

Abstract
With the rapid evolution of next-generation DNA sequencing technologies, the cost of sequencing a human genome has plummeted, and genomics has started to pervade health care across all stages of life — from preconception to adult medicine. Challenges to fully embracing genomics in a clinical setting remain, but some approaches are starting to overcome these barriers such as community-driven data sharing to improve the accuracy and efficiency of applying genomics to patient care.
Society is evolving rapidly in the information age. Portable electronic devices enable access to global knowledge at literally every step: we check the weather on an app, monitor what is happening around the world through online news feeds, and connect with family and friends through social networking services. The immediate availability of a broad range of knowledge sources has led to greater health awareness in the general population. Individuals are increasingly using wearable devices to monitor sleep, activity levels, glucose levels or heart function to identify abnormalities of potential health concern[1–3].
Since the first complete human genome sequences were published 15 years ago[4, 5], technological advances and cost savings in DNA sequencing have enabled millions of people to have their individual genomic sequence analysed, primarily within the settings of research studies or clinical care. Each individual harbours four to five million variants in their genome that collectively differentiate one person from another[6]. Some of these variants have no health effect, whereas others increase or lower the risk of disease. There is widespread recognition that enabling access to an individual’s genomic sequence and other ‘omics’ data can enable a more detailed understanding of our health and disease risks and inform a more precise approach to patient care, a strategy now commonly called ‘precision medicine’[7, 8].
With genomic data now increasingly being used to guide the individual care of patients, our health care systems are evolving, although a number of challenges remain. This Perspective considers how genomics is guiding health care for the individual, illustrating with example scenarios how humans are taking advantage of personal genomic information, ranging from advanced diagnostics to tumour profiling, to genomic risk assessments. These examples are then interweaved with the day-to-day challenges still facing the integration of genomics into clinical practice as well as strategies to overcome these barriers and enable genomics to be a part of ever more aspects of everyday patient care.
Engaging genomics across the lifespan
From conception to elderly care, we now have access to genomic technologies and the information our genomes provide to personalize and inform precise approaches for optimizing our health and combating disease (Figure 1).
Figure 1. The use of genomics throughout an individual’s lifespan.
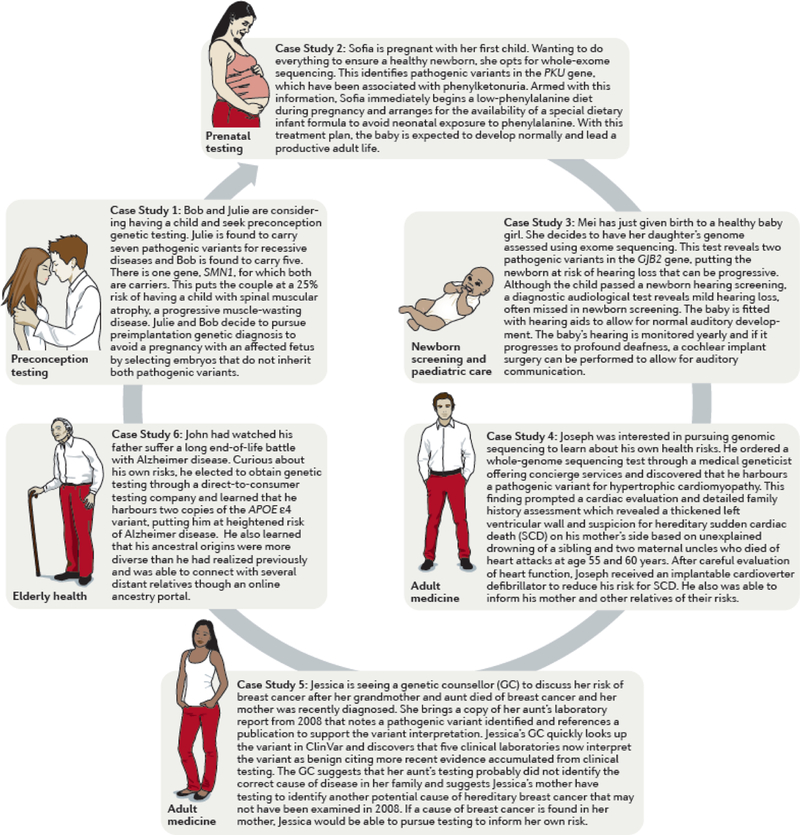
Case studies of the use of genomics to inform patient care at different stages of life.
Case Example 1 - Preconception testing
Bob and Julie are considering having a child and seek preconception genetic testing. Julie is found to carry seven pathogenic variants for recessive diseases and Bob is found to carry five. There is one gene, SMN1, for which both are carriers. This puts the couple at a 25% risk of having a child with spinal muscular atrophy, a progressive muscle-wasting disease. Julie and Bob decide to pursue preimplantation genetic diagnosis to avoid a pregnancy with an affected fetus by selecting embryos that do not inherit both pathogenic variants.
Case Example 2 - Prenatal testing
Sofia is pregnant with her first child. Wanting to do everything to ensure a healthy newborn, she opts for whole-exome sequencing. The sequencing results identify pathogenic variants in the PKU gene, which have been associated with phenylketonuria. Armed with this information, Sofia immediately begins a low-phenylalanine diet during pregnancy and arranges for the availability of a special dietary infant formula to avoid neonatal exposure to phenylalanine. With this treatment plan, the baby is expected to develop normally and lead a productive adult life.
Case Example 3 – Newborn screening and paediatric care
Mei has just given birth to a healthy baby girl. She decides to have her daughter’s genome assessed using exome sequencing. This test reveals two pathogenic variants in the GJB2 gene, putting the newborn at risk of hearing loss that can be progressive. Although the child passed a newborn hearing screening, a diagnostic audiological test reveals mild hearing loss, often missed in newborn screening. The baby is fitted with hearing aids to allow for normal auditory development. The baby’s hearing is monitored yearly and if it progresses to profound deafness, the option for cochlear implantation surgery can be offered to the family.
Case Example 4 – Adult medicine
Joseph was interested in pursuing genomic sequencing to learn about his own health risks. He ordered a whole-genome sequencing test through a medical geneticist offering concierge services and discovered that he harbours a pathogenic variant for hypertrophic cardiomyopathy. This finding prompted a cardiac evaluation which revealed normal cardiac morphology and conduction systems; however, a detailed family history assessment identified suspicion for hereditary sudden cardiac death on his mother’s side based on unexplained drowning of a sibling and two maternal uncles who died of heart attacks at age 55 and 60 years. Given the incomplete penetrance of hypertrophic cardiomyopathy, Joseph’s actual risk of disease is unclear, but with a positive at-risk genotype, he will pursue regular cardiac evaluations and inform family members of their possible risk.
Case Example 5 – Access to shared resources to support patient care
Jessica is seeing a genetic counsellor (GC) to discuss her risk of breast cancer after her grandmother and aunt died of breast cancer and her mother was recently diagnosed. She brings a copy of her aunt’s laboratory report from 2008 that notes a pathogenic variant identified and references a publication to support the variant interpretation. Jessica’s GC quickly looks up the variant in ClinVar and discovers that five clinical laboratories now interpret the variant as benign citing more recent evidence accumulated from clinical testing. The GC suggests that her aunt’s testing probably did not identify the correct cause of disease in her family and suggests Jessica’s mother have testing to identify another potential cause of hereditary breast cancer that may not have been examined in 2008. If a cause of breast cancer is found in her mother, Jessica would be able to pursue testing to inform her own risk.
Case Example 6 – Elderly health
John had watched his father suffer a long end-of-life battle with Alzheimer disease. Curious about his own risks, he elected to obtain genetic testing through a direct-to-consumer testing company and learned that he harbours two copies of the APOE ɛ4 variant, putting him at heightened risk of Alzheimer disease. He also learned that his ancestral origins were more diverse than he had realized previously and was able to connect with several distant relatives though an online ancestry portal.
Dating and preconception
Although primarily confined to certain genetically isolated populations, some cultures embrace genomics in determining compatibility in dating and marriage. For over a decade, it has been common for individuals of Ashkenazi Jewish descent to be screened for carrier status either before a couple begins dating or during prenatal counselling; for example, through specialized organizations such as Dor Yeshorim. The Ashkenazi Jewish population is well known to have a high risk of certain genetic diseases due to high carrier rates such as 1 in 15 for Type 1 Gaucher disease (Online Mendelian Inheritance in Man (OMIM) #230800) and 1 in 27 for Tay-Sachs disease[9](OMIM #272800). For a given couple, if both individuals are found to carry a pathogenic variant in the same gene for a recessive disease, then there is a 25% risk of having an affected child (FIG. 1, Case study 1). In this scenario, the couple may be encouraged to seek other relationships, pursue in vitro fertilization with selected non-affected embryos, or at least be informed of their reproductive risks.
Certain disorders are common to specific ethnic populations, whereas other disorders such as cystic fibrosis (OMIM #219700) are common across all populations, leading to broad recommendations for genetic screening[10]. It is now becoming increasingly common in the United States to screen for a broad array of recessive conditions during preconception counselling to determine risk for a future pregnancy. As such, these tests now routinely include the detection of pathogenic variants in a hundred or more genes. Several companies (such as Counsyl or Good Start Genetics) are primarily focused on this market, whereas many others are adding such testing as one of the many genetic and genomics tests in their arsenal of services. Most services report out carrier status independently for each partner, other companies (such as Gene Peeks) are focused on a joint analysis, confining the report to only disorders in which both couples are positive for carrier status, a much rarer event. Millions of couples have already pursued carrier screening, making it one of the most common indications for genetic testing.
Most preconception genetic testing is focused on detecting carrier status for monogenic diseases such as cystic fibrosis; however, business opportunities in the direct-to-consumer (DTC) market are increasing. Thus, it is unsurprising that online dating services are bringing genomics onto the scene. Primarily based on early studies of the role of the immune system in mate choice[11], companies such as Instant Chemistry or Gene Partner are now offering genetic testing as a companion service to online dating, using genetics to rate compatibility. Although a novelty in the market, there is still little evidence of the validity of genomics in forming successful human relationships.
Prenatal testing
Prenatal genetic testing has been offered for more than 60 years[12]. Testing initially focused on karyotyping, often on the basis of heightened risk of a chromosomal abnormality suggested by ultrasound findings or advanced maternal age. In rarer cases, testing was performed for a known familial pathogenic variant for a disorder previously identified in the family. Although both approaches are still offered, couples are also pursuing routine chromosome microarray (CMA) testing, even without a clinical indication, to screen for genetic abnormalities, including those too small to be detected by conventional karyotyping, such as submicroscopic copy number variants (CNVs). More recently, non-invasive prenatal testing has sky-rocketed in its uptake given its preference over invasive amniocentesis procedures required for CMAs. However, non-invasive prenatal testing is currently only robust for detecting complete chromosomal aneuploidies such as trisomy 21, which is causative for Down syndrome[13, 14]. Although genomic sequencing of fetal DNA has been achieved through non-invasive techniques[15], it is not commonly employed given the very small amount of fetal DNA circulating in the maternal bloodstream and the difficulty differentiating it from maternal DNA. However, it is likely to be only a matter of time before the technology of choice includes genome sequencing, to be as comprehensive as possible in detecting disease risk in a developing fetus. Although personal decisions vary with regard to the potential option to terminate a pregnancy based on genetic testing results, many couples choose prenatal testing not to inform termination but to enable awareness of disease risk and to implement strategies to optimize newborn health. For example, awareness of risk for an inborn error of metabolism, such as phenylketonuria (OMIM #261600), would allow immediate dietary interventions at birth to improve the child’s health outcome[16](Figure 1, case study 2).
The reduced incidence of Tay-Sachs disease owing to carrier screening[17] provides a clear example of how preconception and prenatal testing can reduce the incidence of genetic disease. However, the impact of broader screening on the larger collection of genetic diseases is more difficult to assess. Statistics from the Centers for Disease Control (CDC) indicate a 6% reduction in infant mortality rate from congenital malformations between 2005 and 2011[18], raising the possibility that preconception and prenatal genetic testing may have aided in this reduction. However, it is also possible that improved prenatal imaging diagnostics, medical management and surgical interventions may have played an equal if not larger role.
Newborn screening
With an increasing focus on preventative health, one might consider the newborn period an ideal time for health screens. Most babies are born within a hospital setting and typically stay 48 hours. Newborn screening takes advantage of this setting for performing an individual’s first health screens after birth. Most countries have mandated newborn screening programmes, although the number of disorders for which screening is provided varies, ranging from none (for example, there are no programmes in central Africa, according to the International Society for Neonatal Screening) to over 50 conditions (as in most states of the United States, according to the National Newborn Screening and Global Resource Center). screened in most states in the USA.
The criteria for adding a disorder to newborn screening is based on the availability as well as sensitivity and specificity of a screening test. Moreover, the availability of diagnostic services to make a definitive diagnosis after a screening referral, and the availability and efficacy of treatments to improve the long-term outcomes of diagnosed individuals are considered[19]. For example, the American College of Medical Genetics and Genomics has developed a framework to score each condition being considered for screening based upon the following factors: the clinical characteristics of the condition; the analytical characteristics of the test; and the diagnosis, follow-up, treatment, and management of the condition[19]. There is also a push to include parents as decision-makers and address if any newborn screening tests should be mandatory or should allow an opt-out or opt-in decision by parents[20].
Evidence regarding long-term outcomes in often limited, and experience in the practical aspects of screening is lacking. Thus, when considering the addition of new disorders to a screen, there is also a need to ensure adequate tracking of programmes after implementation to evaluate success in achieving the goals of screening. An example of a success is the screen for phenylketonuria (Fig. 1, Case Study 2). For other disorders, such as cardiomyopathy[21] or retinoblastoma (tumour of the eye)[22], no economical approach to screening is available; however, intervention at birth or over the course of a lifetime could still enable avoidance of adverse outcomes. As such, interest in genomic screening approaches to enable detection of such disorders is increasing[23]. The US National Institutes of Health (NIH) has funded four projects, together forming the Newborn Sequencing in Genomic Medicine and Public Health (NSIGHT) programme, to explore the use of genomic testing in the newborn period[24]. Approaches currently under investigation include using genomics as a rapid diagnostic tool, as well as screening healthy babies for disease risk or to inform decision-making throughout the newborn period as issues arise[25, 26]. Findings from these studies are anticipated over the next few years and will help guide the appropriate use of genomics in the newborn period.
Newborn screening for disease risk is a developing area of genomics; however, it is important to note that one of the most valuable aspects of genetic and genomic testing today is the ability to end the diagnostic odyssey[27–29]. Countless newborns born with multiple congenital anomalies, complex syndromes, or unexplained severe neurological phenotypes are evaluated with extensive biochemical, imaging, and other analytical work-ups. These diagnostic tests are often on an annual basis for many years, along with countless referrals to specialists. The ultimate goal is to not only make a diagnosis but to provide the family with information and tools to understand the cause of disease and enable them to avoid the costly and time-consuming work-ups within the complex distributed health care system. After a diagnosis, physicians can focus care on aspects of the disease that are manageable and intervene to prevent anticipated developments of disease known to be associated with the disorder. In addition, many families, after obtaining a diagnosis, are motivated to build or join patient advocacy organizations, to fundraise or to enter into research programmes and clinical trials. They can also become leading advocates to catalyse advances in disease understanding, diagnosis and treatment[30]; examples of patient advocacy organizations formed by diagnosed individuals include Prion Alliance, the Hypertrophic Cardiomyopathy Association and Hear See Hope.
Paediatric medicine
Although many genetic disorders are apparent at birth, several not revealed until childhood. For example, retinoblastoma is often not present at birth but is a largely paediatric cancer that requires more aggressive screening to be detected[22]. As another example, hearing impairment may not be sufficiently severe for detection during newborn screening or can develop and progress after birth (FIG. 1, Case study 3). Also, intellectual disability and autism are not apparent at birth and require ongoing evaluation during childhood and adolescence to properly diagnose the extent of intellect and behaviour. Growth conditions[31] and other endocrinological disorders[32] also typically manifest in childhood. For these reasons, paediatricians must be adept at detecting paediatric diseases and recognizing when the underlying causes may be genetic, potentially requiring medical intervention, and alert parents regarding the risk to future children.
Adult medicine
Engagement of genomics in the adult population is most often in the context of: diagnostic germline testing for symptomatic individuals; tumour profiling to inform cancer treatment and prognosis; and genomic testing to inform ancestry, health risks and other traits. The first two occur largely within the clinical care setting, whereas the third has been the focused of the DTC market. There are many diseases with adult-onset symptoms for which an individual may seek diagnostic testing. Such diseases include cardiovascular disease (for example, arrhythmia, cardiomyopathy, aortic disease, very high cholesterol; FIG 1, Case study 4), cancer, hearing loss and vision loss, particularly if associated with a known or possible family history of disease. Although an early diagnosis and/or strong family history of cancer may prompt hereditary cancer testing, cancers more often occur sporadically owing to somatically derived genetic alterations that are not inherited but occur in cells of the body[33]. Today, numerous genetic alterations have been identified in tumours that correlate with therapy response, such as epidermal growth factor receptor (EGFR) mutations in lung cancer, which predict response to tyrosine kinase inhibitors[34, 35]. Other examples include anaplastic lymphoma receptor tyrosine kinase (ALK) fusions in lung cancer (which predict response to crizotinib[36]), HER2 (also known as ERBB2) mutations in breast cancer (which predict trastuzumab response[37, 38]), BRAFV600E mutation in melanoma (which confer sensitivity to vemurafenib[39]), and socitrate dehydrogenase (NADP+) 1 (IDH1) mutations in gliomas (which are associated with response to temozolomide[40]).
Widespread uptake of DTC testing to inform ancestry, health risks and other traits began with the introduction of high-throughput genotyping arrays that examine common genetic variation. However, as public awareness of health risks has grown, including monogenic disease risks such as hereditary cancer and inherited cardiovascular diseases, individuals have begun to pursue genomic sequencing to gain a more complete analysis of their own health risks. Some projects have been launched with the aim of data sharing to advance the science (for example, The Personal Genome Project), whereas other projects are integrated into clinical practices and target healthy patients interested in learning about their own personal disease risks[41, 42]. Although DTC services focused initially on wealthy individuals, who were able to afford the high cost of genomic services, the falling costs of genomic sequencing increasingly enable access for the wider population interested in understanding and informing their health[43].
Challenges to clinical genomics
Evolving genetic and genomic test platforms
Diagnostic genetic tests for the many examples presented in this article fall in four main categories (Table 1): genotyping tests for known disease or treatment-associated variants; disease-targeted panel tests for genes known to be associated with the disorder or treatment; exome sequencing tests, which interrogate nearly all protein-coding regions; and genome sequencing tests, which interrogate nearly our entire genetic code including non-coding regions.
Table 1.
A comparison of diagnostic test platforms
| Known variant genotyping tests | Disease-targeted sequencing tests | Exome sequencing tests | Genome sequencing tests | |
|---|---|---|---|---|
| Cost (US$) | <500 | 500–5,000 | 5,000–9,000 | 7,000–10,000 |
| Detection | Low, with exceptions | ~5–50% | ~25% | ~25% |
| Variant types detected | As designed | SVs (+/−) and CNVs | SVs | SVs, CNVs and StrVs |
| Secondary findings | No | No | Yes | Yes |
| Interpretation difficulty | Easy | Moderate to Challenging | Moderate to Challenging | Moderate to Challenging |
| Novel gene discovery | No | No | Yes | Yes |
Abbreviations: SVs, sequence variants; CNVs, copy number variants; StrVs, structural variants
The most straightforward and high-yield tests that are performed today in genetic and genomic testing are diagnostic tests for monogenic diseases and certain cancer diagnostic tests. However, monogenic disease tests typically detect less than half of the aetiologies[44–51] (Fig. 2). In some cases, this is low detection rate is attributed to the existence of both genetic and non-genetic causes of the illness, and a genetic test will never identify the non-genetic causes. However, in most cases, the primary failure is the absence of a complete understanding of the genetic underpinnings of a disease[52]. Indeed, in many families with clearly inherited disease we fail to detect the genetic aetiology owing to insufficient evidence to implicate variation in the gene or due to its occurrence in a regulatory region of the genome that is poorly understood functionally[52]. For somatic cancer testing, a genetic alteration that directs a treatment decision can be found in a subset of tumours[53–55]; however, the cancer may come back with treatment resistance due to continuously evolving genetic alterations that occur as the tumour grows.
Figure 2. Detection rates across a selection of molecular diagnostic tests.
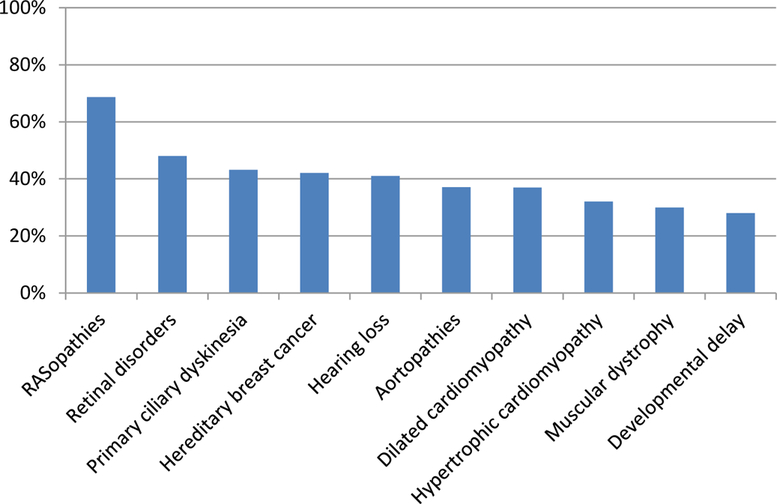
These data are from multiple studies spanning several indications[44–51]. The specific detection rate can vary based upon the specificity of the clinical presentations and the comprehensiveness of the test panel.
Apart from a few rare exceptions (for example, sickle cell anaemia, achondroplasia, and BRAFV600E mutations in skin cancer), most genetic tests for known disease-causing variants have low sensitivity. The reason for this lack of sensitivity is because most conditions are genetically heterogeneous, with multiple possible aetiologies and with many variants being unique to a family. For example, hundreds of variants in over 50 genes, are causative for hypertrophic cardiomyopathy[44]. As a result, most monogenic disease tests require sequencing approaches, either panel-based, exome or genome sequencing. Although more costly and comprehensive, the clinical sensitivities of exome and genome sequencing are not always higher than a disease-focused panel test. In some cases, clinical sensitivities of exome and genome sequencing may be lower depending on the contribution of CNVs to the disease and whether the test includes CNV detection through built-in analysis approaches or adjunct platforms[56, 57].
Given the higher costs compared with conventional genetic tests and the fear of potential downstream health care costs due to the return of secondary genetic findings, exome and genome sequencing services are particularly poorly covered by health plans. This issue has slowed the uptake in adopting these broader tests. However, as the technical and interpretive barriers are gradually overcome and labs offer genomic approaches at similar costs to panel-based tests, these comprehensive approaches will soon dominate the market.
The challenges of variant interpretation
Regardless of the platform chosen or indication for testing, all genetic tests come with one major liability, which is the challenge of interpreting the genetic variants identified during testing (Fig. 1, Case study 5). An analysis of data in ClinVar, a database hosted by the NIH’s National Center for Biotechnology Information[58], quantifies this challenge with 17% of variants interpreted by more than one submitter showing a difference in variant classification[59].
It is unsurprising that variant interpretation is a challenge; our genomes are littered with variation — typically four to five million variants per individual with over ten thousand among gene-coding regions[6] — yet there is no dictionary to define the ‘meaning’ of each variant. Furthermore, many of the variants causative for disease are extremely rare or unique to individuals. Although recent standards for genetic interpretation have been published[60], and will no doubt help prevent the most egregious differences in interpretation, even experts using such a standard differ in their interpretations, particularly before consensus efforts are applied[61]. As of January 2017, more than 250,000 unique interpreted variants had been submitted by over 600 laboratories to ClinVar. However, one-third of those variants were submitted with an uncertain significance classification. More importantly, we know from the Exome Aggregation Consortium (ExAC) that over 7.4 million variants have been identified in gene-coding regions[62], most of which have no entry in ClinVar. Applying a 1% frequency filter, which is typical for monogenic disease analysis, only eliminates 1% of the variants from consideration. Moreover, comparison across individuals shows that each individual harbours over 60 coding variants found only in one in 60,000 individuals[62]. This rarity creates enormous challenges for variant interpretation
So what then is the path to freedom from this deluge of uninterpretable variants? The answer, in my opinion, lies with widespread open data-sharing and the standardization and structuring of data for scaled analyses.
Data sharing to advance genomics
To ensure efficient and accurate diagnosis of patients, it is becoming increasingly clear that the best path to improved DNA variant interpretation is through the sharing of variants in a common database. Such a database can enable crowdsourcing of the labour-intensive effort of gathering data as well as identifying differences in interpretation and resolution of those differences between laboratories and by experts in the relevant fields[61, 63]. With 67% of submitters coming from outside the United States (D. Maglott, personal communication) the ClinVar database[58] robustly serves a data aggregation purpose for the international community. The NIH-funded ClinGen (Clinical Genome Resource) programme is forming and approving panels that convene relevant experts to agree on approaches to variant interpretation and ensure that the relevant variants are being interpreted to those standards[59]. However, this application of expert curation will take time: only ~4% of variants have been reviewed by experts in ClinVar at the time of writing[64], therefore the community must still rely on laboratories to share their knowledge with each other before experts can weigh in.
For each variant found in an individual, curators scour the literature, searching for evidence: a few segregations in a pedigree, detailed phenotyping in a patient, a de novo occurrence in a parent–offspring trio, a single data point from a functional assay. However, such information is often unstructured, non-systematic manner, largely embedded in the unstructured figures and detailed text of published manuscripts. Imagine instead a world where a common data model describes the evidence types for variants, and such evidence is systematically collected in databases connected through a federated network of laboratories and health care systems. Each source shares the individual-level and family data, the results of functional analyses or other data sources useful in the evidence-based interpretation of DNA variation. Imagine no resource barriers to the storage and computing power of an exponentially scaling quantity of genotype and phenotype data, and a plethora of well-trained computational biologists poised to turn raw data into an informed understanding of human health and disease risk. We will get there.
There are many mechanisms to support data sharing of both variant-level data described above and individual-level data including genetic or genomic analyses and phenotype; centralized databases and federated systems are examples of such mechanisms (Fig. 3). In a centralized model, groups submit their data to a single physically located database, such as ClinVar (for variant-level claims), the database of Genotypes and Phenotypes (dbGaP) or the European Genome–Phenome Archive (EGA) (for individual-level data). In a federated model, multiple physical databases exist, located in different sites but connected through an application programming interface. The database schemas in a federated network can be the same or different. What must be agreed upon are the fields and structure of the data sent through the application programming interface, which can be constructed specifically to support a defined need. For example, investigators wishing to solve the causes of rare disease are now using the federated Matchmaker Exchange network, which supports queries between databases on phenotypes and candidate genes in order to match rare disease cases from around the world[65].
Figure 3. Centralized and federated databases.
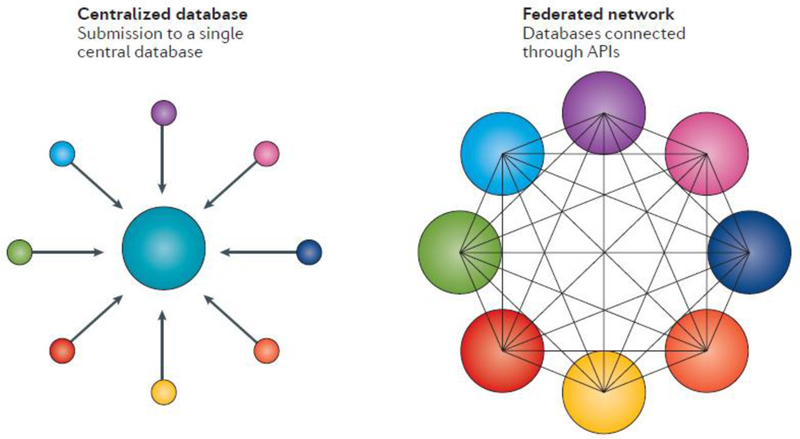
In a centralized database model, submitters send data to a single central database. In a federated system, data remains in the originating database with queries supported through application programming interfaces (APIs).
So which is better: federated or centralized? The answer is not one or the other but depends on the nature of the data and the purpose of the database. If the data being shared is subject to strict patient privacy and security rules, it may be that allowing each group to physically store and protect the data according to their own laws and other restrictions, and then connect those databases in a federated model, is the better solution. Conversely, if such constraints are not a concern and the ability to combine and compare data and display it in a coordinated manner is critically important, such as the sharing of variant-level data in ClinVar or aggregated allele frequency data in ExAC, then a centralized database may make more sense.
A specific example of how centralized and federated systems can be integrated for optimal use is illustrated in defining databases to support variant interpretation. As described above, ClinVar is primarily a variant-level database. The primary accession is a variant with defined attributes (genomic coordinates to describe location, nomenclature recommended by the Human Genome Variation Society to define gene and transcript impacts, interpretations of clinical significance, publication citations and other supporting evidence). As variant-level data are not considered individual data and therefore not subject to the Privacy Rule of HIPAA (the Health Insurance Portability and Accountability Act of 1996), it can easily be shared and deposited in the public domain[66]. And because comparison of multiple clinical interpretations of a variant is helpful to build confidence in a variant interpretation or note discordance, the aggregation of all clinical assertions on a variant is vital for this type of database. By contrast, the individual-level data (genotype and phenotype) from which a variant was originally identified is also very useful to access during the interpretation of a variant. However, one can provide links from variant-level databases to the sources of individual-level data, which could then be accessed only as needed and through secure interfaces designed to protect patient privacy.
Indeed one of the major challenges to advancing our field is the careful balancing of storing and sharing individual genomic and health data in an accessible manner, while protecting the privacy of individuals. Although there is no perfect solution that optimizes sharing yet prevents privacy breaches, the use of federated approaches can reduce concerns over misuse of shared data[67]. Furthermore, proper consent and direct engagement of individuals in sharing their own data is likely to positively influence data sharing as individuals are empowered to make their own decisions about the risks and benefits of giving access to their genomic data[68, 69].
Considerations of the return of results
Although testing for genomic risks is becoming more commonplace, there remains ethical debate over screening children for genetic disease risk[70]. Surveys of parents suggest an interest in using genomics technologies to screen newborn babies[24], although pursuit of such approaches remains uncommon. The low uptake is also due to concerns of genomicists and families over how to deal with the identification of untreatable disease risks that could have negative impacts on the child and their future autonomy; for example, leading to discrimination regarding a child’s eligibility for disability and long-term care insurance policies[71, 72]. The same considerations are relevant when considering paediatric care.
Most children have routine screening visits with a health care provider to assess their health on an annual basis. Care includes, for example, vaccinations, hearing and vision screening, as well as examinations to ensure appropriate physical and behavioural milestones are being achieved. It is a logical extension that the use of genomic screening to inform health risks for a child would have equal merit to that for a newborn baby. For example, retinoblastoma, one of the most common childhood cancers, is largely curable with vision retention if caught early but requires ophthalmological screening under anaesthesia every three to four weeks after birth[22]. Newborn genomic screening would facilitate the identification of babies at risk and enable appropriate intervention to ensure a healthy outcome. However, it is important to note that detection of a pathogenic variant in a newborn, young child, or even an adult, rarely provides an accurate assessment of the likelihood that the individual will develop the disease over their lifetime. Penetrance can range from low to high[73–77] (Fig. 4); however, most data on the penetrance of individual genetic disorders is obtained from families in whom the disease is present. To form an unbiased view on disease penetrance will require extremely large studies of genotype-first approaches followed by clinical follow-up to identify the existence or development of disease over time, such as the All of UsSM research programme of the Precision Medicine Initiative[8]. This initiative will enable a more accurate determination of disease risk and inform appropriate treatment and management strategies based on those risks. Meanwhile, we must proceed forward despite the absence of a completely informed view of penetrance. Indeed, the management strategies for certain disorders may not differ whether disease risk is 90% or 10%. For example, if a newborn baby is diagnosed as having a pathogenic variant in the ELN gene (which encodes elastin), putting the newborn at risk of subaortic stenosis and subsequent sudden cardiac death[78], it is highly unlikely that the awareness of incomplete penetrance will lead to a decision not to have an echocardiogram to detect a potential structural malformation that can be corrected by surgical intervention[79].
Figure 4. Penetrance of genetic disorders.
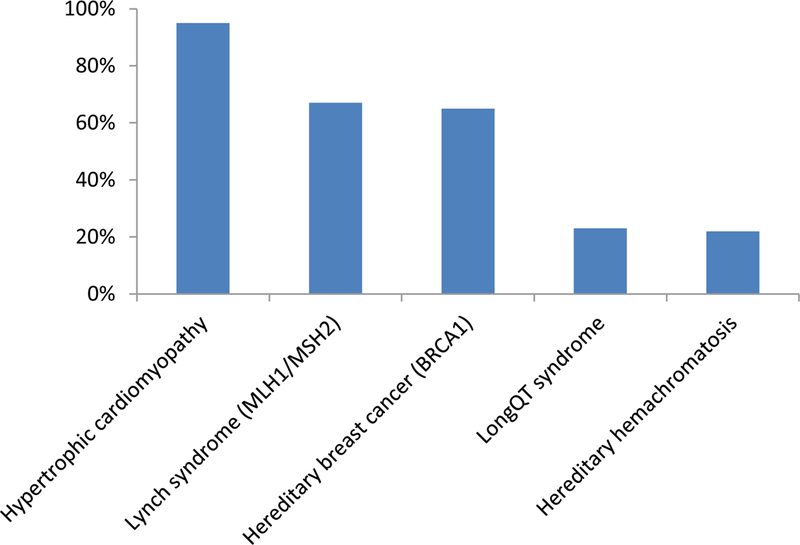
The penetrance of a selection of well-known genetic disorders is shown[73–77]. The midpoint was used for data expressed as a range.
In summary, although we have much to learn in genomics and the true risks of diseases identified, we are increasingly armed with clear examples of the clinical utility of genome sequencing, making it likely for this approach to be embraced as a complementary and expanded method to traditional preventative medicine approaches.
Looking to the future
Although many challenges exist in scaling genomics as a primary tool in health care, these barriers are gradually breaking down as costs are reduced and resources to support the generation and sharing of genomic knowledge are improved. But what else remains to effectively incorporate genomics into the daily practice of medicine at a scale that makes genomics an aspect of every individual’s life?
In addition to the core challenges described above, other secondary challenges are likely to hamper the widespread expansion of precision medicine. These challenges include the need to expand the workforce of professionals trained to understand, deliver and incorporate genetics into the care of patients. Physicians must be more comprehensively trained in genetics, particularly in diseases and relevant testing approaches as it relates to their areas of specialty practice. We must expand training programmes to produce more genetic counsellors able to spend the necessary time with patients to answer complex questions around disease risk, the ever-changing knowledge base that accompanies each variant identified in a patient. Moreover, experts are needed to guide individuals to consider the sharing of the genomic and health information with family members and the broader community to advance knowledge and build evidence. Although genetic counsellors will undoubtedly be critical in this space, it will also become increasingly important to facilitate knowledge-building through online tools and for that individuals take on an active role in educating themselves about genetics. Individuals are already tracking the status of their own variants in ClinVar and actively engaging in data-sharing activities to advance an understanding of their own diseases and genomic variants[80–82]. Finally, we must continue to navigate additional, often country-specific, barriers to widespread use of genomics such as the impact to employment as well as health, life, and long-term disability insurance policies. A detailed review of laws and insurance policies across many countries is beyond the scope of this Essay; however, we must recognize the impact, whether based on real or perceived concerns, that these forces have on uptake of genomics[83].
To use genomics effectively, we must support the delivery of information at the point of care. For example, if a physician receives a high cholesterol level on a patient, the electronic health record system could prompt the physician to either order a genetic test or query genomic data residing directly in the patient’s medical record. If a physician orders a drug that is contraindicated or the dose of which should be adjusted based on an individual’s genetic variants, drug choice or dosage can be automatically adjusted. If a bone marrow transplant is needed, databases of individuals willing to serve as donors could be queried around the world to instantly identify matches. Meanwhile, data on patient phenotypes, medication usage and treatment outcomes could be collected continuously and stored, with access enabled through federated queries and tabulations as our health care system evolves.
An optimized, learning health care system will not happen on its own, particularly given the skyrocketing cost of health care. We must design a system that saves resources while improving outcomes. The two are often at odds with each other, but with a continued focus on evidence-based medicine, and payment for outcomes not services, and the provision of real-time support for decision-making as well as coordinated care delivery, we may be able to achieve cheaper, more cost-effective care, incorporating genomics in a manner that adds value without increasing overall health care costs.
Glossary
- Chromosome microarray (CMA)
A cytogenetic testing platform that uses DNA probes to detect copy number variants of typically 100,000 bp or larger
- Copy number variants (CNVs)
the loss of gain of chromosomal material often resulting from a deletion or duplication event, respectively
- Genotype-first approaches
the use of genetic and genomic testing to enable earlier identification of disease diagnoses compared to first performing detailed clinical tests and evaluations
- Non-invasive prenatal testing (NIPT)
A prenatal screening test to detect chromosome abnormalities in cell-free fetal DNA in maternal blood
- Penetrance
the likelihood that a disease will be expressed in an individual who harbours an at risk genotype
- Precision Medicine Initiative
An NIH funded programme launched in 2016 to advance biomedical research including the aim to enrol of one million individuals who will consent to contribute detailed medical and genetic data
Footnotes
Databases
ClinVar: http://www.ncbi.nlm.nih.gov/clinvar/
dbGAP: http://www.ncbi.nlm.nih.gov/gap
Matchmaker Exchange: http://www.matchmakerexchange.org/
OMIM: http://www.omim.org/
References
- 1.Turakhia MP and Kaiser DW, Transforming the care of atrial fibrillation with mobile health. J Interv Card Electrophysiol, 2016. [DOI] [PMC free article] [PubMed]
- 2.Khan Y, et al. , Monitoring of Vital Signs with Flexible and Wearable Medical Devices. Adv Mater, 2016. 28(22): p. 4373–95. [DOI] [PubMed] [Google Scholar]
- 3.Kolla BP, Mansukhani S, and Mansukhani MP, Consumer sleep tracking devices: a review of mechanisms, validity and utility. Expert Rev Med Devices, 2016. 13(5): p. 497–506. [DOI] [PubMed] [Google Scholar]
- 4.Lander ES, et al. , Initial sequencing and analysis of the human genome. Nature, 2001. 409(6822): p. 860–921. [DOI] [PubMed] [Google Scholar]
- 5.Venter JC, et al. , The sequence of the human genome. Science, 2001. 291(5507): p. 1304–51. [DOI] [PubMed] [Google Scholar]
- 6.Genomes Project, C., et al. , A global reference for human genetic variation. Nature, 2015. 526(7571): p. 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aronson SJ and Rehm HL, Building the foundation for genomics in precision medicine. Nature, 2015. 526(7573): p. 336–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins FS and Varmus H, A new initiative on precision medicine. N Engl J Med, 2015. 372(9): p. 793–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang B, Dearing L, and Amos J, DNA-based carrier screening in the Ashkenazi Jewish population. Expert Rev Mol Diagn, 2004. 4(3): p. 377–92. [DOI] [PubMed] [Google Scholar]
- 10.American College of, O. and G. Gynecologists Committee on, ACOG Committee Opinion No. 486: Update on carrier screening for cystic fibrosis. Obstet Gynecol, 2011. 117(4): p. 1028–31. [DOI] [PubMed] [Google Scholar]
- 11.Wedekind C, et al. , MHC-dependent mate preferences in humans. Proc Biol Sci, 1995. 260(1359): p. 245–9. [DOI] [PubMed] [Google Scholar]
- 12.Fuchs F and Riis P, Antenatal sex determination. Nature, 1956. 177(4503): p. 330. [DOI] [PubMed] [Google Scholar]
- 13.Thung DT, et al. , Implementation of whole genome massively parallel sequencing for noninvasive prenatal testing in laboratories. Expert Rev Mol Diagn, 2015. 15(1): p. 111–24. [DOI] [PubMed] [Google Scholar]
- 14.Lo JO, et al. , Noninvasive prenatal testing. Obstet Gynecol Surv, 2014. 69(2): p. 89–99. [DOI] [PubMed] [Google Scholar]
- 15.Kitzman JO, et al. , Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med, 2012. 4(137): p. 137ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Regier DSG, C. I.Phenylalanine hydroxylase deficiency. 20 Oct 2016, GeneReviews.
- 17.Kaback MM, Population-based genetic screening for reproductive counseling: the Tay-Sachs disease model. Eur J Pediatr, 2000. 159 Suppl 3: p. S192–5. [DOI] [PubMed] [Google Scholar]
- 18.MacDorman MF, Hoyert DL, and Mathews TJ, Recent declines in infant mortality in the United States, 2005–2011. NCHS data brief, no 120 2013, Hyattsville, MD: National Center for Health Statistics. [PubMed] [Google Scholar]
- 19.American College of Medical Genetics Newborn Screening Expert, G., Newborn screening: toward a uniform screening panel and system--executive summary. Pediatrics, 2006. 117(5 Pt 2): p. S296–307. [DOI] [PubMed] [Google Scholar]
- 20.Moyer VA, et al. , Expanding newborn screening: process, policy, and priorities. Hastings Cent Rep, 2008. 38(3): p. 32–9. [DOI] [PubMed] [Google Scholar]
- 21.Cirino AL and Ho CY, Hypertrophic Cardiomyopathy Overview., in GeneReviews® [Internet], Pagon RA, Adam MP, and Ardinger HH, Editors. 2014: Seattle (WA): University of Washington, Seattle. [PubMed] [Google Scholar]
- 22.Lohmann DR and Gallie BL, Retinoblastoma, in GeneReviews® [Internet], Pagon RA, Adam MP, and Ardinger HH, Editors. 2015: Seattle (WA): University of Washington, Seattle. [PubMed] [Google Scholar]
- 23.Kingsmore SF, Newborn testing and screening by whole-genome sequencing. Genet Med, 2016. 18(3): p. 214–6. [DOI] [PubMed] [Google Scholar]
- 24.Waisbren SE, et al. , Parents are interested in newborn genomic testing during the early postpartum period. Genet Med, 2015. 17(6): p. 501–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ceyhan-Birsoy O, et al. , A curated gene list for reporting results of newborn genomic sequencing. Genet Med, 2017. [DOI] [PMC free article] [PubMed]
- 26.Berg JS, et al. , Newborn Sequencing in Genomic Medicine and Public Health. Pediatrics, 2017. 139(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lazaridis KN, et al. , Outcome of Whole Exome Sequencing for Diagnostic Odyssey Cases of an Individualized Medicine Clinic: The Mayo Clinic Experience. Mayo Clin Proc, 2016. 91(3): p. 297–307. [DOI] [PubMed] [Google Scholar]
- 28.Sawyer SL, et al. , Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet, 2016. 89(3): p. 275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thevenon J, et al. , Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet, 2016. 89(6): p. 700–7. [DOI] [PubMed] [Google Scholar]
- 30.Might M and Might C, What happens when N = 1 and you want plus 1? Prenat Diagn, 2016. [DOI] [PubMed]
- 31.Alatzoglou KS, et al. , Isolated growth hormone deficiency (GHD) in childhood and adolescence: recent advances. Endocr Rev, 2014. 35(3): p. 376–432. [DOI] [PubMed] [Google Scholar]
- 32.Forlenza GP, et al. , Next generation sequencing in endocrine practice. Mol Genet Metab, 2015. 115(2–3): p. 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merlo LM, et al. , Cancer as an evolutionary and ecological process. Nat Rev Cancer, 2006. 6(12): p. 924–35. [DOI] [PubMed] [Google Scholar]
- 34.Lynch TJ, et al. , Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med, 2004. 350(21): p. 2129–39. [DOI] [PubMed] [Google Scholar]
- 35.Paez JG, et al. , EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science, 2004. 304(5676): p. 1497–500. [DOI] [PubMed] [Google Scholar]
- 36.Rodig SJ and Shapiro GI, Crizotinib, a small-molecule dual inhibitor of the c-Met and ALK receptor tyrosine kinases. Curr Opin Investig Drugs, 2010. 11(12): p. 1477–90. [PubMed] [Google Scholar]
- 37.Piccart-Gebhart MJ, et al. , Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med, 2005. 353(16): p. 1659–72. [DOI] [PubMed] [Google Scholar]
- 38.Romond EH, et al. , Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med, 2005. 353(16): p. 1673–84. [DOI] [PubMed] [Google Scholar]
- 39.Chapman PB, et al. , Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med, 2011. 364(26): p. 2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Houillier C, et al. , IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology, 2010. 75(17): p. 1560–6. [DOI] [PubMed] [Google Scholar]
- 41.Lupo PJ, et al. , Patients’ perceived utility of whole-genome sequencing for their healthcare: findings from the MedSeq project. Per Med, 2016. 13(1): p. 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vassy JL, et al. , The MedSeq Project: a randomized trial of integrating whole genome sequencing into clinical medicine. Trials, 2014. 15: p. 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meisel SF, et al. , Explaining, not just predicting, drives interest in personal genomics. Genome Med, 2015. 7(1): p. 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alfares AA, et al. , Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med, 2015. [DOI] [PubMed]
- 45.Boaretto F, et al. , Diagnosis of Primary Ciliary Dyskinesia by a Targeted Next-Generation Sequencing Panel: Molecular and Clinical Findings in Italian Patients. J Mol Diagn, 2016. [DOI] [PubMed]
- 46.Cizmarova M, et al. , New Mutations Associated with Rasopathies in a Central European Population and Genotype-Phenotype Correlations. Ann Hum Genet, 2016. 80(1): p. 50–62. [DOI] [PubMed] [Google Scholar]
- 47.Lee H, et al. , Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA, 2014. 312(18): p. 1880–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pugh TJ, et al. , The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med, 2014. 16(8): p. 601–8. [DOI] [PubMed] [Google Scholar]
- 49.Schenkel LC, et al. , Clinical Next-Generation Sequencing Pipeline Outperforms a Combined Approach Using Sanger Sequencing and Multiplex Ligation-Dependent Probe Amplification in Targeted Gene Panel Analysis. J Mol Diagn, 2016. 18(5): p. 657–67. [DOI] [PubMed] [Google Scholar]
- 50.Shearer AE and Smith RJ, Massively Parallel Sequencing for Genetic Diagnosis of Hearing Loss: The New Standard of Care. Otolaryngol Head Neck Surg, 2015. 153(2): p. 175–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang H, et al. , Genetic testing of 248 Chinese aortopathy patients using a panel assay. Sci Rep, 2016. 6: p. 33002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chong JX, et al. , The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am J Hum Genet, 2015. 97(2): p. 199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cree IA, et al. , Guidance for laboratories performing molecular pathology for cancer patients. J Clin Pathol, 2014. 67(11): p. 923–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindeman NI, et al. , Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol, 2013. 8(7): p. 823–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Febbo PG, et al. , NCCN Task Force report: Evaluating the clinical utility of tumor markers in oncology. J Natl Compr Canc Netw, 2011. 9 Suppl 5: p. S1–32; quiz S33. [DOI] [PubMed] [Google Scholar]
- 56.Aradhya S, et al. , Exon-level array CGH in a large clinical cohort demonstrates increased sensitivity of diagnostic testing for Mendelian disorders. Genet Med, 2012. 14(6): p. 594–603. [DOI] [PubMed] [Google Scholar]
- 57.Pugh TJ, et al. , VisCap: inference and visualization of germ-line copy-number variants from targeted clinical sequencing data. Genet Med, 2016. 18(7): p. 712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Landrum MJ, et al. , ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res, 2014. 42(Database issue): p. D980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rehm HL, et al. , ClinGen - The Clinical Genome Resource. N Engl J Med, 2015. 372(23): p. 2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Richards S, et al. , Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015. 17(5): p. 405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amendola LM, et al. , Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet, 2016. 99(1): p. 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lek M, et al. , Analysis of protein-coding genetic variation in 60,706 humans. Nature, 2016. 536(7616): p. 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amendola LM, et al. , Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res, 2015. 25(3): p. 305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Information NC f.B., ClinVar Submissions 2016, ClinVar.
- 65.Philippakis AA, et al. , The Matchmaker Exchange: a platform for rare disease gene discovery. Hum Mutat, 2015. 36(10): p. 915–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thorpe JH and Gray EA, Big data and public health: navigating privacy laws to maximize potential. Public Health Rep, 2015. 130(2): p. 171–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Global Alliance for Genomics and Health. 2015. [cited 2015 May 28, 2015]; Available from: http://genomicsandhealth.org/our-work/working-groups/regulatory-and-ethics-working-group/work-products.
- 68.Baker DB, Kaye J, and Terry SF, Governance Through Privacy, Fairness, and Respect for Individuals. EGEMS (Wash DC), 2016. 4(2): p. 1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Knoppers BM, Framework for responsible sharing of genomic and health-related data. Hugo J, 2014. 8(1): p. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Botkin JR, et al. , Points to Consider: Ethical, Legal, and Psychosocial Implications of Genetic Testing in Children and Adolescents. Am J Hum Genet, 2015. 97(1): p. 6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Botkin JR and Rothwell E, Whole Genome Sequencing and Newborn Screening. Curr Genet Med Rep, 2016. 4(1): p. 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Waisbren SE, et al. , Psychosocial Factors Influencing Parental Interest in Genomic Sequencing of Newborns. Pediatrics, 2016. 137 Suppl 1: p. S30–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alders M and Christiaans I, Long QT Syndrome, in GeneReviews(R), Pagon RA, et al. , Editors. 1993: Seattle (WA). [Google Scholar]
- 74.Charron P, et al. , Penetrance of familial hypertrophic cardiomyopathy. Genet Couns, 1997. 8(2): p. 107–14. [PubMed] [Google Scholar]
- 75.Kohlmann W and Gruber SB, Lynch Syndrome, in GeneReviews(R), Pagon RA, et al. , Editors. 1993: Seattle (WA). [Google Scholar]
- 76.Petrucelli N, Daly MB, and Feldman GL, BRCA1 and BRCA2 Hereditary Breast and Ovarian Cancer, in GeneReviews(R), Pagon RA, et al. , Editors. 1993: Seattle (WA). [PubMed] [Google Scholar]
- 77.Whitlock EP, et al. , Screening for hereditary hemochromatosis: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med, 2006. 145(3): p. 209–23. [DOI] [PubMed] [Google Scholar]
- 78.Metcalfe K, et al. , Elastin: mutational spectrum in supravalvular aortic stenosis. Eur J Hum Genet, 2000. 8(12): p. 955–63. [DOI] [PubMed] [Google Scholar]
- 79.Deo SV, et al. , Late outcomes for surgical repair of supravalvar aortic stenosis. Ann Thorac Surg, 2012. 94(3): p. 854–9. [DOI] [PubMed] [Google Scholar]
- 80.Chong JX, et al. , Gene discovery for Mendelian conditions via social networking: de novo variants in KDM1A cause developmental delay and distinctive facial features. Genet Med, 2016. 18(8): p. 788–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kirkpatrick BE, et al. , GenomeConnect: matchmaking between patients, clinical laboratories, and researchers to improve genomic knowledge. Hum Mutat, 2015. 36(10): p. 974–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lambertson KF, et al. , Participant-driven matchmaking in the genomic era. Hum Mutat, 2015. 36(10): p. 965–73. [DOI] [PubMed] [Google Scholar]
- 83.Wauters A and Van Hoyweghen I, Global trends on fears and concerns of genetic discrimination: a systematic literature review. J Hum Genet, 2016. 61(4): p. 275–82. [DOI] [PubMed] [Google Scholar]