, Britz HM, Hallgrimsson B, et al. Stem cell-derived endochondral cartilage stimulates bone healing by tissue transformation. J Bone Miner Res 2014;29: 1269–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Einhorn TA, Gerstenfeld LC. Fracture healing: mechanisms and interventions. Nat Rev Rheumatol 2015;11:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopas LA, Belkin NS, Mutyaba PL, Gray CF, Hankenson KD, Ahn J. Fracture in geriatric mice show decreased callus expansion and bone volume. Clin Orthop Relat Res 2014;472:3523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer RA, Tsahakis PJ, Martin DF, Banks DM, Harrow ME, Kiebzak GM. Age and ovariectomy impair both the normalization of mechanical properties and the accretion of mineral by the fracture callus in rats. J Orthop Res 2001;19:428–35. [DOI] [PubMed] [Google Scholar]
- 17.Bak B, Andreassen TT. The effect of aging on fracture healing in the rat. Calcif Tissue Int 1989;45:292–7. [DOI] [PubMed] [Google Scholar]
- 18.Bergman RJ, et al. Age-related changes in osteogenic stem cells in mice. J Bone Miner Res 1996;11:568–77. [DOI] [PubMed] [Google Scholar]
- 19.Gruber R, Koch H, Doll BA, Tegtmeier F, Einhorn TA, Hollinger JO. Fracture healing in the elderly patient. Exp Gerontol 2006;41: 1080–93. [DOI] [PubMed] [Google Scholar]
- 20.Baxter M, et al. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells 2004;22:675–82. [DOI] [PubMed] [Google Scholar]
- 21.Nakahara H, Goldberg VM, Caplan AI. Culture-expanded human periosteal-derived cells exhibit osteochondral potential in vivo. J Orthop Res 1991;9:465–76. [DOI] [PubMed] [Google Scholar]
- 22.O’Driscoll SW, Saris DB, Ito Y, Fitzimmons JS. The chondrogenic potential of periosteum decreases with age. J Orthop Res 2001;19: 95–103. [DOI] [PubMed] [Google Scholar]
- 23.Ferretti C, Lucarini G, Andreoni C, Salvolini E, Bianchi N, Vozzi G, et al. Human periosteal derived stem cell potential: the impact of age. Stem Cell Rev 2015;11:487–500. [DOI] [PubMed] [Google Scholar]
- 24.Lu C, Miclau T, Hu D, Hansen E, Tsui K, Puttlitz C, et al. Cellular basis for age-related changes in fracture repair. J Orthop Res 2005;23:1300–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abou-Khalil R, et al. Role of muscle stem cells during skeletal regeneration. Stem Cells 2015;33:1501–11. [DOI] [PubMed] [Google Scholar]
- 26.Brack AS, Muñoz-Cánoves P. The ins and outs of muscle stem cell aging. Skelet Muscle 2016;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marecic O, Tevlin R, McArdle A, et al. Identification and characterization of an injury-induced skeletal progenitor. Proc Natl Acad Sci U S A 2015;112:9920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tevlin R, Walmsley GG, Marecic O, Hu MS, Wan DC, Longaker MT. Stem and progenitor cells: advancing bone tissue engineering. Drug Deliv Transl Res 2016;6:159–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stenderup K, Justesen J, Clausen C, Kassem M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone 2003;33:919–26. [DOI] [PubMed] [Google Scholar]
- 30.Sebastian S, Andrew S, Alexandra S. Aging of mesenchymal stem cells. Ageing Res Rev 2006;5:91–116. [DOI] [PubMed] [Google Scholar]
- 31.Ode A, Duda GN, Geissler S, Pauly S, Ode JE, Perka C, et al. Interaction of age and mechanical stability on bone defect healing: an early transcriptional analysis of fracture hematoma in rat. PLoS One 2014;9:e106462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer RA, Meyer MH, Tenholder M, Wondracek S, Wasserman R, et al. Gene expression in older rats with delayed union of femoral fractures. J Bone Joint Surg Am 2003;85:1243–54. [DOI] [PubMed] [Google Scholar]
- 33.Desai BJ, Meyer MH, Porter S, Kellam JF, Meyer RA Jr. The effect of age on gene expression in adult and juvenile rats following femoral fracture. J Orthop Trauma 2003;17:689–98. [DOI] [PubMed] [Google Scholar]
- 34.Gerstenfeld LC, Cho TJ, Kon T, Aizawa T, Cruceta J, Graves BD, et al. Impaired intramembranous bone formation during bone repair in the absence of tumor necrosis factor-alpha signaling. Cells Tissues Organs 2001;169:285–94. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt-Bleek K, Schell H, Schulz N, Hoff P, Perka C, Buttgereit F, et al. Inflammatory phase of bone healing initiates the regenerative healing cascade. Cell Tissue Res 2012;347:567–73. [DOI] [PubMed] [Google Scholar]
- 36.Thomas MV, Puleo DA. Infection, inflammation, and bone regeneration: a paradoxical relationship. J Dent Res 2011;90:1052–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dishowitz MI, Mutyaba PL, Takacs JD, Barr AM, Engiles JB, Ahn J, et al. Systemic inhibition of canonical notch signaling results in sustained callus inflammation and alters multiple phases of fracture healing. PLoS One 2013;8:e68726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim JC, Ko KI, Mattos M, Fang M, Zhang C, Feinberg D, et al. TNFα contributes to diabetes impaired angiogenesis in fracture healing. Bone 2017;99:26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging: an evolutionary perspective on immunosenescence. Ann N Y Acad Sci 2000;908:244–54. [DOI] [PubMed] [Google Scholar]
- 40.Giunta B, Fernandez F, Nikolic WV, Obregon D, Rrapo E, Town T, et al. Inflammaging as a prodrome to Alzheimer’s disease. J Neuroinflammation 2008;5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boren E, Gershwin ME. Inflamm-aging: autoimmunity, and the immune-risk phenotype. Autoimmun Rev 2004;3:401–6. [DOI] [PubMed] [Google Scholar]
- 42.Lencel P, Magne D. Inflammaging: the driving force in osteoporosis? Med Hypotheses 2011;76:317–21. [DOI] [PubMed] [Google Scholar]
- 43.•.Xia S, Zhang X, Zheng S, Khanabdali R, Kalionis B, Wu J, et al. An update on inflamm-aging: mechanisms, prevention, and treatment. J Immunol Res 2016;2016:8426874.This is an updated and an in-depth review that covers the breadth of the inflammaging field.
- 44.Nathan C, Ding A. Nonresolving inflammation. Cell 2010;140: 871–82. [DOI] [PubMed] [Google Scholar]
- 45.Gruver A, Hudson L, Sempowski G. Immunosenescence of ageing. J Pathol 2007;211:144–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steinmann GG. Changes in the human thymus during aging. Curr Top Pathol 1986;75:43–88. [DOI] [PubMed] [Google Scholar]
- 47.Compston JE. Bone marrow and bone: a functional unit. J Endocrinol 2002;173:387–94. [DOI] [PubMed] [Google Scholar]
- 48.Haynes BF, Markert ML, Sempowski GD, Patel DD, Hale LP. The role of the thymus in immune reconstitution in aging, bone marrow transplantation, and HIV-1 infection. Ann Rev Immunol 2000;18: 529–60. [DOI] [PubMed] [Google Scholar]
- 49.Xing Z, Lu C, Hu D, Yu YY, Wang X, Colnot C, et al. Multiple roles for CCR2 during fracture healing. Dis Model Mech 2010;3: 451–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Melton DW, Roberts AC, Wang H, Sarwar Z, Wetzel MD, Wells JT, et al. Absence of CCR2 results in an inflammaging environment in young mice with age-independent impairments in muscle regeneration. J Leukoc Biol 2016;100:1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xing Z, Lu C, Hu D, Miclau T 3rd, Marcucio RS. Rejuvenation of the inflammatory system stimulates fracture repair in aged mice. J Orthop Res 2010;28:1000–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.•.Baht GS, Silkstone D, Vi L, Nadesan P, Amani Y, Whetstone H, et al. Exposure to a youthful circulation rejuvenates bone repair through modulation of β-catenin. Nat Commun 2015;6:7131.This study demonstrated the significance of hematopoietic cells on fracture healing and the ability to improve healing in old mice with exposure to young hematopoietic cells.
- 53.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature 2013;25:445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferrante CJ, Leibovich SJ. Regulation of macrophage polarization and wound healing. Adv Wound Care 2012;1:10–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity 2010;32:593. [DOI] [PubMed] [Google Scholar]
- 56.Sebastian C, Herrero C, Serra M, Lloberas J, Blasco MA, Celada A. Telomere shortening and oxidative stress in aged macrophages results in impaired STAT5a phosphorylation. J Immunol 2009;183: 2356–64. [DOI] [PubMed] [Google Scholar]
- 57.Ramanathan R, Kohli A, Ingaramo MC, Jain A, Leng SX, Punjabi NM, et al. Serum chitotriosidase, a putative marker of chronically activated macrophages, increases with normal aging. J Gerontol A Biol Sci Med Sci 2013;68:1303–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.•.Duscher D, Rennert RC, Januszyk M, Anghel E, Maan ZN, Whittam AJ, et al. Aging disrupts cell subpopulation dynamics and diminishes the function of mesenchymal stem cells. Sci Rep 2014;4:7144.This paper thoroughly showed age-related disruption of MSC function specifically related to a compromise of angiogenesis in wound healing, via in vitro, in vivo, and single-cell transcriptional analysis.
- 59.Slade Shantz JA, YY Y, Andres W, Miclau T, Marcucio R. Modulation of macrophage activity during fracture repair has differential effects in young adult and elderly mice. J Orthop Trauma 2014;28:S10–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.•.Chang MK, Raggatt LJ, Alexander KA, Kuliwaba JS, Fazzalari NL, Schroder K, et al. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol 2008;181:1232–44.This study was the first to demonstrate a resident tissue macrophage population, osteomacs that are involved in bone homeostasis and regulate osteoblast function.
- 61.Alexander KA, Chang MK, Maylin ER, Kohler T, Müller R, Wu AC, et al. Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. J Bone Miner Res 2011;26:1517–32. [DOI] [PubMed] [Google Scholar]
- 62.Ono T, Takayanagi H. Osteoimmunology in bone fracture healing. Curr Osteoporos Rep 2017;15:367–75. [DOI] [PubMed] [Google Scholar]
- 63.Könnecke I, Serra A, El Khassawna T, et al. T and B cells participate in bone repair by infiltrating the fracture callus in a two-wave fashion. Bone 2014;64:155–65. [DOI] [PubMed] [Google Scholar]
- 64.Sun G, Wang Y, Ti Y, Wang J, Zhao J, Qian H. Regulatory B cell is critical in bone union process through suppressing proinflammatory cytokines and stimulating Foxp3 in Treg cells. Clin Exp Pharmacol Physiol 2017;44:455–62. [DOI] [PubMed] [Google Scholar]
- 65.Al-Sebaei MO, Daukss DM, Belkina AC, et al. Role of Fas and Treg cells in fracture healing as characterized in the Fas-deficient (lpr) mouse model of lupus. J Bone Miner Res 2014;29:1478–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nam D, Mau E, Wang Y, et al. T-lymphocytes enable osteoblast maturation via IL-17F during the early phase of fracture repair. PLoS One 2012;7:e40044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prockop DJ, Oh JY. Mesenchymal stem/stromal cells (MSCs): role as guardians of inflammation. Mol Ther 2012;20:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caplan A, Correa D. The MSC: an injury drugstore. Cell Stem Cell 2011;9:11–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caplan A, Dennis J. Mesenchymal stem cells as trophic mediators. J Cell Biochem 2006;98:1076–84. [DOI] [PubMed] [Google Scholar]
- 70.Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood 2007;110:3499–506. [DOI] [PubMed] [Google Scholar]
- 71.Kim J, Hematti P. Mesenchymal stem cell-educated macrophages: a novel type of alternatively activated macrophages. Exp Hematol 2009;37:1445–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shabbir A, Zisa D, Suzuki G, Lee T. Heart failure therapy mediated by the trophic activities of bone marrow mesenchymal stem cells: a noninvasive therapeutic regimen. Am J Physiol Heart Circ Physiol 2009;296:1888–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol 2008;8:726–32. [DOI] [PubMed] [Google Scholar]
- 74.Le Blanc K, Rasmusson I, Sundberg B, Götherström C, Hassan M, Uzunel M, et al. Treatment of severe acute graft versus-host disease with third party haploidentical mesenchymal stem cells. Lancet 2004;363:1439–41. [DOI] [PubMed] [Google Scholar]
- 75.Tögel F, Hu Z, Weiss K, Isaac J, Lange C, Westenfelder C. Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. Am J Physiol Renal Physiol 2005;289:F31–42. [DOI] [PubMed] [Google Scholar]
- 76.Colnot C, Lu C, Hu D, Helms JA. Distinguishing the contributions of the perichondrium, cartilage, and vascular endothelium to skeletal development. Dev Biol 2004;269:55–69. [DOI] [PubMed] [Google Scholar]
- 77.Lu C, Hansen E, Sapozhnikova A, Hu D, Miclau T, Marcucio RS. Effect of age on vascularization during fracture repair. J Orthop Res 2008;26:1384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jacobsen KA, et al. Bone formation during distraction osteogenesis is dependent on both VEGFR1 and VEGFR2 signaling. J Bone Miner Res 2008;23:596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prisby RD, Ramsey MW, Behnke BJ, Dominguez JM 2nd, Donato AJ, Allen MR, et al. Aging reduces skeletal blood flow, endothelium-dependent vasodilation, and NO bioavailability in rats. J Bone Miner Res 2007;22:1280–8. [DOI] [PubMed] [Google Scholar]
- 80.Prisby RD. Bone marrow blood vessel ossification and “microvascular dead space” in rat and human long bone. Bone 2014;64:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frenkel-Denkberg G, Gershon D, Levy AP. The function of hypoxia-inducible factor 1 (HIF-1) is impaired in senescent mice. FEBS Lett 1999;462:341–4. [DOI] [PubMed] [Google Scholar]
- 82.Wagatsuma A. Effect of aging on expression of angiogenesis related factors in mouse skeletal muscle. Exp Gerontol 2006;41: 49–54. [DOI] [PubMed] [Google Scholar]
- 83.Kosaki N, et al. Impaired bone fracture healing in matrix metalloproteinase-13 deficient mice. Biochem Biophys Res Commun 2007;354:846–51. [DOI] [PubMed] [Google Scholar]