

Abstract
Aims/hypothesis
Empagliflozin (EMPA), an inhibitor of the renal sodium–glucose cotransporter (SGLT) 2, reduces the risk of cardiovascular death in patients with type 2 diabetes. The underlying mechanism of this effect is unknown. Elevated cardiac cytoplasmic Na+ ([Na+]c) and Ca2+ ([Ca2+]c) concentrations and decreased mitochondrial Ca2+ concentration ([Ca2+]m) are drivers of heart failure and cardiac death. We therefore hypothesised that EMPA would directly modify [Na+]c, [Ca2+]c and [Ca2+]m in cardiomyocytes.
Methods
[Na+]c, [Ca2+]c, [Ca 2+]m and Na+/H+ exchanger (NHE) activity were measured fluorometrically in isolated ventricular myocytes from rabbits and rats.
Results
An increase in extracellular glucose, from 5.5 mmol/l to 11 mmol/l, resulted in increased [Na+]c and [Ca2+]c levels. EMPA treatment directly inhibited NHE flux, caused a reduction in [Na+]c and [Ca2+]c and increased [Ca2+]m. After pretreatment with the NHE inhibitor, Cariporide, these effects of EMPA were strongly reduced. EMPA also affected [Na+]c and NHE flux in the absence of extracellular glucose.
Conclusions/interpretation
The glucose lowering kidney-targeted agent, EMPA, demonstrates direct cardiac effects by lowering myocardial [Na+]c and [Ca2+]c and enhancing [Ca2+]m, through impairment of myocardial NHE flux, independent of SGLT2 activity.
Electronic supplementary material
The online version of this article (doi:10.1007/s00125-016-4134-x) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Keywords: Calcium, Cardiac death, Diabetes, Glucose, Heart failure, Sodium
Introduction
The recent Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes (EMPA-REG OUTCOME) study has demonstrated that empagliflozin (EMPA), an inhibitor of renal sodium-glucose cotransporter (SGLT)2, resulted in a 38% reduction in the relative risk of cardiovascular death and a 35% risk reduction of hospitalisation for heart failure in patients with type 2 diabetes [1]. SGLT2 inhibitors also result in increased urinary glucose excretion in diabetic patients. The mechanisms behind these effects are unknown [1, 2], however, since these findings do not suggest any beneficial effects of EMPA on the incidence of myocardial infarction and stroke, it is unlikely that they can be ascribed to a general reduction in risk factors for cardiovascular disease (e.g. glycaemic status, body weight, blood pressure). We hypothesised that EMPA has a direct cardiac effect; since increases in myocardial intracellular Na+ and Ca2+ concentrations are early hallmarks and drivers of cardiovascular death and heart failure [2–6], we investigated whether EMPA (1) directly reduces intracellular cardiac cytoplasmic Na+ ([Na+]c) and Ca2+ ([Ca2+]c) concentration; (2) affect the (upstream) cardiac Na+/H+ exchanger (NHE); and (3) changes (downstream) mitochondrial Ca2+ concentration ([Ca2+]m).
Methods
Animal handling was in accordance with the Institutional Animal Care and Use Committee of the VU Medical Center and Academic Medical Center Amsterdam, and was conducted according to the Guide for the Use and Care of Laboratory Animals.
For detailed methods, see electronic supplementary material [ESM] Methods. Cardiomyocytes were isolated from hearts from healthy rabbits and rats. Cells were left untreated or treated with 1 μmol/l EMPA (MedChem Express, Monmouth Junction, NJ, USA), 5.5 mmol/l or 11 mmol/l glucose (Sigma-Aldrich Chemie, Zwijndrecht, the Netherlands), 10 μmol/l cariporide (Aventis Pharma, Frankfurt, Germany) or 20 mmol NH4Cl (NH+) (Sigma-Aldrich Chemie), either alone or in combination. [Na+]c and [Ca2+]c were fluorometrically measured in rabbit cardiomyocytes using SBF1 and indo-1, respectively, at 2 Hz field stimulation. NHE activity was measured in rabbit cardiomyocytes by recording SNARF fluorescence following an NH4 + pulse. Using adenoviral transfection, a ratiometric mitochondrially targeted fluorescence resonance energy transfer (FRET)-based Ca2+ indicator (4mtD3cpv, MitoCam) was expressed in rat cardiomyocytes and free [Ca2+]m was measured using the fluorescence ratio, yellow fluorescent protein intensity/cyan fluorescent protein intensity (YFP/CFP), in cultured adult rat cardiomyocytes.
Statistics
Data are reported as mean ± SE. Kolmogorov–Smirnov normality testing was applied to decide the use of non-parametric vs parametric testing. Mann–Whitney tests were used to evaluate the effects of glucose on Na+ and Ca2+. ANOVA with Dunnett’s post hoc tests was used to compare group means with the control ([EMPA] 0) group. Additionally, unpaired t tests were used to evaluate EMPA effects on [Ca2+]c and EMPA effects on the NHE flux in the absence of glucose, ANOVA with post hoc testing with Bonferroni corrections were used to compare cariporide and EMPA effects on the NHE flux in the presence of glucose and two-way ANOVA for repeated measures followed by post hoc contrast with Bonferroni correction at one time point were performed to detect EMPA effects on [Ca2+]m.
Results
EMPA decreases [Ca2+]c and [Na+]c and increases [Ca2+]m
First, we examined the acute effects of EMPA in isolated rabbit cardiomyocytes in the presence of 11 mmol/l glucose. EMPA (1 μmol/l) decreased [Na+]c within 10 min (Fig. 1a). Vehicle administration had no effect (data not shown). In addition, similar acute EMPA effects were observed for diastolic and systolic [Ca2+]c (Fig. 1b,c).
Fig. 1.

EMPA effects on [Na+]c, [Ca2+]c and [Ca2+]m. (a–c) EMPA (1 μmol/l) acutely lowers (a) [Na+]c, and (b) diastolic [Ca2+]c and (c) systolic [Ca2+]c in rabbit cardiomyocytes. (d,e) Effects of EMPA (3 h incubation) on [Na+]c at (d) 11 mmol/l and (e) 5.5 mmol/l glucose (Gluc), respectively. *p < 0.05 vs 0 μmol/l EMPA, ANOVA with Dunnett’s post hoc tests. (f) Effects of EMPA (3 h pre-incubation; [EMPA] 1) on diastolic (Dias) and systolic (Sys) [Ca2+]c at 11 mmol/l glucose. *p < 0.05 vs 0 μmol/l EMPA ([EMPA] 0), unpaired t test. (g) [Ca2+]m as determined by the change in fluorescence ratio, YFP/CFP, relative to 0 min (t = 0) during a 15 min incubation with 1 μmol/l EMPA (white bars) or vehicle (black bars). *p < 0.05 vs vehicle at a similar time point, two-way ANOVA for repeated measures followed by post hoc contrast with Bonferroni correction at one time point. For (a–c) n = 6–8 cells from 3 rabbits per investigation; for (d–f), n = 20–30 cells from 3 rabbits per investigation; for (g) n = 8–9 cardiomyocytes from 2–3 rats for each group (EMPA or vehicle)
Extended (3 h at 37°C in 3 ml HEPES buffer) EMPA incubation at clinically relevant concentrations (0.25–1 μmol/l) also reduced [Na+]c, in the presence of both 11 mmol/l (Fig. 1d) and 5.5 mmol/l (Fig. 1e) glucose. Increasing glucose concentration from 5.5 mmol/l to 11 mmol/l significantly increased [Na+]c (6.0 ± 0.2 mmol/l to 7.6 ± 0.3 mmol/l; p < 0.001; Fig. 1d), diastolic [Ca2+]c (68 ± 3 nmol/l to 101 ± 3 nmol/l; p < 0.001) and systolic [Ca2+]c (331 ± 18 nmol/l to 435 ± 20 nmol/l; p < 0.001) (data not shown). EMPA preincubation of cardiomyocytes at 11 mmol/l glucose also significantly lowered diastolic and systolic [Ca2+]c (Fig. 1f). Patch clamp experiments (n = 4 cells) demonstrated that 1 μmol/l EMPA had no effect on action potential duration (data not shown). The downstream effects of the cytosolic changes of [Na+]c and [Ca2+]c on [Ca2+]m were subsequently tested. Acute EMPA (1 μmol/l) administration significantly increased [Ca2+]m (Fig. 1g).
Collectively, these data show that EMPA reduces myocardial [Na+]c and [Ca2+]c and increases [Ca2+]m, whereas increases in glucose are associated with elevated [Na+]c and [Ca2+]c.
Empagliflozin impairs cardiac NHE activity without SGLT involvement
Because the rapid action of EMPA resembled that of the NHE-inhibitor Cariporide on [Na+]c and [Ca2+]c [3], we investigated whether EMPA has NHE-inhibiting properties. First, we examined EMPA and Cariporide interactions on [Na+]c. Following the reduction in [Na+]c with EMPA, additional application of Cariporide had only a minimal effect on [Na+]c (Fig. 2a). Similarly, following the reduction in [Na+]c with 10 min pre-treatment with Cariporide, subsequent EMPA application had a minimal effect on [Na+]c (Fig. 2b). Second, we examined NHE flux, as determined by measuring pH recovery after an acute acidic load via wash-out of NH4 +. In control conditions, pH quickly recovered to normal values after NH4 + wash-out. However, this recovery of pH was totally inhibited by the specific NHE inhibitor Cariporide, reflecting reduced NHE activity (Fig. 2c). In the presence of EMPA, the NHE flux was also strongly reduced by approximately 80% of the reduction observed with Cariporide (Fig. 2c). During NH4 + application, no significant difference in pH recovery was observed between Cariporide and control (data not shown). Finally, we evaluated whether SGLTs were involved in the effects of EMPA on NHE by repeating the experiments in the absence of glucose; in glucose-free conditions, EMPA also reduced [Na+]c (Fig. 2d) and impaired NHE flux (Fig. 2e).
Fig. 2.
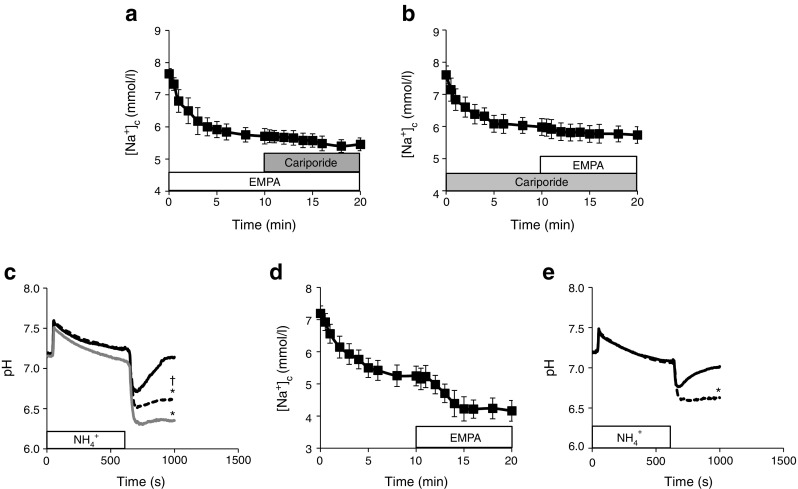
The effects of EMPA on the NHE in the presence and absence of glucose. (a) Cariporide exerted little effect on [Na+]c when preceded by EMPA inhibition. (b) Similarly, EMPA was of little effect on [Na+]c when preceded by Cariporide. (c) pH traces for rabbit cardiomyocytes exposed to an acidic load (NH4 +) and during recovery for control (black solid line), Cariporide-treated (grey line) or EMPA-treated (black dashed line) cells in the presence of 11 mmol/l glucose. *p < 0.05 vs control for pH measured at 1000 s; †p < 0.05 vs Cariporide for pH measured at 1000 s, ANOVA with post hoc testing with Bonferroni corrections. (d) EMPA (1 μmol/l) acutely lowers [Na+ ] c even in the absence of extracellular glucose (n = 6/3 rabbits). (e) EMPA effects on pH recovery in the absence of extracellular glucose (n = 6/3 rabbits). Control (black solid line), EMPA-treuted (black dashed line). *p < 0.05 vs control for pH measured at 1000 s, unpaired t test. n = 6 cells from 3 rabbits for all except for (c), where n = 5–6 cells from 4 rabbits
These data suggest that EMPA affects intracellular ion homeostasis in the cardiomyocyte through direct interaction with the NHE, without the involvement of SGLTs.
Discussion
The present study demonstrates for the first time that the kidney-targeted therapeutic agent, EMPA, directly reduces myocardial intracellular Na2+ through an interaction with the NHE, independent of cardiac SGLT inhibition. The observed decreased [Ca2+]c and increased [Ca2+]m are likely to have occurred secondary to the decrease in [Na+]c via the sarcolemmal and mitochondrial Na+/Ca2+ exchanger (NCX), respectively [6, 7]. Dapagliflozin, another SGLT2 inhibitor with a slightly different chemical composition, was recently shown to reduce cell shortening after 5 min but not after 3 h of treatment. It was also found to reduce systolic but not diastolic Ca2+ in cardiomyocytes from models of type 1 diabetes (but not in control cardiomyocytes) [8]. In comparison with dapagliflozin, the current study indicates a stronger and longer lasting effect of EMPA on cardiomyocytes. It is suggested that further research is required to compare the effects of different classes of SGLT2 inhibitors on cardiomyocytes characteristics.
EMPA: cardiac NHE and SGLT2
Our observations that the effects of EMPA were independent of glucose presence is in agreement with a previous observation that SGLT2 is absent from cardiac tissue [9]. However, SGLT1 is present in the heart [9, 10], potentially explaining the increase in [Na+]c with increased circulating glucose. It was recently reported that SGLT1 within the diseased human heart is mainly localised in capillaries and not in cardiomyocytes [11]. This is in contrast with other observations that SGLT1 is localised in the T tubules of isolated cardiomyocytes [10]. In the current study, the effects of glucose on [Na+]c were detected in isolated cardiomyocytes; therefore, it is unlikely that glucose release from the capillaries plays a role in this effect. However, further research is needed to study the role of SGLT1 within the intact heart. We used EMPA concentrations in the range of clinically measured plasma concentrations (≤1 μmol/l) [1, 12], well below the IC50 of SGLT1 (8.3 μmol/l) for EMPA [13]. Therefore, EMPA does not exert its cardiac effects through SGLT1inhibition.
EMPA raises cardiac [Ca2+]m
Previous studies have demonstrated that increasing [Na+]c results in decreased [Ca2+]m through increased mitochondrial efflux via the mitochondrial NCX [7]. This is very likely to be the mechanism underlying the EMPA-induced elevation in [Ca2+]m, observed in the present study. Mitochondrial Ca2+ is considered to be an important activator of ATP synthesis and of the antioxidant enzymatic network [5, 14]. Knowing that in vivo mitochondrial impairment, energy deficiency and increased oxidative stress [5, 14, 15] are hallmarks of failing hearts, restoring [Ca2+]m (e.g. increasing [Ca2+]m that is otherwise reduced due to Na+ loading) is predicted to be beneficial in this condition. Indeed, in a recent study, it was demonstrated that increasing mitochondrial Ca2+ during heart failure development was associated with the prevention of sudden death and overt heart failure [5]. This is in contrast to conditions where [Ca2+]m is already elevated, such as during reperfusion injury phenomena, in which further increases in [Ca2+]m can be detrimental.
EMPA and heart failure
The propensities for arrhythmias, oxidative stress and heart failure are all associated with, and at least partly driven by, intracellular cardiomyocyte Na+ and Ca2+ loading [2, 3, 5, 6, 14]. Hyperglycaemia (as reported in this study) and diabetes [10] also result in intracellular Na+ and Ca2+ loading, possibly contributing to the reported interaction between hyperglycaemia/diabetes and cardiovascular diseases.
Previous studies have demonstrated that chronic inhibition of NHE prevents or mitigates heart failure in animal models [16, 17]. Although clinical studies of NHE inhibition have been performed in the setting of acute coronary syndromes (and largely show a neutral effect) no clinical studies have been performed using NHE inhibition in the chronic setting of heart failure and diabetes. Therefore, these types of studies are awaited. We surmise that the beneficial cardiovascular effects of EMPA are, at least in part, attributed to NHE inhibition. However, the current results do not allow for the generation of conclusive statements about a positive or negative role on cardiac mitochondrial function following EMPA treatment. The next area that needs to be evaluated is whether EMPA does indeed result in beneficial functional cardiac effects, such as increased ATP production, oxygen consumption and/or antioxidant capacity. Whether the reductions in both cardiac [Na+]c and [Ca2+]c with EMPA treatment, and the downstream effect of elevated [Ca2+]m contribute to the primary mechanisms underlying the cardiovascular benefits observed in the EMPA-REG OUTCOME trial needs to be examined in future research.
In conclusion, our data demonstrate that the kidney-targeted therapeutic agent EMPA has direct cardiac effects, decreases cardiac [Na+]c and [Ca2+]c and increases cardiac [Ca2+]m via inhibition of the cardiac NHE.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 511 kb)
Acknowledgements
We thank J. L. Martin (Loyola University, Chicago, IL, USA) for construction of the adenovirus, and G. A. Marchal (Department of Clinical and Experimental Cardiology, Academic Medical Center, Amsterdam, the Netherlands) for biotechnical assistance.
Abbreviations
- [Ca2+]c
Cytoplasmic Ca2+ concentration
- [Ca2+]m
Mitochondrial Ca2+ concentration
- EMPA
Empagliflozin
- EMPA-REG OUTCOME
Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes
- [Na+]c
Cytoplasmic Na+ concentration
- NCX
Na+/Ca2+ exchanger
- NHE
Na+/H+ exchanger
- SGLT
Sodium-glucose cotransporter
- YFP/CFP
Yellow fluorescent protein intensity/cyan fluorescent protein intensity
Funding
This work was supported, in part, by the Netherlands CardioVascular Research Initiative (CVON2011-11 ARENA).
Data availability
All data is available from the authors upon request.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
AB, CAS, RCIW, and GJMS contributed to the conception and design, planning of the analysis, and acquisition and interpretation of data, and reviewed and edited the manuscript. JWTF contributed to the analysis planning and interpretation of data, and reviewed and edited the manuscript. RC and CJZ contributed to the conception and design, analysis and interpretation of data, drafting and editing of the manuscript. All authors were fully responsible for all content and editorial decisions, and approved the final version. CJZ is the guarantor of this work.
Footnotes
Antonius Baartscheer and Cees A. Schumacher contributed equally to this study and are joint first authors.
Ruben Coronel and Coert J. Zuurbier contributed equally to this study and are joint senior authors.
References
- 1.Zinman B, Wanner C, Lachin JM, et al. EMPA-REG OUTCOME investigators. Empagliflozin, cardiovascular outcomes and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117–2128. doi: 10.1056/NEJMoa1504720. [DOI] [PubMed] [Google Scholar]
- 2.Marx N, McGuire DK. Sodium-glucose cotransporter-2 inhibitor for the reduction of cardiovascular events in high-risk patients with diabetes mellitus. Eur Heart J. 2016 doi: 10.1093/eurheartj/ehw110. [DOI] [PubMed] [Google Scholar]
- 3.Baartscheer A, Schumacher CA, Borren MM, Belterman CN, Coronel R, Fiolet JW. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res. 2003;57:1015–1024. doi: 10.1016/S0008-6363(02)00809-X. [DOI] [PubMed] [Google Scholar]
- 4.Despa S, Islam MA, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure, but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.CIR.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 5.Liu T, Takimoto E, Dimaano VL, et al. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden cardiac death in a guinea pig model of heart failure. Circ Res. 2014;115:44–54. doi: 10.1161/CIRCRESAHA.115.303062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pogwizd SM, Sipido KR, Verdonck F, Bers DM. Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc Res. 2003;57:887–896. doi: 10.1016/S0008-6363(02)00735-6. [DOI] [PubMed] [Google Scholar]
- 7.Liu T, O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–288. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamouda NN, Sydorenko V, Qureshi MA, Alkaabi JM, Oz M, Howarth FC. Dapagliflozin reduces the amplitude of shortening and Ca2+ transient in ventricular myocytes from streptozotocin-induced diabetic rats. Mol Cell Biochem. 2015;400:57–68. doi: 10.1007/s11010-014-2262-5. [DOI] [PubMed] [Google Scholar]
- 9.Van Steenbergen A, Balteau M, Scholasse J et al (2015) Identification of a glucose sensor in the heart. Eur Heart J 36 (Suppl 1):381(abstract)
- 10.Lambert R, Srodulski S, Peng X, et al. Intracellular Na+ concentration ([Na+]i) is elevated in diabetic hearts due to enhanced Na + -glucose cotransport. J Am Heart Assoc. 2015;4:e002183. doi: 10.1161/JAHA.115.002183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vrhovac I, Balen Eror D, Klessen D, et al. Localizations of Na + -D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch. 2015;467:1881–1898. doi: 10.1007/s00424-014-1619-7. [DOI] [PubMed] [Google Scholar]
- 12.Heise T, Seman MS, Macha S. Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple rising doses of Empagliflozin in patients with type 2 diabetes mellitus. Diabetes Ther. 2013;4:331–345. doi: 10.1007/s13300-013-0030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grempler R, Thomas L, Eckhardt M, et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterization and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012;14:83–90. doi: 10.1111/j.1463-1326.2011.01517.x. [DOI] [PubMed] [Google Scholar]
- 14.Kohlhaas M, Liu T, Knopp A, et al. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balestra GM, Mik EG, Eerbeek O, Specht PA, van der Laarse WJ, Zuurbier CJ. Increased in vivo mitochondrial oxygenation with right ventricular failure induced by pulmonary arterial hypertension: mitochondrial inhibition as driver of cardiac failure? Respir Res. 2015;16:6. doi: 10.1186/s12931-015-0178-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baartscheer A, Hardziyenka M, Schumacher CA. Chronic inhibition of the Na+/H+ - exchanger causes regression of hypertrophy, heart failure, and ionic and electrophysiological remodeling. Br J Pharmacol. 2008;154:1266–1275. doi: 10.1038/bjp.2008.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baartscheer A, Schumacher CA, van Borren MM, et al. Chronic inhibition of Na+/H+-exchanger attenuates cardiac hypertrophy and prevents cellular remodeling in heart failure. Cardiovasc Res. 2005;65:83–92. doi: 10.1016/j.cardiores.2004.09.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 511 kb)
Data Availability Statement
All data is available from the authors upon request.