

Abstract
Background
Schizophrenia is a severe mental disorder with a prevalence of about 1% among the general population. It is listed among the top 10 causes of disability‐adjusted life years (DALYs) worldwide. Antipsychotics are the mainstay treatment. Piperacetazine has been reported to be as clinically effective as chlorpromazine, a well established 'benchmark' antipsychotic, for people with schizophrenia. However, the side effect profiles of these antipsychotics differ and it is important that an evidence base is available comparing the benefits, and potential harms of these two antipsychotics.
Objectives
To assess the clinical and side effects of chlorpromazine for people with schizophrenia and schizophrenia‐like psychoses in comparison with piperacetazine.
Search methods
We searched the Cochrane Schizophrenia Group's Trials Register (6 June 2015 and 8 October 2018) which is based on regular searches of CINAHL, CENTRAL, BIOSIS, AMED, Embase, PubMed, MEDLINE, PsycINFO and registries of clinical trials. There are no language, date, document type, or publication status limitations for inclusion of records in the register.
Selection criteria
We included randomised controlled trials (RCTs) focusing on chlorpromazine versus piperacetazine for people with schizophrenia, reporting useable data.
Data collection and analysis
We extracted data independently. For binary outcomes, we calculated risk ratio (RR) and its 95% confidence interval (CI), on an intention‐to‐treat basis. For continuous data, we estimated the mean difference (MD) between groups and its 95% CI. We employed a fixed‐effect model for analyses. We assessed risk of bias for included studies and created 'Summary of findings' tables using GRADE.
Main results
We found 12 records referring to six trials. We included five trials, all from the 1970s, randomising 343 participants. We excluded one trial. The overall methodology and data reporting by the trials was poor. Only short‐term data were available.
Results from the included trials found that, in terms of global state improvement, when rated by a psychiatrist, there was no clear difference between chlorpromazine and piperacetazine (RR 0.90, 95% CI 0.80 to 1.02; participants = 208; studies = 2; very low‐quality evidence). One trial reported change scores on the mental state scale Brief Psychiatric Rating Scale (BPRS); no clear difference was observed (MD ‐0.40, 95% CI ‐1.41 to 0.61; participants = 182; studies = 1; very low‐quality evidence). Chlorpromazine appears no worse or better than piperacetazine regarding adverse effects. In both treatment groups, around 60% of participants experienced some sort of adverse effect (RR 1.00, 95% CI 0.75 to 1.33; participants = 74; studies = 3; very low‐quality evidence), with approximately 40% of these participants experiencing some parkinsonism‐type movement disorder (RR 0.95, CI 0.61 to 1.49; participants = 106; studies = 3; very low‐quality evidence). No clear difference in numbers of participants leaving the study early for any reason was observed (RR 0.50, 95% CI 0.10 to 2.56; participants = 256; studies = 4; very low‐quality evidence). No trial reported data for change in negative symptoms or economic costs.
Authors' conclusions
The results of this review show chlorpromazine and piperacetazine may have similar clinical efficacy, but data are based on very small numbers of participants and the evidence is very low quality. We can not make firm conclusions based on such data. Currently, should clinicians and people with schizophrenia need to choose between chlorpromazine and piperacetazine they should be aware there is no good quality evidence to base decisions. More high quality research is needed.
Plain language summary
Chlorpromazine versus piperacetazine for schizophrenia
Review question
Is the antipsychotic drug, chlorpromazine, better or worse than the antipsychotic drug, piperacetazine, for treating the symptoms of schizophrenia?
Background
Schizophrenia is a serious mental illness that severely disrupts a person's thought processes and affects around 1% of the general population. Schizophrenia is listed among the top 10 causes of disability‐adjusted life years (DALYs) worldwide. People with schizophrenia often have a lower life expectancy and an increased risk of suicide. There are two main types of symptoms, positive and negative. Common positive symptoms include delusions (beliefs that are not based in reality) and hallucinations (seeing or hearing things that are not real). Negative symptoms include social withdrawal and lack of motivation and poor emotional response. Positive symptoms are usually of short duration and the negative symptoms can be long term. Schizophrenia is usually treated with a combination of antipsychotic drugs and psychological therapies. Chlorpromazine and piperacetazine are antipsychotic drugs used to treat schizophrenia, however, they both can also cause unpleasant side effects. This review aims to assess evidence from randomised controlled trials regarding the effectiveness and safety of both of these drugs.
Searching for evidence
A search for randomised controlled trials that could be relevant to this review was carried out on 6 June 2015, and another search was carried out 8 October 2018. This was achieved by searching the Specialised Register of Cochrane Schizophrenia. The 2015 search found six possible trials and we carefully checked these to see if we could include them in the review. The 2018 search found no new trials.
Results
Five trials, randomising a total of 343 participants met the review requirements for inclusion. These trials randomly allocated participants to receive either chlorpromazine or piperacetazine. Data were reported for participants' global and mental state after treatment, incidence of adverse effects and numbers leaving the trial early. However, we did not find any data concerning service use, functioning of participants or economic costs of these treatments. The overall results showed chlorpromazine and piperacetazine may have similar clinical efficacy and side effect profiles. However, these results are based on very low‐quality data.
Conclusions
The number of included studies and the sample size of participants included in this review is small, and the quality of data very low, so the results of this review are not conclusive and must be used with caution. Further research would be needed before decisions can be made regarding which drug is more effective.
Summary of findings
Summary of findings for the main comparison. Chlorpromazine versus piperacetazine for schizophrenia.
| Chlorpromazine versus piperacetazine for schizophrenia | ||||||
| Patient or population: people with schizophrenia Settings: hospital Intervention: chlorpromazine versus piperacetazine for schizophrenia (short term) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control5 | Chlorpromazine versus piperacetazine for schizophrenia | |||||
| Global state: clinically important change ‐ as defined by each of the studies Follow‐up: 8 weeks | 800 per 1000 | 544 per 1000 (208 to 1000) | RR 0.90 (0.80 to 1.02) | 208 (2 studies) | ⊕⊕⊝⊝ Very low1,2 | |
| Mental state: overall mean change score (BPRS total, high = poor)* | See comments | The mean mental state change in total scores (BPRS, high = poor) ‐ short term in the intervention groups was 0.40 lower (1.41 lower to 0.61 higher) | 182 (1 study) | ⊕⊝⊝⊝ Very low1,2,4 | * No trial reported clinically important change in mental state which was our predefined outcome of interest | |
| Mental state: specific ‐ clinically important change in negative symptoms | No trial reported any data which could be used | |||||
| Adverse effects/events: incidence of adverse effects ‐ as defined by each of the studies Follow‐up: mean 10 weeks | 600 per 1000 | 642 per 1000 (504 to 828) | RR 1.00 (0.75 to 1.33) | 74 (3 studies) | ⊕⊝⊝⊝ Very low2,3 | |
| Adverse effects/events: clinically important movement disorder (Parkinsonism) | 400 per 1000 |
392 per 1000 (252 to 608) |
RR 0.95 (0.61 to 1.49) | 106 (3 studies) |
⊕⊝⊝⊝ Very low2,3 | |
| Leaving the study early ‐ for any reason Follow‐up: mean 10 weeks | 5 per 1000 | 15 per 1000 (2 to 129) | RR 0.50 (0.10 to 2.56) | 256 (4 studies) | ⊕⊝⊝⊝ Very low2,3 | |
| Economic costs | No trial reported relevant data | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1Serious imprecision: there are very few participants ‐ downgraded by 1. 2Strongly suspected publication bias: due to small sample size and insignificant results ‐ downgraded by 1. 3Very serious risk of bias: high risk of bias for random sequence generation and allocation concealment. Unclear risk of bias for blinding of outcome assessments and incomplete outcome data ‐ downgraded by 2. 4Serious indirectness: not a direct measure of prespecified outcome ‐ downgraded by 1. 5All control group rates rounded from control group within the relevant studies.
Background
Description of the condition
Schizophrenia is a severe mental disorder with a prevalence of about 1% among the general population. It is listed among the top 10 causes of disability‐adjusted life years (DALYs) worldwide (Rossler 2005). People with schizophrenia have a lower life expectancy and an increased risk of suicide (Palmer 2005; Saha 2007). The illness is characterised by acute positive symptoms, including hallucinations and delusions, and chronic negative symptoms, such as apathy, disorganised thoughts or behaviours, catatonic signs, and lack of motivation (Carpenter 1994). Schizophrenia is usually treated with antipsychotics (Kishimoto 2013).
Description of the intervention
Chlorpromazine is an antipsychotic (class: phenothiazine; subclass: aliphatic side chain; formula: 2‐chloro‐10‐(3‐dimethylaminopropyl) phenothiazine; trade names: many, including Largactil, Hibernal, Megaphen, Solidon, Thorazine; WHO Essential drug; Figure 1). It is a dopamine antagonist, introduced in the 1950s for the treatment of both acute and chronic psychoses, including schizophrenia and the manic phase of bipolar disorder, as well as amphetamine‐induced psychoses.
1.
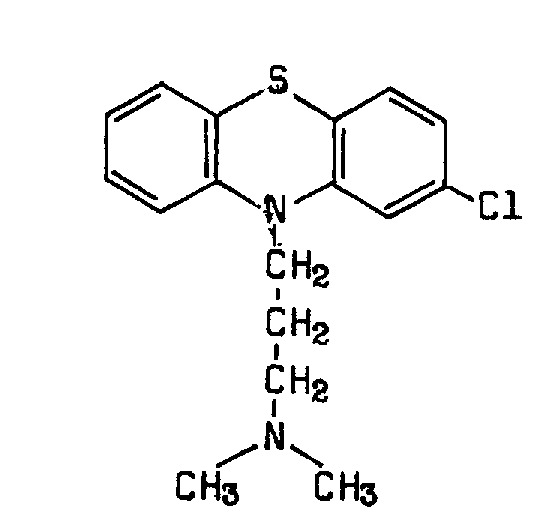
Chlorpromazine structure
Piperacetazine is also an antipsychotic (class: phenothiazine; subclass: piperidine side chain; formula: 10‐[3‐[4‐(2‐hydroxyethyl) piperidino]propyl] phenothiazin‐2‐yl methyl ketone; trade names: Actazine, Quide, Psymod, SC 10.490PC 1421; Figure 2). It is an antipsychotic drug with purportedly minor extrapyramidal symptoms, but anticholinergic effects of postural hypotension and bradycardia.
2.
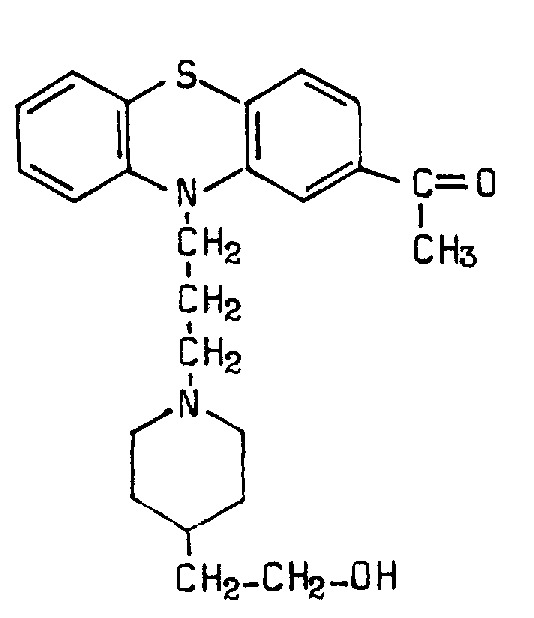
Piperacetazine structure
How the intervention might work
Chlorpromazine works on a variety of receptors in the central nervous system, producing potent anticholinergic, antidopaminergic, antihistaminic, and antiadrenergic effects. Therefore, chlorpromazine causes a variety of side effects, including sedation, constipation, hypotension and extrapyramidal symptoms (Kusumi 2015).
Piperacetazine, a phenothiazine of the piperidine class is considered to be a medium‐dosage, high‐potency antipsychotic drug. Piperacetazine has antidopaminergic , anticholinergic, antihistaminic, and antiadrenergic effects. The untoward reactions most frequently associated with the use of piperacetazine are drowsiness, dizziness, weakness, orthostatic hypotension, syncope, and extrapyramidal symptoms. Euphoria, headache, nausea, vomiting, bradycardia, and changes in libido have occurred occasionally. Reactions that occur rarely include dryness of the mouth, nasal stuffiness, galactorrhoea, amenorrhoea, leukopaenia, thrombocytopenia, urinary retention, pedal oedema, convulsions, and jaundice. All of these effects were reversible when the dose was reduced or administration of the drug was discontinued (Goldstein 1976; Anonymous 1971).
Why it is important to do this review
In an effort to provide the optimal care for people with schizophrenia, careful consideration of the risks and benefits of interventions must be incorporated into a comprehensive treatment plan. Despite the advent of newer antipsychotic drugs considered to be effective for treating the symptoms of schizophrenia with less adverse effects, chlorpromazine is still used commonly and considered a benchmark drug for the treatment of schizophrenia (Adams 2005). Therefore, it is important to compare the clinical effectiveness and side effects of chlorpromazine with other antipsychotic drugs.
Piperacetazine is a phenothiazine derivative with a piperidine side chain in the position 10 and acetyl radical in the position two. Animal studies have demonstrated that piperacetazine was markedly more potent than chlorpromazine as an antiemetic (prevents vomiting.). Several early studies with newly admitted or chronic psychotic inpatients demonstrated effectiveness of piperacetazine in reducing psychotic symptoms. Side effects reported in these studies were considered mild and were reversible following reduction in dosage or the addition of antiparkinsonian compounds (Rada 1972).
To our knowledge, there is no systematic review directly comparing the clinical effects and side effects of chlorpromazine with piperacetazine. This is one of a series of Cochrane Reviews that will build up an overall data set for the effectiveness of chlorpromazine (Table 2).
1. Other Cochrane Reviews in this group.
| Review title | Reference |
| Acetophenazine versus chlorpromazine | Developing protocol |
| Chlorpromazine dose for people with schizophrenia | Liu 2009 |
| Cessation of medication for people with schizophrenia already stable on chlorpromazine | Almerie 2007 |
| Chlorpromazine versus atypical antipsychotic drugs for schizophrenia | Saha 2013 |
| Chlorpromazine versus clotiapine for schizophrenia | Developing protocol |
| Chlorpromazine versus haloperidol* for schizophrenia | Leucht 2008 |
| Chlorpromazine versus metiapine | Developing protocol |
| Chlorpromazine versus penfluridol | Developing protocol |
| Chlorpromazine versus piperacetazine for schizophrenia | This review |
| Chlorpromazine versus placebo for schizophrenia | Adams 2014 |
| Chlorpromazine for psychosis induced aggression or agitation | Ahmed 2010 |
*Since 2015, the title of reviews has been changed to follow the alphabetical order of interventions in the title.
Objectives
To assess the clinical and side effects of chlorpromazine for people with schizophrenia and schizophrenia‐like psychoses in comparison with piperacetazine.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials (RCTs). If a trial had been described as 'double‐blind' but implied randomisation, we included such trials in a sensitivity analysis (see Sensitivity analysis). We excluded quasi‐randomised studies, such as those allocating by alternate days of the week. Where people were given additional treatments within chlorpromazine, we only included data if the adjunct treatment was evenly distributed between groups and it was only the chlorpromazine that was randomised.
Types of participants
Adults, however defined by individual studies, with schizophrenia or related disorders, including schizophreniform disorder, schizoaffective disorder and delusional disorder, again, by any means of diagnosis. If we found trials where there is a range of related disorders, we only included them if the majority of participants (over 50%) have schizophrenia. If we found trials with adolescent participants, we included them if the majority of participants (over 50%) were over 18 years.
We are interested in ensuring that information is relevant to the current care of people with schizophrenia and aimed, where possible, to clearly highlight the current clinical state (acute, early post‐acute, partial remission, remission) as well as the stage (prodromal, first episode, early illness, persistent) and whether the studies primarily focused on people with particular problems (for example, negative symptoms, treatment‐resistant illnesses).
Types of interventions
1. Chlorpromazine
Any dose or form.
2. Piperacetazine
Any dose or form.
Types of outcome measures
We divided all outcomes into short‐term (less than 6 months), medium‐term (7 to 12 months) and long‐term (over 12 months) outcomes.
We reported binary outcomes recording clear and clinically meaningful degrees of change (e.g. global impression of much improved, or more than 50% improvement on a rating scale ‐ as defined within the trials) before any others. Thereafter, we listed other binary outcomes and then those that are continuous.
Primary outcomes
1. Global state ‐ clinically important change.
2. Mental state ‐ overall
2.1 Clinically important change ‐ as defined by each of the studies
3. Adverse effects/events
3.1 Incidence of adverse effects ‐ as defined by each of the studies
Secondary outcomes
1. Global state
1.1 Any change ‐ as defined by each of the studies 1.2 Mean endpoint/change score on global state scale
2. Mental state
2.1 Overall
2.1.1 Any change ‐ as defined by each of the studies 2.2.2 Mean endpoint/change score on mental state scale
2.2 Specific
2.2.1 Clinically important change (e.g. positive, negative, affective symptoms) ‐ as defined by each of the studies 2.2.2 Any change ‐ as defined by each of the studies 2.2.3 Mean endpoint or change score on specific mental state scale
3. Adverse effects/events
3.1 General adverse effects
3.1.1 At least one adverse effect 3.1.2 Mean endpoint or change score on general adverse‐effect scale
3.2 Specific adverse effects ‐ clinically important
3.2.1 Anticholinergic. 3.2.2 Cardiovascular. 3.2.3 Central nervous system. 3.2.4 Gastrointestinal. 3.2.5 Endocrine (e.g. amenorrhoea, galactorrhoea, hyperlipidaemia, hyperglycaemia, hyperinsulinaemia). 3.2.6 Haematology (e.g. haemogram, leukopenia, agranulocytosis/neutropenia). 3.2.7 Hepatitic (e.g. abnormal transaminase, abnormal liver function). 3.2.8 Metabolic. 3.2.9 Movement disorders (including extrapyramidal) 3.2.10 Various other. 3.2.11 Mean endpoint or change score on specific adverse effect scale
3.3 Death
3.3.1 Any cause except suicide and homicide 3.3.2 Suicide 3.3.3 Homicide
4. Leaving the study early
4.1 For any reason 4.2 For specific reason
5. Service use
5.1 Hospital admission, readmission, or both 5.2 Days in hospital
6. Quality of life
6.1 Clinically important change ‐ as defined by each of the studies 7.2 Mean endpoint/change score on quality of life scale
8. Satisfaction with care for either recipients of care or carers
8.1 Clinically important change in satisfaction with care ‐ as defined by each of the studies 8.2 Mean endpoint/change score on satisfaction scale
9. Economic outcomes
'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2011); and used GRADEpro GDT to export data from our review to create a 'Summary of findings' table. The 'Summary of findings' table provides outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
Global state ‐ clinically important change ‐ as defined by each of the studies.
Mental state: overall ‐ clinically important change ‐ as defined by each of the studies.
Mental state: specific ‐ clinically important change in negative symptoms ‐ as defined by each of the studies.
Adverse effects/events ‐ general ‐ incidence of adverse effects ‐ as defined by each of the studies
Adverse effects/events ‐ clinically important movement disorder‐ as defined by each of the studies.
Leaving the study early ‐ for any reason.
Economic costs.
If data were not available for these prespecified outcomes, but were available for ones that were similar, we presented the closest outcome to the prespecified one in the table, but took this into account when grading the finding.
Search methods for identification of studies
Electronic searches
1. Cochrane Schizophrenia Group's Study‐Based Register of Trials
On 6 June 2015 and 8 October 2018, the information specialist searched the register using the following search strategy:
(*Chlorpromazine* AND *Piperacetazine*) in Intervention Field of STUDY
In such study‐based register, searching the major concept retrieves all the synonyms and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics (Shokraneh 2017; Shokraneh 2018).
This register is compiled by systematic searches of major resources (AMED, BIOSIS, CENTRAL, CINAHL, ClinicalTrials.Gov, EMBASE, MEDLINE, PsycINFO, PubMed, WHO ICTRP) and their monthly updates, ProQuest Dissertations and Theses A&I and its quarterly update, Chinese databases (CBM, CNKI, and Wanfang) and their annual updates, hand‐searches, grey literature, and conference proceedings (see Group's website). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
Searching other resources
1. Reference searching
We inspected references of all included studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials. We noted the outcome of this contact in the 'Characteristics of included studies' or 'Characteristics of studies awaiting classification' tables.
Data collection and analysis
Selection of studies
Review authors (MES and LV) independently inspected citations from the searches and identified relevant abstracts. A random 20% sample were independently re‐inspected by RD and RS to ensure reliability. Where disputes arose, the full report was acquired for more detailed scrutiny. Full reports of the abstracts meeting the review criteria were obtained and inspected by MES and LV. Again, a random 20% of reports were re‐inspected by RD and RS in order to ensure reliable selection. Where it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study for clarification and when we could not resolve the disagreement, we did not include the trial but placed it in awaiting assessment until a resolution could be made.
Data extraction and management
1. Extraction
Review authors LV and RD extracted data from all included studies. In addition, to ensure reliability, RS independently extracted data from a random sample of studies, comprising 10% of the total. Again, any disagreement was discussed, decisions documented and, if necessary, we contacted the authors of studies for clarification. With any remaining problems, MES clarified issues and these final decisions were documented. Data presented only in graphs and figures were extracted whenever possible, but included only if two review authors independently had the same result. We attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification, whenever necessary. If studies were multicentred, where possible, we extracted data relevant to each component centre separately.
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if:
the psychometric properties of the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000);
the measuring instrument had not been written or modified by one of the trialists for that particular trial; and
the instrument should have been a global assessment of an area of functioning and not subscores which are not, in themselves, validated or shown to be reliable. However, there are exceptions; we would have included subscores from mental state scales measuring positive and negative symptoms of schizophrenia.
Ideally the measuring instrument should either be i) a self‐report or ii) completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly; in Description of studies we noted if this is the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data: change data can remove a component of between‐person variability from the analysis; however, calculation of change needs two assessments (baseline and endpoint) that can be difficult to obtain in unstable and difficult‐to‐measure conditions, such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. If necessary, we combined endpoint and change data in the analysis, as we preferred to use mean differences (MDs) rather than standardised mean differences (SMDs) throughout (Deeks 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to relevant continuous data before inclusion.
For endpoint data from studies including fewer than 200 participants.
When a scale starts from the finite number zero, we subtracted the lowest possible value from the mean, and divided this by the standard deviation. If this value was lower than one, it strongly suggests that the data are skewed and we would have excluded these data. If this ratio was higher than one but less than two, there is a suggestion that the data were skewed: we would enter these data and test whether their inclusion or exclusion would change the results substantially. If such data change results, we would have entered as 'other data'. Finally, if the ratio was larger than two we would include these data, because it is less likely that they are skewed (Altman 1996; Higgins 2011a).
If a scale starts from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), which can have values from 30 to 210; Kay 1986), we would modify the calculation described above to take the scale starting point into account. In these cases skewed data are present if 2 standard deviations (SDs) > (S − S min), where S is the mean score and 'S min' is the minimum score.
We would have entered all relevant data from studies of more than 200 participants in the analysis irrespective of the above rules, because skewed data pose less of a problem in large studies. We also entered all relevant change data, as when continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether or not data are skewed.
2.5 Common measure
To facilitate comparison between trials, we would have converted variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, efforts were made to convert outcome measures to dichotomous data. This was done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score, such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962), or the PANSS (Kay 1986), this could be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicates a favourable outcome for piperacetazine. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not un‐improved'), we reported data where the left of the line indicates an unfavourable outcome. This was noted in the relevant graphs.
Assessment of risk of bias in included studies
Again, review authors MES and LV worked independently to assess risk of bias using criteria described in the Cochrane Handbook for Systemic reviews of Interventions to assess trial quality (Higgins 2011b). This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the trial, such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
If the raters disagreed, the final rating was made by consensus, with the involvement of another member of the review group (RD). Where inadequate details of randomisation and other characteristics of trials were provided, we contacted trial authors in order to obtain further information. Non‐concurrence in quality assessment was reported, but if disputes arose as to which category a trial was to be allocated, again, we resolved by discussion.
We noted the level of risk of bias in both the text of the review and in the 'Summary of findings' table.
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive than odds ratios (ORs) (Boissel 1999), and that OR tend to be interpreted as RR by clinicians (Deeks 2000). The number Needed to treat/harm (NNT/H) statistic with its CIs is intuitively attractive to clinicians, but is problematic both in its accurate calculation in meta‐analyses and interpretation (Hutton 2009). For binary data presented in the 'Summary of findings' table, where possible, we calculated illustrative comparative risks.
2. Continuous data
For continuous outcomes we estimated MD between groups. We preferred not to calculate effect size measures (SMD). However, if scales of very considerable similarity were used, we would have presumed there is a small difference in measurement, and calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice), but analysis and pooling of clustered data poses problems. Authors often fail to account for intraclass correlation in clustered studies, leading to a unit of analysis error whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated (Divine 1992). This causes type I errors (Bland 1997; Gulliford 1999).
If clustering had been incorporated into the analysis of primary studies, we would have presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
If clustering was not accounted for in primary studies, we would have presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. We would have contacted the first author of studies to obtain intraclass correlation coefficients (ICCs) for their clustered data and adjusted for this by using accepted methods (Gulliford 1999).
We have sought statistical advice and have been advised that the binary data from cluster trials presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC: thus design effect = 1 + (m − 1) * ICC (Donner 2002). If the ICC is not reported we would have assumed it to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed and taken ICCs and relevant data documented in the report into account, synthesis with other studies is possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a washout phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we would have only used data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involves more than two treatment arms, if relevant, we would have presented the additional treatment arms in comparisons. If data are binary, we would simply add these and combine within the 2x2 table. If data are continuous, we would have combined data following the formula in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). Where additional treatment arms are not relevant, we would not reproduce these data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data or use them within analyses. If, however, more than 50% of those in one arm of a study are lost, but the total loss is less than 50%, we addressed this within the 'Summary of findings' table by downgrading quality. Finally, we also downgraded quality within the 'Summary of findings' table when loss was 25% to 50% in total.
2. Binary
In the case where attrition for a binary outcome is between 0% and 50% and where these data are not clearly described, we would present data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat (ITT) analysis). Those leaving the study early are all assumed to have the same rates of negative outcome as those who completed. We would use the rate of those who stay in the study ‐ in that particular arm of the trial ‐ and apply this also to those who did not. We would have undertaken a sensitivity analysis testing how prone the primary outcomes are to change when data only from people who complete the study to that point are compared to the intention‐to‐treat analysis using the above assumptions.
3. Continuous
3.1 Attrition
In the case where attrition for a continuous outcome is between 0% and 50%, and data only from people who complete the study to that point are reported, we reproduced these.
3.2 Standard deviations
If SDs had not been not reported, we would obtain the missing values from the authors. If these are not available, where there are missing measures of variance for continuous data, but an exact standard error (SE) and CIs are available for group means, and either P value or t value are available for differences in mean, we can calculate SDs according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). When only the SE is reported, SDs are calculated by the formula SD = SE * √(n). The Cochrane Handbook for Systematic Reviews of Interventions presents detailed formulae for estimating SDs from P, t or F values, CIs, ranges or other statistics (Higgins 2011a). If these formulae do not apply, we would have calculated the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study's outcome and thus to lose information. Nevertheless, we would have examined the validity of the imputations in a sensitivity analysis that excludes imputed values.
3.3 Assumptions about participants who left the trials early or were lost to follow‐up
Various methods are available to account for participants who left the trials early or were lost to follow‐up. Some trials just present the results of study completers, others use the method of last observation carried forward (LOCF), while more recently methods such as multiple imputation or mixed‐effects models for repeated measurements (MMRM) have become more of a standard. While the latter methods seem to somewhat better than LOCF (Leon 2006), we feel that the high percentage of participants leaving the studies early and differences in the reasons for leaving the studies early between groups is often the core problem in randomised schizophrenia trials. We therefore, did not exclude studies based on the statistical approach used. However, we used the more sophisticated approaches, e.g. we would prefer MMRM or multiple‐imputation to LOCF and we would only present completer analyses if some kind of ITT data were not available at all. Moreover, we addressed this issue in the item 'incomplete outcome data' of the 'Risk of bias' tool.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We inspected all studies for clearly outlying people or situations which we had not predicted would arise. If such situations or participant groups arose, we would fully discuss.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We inspected all studies for clearly outlying methods which we had not predicted would arise. If such methodological outliers arose we would fully discuss.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 P value. I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on i) magnitude and direction of effects and ii) strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a CI for I2). We interpreted an I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic, as evidence of substantial levels of heterogeneity (Deeks 2011). When substantial levels of heterogeneity were found in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in the Cochrane Handbook for Systemic reviews of Interventions (Sterne 2011).
1. Protocol versus full study
We tried to locate protocols of included randomised trials. If the protocol was available, we compared outcomes in the protocol and in the published report . If the protocol was not available, we compared outcomes listed in the methods section of the trial report with actually reported results.
2. Funnel plot
We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We would not use funnel plots for outcomes where there are 10 or fewer studies, or where all studies are of similar size. In other cases, where funnel plots are possible, we will seek statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies, even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model; it puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose to use the fixed‐effect model for analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1 Primary outcomes
We did not anticipate any subgroup analyses.
2. Investigation of heterogeneity
We investigated where inconsistency was high. First, we investigated whether data had been entered correctly. Second, if data were correct, we visually inspected the graph and removed outlying studies to see if homogeneity was restored. For this review we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would present the data. If not, we would not pool data and we would discuss relevant issues. We know of no supporting research for this 10% cut‐off but are investigating use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity were obvious, we would have stated hypotheses regarding these for future reviews or versions of this review. We did not intend to undertake analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
For the primary outcomes, we carried out a sensitivity analysis for a trial that implied randomisation. If adding this trial with implied randomisation study to those with better description of randomisation had made a difference to the result we would not have used data from this trial in the analyses.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption/s and when we used data only from people who completed the study to that point. If there was a substantial difference, we reported results and discussed them, but continued to employ our assumption.
Where assumptions were made regarding missing SDs data (see Dealing with missing data), we would have compared the findings of the primary outcomes when we used our assumption/s and when we used data only from people who completed the study to that point. A sensitivity analysis would have been undertaken to test how prone results are to change when completer‐only data are compared to the imputed data using the above assumption. If there was a substantial difference, we would have reported results and discussed them, but continued to employ our assumption.
3. Risk of bias
We analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains of randomisation (see Assessment of risk of bias in included studies). If the exclusion of trials at high risk of bias for a domain did not substantially alter the direction of effect or the precision of the effect estimates, then we included the data from these trials in the analyses.
4. Imputed values
We would have undertaken a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster‐randomised trials.
If substantial differences were noted in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
5. Fixed‐ and random‐effects models
We synthesised data using a fixed‐effect model, however, we also synthesised data for the primary outcomes using a random‐effects model to evaluate whether this altered the significance of the result. If we found a difference we reported this.
Results
Description of studies
Please see Characteristics of included studies.
Results of the search
See also Figure 3
3.
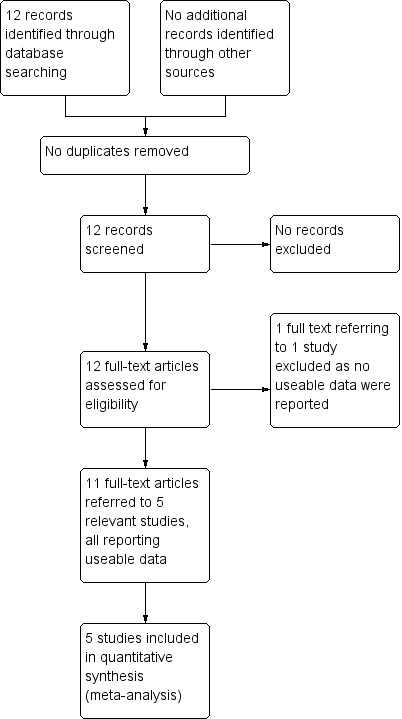
Study flow diagram for searches, up to 2018
The searches identified 12 records that referred to six studies. Five of these studies are included in the review and one is excluded.
Included studies
1. Methods
All included studies are parallel trials. Four were clearly randomised; Kurland 1970 implied randomisation.
2. Length of trial
All five were short‐term (under 6 months) trials, with a duration of eight weeks (Gallant 1970a), or 10 weeks (Gallant 1970b; Johnson 1970; Kurland 1970). Kulkarni 1972 was only 72 hours duration.
3. Participants
343 participants were randomised, with all studies stating people entering the trials were people with schizophrenia. The majority of participants were adults over 18 years. Kulkarni 1972 included a wider age range of 14‐76 years.
4. Setting
All studies were conducted in hospital.
5. Study size
The mean size of trial was 58, with a range of 16 in Gallant 1970b to 182 in Kulkarni 1972.
6. Interventions
6.1 Chlorpromazine
The doses of chlorpromazine in included studies ranged from 30 mg/day in Johnson 1970 to 1350 mg/day in Gallant 1970a. No study provided details on the mean dose of chlorpromazine.
6.2 Piperacetazine
The doses of piperacetazine in included studies ranged from 12 mg/day in Kurland 1970 to 800 mg/day in Gallant 1970b. Again, no study provided details on the mean dose of piperacetazine.
7. Outcomes
The following outcomes were reported by the studies: global state, mental state, adverse effects, and leaving the study early. None of the included studies provided evidence on quality of life, levels of satisfaction, service use or cost of care. Most outcomes reported were dichotomous. Two articles reported ordinary outcomes that could be dichotomised (Gallant 1970a; Kulkarni 1972).
The following scales provided continuous data for the analysis.
7.1 Brief Psychiatric Rating Scale (BPRS)
This scale was developed by Overall and Gorham (Overall 1962). This rating scale is globally used to measure psychiatric symptoms, such as anxiety, hallucinations, depression and unusual behaviour.
7.2 Clinical Global Impression
This rating scale enables clinicians to quantify severity of illness and overall clinical improvement. In this seven‐point scoring system, lower scores indicated decreased severity or better recovery (Busner 2007).
7.3 Target Symptom Rating Scale
This is a brief, multi‐informant measure of commonly observed symptoms in child and adolescent clinical work (Barber 2002).
Excluded studies
Small 1970 is the only excluded study. This seems to be an entirely relevant trial but we could not included it as no useable data were reported in the tiny abstract we identified. The style of this abstract is very similar to that of Johnson 1970, and Small 1970 may well be a further centre involved in one trial, but the Johnson 1970 report does contain some usable data whereas the Small 1970 report does not.
Awaiting classification
There are no studies awaiting classification.
Ongoing studies
We are unaware of any ongoing studies.
Risk of bias in included studies
Please also see Figure 4 and Figure 5.
4.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
5.
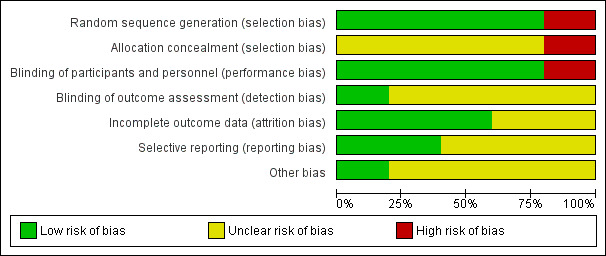
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Four studies have a low risk for selection bias, as they adequately described the methods used for randomisation, reporting that participants were randomised using random numbers. None of the four, however, described how allocation was concealed and we rated them at unclear risk for concealment bias. Kurland 1970 used sequential assignment and we rated it at high risk for both allocation and concealment selection bias.
Blinding
All included studies were double‐blind and we rated them at low risk for blinding of participants and personnel, apart from Kulkarni 1972, which we rated at high risk of bias as it stated that the rating physicians and the nurses were blinded from the identity of the assigned medication.
Incomplete outcome data
We rated Gallant 1970b, Johnson 1970 and Kulkarni 1972 at low risk of bias for incomplete outcome data because there was either no attrition, or attrition was reported. We rated the other studies at unclear risk for this item as information regarding missing values or attrition was not provided (Gallant 1970a; Kurland 1970).
Selective reporting
Only two studies reported all prestated outcomes and had a 'low' risk of reporting bias (Kulkarni 1972; Kurland 1970). The others were at unclear risk for selective reporting as no information regarding prespecified outcomes was provided.
Other potential sources of bias
Only one study reported its source of funding (Gallant 1970a). In this study, the drugs were partly supplied by a pharmaceutical company and we rated this at low risk of bias. The other studies did not report on funding and we assigned these trials an unclear risk of bias for this domain.
Effects of interventions
See: Table 1
1. Chlorpromazine versus piperacetazine (short term)
1.1 Global state: 1. Clinically important change (psychiatrist‐rated)
Two studies involving 208 participants reported clinically important change in global state. There was not a clear difference between chlorpromazine and piperacetazine. For this outcome heterogeneity is high (risk ratio (RR) 0.90, 95% confidence interval (CI) 0.80 to 1.02; participants = 208; studies = 2; I2 = 71%; very low‐quality evidence; Analysis 1.1).
1.1. Analysis.
Comparison 1 Chlorpromazine versus piperacetazine (short term), Outcome 1 Global state: 1. Clinically important change (psychiatrist‐rated).
1.2 Global state: 2a. Any change (improvement after first injection)
One study with 182 participants reported numbers improved after an initial injection of the treatment drug for 26 participants. There was not a clear difference between chlorpromazine and piperacetazine (RR 1.00, 95% CI 0.87 to 1.15; participants = 26; studies = 1; Analysis 1.2).
1.2. Analysis.
Comparison 1 Chlorpromazine versus piperacetazine (short term), Outcome 2 Global state: 2a. Any change (improvement after first injection).
1.3 Global state: 2b. Any change (improved) (CGI, high = poor)
There was not a clear difference between chlorpromazine and piperacetazine for global state improvement using the CGI (RR 0.89, 95% CI 0.80 to 1.01; participants = 182; studies = 1; Analysis 1.3)
1.3. Analysis.
Comparison 1 Chlorpromazine versus piperacetazine (short term), Outcome 3 Global state: 2b. Any change (improved) (CGI, high = poor).
1.4 Mental state: 1a. Overall: mean change score (BPRS total, high = poor)
There was no clear difference between chlorpromazine and piperacetazine for mental state when measured using BPRS total change scores (mean difference (MD) ‐0.40, 95% CI ‐1.41 to 0.61; participants = 182; studies = 1, very low‐quality evidence; Analysis 1.4).
1.4. Analysis.
Comparison 1 Chlorpromazine versus piperacetazine (short term), Outcome 4 Mental state: 1a. Overall: mean change score (BPRS total, high = poor ).
1.5 Mental state: 1b. Overall: mean change score (TSRS total, high = poor)
There was no clear difference between chlorpromazine and piperacetazine for mental state when measured using TSRS total change scores (MD 0.60, 95% CI ‐1.06 to 2.26; participants = 182; studies = 1; Analysis 1.5).
1.5. Analysis.
Comparison 1 Chlorpromazine versus piperacetazine (short term), Outcome 5 Mental state: 1b. Overall: mean change score (TSRS total, high = poor).
1.6 Adverse effects/events: 1. General: incidence of adverse effects
Incidence of adverse effects did not differ between treatment groups (RR 1.00, 95% CI 0.75 to 1.33; participants = 74; studies = 3; very low‐quality evidence; Analysis 1.6).
1.6. Analysis.
Comparison 1 Chlorpromazine versus piperacetazine (short term), Outcome 6 Adverse effects/events: 1. General: incidence of adverse effects.
1.7 Adverse effects/events: 2a. Specific: cardiovascular
1.7.1 Blood pressure, dizziness, syncope, tachycardia
Incidence of cardiac adverse effects did not differ between treatment groups. There was high heterogeneity for this outcome (RR 0.74, 95% CI 0.47 to 1.16; participants = 208; studies = 2; I2 = 80%; Analysis 1.7)
1.7. Analysis.
Comparison 1 Chlorpromazine versus piperacetazine (short term), Outcome 7 Adverse effects/events: 2a. Specific: cardiovascular.
1.8 Adverse effects/events: 2b. Specific: central nervous system
Two studies reported on various cental nervous system adverse effects (Analysis 1.8).
1.8. Analysis.
Comparison 1 Chlorpromazine versus piperacetazine (short term), Outcome 8 Adverse effects/events: 2b. Specific: central nervous system.
1.8.1 Dry mouth
Number of participants experiencing 'dry mouth' was similar between the chlorpromazine and piperacetazine groups (RR 4.89, 95% CI 0.24 to 100.51; participants = 182; studies = 1).
1.8.2 Headache
Number of participants experiencing headache was similar between the chlorpromazine and piperacetazine groups (RR 0.33, 95% CI 0.01 to 7.90; participants = 182; studies = 1).
1.8.3 Nausea/vomiting
Number of participants experiencing nausea/vomiting was similar between the chlorpromazine and piperacetazine groups (RR 1.54, 95% CI 0.42 to 5.73; participants = 208; studies = 2).
1.8.4 Sleepiness
There was a clear difference in the number of participants experiencing sleepiness in the chlorpromazine group compared to the piperacetazine group. Fewer participants experienced this adverse effect in the piperacetazine group (RR 2.87, 95% CI 1.13 to 7.31; participants = 208; studies = 2).
1.8.5 Weakness
Number of participants experiencing weakness was similar between the chlorpromazine and piperacetazine groups (RR 0.82, 95% CI 0.23 to 2.95; participants = 182; studies = 1).
1.9 Adverse effects/events: 2c. Specific: hepatitic
One study reported on hepatic adverse effects (Analysis 1.9).
1.9. Analysis.
Comparison 1 Chlorpromazine versus piperacetazine (short term), Outcome 9 Adverse effects/events: 2c. Specific: hepatitic.
1.9.1 Liver problems (changes in alkaline phosphatase and SGPT levels)
Number of participants experiencing changes in alkaline phosphatase and SGPT was similar between the chlorpromazine and piperacetazine groups (RR 1.00, 95% CI 0.16 to 6.07; participants = 26; studies = 1).
1.10 Adverse effects/events: 2d. Specific: movement disorders
Four studies reported on various movement disorders (Analysis 1.10).
1.10. Analysis.
Comparison 1 Chlorpromazine versus piperacetazine (short term), Outcome 10 Adverse effects/events: 2d. Specific: movement disorders.
1.10.1 Akathisia
The number of participants experiencing akathisia was similar between the chlorpromazine and piperacetazine groups (RR 0.59, 95% CI 0.08 to 4.36; participants = 208; studies = 2).
1.10.2 Dystonia
The number of participants experiencing dystonia was similar between the chlorpromazine and piperacetazine groups (RR 1.96, 95% CI 0.18 to 21.20; participants = 182; studies = 1).
1.10.3 Parkinsonism
The number of participants experiencing parkinsonism effects was similar between the chlorpromazine and piperacetazine groups (RR 0.95, 95% CI 0.61 to 1.49; participants = 106; studies = 3; very low‐quality evidence).
1.10.4 Rigidity
The number of participants experiencing rigidity was similar between the chlorpromazine and piperacetazine groups (RR 1.01, 95% CI 0.15 to 7.00; participants = 208; studies = 2).
1.10.5 Tremor
The number of participants experiencing tremor was similar between the chlorpromazine and piperacetazine groups (RR 1.02, 95% CI 0.15 to 7.10; participants = 182; studies = 1).
1.11 Leaving the study early
Three trials reported participants leaving the study early (Analysis 1.11).
1.11. Analysis.
Comparison 1 Chlorpromazine versus piperacetazine (short term), Outcome 11 Leaving the study early.
1.11.1 For any reason
There was no clear difference in number of participants leaving the study early for any reason (RR 0.50, 95% CI 0.10 to 2.56; participants = 256; studies = 4; very low‐quality evidence).
1.12 Due to adverse effects
There was no clear difference in number of participants leaving the study early because of adverse effects (RR 0.98, 95% CI 0.06 to 15.40; participants = 182; studies = 1).
Missing outcomes
No data were reported for service use, economic outcomes or quality of life.
Discussion
Summary of main results
The summary below indicates the outcomes selected for Table 1 and highlights the other findings of this review for evidence‐based decision making.
1. Global state: clinically important change
Very low‐quality evidence from two studies suggested no real difference between chlorpromazine and piperacetazine for this global effect. The studies are heterogenous for this outcome (71%) but we could not found any source for it. This would fit logically since these drugs are both from the phenothiazines family.
2. Mental state
2.1 Overall and specific: Clinically important change/changes in the mean total scores
Our findings indicated no clear difference between chlorpromazine and piperacetazine according to Brief Psychiatric Rating Scale (BPRS) scores. We, however, prestated that a binary outcome was desirable. The studies did not report such an outcome. This could be a function of trial design. Fine‐grain measures are often desired over more clinically interpretable outcomes. In this case continuous data seem to indicate that there is no difference between groups. There should be clear binary outcomes but, if not available we have to settle for continuous findings as second best. As for the global state outcome, mental state findings do not indicate a difference between the two drugs.
2.2 Missing data for negative symptoms
We did not identify relevant data for negative symptoms. All trials are old. The more focused approach to categorisation of symptoms into positive and negative groups is predated by all studies. Sometimes old data are so well reported that we use them in a way as to present effects on symptoms that might now be categorised as 'negative' ‐ but this was not the case for these trials.
3. Adverse effects
3.1 General
The total number of people experiencing some sort of adverse effect is about 66% in both groups ‐ with no difference between the drugs. When it comes to specific effects, piperacetazine produced higher rates of sleepiness in people with schizophrenia than chlorpromazine.
3.2 Specific: movement disorders
We did identify data for 'parkinsonism'; there was no difference between the two groups in data derived from three studies. Although these data are heterogenous for this outcome (80%) without specific sources and very low quality for the reasons outlined in Table 1 they are, nevertheless, the best available. There is no difference between the more unfamiliar piperacetazine and chlorpromazine but around 40% of both groups experienced this disfiguring and disabling adverse effect. The doses of medication in these old studies was, perhaps, for some of the trials, high and modern treatment would probably avoid such doses ‐ especially those in Gallant 1970a and Gallant 1970b ‐ but parkinsonism is an important and off‐putting adverse effect.
4. Leaving the study early
Four studies reported data with no difference between groups (Analysis 1.11). We had to rate these data as being of very low‐quality but, again, they are the best we are likely to find. However, just over 1% left each group. These are very low rates indeed. It could be a testimony to good trial design, a compliant participant group or limited opportunities to leave studies. We are not sure which, if any is important. We do not really believe this reflects the situation that would happen in the 'real world'.
5. Economic costs
It is not really surprising that no data are available and we may have been overly hopeful to stipulate it as one of our 'Summary of findings' outcomes ‐ nevertheless it remains important. Proxy outcomes such as relapse and time in hospital can be used in place of direct cost data ‐ but we also lack these data for this particular comparison.
Overall completeness and applicability of evidence
1. Completeness
Of the five studies included, only two reported data for the primary outcomes of global state and mental state. Studies reported other useful effects but evidence on all outcomes is incomplete. There were no data available for behaviour, service use, relapse, level of satisfaction and cost of care.
2. Applicability
All included studies were from the 1970s, which could lead to issues of applicability. No study reported their diagnostic criteria and the definition of schizophrenia has evolved during this period. It is difficult to know how this effects applicability. The people within the trials were recognisable by descriptions given within the trials and may not be that different to people with the illness today.
Doses of drugs were also reasonably recognisable by today's standards ‐ although the two Gallant studies did have high maximums (Gallant 1970a; Gallant 1970b).
Although data from these trials is nearly half a century old ‐ we do not feel they are entirely inapplicable to today's practice.
Quality of the evidence
The quality of evidence, based on GRADE is very low. Four studies reported the methods of random sequence generation but only one of these reported methods of allocation concealment. One study implied randomisation and was at high risk of bias for random sequence generation. Two studies had unclear risk of bias for blinding assessments and incomplete outcome data. Only one study stated that physicians and rating nurses were blinded to the treatments. For the one study that reported its source of funding, we rated it at low risk of bias overall (Gallant 1970a). The other studies did not report on funding at all and, taking others issues into account, we had to assign these trials as being of 'unclear' risk of bias. Studies often reported no usable data on important outcomes as they solely reported statistical measures of probability (P value) or means without any standard deviation or standard error. We also detected imprecision and publication bias for three included studies, leading to very low‐quality in Table 1 table.
We realise we are judging trials of the far past by standards of today. The trials in this review are imperfect but important and pioneering. They partially tell a story of evaluation of a now old and largely unused drug. At least these trials took place. Many treatments practiced in modern mental health care have no trials at all to support their use.
Potential biases in the review process
For this review, the search was based on Cochrane Schizophrenia's Trials Register. We are unaware of any other unpublished trials related to this review. We had limited reports of a few small trials. Publication bias and reporting bias could well be an issue in what we have done. We have no reason to skew results in favour of either compound. We have no reason to believe that searches for this review are any more prone to biases than other reviews.
Agreements and disagreements with other studies or reviews
We are not aware of any other systematic reviews on the efficacy of chlorpromazine versus piperacetazine for schizophrenia.
Authors' conclusions
Implications for practice.
1. For people with schizophrenia
Given a choice between taking chlorpromazine and piperacetazine, there is much more information and evidence concerning the effects of chlorpromazine than for piperacetazine (Table 2). People do have idiosyncratic responses to one drug in favour of another. Evidence for a clear average difference between chlorpromazine and piperacetazine is lacking but differences from one person to the next can happen. What limited trial‐based evidence we have suggests piperacetazine is an antipsychotic drug that may have similar effects to chlorpromazine.
2. For clinicians
There is no evidence to use piperacetazine before chlorpromazine. All data are poor quality and based on very small, short studies. Should, however, chlorpromazine be impossible to use, and a very similar drug indicated, then piperacetazine could be a viable choice.
3. For policy makers
There is no obvious reason from the data we have identified that piperacetazine should not be an available antipsychotic drug.
Implications for research.
1. General
We think it important to ensure that all reporting meets the highest CONSORT standards and that all data are available for future researchers (AllTrials). Asking this, retrospectively, of trialists of the 1970s could be seen as overly‐optimistic, but there are examples of studies well before this time which have reported in ways that have fully anticipated standards of the future. Certainly, any future trials relevant to the question focused upon in this review should comply with the highest conduct and reporting standards. The one excluded study asked consent of people to be included but then reported data in such a way as to make them unusable (Small 1970). This is wasteful and unethical.
2. Specific
2.1 Reviews
Sometimes, excluded trials suggest studies that can be included in other existing reviews or generate related questions for new reviews. The search for this review was so specific that we did not identify any trials beyond the direct scope of this review.
2.2 Trials
In terms of missing data about the particular question of the comparative effects of these two old drugs, it is not difficult to justify the idea of undertaking another definitive study. If clinicians and patients are using and choosing between the two drugs then there could be an opportunity for good quality evaluation. We realise that such design needs great attention to detail and that there are many calls on research funding and energies ‐ but we have given this question some thought and provide an outline for such a study in Table 3. Certainly, studies with much rigorous methodologies, larger sample sizes and with better outcome reporting are needed to explore the role of chlorpromazine and piperacetazine in different patient subgroups.
2. Design of a future study.
| Methods | Allocation: randomised (clearly described). Blinding: single‐blind (outcomes assessor). Duration: up to 1 year. Design: parallel. |
| Participants | Diagnosis: anyone with schizophrenia for whom there is a dilemma of which drugs to use. N = 300. Age: > 18 years. Sex: all. Exclusion criteria: specific contraindication to evaluated treatments. |
| Interventions | 1. Chlorpromazine: dose of choice. N = 150. 2. Piperacetazine: dose of choice. N = 150. |
| Outcomes | Global state ‐ clinically important change in global state as defined by each study. Mental state ‐ clinically important change in mental state as defined by each study. Mental state ‐ average change in negative symptoms. Adverse events/effects ‐ incidence of serious adverse events/effects. Adverse events/effects ‐ clinically significant extrapyramidal symptoms. Leaving the study early ‐ for any reason. Costs: cost of services, cost of care. Service outcomes: days in hospital, discharged. |
Acknowledgements
The Cochrane Schizophrenia Group Editorial Base in Nottingham produces and maintains standard text for use in the Methods section of their reviews. We have used this text as the basis of what appears here and adapted it as required. We would like to thank Professor Clive Adams (Co‐ordinating editor of the Cochrane Schizophrenia Group, University of Nottingham) and Farhad Shokraneh (Information Specialist, Cochrane Schizophrenia Group) for providing advice, guidance and support throughout the undertaking of this review. We would also like to thank Claire Irving for providing additional support.
Dr Sunitha Muniyappl Hanumanthappa Gemma Andrew kindly peer reviewed this protocol.
Parts of this review were generated using RevMan HAL.
Data and analyses
Comparison 1. Chlorpromazine versus piperacetazine (short term).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. Clinically important change (psychiatrist‐rated) | 2 | 208 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.80, 1.02] |
| 1.1 Short term | 2 | 208 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.80, 1.02] |
| 2 Global state: 2a. Any change (improvement after first injection) | 1 | 26 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.87, 1.15] |
| 3 Global state: 2b. Any change (improved) (CGI, high = poor) | 1 | 182 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.80, 1.01] |
| 4 Mental state: 1a. Overall: mean change score (BPRS total, high = poor ) | 1 | 182 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐1.41, 0.61] |
| 5 Mental state: 1b. Overall: mean change score (TSRS total, high = poor) | 1 | 182 | Mean Difference (IV, Fixed, 95% CI) | 0.60 [‐1.06, 2.26] |
| 6 Adverse effects/events: 1. General: incidence of adverse effects | 3 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.75, 1.33] |
| 7 Adverse effects/events: 2a. Specific: cardiovascular | 2 | 208 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.47, 1.16] |
| 7.1 Blood pressure, dizziness, syncope, tachycardia | 2 | 208 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.47, 1.16] |
| 8 Adverse effects/events: 2b. Specific: central nervous system | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Dry mouth | 1 | 182 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.89 [0.24, 100.51] |
| 8.2 Headache | 1 | 182 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.90] |
| 8.3 Nausea/vomiting | 2 | 208 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.54 [0.42, 5.73] |
| 8.4 Sleepiness | 2 | 208 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.87 [1.13, 7.31] |
| 8.5 Weakness | 1 | 182 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.23, 2.95] |
| 9 Adverse effects/events: 2c. Specific: hepatitic | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 Liver problems (changes in alkaline phosphatase and SGPT levels) | 1 | 26 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.16, 6.07] |
| 10 Adverse effects/events: 2d. Specific: movement disorders | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 Akathisia | 2 | 208 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.08, 4.36] |
| 10.2 Dystonia | 1 | 182 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.96 [0.18, 21.20] |
| 10.3 Parkinsonism | 3 | 106 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.61, 1.49] |
| 10.4 Rigidity | 2 | 208 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.15, 7.00] |
| 10.5 Tremor | 1 | 182 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.15, 7.10] |
| 11 Leaving the study early | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 For any reason | 4 | 256 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.50 [0.10, 2.56] |
| 11.2 Due to adverse effects | 1 | 182 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.06, 15.40] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Gallant 1970a.
| Methods | Allocation: random allocation (by random numbers). Blindness: double‐blind. Duration: 8 weeks (2 weeks of baseline evaluation + 6 weeks of medication + 2 weeks of drying out period). Setting: hospital. |
|
| Participants | Diagnosis: severe chronic schizophrenia. N = 26. Age: mean ˜ 44.7 years. Sex: male. History: free of significant disabilities of the cardiovascular, renal, and hepatic systems. Prior psychotherapeutic drug agents were discontinued for a minimum period of 4 weeks prior to initial doses of medication. |
|
| Interventions | 1. Chlorpromazine (liquid): initial dose of 120 mg/day, maximum dose: 1350 mg/day (N = 13). 2. Piperacetazine (liquid): initial dose 20 mg/day, maximum dose: 360 mg/day (N = 13). |
|
| Outcomes | Global state: clinically important change (psychiatrist‐rated). Adverse effects: specific effects. Unable to use: Mental state: BPRS, NOSIE score (no mean or SD). Global state: CGI score (no mean or SD). Behaviour: not outcome of interest in protocol, plus no mean or SD. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Subjects were selected and/or assigned to treatment by random numbers" |
| Allocation concealment (selection bias) | Unclear risk | Further information regarding allocation concealment was not provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "in a controlled, double blind study" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information was provided regarding blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information was provided for missing values or attrition. |
| Selective reporting (reporting bias) | Unclear risk | No information was provided for prespecified outcomes. |
| Other bias | Low risk | The study was partially supported by a research grant. The drugs were supplied by a pharmaceutical company. |
Gallant 1970b.
| Methods | Allocation: randomised (by random numbers). Blindness: double blind. Duration: 10 weeks (6 weeks treatment with a 4‐week drying out period). Setting: hospital. |
|
| Participants | Diagnosis: chronic schizophrenia. N = 16. Age: 27‐59 years. Sex: female. History: not reported. |
|
| Interventions | 1. Chlorpromazine (tablet): minimum dose: 240 mg/day, maximum dose: 1170 mg/day (N = 8). 2. Piperacetazine (tablet): minimum dose: 40 mg/day, maximum dose: 800 mg/day (N = 8). |
|
| Outcomes | Adverse effects: general‐total number of events, specific effects. Leaving the study early. Unable to use: Global state: BPRS (no mean or SD). Mental state: CGI, NOSIE (no mean or SD). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "subjects were selected and/or assigned to treatment by random numbers". |
| Allocation concealment (selection bias) | Unclear risk | Further information regarding allocation concealment was not provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "this was a double bind, multidrug study" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided regarding blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no losses to follow‐up. |
| Selective reporting (reporting bias) | Unclear risk | No information provided |
| Other bias | Unclear risk | Source of funding not reported |
Johnson 1970.
| Methods | Allocation: randomised. Blindness: double blind. Duration: 10 weeks (6 weeks of treatment + 4 weeks of drying out period). Setting: hospital. |
|
| Participants | Diagnosis: acute and chronic schizophrenia. N = 26. Age: 26‐59 years. Sex: male. History: not reported. |
|
| Interventions | 1. Chlorpromazine (liquid form): minimum dose of 30 mg/day, maximum dose of 96 mg/day (N = 13). 2. Piperacetazine (liquid form): minimum dose of 15 mg/day, maximum dose of 60 mg/day (N = 13). |
|
| Outcomes | Adverse effects: general‐total number of events Leaving the study early. Unable to use: Global state: BPRS (no mean or SD). Mental state: CGI, NOSIE (no mean or SD). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Subjects were selected and/or assigned to treatment by random numbers". |
| Allocation concealment (selection bias) | Unclear risk | Further information regarding allocation concealment was not provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "This was a double blind, multidrug study" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information was provided regarding blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No attrition from trial |
| Selective reporting (reporting bias) | Unclear risk | Unclear if all data were reported as no information available regarding prespecified outcomes |
| Other bias | Unclear risk | Source of funding not reported |
Kulkarni 1972.
| Methods | Allocation: randomised. Blindness: double‐blind (but it was not possible to disguise the drugs). Duration: 72 hours. Setting: hospital. |
|
| Participants | Diagnosis: schizophrenia. N = 182. Age:14‐76 years. Sex: male and female. History: uncooperative, aggressive, agitated, hyperactive, and unmanageable behaviour. Excluded: condition is a result of acute alcoholism or drug addiction were excluded. Evidence or a history of kidney or liver dysfunctions. |
|
| Interventions | 1. Chlorpromazine (liquid form): initial dose of 25 mg/ml (N = 92). 2. Piperacetazine (liquid form): initial dose of 2 mg/ml (N = 90). |
|
| Outcomes | Global state: clinically important change (psychiatrist‐rated), improved (CGI). Global state: improvement due to the first injection (this outcome was reported for a sample of 26 participants). Mental state: improved (BPRS, TSRS). Leaving the study early due to adverse effects. Adverse effects: specific. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “using a randomization scheme” |
| Allocation concealment (selection bias) | Unclear risk | Further information regarding allocation concealment not provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: “all other measures were taken to maintain a double‐blind status. The rating physicians and the nurses were blinded from the identity of the assigned medication, but the patients were not blind because the drugs were not disguisable.” |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “all other measures were taken to maintain a double‐blind status. The rating physicians and the nurses were blinded from the identity of the assigned medication.” |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data and attrition addressed and reported |
| Selective reporting (reporting bias) | Low risk | All prestated outcomes are reported. |
| Other bias | Unclear risk | Source of funding not reported |
Kurland 1970.
| Methods | Allocation: sequential assignment. Blindness: double‐blind. Duration: 10 weeks (6 weeks of treatment + 4 weeks of drying out period). Setting: hospital |
|
| Participants | Diagnosis: acute and chronic schizophrenia. N = 64. Age:19‐52 years. Sex: male and female History: not reported. |
|
| Interventions | 1. Chlorpromazine (liquid form): minimum dose of 75 mg/day, maximum dose of 108 mg/day (N = 32). 2. Piperacetazine (liquid form): minimum dose of 12 mg/day, maximum dose of 160 mg/day (N = 32). |
|
| Outcomes | Adverse effects: general‐total number of events, specific effects. Unable to use Global/mental state‐ CGI, NOSIE, BPRS (no mean or SD). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Subjects were selected and/or assigned to treatment by sequential assignment" |
| Allocation concealment (selection bias) | High risk | Unclear if allocation was randomised and no further information regarding allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "This was a double blind, multidrug study" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided regarding blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information provided for missing values and attrition |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Unclear risk | Source of funding not reported |
BPRS ‐ Brief Psychiatric Rating Scale CGI ‐ Clinical Global Impression NOSIE ‐ Nurses' Observation of Scale for Inpatient Evaluation SD ‐ standard deviation TSRS ‐ Target Symptom Rating Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Small 1970 | Allocation: unknown. Participants: adults with acute and chronic schizophrenia, N = 29. Interventions: chlorpromazine (liquid form; minimum dose of 60 mg/day, maximum dose of 1200 mg/day) versus piperacetazine (liquid form; minimum dose of 20 mg/day, maximum dose of 160 mg/day). Outcomes: all data impossible to use ‐ Mental state: CGI, NOSIE (no mean or SD). Global state – BPRS (no mean or SD). Adverse effects (total numbers in each group not given). Leaving the study early (total numbers in each group not given). |
BPRS ‐ Brief Psychiatric Rating Scale CGI ‐ Clinical Global Impression NOSIE ‐ Nurses' observation of Scale for Inpatient Evaluation SD ‐ Standard Deviation
Differences between protocol and review
In our protocol we stated we would include adults with schizophrenia; we did not clarify the age specifically as we did not want to exclude trials that included a few participants under 18 years but where the majority of participants were aged 18 years or above. We have amended our inclusion criteria to clarify this.
We have updated some sections of the methods to reflect the latest changes to Cochrane Schizophrenia's methods template. These changes either expand and clarify previous methods or are improvements in the wording of the text. They are no major changes to the methods of the published protocol.
We have renamed and reordered outcomes to standardise them with Cochrane Schizophrenia's template. For example, we have changed significant improvement to important change, average change/score to mean endpoint/change score.We also decided that global state would be a more relevant primary outcome for this review than service use and added an adverse effect to the primary outcomes as required by MECIR. .
Contributions of authors
Mahin Eslami Shahrbabaki: defined the objective, identified primary and secondary outcomes, identified 'Summary of findings' outcomes, discussed allocation of responsibility for the tasks: selection of studies, data extraction, assessment of risk of bias in included studies, wrote the sensitivity analysis section, edited Cochrane Schizophrenia Group's template for methods section.
Rahim Sharafkhani: defined the objective, identified primary and secondary outcomes, identified 'Summary of findings' outcomes, discussed allocation of responsibility for the tasks: selection of studies, data extraction, assessment of risk of bias in included studies, edited Cochrane Schizophrenia Group's template for methods section.
Reza Dehnavih: defined the objective, identified primary and secondary outcomes, identified 'Summary of findings' outcomes, discussed allocation of responsibility for the tasks: selection of studies, data extraction, assessment of risk of bias in included studies, edited Cochrane Schizophrenia Group's template for methods section.
Leila Vali: defined the objective, wrote the background section, identified primary and secondary outcomes, identified 'Summary of findings' outcomes, discussed allocation of responsibility for the tasks: selection of studies, data extraction, assessment of risk of bias in included studies, wrote sensitivity analysis section, edited Cochrane Schizophrenia Group's template for methods section.
Sources of support
Internal sources
-
Kerman University of Medical Sciences, Kerman, Iran.
Employs review authors
External sources
No sources of support supplied
Declarations of interest
Mahin Eslami Shahrbabaki: none known Rahim Sharafkhani: none known Reza Dehnavieh: none known Leila Vali: none known
New
References
References to studies included in this review
Gallant 1970a {published data only}
- Gallant DM, Bishop MP. Piperacetazine (quide): a controlled evaluation of the elixir in chronic schizophrenic patients. Current Therapeutic Research, Clinical and Experimental 1970;12(6):387‐9. [PubMed] [Google Scholar]
- Gallant DM, Bishop MP. Piperacetazine versus chlorpromazine (liquid). Psychopharmacology Bulletin 1970;7(1):38‐40. [Google Scholar]
Gallant 1970b {published data only}
- Gallant DM, Bishop MP. Piperacetazine versus chlorpromazine (tablet). Psychopharmacology Bulletin 1970;6(4):101‐3. [Google Scholar]
Johnson 1970 {published data only}
- Johnson AC. Piperacetazine (liquid) versus chlorpromazine. Psychopharmacology Bulletin 1970;7(1):55‐7. [Google Scholar]
Kulkarni 1972 {published data only}
- Johnson AC, Kulkarni AS. Piperacetazine and chlorpromazine: a comparison. American Journal of Psychiatry 1973;130(5):603‐5. [DOI] [PubMed] [Google Scholar]
- Kiev A, Guclu B, Kulkarni AS. Evaluation of piperacetazine (quide) injection in acute schizophrenics. Current Therapeutic Research, Clinical and Experimental 1972;14(7):376‐80. [PubMed] [Google Scholar]
- Kulkarni AS. Clinical effectiveness of piperacetazine injection in schizophrenic patients: a controlled study. Advances in Biochemical Psychopharmacology 1974;9:691‐9. [PubMed] [Google Scholar]
- McLaughlin B, Kulkarni AS. Comparative evaluation of injectable chlorpromazine and piperacetazine. Psychosomatics 1973;14:220‐1. [DOI] [PubMed] [Google Scholar]
- Simeon J, Wadud A, Itil T. A comparison of phenothiazines in managing aggressive episodes in schizophrenic patients. Hospital and Community Psychiatry 1975;26(9):574. [DOI] [PubMed] [Google Scholar]
Kurland 1970 {published data only}
- Kurland O. Piperacetazine versus Chlorpromazine‐revision of summary reported on 2/13/70 with additional subjects. Psychopharmacology Bulletin 1970;7(1):57‐60. [Google Scholar]
- Kurland O. Piperacetazine versus chlorpromazine. Psychopharmacology Bulletin 1970;6(4):107‐9. [Google Scholar]
References to studies excluded from this review
Small 1970 {published data only}
- Small S. Piperacetazine (liquid) versus chlorpromazine. Psychopharmacology Bulletin 1970;7(1):52‐4. [Google Scholar]
Additional references
Adams 2005
- Adams CE, Rathbone J, Thornley B, Clarke M, Borrill J, Wahlbeck K, et al. Chlorpromazine for schizophrenia: a Cochrane systematic review of 50 years of randomised controlled trials. BMC Medicine 2005;3:15. [PUBMED: 16229742] [DOI] [PMC free article] [PubMed] [Google Scholar]
Adams 2014
- Adams CE, Awad GA, Rathbone J, Thornley B, Soares‐Weiser K. Chlorpromazine versus placebo for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD000284.pub3; CD000284] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ahmed 2010
- Ahmed U, Jones H, Adams CE. Chlorpromazine for psychosis induced aggression or agitation. Cochrane Database of Systematic Reviews 2010, Issue 4. [DOI: 10.1002/14651858.CD007445.pub2; CD007445] [DOI] [PubMed] [Google Scholar]
Almerie 2007
- Almerie MQ, Alkhateeb H, Essali A, Matar HE, Rezk E. Cessation of medication for people with schizophrenia already stable on chlorpromazine. Cochrane Database of Systematic Reviews 2007, Issue 1. [DOI: 10.1002/14651858.CD006329; CD006329] [DOI] [PMC free article] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Anonymous 1971
- Anonymous. Evaluation of a new antipsychotic agent. Piperacetazine (quide). JAMA 1971;215(5):783‐4. [DOI] [PubMed] [Google Scholar]
Barber 2002
- Barber CC, Neese DT, Coyne L, Fultz J, Fonagy P. The target symptom rating: A brief clinical measure of acute psychiatric symptoms in children and adolescents. Journal of Clinical Child and Adolescent Psychology 2002 May 1;31(2):181‐92. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Busner 2007
- Busner J, Targum SD. The clinical global impressions scale: applying a research tool in clinical practice. Psychiatry (Edgmont) 2007 Jul;4(7):28. [PMC free article] [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WTJ, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;330(10):681‐90. [PUBMED: 8107719] [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Eight International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Deeks 2011
- Deeks JJ, Higgins JP, Altman DG, editor(s). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JP, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JP, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Goldstein 1976
- Goldstein SE, Birnbom F. Piperacetazine versus thioridazine in the treatment of organic brain disease: a controlled double‐blind study. Journal of the American Geriatrics Society 1976;24(8):355‐8. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JP, Deeks JJ, editor(s). Chapter 7: Selecting studies and collecting data. In: Higgins JP, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JP, Altman DG, Sterne JA, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British Journal of Haematology 2009;146(1):27‐30. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kishimoto 2013
- Kishimoto T, Agarwal V, Kishi T, Leucht S, Kane JM, Correll CU. Relapse prevention in schizophrenia: a systematic review and meta‐analysis of second‐generation antipsychotics versus first‐generation antipsychotics. Molecular Psychiatry 2013;18(1):53‐66. [PUBMED: 22124274] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kusumi 2015
- Kusumi I, Boku S, Takahashi Y. Psychopharmacology of atypical antipsychotic drugs: From the receptor binding profile to neuroprotection and neurogenesis. Psychiatry and Clinical Neurosciences 2015;69(5):243‐58. [PUBMED: 25296946] [DOI] [PubMed] [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59(11):1001‐5. [PUBMED: 16905632] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2008
- Leucht C, Kitzmantel M, Chua WL, Kane J, Leucht S. Haloperidol versus chlorpromazine for schizophrenia. Cochrane Database of Systematic Reviews 2008, Issue 1. [DOI: 10.1002/14651858.CD004278.pub2; CD004278] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2009
- Liu X, Han S. Chlorpromazine dose for people with schizophrenia. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD007778; CD007778] [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Palmer 2005
- Palmer BA, Pankratz VS, Bostwick JM. The lifetime risk of suicide in schizophrenia: a re‐examination. Archives of General Psychiatry 2005;62:247‐53. [DOI] [PubMed] [Google Scholar]
Rada 1972
- Rada RT, Donlon PT. Piperacetazine vs. thioridazine for the control of schizophrenia in outpatients. Psychosomatics 1972;13(6):373‐6. [PUBMED: 4153002] [DOI] [PubMed] [Google Scholar]
Rossler 2005
- Rossler W, Salize HJ, Os J, Riecher‐Rossler A. Size of burden of schizophrenia and psychotic disorders. European neuropsychopharmacology 2005;15(4):399‐409. [PUBMED: 15925493] [DOI] [PubMed] [Google Scholar]
Saha 2007
- Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: Is the differential mortality gap worsening over time?. Archives of General Psychiatry 2007;64:1123‐31. [DOI] [PubMed] [Google Scholar]
Saha 2013
- Saha KB, Sampson S, Zaman RU. Chlorpromazine versus atypical antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2013, Issue 7. [DOI: 10.1002/14651858.CD010631; CD010631] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JP, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In Higgins JP, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Shokraneh 2017
- Shokraneh F, Adams CE. Study‐based registers of randomized controlled trials: Starting a systematic review with data extraction or meta‐analysis. BioImpacts 2017;7(4):209‐17. [DOI: 10.15171/bi.2017.25] [DOI] [PMC free article] [PubMed] [Google Scholar]
Shokraneh 2018
- Shokraneh F, Adams CE. Gallstone, snake venom and witchcraft for schizophrenia: the challenges of classifying [schizophrenia] trials. Evidence‐Based Medicine 2018;23(Suppl. 1):A18. [DOI: 10.1136/bmjebm-2018-111024.36] [DOI] [Google Scholar]
Sterne 2011
- Sterne JA, Egger M, Moher D, editor(s). Chapter 10: Addressing reporting biases. In: Higgins JP, Green S editor(s). Cochrane Handbook for Systematic Reviews of Intervention Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JA, Burney PG. Methods for evaluating area‐wide and organisation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
References to other published versions of this review
Eslami Shahrbabaki 2015
- Eslami Shahrbabaki M, Sharafkhani R, Dehnavieh R, Vali L. Chlorpromazine versus piperacetazine for schizophrenia. Cochrane Database of Systematic Reviews 2015, Issue 6. [DOI: 10.1002/14651858.CD011709] [DOI] [PMC free article] [PubMed] [Google Scholar]