

Abstract
Cellular tubes have diverse morphologies, including multicellular, unicellular and subcellular architectures. Subcellular tubes are found prominently within the vertebrate vasculature, the insect breathing system and the nematode excretory apparatus, but how such tubes form is poorly understood. To characterize the cellular mechanisms of subcellular tube formation, we have refined methods of high pressure freezing/freeze substitution to prepare Drosophila larvae for transmission electron microscopic (TEM) analysis. Using our methods, we have found that subcellular tube formation may proceed through a previously undescribed multimembrane intermediate composed of vesicles bound within a novel subcellular compartment. We have also developed correlative light/TEM procedures to identify labeled cells in TEM-fixed larval samples. Using this technique, we have found that Vacuolar ATPase (V-ATPase) and the V-ATPase regulator Rabconnectin-3 are required for subcellular tube formation, probably in a step resolving the intermediate compartment into a mature lumen. In general, our ultrastructural analysis methods could be useful for a wide range of cellular investigations in Drosophila larvae.
KEY WORDS: Drosophila, TEM, Lumenogenesis, HPF, Freeze substitution, CLEM
Highlighted article: A previously undescribed intermediary membrane-lined multivesicular compartment is required for subcellular lumen formation in maturing Drosophila tracheal cells.
INTRODUCTION
Understanding how complex organ structure is generated within multicellular organisms is a major unsolved problem in developmental biology. A common form of tissue architecture is a network of cellular tubes, which functions in the transportation of liquids and gases throughout the body. The cellular structures of the tubes that make up these networks are diverse, with tubes made of multiple cells, tubes generated by single cells, and tubes that form on a subcellular level. Considerable progress has been made to understand the cellular processes that are used to form many of these different architectures (Iruela-Arispe and Beitel, 2013; Sigurbjörnsdóttir et al., 2014), but probably least well understood are the mechanisms used to form subcellular tubes, despite the fact that such tubes are a major component of the mammalian vasculature (Bär et al., 1984; Davis and Bayless, 2003).
We are dissecting the mechanisms of subcellular tubulogenesis by identifying the molecular and cellular processes required for the morphogenesis of a specific type of subcellular tube: those found within the insect breathing system. Insect respiratory systems, called tracheal systems, function as a combined lung and vasculature system (Maina, 2002). Diverse tube architectures are present within the tracheal system, including numerous subcellular tubes generated in a particular cell type, tracheal terminal cells (Guillemin et al., 1996; Manning and Krasnow, 1993). Terminal cells are an attractive system for characterizing subcellular tubulogenesis as we can use the genetic and cellular tools available in Drosophila melanogaster to study their development.
Terminal cells initiate morphogenesis during embryonic stages by extending a single, thin, non-branched process from their distal ends (Gervais and Casanova, 2010; Samakovlis et al., 1996). During larval stages, in response to hypoxia in surrounding tissues and within the cell itself (Centanin et al., 2008; Jarecki et al., 1999), terminal cells undergo extensive morphogenesis during which the single embryonically generated process undergoes iterative outgrowth and branching events, eventually producing a tree-like network emanating from each cell. By the end of the final larval stage, a typical terminal cell has between 30 and 100 subcellular branches, the longest of which extends over 200 µm (Fig. 1A). Although the diameter of the branches does increase slightly during larval development, they remain very thin, with diameters ranging from 0.8 to 2 µm.
Fig. 1.

Drosophila tracheal terminal cell structure observed by light microscopy. (A-C′) Fluorescence (A,C) and brightfield (B,C′) images of a single, GFP-labeled Drosophila tracheal terminal cell in an L3 larvae. The fluorescence images show the pattern of cell branches and the brightfield image highlights gas running within the branch subcellular lumens; this contrasts strongly owing to the refractive index difference between the gas- and fluid-filled tissues. Arrows indicate examples of proximal portions of newly outgrown branches that are not yet gas-filled. The cell is joined at its proximal end (arrowhead) with the rest of the tracheal system. As a mosaic approach was used to label the single cell with GFP, the adjoining cell does not show fluorescence; however, its gas-filled lumen is observable in the brightfield image and is continuous with the terminal cell lumen (inset in B). (C,C′) Close up of a portion of the terminal cell indicated by the dashed boxes in A and B, highlighting the relationship between a cytoplasmic branch; a gas-filled portion of lumen; a non-gas-filled portion of lumen, which is just visible by brightfield microscopy; and the more distal portion of the branch in which there is no visual evidence of a lumen. Scale bars: 50 µm (A,B); 10 µm (C,C′).
While they are growing, terminal cell branches are filled with cytoplasm. To function in respiration, these nascent branches must undergo a process of tubulogenesis, forming a membrane-bound intracellular lumen of ∼500 nm in diameter. Inside the membrane, the lumen is lined with an elaborate cuticle organized into helical ridges known as taenidia (larger taenidia are also observed in other tube types within the tracheal system and are highly characteristic of insect trachea) (Noirot and Noirot-Timothee, 1982). Like the cuticle covering the larval surface, the tracheal cuticle consists of a layer of protein and lipids (epicuticle) overlying a layer comprised primarily of polymerized chitin (procuticle). Once mature, the subcellular tubular branches emanating from terminal cells are referred to as tracheoles.
When first formed, the lumen is filled with liquid, but at each larval molt this liquid, along with old cuticular lining, is excreted, a new cuticle is formed, and the lumen is filled with gas (Camp et al., 2014; Snelling et al., 2011). The presence of a mature, gas-filled lumen can be monitored by brightfield microscopy, by which the gas-filled tube contrasts strongly with the surrounding fluid-filled tissues (Fig. 1B,C′). The mechanism of gas filling in terminal branches is not well understood, but in terminal cells mutant for genes involved in chitin synthesis, gas filling does not occur (Ghabrial et al., 2011), suggesting that normal cellular architecture is required for either gas production or for supplying gas to the lumen.
Investigation into the cellular and molecular mechanisms required to form the terminal cell subcellular lumen is a field of active research. The predominant model suggests that the luminal membrane forms by processes of vesicle trafficking. This model specifically proposes that vesicles are generated within the terminal cell cytoplasm, move to the center of each subcellular branch, and undergo fusion to form the continuous luminal membrane (Lubarsky and Krasnow, 2003). A role for vesicle trafficking in terminal cell lumen formation is bolstered by observations showing that a number of vesicle fusion and trafficking genes are required for the lumen formation process (Baer et al., 2013; Ghabrial et al., 2011; Jarecki et al., 1999; JayaNandanan et al., 2014; Jones et al., 2014; Schottenfeld-Roames and Ghabrial, 2012; Schottenfeld-Roames et al., 2014). However, the sources of the vesicles required for lumen formation are not known, nor have vesicles been directly observed fusing to form the mature membrane. Least well understood are the identities of the molecules that function to drive lumenogenesis in naïve branches.
One major impediment to analysis of subcellular tubulogenesis in terminal branches is the small size of the structures involved. At ∼500 nm, the subcellular lumen is smaller in diameter than most bacteria, and close to the resolution limit of conventional light microscopy (∼200 nm under optimal conditions). Here, we investigate membrane trafficking during early tracheal terminal cell lumen formation by using transmission electron microscopy (TEM) to analyze cells in developing tracheoles at the ultrastructural level. To do this, we have adapted high pressure freezing (HPF)/freeze substitution (FS) techniques for Drosophila larvae. HPF/FS is a procedure involving fixation of tissues by rapid freezing while subjecting the samples to high pressures, thus preventing the formation of tissue-disruptive water ice crystals (McDonald, 2009). Water in the frozen specimens is then substituted with other solvents, such as ethanol or acetone, thus avoiding water ice crystal damage that would occur while returning the samples to ambient temperatures. Finally, the fixed material is embedded in resin for sectioning and observation by TEM. HPF is considered to be superior to chemical cross-linking because of the preservation of cellular membranes in their native state (Hurbain and Sachse, 2011; Studer et al., 2008). Using our HPF/FS technique, we have obtained evidence that intracellular lumen formation is not driven by direct fusion of cytoplasmic vesicles, but instead goes through an intermediate comprising a large membrane-bound cellular compartment, within which vesicles appear to assemble the nascent lumen.
Additionally, we have developed a modified version of our HPF protocol in which fluorescent-protein markers are maintained through the fixation and embedding procedures, allowing us to perform correlative light and electron microscopy (CLEM). This protocol is particularly useful for identifying cells in mosaic mutant animals, in order to analyze them by TEM. We use our CLEM procedure to examine terminal cell ultrastructure in cells mutant for Rabconnectin-3 alpha (Rbcn-3A), which are unable to form functional lumens. We have found that Rbcn-3A-mutant terminal cells contain multi-membranous subcellular structures in place of the mature lumen, further indicating that subcellular lumen formation goes through a complex membrane intermediate. Rbcn-3 functions to regulate the activity of Vacuolar ATPase (V-ATPase), an ATP-dependent proton pump that performs numerous roles within the cell, particularly in acidification of subcellular compartments, such as endosomes, lysosomes and synaptic vesicles (Forgac, 2007). We find that terminal cell ultrastructure when V-ATPase is partially knocked down phenocopies the loss of Rbcn-3A. Our results suggest that V-ATPase-mediated acidification could be a crucial step in resolution of a multi-membranous luminal precursor into a mature, functional lumen.
RESULTS
Optimization of high pressure freezing/freeze substitution for Drosophila larvae
Nearly all terminal branch tubulogenesis events in Drosophila take place during larval development (Jarecki et al., 1999). Owing to the small diameter of terminal cell branches, it is necessary to use electron microscopy (EM) to characterize the membrane movements required for lumen formation. Previous ultrastructural studies of tracheal development in larvae have been carried out using chemical fixation of dissected samples (Levi et al., 2006). However, such fixation techniques are known to result in a number of artifacts. In particular, intracellular membranes, the focus of our investigation, are frequently distorted by chemical fixation procedures (Murk et al., 2003). Additionally, the tracheal system is dispersed in the body of the animal, so dissection entails widespread disruption of trachea. Terminal cell branches, which strongly adhere to internal tissues, are particularly sensitive to dissection.
To overcome these difficulties, we adapted the technique of HPF/FS to intact Drosophila larvae (McDonald, 2014). HPF/FS has previously been performed in Drosophila embryonic stages and in dissected tissues from larvae and adult animals (Jiao et al., 2010; Kolotuev et al., 2010; McDonald et al., 2012; Moussian et al., 2006; Zhang and Chen, 2008). Use of HPF/FS in intact larval stages has been carried out (Kolotuev et al., 2010; Moussian and Schwarz, 2010), but systematic identification of optimal conditions has not been previously reported. Using our optimized conditions (see Materials and Methods), we can reliably produce samples embedded in epoxy resins that have excellent preservation of cell membranes, including the plasma membrane, nuclear envelope, and organelles, such as mitochondria and endoplasmic reticulum (Fig. 2). Other subcellular structures, such as nuclear pores, cytoskeleton, ribosomes and chromatin are also well preserved (Fig. 2). Of particular note is preservation of the cuticle, including both the protein/lipid epicuticle and the chitinous procuticle (Fig. 2B). Apical microvilli on epidermal cells, which are thought to function in cuticle deposition (Locke, 1991), can also be observed; such microvilli are generally lost using chemical fixation techniques (Galko and Krasnow, 2004).
Fig. 2.
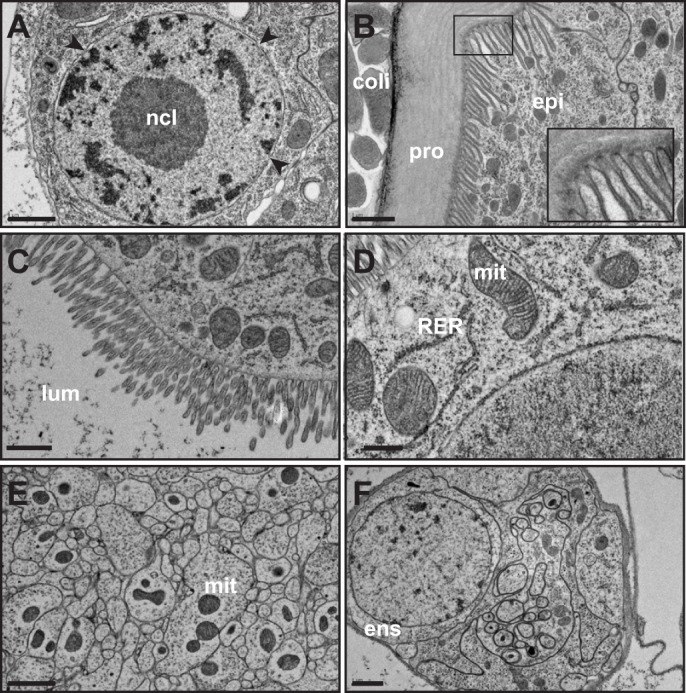
Examples of structure preservation using high pressure freezing and freeze substitution fixation in Drosophila larvae. (A) Cell nucleus. The nuclear membrane appears smooth, indicative of minimal tissue shrinkage and protein aggregation during fixation. Heterochromatin (dark staining) and euchromatin (light staining) are distinct and nuclear pores (arrowheads) are preserved. ncl, nucleolus. (B) Larval external cuticle and epidermal cell (epi). Cuticular chitin (procuticle, pro) shows layers characteristic of chitin microfibrils arranged in helical stacks of parallel fibers (Moussian et al., 2006). The epidermal cells have many apical microvilli, thought to be important for the orientation the chitin fibrils (Locke, 1991). Inset: close up of microvilli showing the darkly staining tips that are the proposed site of chitin biosynthesis. External to the animal, Escherichia coli cells, used as a loading medium for HPF are visible (coli). (C) Gut brush border showing microvilli. lum, gut lumen. (D) Mitochondria (mit) and rough endoplasmic reticulum (RER) in gut cells. Mitochondria morphology is well preserved as evidenced by intact cristae. (E) Nerve bundle. Mitochondria (mit) are visible in some neurites. (F) Ensheathed nerve bundle and ensheathing cell (ens). Scale bars: 1 µm (A-C,E,F); 0.5 µm (D).
Ultrastructural analysis of developing tracheoles in HPF/FS-fixed Drosophila larvae
Wild-type tracheoles display a highly consistent morphology using HPF/FS (Fig. 3A,B; illustrated diagrammatically in Fig. 3C). Tracheoles are readily identified based on morphology, position and, in particular, a lumen lined by cuticle arranged into taenidial folds. Tracheoles either run along tissues, commonly muscles and epidermal cells for superficial terminal cells (Fig. 3B), or traverse the haemolymph, the fluid that fills the insect internal body cavity (Fig. 3A). A thick basement lamina covers the tracheolar basal membrane where the branch is not in direct contact with tissues (Fig. 3A); this lamina appears to be absent when the branch is in direct apposition to tissues. The subcellular lumen is located centrally within the cytoplasm of the branch and is observed to be surrounded by an approximately circular apical membrane (which stains weakly in our fixation procedure). Distinct from other types of tracheal tubes, tracheolar tubes are seamless and thus show no membranes extending between the basal and apical plasma membranes, nor are intracellular junctions present. Within the lumen, taenidial folds are well preserved and are particularly obvious when examined in longitudinal section (Fig. 3B). Taenidial ridges in tracheoles are ∼50 nm in height and spaced ∼200 nm apart. They show the multiple layers typical of cuticle, with a darkly stained epicuticle, presumably composed of lipids and proteins, overlying a more weakly stained procuticle chitin layer (Fig. 3B, inset). The epicuticle appears to be of a relatively consistent thickness along the tracheal lumen, with taenidial ridging as a result of variation in procuticle thickness. The branch cytoplasm is rich in microtubules running along the axis of branch extension (Fig. 3A,B). Although there is evidence that actin matrices underlie organization of both the basal and apical plasma membranes in terminal cells (Gervais and Casanova, 2010; JayaNandanan et al., 2014; Levi et al., 2006; Schottenfeld-Roames et al., 2014; Ukken et al., 2014), we do not observe such matrices, probably because our fixation methods do not preserve these particular cytoskeletal elements. The features we observe are consistent with previous reports of tracheolar ultrastructure, observed in Drosophila and other insects fixed using standard chemical techniques, but with an increased resolution of cellular membranes (Levi et al., 2006; Manning and Krasnow, 1993; Noirot and Noirot-Timothee, 1982; Snelling et al., 2011).
Fig. 3.
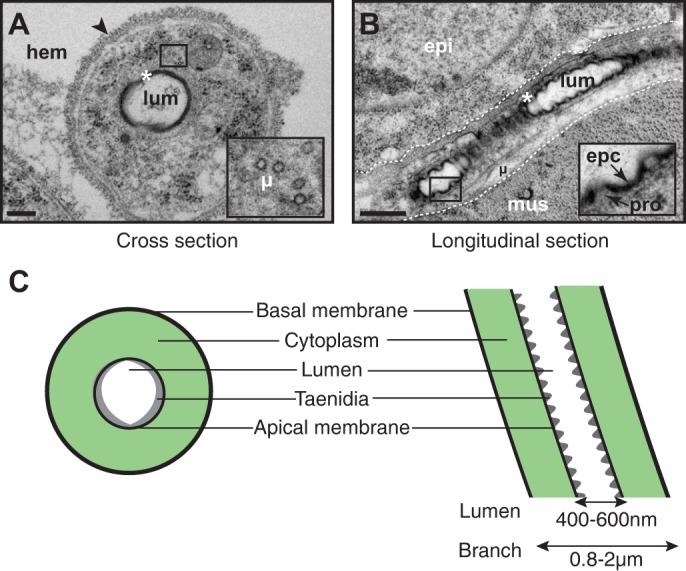
Tracheole ultrastructure in HPF-fixed/freeze substituted larvae. (A-C) Mature tracheole in cross-section (A), longitudinal section (B; tracheole is demarked by dashed lines), and illustrated diagrammatically (C). Insets are higher magnifications of the boxed regions in A and B. Asterisks, taenidia; arrowhead, basal lamina; epc, epicuticle; epi, epidermal cell; hem, hemolymph; µ, microtubules; mus, muscle; pro, procuticle. Scale bars: 200 nm (A); 500 nm (B).
Identification of candidate luminal intermediates in developing tracheoles
To identify immature terminal cell branches, we had to rely on characteristics independent of a mature lumen. The first characteristic is that terminal branches are elongated, and thus traverse many planes of section; second, the branches are circular in cross-section and 500-1000 nm in diameter; third, they are enriched in microtubules running in parallel to the long axis; and fourth, when not tightly apposed to other tissues, they possess a thick basement lamina. Using these criteria, we could identify tracheoles in which lumens had not yet fully matured.
Based on current models, described above, we expected to observe cytoplasmic vesicles accumulating in the center of immature branches where the luminal membrane will be formed. However, we were unable to identify such accumulations in any of our sections. What we did observe were unusual membrane-circled subcellular structures of ∼300 nm diameter (Fig. 4A,B). Ultrathin serial sectioning revealed these structures to be cross-sections of discrete, slightly elongated (∼400 nm in length) cellular compartments (Fig. 4C,D). These compartments contained a variety of other structures, most obviously a membrane-bound subcompartment of ∼200-300 nm in diameter (Fig. 4A,C), or a folded, darkly staining lamina, reminiscent of the cuticular lining found in mature tracheolar lumens (Fig. 4B), and multiple, small vesicles with diameters of 40-60 nm, a size typical for transport vesicles (Fig. 4A-C).
Fig. 4.

Characterization of candidate subcellular lumen precursors. (A,B) Examples of immature tracheoles with luminal precursors. The precursor appears as a membrane (asterisk) delimiting a compartment of ∼400 nm diameter and contains a number of ∼50-100 nm vesicular substructures (arrowheads in insets). In A, a larger vesicular structure is observed (arrow). In B, cuticle with rudimentary taenidial folds appears to be forming (arrow). (C) Serial sections (1-8; 50 nm) of a luminal precursor in the terminal cell shown in A. The compartment forms a slightly elongated spheroid, ∼300 nm in diameter and 400 nm long. (D) Diagrammatic representation of luminal precursor. Gray is the terminal branch plasma membrane; green is the outer bounding membrane of the precursor compartment; red are membranous structures within the compartment. Scale bars: 200 nm.
Based on their size, position, and presence only in lumenless (thus immature) tracheoles, we propose that the membrane-bound compartments are nascent lumens developing within tracheoles. This proposal suggests an alternative model of subcellular lumen formation: tracheole luminal membrane formation proceeds not by a process of direct fusion of cytoplasmic vesicles but rather by lumen assembly and maturation within a larger, membrane-bound, subcellular compartment.
Development of correlative light-electron microscopy
A significant hurdle in characterizing cell ultrastructure in a multicellular organism is the identification of cells of interest within embedded samples or sections. In particular, as specific ultrastructure is typically used to identify a specific cell type, locating cells for which ultrastructure has been disrupted by mutation or other perturbations can be a considerable challenge. A related problem is in locating one specific cell within a field of cells of similar morphology. These hurdles are typified in experiments using organisms such as Drosophila, in which it is possible to generate genetically mosaic individuals that contain small numbers of mutant cells within otherwise wild-type animals. When using light microscopy, specific cells can be identified by expression of cell-autonomous visible markers, such as fluorescent proteins. However, although such markers are very effective for cell identification, they cannot be directly observed using EM. As an adjunct to visible markers, it is possible to use transgenes that encode for enzymes, such as horseradish peroxidase, activity of which can be used to produce depositions of electron-dense material (Watts et al., 2004). Additionally, it has been demonstrated that stimulation of fluorescent proteins can be used to generate reactive oxygen species, which can then be harnessed to deposit electron-dense material locally (Grabenbauer et al., 2005; Shu et al., 2011). In both cases, electron-dense stains can be directly observed by EM, and thus can be used for identification of cells of interest. However, such enzymatic and photoconversion techniques have the disadvantage of requiring considerable sample processing under conditions that maintain enzymatic and/or fluorescent protein activity. Such conditions can lead to significant ultrastructural artifacts, and are also generally not compatible with HPF/FS methods of sample fixation.
An alternative to using enzymatic or fluorescence-based methods to deposit electron-dense stains is the use of fixation procedures that maintain activity of expressed fluorescent proteins (Brown et al., 2009). In such experiments, the visible label serves two purposes. First, the fluorescent signal allows labeled cells to be identified in embedded samples, facilitating sectioning at appropriate locations. This is a major advantage in identifying cells of interest within large samples. Second, specific cells can be identified within sections and then directly imaged by EM to determine ultrastructure. Using such CLEM techniques allows for definitive identification of a particular cell or tissue types in samples in which it would be hard to identify the cells by ultrastructural criteria alone. In particular, CLEM is very well suited for identifying homozygous mutant cells in genetically mosaic organisms in which mutant cells are specifically labeled with fluorescent markers.
CLEM has been performed in chemically fixed Drosophila larval samples (Urwyler et al., 2015; Vasin et al., 2014) and HPF-fixed embryos (Fabrowski et al., 2013), but not previously in HPF/FS-fixed larvae. Therefore, we adapted an HPF/FS CLEM protocol developed by Watanabe et al. for Caenorhabditis elegans (Watanabe et al., 2010). This protocol uses low concentrations of osmium tetroxide along with potassium permanganate as a fixative and has been shown to maintain activity of a number of fluorescent proteins. After extensive optimization (see Materials and Methods), we established conditions in which DsRed protein fluorescence was maintained and membrane structure effectively preserved (Fig. 5A-C; Fig. S1). Although ultrastructural preservation is not quite as good as our HPF/FS/epoxy resin method, we still obtained high resolution images of cell and organelle membranes, as well as other cellular structures. Importantly for our analysis, tracheal terminal cell morphology was well preserved in our CLEM procedure (Fig. 5C), and when we used the MARCM (mosaic analysis with a repressible cell marker) positive-marking system (Lee and Luo, 1999) to generate mosaic larvae in which homozygous mutant cells were labeled with DsRed in an otherwise unlabeled animal, we found these homozygous DsRed-labeled cells were easily visible in embedded samples and sections (Fig. 5D-F), demonstrating the utility of our CLEM procedure for characterizing mutant cells in genetic mosaics.
Fig. 5.

Examples of CLEM in Drosophila larvae. (A) Fluorescent image of an L2 Drosophila larva expressing DsRed throughout the tracheal system that has been fixed and embedded for EM (the animal is within the resin block). (B) Thin section from tracheal DsRed-expressing larva observed using total internal reflection (TIRF) microscopy. Cross-sections of labeled tracheoles appear as small ovals (labeled 1-4). (B′) EM image of the neighboring section to that shown in B showing the correlation between DsRed signal and tracheolar profiles. (C) Close up of tracheolar profile (number 4 in B′). This particular section is through a branch point and two lumens are observed (a, b). Arrows, taenidial folds; arrowheads, luminal membrane; asterisks, rough endoplasmic reticulum . (D) Mosaic Rbcn-3A11L L1 larva with homozygous mutant cells labeled with DsRed. (E) Brightfield image of the same larva as in D. (F) The area boxed in D and E is shown as a close-up merged image of labeled terminal cells (pseudocolored red) with brightfield image. Note that as this is an earlier stage larva, terminal cell branches have not yet grown to the 100-200 µm lengths observed in the L3 larva, as shown in Fig. 1. Scale bars: 0.5 mm (A); 5 µm (B,B′); 500 nm (C); 100 µm (D,E); 30 µm (F).
Characterization of mutants with defects in intracellular lumen development using HPF/FS and CLEM
To test our HPF/FS and CLEM techniques and to gain further insight into terminal cell lumen formation, we used our procedures to analyze mutants with defects in terminal cell lumen formation. One such class of mutants are those affecting the Rabconnectin-3 complex, a heterodimer of the proteins Rabconnectin-3α and Rabconnectin-3β (Rbcn-3A and Rbcn-3B in Drosophila) (Kawabe et al., 2003; Nagano et al., 2002). Preliminary data indicated that loss of Rbcn-3A in terminal cells leads to defects in terminal cell lumen formation (M.M.M. and M. A. Krasnow, unpublished). To analyze the role of Rbcn-3 in tracheal terminal cells, we used MARCM to generate fluorescent protein-marked homozygous mutant terminal cells in otherwise unmarked, heterozygous animals; using this genetic mosaic approach bypasses the embryonic lethality associated with Drosophila Rbcn-3 mutations (Yan et al., 2009). We found that terminal cells homozygous for a putative null allele of Rbcn-3A, 11L, have mostly normal outgrowth and branching (Fig. 6A), but do not form a gas-filled lumen (Fig. 6A′). Rbcn-3A is specifically required for terminal cell lumenogenesis: unicellular tubes and fusion cells (toroidal tracheal cells that join adjacent tracheal segments) homozygous for Rbcn-3A11L appear completely normal (Fig. 6B,B′) and individual Rbcn-3A11L homozygous mutant cells in the multicellular tracheal dorsal trunks appear indistinguishable from wild-type cells (data not shown). We found that other Rbcn-3A alleles, obtained in a screen for mutations affecting embryonic development (Yan et al., 2009), have very similar or identical phenotypes to 11L (Fig. S2A,A′; data not shown). From these experiments, we conclude that Rbcn-3A is required for normal tracheolar lumen formation, but is not required for other tracheal tube architectures. Essentially identical results were obtained when we analyzed mutants for Rbcn-3B (Fig. S2B,B′). Thus, we conclude that the heterodimeric Rbcn-3 complex is required for terminal cell lumen formation. The Rbcn-3 defects are 100% penetrant in that all mutant terminal cells show a complete lack of a mature lumen in tracheoles. In some cells, a portion of gas-filled lumen can be observed in the proximal portion of the cell where it joins the rest of the tracheal system, probably representing compensatory growth of the neighboring unicellular tube (Francis and Ghabrial, 2015).
Fig. 6.

Rbcn-3A is required for subcellular lumen formation in tracheal terminal cells. (A-B′) Fluorescence (A,B) and brightfield (A′,B′) images of homozygous mutant DsRed-labeled Rbcn-3A11L tracheal cells in mosaic L3 larva. Rbcn-3A11L terminal cells have grown and branched extensively (A), but no contrasting gas-filled lumen is observed (A′). Dashed line indicates where the mutant cell joins other cells of the tracheal system. (B,B′) Homozygous mutant unicellular tracheal branch (arrow) and fusion cell (asterisk) appear normal, and contain a gas-filled lumen contiguous with neighboring (unlabeled) tracheal cells. Arrowheads indicate the junctions between mutant and non-mutant cells. (C-D′) EM analysis of a Rbcn-3A11L mutant terminal cells reveal a structure in the position typical of a lumen, but instead contain multiple disorganized membranes, but no cuticle nor a cleared lumen. (C) Sample fixed using CLEM procedure. (D) Sample fixed using HPF/FS procedure. (D′) Higher magnification view of the defective lumen (boxed region in D). (E) EM analysis of tracheole with V-ATPase component Vha68-2 inhibited by bs500-GAL4-driven RNAi. Scale bars: 100 µm (A); 50 µm (B); 250 nm (C,D,E); 150 nm (D′).
Next, we generated mosaic larvae in which Rbcn-3A mutant cells were marked by expression of DsRed and fixed these larvae using our CLEM protocol (Fig. 5D-F). We cut sections from these fixed larvae using the fluorescent signal to identify the appropriate position of sectioning, identified DsRed expressing profiles on the sections, and imaged these profiles using TEM. We found that Rbcn-3A mutant cells show an abnormal, multi-membranous structure in place of the normal lumen (Fig. 6C). Next, because our regular HPF/FS protocol preserves ultrastructure better than the CLEM technique, we examined Rbcn-3A mutant cells using HPF/FS. To find mutant cells without a TEM-detectable label within the otherwise wild-type animals, we generated MARCM-mosaic animals, identified mutant cells by fluorescence, and noted the position of the labeled cell within the animal by recording the distance of the cell from the anterior and posterior ends, the dorsal and ventral surfaces, and the left and right sides of the larvae, before fixation. Mosaic animals were then fixed and processed individually, so that we could keep track of the position of the mutant cell within each specimen. The fixed samples were then sectioned at the approximate position of the mutant cell and sections examined by TEM for tracheolar profiles that appeared similar to the morphology observed in our CLEM samples. This analysis confirmed that Rbcn-3A mutant terminal cells contain abnormal membranous structures in the positions typically occupied by the lumen. These abnormal structures typically appeared as multiple membranous swirls (Fig. 6D,D′; Fig. S3A), but also appeared as multiple vacuoles separated from the cytoplasm by a limiting membrane (Fig. S3A). The abnormal membrane structures were never distributed across the width of the branch, but always occupied a discrete, membrane-bound region approximately in the middle of the branch.
As a further confirmation that the cells observed in both the HPF/FS experiments were indeed Rbcn-3A mutants, we used the MARCM system to generate mutant cells co-expressing green fluorescent protein (GFP) and membrane-tethered HRP (HRP::CD2) (Watts et al., 2004). HRP activity was used to specifically label mutant cells with an electron dense DAB/nickel stain. Although this technique has been used previously to identify mutant tracheal terminal cells (Levi et al., 2006), owing to the conditions required to preserve HRP activity, it tends to result in artifacts, such as abnormal vacuolation of the cell cytoplasm. Despite these artifacts, our analysis clearly revealed that mutant tracheoles contain multi-membrane structures in place of the normal lumens found in wild-type cells (Fig. S3B), confirming our identification of Rbcn-3A mutant terminal cells in our HPF-fixed samples.
V-ATPase is required for intracellular lumen formation
Rabconnectin-3 functions in regulated assembly of V-ATPase (Einhorn et al., 2012; Seol et al., 2001; Sethi et al., 2010; Yan et al., 2009). One prediction from our analysis of Rbcn-3 is that V-ATPase would be required for intracellular lumen formation. In addition, a recent report analyzing two V-ATPase subunits, Vha13 and Vha26, has directly shown a role in maturation of the intracellular lumen through regulation of cell polarity (Francis and Ghabrial, 2015). To test whether loss of the V-ATPase phenocopies the ultrastructural defects we found in Rbcn-3A mutant terminal cells, we first attempted to make terminal cells mutant for V-ATPase subunit genes VhaAC39-1 and Vha55 using the MARCM system. However, we found that mutant cells were present very infrequently, suggesting a cell survival defect, and even surviving homozygous mutant terminal cells had extremely severe outgrowth and branching defects, making assessment of lumen formation impossible.
Next, we used RNA interference (RNAi) to downregulate the V-ATPase gene Vha68-2, which encodes the only V-ATPase V1 A subunit prominently expressed in larvae (Allan et al., 2005), throughout the tracheal system with the strong btl-GAL4 driver (Shiga et al., 1996). However, we found that this resulted in early larval lethality. To bypass this lethality, we developed a new terminal cell-specific driver, bs500-GAL4, with weaker expression than btl-GAL4 (see Materials and Methods; Fig. S4A-C). When we expressed Vha68-2 RNAi using bs500-GAL4, we found that although terminal cell development was reduced and abnormal, there was significantly more growth and branching than in cells in which Vha68-2 RNAi was driven by btl-GAL4 (Fig. S4D). Importantly, in bs500-GAL4>Vha68-2 RNAi cells, lumen formation, as judged by brightfield microscopy to observe gas-filled lumens, was highly defective, with lumens only visible in the central cell branch, but completely absent in any side branches (Fig. S4E,F).
We next performed ultrastructural analysis of bs500-GAL4>Vha68-2 RNAi terminal cells, using our HPF/FS technique (because all terminal cells were defective in these animals, we did not have to use CLEM to identify specific cells). We found that terminal cell branches expressing Vha68 RNAi showed defective lumen development with what appeared to be nascent lumens filled with swirls of membranes (Fig. 6E), highly reminiscent of those observed in Rbcn-3 mutant terminal cells. These data suggest that the role for Rbcn-3 in terminal cell lumen formation is in regulation of V-ATPase activity.
DISCUSSION
The ability of light microscopy to resolve cell ultrastructure has been dramatically improving with the development of sub-diffraction limit methods of imaging (Fornasiero and Opazo, 2015; Requejo-Isidro, 2013), but for most subcellular structures EM still affords significantly higher resolving power. A limitation for EM studies, however, is the challenge of preserving tissues in as native a state as possible during the processing needed for making them observable in the electron microscope. Furthermore, the identification of specific subcellular structures during EM imaging is difficult, especially in large complex samples, such as dissected tissues or entire multicellular organisms. In this paper, we report on two methods that can be used to overcome these experimental limitations in Drosophila larvae. First, we have identified conditions for high pressure freezing and freeze substitution that lead to excellent ultrastructural preservation and that can be applied to intact larvae. Second, we have identified EM fixation conditions in which reporter protein fluorescence is maintained. This fluorescence allows specific cells to be identified, and has proven to be particularly useful for analysis of genetic mosaics in which homozygous mutant cells are engineered to express a fluorescent protein but wild-type cells remain unlabeled.
Using our EM fixation methods, we have studied the development of tracheal terminal cells and have identified a novel multimembrane subcellular structure that appears to be a precursor to the mature subcellular lumen. A number of lines of evidence support this interpretation. First, we only observed such multimembrane structures within immature tracheal terminal branches. Second, the structures do not form a continuous domain in tracheal branches, as would be expected for a mature lumen, including lumens in molting animals (Snelling et al., 2011). Third, we have analyzed three genes (Rbcn-3A, Rbcn-3B and Vha68-2) that are required for subcellular lumen formation and find that cells defective for any of them accumulate structures with multiple membrane layers within the terminal cell branches, suggesting that resolving a multimembrane intermediate is a key step in subcellular lumen formation. We propose a new model for intracellular lumen formation that involves formation and resolution of a multimembrane intermediate (Fig. 7). Intriguingly, EM images of early steps in the development of the C. elegans excretory cell show a structure with striking similarity to the multimembrane compartment we have identified (Berry et al., 2003). Furthermore, loss of the V-ATPase leads to severe defects in tubulogenesis and other morphogenetic processes within the excretory cells (Hahn-Windgassen and Van Gilst, 2009; Liégeois et al., 2007). Thus, the processes we have identified in Drosophila terminal cell branches might be a general feature of subcellular lumen formation.
Fig. 7.

Model for lumen formation in terminal cells. Green represents intracellular (cytoplasmic) topology; blue represents extracellular topology; purple represents cuticle. We propose that in terminal cells a pinocytic mechanism forms a compartment with extracellular topology, within the cell. Cytoplasmic vesicles containing chitin precursors bud into this compartment, and chitin is secreted into the compartment from the vesicles. This is followed by acidification of the compartment by Rbcn-3-dependent V-ATPase activity, triggering breakdown of the membranes of the cytoplasmic vesicles and cuticle assembly. Further processes to make the mature lumen include fusion of precursor compartments to make a continuous tube, and clearing of the liquid contents and gas filling. By contrast, in unicellular tubes, which form by a cell-wrapping process, chitin is secreted directly from the cytoplasm into a previously existing extracellular space. Rbcn-3 is not required for lumen formation in these tubes.
We do not yet understand how the compartment initially forms during tracheole development, but one interesting possibility is that it is generated by a pinocytic mechanism, which has also been proposed to be a key step in intracellular lumen formation in endothelial cells (Davis and Bayless, 2003). One attractive feature of this model is that the lumen of a pinocytic compartment is topologically equivalent to the outside of the cell (Fig. 7), and this may be key to generating tracheolar cuticle. Chitin, the major constituent of cuticle, is polymerized from soluble monomer precursors by transmembrane chitin synthases (Merzendorfer, 2011). The active site of chitin synthase faces the cytoplasm, and polymerized chitin is excreted through channels (possibly formed by chitin synthase itself) out of the cell. There, the individual chitin chains are assembled into microfibrils, and the microfibrils then organize into laminar arrays, creating the chitin sheets that are part of the cuticle. These final assembly steps are thought to require an extracellular environment to occur; as described above, such an environment would be the natural outcome of a pinocytic mechanism forming a luminal precursor compartment.
As an extension to this model, the vesicles found in the novel subcellular compartment are predicted to contain chitin precursor molecules, chitin synthase, and chitin excretion machinery; chitin would be polymerized within the vesicles, excreted into the compartment lumen, and then undergo the final steps of assembly and deposition to line the lumen (Fig. 7). Intriguingly, vesicles, called chitosomes, with very similar properties of chitin and assembly machinery storage are known to exist in most fungi (Bartnicki-Garcia, 2006). A final, interesting possibility is that the positions of vesicle entry into the compartment are spatially regulated so as to organize the taenidia; this spatial regulation could be accomplished by the actin cytoskeleton, as has been described for taenidia formation in other tracheal branches (Matusek et al., 2006).
The observation that subcellular lumen formation proceeds through a multimembrane intermediate leads to a significant question: how are the multiple membranes resolved into a single membrane observed lining the mature lumen? We propose that this is the role played by Rbcn-3-regulated V-ATPase activity. This is analogous to the requirement for V-ATPase in lysosome function. So-called multivesicular bodies (MVBs) consist of a limiting membrane containing numerous transport vesicles that contain endocytosed proteins and other macromolecules (Babst, 2006). To recycle these macromolecules, MVBs fuse with lysosomes, thus exposing the vesicles to the lysosomal lumen, which is acidic as a result of the activity of V-ATPase. The low pH allows for the activity of acid-dependent hydrolases stored in the lysosome, including lipases responsible for breakdown of the membranes and contents of the vesicles. We propose a related function for the V-ATPase in intracellular lumen formation: acidification of the multimembrane subcompartment activates lipases that break down the internal membranes, leaving the single membrane observed in the mature lumen (Fig. 7). In the absence of V-ATPase activity the internal membranes persist, resulting in the occluded lumens observed in mutants. It is interesting to note that although V-ATPase is generally required for tracheal cell development (Francis and Ghabrial, 2015), Rbcn-3 has a much more specific role in only being required for terminal cell lumen formation. This suggests that Rbcn-3 controls stage- and process-specific activity of V-ATPase; such stage specificity of V-ATPase activity by regulated assembly has been previously described for other aspects of insect physiology (Sumner et al., 1995). Finally, although acidification of the compartment is our favored model for the requirement for V-ATPase in lumen formation, it is important to point out that V-ATPase is required for multiple trafficking processes within cells (Marshansky and Futai, 2008), as well as having a direct function in membrane fusion (Bayer et al., 2003), so the defects we observe on intracellular lumen formation might be due to indirect effects on general membrane movement throughout the cell.
Although we developed our EM methods for studying the development of tracheal terminal cells, these methods could be applied to the analysis of any larval tissue of interest. The Drosophila larva has become an important model for studying diverse aspects of physiology and behavior, including metabolism (Palanker et al., 2009), wound healing (Galko and Krasnow, 2004), locomotion and feeding (Schoofs et al., 2014), and sensory perception (Zhang et al., 2013). Studies of any of these processes could benefit from ultrastructural analysis. For instance, the function and development of peripheral sensory dorsal arborization neurons has become a choice genetic model for understanding dendritic branching and its role in nociception (Bagley et al., 2014; Robertson et al., 2013). Our ultrastructural analysis techniques will provide a valuable tool for illuminating the development and function of these cells, both in wild-type and mutant animals.
MATERIALS AND METHODS
High pressure freezing, freeze substitution, and electron microscopy
Detailed protocols for HPF, FS and microscopy are described in supplementary materials and methods. Briefly, we collected L1 and early L2 larvae and froze them using a BAL-TEC HPM 010 freezer (BAL-TEC). For epoxy embedding, larvae were fixed and freeze substituted with 1-2% osmium tetroxide (OsO4) and 0.1% uranyl acetate in 97% acetone (McDonald and Müller-Reichert, 2002) and imbedded in Durcupan resin. For correlative light/electron microscopy, larvae were fixed and freeze substituted in 95% acetone, 5% distilled water, 0.1% potassium permanganate and 0.001% osmium tetroxide (Watanabe et al., 2010) and embedded in glycidyl methacrylate resin. For sectioning and imaging, 45-70 nm-thick sections were obtained using a diamond knife and imaged at 125 keV using a Hitachi 7200 electron microscope.
HRP labeling
To label terminal cells with HRP we used the protocol described by Watts et al. (2004), as modified in Levi et al. (2006).
Light microscopy
Terminal cell branch outgrowth and lumen formation was examined using a Zeiss AxioImager M1 with a 40× PLAN NEOFLUAR objective and images were recorded on a AxioCam MRm using Axiovision software, using established protocols (Jones and Metzstein, 2013). Brightness and contrast were adjusted using ImageJ (Schneider et al., 2012) and Photoshop software (Adobe).
Fly stocks and genetics
Flies were reared on standard cornmeal/dextrose media and larvae to be scored were raised at 25°C. The control chromosome used in experiments was y w FRT19A. Alleles and transgenes analyzed were Rbcn-3A11L (this work); Rbcn-3AFE6, Rbcn-3AFU25, Rbcn-3AFV10, Rbcn-3BFC70, Rbcn-3BFK39, VhaAC39FZ29 (Yan et al., 2009). 11L alters codon 1027 of Rbcn-3A from a CAG (glutamine) codon to a TAG termination codon, truncating the 3426 amino acid protein approximately one-third of the way through the coding sequence and appears to be a null allele. For MARCM mosaic analysis (Lee and Luo, 1999), we used the tracheal-specific breathless (btl) driver (Shiga et al., 1996) in the stock y w P{w+, btl-Gal80} FRT19A, hsFLP122 ; btl-Gal4 UAS-GFP, the stock y w P{w+, btl-Gal80} FRT19A, hsFLP122 ; btl-Gal4 UAS-GFP UAS-CD2::HRP and the stock y w P{w+, btl-Gal80} FRT19A, hsFLP122 ; btl-Gal4 UAS-DsRed. To generate mosaics, 0-6 h embryos were collected in fly food vials at 25° and heat shocked at 38° for 45 min in a circulating water bath. For Vha68-2 RNAi transgene, we used the insertion P{TRiP.HMS01056}attP2, generated by the TRiP at Harvard Medical School. Other UAS-RNAi transgenes were obtained from the Vienna Drosophila RNAi Center and the National Institute of Genetics Fly Stock Center, Japan.
Construction of the bs500 driver
This driver is based on the blistered (bs) gene, which is expressed in a number of tissue types, including terminal cells, as well as muscles and wing veins (Nussbaumer et al., 2000). Within the bs enhancer, a 500-bp element has been shown to drive expression specifically in terminal cells (Nussbaumer et al., 2000). This 500-bp element, from construct pCβ 0.5 (Nussbaumer et al., 2000), was 4× multimerized using a PCR strategy and placed upstream of a minimal hsp70 promoter in vector pKG4021 (Guillemin et al., 2001), which also contains GAL4 and an hsp70 terminator. This combined expression unit was then subcloned into pCasper4 and integrated into the Drosophila genome using standard P element-mediated transformation. To make a GFP directly driven by bs500, we replaced the GAL4 with a previously described GAP:GFP (Moriyoshi et al., 1996).
Acknowledgements
We are very grateful to Kent MacDonald, Shigeki Watanabe and Erik Jorgensen, for extensive discussion of electron microscopy and fixation protocols. We thank Trudi Schüpbach and Yan Yan for fly stocks and discussion of unpublished results; Liqun Lo and Ryan Watts for CD2::HRP fly stocks; Markus Affolter for the pCβ 0.5 construct; John Perrino, Boaz Levi and Molly Weaver for help with the HRP/EM experiments; Markus Babst for helpful discussions; and Kristen Kwan, Jonathan Nelson and Gillian Stanfield for comments on the manuscript. We thank the TRiP at Harvard Medical School (NIH/NIGMS R01-GM084947) for providing transgenic RNAi fly stocks. Other fly stocks were obtained from the Bloomington Drosophila Stock Center, the Vienna Drosophila RNAi Center, and the National Institute of Genetics Fly Stock Center, Japan. We thank Mark Krasnow, in whose lab this work was initiated, and Andrea Stewart for technical support there.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
L.S.N. performed all electron microscopy sample preparation and imaging. L.S.N. and M.M.M. performed genetic experiments and wrote the manuscript.
Funding
This work was supported by a University of Utah seed grant (to M.M.M.). M.M.M. was supported by the Helen Hay Whitney foundation post-doctoral fellowship for a portion of this work.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.127902/-/DC1
References
- Allan A. K., Du J., Davies S. A. and Dow J. A. T. (2005). Genome-wide survey of V-ATPase genes in Drosophila reveals a conserved renal phenotype for lethal alleles. Physiol. Genomics 22, 128-138. 10.1152/physiolgenomics.00233.2004 [DOI] [PubMed] [Google Scholar]
- Babst M. (2006). A close-up of the ESCRTs. Dev. Cell 10, 547-548. 10.1016/j.devcel.2006.04.013 [DOI] [PubMed] [Google Scholar]
- Baer M. M., Palm W., Eaton S., Leptin M. and Affolter M. (2013). Microsomal triacylglycerol transfer protein (MTP) is required to expand tracheal lumen in Drosophila in a cell-autonomous manner. J. Cell Sci. 125, 6038-6048. 10.1242/jcs.110452 [DOI] [PubMed] [Google Scholar]
- Bagley J. A., Yan Z., Zhang W., Wildonger J., Jan L. Y. and Jan Y. N. (2014). Double-bromo and extraterminal (BET) domain proteins regulate dendrite morphology and mechanosensory function. Genes Dev. 28, 1940-1956. 10.1101/gad.239962.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bär T., Güldner F. H. and Wolff J. R. (1984). “Seamless” endothelial cells of blood capillaries. Cell Tissue Res. 235, 99-106. 10.1007/BF00213729 [DOI] [PubMed] [Google Scholar]
- Bartnicki-Garcia S. (2006). Chitosomes: past, present and future. FEMS Yeast Res. 6, 957-965. 10.1111/j.1567-1364.2006.00158.x [DOI] [PubMed] [Google Scholar]
- Bayer M. J., Reese C., Bühler S., Peters C. and Mayer A. (2003). Vacuole membrane fusion: V0 functions after trans-SNARE pairing and is coupled to the Ca2+-releasing channel. J. Cell Biol. 162, 211-222. 10.1083/jcb.200212004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry K. L., Bülow H. E., Hall D. H. and Hobert O. (2003). A C. elegans CLIC-like protein required for intracellular tube formation and maintenance. Science 302, 2134-2137. 10.1126/science.1087667 [DOI] [PubMed] [Google Scholar]
- Brown E., Mantell J., Carter D., Tilly G. and Verkade P. (2009). Studying intracellular transport using high-pressure freezing and Correlative Light Electron Microscopy. Semin. Cell Dev. Biol. 20, 910-919. 10.1016/j.semcdb.2009.07.006 [DOI] [PubMed] [Google Scholar]
- Camp A. A., Funk D. H. and Buchwalter D. B. (2014). A stressful shortness of breath: molting disrupts breathing in the mayfly Cloeon dipterum. Freshw. Sci. 33, 695-699. 10.1086/677899 [DOI] [Google Scholar]
- Centanin L., Dekanty A., Romero N., Irisarri M., Gorr T. A. and Wappner P. (2008). Cell autonomy of HIF effects in Drosophila: tracheal cells sense hypoxia and induce terminal branch sprouting. Dev. Cell 14, 547-558. 10.1016/j.devcel.2008.01.020 [DOI] [PubMed] [Google Scholar]
- Davis G. E. and Bayless K. J. (2003). An integrin and Rho GTPase-dependent pinocytic vacuole mechanism controls capillary lumen formation in collagen and fibrin matrices. Microcirculation 10, 27-44. 10.1080/713773584 [DOI] [PubMed] [Google Scholar]
- Einhorn Z., Trapani J. G., Liu Q. and Nicolson T. (2012). Rabconnectin3α promotes stable activity of the H+ pump on synaptic vesicles in hair cells. J. Neurosci. 32, 11144-11156. 10.1523/JNEUROSCI.1705-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrowski P., Necakov A. S., Mumbauer S., Loeser E., Reversi A., Streichan S., Briggs J. A. G. and De Renzis S. (2013). Tubular endocytosis drives remodelling of the apical surface during epithelial morphogenesis in Drosophila. Nat. Commun. 4, 2244 10.1038/ncomms3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgac M. (2007). Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 917-929. 10.1038/nrm2272 [DOI] [PubMed] [Google Scholar]
- Fornasiero E. F. and Opazo F. (2015). Super-resolution imaging for cell biologists: concepts, applications, current challenges and developments. Bioessays 37, 436-451. 10.1002/bies.201400170 [DOI] [PubMed] [Google Scholar]
- Francis D. and Ghabrial A. S. (2015). Compensatory branching morphogenesis of stalk cells in the Drosophila trachea. Development 142, 2048-2057. 10.1242/dev.119602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galko M. J. and Krasnow M. A. (2004). Cellular and genetic analysis of wound healing in Drosophila larvae. PLoS Biol. 2, e239 10.1371/journal.pbio.0020239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais L. and Casanova J. (2010). In vivo coupling of cell elongation and lumen formation in a single cell. Curr. Biol. 20, 359-366. 10.1016/j.cub.2009.12.043 [DOI] [PubMed] [Google Scholar]
- Ghabrial A. S., Levi B. P. and Krasnow M. A. (2011). A systematic screen for tube morphogenesis and branching genes in the Drosophila tracheal system. PLoS Genet. 7, e1002087 10.1371/journal.pgen.1002087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabenbauer M., Geerts W. J. C., Fernadez-Rodriguez J., Hoenger A., Koster A. J. and Nilsson T. (2005). Correlative microscopy and electron tomography of GFP through photooxidation. Nat. Methods 2, 857-862. 10.1038/nmeth806 [DOI] [PubMed] [Google Scholar]
- Guillemin K., Groppe J., Ducker K., Treisman R., Hafen E., Affolter M. and Krasnow M. A. (1996). The pruned gene encodes the Drosophila serum response factor and regulates cytoplasmic outgrowth during terminal branching of the tracheal system. Development 122, 1353-1362. [DOI] [PubMed] [Google Scholar]
- Guillemin K., Williams T. and Krasnow M. A. (2001). A nuclear lamin is required for cytoplasmic organization and egg polarity in Drosophila. Nat. Cell Biol. 3, 848-851. 10.1038/ncb0901-848 [DOI] [PubMed] [Google Scholar]
- Hahn-Windgassen A. and Van Gilst M. R. (2009). The Caenorhabditis elegans HNF4alpha Homolog, NHR-31, mediates excretory tube growth and function through coordinate regulation of the vacuolar ATPase. PLoS Genet. 5, e1000553 10.1371/journal.pgen.1000553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurbain I. and Sachse M. (2011). The future is cold: cryo-preparation methods for transmission electron microscopy of cells. Biol. Cell 103, 405-420. 10.1042/BC20110015 [DOI] [PubMed] [Google Scholar]
- Iruela-Arispe M. L. and Beitel G. J. (2013). Tubulogenesis. Development 140, 2851-2855. 10.1242/dev.070680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarecki J., Johnson E. and Krasnow M. A. (1999). Oxygen regulation of airway branching in Drosophila is mediated by branchless FGF. Cell 99, 211-220. 10.1016/S0092-8674(00)81652-9 [DOI] [PubMed] [Google Scholar]
- JayaNandanan N., Mathew R. and Leptin M. (2014). Guidance of subcellular tubulogenesis by actin under the control of a synaptotagmin-like protein and Moesin. Nat. Commun. 5, 3036 10.1038/ncomms4036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao W., Masich S., Franzén O. and Shupliakov O. (2010). Two pools of vesicles associated with the presynaptic cytosolic projection in Drosophila neuromuscular junctions. J. Struct. Biol. 172, 389-394. 10.1016/j.jsb.2010.07.007 [DOI] [PubMed] [Google Scholar]
- Jones T. A. and Metzstein M. M. (2013). Examination of Drosophila larval tracheal terminal cells by light microscopy. J. Vis. Exp., e50496 10.3791/50496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T. A., Nikolova L. S., Schjelderup A. and Metzstein M. M. (2014). Exocyst-mediated membrane trafficking is required for branch outgrowth in Drosophila tracheal terminal cells. Dev. Biol. 390, 41-50. 10.1016/j.ydbio.2014.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe H., Sakisaka T., Yasumi M., Shingai T., Izumi G., Nagano F., Deguchi-Tawarada M., Takeuchi M., Nakanishi H. and Takai Y. (2003). A novel rabconnectin-3-binding protein that directly binds a GDP/GTP exchange protein for Rab3A small G protein implicated in Ca2+-dependent exocytosis of neurotransmitter. Genes Cells 8, 537-546. 10.1046/j.1365-2443.2003.00655.x [DOI] [PubMed] [Google Scholar]
- Kolotuev I., Schwab Y. and Labouesse M. (2010). A precise and rapid mapping protocol for correlative light and electron microscopy of small invertebrate organisms. Biol. Cell 102, 121-132. 10.1042/BC20090096 [DOI] [PubMed] [Google Scholar]
- Lee T. and Luo L. (1999). Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451-461. 10.1016/S0896-6273(00)80701-1 [DOI] [PubMed] [Google Scholar]
- Levi B. P., Ghabrial A. S. and Krasnow M. A. (2006). Drosophila talin and integrin genes are required for maintenance of tracheal terminal branches and luminal organization. Development 133, 2383-2393. 10.1242/dev.02404 [DOI] [PubMed] [Google Scholar]
- Liégeois S., Benedetto A., Michaux G., Belliard G. and Labouesse M. (2007). Genes required for osmoregulation and apical secretion in Caenorhabditis elegans. Genetics 175, 709-724. 10.1534/genetics.106.066035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke M. (1991). Insect epidermal cells. In Physiology of the Insect Epidermis. (ed. Binnington K. and Retnakaran A.), pp. 1-22. Melbourne: CSIRO Publications. [Google Scholar]
- Lubarsky B. and Krasnow M. A. (2003). Tube morphogenesis: making and shaping biological tubes. Cell 112, 19-28. 10.1016/S0092-8674(02)01283-7 [DOI] [PubMed] [Google Scholar]
- Maina J. N. (2002). Structure, function and evolution of the gas exchangers: comparative perspectives. J. Anat. 201, 281-304. 10.1046/j.1469-7580.2002.00099.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G. and Krasnow M. A. (1993). Development of the Drosophila tracheal system. In The Development of Drosophila Melanogaster (ed. Bate M. and Martinez Arias A.), pp. 609-685. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Marshansky V. and Futai M. (2008). The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr. Opin. Cell Biol. 20, 415-426. 10.1016/j.ceb.2008.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matusek T., Djiane A., Jankovics F., Brunner D., Mlodzik M. and Mihály J. (2006). The Drosophila formin DAAM regulates the tracheal cuticle pattern through organizing the actin cytoskeleton. Development 133, 957-966. 10.1242/dev.02266 [DOI] [PubMed] [Google Scholar]
- McDonald K. L. (2009). A review of high-pressure freezing preparation techniques for correlative light and electron microscopy of the same cells and tissues. J. Microsc. 235, 273-281. 10.1111/j.1365-2818.2009.03218.x [DOI] [PubMed] [Google Scholar]
- McDonald K. L. (2014). Out with the old and in with the new: rapid specimen preparation procedures for electron microscopy of sectioned biological material. Protoplasma 251, 429-448. 10.1007/s00709-013-0575-y [DOI] [PubMed] [Google Scholar]
- McDonald K. and Müller-Reichert T. (2002). Cryomethods for thin section electron microscopy. Methods Enzymol. 351, 96-123. 10.1016/S0076-6879(02)51843-7 [DOI] [PubMed] [Google Scholar]
- McDonald K. L., Sharp D. J. and Rickoll W. (2012). Preparation of Drosophila specimens for examination by transmission electron microscopy. Cold Spring Harb. Protoc. 2012, 1044-1048. 10.1101/pdb.top068452 [DOI] [PubMed] [Google Scholar]
- Merzendorfer H. (2011). The cellular basis of chitin synthesis in fungi and insects: common principles and differences. Eur. J. Cell Biol. 90, 759-769. 10.1016/j.ejcb.2011.04.014 [DOI] [PubMed] [Google Scholar]
- Moriyoshi K., Richards L. J., Akazawa C., O'Leary D. D. and Nakanishi S. (1996). Labeling neural cells using adenoviral gene transfer of membrane-targeted GFP. Neuron 16, 255-260. 10.1016/S0896-6273(00)80044-6 [DOI] [PubMed] [Google Scholar]
- Moussian B. and Schwarz H. (2010). Preservation of plasma membrane ultrastructure in Drosophila embryos and larvae prepared by high-pressure freezing and freeze- substitution. Drosophila Inf. Serv. 93, 215-219. [Google Scholar]
- Moussian B., Seifarth C., Müller U., Berger J. and Schwarz H. (2006). Cuticle differentiation during Drosophila embryogenesis. Arthropod Struct. Dev. 35, 137-152. 10.1016/j.asd.2006.05.003 [DOI] [PubMed] [Google Scholar]
- Murk J. L. A. N., Posthuma G., Koster A. J., Geuze H. J., Verkleij A. J., Kleijmeer M. J. and Humbel B. M. (2003). Influence of aldehyde fixation on the morphology of endosomes and lysosomes: quantitative analysis and electron tomography. J. Microsc. 212, 81-90. 10.1046/j.1365-2818.2003.01238.x [DOI] [PubMed] [Google Scholar]
- Nagano F., Kawabe H., Nakanishi H., Shinohara M., Deguchi-Tawarada M., Takeuchi M., Sasaki T. and Takai Y. (2002). Rabconnectin-3, a novel protein that binds both GDP/GTP exchange protein and GTPase-activating protein for Rab3 small G protein family. J. Biol. Chem. 277, 9629-9632. 10.1074/jbc.C100730200 [DOI] [PubMed] [Google Scholar]
- Noirot C. and Noirot-Timothee C. (1982). The structure and development of the tracheal system. In Insect Ultrastructure (ed. Akai H. and King C. R.), pp. 351-381. US: Springer. [Google Scholar]
- Nussbaumer U., Halder G., Groppe J., Affolter M. and Montagne J. (2000). Expression of the blistered/DSRF gene is controlled by different morphogens during Drosophila trachea and wing development. Mech. Dev. 96, 27-36. 10.1016/S0925-4773(00)00373-7 [DOI] [PubMed] [Google Scholar]
- Palanker L., Tennessen J. M., Lam G. and Thummel C. S. (2009). Drosophila HNF4 regulates lipid mobilization and beta-oxidation. Cell Metab. 9, 228-239. 10.1016/j.cmet.2009.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Requejo-Isidro J. (2013). Fluorescence nanoscopy. Methods and applications. J. Chem. Biol. 6, 97-120. 10.1007/s12154-013-0096-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson J. L., Tsubouchi A. and Tracey W. D. (2013). Larval defense against attack from parasitoid wasps requires nociceptive neurons. PLoS ONE 8, e78704 10.1371/journal.pone.0078704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samakovlis C., Hacohen N., Manning G., Sutherland D. C., Guillemin K. and Krasnow M. A. (1996). Development of the Drosophila tracheal system occurs by a series of morphologically distinct but genetically coupled branching events. Development 122, 1395-1407. [DOI] [PubMed] [Google Scholar]
- Schneider C. A., Rasband W. S. and Eliceiri K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671-675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoofs A., Hückesfeld S., Schlegel P., Miroschnikow A., Peters M., Zeymer M., Spieß R., Chiang A.-S. and Pankratz M. J. (2014). Selection of motor programs for suppressing food intake and inducing locomotion in the Drosophila brain. PLoS Biol. 12, e1001893 10.1371/journal.pbio.1001893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schottenfeld-Roames J. and Ghabrial A. S. (2012). Whacked and Rab35 polarize dynein-motor-complex-dependent seamless tube growth. Nat. Cell Biol. 14, 386-393. 10.1038/ncb2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schottenfeld-Roames J., Rosa J. B. and Ghabrial A. S. (2014). Seamless tube shape is constrained by endocytosis-dependent regulation of active moesin. Curr. Biol. 24, 1756-1764. 10.1016/j.cub.2014.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seol J. H., Shevchenko A., Shevchenko A. and Deshaies R. J. (2001). Skp1 forms multiple protein complexes, including RAVE, a regulator of V-ATPase assembly. Nat. Cell Biol. 3, 384-391. 10.1038/35070067 [DOI] [PubMed] [Google Scholar]
- Sethi N., Yan Y., Quek D., Schüpbach T. and Kang Y. (2010). Rabconnectin-3 is a functional regulator of mammalian Notch signaling. J. Biol. Chem. 285, 34757-34764. 10.1074/jbc.M110.158634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiga Y., Tanaka-Matakatsu M. and Hayashi S. (1996). A nuclear GFP/β-galactosidase fusion protein as a marker for morphogenesis in living Drosophila. Dev. Growth Differ. 38, 99-106. 10.1046/j.1440-169X.1996.00012.x [DOI] [Google Scholar]
- Shu X., Lev-Ram V., Deerinck T. J., Qi Y., Ramko E. B., Davidson M. W., Jin Y., Ellisman M. H. and Tsien R. Y. (2011). A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 9, e1001041 10.1371/journal.pbio.1001041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurbjörnsdóttir S., Mathew R. and Leptin M. (2014). Molecular mechanisms of de novo lumen formation. Nat. Rev. Mol. Cell Biol. 15, 665-676. 10.1038/nrm3871 [DOI] [PubMed] [Google Scholar]
- Snelling E. P., Seymour R. S. and Runciman S. (2011). Moulting of insect tracheae captured by light and electron-microscopy in the metathoracic femur of a third instar locust Locusta migratoria. J. Insect Physiol. 57, 1312-1316. 10.1016/j.jinsphys.2011.06.006 [DOI] [PubMed] [Google Scholar]
- Studer D., Humbel B. M. and Chiquet M. (2008). Electron microscopy of high pressure frozen samples: bridging the gap between cellular ultrastructure and atomic resolution. Histochem. Cell Biol. 130, 877-889. 10.1007/s00418-008-0500-1 [DOI] [PubMed] [Google Scholar]
- Sumner J.-P., Dow J. A. T., Earley F. G., Klein U., Jäger D. and Wieczorek H. (1995). Regulation of plasma membrane V-ATPase activity by dissociation of peripheral subunits. J. Biol. Chem. 270, 5649-5653. 10.1074/jbc.270.10.5649 [DOI] [PubMed] [Google Scholar]
- Ukken F. P., Aprill I., JayaNandanan N. and Leptin M. (2014). Slik and the receptor tyrosine kinase breathless mediate localized activation of moesin in terminal tracheal cells. PLoS ONE 9, e103323 10.1371/journal.pone.0103323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urwyler O., Izadifar A., Dascenco D., Petrovic M., He H., Ayaz D., Kremer A., Lippens S., Baatsen P., Guérin C. J. et al. (2015). Investigating CNS synaptogenesis at single-synapse resolution by combining reverse genetics with correlative light and electron microscopy. Development 142, 394-405. 10.1242/dev.115071 [DOI] [PubMed] [Google Scholar]
- Vasin A., Zueva L., Torrez C., Volfson D., Littleton J. T. and Bykhovskaia M. (2014). Synapsin regulates activity-dependent outgrowth of synaptic boutons at the Drosophila neuromuscular junction. J. Neurosci. 34, 10554-10563. 10.1523/JNEUROSCI.5074-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S., Punge A., Hollopeter G., Willig K. I., Hobson R. J., Davis M. W., Hell S. W. and Jorgensen E. M. (2010). Protein localization in electron micrographs using fluorescence nanoscopy. Nat. Methods 8, 80-84. 10.1038/nmeth.1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts R. J., Schuldiner O., Perrino J., Larsen C. and Luo L. (2004). Glia engulf degenerating axons during developmental axon pruning. Curr. Biol. 14, 678-684. 10.1016/j.cub.2004.03.035 [DOI] [PubMed] [Google Scholar]
- Yan Y., Denef N. and Schüpbach T. (2009). The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Dev. Cell 17, 387-402. 10.1016/j.devcel.2009.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. and Chen E. H. (2008). Ultrastructural analysis of myoblast fusion in Drosophila. Methods Mol. Biol. 475, 275-297. 10.1007/978-1-59745-250-2_16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Yan Z., Jan L. Y. and Jan Y. N. (2013). Sound response mediated by the TRP channels NOMPC, NANCHUNG, and INACTIVE in chordotonal organs of Drosophila larvae. Proc. Natl. Acad. Sci. USA 110, 13612-13617. 10.1073/pnas.1312477110 [DOI] [PMC free article] [PubMed] [Google Scholar]