

Abstract
This is a protocol for a Cochrane Review (Diagnostic test accuracy). The objectives are as follows:
To estimate the diagnostic accuracy of point‐of‐care tests to detect high viral load levels in HIV‐positive people on ART.
Background
Access to antiretroviral therapy (ART) for HIV‐positive people has increased significantly over the years. It is estimated that 53% of HIV‐positive people globally were on ART in 2016 (UNAIDS 2017), up from 23% in 2010 (UNAIDS 2015). In sub‐Saharan Africa, about 12 million HIV‐positive people (47%) were on ART in 2015 (UNAIDS 2016), up from less than 100,000 in 2002 (UNAIDS 2015). In order to effectively sustain treatment for people on ART, it is essential to know the HIV viral load (VL) levels in those who are on treatment. VL (the number of HIV viral ribonucleic acid (RNA) particles per millilitre of blood) is the recommended monitoring approach to diagnose and confirm ART treatment failure (WHO 2016). VL is usually measured in plasma but some technologies use whole blood (UNITAID 2015). In Africa, it is estimated that less than 20% of people on ART received routine VL testing in Africa in 2013 (ASLM 2013). This could be partly be explained by poor access to VL testing services. Currently, VL testing is largely done on laboratory‐based platforms that involve sophisticated equipment requiring dedicated laboratory space, substantial financial resources, and trained laboratory technicians. These laboratory tests require venous blood collection, cold chain storage of collected samples, and instrument‐based sample processing techniques. With transport shortcomings being a common challenge in resource‐limited settings, delays in transporting samples to the laboratory and relaying test results back to the health centre lead to delays in changing therapy in cases of treatment failure. To overcome this challenge, point‐of‐care tests are increasingly being developed because they are potentially easy to use, cost‐effective, require minimal laboratory infrastructure, and may minimize the need for transporting samples to the laboratory. They could also potentially reduce patient waiting time and therefore reduce loss to follow‐up cases (UNITAID 2014; UNITAID 2015; WHO 2014).
Target condition being diagnosed
The target condition of this review is high HIV VL levels in blood or plasma of people infected with either HIV‐1 or HIV‐2 on ART. The main objective of ART is to reduce HIV VL to undetectable levels meaning that the concentration HIV RNA cannot be detectable by a test. In HIV‐infected people, it is therefore essential to know the VL levels after ART initiation. The higher the VL, the higher the increased risk of transmission when VL is detectable and the faster the CD4 cells and body's immune system are destroyed. Detectable VL can be a reflection of poor adherence to treatment or treatment failure once poor adherence is ruled out. Intermittent low level viraemia (50 copies/mL to 1000 copies/mL) not associated with treatment failure may also occur during effective treatment (Havlir 2001). Current World Health Organization (WHO) guidelines on ART define a high or detectable VL level to be 1000 copies/mL or greater and treatment failure as a persistently high VL concentration (1000 copies/mL or greater) in two consecutive measurements (with adherence support between measurements) (WHO 2016). Treatment failure should trigger evaluation or changing the antiretroviral drugs included in ART. Delayed detection of treatment failure may therefore lead to progression of HIV infection to AIDS or the resistance of the infection to ART or increase the risk of HIV transmission (UNITAID 2015; WHO 2013).
Index test(s)
In this Cochrane Review, we will estimate the accuracy of molecular point‐of‐care (POC) tests in detecting high VL levels (POC VL) after ART initiation. Molecular POC VL include semi‐quantitative and quantitative tests that quantify the copies of HIV virus in plasma or whole blood (UNITAID 2014; UNITAID 2015). Results are reported as HIV copies in a millilitre (copies/mL). There is no established optimal threshold for detecting VL concentration or defining virological failure (Fox 2012; Ritchie 2014; WHO 2013; WHO 2016). In 2013, the WHO lowered the threshold for detecting high VL levels from 5000 copies/mL to 1000 copies/mL based on evidence that below 1000 copies/mL intermittent low‐level viraemia (50 copies/mL to 1000 copies/mL) not associated with treatment failure can occur during effective treatment (Ritchie 2014; WHO 2013). Also, the risk of HIV transmission and progression of disease is minimal when VL concentration is less than 1000 copies/mL. Nonetheless, the lower limit of VL detection depends on the test and sample used. For example, a capillary sample from a finger prick may not accurately detect a VL level below 5000 copies/mL (ASLM 2013; UNITAID 2015).
Various definitions of POC testing have been proposed with no universally accepted definition (Drain 2014; UNITAID 2015). Some definitions consider technical characteristics of the test (rapid test with minimal infrastructure requirements) (Wu 2012), or its effect on management (linking to decision making at the same patient visit) (Pai 2012), or its location (at the patient site or near the treatment facility) (Drain 2014). A general definition of a POC test would be a diagnostic test that is administered near the patient or at a health facility, with a fast turnaround time, leading to a change in patient management (Schito 2012). In this review, we will include studies with VL tests that meet the ASSURED (Affordable, Sensitive, Specific, User‐friendly, Robust & Rapid, Equipment free, and Deliverable to end‐users) criteria developed by the WHO for the ideal rapid test for resource limited settings (Wu 2012). In resource‐limited settings, testing locations are often blurred as POC tests have been evaluated and implemented across a variety of healthcare and laboratory settings ranging from primary level next to patients (Level 1 facilities) to district (Level 2) and provincial levels (Level 3) (UNITAID 2015). Ideally, true POC tests are conducted on patient samples next to the patient or at the bedside in settings with minimaI laboratory and training requirements (Level 1 facilities). Tests otherwise referred to as near‐POC tests are conducted on patient samples away from the patient by technicians in slightly more developed laboratories (Level 2 and 3 facilities). For example, some POC tests detect VL in plasma (Ritchie 2014), and some settings may not have the infrastructure required for plasma separation from whole blood at the point of care (UNITAID 2015). To maximize the utility of our review, we will evaluate all forms of POC tests for VL regardless of the health facility setting in which the test was conducted.
Clinical pathway
The role of POC VL for monitoring response to ART will be to act as a replacement for laboratory‐based VL testing platforms in the current testing algorithms outlined in Figure 1.
Figure 1.
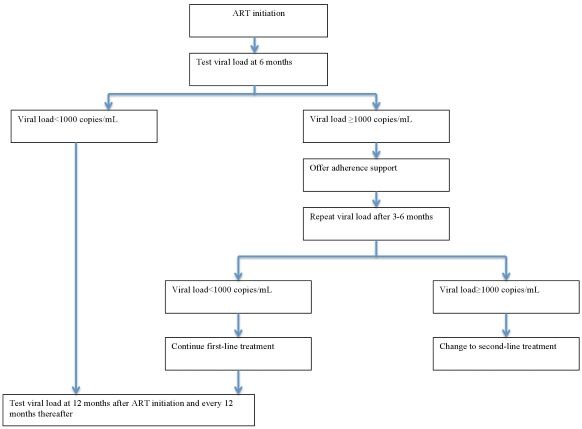
Routine viral load testing algorithm. Abbreviations: ART: antiretroviral therapy.
In routine care, current WHO guidelines recommend that VL testing be done at six and 12 months after initiation of ART and repeated every 12 months thereafter. If the VL is detectable at any time (1000 copies/mL or greater), it is recommended that a patient undergoes intensive adherence support and repeat VL testing three to six months later. If the VL is still detectable and non‐adherence can be ruled out, a clinician may then decide to change to second‐line therapy (WHO 2013; WHO 2016).
In this review, we will focus on the accuracy of a single POC VL test done at one time point (for example, at six or 12 months after ART initiation). We believe that this evidence may be extrapolated to provide evidence that may guide the use of the test at other time points.
Alternative test(s)
Alternative HIV VL tests include non‐nucleic acid tests (non‐molecular tests) that detect HIV viral enzymes (reverse transcriptase) and HIV viral proteins (p24 antigen); markers that can be correlated to HIV RNA. These tests indirectly reflect VL concentration and are currently not commonly used (UNITAID 2015).
Alternative methods for monitoring response to ART include immunological monitoring through CD4 testing and clinical monitoring through WHO clinical staging. For example, in adults, a persistent CD4 count less than 100 cells/mm3 or a new or recurrent clinical condition indicative of WHO clinical stage 4 after six months of treatment is regarded as treatment failure. However, these methods are less sensitive and specific than VL testing and are not recommended as the first‐line approach for monitoring response to ART (Rutherford 2014). This may lead to delayed detection of treatment failure or lead to unnecessary therapy switches. In addition, the WHO revised its guidelines in 2013 to recommend that all HIV‐positive people be started on ART regardless of CD4 count and clinical status (WHO 2013). In this regard, using these criteria to monitor response to therapy will not be an accurate measure of treatment failure. Nonetheless, these alternative tests may still be used in areas that do not have access to VL testing (WHO 2013).
Rationale
In 2014, the Joint United Nations Programme on HIV/AIDS (UNAIDS) declared the 90‐90‐90 target; it aimed to have at least 90% of HIV‐positive people diagnosed, at least 90% of those diagnosed receiving ART, and at least 90% of those receiving ART having suppressed viral replication by 2020 (WHO 2016). POC VL tests being developed to detect HIV RNA and treatment failure in HIV‐positive people on ART in resource‐limited settings will be instrumental in checking if the third target will be met effectively. If these POC VL tests have a high level of accuracy, they can replace or complement laboratory‐based testing platforms because they are quicker to use and may minimize delays in initiating therapy or changing therapy in cases of treatment failure (UNITAID 2014; UNITAID 2015). A high sensitivity is required because false‐negative results will lead to a delay in detecting treatment failure or adherence concerns related to treatment. This will ultimately lead to progression to AIDS and mortality. A high specificity is also required because false‐positive results will lead to unnecessary switching to costly second‐line therapy. A test with an optimal combination of sensitivity and specificity is thus needed.
Objectives
To estimate the diagnostic accuracy of point‐of‐care tests to detect high viral load levels in HIV‐positive people on ART.
Secondary objectives
To investigate sources of heterogeneity in test accuracy estimates including age (children versus adults), test type (commercially available versus in‐house assays), sample type (whole blood versus plasma), test threshold (1000 copies/mL or greater versus other thresholds), location of testing (true‐POCs versus near‐POCs), geographical location (sub‐Saharan Africa versus other regions), and methodological quality (high versus low risk of bias).
Methods
Criteria for considering studies for this review
Types of studies
We will include any primary study that compares the results of the index test to that of a reference standard (cross‐sectional, prospective, and retrospective study designs or diagnostic accuracy studies performed within randomized trials), and those that provide sufficient data to create the 2 × 2 table to calculate sensitivity, specificity, and negative and positive predictive values.
We will exclude ecological studies and diagnostic case‐control studies in which the test performance was compared in participants with the target condition versus healthy controls, as specificity will be overestimated (Macaskill 2010).
We will exclude studies without a reference standard, case reports and case‐series studies, animal or laboratory studies, reviews, discussion papers, non‐research letters, commentaries, or editorials.
Participants
People infected with either HIV‐1 or HIV‐2 on ART irrespective of age and gender, from any healthcare or geographical setting. Though POC VL tests are mainly applicable to resource‐limited settings where the burden of HIV is high, we will not exclude evaluations conducted in resource‐rich settings in order to maximize utility of our review.
Index tests
We will include studies evaluating the accuracy of molecular POC VL. We will consider the current WHO recommended threshold (1000 copies/mL or greater) as the main threshold to define test positivity (WHO 2013; WHO 2016). We will also consider the previous WHO recommended threshold (5000 copies/mL or greater) (WHO 2010), and other thresholds that may have been used for test evaluations in the subgroup analyses.
Examples of POC VL include (but not limited to):
SAMBA I HIV‐1 Semi‐Quantitative Test;
SAMBA II HIV‐1 Semi‐Quantitative Test;
Alere q Analyser and Alere q HIV‐1/2 assay (quantitative whole blood assay);
Savanna RealTime HIV‐1 Viral Load assay (Quidel);
Cobas Liat analyser (Roche);
Xpert HIV‐1 Viral Load (Cepheid);
ZIVA (Cavidi);
Liat Analyser (IQuum Inc);
EOSCAPE HIV Rapid RNA Assay System;
True lab Real Time Micro PCR system (Molbio);
RT CPA HIV‐1 viral load.
Most of these tests are still in the pipeline except the SAMBA VL assay, which is available. The SAMBA‐Semiquantitative HIV tests use 200 µL of plasma or 120 µL of whole blood and have a total assay time of 90 minutes. They have been designed to detect VL concentrations of 1000 copies/mL or greater. SAMBA I is a semi‐automated test with a daily throughput of 16 to 48 samples whereas SAMBA II is fully automated with a daily throughput of four to 32 samples. SAMBA II is better suited for facilities with technicians and electricity (UNITAID 2014; UNITAID 2015).
Target conditions
A high HIV VL level in people infected with HIV‐1‐ or HIV‐2 on ART.
Reference standards
Laboratory‐based testing platforms to detect high VL levels taken at the same time (within 24 hours) as the sample for POC VL tests. Most laboratory‐based VL platforms are designed to detect the HIV virus in plasma that is extracted from a venous blood sample though centrifugation. Typical laboratories for VL technologies involve sophisticated equipment and have three rooms for sample extraction, reagent preparation, and amplification (and detection) of the HIV virus (UNITAID 2015). Examples of laboratory‐based platforms for VL include: nucleic acid‐based tests (NAT) including five commercially available reverse transcriptase polymerase chain reaction (RT‐PCR)‐based VL assays:
COBAS AmpliPrep/ COBAS TaqMan v2.0 (Roche);
RealTime HIV‐1 (Abbott);
VERSANT HIV RNA 1.0 (kPCR) (Siemens);
Artus HIV‐1 QS‐RGQ (QIAGEN);
RT‐TMA technology for Panther system (Hologic).
Current and previous WHO recommended thresholds to detect high HIV VL levels in plasma and classify a patient as having treatment failure include 1000 copies/mL or greater (WHO 2013; WHO 2016), and 5000 copies/mL or greater (WHO 2010). We will include data where the threshold of 1000 copies/mL were presented but also collect data of the 5000 copies/mL threshold.
Where studies have used a tie‐breaker approach (where a second test/polymerase chain reaction (PCR) is used for discordant results, we will include results for the first test/PCR only in the 2 × 2 tables to avoid inflation of sensitivity and specificity (Ritchie 2014).
Search methods for identification of studies
Electronic searches
We will conduct the search in the electronic databases; Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, Embase, LILACS, the WHO International Clinical Trials Registry Platform (WHO ICTRP), Science Citation Index Expanded (SCI‐EXPANDED)/Conference Proceedings Citation Index‐ Science (CPCI‐S), and WHO Global Index Medicus from March 2015 without language, document type, or publication status limitations (Appendix 1).
Searching other resources
We will track reference lists of included studies, relevant systematic reviews, and conference proceedings (Conference on Retroviruses and Opportunistic Infections, International AIDS Society Conference and African Society for Laboratory Medicine). We will consult experts in the field such as the WHO HIV Department for potentially relevant studies.
Data collection and analysis
Selection of studies
We will deduplicate search results in EndNote X7. Two review authors (EO and AK) will independently screen the titles and abstracts of the search results to identify eligible articles. They will initially remove reports that are obviously not relevant based on title and abstract and remove duplicates as well. The two review authors (EO and AK) will then independently assess full texts of journal articles or conference proceedings for eligibility based on our a priori inclusion criteria. We will resolve any disagreements by consensus or by consulting a third review author (SM or JD). Justifications for excluding articles from the review will be documented in the 'Characteristics of excluded studies' table. We will present details of included studies in the 'Characteristics of included studies' table. We will present the study selection process in a PRISMA flow diagram.
Data extraction and management
We will extract information on study characteristics including: study design; demographic and participant characteristics; methods of collecting and preparing blood specimen; time point at which VL testing is done after ART initiation; index test and reference standard characteristics; test cut‐off and performance; main outcome data or results; number of true‐positive, false‐positive, false‐negative, and true‐negative results (Appendix 2).
Two review authors (EO and AK) will independently extract data. We will resolve any disagreements by discussion and will document all decisions. If we cannot reach a consensus, a third review author (SM or JD) will make the final decision on inclusion.
If we identify more than one publication for the same included study, we will consider the main publication as the one with more information; all others will be considered companion publications and we will only collect data if they had not been provided in the main publication to avoid double‐counting. We will collate the companion publications of the same study, so that each study rather than each report, is the unit of interest in the review.
Assessment of methodological quality
We will use the QUADAS‐2 (Quality Assessment of Diagnostic Accuracy Studies) tool to assess the risk of bias and applicability concerns of the included studies (Whiting 2011). We will tailor the tool in line with the context of our review question (Appendix 3). Two review authors (EO and AK) will independently assess included studies using the outlined tool in Appendix 3. We will resolve any disagreements by consensus or by consulting a third review author (SM or JD).
Statistical analysis and data synthesis
Our unit of analysis will be individual participants. For each study, we will identify the threshold(s) used to define test positivity and construct 2 × 2 tables (true positive, false positive, false negative, true negative) at the presented thresholds. We will perform the main analysis with study data using the current WHO recommended threshold (1000 copies/mL or greater) definition of test positivity (WHO 2016). We will undertake subgroup analyses separately at other commonly presented thresholds. Preliminary exploratory analyses on diagnostic accuracy will be conducted by plotting estimates of sensitivity and specificity from each study on Forest plots and in receiver operating characteristic (ROC) space. These analyses will enable visual assessment of the variation between studies, and will also facilitate investigations of heterogeneity for exploring the effect of certain characteristics on test performance.
If there are sufficient data (for example, four or more studies), we will use the bivariate model to estimate the summary sensitivity and specificity at the current WHO threshold (1000 copies/mL) for the main meta‐analysis and compare the accuracy of two or more tests. The bivariate model with random effects accounts for within‐study variability and correlation of sensitivity and specificity. This method models sensitivity and specificity directly at a common threshold (Macaskill 2010; Reitsma 2005). If bivariate models do not converge and so cannot give a model estimate, we will fit simplified univariable models for sensitivity and specificity separately, using a random‐effects model (Takwoingi 2017). If there are no false positives across all studies in a meta‐analysis (i.e. specificity is estimated at 100% in all studies), we will undertake a univariate random‐effects meta‐analysis of sensitivity, and compute the exact 95% confidence interval for the 100% specificity estimate using the total number of participants without disease across all studies as the denominator.
For comparisons between tests (if there is sufficient data), we will initially include all studies in the analysis (indirect comparison). Subsequently, if data are available, we will restrict the analyses to only studies that have compared tests in the same population, either within participants or between randomized groups (direct comparison). Such analyses are likely to produce results not confounded by differences in study or participant characteristics.
We will perform analyses using Review Manager 5 (RevMan 5) (RevMan 2014), and the meta‐analysis using STATA (STATA 2017), or SAS (SAS 2017).
Investigations of heterogeneity
If there are sufficient data, we will investigate sources of heterogeneity in estimates of test accuracy. We will add the following covariates to the bivariate model to assess its influence on test performance: age (children versus adults), test type (commercially available versus in‐house assays), sample (whole blood versus plasma), and location of testing (true‐POC versus near‐POC). We will flag studies that report mixed‐age populations (unclear data on data per age group) as age 'not reported' in the statistical models. If there are sufficient data we will estimate accuracy for individual tests as per manufacturer type. We will estimate sensitivity and specificity at other commonly used thresholds separately in the subgroup analyses as well.
Sensitivity analyses
If there are sufficient data, we will use sensitivity analyses to explore the effect of geographical setting and study quality. We will restrict analysis to studies conducted in sub‐Saharan Africa and to studies at low risk of bias for participant selection and high applicability for index test conduct.
Assessment of reporting bias
We will not assess reporting bias.
Assessment of strength of the evidence
We will summarize the main findings from the review, reporting the numbers of true positives, true negatives, false positive and false negatives per 1000 people tested in the 'Summary of findings' table. GRADE for diagnostic test accuracy reviews is still under development (Gopalakrishna 2014; Gopalakrishna 2016), and rather than following any formal process for downgrading evidence, we will fully describe the following concepts which constitute an assessment of strength of evidence.
Precision of the study estimates.
Heterogeneity in study findings.
Risk of bias.
Concerns about applicability.
Indirect comparisons between tests.
These issues cover the key domains of GRADE (GRADE 2013), except publication bias which cannot be assessed, and would allow the evidence to be included in a GRADE assessment should a guideline developer wish to do so.
Acknowledgements
We acknowledge the contribution of Dr Karla Soares‐Weiser in developing the protocol and writing the final report submitted to the WHO ART Guideline Committee in 2015.
Eleanor A Ochodo is supported by a grant from the Wellcome Trust Foundation (grant number: 109939). The Wellcome Trust has no role in the design, conduct, and interpretation of this protocol or review.
The editorial base of the Cochrane Infectious Diseases Group is funded by UK aid from the UK government for the benefit of low‐ and middle‐income countries (project number 300342‐104). The views expressed do not necessarily reflect the UK government’s official policies.
Appendices
Appendix 1. Search resources and strategies
| Resource | Search interface |
| Cochrane Central Register of Controlled Trials (CENTRAL) The Cochrane Register of Diagnostic Test Accuracy Studies | Wiley Online Library |
| MEDLINE | OvidSP |
| Embase | OvidSP |
| International Clinical Trials Registry Platform (ICTRP) | WHO Portal |
| LILACS | VHL Search Portal |
| Science Citation Index Expanded (SCI‐EXPANDED)/Conference Proceedings Citation Index‐ Science (CPCI‐S) | Web of knowledge |
| WHO Global Index Medicus | Global Health Library |
| International AIDS Society | www.iasociety.org/Conferences |
| Conference on Retroviruses and Opportunistic Infections (CROI) | www.croiconference.org |
| Conference for African Society for Laboratory Medicine |
www.aslm2014.org/ www.aslm2016.org/ |
Cochrane Central Register of Controlled Trials (CENTRAL)
#1 MeSH descriptor: [HIV] explode all trees
#2 MeSH descriptor: [Acquired Immunodeficiency Syndrome] explode all trees
#3 MeSH descriptor: [HIV Infections] explode all trees
#4 (Acquired Immunodeficiency Syndrome* or Acquired Immunologic Deficiency Syndrome* or Acquired Immun* Deficiency Syndrome* or Human Immunodeficiency Virus* or Human T Cell Lymphotropic Virus* or Human T Lymphotropic Virus* or Human T Cell Leukemia Virus* or LAV HTLV III or Lymphadenopathy Associated Virus* or HIV or "HIV 1" or "HIV 2" or "HIV/AIDS" or HIV I or "LAV 2" or LAV HTLV III or HIV II or HTLV III or HTLV IV or "SBL 6669" or AIDS):ti,ab
#5 #1 or #2 or #3 or #4
#6 MeSH descriptor: [Viral Load] explode all trees
#7 (Viral Load* or Virus* Load* or Viral Burden* or Virus* Burden* or Virus Titer* or Viral Titer* or VL*):ti,ab
#8 MeSH descriptor: [Nucleic Acid Amplification Techniques] explode all trees
#9 MeSH descriptor: [Nucleic Acid Hybridization] explode all trees
#10 MeSH descriptor: [Self‐Sustained Sequence Replication] explode all trees
#11 MeSH descriptor: [Polymerase Chain Reaction] explode all trees
#12 MeSH descriptor: [Reverse Transcriptase Polymerase Chain Reaction] explode all trees
#13 MeSH descriptor: [Branched DNA Signal Amplification Assay] explode all trees
#14 (NAT or NATs or NAAT or NAATs or Nucleic Acid Amplif* or DNA Amplif* or RNA Amplif* or nucleic acid sequence based amplification or NASBA or nucleic acid hybridization or nucleic acid hybridization or nucleic acid test* or nucleic acid based test* or transcription‐mediated amplification or self‐sustained sequence replication or polymerase chain reaction or PCR or RT‐PCR or RTPCR or bDNA or b‐DNA or branched DNA or branched‐chain DNA):ti,ab
#15 #6 or #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14
#16 MeSH descriptor: [Point‐of‐Care Systems] explode all trees
#17 (Point of Care or Care Technolog* Point* or Bedside Test* or Bedside Comput* or Bedside Technolog* or Rapid Test* or Rapid Diagnos* or RDT):ti,ab
#18 #16 or #17
#19 #5 and #15 and #18 Publication Year from 1990, in Trials
Embase
Exp Human immunodeficiency virus/ or exp acquired immune deficiency syndrome/ or exp human immunodeficiency virus infection/ or exp human immunodeficiency virus 1/ or exp human immunodeficiency virus 2/ or (Acquired Immunodeficiency Syndrome? or Acquired Immunologic Deficiency Syndrome? or Acquired Immun? Deficiency Syndrome? or Human Immunodeficiency Virus$ or Human T Cell Lymphotropic Virus$ or Human T Lymphotropic Virus$ or Human T Cell Leukemia Virus$ or LAV HTLV III or Lymphadenopathy Associated Virus$ or HIV or HIV 1 or HIV 2 or HIV/AIDS or HIV I or LAV 2 or LAV HTLV III or HIV II or HTLV III or HTLV IV or SBL 6669 or AIDS).ti,ab.
Viral Load/ or nucleic acid amplification/ or nucleic acid hybridization/ or nucleic acid sequence based amplification/ or polymerase chain reaction/ or reverse transcription polymerase chain reaction/ or branched DNA signal amplification assay/ or (Viral Load$ or Virus$ Load$ or Viral Burden? or Virus$ Burden? or Virus Titer$ or Viral Titer$ or VL$ or NAT or NATs or NAAT or NAATs or Nucleic Acid Amplif$ or DNA Amplif$ or RNA Amplif$ or nucleic acid sequence based amplification or NASBA or nucleic acid hybridization or nucleic acid hybridization or nucleic acid test$ or nucleic acid based test$ or transcription‐mediated amplification or self‐sustained sequence replication or polymerase chain reaction or PCR or RT‐PCR or RTPCR or bDNA or b‐DNA or branched DNA or branched‐chain DNA).ti,ab.
Point of care testing/ or exp rapid test/ or (Point of Care or Care Technolog$ Point$ or Bedside Test$ or Bedside Comput$ or Bedside Technolog$ or Rapid Test$ or Rapid Diagnos$ or RDT).ti,ab.
exp animals/ or exp invertebrate/ or animal experiment/ or animal model/ or animal tissue/ or animal cell/ or nonhuman/
human/ or normal human/ or human cell/
4 and 5
4 not 6
1 and 2 and 3
8 not 7
limit 9 to yr="1990 ‐Current"
limit 10 to exclude medline journals
World Health Organization International Clinical Trials Registry Platform (WHO ICTRP)
(Acquired Immunodeficiency Syndrome* OR Acquired Immunologic Deficiency Syndrome* OR Acquired Immun* Deficiency Syndrome* OR Human Immunodeficiency Virus* OR HIV* OR AIDS*) in the Condition
(Viral Load* or Virus* Load* or Viral Burden* or Virus* Burden* or Virus Titer* or Viral Titer* or VL* or Point of Care OR Care Technolog* Point* OR Bedside Test* OR Bedside Comput* OR Bedside Technolog* OR Rapid Test* OR Rapid Diagnos* OR RDT) in the Intervention
MEDLINE
exp HIV/ or exp HIV Infections/ or Acquired Immunodeficiency Syndrome/ or (Acquired Immunodeficiency Syndrome? or Acquired Immunologic Deficiency Syndrome? or Acquired Immun? Deficiency Syndrome? or Human Immunodeficiency Virus$ or Human T Cell Lymphotropic Virus$ or Human T Lymphotropic Virus$ or Human T Cell Leukemia Virus$ or LAV HTLV III or Lymphadenopathy Associated Virus$ or HIV or HIV 1 or HIV 2 or HIV/AIDS or HIV I or LAV 2 or LAV HTLV III or HIV II or HTLV III or HTLV IV or SBL 6669 or AIDS).ti,ab.
Viral Load/ or Exp Nucleic Acid Amplification Techniques/ or nucleic acid hybridization/ or self‐sustained sequence replication/ or polymerase chain reaction/ or reverse transcriptase polymerase chain reaction/ or Branched DNA signal amplification assay/ or (Viral Load$ or Virus$ Load$ or Viral Burden? or Virus$ Burden? or Virus Titer$ or Viral Titer$ or VL$ or NAT or NATs or NAAT or NAATs or Nucleic Acid Amplif$ or DNA Amplif$ or RNA Amplif$ or nucleic acid sequence based amplification or NASBA or nucleic acid hybridization or nucleic acid hybridization or nucleic acid test$ or nucleic acid based test$ or transcription‐mediated amplification or self‐sustained sequence replication or polymerase chain reaction or PCR or RT‐PCR or RTPCR or bDNA or b‐DNA or branched DNA or branched‐chain DNA).ti,ab.
Point‐of‐Care Systems/ or (Point of Care or Care Technolog$ Point$ or Bedside Test$ or Bedside Comput$ or Bedside Technolog$ or Rapid Test$ or Rapid Diagnos$ or RDT).ti,ab.
1 and 2 and 3
limit 4 to yr="1990 ‐Current"
(animals not (humans and animals)).sh.
5 not 6
SCI‐EXPANDED and SPCI‐S (via Web of Knowledge)
TITLE: (Acquired Immunodeficiency Syndrome* OR Acquired Immunologic Deficiency Syndrome* OR Acquired Immun* Deficiency Syndrome* OR Human Immunodeficiency Virus* OR Human T Cell Lymphotropic Virus* OR Human T Lymphotropic Virus* OR Human T Cell Leukemia Virus* OR LAV HTLV III OR Lymphadenopathy Associated Virus* OR HIV OR HIV 1 OR HIV 2 OR HIV/AIDS OR HIV I OR LAV 2 OR LAV HTLV III OR HIV II OR HTLV III OR HTLV IV OR SBL 6669 OR AIDS) AND TITLE: (NAT OR NATs OR NAAT OR NAATs OR Nucleic Acid Amplif* OR DNA Amplif* OR RNA Amplif* OR nucleic acid sequence based amplification OR NASBA OR nucleic acid hybridization OR nucleic acid hybridization OR nucleic acid test* OR nucleic acid based test* OR transcription‐mediated amplification OR self‐sustained sequence replication OR polymerase chain reaction OR PCR OR RT‐PCR OR RTPCR OR bDNA OR b‐DNA OR branched DNA OR branched‐chain DNA) AND TITLE: (Viral Load* or Virus* Load* or Viral Burden* or Virus* Burden* or Virus Titer* or Viral Titer* or VL or Point of Care OR Care Technolog* Point* OR Bedside Test* OR Bedside Comput* OR Bedside Technolog* OR Rapid Test* OR Rapid Diagnos* OR RDT)
Timespan: 1990‐2016. Indexes: SCI‐EXPANDED, CPCI‐S.
WHO Global Index Medicus
((tw:(Acquired Immunodeficiency Syndrome$)) OR (tw:(Acquired Immunologic Deficiency Syndrome$)) OR (tw:(Acquired Immun$ Deficiency Syndrome$)) OR (tw:(Human Immunodeficiency Virus$)) OR (tw:(HIV)) OR (tw:(HIV/AIDS)) OR (tw:(AIDS))) AND ((tw:(Viral Load$)) OR (tw:(Virus$ Load$)) OR (tw:(Viral Burden$)) OR (tw:(Virus$ Burden$)) OR (tw:(Virus Titer$)) OR (tw:(Viral Titer$)) OR (tw:(VL$)) OR (tw:(Point of Care)) OR (tw:(Care Technolog$ Point$)) OR (tw:(Bedside Test$)) OR (tw:(Bedside Comput$)) OR (tw:(Bedside Technolog$)) OR (tw:(Rapid Test$)) or (tw:(Rapid Diagnos$)) or (tw:(RDT))) AND ((tw:(NAT)) OR (tw:(NATs)) OR (tw:(NAAT)) OR (tw:(NAATs)) OR (tw:(Nucleic Acid Amplif$)) OR (tw:(DNA Amplif$)) OR (tw:(RNA Amplif$)) OR (tw:(nucleic acid sequence based amplification)) OR (tw:(NASBA)) OR (tw:(nucleic acid hybridization)) OR (tw:(nucleic acid hybridization)) OR (tw:(nucleic acid test$)) OR (tw:(nucleic acid based test$)) OR (tw:(transcription‐mediated amplification)) OR (tw:(self‐sustained sequence replication)) OR (tw:(polymerase chain reaction)) OR (tw:(PCR)) OR (tw:(RT‐PCR)) OR (tw:(RTPCR)) OR (tw:(bDNA)) OR (tw:(b‐DNA)) OR (tw:(branched DNA)) OR (tw:(branched‐chain DNA)))
Appendix 2. Data to be extracted
We will extract the following information for cross‐sectional, cohort, and case‐control studies.
Study ID: studies by the name of the first author and the year in which the study was first published.
Eligibility: study design, population, HIV status, details of antiretroviral therapy used.
Study details: aim/objective of the study, inclusion and exclusion criteria, study design, prospective/retrospective, whether study was restricted to a subgroup of a larger cohort, how sample size was determined, region and country, setting (inpatients, outpatients), study start and end dates, duration of follow‐up, and sponsor/source of funding.
Study population: description of the participants included in the study (age, gender), predefined inclusion or exclusion criteria (or both), special populations, number of participants recruited/included in the study, how participants were allocated to groups. ART used (first or second line);
Interventions: details of POC VL test used, manufacturer/brand name, conduct of the test, test cut‐off and performance, staff performing test, specimens or sample type, time point at which VL testing was done after ART initiation.
Accuracy estimates: true positives, false positives, false negatives, true negatives.
Study aim and comments: short description of the overall aim of the study, and any additional comments on the study.
Appendix 3. QUADAS‐2; list of signalling questions, risk of bias and applicability
| Domain | Participant selection | *Index test (IT) | Reference standard (RS) | Flow and timing |
| Description | Methods of participant selection | How IT was conducted and reported | How RS was conducted and reported | Describe participants who did not receive and time interval between IT or RS |
| Signalling questions (yes, no, unclear) | Consecutive or random sample of participants? Yes: when the authors reported random participant sampling or consecutive enrolment. No: when participants were selected, for example, based on previous (reference or index) test results. Unclear: if there was insufficient information on study sampling. |
IT results interpreted without knowledge of the results of RS? Yes: when study reported that results of the ITs were interpreted without knowledge of RS results or when ITs were done before the RS. No: when study reported that results of the ITs were interpreted with knowledge of RS results or in cases when RS were used before the index tests. Unclear: when there was insufficient information on when the IT and RS were interpreted. |
RS likely to correctly classify the target condition? Yes: if the RS threshold was clearly reported as > 1000 copies/mL or > 5000 copies/mL. No: if the RS threshold was not reported or if other thresholds used without justification. Unclear: if there was insufficient information to make a judgement. |
Appropriate interval between IT and RS? Yes: if samples tested by both the RS and IT were taken at the same time or within 24 hours. No: if samples tested by both the RS and IT were taken at the same time or within 24 hours. Unclear: when there was no or insufficient information on time period. |
| Was a case‐control design avoided? Yes: if a case‐control design was not used. No: if a case‐control design was used. Unclear: if there was insufficient information on study design. |
Prespecified threshold used? Yes: when the authors reported the use of 1, prespecified, cut‐off value. A prespecified threshold also included statements such as, "the test was scored according to manufacturer’s instructions." No: when multiple cut‐off values were tested and the best 1 chosen afterwards. Unclear: when only a cut‐off value was used, but this was not explicitly stated in the methods section. |
RS results interpreted without knowledge of the results of IT? Yes: when study reported that results of the RS were interpreted without knowledge of IT results or in cases when RS were used before the IT. No: when study reported that results of the RS were interpreted with knowledge of the IT results in cases when IT were used before the RS. Unclear: when there was insufficient information on when the IT and RS were interpreted. |
Number of participants receiving a RS, and included in the analysis? Yes: when the whole sample or a random selection of the sample or a selection of the sample with consecutive series received verification using a RS. No: when a part of the sample that was non‐randomly or non‐consecutively selected receives verification with the RS. Unclear: when there was no or insufficient information to ascertain if the whole sample or a random selection of the sample received verification with a RS. |
|
| Did the study avoid inappropriate exclusions? Yes: no participants were excluded after inclusion. No: for example, when specific participants were excluded (for example, those with mild disease because they are more difficult to detect). Unclear: if there was insufficient information on inclusion/exclusion criteria. |
Number of participants receiving same RS, and included in the analysis? Yes: when study participants were tested with the same reference standard RS regardless of index test result. No: when different RS were used. Unclear: when there was no or insufficient information the different RS used. |
|||
| Were all participants included in the analysis? Yes: when the participants who were included in the study, were also included in the analysis. No: when some participants/results were missing. Unclear: when there was no or insufficient information to make a judgement. | ||||
| Risk of bias (high, low, unclear) | Could the selection of participants have introduced bias? | Could the conduct or interpretation of the IT have introduced bias? | Could the RS, its conduct, or its interpretation has introduced bias? | Could the participant flow have introduced bias? |
| Applicability concerns (high, low, unclear) | Are there concerns that the included participants do not match the review question? High: if some included participants were not on ART. Low: if all participants were on ART. Unclear: if there was insufficient information to make a judgement. |
Are there concerns that the IT, its conduct, or interpretation differs from the review question? High if IT was not a true POC, i.e. required ancillary laboratory equipment or staff or testing done on frozen samples or if IT was not commercially available (a prototype). Low: if IT was a true POC and commercially available. Unclear: if there was insufficient information to make a judgement. |
Are there concerns that the target condition as defined by the RS does not match the review question? High if the RS threshold was not reported or if other thresholds used without justification. Low if the RS threshold was clearly reported as > 1000 copies/mL or > 5000 copies/mL. Unclear if there was insufficient information to make a judgement. |
— |
Scoring risk of bias assessment.
IT: index test; POC: point of care; RS: reference standard. | ||||
Contributions of authors
All review authors contributed to drafting the protocol, and approved the final protocol version.
Sources of support
Internal sources
Liverpool School of Tropical Medicine, UK.
External sources
-
World Health Organization (WHO), Switzerland.
- The WHO funded the preliminary findings of this review that were presented to the guideline development group meeting in Geneva in June 2015
-
Department for International Development (DFID), UK.
Project number 300342‐104
Declarations of interest
We presented preliminary findings of this review to the WHO Guideline Meeting Group in Geneva in June 2015.
EO has no known conflicts of interest.
AK has no known conflicts of interest.
SM received funding from the WHO to complete this review.
JD received funding from the WHO to complete the review and present it to the WHO Guideline Meeting Group.
New
References
Additional references
- African Society for Laboratory Medicine. Viral load monitoring in African HIV Treatment Programmes. Expert consultation; Cape Town South Africa; 2013 18‐20 April 2013. Available from http://fr.saafrica.org/viral‐load‐testing‐in‐african‐hiv‐treatment‐programmes/ (accessed prior to 1st October 2018).
- Drain PK, Hyle EP, Noubary F, Freedberg KA, Wilson D, Bishai W, et al. Evaluating diagnostic point‐of‐care tests in resource‐limited settings. Lancet Infectious Diseases 2014;14(3):239‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MP, Cutsem GV, Giddy J, Maskew M, Keiser O, Prozesky H, et al. IeDEA‐SA collaboration. Rates and predictors of failure of first‐line antiretroviral therapy and switch to second‐line ART in South Africa. Journal of Acquired Immune Deficiency Syndromes 2012;60(4):428‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishna G, Mustafa RA, Davenport C, Scholten RJ, Hyde C, Brozek J, et al. Applying Grading of Recommendations Assessment, Development and Evaluation (GRADE) to diagnostic tests was challenging but doable. Journal of Clinical Epidemiology 2014;67(7):760‐8. [DOI] [PubMed] [Google Scholar]
- Gopalakrishna G, Leeflang MM, Davenport C, Sanabria AJ, Alonso‐Coello P, McCaffery K, et al. Barriers to making recommendations about medical tests: a qualitative study of European guideline developers. BMJ Open 2016;6(9):e010549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schünemann H, Brożek J, Oxman A, editor(s). GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. gdt.guidelinedevelopment.org/app/handbook/handbook.html. The Grade Working Group, (accessed 26 September 2017).
- Havlir DV, Bassett R, Levitan D, Gilbert P, Tebas P, Collier AC, et al. Prevalence and predictive value of intermittent viremia with combination HIV therapy. JAMA 2001;286(2):171‐9. [DOI] [PubMed] [Google Scholar]
- Macaskill P, Gatsonis C, Deeks JJ, Harbord R, Takwoingi Y. Chapter 10: Analysing and Presenting results. In: Deeks JJ, Bossuyt PM, Gatsonis C, editor(s). Cochrane Handbook for Systematic Reviews of Diagnostic Test Accuracy Version 1.0.0. The Cochrane Collaboration, 2010. Available from srdta.cochrane.org.
- Pai NP, Vadnais C, Denkinger C, Engel N, Pai M. Point‐of‐care testing for infectious diseases: diversity, complexity, and barriers in low‐ and middle‐income countries. PLoS Medicine 2012;9(9):e1001306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitsma JB, Glas AS, Rutjes AW, Scholten RJ, Bossuyt PM, Zwinderman AH. Bivariate analysis of sensitivity and specificity produces informative summary measures in diagnostic reviews. Journal of Clinical Epidemiology 2005;58(10):982‐90. [DOI] [PubMed] [Google Scholar]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
- Ritchie AV, Ushiro‐Lumb I, Edemaga D, Joshi HA, Ruiter A, Szumilin E, et al. SAMBA HIV semiquantitative test, a new point‐of‐care viral‐load‐monitoring assay for resource‐limited settings. Journal of Clinical Microbiology2014; Vol. 52:3377‐83. [DOI] [PMC free article] [PubMed]
- Rutherford GW, Anglemyer A, Easterbroo, PJ, Horvath T, Vitoria M, Penazzato M, et al. Predicting treatment failure in adults and children on antiretroviral therapy: a systematic review of the performance characteristics of the 2010 WHO immunologic and clinical criteria for virologic failure. AIDS (London, England) 2014;28(Suppl 2):S161‐9. [DOI] [PubMed] [Google Scholar]
- SAS Institute Inc. SAS Software. Version 9.4. Cary (NC): SAS Institute Inc., 2013.
- Schito M, Peter TF, Cavanaugh S, Piatek AS, Young GJ, Alexander H, et al. Opportunities and challenges for cost‐efficient implementation of new point‐of‐care diagnostics for HIV and tuberculosis. Journal of Infectious Diseases 2012;205(Suppl 2):S169‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- StataCorp LLC. Stata Statistical Software: Release 15. College Station (TX): StataCorp LLC, 2017.
- Takwoingi Y, Guo B, Riley RD, Deeks JJ. Performance of methods for meta‐analysis of diagnostic test accuracy with few studies or sparse data. Statistical Methods in Medical Research 2017;26(4):1896‐911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UNAIDS. Fact sheet, 2015. www.unaids.org/sites/default/files/media_asset/20150901_FactSheet_2015_en.pdf (accessed prior to 12 September 2018).
- UNAIDS. Global AIDS update 2016. www.unaids.org/en/resources/documents/2016/Global‐AIDS‐update‐2016 (accessed prior to 12 September 2018).
- UNAIDS. Ending AIDS: progress towards the 90‐90‐90 targets. The Global AIDS update 2017. www.unaids.org/en/resources/documents/2017/20170720_Global_AIDS_update_2017 (accessed prior to 12 September 2018).
- UNITAID. HIV/AIDS diagnostics technology landscape – 4th edition, 2014. unitaid.org/assets/UNITAID‐HIV_Diagnostic_Landscape‐4th_edition.pdf (accessed prior to 12 September 2018).
- UNITAID. HIV/AIDS diagnostics technology landscape – 5th edition, 2015. www.unitaid.org/assets/UNITAID_HIV_Nov_2015_Dx_Landscape‐1.pdf (accessed prior to 12 September 2018).
- Whiting PF, Rutjes AW, Westwood ME, Mallett S, Deeks JJ, Reitsma JB, et al. QUADAS‐2: a revised tool for the quality assessment of diagnostic accuracy studies. Annals of Internal Medicine 2011;155(8):529‐36. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Antiretroviral therapy for HIV infection in adults and adolescents: recommendations for a public health approach revision, 2010. www.who.int/hiv/pub/arv/adult2010/en (accessed prior to 12 September 2018). [PubMed]
- World Health Organization. Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection. Recommendations for a public health approach, 2013. www.who.int/hiv/pub/guidelines/arv2013/en/ (accessed prior to 12 September 2018). [PubMed]
- World Health Organization. Technical and operational considerations for implementing HIV viral load testing: interim technical update, 2014. apps.who.int/iris/handle/10665/128121 (accessed prior to 10 September 2018).
- World Health Organization. Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: recommendations for a public health approach – 2nd edition, 2016. www.who.int/hiv/pub/arv/arv‐2016/en/ (last accessed 12 September 2018). [PubMed]
- Wu G, Zaman MH. Low‐cost tools for diagnosing and monitoring HIV infection in low‐resource settings. Bulletin of the World Health Organization 2012;90:914‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]