

Summary
Parkin and DJ-1 are two multi-functional proteins linked to autosomal recessive early-onset Parkinson's disease (PD) that have been shown to functionally interact by as-yet-unknown mechanisms. We have delineated the mechanisms by which parkin controls DJ-1. Parkin modulates DJ-1 transcription and protein levels via a signaling cascade involving p53 and the endoplasmic reticulum (ER)-stress-induced active X-box-binding protein-1S (XBP-1S). Parkin triggers the transcriptional repression of p53 while p53 downregulates DJ-1 protein and mRNA expressions. We show that parkin-mediated control of DJ-1 is fully p53-dependent. Furthermore, we establish that p53 lowers the protein and mRNA levels of XBP-1S. Accordingly, we show that parkin ultimately upregulates XBP-1 levels. Subsequently, XBP-1S physically interacts with the DJ-1 promoter, thereby enhancing its promoter trans-activation, mRNA levels and protein expression. This data was corroborated by the examination of DJ-1 in both parkin- and p53-null mice brains. This transcriptional cascade is abolished by pathogenic parkin mutations and is independent of its ubiquitin-ligase activity. Our data establish a parkin-dependent ER-stress-associated modulation of DJ-1 and identifies p53 and XBP-1 as two major actors acting downstream of parkin in this signaling cascade in cells and in vivo. This work provides a mechanistic explanation for the increase in the unfolded protein response observed in PD pathology, i.e. that it is due to a defect in parkin-associated control of DJ-1.
Key words: Parkinson's disease, DJ-1, Parkin, Mutations, p53, XBP-1, UPR
Introduction
Parkinson's disease (PD) is a neurodegenerative disorder that is characterized by a severe loss of dopaminergic neurons of the substantia nigra and by the presence of intra-cytoplasmic inclusions named Lewy bodies (Moore et al., 2005a). This syndrome is mainly sporadic but a few percent of the cases are linked to a genetic origin (familial PD, FPD) and are either associated with an autosomal dominant or recessive mode of inheritance. The latter forms of the disease are usually characterized by an early onset and are mostly linked to mutations in the genes of parkin, Pink-1 and DJ-1 (Shulman et al., 2011).
DJ-1 is implicated in ∼1% of autosomal recessive FPD (Abou-Sleiman et al., 2003) and has been documented as a ubiquitously expressed and highly conserved protein present as a homodimeric complex in the cytoplasm (Bandopadhyay et al., 2004). Several lines of evidence indicate that DJ-1 could act as an antioxidant (Taira et al., 2004) and could control mitochondrial function (Thomas et al., 2011), autophagy (Thomas et al., 2011) and pro-survival pathways (Giaime et al., 2010). However, only fragmentary data concern the mechanisms by which DJ-1 transcription/expression is regulated and whether such regulation could be controlled by physiopathological stresses or cellular challenges.
Parkin is another important FPD-associated gene product that has been characterized as an ubiquitin-ligase (Shimura et al., 2000; Tanaka et al., 2001). Interestingly, parkin was reported to interact physically with DJ-1 monomers in oxidative stress conditions (Moore et al., 2005b). However, unlike expected from its ubiquitin ligase activity, parkin did not promote DJ-1 proteasomal degradation but instead, increased its cellular stability (Moore et al., 2005b). This suggests that parkin-mediated control of DJ-1 expression could indeed reflect a functional cross-talk by which parkin could modulate DJ-1-associated functions.
Several works evidenced a major role of parkin and DJ-1 in the mitochondria, one of the major organelle targets affected in PD pathology (Dodson and Guo, 2007). However, Thomas and co-workers showed very recently that DJ-1 depletion triggers loss of mitochondrial polarization, fragmentation and increased autophagy in dopaminergic M17 cells, independently of the parkin–Pink-1 pathway (Thomas et al., 2011). Therefore, although parkin and DJ-1 could coordinately but independently contribute to mitochondrial function, the ability of parkin to interact with DJ-1 may likely have other functional consequences.
In this context, it is noteworthy that both parkin and DJ-1 have been associated to the ER-stress response also called unfolded protein response (UPR). Thus, parkin controls both endoplasmic reticulum and oxidative stresses (Takahashi et al., 2003) and interacts with Hsp70 and some members of the ERAD (ER-associated degradation) machinery, Ubc7 and Ubc8 (Tanaka et al., 2001). Directly linked to parkin-associated ligase activity, Imai and colleagues showed that the Pael receptor that tends to be poly-ubiquitinated, unfolded and poorly soluble, in vivo, behaves as a parkin substrate (Imai et al., 2001). However, unrelated to parkin enzymatic properties, oxidative stress indeed enhances the physical interaction between parkin and DJ-1 (Moore et al., 2005b). We therefore questioned whether parkin could be functionally linked to DJ-1 in basal and ER-stress-induced conditions.
We show here that parkin upregulates DJ-1 promoter trans-activation, mRNA levels and protein expression via a transcriptional cascade involving p53 repression and subsequent activation of ER-stress induced X-box-binding protein-1S (XBP-1S). Subsequently, XBP-1S physically interacts with DJ-1 promoter and thereby raises DJ-1 mRNA and protein levels. Interestingly, parkin-mediated control of DJ-1 remained independent of its ubiquitin-ligase activity and is abolished by PD-related pathogenic mutations. Overall, our study is the first delineation of the molecular events linking parkin and DJ-1 and unravels a functional dialogue by which these two proteins could control ER-stress response in physiopathological conditions.
Results
Parkin controls DJ-1 in cells and in mice brain
We have examined the influence of the modulation of parkin (PK) levels on DJ-1. Stably transfected TSM1 neurons overexpressing wild-type PK exhibit increased DJ-1 expression (+82±23.4%, Fig. 1A), promoter trans-activation (+41.6±5.9%, Fig. 1B) and mRNA levels (+45.2±10.4, Fig. 1C). The PK-associated upregulation of DJ-1 has been corroborated by the examination of the influence of endogenous PK depletion on DJ-1 expression. Thus, parkin-null fibroblasts (PK−/−) display lower DJ-1 expression (−49±4.7% compared to control PK+/+ fibroblasts, Fig. 1D), promoter transactivation (−77±4.9%, Fig. 1E) and mRNA levels (−30±0.8%, Fig. 1F). These results were similar in two distinct PK-deficient fibroblastic cell lines from distinct origins (data not shown). In order to rule out any artifactual effect linked to genetic manipulation, we have confirmed the latter set of data by the analysis of parkin depletion by a shRNA approach. As demonstrated in supplementary material Fig. S1, two distinct parkin-directed shRNAs (KD1 and KD2, see Materials and Methods) that significantly decrease parkin expression [−53±0.3% of scrambled (SC)-treated cells, for KD1 and −35±1.3%, for KD2, supplementary material Fig. S1A,B] concomitantly lower DJ-1 expression (−38±2.1% of control value for KD1 and −41±0.1% for KD2, supplementary material Fig. S1B) and promoter transactivation (−25±1.2% for KD1, supplementary material Fig. S1C). In order to examine the influence of parkin, in vivo, we have analyzed DJ-1 expression in brain samples derived from wild-type or parkin-null mice (Itier et al., 2003). The depletion of parkin leads to the reduction of DJ-1 expression (−33±2% of control PK+/+ brains, Fig. 1G), indicating that PK controls DJ-1 in both cellular models and in vivo.
Fig. 1.
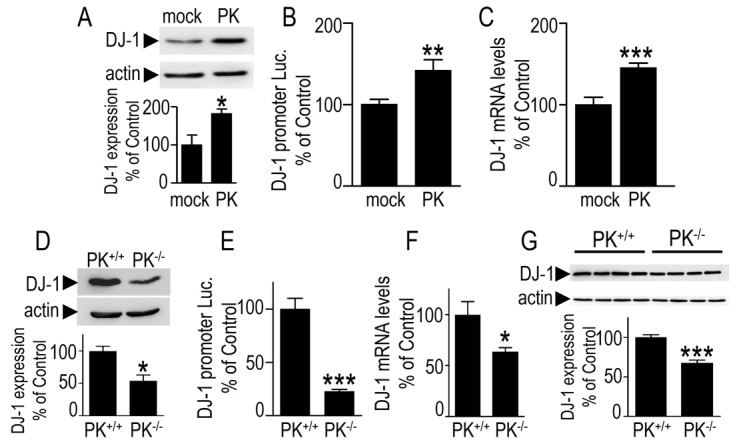
Parkin modulates DJ-1 expression, promoter transactivation and mRNA levels. (A–C) TSM1 neurons were stably transfected with pcDNA3 (mock) or human parkin cDNA (PK) then DJ-1 expression (A), promoter transactivation (B) and mRNA levels (C) were analyzed. Bars represent the means ± s.e.m. of 4–11 independent experiments performed in triplicate (A,C) or in 6 replicates (B) and are expressed as a percentage of control mock-transfected cells. Actin was used as a loading control. (D–F) Wild-type (PK+/+) or parkin-null (PK−/−) mouse fibroblasts were analyzed for DJ-1 expression (D), promoter transactivation (E) and mRNA levels (F). Bars represent the means ± s.e.m. of 3 or 4 independent experiments performed in 3–6 replicates and are expressed as a percentage of control wild-type fibroblasts. (G) DJ-1 and actin immunoreactivities were determined in homogenates derived from PK+/+ and PK−/− mice brains. Bars represent the densitometric analyses of DJ-1 expression and are means ± s.e.m. of 4 brains. *P<0.05, **P<0.01, ***P<0.001 compared with controls.
The number of yet available parkin-associated human brain samples is extremely low and corresponds to a maximum of six worldwide. It is therefore very difficult to assess whether PK mutations responsible for FPD also trigger decreased DJ-1 expression in pathogenic human brains. However, we had the opportunity to examine two of these samples in which a parkin missense mutation and deletion (FPD) have been identified (da Costa et al., 2009). We observed a diminution of DJ-1 expression (about 25% reduction versus control brains, data not shown). Although it is obviously hard to draw any firm conclusion, it remains that loss of function parkin mutation could also trigger DJ-1 reduction in human PD-affected brains.
PD-associated parkin mutations abolish PK-mediated control of DJ-1
The human SH-SY5Y neuroblastoma cell line is a relevant dopaminergic cell model widely used in cell biology approaches of PD pathology. We have first confirmed that transient overexpression of PK in these cells also triggers increased DJ-1 expression (+44.4±7.8% of control EV-transfected cells, Fig. 2A,B), promoter transactivation (+21±1.5%, Fig. 2C) and mRNA levels (+31±1.7%, Fig. 2D). Interestingly, two familial PD-associated mutations known to abolish (C418R) or preserve (K161N) the PK-linked ubiquitin-ligase activity both block these augmentations (Fig. 2A–D). It should be noted that the transient transfection of PK mutant cDNAs led to various expressions (Fig. 2A) that are not due to variability of transfection efficiency as evidenced by similar GFP expression (GFP coding sequence is harbored by the vector bearing PK sequences as described in the Materials and Methods and under the control of the same promoter), but rather to faster catabolic rates of PK mutants as was described previously (da Costa et al., 2009; Hampe et al., 2006). Therefore, we confirmed the above data in stably transfected HEK293 cells overexpressing wild-type, K161N-PK as well as another ubiquitin-ligase-deficient C441R-PK mutant. As was observed with transiently transfected cells (Fig. 2A–D), wild-type PK expression elicits an increase of DJ-1 expression (+86.7±18.7% of mock-transfected cells, Fig. 2E,F), promoter transactivation (+123±19.3% of mock-transfected cells, Fig. 2G) and mRNA levels (+72±12.8%, Fig. 2H), the increments of which were fully prevented by both mutations (Fig. 2E–H). Overall, the above data indicate that PK-mediated control of DJ-1 is not cell specific, occurs in dopaminergic cells as well as in vivo, and remains unrelated to PK-associated ubiquitin ligase activity.
Fig. 2.
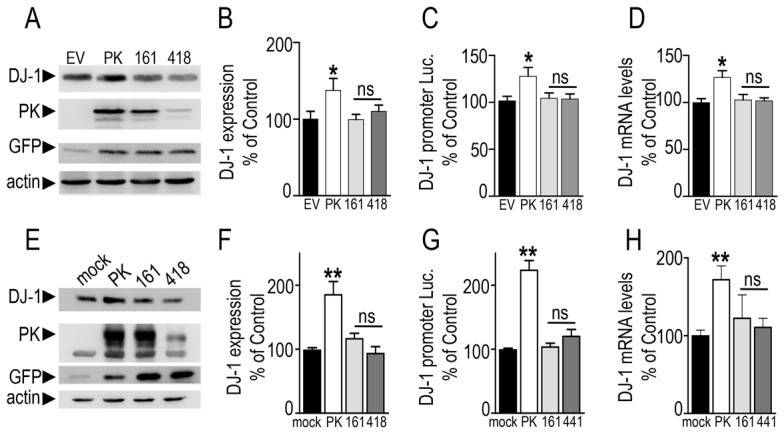
Familial Parkinson's disease-associated mutations abolish parkin-mediated regulation of DJ-1. (A–D) SH-SY5Y human neuroblastoma cells were transiently transfected with the indicated cDNAs (EV, empty pIRES2-EGFP vector; PK, wild-type parkin; 161, K161-PK; 418, C418R-PK). Twenty-four hours after transfection, cells were examined for DJ-1, parkin, GFP and actin-like immunoreactivities (A,B), human DJ-1 promoter transactivation (C) and DJ-1 mRNA levels (D). Bars represent the means ± s.e.m. of 6–12 independent experiments and are expressed as a percentage of control EV-transfected cells. (E–H) HEK293 human embryonic kidney cells were stably transfected with the indicated cDNAs and analyzed for DJ-1 expression (E,F), promoter transactivation (G) and mRNA levels (H). Bars represent means ± s.e.m. of 2 independent experiments performed in 3–6 replicates and are expressed as a percentage of control mock-transfected cells. *P<0.05, **P<0.01, compared with controls; ns, not statistically significant.
DJ-1 is controlled by p53
We recently documented a novel ubiquitin-ligase-independent function of PK that could act as a transcriptional repressor of the tumor suppressor p53 (da Costa et al., 2009). Since the control of DJ-1 by PK was also independent of its ubiquitin-ligase activity, we have examined whether PK could control DJ-1 via p53. To address this question, we first examined the putative ability of p53 to control DJ-1. We took advantage of two cell models in which either p19Arf or p19Arf and p53 genes had been invalidated. The depletion of p19Arf allows the analysis of the involvement of p53 in cell death without interference with p53-associated contribution to cell cycle control (Weber et al., 2000). In Fig. 3, the comparison of p19Arf−/− (white bars) and p19Arf−/−p53−/− (black bars) indicates that the depletion of endogenous p53 (compare lanes marked −) induces an increase of DJ-1 expression (+43.7±13.6% of control EV-transfected p19Arf−/− cells, Fig. 3A,B), promoter transactivation (+149.3±37.4%, Fig. 3C) and mRNA levels (+147±32%, Fig. 3D). Complementation of both p19Arf−/− and p19Arf−/−p53−/− fibroblasts by p53 cDNA transfection (lanes marked + in Fig. 3A–D) leads to a significant diminution of DJ-1 expression (−30.8±3.7% of control EV-transfected p19Arf−/− cells, and −33.8±4.1% of control EV-transfected p19Arf−/−p53−/− cells, Fig. 3A,B), promoter transactivation (−37.1±3.3%, and −30.9±8.1%, Fig. 3C), and mRNA levels (−53±2.1%, and −57±7.4%, Fig. 3D). Interestingly, analysis of brain samples from age-matched mice in which the gene of p53 has been invalidated (p53−/−) or not (p53+/+), indicates an increase of DJ-1 protein expression (+95±8.9% of control p53+/+ brains, Fig. 3E) and mRNA levels (+27.6±9.3%, Fig. 3F).
Fig. 3.
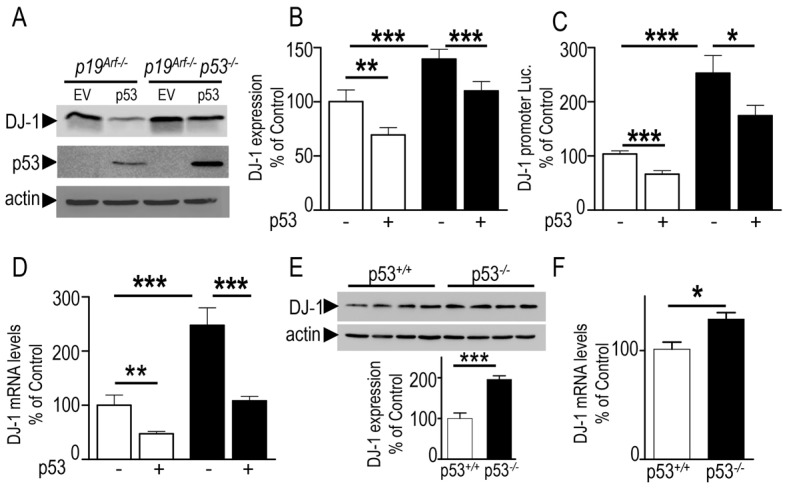
Endogenous p53 modulates DJ-1 expression and mRNA levels in vitro and in vivo. (A–D) p19Arf−/− (white bars) and p19Arf−/−p53−/− (black bars) mouse embryonic fibroblasts were transiently transfected with pcDNA3 empty vector (EV, bars marked −) or with p53 cDNA (p53, bars marked +). Twenty-four hours after transfection, cells were examined for DJ-1, p53 and actin-like immunoreactivities (A,B), human DJ-1 promoter transactivation (C) and mRNA levels (D). Bars are the means ± s.e.m. of 3 or 4 independent experiments (each performed in 5 replicates) and are expressed as a percentage of control EV-transfected p19Arf−/− cells. Brain homogenates derived from p53+/+ and p53−/− mice were analyzed for their DJ-1 and actin immunoreactivities (E) and DJ-1 mRNA levels (F). Bars represent the means ± s.e.m. of 4–8 independent experiments and are expressed as a percentage of control p53+/+ brains. *P<0.05, **P<0.01, ***P<0.001.
In order to further confirm the influence of p53 on DJ-1, we took advantage of another cell system in which p53 expression was altered to examine DJ-1 expression. Thus, the human colorectal carcinoma cell line (HCT116-p53−/−) is a p53 knockout cell line derived from HCT116-p53+/+ by homologous recombination (Bunz et al., 1998). Interestingly, here again, we observed an upregulation of DJ-1 expression (+49.1±12.7% of HCT116-p53+/+ cells, supplementary material Fig. S2A) and promoter transactivation (+116±13.8% of control HCT116-p53+/+ cells, supplementary material Fig. S2B). The above set of data first excludes a cell-specific regulation of DJ-1 by p53 and clearly establishes that p53 downregulates DJ-1 protein expression and mRNA levels both in vitro and in vivo.
PK-mediated control of DJ-1 is p53-dependent
The above data showing the upregulation of DJ-1 by PK and its downregulation by p53, together with our previous demonstration that parkin transcriptionally repressed p53 (da Costa et al., 2009) led us to envision that PK-associated increase of DJ-1 expression could occur through PK-mediated repression of p53. As previously established (da Costa et al., 2009), stably transfected TSM1 neurons expressing wild-type PK exhibit drastically lower p53 expression (−81±3% of control mock-transfected neurons, Fig. 4A). Thus, we examined the contribution of p53 by assessing the influence of endogenous p53 depletion on PK-mediated increase of DJ-1. We first observed that p19Arf−/− fibroblasts expressing parkin cDNA exhibit an increased DJ-1 expression [+90.5±23% of EV-transfected p19Arf−/− cells (compare white bars), Fig. 4B,C], promoter transactivation (+56±11%, Fig. 4D) and mRNA levels (+91.9±57.7%, Fig. 4E). This shows that fibroblasts containing endogenous p53 respond to parkin cDNA transfection and exhibit expected PK-mediated augmentation of DJ-1. As expected from above-described data, we confirmed that p19Arf−/− p53−/− cells display increased DJ-1 expression, promoter transactivation and mRNA levels [Fig. 4B and compare white and black bars marked (−) in Fig. 4C–E, respectively], and that they remained totally unaffected by parkin cDNA transfection [see Fig. 4B and compare black bars marked (+) and (−) in Fig. 4C–E]. This set of experiments demonstrates that parkin-mediated regulation of DJ-1 is abolished by p53 depletion and thus, fully dependent of p53.
Fig. 4.
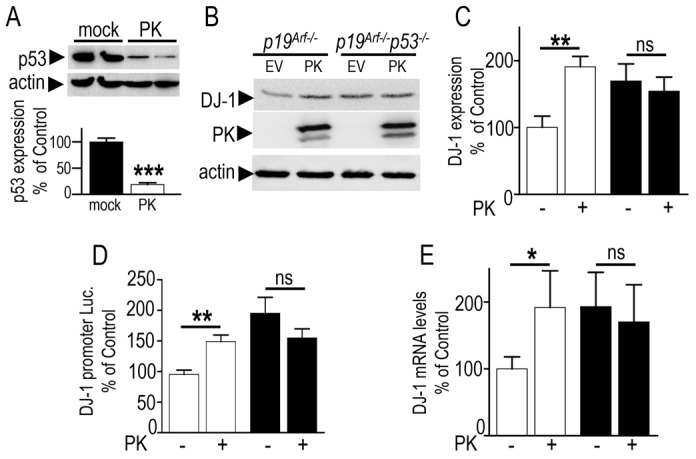
Parkin-mediated regulation of DJ-1 is fully p53-dependent. (A) TSM1 neurons stably transfected with an empty vector (mock) or with wild-type parkin (PK) cDNA were examined for their p53- and actin-like immunoreactivities. Bars represent the densitometric analysis expressed as a percentage of control mock-transfected cells and are the means ± s.e.m. of 4 independent experiments. (B–E) p19Arf−/− (white bars) and p19Arf−/−p53−/− (black bars) fibroblasts were transiently transfected with an empty vector (EV) or with wild-type parkin (PK) cDNAs. Twenty-four hours after transfection, cells were examined for DJ-1-, parkin- and actin-like immunoreactivities (B,C), human DJ-1 promoter transactivation (D) and mRNA levels (E). Bars indicate results as a percentage of control EV-transfected p19Arf−/− cells and are the means ± s.e.m. of 4–11 independent experiments (6 replicates each). *P<0.05, **P<0.01; ns, not statistically significant.
DJ-1 transcription is controlled by ER stress via the transcription factor XBP-1
Based on our observation that p53 lowers DJ-1 promoter activity and mRNA levels (Fig. 3), and sticking with the well characterized function of p53 as a transcription factor, we have performed an in silico study to search for putative p53 consensus binding sites in the human DJ-1 promoter. We did not delineate any fully conserved consensus site recognized by p53. We identified some putative partially conserved sites that we have mutated. However, none of the mutations tested affected p53-mediated control of DJ-1 promoter transactivation (data not shown), meaning that an additional molecular intermediate in the cascade linking PK and DJ-1 remained to be identified.
Interestingly, we identified a highly conserved XBP-1 binding site motif close to the transcription start site of different DJ-1 promoter sequences including human (h.DJ-1) and mice (m.DJ-1) species (see Fig. 5A). XBP-1 is a well-characterized protein activated by the unfolded protein response (UPR or ER stress) (Yoshida et al., 2001). Therefore, we envisioned the possibility that XBP-1 could be the missing link of the cellular machinery functionally connecting PK and DJ-1. As a first attempt to study XBP-1 contribution to DJ-1 expression, we have first examined parkin, p53 and DJ-1 expressions responsiveness to an ER-stress. The treatment with a classical ER stress-inducer, thapsigargin (TPS), triggers an increase of parkin (+151±24.2% of control untreated cells, Fig. 5B) and DJ-1 protein levels (+52±19%, Fig. 5C) as well as a reduction of the promoter activity of p53 (−49±2.3%, supplementary material Fig. S3). These observations were fully in line with our hypothesis of an involvement of XBP-1 as a cellular intermediate between PK and DJ-1.
Fig. 5.
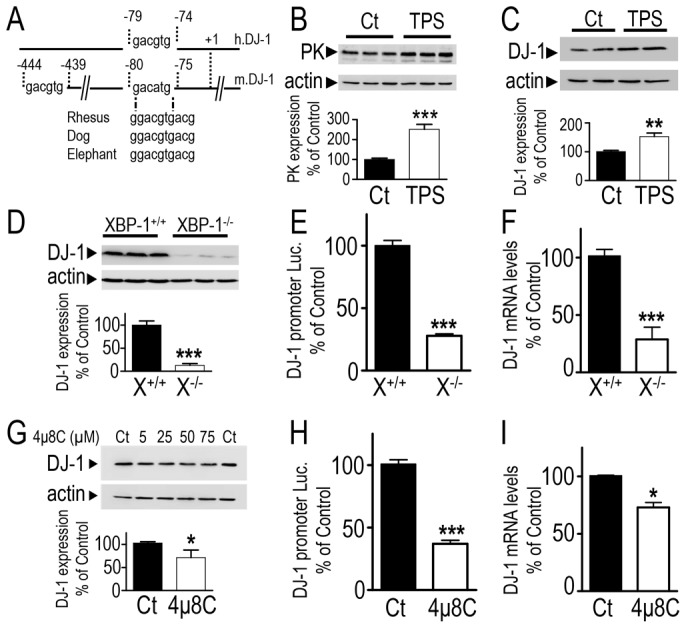
ER stress-associated transcriptional regulation of DJ-1 by XBP-1. (A) Representation of DJ-1 promoter regions harboring potential XBP-1 binding sites. (B,C) SH-SY5Y cells were treated for 16 hours with thapsigargin (TPS, 1 µM) or vehicle (DMSO) then harvested and analyzed for parkin (PK, B), DJ-1 (C) and actin (B,C) immunoreactivities. Bars correspond to densitometric analyses expressed as a percentage of control untreated cells (Ct) and are the means ± s.e.m. of 3 independent experiments performed in 3 replicates. (D–F) XBP-1+/+ (X+/+, black bars) and XBP-1−/− (X−/−, white bars) mouse fibroblasts were analyzed for their DJ-1 expression (D), promoter transactivation (E) and mRNA (F) levels. Bars are expressed as a percentage of control XBP-1+/+-treated cells and represent the means ± s.e.m. of 3 or 4 independent experiments performed in 3 or 4 replicates. (G–I) SH-SY5Y cells were treated with TPS (1 µM, overnight, Ct) or with 4µ8C at the indicated concentrations (G) or with 50 µM of the Ire-1 inhibitor 4μ8C (H,I). Then, DJ-1 expression (G), promoter transactivation (H) and mRNA levels (I) were analyzed. Bars express results as a percentage of control TPS-treated cells and represent means ± s.e.m. of 4–6 replicates. **P<0.01, ***P<0.001 compared with controls.
We have taken advantage of a cell model in which the Xbp-1 gene has been genetically invalidated (see XBP-1 expression in XBP-1−/− cells in supplementary material Fig. S4) to directly test the capability of XBP-1 to control DJ-1. XBP-1 depletion triggers a decrease of DJ-1 protein expression (−87±0.6% of control XBP-1+/+ cells, Fig. 5D), promoter transactivation (−72±0.6%, Fig. 5E) and mRNA levels (−76±3.3%, Fig. 5F). The depletion of XBP-1 by a siRNA approach confirmed the control of DJ-1 by XBP-1. Thus, two sets of siRNA (si1 and to a lesser extent, si2 siRNA, see supplementary material Fig. S4) that lower XBP-1 also reduce DJ-1 expressions by about 26%.
Data derived from XBP-1 genetic manipulation experiments were further confirmed by a pharmacological approach. Thus, XBP-1 is activated by an unconventional mRNA splicing by the endonuclease IRE-1α in response to ER stress (Yoshida et al., 2001). Recently, one compound (referred to as 4μ8C thereafter) aimed at blocking IRE1-mediated splicing of XBP-1 and, as a consequence, interfering with XBP-1 function has been designed (Cross et al., 2012). As shown in supplementary material Fig. S5, TPS-induced cellular stress reveals a XBP-1 spliced-like immunoreactive protein (supplementary material Fig. S5A), the production of which is abolished in XBP−/− cells and interestingly, totally prevented by 4μ8C in XBP+/+ cells (supplementary material Fig. S5A). It should be noted that 4μ8C did not affect unspliced XBP-1 (XBP-1U) in basal conditions (supplementary material Fig. S5B, middle gel). However, 4μ8C abolishes TPS-induced specific XBP-1 spliced transcript (supplementary material Fig. S5B, upper gel) and concomitantly, lowers the expression of XBP-1 spliced mRNA levels (supplementary material Fig. S5C) and drastically reduces associated promoter auto-activation (Acosta-Alvear et al., 2007) (supplementary material Fig. S5D). Interestingly, 4μ8C also dose-dependently reduces DJ-1 protein expression (−31.6±10.3, Fig. 5G) and lowers DJ-1 promoter transactivation (−63.7±4.5, Fig. 5H) and mRNA levels (−27.5±5.4, Fig. 5I).
In order to definitely prove the direct transcriptional regulation of DJ-1 by XBP-1, we have mutated the human DJ-1 promoter consensus binding motif of XBP-1 and analyzed the effect of the overexpression of unspliced (transcriptionally inactive, XBP-1U) and spliced (transcriptionally active, XBP-1S) forms of XBP-1 on wild-type and mutated DJ-1 promoter activity in SH-SY5Y cells. It should be noted first that EV-transfected cells display lower transactivation of mutated promoter, indicating that an endogenous active form of XBP-1 that appears to control positively DJ-1 promoter transactivation, occurs in these cells (Fig. 6A, compare white bars). This agrees with the faint but detectable spliced transcript observed in supplementary material Fig. S5B, upper gel, and the 4μ8C-sensitive XBP-1 mRNA auto-activation in basal conditions (not shown). As expected from above-described data, the spliced form (Fig. 6A, black bar, wt lane) but not the inactive form (Fig. 6A, gray bar, wt lane) of XBP-1 increased the transactivation of DJ-1 promoter (+157±30.9%, n = 12, P<0.001; Fig. 6A). Interestingly, the mutation disrupting the consensus binding site of XBP-1 completely abolished the responsiveness of the DJ-1 promoter to spliced XBP-1-mediated activation (Fig. 6A, compare black bars).
Fig. 6.
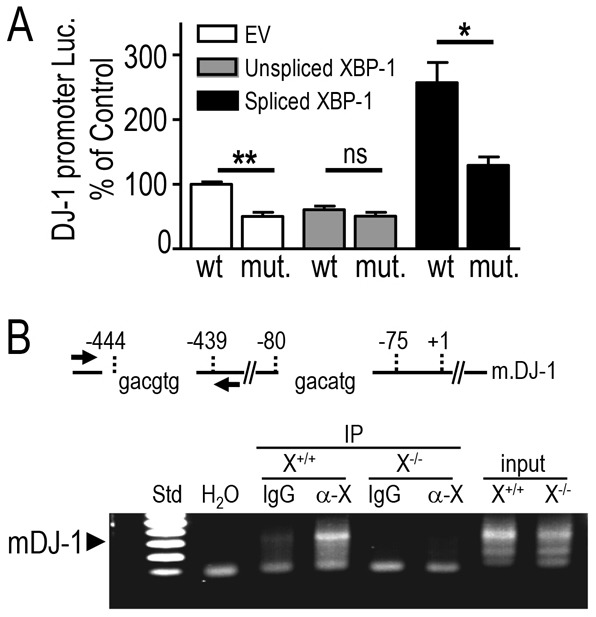
XBP-1 physically interacts with DJ-1 promoter. (A) SH-SY5Y human neuroblastoma cells were transiently co-transfected with human wild-type (wt) or mutated (XBP-1 binding site deleted, mut) DJ-1-luciferase promoter construct and either empty vector (EV, white bars) or cDNAs encoding XBP-1U (gray bars) or XBP-1S (black bars). Data were normalized by additional transfection of a CMV-β galactosidase vector. Twenty-four hours after transfection, DJ-1 promoter transactivation was monitored as described in the Materials and Methods. Bars express results as a percentage of control EV and wild-type DJ-1 promoter co-transfected cells and are the means ± s.e.m. of 4 independent experiments performed in 3 replicates. (B) Top: Representation of mouse DJ-1 promoter region including the sequence and the position of two putative XBP-1 binding sites at positions −444 to −439 and −80 to −75 from the transcription start site (denoted +1). The two arrows indicate the approximate position of the primers used in the ChIP experiment on mouse fibroblasts. (B) Bottom: ChIP analysis of the interaction between endogenous mouse XBP-1 and mouse DJ-1 promoter in wild-type (X+/+) and XBP-1-deficient (X−/−) fibroblasts reveals a PCR product (300-nucleotides long) obtained with the mouse PCR primers covering the −565 to −266 region of the DJ-1 mouse promoter. α-X and IgG represent ChIP analyses carried out after immunoprecipitation with specific XBP-1 antibody (M-186; sc-7160) or with unrelated control antibodies. DNA inputs in XBP-1+/+ and XBP-1−/− cells are indicated on the right-hand lanes. H2O is a negative control in which the DNA is replaced by water. The left lane of the gel (Std) shows a portion (from 600 bp to 100 bp) of the 100 bp DNA Ladder from Promega (G2101). The gel presented here is representative of 3 independent experiments. *P<0.05, **P<0.01; ns, not statistically significant.
Finally, we performed chromatin immunoprecipitation (ChIP) experiments to determine if XBP-1 physically interacted with the mouse DJ-1 promoter. The motif corresponding to the XBP-1 consensus binding site is located at position −444 to −439 from the TSS of DJ-1 promoter (see Fig. 6B, upper scheme). We thus designed a couple of primers covering the region −444 and −439 to examine the interaction of endogenous XBP-1 with the mouse DJ-1 promoter. As indicated in Fig. 6B (lower panel), selective immunoprecipitation of XBP-1 leads to the amplification of a specific DJ-1-associated fragment in XBP-1+/+ (X+/+) cells (Fig. 6B, compare lanes IgG control versus α-X). This fragment was absent when the immunoprecipitation was performed in XBP-1−/− (X−/−) cells (Fig. 6B, compare lanes α-X in X+/+ and X−/− cells). Overall, the above-data all concur to firmly support the view that endogenous XBP-1 directly and specifically interacts with, and upregulates DJ-1 promoter.
XBP-1 is controlled by p53 and parkin
In order to reconcile the fact that PK and p53 inversely modulate DJ-1 together with our observation that XBP-1 could upregulate DJ-1, we have further examined the ability of parkin and p53 to control XBP-1. First, we show that the overexpression of p53 in SH-SY5Y cells represses the transactivation of the XBP-1 promoter (−41.6±4.5%, supplementary material Fig. S6). Conversely, p53 depletion leads to increased XBP-1S expression in both basal (Fig. 7A, compare lanes p19Arf−/− and p19Arf−/−p53−/− in upper gel) and TPS-induced (Fig. 7B, compare lanes p19Arf−/− and p19Arf−/−p53−/− in lower gel; +133±34.1% of control p19Arf−/− cells) conditions. Clearly, the lack of p53 also increases XBP-1 promoter transactivation (+576±81.8%, Fig. 7C, Prom) and mRNA levels (+61±24%, Fig. 7C). The regulation of XBP-1 promoter by p53 was confirmed in HCT116-p53−/− cells that also display higher XBP-1 promoter activity (+32±6.4% of control HCT116-p53+/+ cells, supplementary material Fig. S2C). Finally, we have also established that the mRNA levels of XBP-1 are increased in p53 knockout mice brains (+80.8±22% of control wild-type brains, Fig. 7D). Therefore, p53 behaves as a repressor of XBP-1 promoter transactivation in cells and in vivo.
Fig. 7.
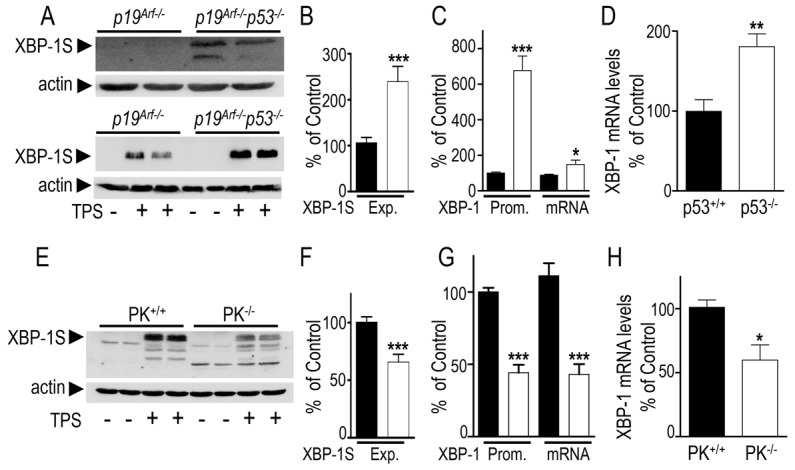
Influence of endogenous p53 and parkin on XBP-1 expression. (A) Expression of endogenous XBP-1S in p19Arf−/− and p19Arf−/−p53−/− cells in basal (top panel) or thapsigargin (TPS)-treated (1 µM, 16 hours; lower panel) conditions. (B) Densitometric analysis of the XBP-1S-like immunoreactivity (Exp) in TPS-treated p19Arf−/− (black bars) and p19Arf−/−p53−/− (white bars) cells (normalized by actin expression). Values are the means ± s.e.m. of 3 independent experiments performed in 2–3 replicates. (C) Mouse XBP-1 promoter transactivation (Prom) and mRNA levels were measured in p19Arf−/− (black bars) and p19Arf−/−p53−/− (white bars) cells. Values are expressed as a percentage of control values obtained in p19Arf−/− cells and are the means ± s.e.m. of 3 independent experiments performed in 3 replicates. (D) XBP-1 mRNA levels detected in brain samples derived from p53+/+ and p53−/− mice measured by RT-PCR and expressed as a percentage of control p53+/+ brains. Bars represent means ± s.e.m. of 4 or 5 brains. (E) Analysis of the expression of endogenous XBP-1 in PK+/+ and PK−/− cells treated with TPS (1 µM, 16 hours). (F) Densitometric analysis of the XBP-1S-like immunoreactivity (Exp) in TPS-treated PK+/+ (black bars) and PK−/− (white bars) cells expressed as a percentage of control TPS-treated PK+/+ cells. Values are the means ± s.e.m. of 5 independent experiments performed in 2–3 replicates. (G) Mouse XBP-1 promoter transactivation (Prom) and mRNA levels were measured in PK+/+ (black bars) and PK−/− (white bars) cells. Values are expressed as a percentage of control values obtained in PK+/+ cells and are the means ± s.e.m. of 3 or 4 independent experiments performed in 4 to 6 replicates. (H) Densitometric analysis of XBP-1 mRNA levels detected in brain samples from PK+/+ and PK−/− mice measured by RT-PCR. Results are expressed as a percentage of control PK+/+ brains and are the means ± s.e.m. of 5 mouse brains of each. *P<0.05, **P<0.01, ***P<0.001.
In agreement with the above-described data, the invalidation of the parkin gene triggers a significant reduction of XBP-1S expression (−31.9±3.6% of control PK+/+ cells, Fig. 7E,F), promoter transactivation (−56±2.4%, Fig. 7G) and mRNA levels (−68±3.3%, Fig. 7G). We have also demonstrated that mRNA levels of XBP-1 are lowered in PK-deficient mice brain (−41±18%, Fig. 7H). The influence of PK on XBP-1S is therefore similar to the effect of PK on DJ-1. As a matter of fact, wild-type PK increases XBP-1 promoter transactivation (+143.1±7%, supplementary material Fig. S7A) and mRNA levels (+41.6±4.3%, supplementary material Fig. S7B). Interestingly, this phenotype was fully abolished by both ubiquitin-deficient and ubiquitin-active PK mutants.
In order to examine whether parkin-associated control of DJ-1 was fully dependent of XBP-1S, we pharmacologically blocked the activity of XBP-1S in cells devoid of endogenous parkin. We show that in both basal (Fig. 8A) and thapsigargin-treated (Fig. 8B) conditions, 4μ8C treatment triggers reduced DJ-1 expression (−32.4±4.1% and −53.6±7.5%, respectively; Fig. 8, black bars) in wild-type but not in PK−/− cells. As expected, parkin depletion drastically reduces DJ-1 expression [−69.7±3.3% and −85±4.7%; Fig. 8, compare black and empty bars labeled (−), respectively] the levels of which remained unaffected by 4μ8C (Fig. 8, open bars). Overall, this indicates that most of parkin-mediated control of DJ-1 indeed requires XBP-1.
Fig. 8.
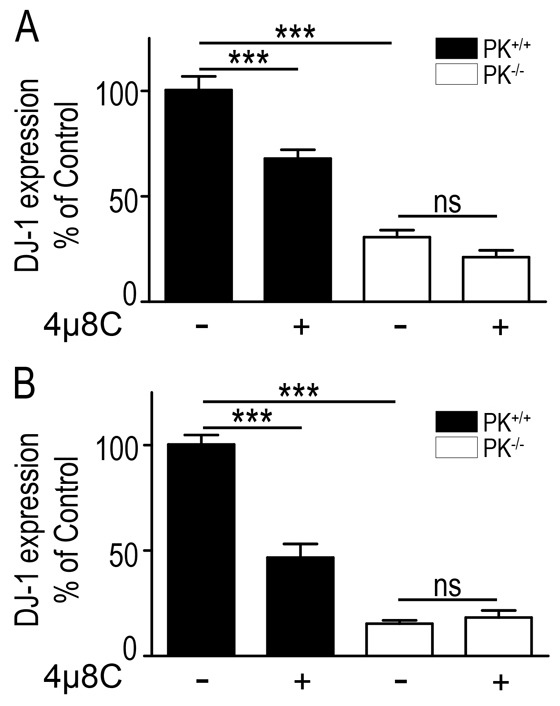
Parkin-mediated control of DJ-1 is XBP-1 dependent. (A,B) MEF cells invalidated (PK−/−) or not (PK+/+) for the parkin gene were treated with vehicle (−) or with the IRE-1 inhibitor 4μ8C (+), in basal (A) and thapsigargin (B) conditions as described in Materials and Methods. Bars correspond to western blot analysis and quantification is expressed as a percentage of the DJ-1 expression in untreated wild-type fibroblasts. Values are the means ± s.e.m. of 12–15 replicates. ***P<0.001; ns, not statistically significant.
Discussion
The etiology of PD is complex and several cellular dysfunctions appear to underlie the neurodegenerative process occurring in this disease. Particularly significant is the observation of PD-related alterations of the ubiquitin-proteasome system that likely account for aberrant protein degradation and therefore, subsequent accumulation and aggregation as revealed by intracytoplasmic inclusions called Lewy bodies. Directly linked to these observations, the activation of the unfolded protein response (UPR) also called endoplasmic reticulum-stress (ER-stress) has been consistently documented in PD-related cellular model (Ryu et al., 2002) as well as in PD-affected brain (Hoozemans et al., 2007). Thus, this cellular response aims at regulating the overload of toxic abnormally folded proteins by inducing ER-resident chaperone proteins and by promoting their degradation (Ron and Walter, 2007). The latter process is therefore a key event and alterations of proteins or enzymes linked to ubiquitin-mediated proteasomal degradation are likely detrimental and central to PD development.
Parkin is a 52 kDa protein responsible for most of juvenile autosomal recessive forms (AR-JP) of PD. The structural features of this protein indicated that parkin could be involved in proteasomal degradation of ubiquitinated substrates. Thus, parkin harbors a N-terminal domain that displays homology with ubiquitin while its C-terminal moiety harbors RING motifs that are a structural hallmark generally exhibited by ubiquitin-ligases (Morett and Bork, 1999). Indeed, parkin has been characterized as an E3-ubiquitin ligase, the activity of which is abolished by mutations associated with AR-JP (Shimura et al., 2000; Zhang et al., 2000). Interestingly, the ability of parkin to prevent ER-stress has been linked to its capacity to trigger the degradation of a usually unfolded protein called Pael receptor (Imai et al., 2001). Parkin ubiquitinates Pael receptor in the ER and Pael receptor accumulates in AR-JP (Imai et al., 2001). Thus, parkin-associated control of the UPR could be linked to its enzymatic properties and it was proposed that its defect could account for selective dopaminergic neurons death. However, it is noteworthy that it was recently documented that parkin-associated protection against ER-stress-induced cell death could be independent of the proteasomal machinery, indicating that parkin-related neuroprotection against ER-stress could not necessarily require its ubiquitin-ligase activity (Bouman et al., 2011). In this context, it is interesting that we recently documented an additional parkin-associated function as a transcription factor (da Costa et al., 2009). This agreed well with the preliminary description of a C-terminal IBR (In Between Ring) domain that indeed predicted a potential transcription factor activity for parkin (Morett and Bork, 1999). Thus, we established that parkin transrepressed the transcription of the pro-apoptotic tumor suppressor p53 and that this led to neuronal neuroprotection (da Costa et al., 2009). Whether this novel function could account for part of parkin-associated control of the UPR was therefore assessed.
Our study demonstrates that parkin controls the protein expression and promoter trans-activation of DJ-1, another protein which, when mutated, accounts for a subset of autosomal recessive cases of PD (Bonifati et al., 2003). By means of selective protein overexpression or gene inactivation in vitro and in vivo, we delineate a transcriptional cascade involving two intermediate transcription factors, namely p53 and the X-box-binding protein 1 (XBP-1). We establish that parkin represses p53 that acts as a repressor of XBP-1. Thereby, parkin activates XBP-1 that subsequently behaves as a strong transactivator of DJ-1 promoter transcription. We show that human and mouse DJ-1 harbors a putative XBP-1-targeted consensus sequence, the mutation of which abolishes XBP-1-mediated DJ-1 promoter transactivation and we document a physical interaction of XBP-1 with the human (data not shown) and mouse DJ-1 promoter by ChIP approach (Fig. 7B). Interestingly, interfering with any of the actors of this molecular cascade modulates parkin-associated control of DJ-1.
The present study unravels several novel cellular processes and has strong implications for ER-stress-mediated cell death in PD. First, this is the initial demonstration of a UPR-related functional link between parkin and DJ-1, two proteins involved in autosomal recessive cases of PD. It is very interesting to note that this control occurs besides the canonical ubiquitin-ligase activity of parkin. This agrees well with a recent study showing that parkin could control DJ-1 but failed to promote its ubiquitination (Moore et al., 2005a). Conversely, parkin appeared to trigger DJ-1 protein stabilization. Therefore, parkin could likely exert a dual control of DJ-1, either by directly interacting physically with the protein (Moore et al., 2005a) or indirectly functionally at a transcriptional level (present study).
The second important point of our study consists in the delineation of the mechanisms by which parkin protects against the UPR. Clearly, parkin triggers p53-mediated control of XBP-1, a protein involved in plasma cellular differentiation as well as in liver development (Sha et al., 2009). More related to PD, XBP-1 mRNA splicing/activation by the ER-resident endonuclease IRE1 is triggered by ER stress (Yoshida et al., 2001). Interestingly, the activation of XBP-1 indeed protects against several toxin-associated PD-related phenotypes in dopaminergic cells (Sado et al., 2009).
The demonstration that XBP-1 could directly transactivate DJ-1 promoter delineates a novel mechanism by which XBP-1 could interfere with ER-stress-associated cell death. Thus, XBP-1 was previously shown to regulate the mRNA levels of several ER chaperone proteins, the induction of which corresponds to one of the main response elicited by the UPR. Interestingly, DJ-1 has been shown to protect against both oxidative and reticular stress (Taira et al., 2004; Yokota et al., 2003) via the control of oxidative stress associated genes, like SOD (Nishinaga et al., 2005), and key apoptotic mediators like p53 and Pten (Giaime et al., 2010; Kim et al., 2005) and to behave as a chaperone protein (Lee et al., 2003). Thus the present work also contributes to the understanding of one of the cellular responses leading to the enhancement of its chaperone, and thereby folding capacities, in response to ER-stress.
UPR is initially triggered by the binding of GRP78 to misfolded proteins. This leads to the release of PERK, a pancreatic ER kinase that, when freely recovered in the cells, is activated by homodimerization and catalytic auto-phosphorylation (Merksamer and Papa, 2010). Phopho-PERK then phosphorylates the initiation factor peIF2a that increases the transcription of ATF4 (Zhang and Kaufman, 2006). Interestingly, parkin was recently shown to be transcriptionally modulated by ATF4 (Bouman et al., 2011). Therefore, overall, our work identifies a cellular response to ER-stress implying a transcriptional cascade by which parkin-mediated p53-dependent activation of XBP-1 could prevent ER-stress-mediated cell death by increasing DJ-1 levels. Our demonstration that parkin-associated pathogenic mutations abolish the parkin-mediated effects on p53, XBP-1 and DJ-1 suggest that this cellular cascade could be altered in some cases of Parkinson's disease.
Materials and Methods
Cell systems and transfections
Telencephalon specific mouse 1 (TSM1), mouse embryonic fibroblasts (MEF), human colorectal adenocarcinoma (HCT116), human embryonic kidney 293 (HEK293) and SH-SY5Y human neuroblastoma cells were cultured in 5% CO2, in DMEM supplemented with 10% fetal calf serum containing penicillin (100 U/ml) and streptomycin (50 µg/ml). TSM1 neurons expressing empty vector or wild-type HA-tagged parkin were obtained as described (da Costa et al., 2003). HEK293 cells stably expressing wild-type or mutated parkin were obtained after jetPRIME agent transfection (Polyplus) and geneticin selection for ten days. Immortalized mouse embryonic fibroblasts invalidated for the parkin, XBP-1, p19arf and both p19arf and p53 genes and HCT116 have been generously provided by Drs T. Dawson, L. Glimsher, M. Roussel and B. Vogelstein. Transient transfection of SH-SY5Y, HEK293 were carried by means of either lipofectamine 2000 (Invitrogen) or jetPRIME reagent according to the manufacturer's instructions while parkin, p19Arf−/−, p19Arf−/−p53−/− fibroblasts were transfected by means of NucleofactorTM kit (Amaxa Biosystems) as previously described (Pardossi-Piquard et al., 2005).
Constructs and site-directed mutagenesis
The cDNA of human parkin has been cloned in the pIRES2-EGFP-NEO vector (Clonetech). Wild-type human DJ-1 and p53 cDNAs were gifts from Dr P. Heutink, and M. Oren. Parkin mutagenesis was performed with the QuikChange II kit (Stratagene). 5′-end truncated mouse XBP-1 promoter-luciferase constructs have been generated from the mouse XBP-1 full-length promoter-luciferase used as a template in PCR reactions. Primers used for the obtention of the 581, 438, 285 and 138 nucleotides promoter fragments are described in supplementary material Table S2. These fragments were digested with SacI and HinDIII and subcloned in the pGL3 basic vector (Promega). The deletion of the putative p53 binding site on the wild-type mouse XBP-1 promoter was achieved with the XBP-1 forward and reverse primers described in supplementary material Table S2.
The deletion of the XBP-1 binding site of hDJ-1 promoter was obtained by three PCR steps: the first one using the GLprimer1 forward primer and the human DJ-1 deleted of XBP-1 binding site reverse primer. The second fragment was obtained using the GLprimer2 reverse primer with the human DJ-1 deleted of XBP-1 binding site forward primer. The right size products of the two first PCR were combined and mixed, then 10 PCR cycles were performed, the GLprimer1 and GLprimer2 were added and 25 more cycles were realized. Finally the human DJ-1 promoter deleted for the XBP-1 binding site was cloned in the pGL2-basic vector (Promega).
The p53 and DJ-1 promoter-luciferase constructs have been previously described (Ginsberg et al., 1990; Taira et al., 2001). The unspliced and spliced mouse XBP-1 cDNA in pcDNA3-FLAG, the pGL3-mouse XBP-1 promoter-luciferase, and the four XBP-1 shRNA-pSuper-puro were from Dr L. Qi. shRNA (KD1, KD2 and SC) targeting human parkin have been engineered in our laboratory based on the published siRNA sequences (Veeriah et al., 2010) and inserted in the pSuper-GFP-neo vector according to the manufacturer's instructions (Oligoengine). All the primers (Eurogentec) used are described in supplementary material Tables S1 and S2. All the constructs were verified by sequencing.
Western blot analysis
Cells were homogenized in lysis buffer (Tris-HCl, 10 mM; pH 7.5, containing NaCl (150 mM), Triton X-100 (0.5%), deoxycholate (0.5%) and EDTA (5 mM) and resolved on 12% SDS-PAGE gel and wet-transferred to Hybond-C (Amersham Life science) membranes. Immunoblottings were performed by means of the following antibodies: rabbit polyclonal anti-DJ-1 antibodies (MAB18257, Abcam), mouse monoclonal anti-parkin antibodies (MAB5512, Abcam), mouse monoclonal anti-p53 antibodies (Santa-Cruz Biotechnology), mouse monoclonal anti-GFP antibodies (Roche Applied Science), anti-XBP-1 antibodies (M-186, Santa-Cruz Biotechnology) or mouse monoclonal anti-actin antibodies (Sigma). Protein immunoreactivities were revealed with either an anti-rabbit peroxidase or an anti-mouse peroxidase (Jackson Immunoresearch) by electrochemoluminescence method (Roche Diagnostics S.A.S.). Chemiluminescence was recorded using a luminescence image analyzer LAS-3000 (Raytest), and quantification of images was performed using the FUJI FILM Multi Gauge image analyzer software.
DJ-1, p53 and XBP-1 promoter transactivations
The transcriptional activations of the human DJ-1, human p53 and mouse XBP-1 promoters in frame with luciferase (provided by Dr H. Ariga; Dr M. Oren and Dr L. Qi, respectively) were measured after co-transfection of β-galactosidase cDNA, in order to normalize transfection efficiencies (Promega). The protocol has been previously described (da Costa et al., 2003).
Real-time quantitative PCR
Total RNA from cells and mice brains were respectively extracted using the Nucleospin RNAII kit and RNeasy Lipid Tissue kit following instructions of the manufacturer (Macherey-Nagel GmbH and Qiagen, respectively). After treatment with DNase I, total RNA (2 µg) were reverse-transcribed with AMV-transcriptase (Promega, Madison, WI) using oligo-dT priming. Real-time PCR were performed in the Light Cycler 480 (Roche) or Rotor-Gene6000 (Qiagen), using the SYBR Green detection protocol recommended by the manufacturer. Specific-gene primers were designed by means of the Universal Probe Library Assay Design Center software (Roche Applied Science). Relative expression levels of mouse and human DJ-1 mRNA were normalized with mouse γ-actin and human GAPDH genes while mouse and human Top1 gene were used to normalize for RNA concentration the relative expression levels of mouse and human XBP-1 gene. All primers are described in supplementary material Table S3.
Semi-quantitative RT-PCR
Two µg of total RNA were reverse transcribed and 1/40 of the resulting cDNA was then used as a template for semi-quantitative PCR. In each case, we checked that the PCR reaction was in the linear phase of the amplification. Based on primers already reported to detect mouse XBP-1 spliced isoform (Acosta-Alvear et al., 2007), we have designed primers capable of specifically amplifying the spliced form of human XBP-1. To amplify both isoforms of human XBP-1, we designed primers according to what had been already described for mouse XBP-1 (Iwawaki et al., 2004). Primers are described in supplementary material Table S3. The same primers were used in our quantitative PCR experiments.
Chromatin immunoprecipitation assay
Wild-type and knockout XBP-1 MEF cells were seeded in three 150-mm dishes and allowed to reach 70–80% confluence. Cells were fixed with formaldehyde, cross-linked and processed for chromatin preparation according to the manufacturer's recommendations. DNA was digested with the shearing enzyme provided in the kit, yielding chromatin fragments of about 200–500 bp in size. Each chromatin immunoprecipitation assay (ChIP) was performed on 50 µl of chromatin in the ChIP immunoprecipitation buffer supplied and incubated overnight with 2 µg of anti-XBP-1 M-186 antibody (Santa Cruz Biotechnology) or with irrelevant antibodies as negative controls. Immune complexes were then incubated (1.5 hours at 4°C) with 100 µl of a solution of protein G-Sepharose. After elution of the immune complexes, cross-links were reversed and RNA digested using RNase (100 mg/ml). To eliminate proteins, proteinase K (100 mg/ml) was added and the samples were incubated for 2 hours at 42°C. DNA was purified on columns provided in the kit and PCR amplifications were performed using primers specific for the −444/−439 bp region of the mouse DJ-1 promoter (forward primer: 5′-GGATGCTGGGAACCGAACCTGGGTC-3′ and reverse primer: 5′-CTGGGCTACAGCCTGAACCCCACAC-3′).
Statistical analysis
Statistical analyses were performed with GraphPad Prism software (GraphPad Software, San Diego, CA) by using either the unpaired Student's t-test for pairwise comparison or Newmann–Keuls multiple comparison tests for one-way ANOVA analysis of variance. *P<0.05; **P<0.01; ***P<0.001; ns, not statistically significant.
Acknowledgments
Drs B. Vogelstein, M. Roussel, L. Glimcher, J. Shen T. and Dawson are warmly thanked for providing us with the p53, XBP-1 and parkin knockout cells, respectively. Drs M. Oren and P. Heutink are thanked for providing us the p53 and DJ-1 constructs, respectively. Dr J.C. Bourdon is warmly thanked for providing p53 antibodies. Dr D. Ron is thanked for the Ire-1α inhibitor (4μ8C).
Footnotes
Author contributions
E.D., E.G., J.V. and J.S. performed the experiments. O.C., A.B., H.A. and L.Q. provided materials and corrected the writing. E.D., F.C. and C.A.d.C. designed the experiments, discussed the data and wrote the paper.
Funding
J.V. is supported by the Association Monégasque pour la recherche contre la Maladie d'Alzheimer and E.D. is funded by the Fondation pour la Recherche Médicale. This work was supported by the Fondation pour la Recherche Médicale and by the Conseil Général des Alpes Maritimes. This work has been developed and supported through LABEX (excellence laboratory, program investment for the future) and DISTALZ (development of innovative strategies for a transdisciplinary approach to Alzheimer's disease).
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.127340/-/DC1
References
- Abbas N., Lücking C. B., Ricard S., Dürr A., Bonifati V., De Michele G., Bouley S., Vaughan J. R., Gasser T., Marconi R., et al. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease(1999). A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. Hum. Mol. Genet. 8, 567–574. 10.1093/hmg/8.4.567 [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman P. M., Healy D. G., Quinn N., Lees A. J., Wood N. W. (2003). The role of pathogenic DJ-1 mutations in Parkinson's disease. Ann. Neurol. 54, 283–286. 10.1002/ana.10675 [DOI] [PubMed] [Google Scholar]
- Acosta-Alvear D., Zhou Y., Blais A., Tsikitis M., Lents N. H., Arias C., Lennon C. J., Kluger Y., Dynlacht B. D. (2007). XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66. 10.1016/j.molcel.2007.06.011 [DOI] [PubMed] [Google Scholar]
- Bandopadhyay R., Kingsbury A. E., Cookson M. R., Reid A. R., Evans I. M., Hope A. D., Pittman A. M., Lashley T., Canet-Aviles R., Miller D. W. et al. (2004). The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson's disease. Brain 127, 420–430. 10.1093/brain/awh054 [DOI] [PubMed] [Google Scholar]
- Bertoli-Avella A. M., Giroud-Benitez J. L., Akyol A., Barbosa E., Schaap O., van der Linde H. C., Martignoni E., Lopiano L., Lamberti P., Fincati E. et al. (2004). Novel parkin mutations detected in patients with early-onset Parkinson's disease. Mov. Disord. 20, 424–41. [DOI] [PubMed] [Google Scholar]
- Bonifati V., Rizzu P., van Baren M. J., Schaap O., Breedveld G. J., Krieger E., Dekker M. C., Squitieri F., Ibanez P., Joosse M. et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. 10.1126/science.1077209 [DOI] [PubMed] [Google Scholar]
- Bouman L., Schlierf A., Lutz A. K., Shan J., Deinlein A., Kast J., Galehdar Z., Palmisano V., Patenge N., Berg D. et al. (2011). Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 18, 769–782. 10.1038/cdd.2010.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J. P., Sedivy J. M., Kinzler K. W., Vogelstein B. (1998). Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501. 10.1126/science.282.5393.1497 [DOI] [PubMed] [Google Scholar]
- Cross B. C., Bond P. J., Sadowski P. G., Jha B. K., Zak J., Goodman J. M., Silverman R. H., Neubert T. A., Baxendale I. R., Ron D. et al. (2012). The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 109, E869–E878. 10.1073/pnas.1115623109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Costa C. A., Masliah E., Checler F. (2003). β-synuclein displays an antiapoptotic p53-dependent phenotype and protects neurons from 6-hydroxydopamine-induced caspase 3 activation: cross-talk with alpha-synuclein and implication for Parkinson's disease. J. Biol. Chem. 278, 37330–37335. 10.1074/jbc.M306083200 [DOI] [PubMed] [Google Scholar]
- da Costa C. A., Sunyach C., Giaime E., West A., Corti O., Brice A., Safe S., Abou-Sleiman P. M., Wood N. W., Takahashi H. et al. (2009). Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson's disease. Nat. Cell Biol. 11, 1370–1375. 10.1038/ncb1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson M. W., Guo M. (2007). Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson's disease. Curr. Opin. Neurobiol. 17, 331–337. 10.1016/j.conb.2007.04.010 [DOI] [PubMed] [Google Scholar]
- Giaime E., Sunyach C., Druon C., Scarzello S., Robert G., Grosso S., Auberger P., Goldberg M. S., Shen J., Heutink P. et al. (2010). Loss of function of DJ-1 triggered by Parkinson's disease-associated mutation is due to proteolytic resistance to caspase-6. Cell Death Differ. 17, 158–169. 10.1038/cdd.2009.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg D., Oren M., Yaniv M., Piette J. (1990). Protein-binding elements in the promoter region of the mouse p53 gene. Oncogene 5, 1285–1290. [PubMed] [Google Scholar]
- Hampe C., Ardila-Osorio H., Fournier M., Brice A., Corti O. (2006). Biochemical analysis of Parkinson's disease-causing variants of Parkin, an E3 ubiquitin-protein ligase with monoubiquitylation capacity. Hum. Mol. Genet. 15, 2059–2075. 10.1093/hmg/ddl131 [DOI] [PubMed] [Google Scholar]
- Hoozemans J. J., van Haastert E. S., Eikelenboom P., de Vos R. A., Rozemuller J. M., Scheper W. (2007). Activation of the unfolded protein response in Parkinson's disease. Biochem. Biophys. Res. Commun. 354, 707–711. 10.1016/j.bbrc.2007.01.043 [DOI] [PubMed] [Google Scholar]
- Imai Y., Soda M., Inoue H., Hattori N., Mizuno Y., Takahashi R. (2001). An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105, 891–902. 10.1016/S0092-8674(01)00407-X [DOI] [PubMed] [Google Scholar]
- Itier J-M., Ibanez P., Mena M. A., Abbas N., Cohen-Salmon C., Bohme G. A., Laville M., Pratt J., Corti O., Pradier L. et al. (2003). Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum. Mol. Genet. 12, 2277–2291. 10.1093/hmg/ddg239 [DOI] [PubMed] [Google Scholar]
- Iwawaki T., Akai R., Kohno K., Miura M. (2004). A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat. Med. 10, 98–102. 10.1038/nm970 [DOI] [PubMed] [Google Scholar]
- Kim R. H., Peters M., Jang Y., Shi W., Pintilie M., Fletcher G. C., DeLuca C., Liepa J., Zhou L., Snow B. et al. (2005). DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer. Cell 7, 263–273. [DOI] [PubMed] [Google Scholar]
- Lee S. J., Kim S. J., Kim I. K., Ko J., Jeong C. S., Kim G. H., Park C., Kang S. O., Suh P. G., Lee H. S. et al. (2003). Crystal structures of human DJ-1 and Escherichia coli Hsp31, which share an evolutionarily conserved domain. J. Biol. Chem. 278, 44552–44559. 10.1074/jbc.M304517200 [DOI] [PubMed] [Google Scholar]
- Merksamer P. I., Papa F. R. (2010). The UPR and cell fate at a glance. J. Cell Sci. 123, 1003–1006. 10.1242/jcs.035832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore D. J., West A. B., Dawson V. L., Dawson T. M. (2005a). Molecular pathophysiology of Parkinson's disease. Annu. Rev. Neurosci. 28, 57–87. 10.1146/annurev.neuro.28.061604.135718 [DOI] [PubMed] [Google Scholar]
- Moore D. J., Zhang L., Troncoso J., Lee M. K., Hattori N., Mizuno Y., Dawson T. M., Dawson V. L. (2005b). Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum. Mol. Genet. 14, 71–84. 10.1093/hmg/ddi007 [DOI] [PubMed] [Google Scholar]
- Morett E., Bork P. (1999). A novel transactivation domain in parkin. Trends Biochem. Sci. 24, 229–231. 10.1016/S0968-0004(99)01381-X [DOI] [PubMed] [Google Scholar]
- Nishinaga H., Takahashi-Niki K., Taira T., Andreadis A., Iguchi-Ariga S. M., Ariga H. (2005). Expression profiles of genes in DJ-1-knockdown and L 166 P DJ-1 mutant cells. Neurosci. Lett. 390, 54–59. 10.1016/j.neulet.2005.07.053 [DOI] [PubMed] [Google Scholar]
- Pardossi-Piquard R., Petit A., Kawarai T., Sunyach C., Alves da Costa C., Vincent B., Ring S., D'Adamio L., Shen J., Müller U. et al. (2005). Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron 46, 541–554. 10.1016/j.neuron.2005.04.008 [DOI] [PubMed] [Google Scholar]
- Ron D., Walter P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- Ryu E. J., Harding H. P., Angelastro J. M., Vitolo O. V., Ron D., Greene L. A. (2002). Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J. Neurosci. 22, 10690–10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sado M., Yamasaki Y., Iwanaga T., Onaka Y., Ibuki T., Nishihara S., Mizuguchi H., Momota H., Kishibuchi R., Hashimoto T. et al. (2009). Protective effect against Parkinson's disease-related insults through the activation of XBP1. Brain Res. 1257, 16–24. 10.1016/j.brainres.2008.11.104 [DOI] [PubMed] [Google Scholar]
- Sha H., He Y., Chen H., Wang C., Zenno A., Shi H., Yang X., Zhang X., Qi L. (2009). The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 9, 556–564. 10.1016/j.cmet.2009.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H., Hattori N., Kubo S., Mizuno Y., Asakawa S., Minoshima S., Shimizu N., Iwai K., Chiba T., Tanaka K. et al. (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305. 10.1038/77060 [DOI] [PubMed] [Google Scholar]
- Shulman J. M., De Jager P. L., Feany M. B. (2011). Parkinson's disease: genetics and pathogenesis. Annu. Rev. Pathol. 6, 193–222. 10.1146/annurev-pathol-011110-130242 [DOI] [PubMed] [Google Scholar]
- Taira T., Takahashi K., Kitagawa R., Iguchi-Ariga S. M., Ariga H. (2001). Molecular cloning of human and mouse DJ-1 genes and identification of Sp1-dependent activation of the human DJ-1 promoter. Gene 263, 285–292. 10.1016/S0378-1119(00)00590-4 [DOI] [PubMed] [Google Scholar]
- Taira T., Saito Y., Niki T., Iguchi-Ariga S. M., Takahashi K., Ariga H. (2004). DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 5, 213–218. 10.1038/sj.embor.7400074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi R., Imai Y., Hattori N., Mizuno Y. (2003). Parkin and endoplasmic reticulum stress. Ann. New York Acad. Sci. 991, 101–106. 10.1111/j.1749-6632.2003.tb07467.x [DOI] [PubMed] [Google Scholar]
- Tanaka K., Suzuki T., Chiba T., Shimura H., Hattori N., Mizuno Y. (2001). Parkin is linked to the ubiquitin pathway. J. Mol. Med. 79, 482–494. 10.1007/s001090100242 [DOI] [PubMed] [Google Scholar]
- Thomas K. J., McCoy M. K., Blackinton J., Beilina A., van der Brug M., Sandebring A., Miller D., Maric D., Cedazo-Minguez A., Cookson M. R. (2011). DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 20, 40–50. 10.1093/hmg/ddq430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeriah S., Taylor B. S., Meng S., Fang F., Yilmaz E., Vivanco I., Janakiraman M., Schultz N., Hanrahan A. J., Pao W. et al. (2010). Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet. 42, 77–82. 10.1038/ng.491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J. D., Jeffers J. R., Rehg J. E., Randle D. H., Lozano G., Roussel M. F., Sherr C. J., Zambetti G. P. (2000). p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev. 14, 2358–2365. 10.1101/gad.827300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A., Periquet M., Lincoln S., Lücking C. B., Nicholl D., Bonifati V., Rawal N., Gasser T., Lohmann E., Deleuze J. F., et al. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility on Parkinson's Disease(2002). Complex relationship between Parkin mutations and Parkinson disease. Am. J. Med. Genet. 114, 584–591. 10.1002/ajmg.10525 [DOI] [PubMed] [Google Scholar]
- Yokota T., Sugawara K., Ito K., Takahashi R., Ariga H., Mizusawa H. (2003). Down regulation of DJ-1 enhances cell death by oxidative stress, ER stress, and proteasome inhibition. Biochem. Biophys. Res. Commun. 312, 1342–1348. 10.1016/j.bbrc.2003.11.056 [DOI] [PubMed] [Google Scholar]
- Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. 10.1016/S0092-8674(01)00611-0 [DOI] [PubMed] [Google Scholar]
- Zhang K., Kaufman R. J. (2006). The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology 66 Suppl. 1, S102–S109. 10.1212/01.wnl.0000192306.98198.ec [DOI] [PubMed] [Google Scholar]
- Zhang Y., Gao J., Chung K. K., Huang H., Dawson V. L., Dawson T. M. (2000). Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. USA 97, 13354–13359. 10.1073/pnas.240347797 [DOI] [PMC free article] [PubMed] [Google Scholar]