

Abstract
Cells govern tissue shape by exerting highly regulated forces at sites of matrix adhesion. As the major force-bearing adhesion-receptor protein, integrins have a central role in how cells sense and respond to the mechanics of their surroundings. Recent studies have shown that a key aspect of mechanotransduction is the cycle by which integrins bind to the matrix at the leading cell edge, attach to the cytoskeleton, transduce mechanical force, aggregate in the plasma membrane as part of increasingly strengthened adhesion complexes, unbind and, ultimately, are recycled. This mechanical cycle enables the transition from early complexes to larger, more stable adhesions that can then rapidly release. Within this mechanical cycle, integrins themselves exhibit intramolecular conformational change that regulates their binding affinity and may also be dependent upon force. How the cell integrates these dynamic elements into a rigidity response is not clear. Here, we focus on the steps in the integrin mechanical cycle that are sensitive to force and closely linked to integrin function, such as the lateral alignment of integrin aggregates and related adhesion components.
Key words: Cell-matrix adhesion, Integrin dynamics, Mechanotransduction, Vinculin, Talin, Integrin seregation, Adhesion remodelling
Introduction
Cells shape tissues by pulling on neighboring cells and extracellular matrices (ECMs), creating specific levels of tension. In turn, cells are finely attuned to the forces and rigidity of their surroundings. As rigidity is defined by the force per unit displacement, rigidity-sensing cells must measure both force and displacement. Across different tissue types, rigidities are in the range of 1-100 kPa, from the softness in which fat cells or neurons thrive, to the relative stiffness that is home for chrondrocytes (Discher et al., 2005). Yet, despite differences in tissue and cell types, force and position are crucial aspects of many, if not most, cell-matrix interactions. For example, fibroblasts and endothelial cells periodically contract fibronectin to test its rigidity (Giannone et al., 2004). When that process is analyzed in detail, it appears to be controlled by a series of mechanical steps that result in periodic rows of αVβ3-integrin aggregates together with early adhesion components (Giannone et al., 2007). Similarly, to produce a uniform displacement on substrates of increasing rigidity, epithelial cells recruit additional motor proteins to generate higher forces (Saez et al., 2005). This implies a rapid feedback mechanism at sites of integrin-mediated attachment, between the rigidity-sensing system and the force-producing machinery.
A cell, probing its environment, initiates matrix adhesion through actin-dependent protrusions that bring integrins at the leading edge in contact with the matrix where they can bind (Fig. 1A). The binding of integrin to the ECM is rapidly followed by integrin binding to the actin cytoskeleton, which is typically moving inwards from the site of assembly at the leading edge towards the cell center. Thus, a pulling force is quickly generated across nascent integrin-matrix linkages (Fig. 1B). Within seconds, these initial sites of integrin-ECM linkage begin to strengthen as additional components are recruited under force (Fig. 1C) (Galbraith et al., 2002; von Wichert et al., 2003b). On a larger scale, the pulling force also acts to either pull the matrix over the cell or pull the cell over the matrix. Following this matrix movement or, in the case of cell migration, following this cell movement, the integrins then release from the matrix. When forces are sufficiently high – that is, when the substrate is sufficiently rigid – sites of integrin-mediated adhesion undergo further maturation, extending anisotropically several μm in length as additional proteins are recruited. These centripetally polarized supramolecular structures have been termed `focal adhesions'. In supplementary material Fig. S1, a single integrin is highlighted during each step of this mechanical cycle. The mechanism of cell-derived tension that drives this mechanical cycle is described in Box 1.
Fig. 1.
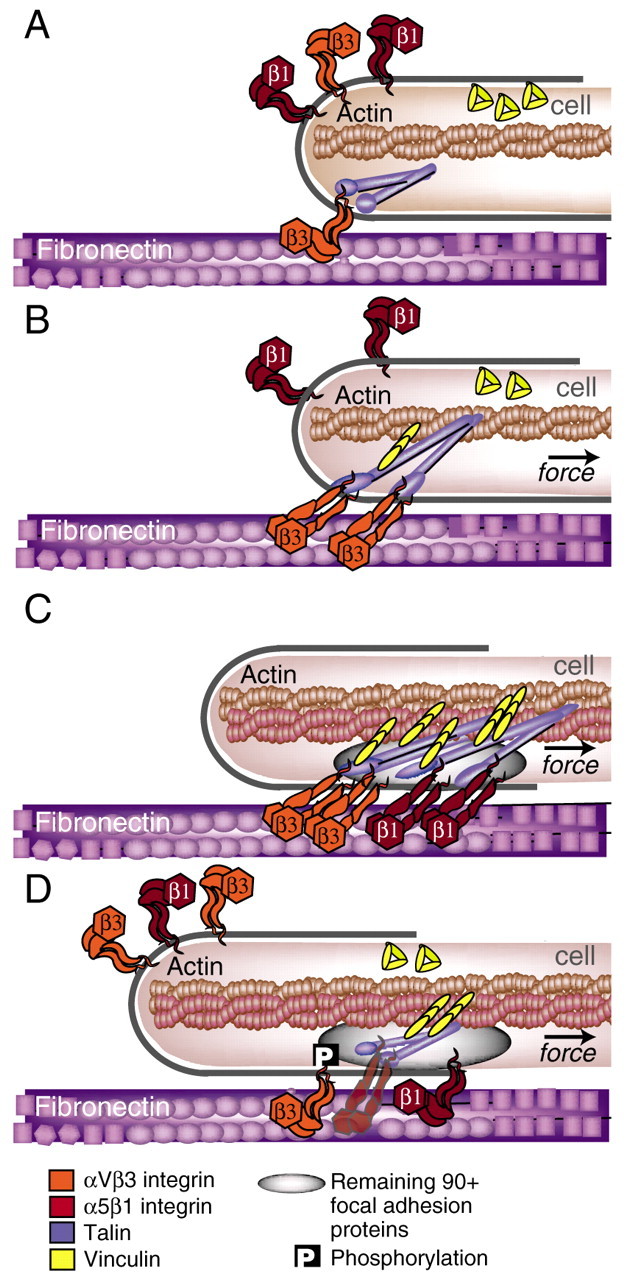
The mechanical integrin cycle. (A) Cell-ECM adhesion occurs when actin-dependent protrusions bring integrins at the leading edge (orange) in contact with the matrix (purple) where they can bind. (B) Next, the integrins link to the actin cytoskeleton through adaptor proteins, such as talin (blue), Shp2, filamin or α-actinin. Integrins bind to these adaptor proteins through their β-tails. Rearward actin flow, generated by actin polymerization and actomyosin contractions (see Box 1) induces a pulling force on the integrin-ECM linkage. On sufficiently rigid substrates, this may serve to accelerate an integrin-activating conformational change, as well as a talin stretch, which may expose buried vinculin-binding sites (yellow). Although the bent conformation of the ligand-bound αVβ3-integrin crystal structure produced much controversy in the integrin field (Liddington and Ginsberg, 2002; Mould et al., 2003), it has subsequently been shown in electron microscopy experiments to stably bind fibronectin (Adair et al., 2005). Force might accelerate the switch to high-binding affinity by freeing the ligand-bound integrin head from the constraints of neighboring domains, which would essentially accelerate the allosteric pathway to the activated state (Puklin-Faucher et al., 2006). (C) The cell begins to pull itself over the site of adhesion. Intramolecular conformational changes in α5β1 integrins facilitate their inward translocation, whereas αVβ3 integrins remain anchored at the edge. This segregation of integrins may further facilitate the talin stretch. At this stage of adhesion, a wide variety of intracellular focal-adhesion proteins are accumulated in the adhesive plaque (grey oval). (D) Ultimately, highly clustered integrins switch from high- to low-binding affinity, possibly catalyzed by the phosphorylation of β3-integrin tails. Membrane exocytosis places recycled, low-affinity integrins at the end of microtubules, often 2-4 μm away from the leading edge. The integrin turnover in focal adhesions (from C to D) is ∼1-3 minutes (Hu et al., 2007). For the description of a single integrin see supplementary material Fig. S1.
Force-dependent strengthening of adhesion sites is remarkable, because all receptor-ligand bonds eventually break under high forces. For example, the lifetime of the notoriously strong avidin-biotin bond is reduced from more than a day to ∼1 minute under a force of 5 pN (Merkel et al., 1999). Two logical explanations for force-dependent adhesion strengthening are increased recruitment of integrin receptors, which is known to occur when integrin-mediated adhesions exhibit physical growth under force, and/or catch bonds, receptor-ligand complexes that exhibit an increased lifetime under mechanical load. Moreover, as the proteins in these force-regulated adhesion sites turn over rapidly to enable cell spreading and migration, their strength and position are also remodeled under force. Cells clearly control this cycle of integrin-dependent attachment, force production and release to generate precise tissue morphologies. A guiding question at the forefront of current cell science is how these sites of integrin-mediated adhesion participate in this process of cell-tissue morphodynamics.
In an effort to understand how integrin-mediated force regulates protein recruitment and the strengthening of focal-adhesions, physical models have been derived. For example, elastic strain is proposed to induce anisotropic protein aggregation under force because of the resulting asymmetric extension and compression of the focal-adhesion entity (Besser and Safran, 2006; Nicolas et al., 2004). Alternatively, protein aggregation under force has been proposed to be the result of a purely thermodynamic process, whereby stress-induced changes in the chemical potentials of focal-adhesion proteins are compensated for by the binding of additional focal-adhesion proteins (Shemesh et al., 2005). Although these models describe general physical mechanisms of how force governs focal-adhesion morphodynamics, recent experimental and computational findings have revealed how mechanical stress directly regulates the function of integrins and several other associated molecules, such as talin and vinculin. Further, force-dependent tyrosine phosphorylation can result in dramatic changes in signaling pathways that alter integrin-ligand binding (Tamada et al., 2004; Sawada et al., 2006). Together, an understanding of how different signals affect the distinct molecular dynamics of the mechanical integrin adhesion cycle can illuminate the basis of changes in cell and tissue shape.
Box 1. Cells steer adhesion-site maturation by forces that are generated through actin assembly and actomyosin contractions
In the most peripheral, 1-4-μm region of a spreading cell, termed the lamellipodium, relatively fast rates of actin assembly and disassembly generate force on the cell membrane. This force is comparatively small (Dubin-Thaler et al., 2008; Raucher and Sheetz, 2000) and has been linked to the initiation of integrin-mediated adhesion (Alexandrova et al., 2008; DeMali et al., 2002).
Just behind the lamellipodium, in a region termed the lamellum, the retrograde actin flow is slower. The boundary between the lamellipodium and the lamellum appears to be regulated by the development of nascent adhesion sites into mature focal adhesions (Alexandrova et al., 2008). The contraction of actomyosin filament bundles, called stress fibers, drives the growth of focal complexes into focal adhesions. Myosin II A and B are the isoforms responsible for generating this force (Cai et al., 2006). Whereas myosin II inhibition blocks the conversion from focal complexes to focal adhesions, the application of a local, external pulling force can replace the role of actomyosin contractility and restore focal-adhesion formation (Riveline et al., 2001).
Force generated from actomyosin contractions is also thought to contribute to early adhesion-site formation in the lamellopodium (Galbraith et al., 2002). In support of this notion, adhesion-site formation at the cell edge has been found to have the same periodicity as myosin-mediated contractions (Giannone et al., 2004). Recently, lamellopodium actin has been shown to form an adaptable, mechanical link between the site of an actin polymerization at the leading edge and the myosin motor activity in the lamellum (Giannone et al., 2007). Thus, the interplay between forces generated by actin polymerization and by myosin-mediated contractility work cooperatively to drive the integrin mechanical cycle (Dubin-Thaler et al., 2008).
In this Commentary, we will focus on the integrin-dependent motility that has been studied extensively in primarily mammalian fibroblastic, immune and endothelial cells, with many corresponding features also evident in stem cells and cancer cells. First, we will discuss intramolecular integrin dynamics, which govern integrin activation. Second, we will consider focal-adhesion assembly in terms of integrin aggregation under force, which exhibits a linear directionality that may regulate intracellular signaling pathways.
Integrin activation under force
An understanding of integrin-mediated mechanosensing begins with integrin activation, which governs integrin-binding kinetics and clustering (Cluzel et al., 2005; Kim et al., 2004). Integrin activation occurs allosterically, involving long-range intramolecular conformational changes that can originate from the extracellular or cytoplasmic end of the integrin heterodimer. Integrin heterodimers comprise non-covalently bound α- and β-subunits, which associate to form the extracellular ligand-binding head, two multi-domain `legs', two single-pass transmembrane helices and two short cytoplasmic tails. All known integrin heterodimers contain the βA domain (also called the I-like or βI domain), which is located at the extracellular end of the β-subunit. Mutational and monoclonal-antibody experiments have shown that the switch from low- to high-binding affinity in the ECM-binding integrin headpiece involves an increase in the hinge angle between the βA- and hybrid-domains (Luo et al., 2003; Luo et al., 2004; Mould et al., 2003). X-ray crystallographic structures provide the stationary endpoints of this conformational switch in the unliganded closed-hinge and the ligand-bound open-hinge β3-integrin headpiece domains (Xiao et al., 2004; Xiong et al., 2001). Molecular dynamics (MD) simulations of the β3-integrin headpiece domains have illustrated the Ångstrom-level structural pathway of ligand-induced hinge-angle opening (Puklin-Faucher et al., 2006).
One hallmark of allosteric proteins such as integrins is their bi-directionality, which means that the same activating structural pathway can be induced by extracellular (`outside in') or intracellular (`inside out') factors (Hynes, 2002). In vivo events that are known to activate integrins are the ligand binding by the extracellular head (Takagi et al., 2002) or the talin binding by the intracellular tail of the β-subunit (Tadokoro et al., 2003). In the absence of force, integrin activation occurs within seconds. However, there are clearly mechanical signals that can induce events downstream of activation in seconds, such as integrin aggregation (Giannone et al., 2004) and adhesion-protein assembly (Galbraith et al., 2002; Riveline et al., 2001; von Wichert et al., 2003b). In the case of T cells, firm integrin adhesiveness was shown to be tightly regulated by mechanical signals that involve the combination of force from shear-fluid flow and immobilized chemokines (Woolf et al., 2007). Consistent with these observations, when the application of ligand-mediated mechanical force was simulated in steered MD (SMD) investigations, it was shown to accelerate the allosteric pathway to activation in the integrin headpiece to the sub-microsecond timeframe (Puklin-Faucher et al., 2006). As binding to ECM ligands is known to activate integrins under equilibrium conditions (Takagi et al., 2002) and binding is needed for force to be transduced across integrins, the major effect of force on integrin activation may be in accelerating the allosteric activation pathway and, thereby, in stabilizing bonds that would otherwise dissociate within sub-seconds.
It is logical to postulate that the application of a force vertical to the membrane would induce an activating conformational change in integrins (as shown in Fig. 1). Such a force could be generated even though the force vector is often almost parallel to the membrane, rather than perpendicular. αVβ3 integrins can stably bind fibronectin with only a modest (∼11°) increase in the angle of their headpiece hinge and with a severe bend (of ∼135°, based on crystallographic data) in their extracellular legs (Adair et al., 2005). After matrix is bound, this bond could potentially be stabilized by force-induced conformational change. For example, with only the β- and not the α-cytoplasmic tail linked to the cytoskeleton, force could vary the interdomain headpiece hinge via separation of the heterodimer legs as the β-subunit becomes aligned along the force vector.
Intracellularly, binding of the talin head to the β-subunit of the integrin tail has been shown to activate integrins by disrupting membrane-proximal and transmembrane associations with the neighboring α-subunit domains (Tadokoro et al., 2003; Wegener et al., 2007). Recently, the structurally homologous kindlin family of proteins has been shown to interact directly with β3- and β1-integrin tails and to catalyze (kindlin-2, also known as FERMT2) or even supersede (kindlin-3, also known as FERMT3) integrin activation by talin (Ma et al., 2008; Moser et al., 2008). To influence the ECM-binding affinity of the integrin head, the structural change induced by kindlin and talin at the integrin tails must propagate across the multiple leg domains of the ∼28-nm-long integrin molecule. As described above, ligand-mediated force may accelerate this allosteric structural change (Alon and Dustin, 2007; Puklin-Faucher et al., 2006).
As the highly flexible integrin β-tails provide a scaffold for a wide range of cytoskeletal proteins (Calderwood et al., 2003) and are extremely flexible, there is also the possibility that ligand-mediated force could accelerate binding to kindlin and talin by making the binding sites more accessible through disruption of the membrane-proximal and transmembrane integrin-heterodimer associations. In support of this, the presence of the head part of talin – but not the rod – appears to stabilize integrin binding to fibronectin even in the absence of actin binding (Zhang et al., 2008). The binding of single fibronectin trimers is highly dependent upon talin, as is a weak slip bond with the actin cytoskeleton (Jiang et al., 2003). Also, swapping α- and β-tails blocked lateral integrin aggregation, but moving the β1 tail further from the membrane by lengthening the membrane-proximal domain of the α5-chimera with a spacer restored the lateral aggregation that was dependent upon the β1 tail (Partridge et al., 2006). This result implies that allowing the distal β-cytoplasmic domain to adopt a distinctive conformation by freeing it from the proximal α-cytoplasmic domain is the structural event that drives aspects of ligand-dependent integrin signalling such as lateral aggregation. Together, these findings imply that the physical unmasking of kindlin- and talin-binding sites on the integrin β-tail can stabilize their structural and functional state (Ulmer et al., 2003). Although there is considerable evidence that the early linkages between integrins and the cytoskeleton depend upon kindlin-2, kindlin-3 and talin, there are other integrin-tail-binding partners that can also link to the contractile actin cytoskeleton in other adhesion processes. These include filamin, α-actinin, melusin, SH2-domain-containing protein-tyrosine phosphatase (Shp2), skelemin, integrin-linked kinase and, possibly, myosin (Phillips et al., 2001; Critchley and Ginggras, 2008; Kiema et al., 2006; Pavalko et al., 1991; von Wichert et al., 2003a).
Force generation, integrin segregation and adhesion growth
Nascent focal adhesions, termed `focal complexes', mediate high forces relative to mature focal adhesions (Beningo et al., 2001) and grow in size in linear proportion to the traction forces exerted upon them (Balaban et al., 2001). These traction forces are, in turn, directly proportional to the rigidity of the extracellular substrate (Saez et al., 2005). During mechanosensing, the density of integrins that surround these sites of attachment increases in a directional fashion. As shown schematically in Fig. 1C and with antibody staining in Fig. 2, αVβ3 integrins remain anchored at the distal end, closer to the leading edge, and α5β1 integrins translocate to the proximal end, closer to the center of the cell. The spatial segregation of α5β1- from αVβ3-integrins has been well established during the transition from focal to fibrillar adhesions, when cells create extracellular fibrils from plasma fibronectin (Pankov et al., 2000; Zamir et al., 2000). During this process, termed `fibrillogenesis', α5β1 integrins translocate inwards by a distance of ∼10 μm. This movement along the actin cytoskeleton serves to elongate and organize newly formed fibrils into the ECM. In the absence of fibrillogenesis, for example, when the fibronectin matrix is non-deformable (Katz et al., 2000), α5β1 integrins also aggregate in a linear fashion inside focal adhesions. In this fashion, mature focal adhesions exhibit a linear, centripetal spatial segregation of αVβ3- and α5β1-integrins that extends ∼5-6 μm along contractile actomyosin bundles (Felsenfeld et al., 1999). As this highly regulated integrin activity is governed by a force-dependent structural change and follows the primary force vector, it is logical to propose that force and rigidity choreograph this directionality in integrin dynamics.
Fig. 2.
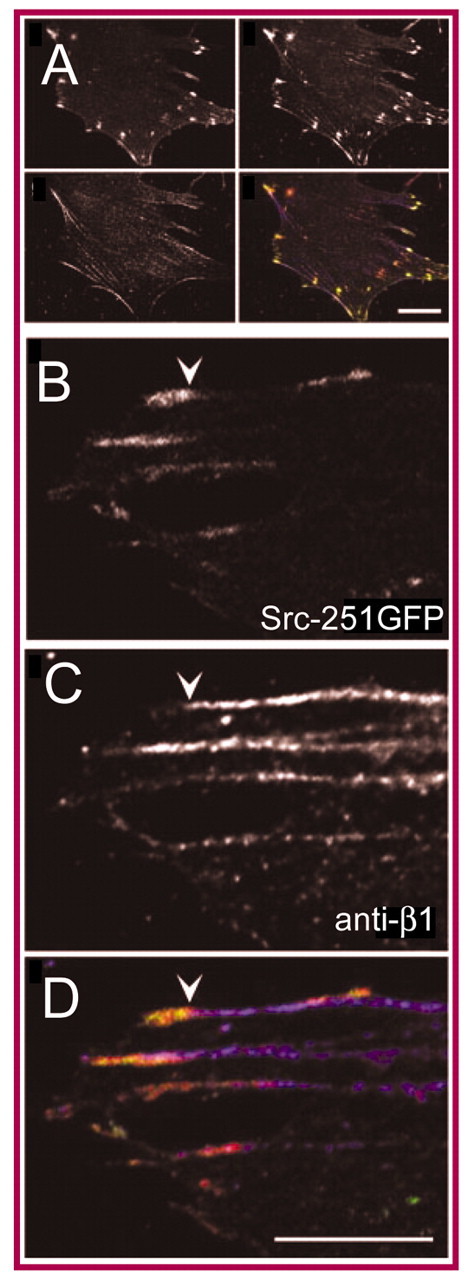
Segregation of αVβ3- and α5β1-integrins in focal adhesions. The segregation of αVβ3- and α5β1-integrins in focal adhesions (depicted schematically in Fig. 1D) is shown here by antibody staining (Felsenfeld et al., 1999). (A) Src-deficient cells that expressing a truncated form of Src tagged with GFP (Src-251GFP) were fixed and stained with antibodies recognizing β1 integrins, αVβ3 integrins and vinculin. (B) In higher-magnification views, the αVβ3 subunit can be seen to colocalize at the periphery of focal adhesions, with vinculin and Src-251GFP. Vinculin staining was co-distributed with that of Src-251GFP in all cases. (C). By contrast, β1 integrins distributed in longer peripheral stripes (consistent with the distribution of stress fibers) that did not overlap with the distribution of αVβ3–Src-251GFP. Arrowhead indicates the boundary of staining. (D) Overlap of GFP and vinculin staining without β1 integrins is indicated by yellow pixels. Scale bars: 10 μm (A) and 5 μm (D). Images reproduced with permission (Felsenfeld et al., 1999).
At the leading edge of mechanosensing cells, αVβ3 integrins form the nascent cell-ECM contacts and mediate initial force-accelerated adhesion-strengthening events (von Wichert et al., 2003b). For example, during the early stages of cell-adhesion-contact formation, activation of the Src family kinase Fyn requires that surface-bound αVβ3 integrins interact with receptor-like protein tyrosine phosphatase α (RPTPα) (Su et al., 1999; von Wichert et al., 2003b). This RPTPα-induced activation of Fyn at the leading edge of rigidity-sensing cells is necessary for the force-dependent strengthening of αVβ3-integrin–cytoskeleton connections (Jiang et al., 2006; von Wichert et al., 2003b).
Similar to αVβ3 integrins, α5β1 integrins have also been shown to bind to the ECM at the leading edge of the cell and translocate inwards (Nishizaka et al., 2000). Release of α5β1 integrins from fibronectin-coated beads at the back of the lamellipodium, where adhesions often end, was observed in experiments using optical tweezers (Nishizaka et al., 2000). Humphries and co-workers have now shown that the inwards translocation of α5β1 integrins within focal adhesions corresponds to a series of distinctive conformational changes in the extracellular domains of α5β1 integrins, namely from bent to straight and then to separated (Clark et al., 2005). Similarly, a recent study shows that the inwards translocation of α5β1 integrins causes centripetal focal-adhesion orientation but relies upon the extracellular binding interactions and the subsequent conformational changes of α5β1- but not αVβ3-integrins (Huveneers et al., 2008). Alternative explanations for the spatial segregation of αVβ3- and α5β1-integrins include integrin-recycling pathways (White et al., 2007) and retrograde flux of actin filaments (Guo and Wang, 2007).
Talin
Talin binds directly to β1-, β2- and β3-integrins. Recently, differences in the way that talin interacts with integrins have come to light. For instance, although a small fragment of the N-terminal talin head domain, the F3 sub-domain, is sufficient to activate β3 integrins (Calderwood et al., 2003; Garcia-Alvarez et al., 2003), this sub-domain is not sufficient to activate β1 integrins. To produce detectable β1-integrin activation, the entire ∼50 kDa talin head is required (Bouaouina et al., 2008).
Similar to activation, clustering has structural origins. Clustering of αVβ3- and α5β1-integrins, which follows activation (Kim et al., 2004), is driven by interactions with the talin head domain (Cluzel et al., 2005; Zhang et al., 2008). Talin also contains a second integrin-binding site in its C-terminal rod domain, which directly interacts with both β3- and β1-integrin tails (Parsons et al., 2008; Tremuth et al., 2004; Xing et al., 2001). Interaction with the talin rod, however, does not lead to integrin activation; rather, it provides the link to the cytoskeleton, thus enabling the substrate traction forces that are necessary for sustained cell spreading (Moes et al., 2007; Zhang et al., 2008). It has been suggested that the integrin-activating conformational change that is induced by binding of the talin head may result in the exposure of a de novo high-affinity integrin-binding site for the talin rod (Moes et al., 2007). In line with this idea, a recent fluorescence resonance energy transfer (FRET) analysis identified specific interactions between β1 integrins and the talin rod domain but not the talin head domain, leading the authors to suggest that interactions between the β1-integrin and the talin head domain may be transient and/or restricted to early adhesion complexes (Parsons et al., 2008).
The initial binding of β1 integrins to talin is linked to their synchronous sideways movement when clustered at the leading edge by polymerizing actin fibers (Galbraith et al., 2007). Clustering of α5β1 integrins has been linked with the transition from focal adhesions into fibrillar adhesions (Clark et al., 2005). When ligand-dependent translocation of α5β1 integrins in human fibroblasts was blocked and integrin clustering was then induced with monoclonal antibodies, movement of α5β1 integrins out of focal adhesions into fibrillar adhesions was observed. Thus, clustering of α5β1 integrins can independently drive their directional, centripetal translocation into fibrillar adhesions (Clark et al., 2005), where talin is replaced by the adaptor protein tensin (Pankov et al., 2000). Together, these findings point to a model in which talin, which is ∼60 nm long, may ultimately be oriented inside focal adhesions by head and rod contacts with centripetally organized αVβ3- and α5β1-integrins. Force-induced deformation of the integrin-talin linkage may then facilitate exchange to the adapter protein tensin.
Although we are focused here on mammalian integrin interactions, it is interesting to note the similarities and differences relative to Drosophila melanogaster that have recently come to light. Although the integrin link to the cytoskeleton via the talin rod is conserved in both species (Tanentzapf and Brown, 2006), talin is not sufficient to activate the β1-integrin orthologue in Drosophila (termed βPS) (Helsten, 2008). Whereas the integrin link to the cytoskeleton through the talin rod is conserved in both species (Tanentzapf and Brown, 2006), talin is not sufficient to activate Drosophila βPS integrins (Helsten et al., 2008). The authors of this recent finding speculate that the regulation of integrin activation by talin in mammalian cells may have developed “...later in vertebrate evolution to provide exquisite regulation of integrin affinity in highly motile cells” (Helsten et al., 2008).
Vinculin
The talin rod contains multiple binding sites for the adaptor protein vinculin, which is known to be involved in focal-adhesion dynamics (Coll et al., 1995; Volberg et al., 1995) and is recruited to sites of talin-integrin adhesion under tension (Galbraith et al., 2002; Zaidel-Bar et al., 2003). Importantly, talin's binding sites for vinculin are buried inside α-helix bundles under equilibrium conditions and have been shown in silico to be exposed by stretching force in recent SMD simulations (Hytonen and Vogel, 2008). Indeed, traction forces on integrin-mediated adhesions have been shown to result in recruitment of vinculin within tens of seconds (Galbraith et al., 2002) and vinculin activity, in turn, regulates both paxillin recruitment and integrin turnover (Humphries et al., 2007). Currently, direct experimental verification of vinculin binding-site exposure by talin mechanical stretch, as illustrated in Fig. 1A-C, is lacking.
Notably, a decisive factor in adhesion strengthening is not simply the increase in integrin density, but rather the distance between individual integrin molecules. As revealed in a recent study of integrin adhesion strengthening, cyclic Arg-Gly-Asp (RGD) ligands, which preferentially bind αVβ3 integrins, need to be spaced at a distance of 55 nm or less for adhesions to be reinforced (Selhuber-Unkel et al., 2008) and vinculin molecules to bind (Cavalcanti-Adam et al., 2006). Vinculin also assists α5β1-integrin clustering through a tight association with talin (Humphries et al., 2007). Similar to mammalian talin, the N-terminal head and C-terminal tail domains of mammalian vinculin have been pinpointed as being the locations that support clustering and mechanical linkage to the actin cytoskeleton, respectively (Humphries et al., 2007). Together, these findings portray a model of focal-adhesion dynamics in which clustered cytoskeletal-integrin-ECM linkages are crosslinked by talin homodimers, which then regulate integrin dynamics by recruiting vinculin when stretched in a directional fashion (Fig. 1C).
Force acceleration of biochemical pathways
In addition to spatial arrangements, temporal changes are a crucial component of integrin-mediated mechanotransduction. This is illustrated on the intramolecular scale in MD and SMD simulations of the integrin headpiece, which show that mechanical force accelerates the same allosteric pathway to hinge-angle opening that is induced by ligand binding under equilibrium conditions (Puklin-Faucher et al., 2006). Currently, direct experimental verification of integrin activation by mechanical force is lacking, owing in part to the tendency of integrins to cluster upon adhesion and thus obscure measurements of monovalent substrate interactions. On the intermolecular scale, vinculin and talin can drive integrin clustering in focal adhesions independently of tensile force (Humphries et al., 2007). Similarly, in mature focal complexes, αVβ3 integrins interact with Src family kinases directly (Arias-Salgado et al., 2003), versus interacting through RPTPα in nascent integrin-ECM linkages under force (von Wichert et al., 2003b).
In RPTPα (RPTPA)-knockout cells, as well as talin 1 (TLN1)-knockout cells, integrin-ECM adhesion sites eventually form and become strengthened in a manner similar to that observed on a faster timescale under force (Kostic et al., 2007; Priddle et al., 1998). This indicates that other proteins (probably talin 2) can substitute for talin 1 in building integrin-cytoskeleton connections (Giannone et al., 2003; Zhang et al., 2008). Correspondingly, a model of force-accelerated integrin activation has recently been proposed in which integrins that are not anchored to the ECM are more mobile under force and are thus readily recruited to sites that have already been stabilized by anchored integrins (Rose et al., 2007).
Together, these observations point to a common mechanosensing mechanism: force-induced acceleration of biochemical events that are likely to occur, albeit on slower timescales, in a diffusion-controlled fashion in the absence of force. In this scenario, force strengthens adhesions by accelerating reactions and governing the spatial pattern of the cell-ECM interface, whereas relaxation of force inhibits strengthening and accelerates dissociation.
Force, integrin detachment and adhesion remodeling
Although detachment has received less attention, it is an extremely important, tension-dependent step in the integrin mechanical cycle, because cells must release to move and restructure their environment. The lifetime of focal adhesions as distinct entities is in the order of 5-10 minutes (Ren et al., 2000). Within focal adhesions, integrins that are directly linked to the ECM exhibit the slowest dynamics relative to other focal-adhesion proteins, with exchange rates in the order of 1-3 minutes (Ballestrem et al., 2001; Hu et al., 2007). Vinculin, focal adhesion kinase (FAK) and talin have much faster exchange rates than integrins (Hu et al., 2007; von Wichert et al., 2003a), and integrin turnover is preceded by the switch from high to low integrin-binding affinity (Cluzel et al., 2005). What drives this hierarchy in protein exchange rates, and how does that govern integrin-binding affinity under force? More generally, how is adhesion-site remodeling controlled? To address this larger question, which is crucial for understanding tissue homeostasis, we will consider the role of integrin segregation in adhesion remodeling in terms of syndecans, phosphorylation and recycling.
Syndecans
Syndecans, similar to integrins, are a family of transmembrane ECM-adhesion receptors. Whereas integrins bind to peptide motifs in their ECM ligand (e.g. the RGD loop on the tenth fibronectin type III module), syndecans bind to heparin-binding motifs in their ECM ligand (e.g. the heparin-binding motif on the thirteenth type III fibronectin module). Together, syndecans and integrins have been shown to facilitate the transduction of multiple signaling pathways (for a review, see Morgan et al., 2007). In particular, syndecan 1 has been shown to modulate αVβ3-integrin-binding affinity (Beauvais et al., 2004), whereas syndecan 4 interacts, albeit indirectly (Zimmermann et al., 2005), with α5β1 integrin in focal adhesions. The engagement of the ECM with syndecan 4 is linked with activation of FAK and Src, two non-receptor kinases that have important roles in the weakening of integrin-cytoskeletal linkages under force (Felsenfeld et al., 1999; Galbraith et al., 2002; von Wichert et al., 2003a). In vivo, FAK inhibition occurs in a rigidity-dependent fashion as its expression is required for durotaxis on collagen (Wang et al., 2001). It also occurs in a temporal fashion, as the prolonged association of FAK within focal adhesions is linked with increased FAK activity and increased focal-adhesion disassembly (Giannone et al., 2004). When activated, FAK and Src have been shown to form a complex with one another, which extends the lifetime of their active states (Lietha et al., 2007). Whereas Src has been shown to colocalize with αVβ3- but not α5β1-integrins (Felsenfeld et al., 1999), evidence of regulation of syndecan 4 by FAK is robust (reviewed by Morgan et al., 2007). Together, these findings suggest that, similar to focal-adhesion strengthening through talin crosslinking across linearly arranged αVβ3 and α5β1 integrins, focal-adhesion turnover can be similarly regulated by the linear arrangement of αVβ3- and α5β1-integrin, in synergy with syndecan 1 and syndecan 4, and crosslinked inside and outside the cell by the FAK-Src complex and fibronectin module, respectively.
Integrin phosphorylation
The dissociation of talin from β3-integrin tails is probably an important event along the pathway to force-induced focal-adhesion turnover. When the localization of talin to adhesion sites is altered by the injection of antibody (Nuckolls et al., 1992) or sequestration of phosphoinositides (Martel et al., 2001), there is no simultaneous disruption of mature adhesion sites. However, antibody injection disrupts newly formed adhesion sites or prevents their formation, indicating the crucial role of talin in early rather than mature adhesion sites. As talin cannot bind to phosphorylated β3-integrin tails, one possible mechanism of force-accelerated integrin turnover comes from the tension-induced increase in kinase and phosphatase activity that has been shown to occur in the vicinity of the β3-integrin tail under force (Giannone and Sheetz, 2006; Tamada et al., 2004).
Talin activates β3 integrins by influencing specific interactions in the membrane-proximal region of the integrin tail (Wegener et al., 2007). In contrast to talin, binding of the signaling adaptor Shc (Cowan et al., 2000) requires phosphorylation of the β3-integrin tails. Interestingly, Shc requires phosphorylation of only the membrane-distal and not the membrane-proximal tyrosine of the β3 tail (Cowan et al., 2000), thus leaving the membrane-proximal region free to renew its association with the α-subunit, which could switch the integrin from the high-affinity to the low-affinity state. Perhaps the binding of Shc to the β3-integrin tail promotes the switch from high to low integrin-binding affinity by promoting renewed interactions between integrin transmembrane domains and membrane-proximal segments of the cytoplasmic tails. In support of this hypothesis, Shc association with αVβ3 integrins has been shown to be induced under shear-fluid flow in endothelial cells, in which dynamic remodeling of the adhesion plaques to allow strategic positioning for sustaining force requires constant integrin association with and dissociation from the ECM (Chen et al., 1999).
It has been previously proposed that tyrosine phosphorylation functions as a `molecular switch' for the binding interactions of β3-integrin tails following adhesion, activation and clustering (Calderwood et al., 2003). Although a recent in vivo study has revealed that cytoplasmic regulation of β1-integrin function is phosphorylation independent – with the hydrophobic interactions that the tyrosines support having the key role instead (Chen et al., 2006) – phosphorylation of the cytoplasmic tail of β3 integrins is crucial for platelet aggregation (Blystone et al., 1997; Jenkins et al., 1998; Law et al., 1999). Knock-in mice, in which each of the tyrosines (Y) in the two NPxY motifs on the β3-integrin tail was replaced by phenylalanine, displayed platelet clotting deficiencies (Law et al., 1999). These mutational studies showed that disruption of β3-tail phosphorylation does not disrupt initial aggregation rates but, rather, disrupts the ability of β3 integrins to maintain aggregation. Interestingly, fluorescence recovery after photobleaching (FRAP) experiments have shown that exchange rates are faster in high-density β3-integrin clusters and slower in low-density β3-integrin clusters (Ballestrem et al., 2001). Intracellular tension that is induced by RhoA or blocked by the protein-kinase inhibitor staurosporine correlates with the formation and maintenance of high-density or low-density αVβ3-integrin focal adhesions, respectively (Ballestrem et al., 2001). Increased integrin density under force has thus been proposed to lead to lower-affinity integrin-ECM binding (Ballestrem et al., 2001). Together, these findings suggest that clustering of αVβ3 integrins, and the subsequent competition for β3-integrin tails by force-increased kinase activity, may be a key event in focal-adhesion turnover.
Recycling of integrins to the leading edge
Although it is possible for the integrins to diffuse back to the leading edges of active cells, observations of the diffusion of unliganded integrins have revealed that they often undergo active movements towards the leading edge (Schmidt et al., 1993). Similar to the sites of integrin release from adhesions, the major sites of membrane exocytosis are located at the ends of microtubules, often 2-4 μm back from the leading edge, and it takes integrins nearly 10 minutes to diffuse to the leading edge at the observed diffusion rates (D= (0.1 micrometer)2/second) (Schmidt et al., 1995; Schmidt et al., 1993). Recently, recycling of αVβ3 integrins to the leading edge has been proposed to resensitize the integrins to ligand occupation, by acting to return them to the membrane that is competent to bind and promote migration (White et al., 2007). Thus, recycling of integrins to the leading edge through the active transport mechanism is likely to be an important component of adhesion remodeling during rapid cell motility.
Conclusions and perspectives
The integrin mechanical cycle (supplementary material Fig. S1) is crucial for cellular function and depends upon the biophysical and biochemical changes in integrin structure and post-translational modifications of both integrins and associated proteins. In the initial binding to matrix, both intracellular and extracellular factors can alter the level of integrin binding, in terms of affinity (e.g. talin-head activation or matrix binding) as well as avidity (e.g. edge localization and lateral interactions with externally activated proteins such as syndecans). Stabilization of integrin-matrix binding and the formation of an adhesion complex by lateral recruitment of more integrins is probably catalyzed at early times by force-dependent alterations in talin and, possibly, in the integrin itself. As the lifetimes of components in the adhesion complexes are much shorter than the lifetimes of focal adhesions themselves, there must be a self-renewal process that supports tension while enabling turnover. Recruitment of new actin filaments and actin-binding components in overlapping arrays through radial or simply lateral aggregation could facilitate this.
The local and directional growth of focal adhesions under force can be explained in terms of a network of mechanotransduction pathways. Force probably accelerates integrin activation, both by extracellular and intracellular rearrangements, induces protein recruitment through protein stretching and accelerates integrin clustering, leading to deactivation. Dispersal of adhesion contacts is favored by the loss of tension. The recycling of integrins to the leading edge of moving cells allows the integrin cycle to begin again. Force clearly alters the biochemical steps in the integrin cycle, which enables cellular signaling pathways to affect the strength of the adhesions indirectly by altering cell-force generation, or directly by altering adhesion protein dynamics.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/2/179/DC1
Thanks to Pere Roca-Cusachs, Olivier Rossier, Nils Gauthier, Xian Zhang and Viola Vogel for helpful discussions. This work was supported by NSF funds and the Nanotechnology Center for Mechanics in Regenerative Medicine.
References
- Adair, B., Xiong, J.-P., Maddock, C., Goodman, S., Arnaout, M. A. and Yeager, M. (2005). Three-dimensional EM structure of the ectodomain of integrin alphaVbeta3 in a complex with fibronectin. J. Cell Biol. 168, 1109-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrova, A. Y., Arnold, K., Schaub, S., Vasiliev, J. M., Meister, J. J., Bershadsky, A. D. and Verkhovsky, A. B. (2008). Comparative dynamics of retrograde actin flow and focal adhesions: formation of nascent adhesions triggers transition from fast to slow flow. PLoS ONE 3, e3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon, R. and Dustin, M. L. (2007). Force as a facilitator of integrin conformational changes during leukocyte arrest on blood vessels and antigen-presenting cells. Immunity 26, 17-27. [DOI] [PubMed] [Google Scholar]
- Arias-Salgado, E. G., Lizano, S., Sarkar, S., Brugge, J., Ginsberg, M. H. and Shattil, S. J. (2003). Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc. Natl. Acad. Sci. USA 100, 13298-13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban, N. Q., Schwarz, U. S., Riveline, D., Goichberg, P., Tzur, G., Sabanay, I., Mahalu, D., Safran, S., Bershadsky, A., Addadi, L. et al. (2001). Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat. Cell Biol. 3, 466-472. [DOI] [PubMed] [Google Scholar]
- Ballestrem, C., Hinz, B., Imhof, B. A. and Wehrle-Haller, B. (2001). Marching at the front and dragging behind: differential alphaVbeta3-integrin turnover regulates focal adhesion behavior. J. Cell Biol. 155, 1319-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauvais, D. M., Burbach, B. J. and Rapraeger, A. C. (2004). The syndecan-1 ectodomain regulates alphavbeta3 integrin activity in human mammary carcinoma cells. J. Cell Biol. 167, 171-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beningo, K. A., Dembo, M., Kaverina, I., Small, J. V. and Wang, Y. L. (2001). Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J. Cell Biol. 153, 881-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besser, A. and Safran, S. A. (2006). Force-induced adsorption and anisotropic growth of focal adhesions. Biophys. J. 90, 3469-3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blystone, S. D., Williams, M. P., Slater, S. E. and Brown, E. J. (1997). Requirement of integrin beta3 tyrosine 747 for beta3 tyrosine phosphorylation and regulation of alphavbeta3 avidity. J. Biol. Chem. 272, 28757-28761. [DOI] [PubMed] [Google Scholar]
- Bouaouina, M., Lad, Y. and Calderwood, D. A. (2008). The N-terminal domains of talin cooperate with the phosphotyrosine binding-like domain to activate beta1 and beta3 integrins. J. Biol. Chem. 283, 6118-6125. [DOI] [PubMed] [Google Scholar]
- Cai, Y., Biais, N., Giannone, G., Tanase, M., Jiang, G., Hofman, J. M., Wiggins, C. H., Silberzan, P., Buguin, A., Ladoux, B. et al. (2006). Nonmuscle myosin IIA-dependent force inhibits cell spreading and drives F-actin flow. Biophys. J. 91, 3907-3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood, D. A., Fujioka, Y., de Pereda, J. M., Garcia-Alvarez, B., Nakamoto, T., Margolis, B., McGlade, C. J., Liddington, R. C. and Ginsberg, M. H. (2003). Integrin beta cytoplasmic domain interactions with phosphotyrosine-binding domains: a structural prototype for diversity in integrin signaling. Proc. Natl. Acad. Sci. USA 100, 2272-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalcanti-Adam, E. A., Micoulet, A., Blummel, J., Auernheimer, J., Kessler, H. and Spatz, J. P. (2006). Lateral spacing of integrin ligands influences cell spreading and focal adhesion assembly. Eur. J. Cell Biol. 85, 219-224. [DOI] [PubMed] [Google Scholar]
- Chen, H., Zou, Z., Sarratt, K. L., Zhou, D., Zhang, M., Sebzda, E., Hammer, D. A. and Kahn, M. L. (2006). In vivo beta1 integrin function requires phosphorylation-independent regulation by cytoplasmic tyrosines. Genes Dev. 20, 927-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, K. D., Li, Y. S., Kim, M., Li, S., Yuan, S., Chien, S. and Shyy, J. Y. (1999). Mechanotransduction in response to shear stress: roles of receptor tyrosine kinases, integrins, and Shc. J. Biol. Chem. 274, 18393-18400. [DOI] [PubMed] [Google Scholar]
- Clark, K., Pankov, R., Travis, M. A., Askari, J. A., Mould, A. P., Craig, S. E., Newham, P., Yamada, K. M. and Humphries, M. J. (2005). A specific alpha5beta1-integrin conformation promotes directional integrin translocation and fibronectin matrix formation. J. Cell Sci. 118, 291-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluzel, C., Saltel, F., Lussi, J., Paulhe, F., Imhof, B. A. and Wehrle-Haller, B. (2005). The mechanisms and dynamics of (alpha)v(beta)3 integrin clustering in living cells. J. Cell Biol. 171, 383-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll, J. L., Ben-Ze'ev, A., Ezzell, R. M., Rodriguez Fernandez, J. L., Baribault, H., Oshima, R. G. and Adamson, E. D. (1995). Targeted disruption of vinculin genes in F9 and embryonic stem cells changes cell morphology, adhesion, and locomotion. Proc. Natl. Acad. Sci. USA 92, 9161-9165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan, K. J., Law, D. A. and Phillips, D. R. (2000). Identification of shc as the primary protein binding to the tyrosine-phosphorylated beta 3 subunit of alpha IIbbeta 3 during outside-in integrin platelet signaling. J. Biol. Chem. 275, 36423-36429. [DOI] [PubMed] [Google Scholar]
- Critchley, D. R. and Gingras, A. R. (2008). Talin at a glance. J. Cell Sci. 121, 1345-1347. [DOI] [PubMed] [Google Scholar]
- DeMali, K. A., Barlow, C. A. and Burridge, K. (2002). Recruitment of the Arp2/3 complex to vinculin: coupling membrane protrusion to matrix adhesion. J. Cell Biol. 159, 881-891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher, D. E., Janmey, P. and Wang, Y. L. (2005). Tissue cells feel and respond to the stiffness of their substrate. Science 310, 1139-1143. [DOI] [PubMed] [Google Scholar]
- Dubin-Thaler, B. J., Hofman, J. M., Cai, Y., Xenias, H., Spielman, I., Shneidman, A. V., David, L. A., Dobereiner, H. G., Wiggins, C. H. and Sheetz, M. P. (2008). Quantification of cell edge velocities and traction forces reveals distinct motility modules during cell spreading. PLoS ONE 3, e3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenfeld, D. P., Schwartzberg, P. L., Venegas, A., Tse, R. and Sheetz, M. P. (1999). Selective regulation of integrin-cytoskeleton interactions by the tyrosine kinase Src. Nat. Cell Biol. 1, 200-206. [DOI] [PubMed] [Google Scholar]
- Galbraith, C. G., Yamada, K. M. and Sheetz, M. P. (2002). The relationship between force and focal complex development. J. Cell Biol. 159, 695-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith, C. G., Yamada, K. M. and Galbraith, J. A. (2007). Polymerizing actin fibers position integrins primed to probe for adhesion sites. Science 315, 992-995. [DOI] [PubMed] [Google Scholar]
- Garcia-Alvarez, B., de Pereda, J. M., Calderwood, D. A., Ulmer, T. S., Critchley, D., Campbell, I. D., Ginsberg, M. H. and Liddington, R. C. (2003). Structural determinants of integrin recognition by talin. Mol. Cell 11, 49-58. [DOI] [PubMed] [Google Scholar]
- Giannone, G. and Sheetz, M. P. (2006). Substrate rigidity and force define form through tyrosine phosphatase and kinase pathways. Trends Cell Biol. 16, 213-223. [DOI] [PubMed] [Google Scholar]
- Giannone, G., Jiang, G., Sutton, D. H., Critchley, D. R. and Sheetz, M. P. (2003). Talin1 is critical for force-dependent reinforcement of initial integrin-cytoskeleton bonds but not tyrosine kinase activation. J. Cell Biol. 163, 409-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone, G., Dubin-Thaler, B. J., Dobereiner, H. G., Kieffer, N., Bresnick, A. R. and Sheetz, M. P. (2004). Periodic lamellipodial contractions correlate with rearward actin waves. Cell 116, 431-443. [DOI] [PubMed] [Google Scholar]
- Giannone, G., Dubin-Thaler, B. J., Rossier, O., Cai, Y., Chaga, O., Jiang, G., Beaver, W., Dobereiner, H. G., Freund, Y., Borisy, G. et al. (2007). Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 128, 561-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W. H. and Wang, Y. L. (2007). Retrograde fluxes of focal adhesion proteins in response to cell migration and mechanical signals. Mol. Biol. Cell 18, 4519-4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsten, T. L., Bunch, T. A., Kato, H., Yamanouchi, J., Choi, S. H., Jannuzi, A. L., Feral, C. C., Ginsberg, M. H., Brower, D. L. and Shattil, S. J. (2008). Differences in regulation of Drosophila and vertebrate integrin affinity by talin. Mol. Biol. Cell 19, 3589-3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, K., Ji, L., Applegate, K. T., Danuser, G. and Waterman-Storer, C. M. (2007). Differential transmission of actin motion within focal adhesions. Science 315, 111-115. [DOI] [PubMed] [Google Scholar]
- Humphries, J. D., Wang, P., Streuli, C., Geiger, B., Humphries, M. J. and Ballestrem, C. (2007). Vinculin controls focal adhesion formation by direct interactions with talin and actin. J. Cell Biol. 179, 1043-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huveneers, S., Truong, H., Fassler, R., Sonnenberg, A. and Danen, E. H. (2008). Binding of soluble fibronectin to integrin {alpha}5{beta}1-link to focal adhesion redistribution and contractile shape. J. Cell Sci. 121, 2452-2462. [DOI] [PubMed] [Google Scholar]
- Hynes, R. O. (2002). Integrins: bidirectional, allosteric signaling machines. Cell 110, 673-687. [DOI] [PubMed] [Google Scholar]
- Hytonen, V. P. and Vogel, V. (2008). How force might activate talin's vinculin binding sites: SMD reveals a structural mechanism. PLoS Comput. Biol. 4, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, G., Giannone, G., Critchley, D. R., Fukumoto, E. and Sheetz, M. P. (2003). Two-piconewton slip bond between fibronectin and the cytoskeleton depends on talin. Nature 424, 334-337. [DOI] [PubMed] [Google Scholar]
- Jiang, G., Huang, A. H., Cai, Y., Tanase, M. and Sheetz, M. P. (2006). Rigidity sensing at the leading edge through alphavbeta3 integrins and RPTPalpha. Biophys. J. 90, 1804-1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz, B. Z., Zamir, E., Bershadsky, A., Kam, Z., Yamada, K. M. and Geiger, B. (2000). Physical state of the extracellular matrix regulates the structure and molecular composition of cell-matrix adhesions. Mol. Biol. Cell 11, 1047-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiema, T., Lad, Y., Jiang, P., Oxley, C. L., Baldassarre, M., Wegener, K. L., Campbell, I. D., Ylanne, J. and Calderwood, D. A. (2006). The molecular basis of filamin binding to integrins and competition with talin. Mol. Cell 21, 337-347. [DOI] [PubMed] [Google Scholar]
- Kim, M., Carman, C. V., Yang, W., Salas, A. and Springer, T. A. (2004). The primacy of affinity over clustering in regulation of adhesiveness of the integrin {alpha}L{beta}2. J. Cell Biol. 167, 1241-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic, A., Sap, J. and Sheetz, M. P. (2007). RPTPalpha is required for rigidity-dependent inhibition of extension and differentiation of hippocampal neurons. J. Cell Sci. 120, 3895-3904. [DOI] [PubMed] [Google Scholar]
- Law, D. A., DeGuzman, F. R., Heiser, P., Ministri-Madrid, K., Killeen, N. and Phillips, D. R. (1999). Integrin cytoplasmic tyrosine motif is required for outside-in alphaIIbbeta3 signalling and platelet function. Nature 401, 808-811. [DOI] [PubMed] [Google Scholar]
- Liddington, R. C. and Ginsberg, M. H. (2002). Integrin activation takes shape. J. Cell Biol. 158, 833-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lietha, D., Cai, X., Ceccarelli, D. F., Li, Y., Schaller, M. D. and Eck, M. J. (2007). Structural basis for the autoinhibition of focal adhesion kinase. Cell 129, 1177-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, B. H., Springer, T. A. and Takagi, J. (2003). Stabilizing the open conformation of the integrin headpiece with a glycan wedge increases affinity for ligand. Proc. Natl. Acad. Sci. USA 100, 2403-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, B. H., Strokovich, K., Walz, T., Springer, T. A. and Takagi, J. (2004). Allosteric beta1 integrin antibodies that stabilize the low affinity state by preventing the swing-out of the hybrid domain. J. Biol. Chem. 279, 27466-27471. [DOI] [PubMed] [Google Scholar]
- Ma, Y. Q., Qin, J., Wu, C. and Plow, E. F. (2008). Kindlin-2 (Mig-2): a co-activator of beta3 integrins. J. Cell Biol. 181, 439-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel, V., Racaud-Sultan, C., Dupe, S., Marie, C., Paulhe, F., Galmiche, A., Block, M. R. and Albiges-Rizo, C. (2001). Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J. Biol. Chem. 276, 21217-21227. [DOI] [PubMed] [Google Scholar]
- Merkel, R., Nassoy, P., Leung, A., Ritchie, K. and Evans, E. (1999). Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature 397, 50-53. [DOI] [PubMed] [Google Scholar]
- Moes, M., Rodius, S., Coleman, S. J., Monkley, S. J., Goormaghtigh, E., Tremuth, L., Kox, C., van der Holst, P. P., Critchley, D. R. and Kieffer, N. (2007). The integrin binding site 2 (IBS2) in the talin rod domain is essential for linking integrin beta subunits to the cytoskeleton. J. Biol. Chem. 282, 17280-17288. [DOI] [PubMed] [Google Scholar]
- Morgan, M. R., Humphries, M. J. and Bass, M. D. (2007). Synergistic control of cell adhesion by integrins and syndecans. Nat. Rev. Mol. Cell Biol. 8, 957-969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser, M., Nieswandt, B., Ussar, S., Pozgajova, M. and Fassler, R. (2008). Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 14, 325-330. [DOI] [PubMed] [Google Scholar]
- Mould, A. P., Barton, S. J., Askari, J. A., McEwan, P. A., Buckley, P. A., Craig, S. E. and Humphries, M. J. (2003). Conformational changes in the integrin betaA domain provide a mechanism for signal transduction via hybrid domain movement. J. Biol. Chem. 278, 17028-17035. [DOI] [PubMed] [Google Scholar]
- Nicolas, A., Geiger, B. and Safran, S. A. (2004). Cell mechanosensitivity controls the anisotropy of focal adhesions. Proc. Natl. Acad. Sci. USA 101, 12520-12525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizaka, T., Shi, Q. and Sheetz, M. P. (2000). Position-dependent linkages of fibronectin-integrin-cytoskeleton. Proc. Natl. Acad. Sci. USA 97, 692-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuckolls, G. H., Romer, L. H. and Burridge, K. (1992). Microinjection of antibodies against talin inhibits the spreading and migration of fibroblasts. J. Cell Sci. 102, 753-762. [DOI] [PubMed] [Google Scholar]
- Pankov, R., Cukierman, E., Katz, B. Z., Matsumoto, K., Lin, D. C., Lin, S., Hahn, C. and Yamada, K. M. (2000). Integrin dynamics and matrix assembly: tensin-dependent translocation of alpha(5)beta(1) integrins promotes early fibronectin fibrillogenesis. J. Cell Biol. 148, 1075-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons, M., Messent, A. J., Humphries, J. D., Deakin, N. O. and Humphries, M. J. (2008). Quantification of integrin receptor agonism by fluorescence lifetime imaging. J. Cell Sci. 121, 265-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge, M. A., David, F. S. and Marcantonio, E. E. (2006). Displacement of the {beta} cytoplasmic domain recovers focal adhesion formation, cytoskeletal organization and motility in swapped integrin chimeras. J. Cell Sci. 119, 1175-1183. [DOI] [PubMed] [Google Scholar]
- Pavalko, F. M., Otey, C. A., Simon, K. O. and Burridge, K. (1991). Alpha-actinin: a direct link between actin and integrins. Biochem. Soc. Trans. 19, 1065-1069. [DOI] [PubMed] [Google Scholar]
- Phillips, D. R., Prasad, K. S., Manganello, J., Bao, M. and Nannizzi-Alaimo, L. (2001). Integrin tyrosine phosphorylation in platelet signaling. Curr. Opin. Cell Biol. 13, 546-554. [DOI] [PubMed] [Google Scholar]
- Priddle, H., Hemmings, L., Monkley, S., Woods, A., Patel, B., Sutton, D., Dunn, G. A., Zicha, D. and Critchley, D. R. (1998). Disruption of the talin gene compromises focal adhesion assembly in undifferentiated but not differentiated embryonic stem cells. J. Cell Biol. 142, 1121-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puklin-Faucher, E., Gao, M., Schulten, K. and Vogel, V. (2006). How the headpiece hinge angle is opened: new insights into the dynamics of integrin activation. J. Cell Biol. 175, 349-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucher, D. and Sheetz, M. P. (2000). Cell spreading and lamellipodial extension rate is regulated by membrane tension. J. Cell Biol. 148, 127-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, X. D., Kiosses, W. B., Sieg, D. J., Otey, C. A., Schlaepfer, D. D. and Schwartz, M. A. (2000). Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 113, 3673-3678. [DOI] [PubMed] [Google Scholar]
- Riveline, D., Zamir, E., Balaban, N. Q., Ishizaki, T., Narumiyq, S., Kam, Z., Geiger, B. and Bershadsky, A. (2001). Fcoal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J. Chem. Biol. 153, 1175-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, D. M., Alon, R. and Ginsberg, M. H. (2007). Integrin modulation and signaling in leukocyte adhesion and migration. Immunol. Rev. 218, 126-134. [DOI] [PubMed] [Google Scholar]
- Saez, A., Buguin, A., Silberzan, P. and Ladoux, B. (2005). Is the mechanical activity of epithelial cells controlled by deformations or forces? Biophys. J. 89, L52-L54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada, Y., Tamada, M., Dubin-Thaler, B. J., Cherniavskaya, O., Sakai, R., Tanaka, S. and Sheetz, M. (2006). Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell 127, 1015-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, C. E., Horwitz, A. F., Lauffenburger, D. A. and Sheetz, M. P. (1993). Integrin-cytoskeletal interactions in migrating fibroblasts are dynamic, asymmetric, and regulated. J. Cell Biol. 123, 977-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, C. E., Dai, J., Lauffenburger, D. A., Sheetz, M. P. and Horwitz, A. F. (1995). Integrin-cytoskeletal interactions in neuronal growth cones. J. Neurosci. 15, 3400-3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selhuber-Unkel, C., Lopez-Garcia, M., Kessler, H. and Spatz, J. P. (2008). Cooperativity in adhesion cluster formation during initial cell adhesion. Biophys. J. 95, 5424-5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemesh, T., Geiger, B., Bershadsky, A. D. and Kozlov, M. M. (2005). Focal adhesions as mechanosensors: a physical mechanism. Proc. Natl. Acad. Sci. USA 102, 12383-12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, J., Muranjan, M. and Sap, J. (1999). Receptor protein tyrosine phosphatase alpha activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr. Biol. 9, 505-511. [DOI] [PubMed] [Google Scholar]
- Tadokoro, S., Shattil, S. J., Eto, K., Tai, V., Liddington, R. C., de Pereda, J. M., Ginsberg, M. H. and Calderwood, D. A. (2003). Talin binding to integrin beta tails: a final common step in integrin activation. Science 302, 103-106. [DOI] [PubMed] [Google Scholar]
- Takagi, J., Petre, B. M., Walz, T. and Springer, T. A. (2002). Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110, 599-611. [DOI] [PubMed] [Google Scholar]
- Tamada, M., Sheetz, M. P. and Sawada, Y. (2004). Activation of a signaling cascade by cytoskeleton stretch. Dev. Cell 7, 709-718. [DOI] [PubMed] [Google Scholar]
- Tanentzapf, G. and Brown, N. H. (2006). An interaction between integrin and the talin FERM domain mediates integrin activation but not linkage to the cytoskeleton. Nat. Cell Biol. 8, 601-606. [DOI] [PubMed] [Google Scholar]
- Tremuth, L., Kreis, S., Melchior, C., Hoebeke, J., Ronde, P., Plancon, S., Takeda, K. and Kieffer, N. (2004). A fluorescence cell biology approach to map the second integrin-binding site of talin to a 130-amino acid sequence within the rod domain. J. Biol. Chem. 279, 22258-22266. [DOI] [PubMed] [Google Scholar]
- Ulmer, T. S., Calderwood, D. A., Ginsberg, M. H. and Campbell, I. D. (2003). Domain-specific interactions of talin with the membrane-proximal region of the integrin beta3 subunit. Biochemistry 42, 8307-8312. [DOI] [PubMed] [Google Scholar]
- Volberg, T., Geiger, B., Kam, Z., Pankov, R., Simcha, I., Sabanay, H., Coll, J. L., Adamson, E. and Ben-Ze'ev, A. (1995). Focal adhesion formation by F9 embryonal carcinoma cells after vinculin gene disruption. J. Cell Sci. 108, 2253-2260. [DOI] [PubMed] [Google Scholar]
- von Wichert, G., Haimovich, B., Feng, G. S. and Sheetz, M. P. (2003a). Force-dependent integrin-cytoskeleton linkage formation requires downregulation of focal complex dynamics by Shp2. EMBO J. 22, 5023-5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Wichert, G., Jiang, G., Kostic, A., De Vos, K., Sap, J. and Sheetz, M. P. (2003b). RPTP-alpha acts as a transducer of mechanical force on alphav/beta3-integrin-cytoskeleton linkages. J. Cell Biol. 161, 143-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. B., Dembo, M., Hanks, S. K. and Wang, Y. (2001). Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc. Natl. Acad. Sci. USA 98, 11295-11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener, K. L., Partridge, A. W., Han, J., Pickford, A. R., Liddington, R. C., Ginsberg, M. H. and Campbell, I. D. (2007). Structural basis of integrin activation by talin. Cell 128, 171-182. [DOI] [PubMed] [Google Scholar]
- White, D. P., Caswell, P. T. and Norman, J. C. (2007). alpha v beta3 and alpha5beta1 integrin recycling pathways dictate downstream Rho kinase signaling to regulate persistent cell migration. J. Cell Biol. 177, 515-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf, E., Grigorova, I., Sagiv, A., Grabovsky, V., Feigelson, S. W., Shulman, Z., Hartmann, T., Sixt, M., Cyster, J. G. and Alon, R. (2007). Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat. Immunol. 8, 1076-1085. [DOI] [PubMed] [Google Scholar]
- Xiao, T., Takagi, J., Coller, B. S., Wang, J. H. and Springer, T. A. (2004). Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 432, 59-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, B., Jedsadayanmata, A. and Lam, S. C. (2001). Localization of an integrin binding site to the C terminus of talin. J. Biol. Chem. 276, 44373-44378. [DOI] [PubMed] [Google Scholar]
- Xiong, J.-P., Stehle, T., Zhang, R., Joachimiak, A., Frech, M., Goodman, S. L. and Arnaout, M. A. (2001). Crystal structure of the extracellular segment of integrin alpha Vbeta 3. Science 294, 339-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, J.-P., Stehle, T., Zhang, R., Joachimiak, A., Frech, M., Goodman, S. L. and Arnaout, M. A. (2002). Crystal structure of the extracellular segment of integrin alpha Vbeta 3 in complex with an Arg-Gly-Asp ligand. Science 296, 151-155. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar, R., Ballestrem, C., Kam, Z. and Geiger, B. (2003). Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J. Cell Sci. 116, 4605-4613. [DOI] [PubMed] [Google Scholar]
- Zamir, E., Katz, M., Posen, Y., Erez, N., Yamada, K. M., Katz, B. Z., Lin, S., Lin, D. C., Bershadsky, A., Kam, Z. et al. (2000). Dynamics and segregation of cell-matrix adhesions in cultured fibroblasts. Nat. Cell Biol. 2, 191-196. [DOI] [PubMed] [Google Scholar]
- Zhang, X., Jiang, G., Cai, Y., Monkley, S. J., Critchley, D. R. and Sheetz, M. P. (2008). Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat. Cell Biol. 10, 1062-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann, P., Zhang, Z., Degeest, G., Mortier, E., Leenaerts, I., Coomans, C., Schulz, J., N'Kuli, F., Courtoy, P. J. and David, G. (2005). Syndecan recycling [corrected] is controlled by syntenin-PIP2 interaction and Arf6. Dev. Cell 9, 377-388. [DOI] [PubMed] [Google Scholar]