

Abstract
Amyloid β is an inherently disordered peptide that can form diverse neurotoxic aggregates, and its 42 amino acid long isoform is the believed culprit of Alzheimer’s Disease. Cellular uptake of the peptide is a pivotal step for it to be able to exert many of its toxic actions. The cellular uptake process is complex, and numerous competing internalization pathways have been proposed. To date, it remains unclear, which of the uptake mechanisms are particularly important for the overall process, and improving this understanding is needed, so that better molecular therapeutics of Alzheimer’s Disease can be designed. Chirality can be used as a unique tool to study this process, because some of the proposed mechanisms are expected to proceed in stereoselective fashion, whilst others are not. To shed light on this important issue, we synthesized fluorescently labelled enantiomers of Amyloid β and quantified their cellular uptake, finding that uptake occurs in stereoselective fashion, with a typical preference for the L-stereoisomer of ~5:1. This suggests that the process is predominantly receptor-mediated, with likely minor contribution of non-stereoselective mechanisms.
Keywords: Mirror-image peptides, Cellular uptake mechanism, Amyloid β, Stereoselective biomolecular processes
Graphical Abstract
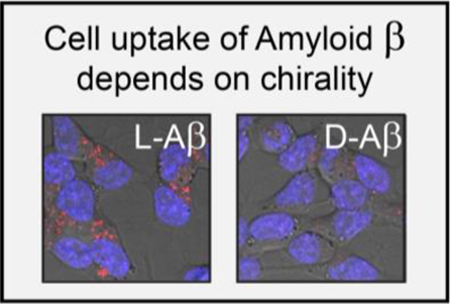
We made fluorescently tagged Aβ in its natural and mirror-image form and used the chiral reagents as tools to study cellular internalization. A significant preference for the uptake of the L-stereoisomer was observed, which we interpret as evidence for receptor intermediacy. D-Aβ also being internalized, albeit less efficiently, indicates that other, non-stereoselective mechanisms, such as membrane poration and macropincytosis, likely contribute to the process to a degree.
Amyloid β (Aβ) is an aggregation-prone, inherently disordered peptide that is produced by neurons through two sequential cleavage events from its trans-membrane protein precursor, APP, and can vary in length. The 40 amino acid variant (Aβ40) is produced predominantly, but the 42 amino acid isoform (Aβ42) is substantially more aggregation-prone and neurotoxic, and is believed to be responsible for some of the key actions that lead to synaptic dysfunction and neuronal death that is observed in Alzheimer’s Disease (AD).[1]
Although recent studies strongly suggest that intracellular accumulation of Aβ is required for the peptide to exert some of its key toxic actions,[2] there are substantial gaps in our knowledge of the mechanism of Aβ cellular uptake. Advances in our understanding of the phenomenon are urgently needed so that molecular therapeutics of AD targeting cellular uptake of Aβ can be rationally designed. Diverse biomolecules that could act as Aβ cellular binding partners and may lead to its internalization (i.e., receptors) have been proposed, including transmembrane proteins, lipids and other biomacromolecules.[3] However, receptor binding is not a strict pre-requisite for cellular uptake. For example, cells employ the macropinocytosis process to internalize molecules without the requirement of providing discrete molecular binding sites for them.[4] Membrane pore assembly is another mechanism, through which antimicrobial, aggregation-prone peptides can enter cells in a receptor-independent fashion.[5] All of the above have been proposed to contribute to Aβ cellular uptake to some degree, yet it remains unclear, which of the many postulated mechanisms are the dominant ones.
Chirality can be employed as a unique tool to distinguish between the two classes of peptide internalization mechanisms: receptor-mediated cell uptake events are likely to occur with a certain degree of stereoselectivity (which is referred to as uptake mechanism class I or the chirality-dependent uptake component), whereas receptor-independent processes lack the chiral interactions between the molecule that is being taken up and the cell and are not expected to proceed in a stereoselective fashion (which is referred to as uptake mechanism class II or the chirality-independent uptake component). Aβ42 cellular toxicity has been found to depend on chirality by us and others, with the natural (L-)Aβ42 stereoisomer being significantly more toxic than the mirror image (D-)peptide.[6] This led us to hypothesize that Aβ cellular uptake is a stereoselective process. To test this hypothesis, we synthesized fluorescently tagged Aβ enantiomers and used them as tools to study the stereoselectivity of the Aβ cellular uptake process.
Using microwave-assisted, Fmoc-based solid phase peptide synthesis, we made the two enantiomers (L/D) of Aβ(40/42). The peptides were labeled with 5(6)-Carboxytetramethylrhodamine (TAMRA), following our published methods (see also SI experimental description as well as Figures S1-S4).[6a] Thioflavin T (ThT) fibril formation assays were conducted and showed L- and D-Aβ42 to form fibrils with T1/2 values that were consistent with our previous studies and, as expected, within experimental error between the two enantiomers (Figure 1; T1/2;L-Aβ42 = 21.0 ± 0.7 min; T1/2;D-Aβ42 = 20.2 ± 0.2 min).
Figure 1.
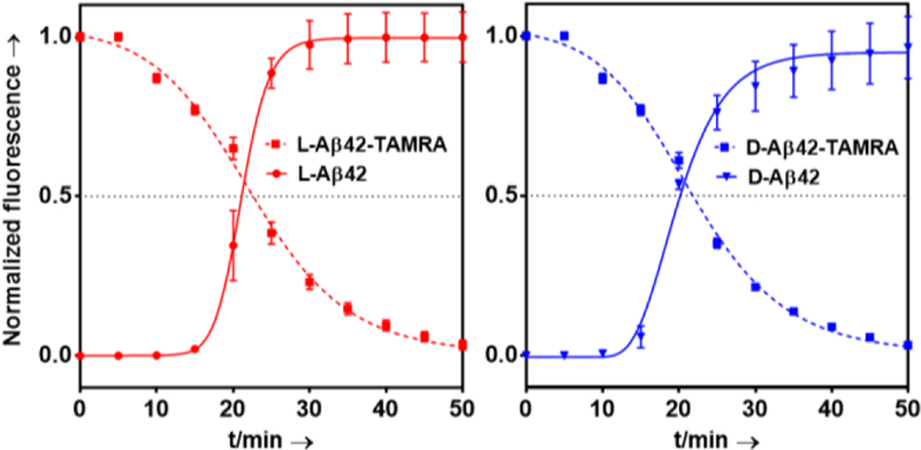
Fibril formation kinetics of (L/D)-Aβ42 monitored by Thioflavin T (ThT, 20 µM) fluorescence and (L/D)-Aβ42-TAMRA monitored by TAMRA self-quenching at 37 ˚C in PBS (pH 7.4; Aβ at 20 µM in all cases). Each data point is an average of five replicates with error bars representing the standard deviations.
We found the N-terminal TAMRA derivatization not to affect fibril formation properties in any significant fashion (Figure 1; T1/2;L-Aβ42-TAMRA = 22.6 ± 0.7 min; T1/2;D-Aβ42-TAMRA = 21.8 ± 0.4 min), which is consistent with previous reports.[7] Fibril formation of TAMRA-labeled Aβ (i.e, (L/D)-Aβ42-TAMRA)) was monitored through TAMRA fluorescence quenching upon fibril assembly.[7a] Fibril formation was confirmed via TEM (Figure S5).i We found this modification to not influence cellular toxicity to any significant degree (Figure S6).
To determine the dependence of cellular uptake on Aβ chirality, confocal imaging experiments were conducted. SH-SY5Y human neuroblastoma cells were incubated with TAMRA labeled peptide solutions (5 µM, 2 h), counter-stained with the Hoechst nuclear stain, washed with DPBS (i.e., PBS containing calcium and magnesium) and imaged (see SI for a detailed procedure). A significant and consistent difference in uptake quantity was noted between L- and D-Aβ, which was observable for both the 40 and the 42 amino acid long isoforms (Figure 2). Bright intracellular puncta were clearly visible, which is in agreement with a recent study that probed intracellular localization of L-Aβ42 to SH-SY5Y and reported the majority of it to localize to cell lysosomes.[8] We confirmed this further by conducting lysotracker co-staining experiments for both Aβ stereoisomers and isoforms (Figure S7). Prolonged incubation of SH-SY5Y cells with (L/D)-Aβ42-TAMRA (15 h instead of 2 h) revealed a consistent preference for the internalization of the L-stereoisomer (Figure S8), which was also re-capitulated in PC12 cells (Figure S9).
Figure 2.
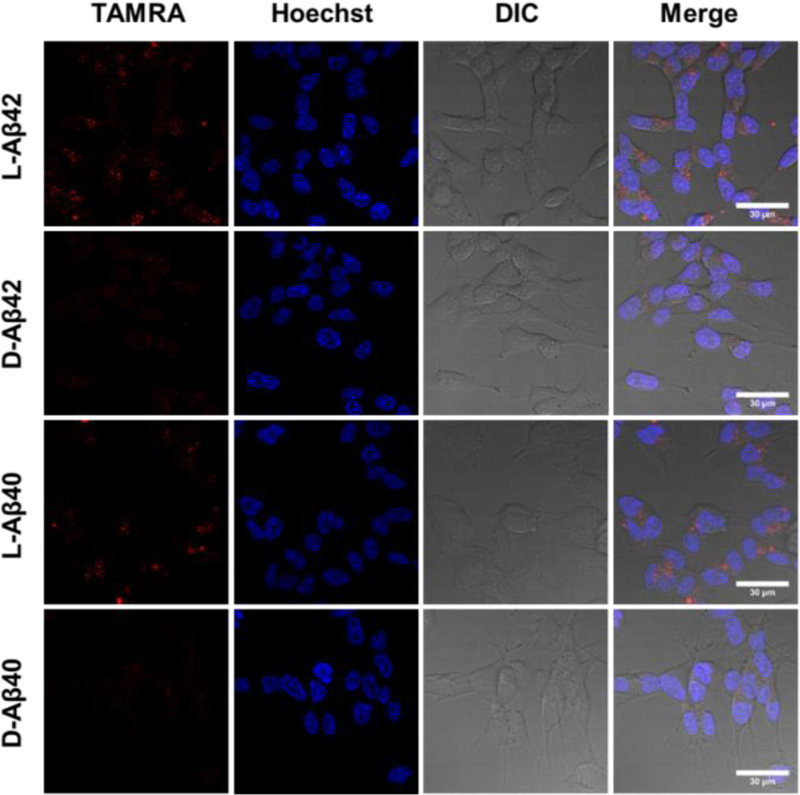
Confocal microscopy imaging of SH-SY5Y cells incubated with (L/D)-Aβ(40/42), as indicated, at 5 μM for 2 h and rinsed with DPBS prior to imaging. TAMRA (TAMRA-Aβ uptake): excitation at 543 nm, emission over 590–720 nm; Hoechst (nuclear staining): excitation at 405 nm; emission over 415–485 nm. Scalebar: 30 µm.
To further examine Aβ cellular uptake differences in a more quantitative, unbiased fashion, flow cytometry experiments were performed (Figure 3). Cells were incubated with 5 µM (L/D)-Aβ(40/42) peptide for 2 h, which is identical to conditions that were employed in confocal imaging experiments (Figure 2). Following this incubation, cells were rinsed with PBS, trypsinized, pelleted, re-suspended in PBS and subjected to flow cytometry analysis (see SI for a more detailed description). A 5.45-fold difference (p<0.01) in SH-SY5Y cellular peptide was observed between L-Aβ42 and D-Aβ42. Stereodifferentiation was still highly significant, albeit less pronounced, for the Aβ40 isoform, with L-Aβ40 localizing to SH-SY5Y cells 2.9-fold more efficiently (p<0.001) than D-Aβ40. A 1.85-fold stronger signal (p<0.05) was measured for L-Aβ42 than for L-Aβ40, which is in good agreement with an ~2-fold difference that was reported previously.[9] We also found this trend to hold true over a range of (L/D)-Aβ42 concentrations (Figure S10). In further support of stereoselectivity of the cellular uptake process, a comparable preference for the L-stereoisomer was observed via flow cytometry for the Aβ42 isoform in PC12 cells and in rat primary hippocampal neurons (Figure S11). In a separate experiment, SH-SY5Y cells were incubated with (L/D)-Aβ42-TAMRA for 2 h at 4 °C (Figure S12) and, in agreement with a previous study,[9] showed low fluorescence, indicating limited membrane binding. This further corroborates our interpretation that the fluorescence measured by flow cytometry for cells incubated with Aβ at 37 °C (Figure 3, Figure S10 and Figure S11) stems from intracellular and not membrane-bound peptide, and is also consistent with the confocal microscopy experiments (Figure 2 and Figure S7).
Figure 3.
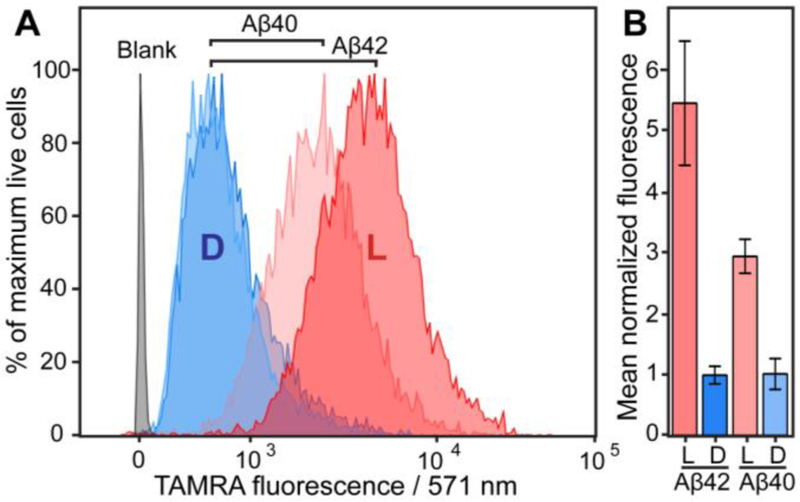
A) Flow cytometry quantitation of TAMRA-labeled (L/D)-Aβ(40/42) uptake by SH-SY5Y (human neuroblastoma) cells, as indicated; representative biological replicate shown. Cells were exposed to 5 μM Aβ for 2 h, and then analyzed by flow cytometry (10,000 cells sampled per condition; only live cells selected for analysis; data analyzed using the FlowJo software package). B) Relative uptake levels of (L/D)-Aβ(40/42)-TAMRA, obtained through averaging of three biological replicates (D-Aβ42 uptake was normalized to 1). Live cells were selected via LIVE/DEAD Fixable Violet Dead Cell Staining (see SI for descriptive protocols and individual biological replicates of the experiments).
Experiments presented above demonstrate that molecular chirality is an important determinant of cellular uptake of Aβ. Consistent trends were observed for both the Aβ40 and the Aβ42 peptide isoforms by confocal microscopy and flow cytometry in two model cell lines (SH-SY5Y cells and PC12 cells) at various incubation times (2 and 15 h) over a range of concentrations (0.1 μM to 5 μM). The trend was also found to hold true with primary neurons. The level of generality observed suggests that intracellular (e.g., lysosomal) peptide degradation, which may occur at different rates for the two Aβ40/42 enantiomers, is unlikely to contribute to more than a minor degree to the overall picture that emerged from our study. Because chiral interactions between the cell and the peptide are a pre-requisite for stereodifferentiation, our findings strongly suggest cellular uptake of Aβ to be receptor-mediated to a substantial degree. Cellular uptake of the D-enantiomer, whilst significantly less pronounced, was still clearly and consistently detectable in all experiments, pointing towards the complexity of the process and suggestive of multiple mechanisms of intracellular localization being at play simultaneously. The ~5:1 ratio, which is a representative stereoselectivity ratio for cellular uptake of the two Aβ42 enantiomers, can be interpreted as follows: approximately 80 % of L-Aβ42 is taken up by cells through chirality-dependent mechanisms (i.e., uptake mechanism class I), whereas the residual ~20 % enter the cells by a chirality-independent (i.e., class II) mechanism. Some likely contributors are discussed below.
Class I:
The stereoselective component of Aβ cellular uptake has to arise as a consequence of interactions of the peptide with cell-associated chiral binding partners. Diverse biomolecules have been proposed to contribute to intracellular Aβ localization, including the transmembrane proteins PrP and RAGE, as well as the soluble protein APOE, inter alia.[3a,b,10] Head groups of certain lipids, such as the GM1 ganglioside and phosphatidylserine, have also been found to play roles in physiologically relevant interactions between Aβ and cellular membranes.[3c,d,11] Because binding of Aβ to membranes can affect its aggregation properties and cellular internalization capacity, interactions between Aβ and chiral lipid head groups may also make contributions to stereoselectivity of Aβ cellular uptake by providing chiral binding sites on cell membranes.
Class II:
The non-stereoselective component of Aβ uptake likely arises through mechanisms of action that does not involve the establishement of chiral interactions between the peptide and the cell. For example, macropinocytosis can be expected to operate with no chiral preference, and has been suggested to play a role in cellular uptake of Aβ.[9] Membrane poration constitutes another mechanism that has been discussed as a potential contributor to Aβ uptake and toxicity.[12] This mechanism of action is perhaps most well-established in the context of antimicrobial peptides. As such, in the seminal study published by Merrifield and co-workers in 1990, it was found that L- and D-enantiomers of three α-helical peptides (cecropin, magainin, and melittin) were equally potent as pore-forming antibacterial peptides.[5b] It should be noted that there is evidence for Aβ to have antimicrobial activity,[13] which is likely related to its ability to permeabilize membranes of pathogens.
In summary, we made fluorescently tagged Amyloid β enantiomers of the 40 and 42 amino acid long isoforms, and were able to use them as mechanistic tools to gain unique insights into the cellular uptake process, which is believed to be a critical step for various toxic actions of the peptide to become manifest.[2] A conundrum for the field for over two decades, innumerable uptake mechanisms have been proposed, often with contradictory implications.[6b,14] We find that Aβ uptake depends on chirality of the peptide, with a typical preference ~5:1 for the L-stereoisomer. Consistent observations are made for both Aβ40 and Aβ42 in SH-SY5Y and PC12 cells, using flow cytometry and confocal microscopy. Our results provide a new perspective on the cellular Aβ uptake process, through which it emerges as a multi-mechanism process that is dominated by a stereoselective, receptor-mediated component.
Supplementary Material
Acknowledgements
JAR thanks UCSC for the flexible start-up funds and the NIH for the award of the grant R21AG058074. We are grateful to The Institute for the Biology of Stem Cells (IBSC) Shared FACS Facility for assistance with flow cytometry experiments.
Footnotes
SI for this article is given via a link at the end of the document.
Analogous ThT fibril formation assays were also performed with (L/D)-Aβ40 and (L/D)-Aβ40-TAMRA, but the T1/2 variance between experiments was too large, rendering the outcome of those measurements inconclusive.
References
- [1]. a)Selkoe DJ, Hardy J, EMBO Mol. Med 2016, 8, 595–608; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Querfurth HW, LaFerla FM, Engl N. J. Med 2010, 362, 329–344; [DOI] [PubMed] [Google Scholar]; c) Haass C, Selkoe DJ, Nat. Rev. Mol. Cell. Biol 2007, 8, 101–112; [DOI] [PubMed] [Google Scholar]; d) Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB, Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]. a)Jin S, Kedia N, Illes-Toth E, Haralampiev I, Prisner S, Herrmann A, Wanker EE, Bieschke J, J. Biol. Chem 2016, 291, 19590–19606; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) LaFerla FM, Green KM, Oddo S, Nat. Rev. Neurosci 2007, 8, 499–509. [DOI] [PubMed] [Google Scholar]
- [3]. a)Jarosz-Griffiths HH, Noble E, Rushworth JV, Hooper NM, J. Biol. Chem 2016, 291, 3174–3183; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zlokovich BV, Nat. Rev. Neurosci 2011, 12, 723–738; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gibson Wood W ,Eckert GP, Igbavboa U, Müller WE, Biochim. Biophys. Acta 2003, 1610, 281–290; [DOI] [PubMed] [Google Scholar]; d) Terzi E, Hölzemann G, Seelig J, Biochemistry 1997, 36, 14845–14852. [DOI] [PubMed] [Google Scholar]
- [4].Lim JP, Gleeson PA, Immunol. Cell Biol 2011, 89, 836–843. [DOI] [PubMed] [Google Scholar]
- [5]. a)Wang CK, King GJ, Conibear AC, Ramos MC, Chaousis S, Henriques ST, Craik DJ, J. Am. Chem. Soc 2016, 138, 5706–5713; [DOI] [PubMed] [Google Scholar]; b) Wade D, Boman A, Wåhlin B, Drain CM, Andreu D, Boman HG, Merrifield RB, Proc. Natl. Acad. Sci. USA 1990, 87, 4761–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6]. a)Dutta S, Foley AR, Warner CJA, Zhang X, Rolandi M, Abrams B, Raskatov JA, Angew. Chem. Int. Ed 2017, 56, 11506–11510; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ciccotosto GD, Tew DJ, Drew SC, Smith DG, Johanssen T, Lal V, Lau TL, Perez K, Curtain CC, Wade JD, Separovic F, Masters CL, Smith JP, Barnham KJ, Cappai R, Neurobiol. Aging 2011, 32, 235–248. [DOI] [PubMed] [Google Scholar]
- [7]. a)Garai K, Frieden C, Proc. Natl. Acad. Sci. USA 2013, 110, 3321–3326; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Walsh DM, Thulin E, Minogue AM, Gustavsson N, Pang E, Teplow DB, Linse S, FEBS J 2009, 276, 1266–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee J-M, Proc. Natl. Acad. Sci. USA 2009, 106, 20324–20329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wesén E, Jeffries GDM, Dzebo MM, Esbjörner EK, Sci. Rep 2017, 7, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10]. a)Liu C-C, Kanekiyo T, Xu H, Bu G, Nat. Rev. Neurol 2013, 9, 106–118; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, Marky A, Lenting PJ, Wu Z, Zarcone T, Goate A, Mayo K, Perlmutter D, Coma M, Zhong Z, Zlokovic BV, Nat. Med 2007, 13, 1029–1031; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Deane R, Yan SD, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B, Nat. Med 2003, 9, 907–913. [DOI] [PubMed] [Google Scholar]
- [11]. a)Hong S, Ostaszewski BL, Yang T, O’Malley TT, Jin M, Yanagisawa K, Li S, Bartels T, Selkoe DJ, Neuron 2014, 82, 308–319; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee G, Pollard HB, Arispe N, Peptides 2002, 23, 1249–1263; K. Yanagisawa, A. Odaka, N. Suzuki, Y. Ihara, Nat. Med. 1995, 1062–1066. [DOI] [PubMed] [Google Scholar]
- [12]. a)DiScala C, Yahi N, Boutemeur S, Flores A, Rodriguez L, Chahinian H, Fantini J, Sci. Rep 2016, 6, 28781; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Serra-Batiste M, Ninot-Pedrosa M, Bayoumi M, Gairi M, Maglia G, Carulla N, Proc. Natl. Acad. Sci. USA 2016, 113, 10866–10871; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kreutzer AG, Hamza IL, Spencer RK, Nowick JS, J. Am. Chem. Soc 2016, 138, 4634–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, Moir RD, Sci. Transl. Med 2016, 8, 340ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cribbs DH, Pike CJ, Weinstein SL, Velazquez P, Cotman CW, J. Biol. Chem 1997, 272, 7431–7436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.