

ABSTRACT
The APC tumor suppressor regulates diverse stem cell processes including gene regulation through Wnt–β-catenin signaling and chromosome stability through microtubule interactions, but how the disparate functions of APC are controlled is not well understood. Acting as part of a Wnt–β-catenin pathway that controls asymmetric cell division, Caenorhabditis elegans APC, APR-1, promotes asymmetric nuclear export of the β-catenin WRM-1 by asymmetrically stabilizing microtubules. Wnt function also depends on a second β-catenin, SYS-1, which binds to the C. elegans TCF POP-1 to activate gene expression. Here, we show that APR-1 regulates SYS-1 levels in asymmetric stem cell division, in addition to its known role in lowering nuclear levels of WRM-1. We demonstrate that SYS-1 is also negatively regulated by the C. elegans homolog of casein kinase 1α (CKIα), KIN-19. We show that KIN-19 restricts APR-1 localization, thereby regulating nuclear WRM-1. Finally, the polarity of APR-1 cortical localization is controlled by PRY-1 (C. elegans Axin), such that PRY-1 controls the polarity of both SYS-1 and WRM-1 asymmetries. We propose a model whereby Wnt signaling, through CKIα, regulates the function of two distinct pools of APC – one APC pool negatively regulates SYS-1, whereas the second pool stabilizes microtubules and promotes WRM-1 nuclear export.
KEY WORDS: APC, C. elegans, SYS-1, WRM-1, Wnt, β-catenin
INTRODUCTION
Asymmetric cell division (ACD) is a fundamental mechanism for controlling cell fate specification and increasing the diversity of cell types during development and adult tissue homeostasis, including stem cell specification (Horvitz and Herskowitz, 1992; Neumüller and Knoblich, 2009). Stem cells self-renew by undergoing ACD to generate a new stem cell and a more differentiated daughter cell (Knoblich, 2008). Consistent with a role in stem cell homeostasis, conserved regulators of ACD are tumor suppressors in mammals, and ACD defects are broadly implicated in human cancers (Cicalese et al., 2009; Morrison and Kimble, 2006; Powell et al., 2010; Quyn et al., 2010). Elucidating the cell signaling pathways that govern ACD will have broad impacts on our understanding of cell fate acquisition and tumorigenesis.
The Wnt–β-catenin pathway has emerged as an important regulator of cell polarity and ACD of stem cells. In this pathway, the secreted Wnt ligand regulates target gene expression by stabilizing the transcriptional coactivator β-catenin, which then translocates into the nucleus to activate the TCF family of transcription factors (MacDonald et al., 2009). When ACD or cell polarity is perturbed in Wnt-regulated stem cells (e.g. mammary, hematopoietic and intestinal stem cells), defects in cell fate and/or tumorigenesis can occur (Cicalese et al., 2009; Quyn et al., 2010; Khramtsov et al., 2010; Reya and Clevers, 2005; Bellis et al., 2012). By contrast, when Wnt is asymmetrically presented to stem cells in vitro, the daughter cells of the resultant ACD are asymmetric with respect to the expression of Wnt target genes (Habib et al., 2013), providing evidence that Wnt is sufficient to drive ACD in mammals. A commonly mutated Wnt signaling gene found in familial and sporadic colon cancers is adenomatous polyposis coli (APC). APC is a multifunctional protein that is well known for its role in the negative regulation of β-catenin, although APC has other roles in the regulation of microtubule stability that are also perturbed in colon cancer (Clevers and Nusse, 2012; Brocardo and Henderson, 2008; Green and Kaplan, 2003; Barth et al., 2008; Zumbrunn et al., 2001; Nakamura et al., 2001). Although much is known about APC function in the regulation of β-catenin stability during Wnt signaling, the mechanisms that control the multiple functions of APC during asymmetric stem cell divisions are unclear.
Caenorhabditis elegans is well suited for elucidating the mechanisms of Wnt–β-catenin signaling in in vivo asymmetric stem cell divisions, because a Wnt–β-catenin signaling pathway, known as the Wnt–β-catenin asymmetry (WβA) pathway, induces serial asymmetric divisions throughout C. elegans invariant development (for reviews see Phillips and Kimble, 2009; Sawa, 2012) and because the cells in question are amenable to in vivo observations and perturbations. WβA directs differential cell fate determination during ACD by regulating the asymmetric distribution of Wnt signaling components before and during cell division. A primary target of the WβA pathway is SYS-1, a transcriptionally active β-catenin. SYS-1 levels are downregulated in Wnt-independent daughter cells and upregulated in Wnt-dependent daughters (Kidd et al., 2005; Phillips et al., 2007; Huang et al., 2007). SYS-1 asymmetry is a crucial polarity readout that results in asymmetric cell fate. Therefore, understanding how SYS-1 is regulated is crucial for understanding the output of the WβA pathway. Several lines of evidence suggest that SYS-1 protein is asymmetrically degraded: (1) a SYS-1 transcriptional reporter localizes symmetrically to daughter cells, indicating that SYS-1 expression is not regulated at the level of transcription or mRNA processing (Phillips et al., 2007); (2) SYS-1 localizes symmetrically to the centrosomes during metaphase and anaphase, indicating that SYS-1 protein is initially loaded equally into anterior and posterior regions of the dividing mother cell (Phillips et al., 2007; Huang et al., 2007); and (3) RNAi depletion of a proteasome component results in increased SYS-1 levels in an anterior embryonic daughter cell (Huang et al., 2007), indicating that protein degradation is required for the loss of SYS-1 in Wnt pathway-inactive cells. Therefore, SYS-1 and mammalian β-catenin appear to be regulated by the same general processes. However, for SYS-1 regulation, the role of the destruction complex including APC is largely unknown.
During C. elegans ACD, APC indirectly regulates the C. elegans TCF POP-1, by stabilizing microtubules and promoting the nuclear export of a second β-catenin, WRM-1 (Fig. 1; Sugioka et al., 2011; Rocheleau et al., 1999; Lo et al., 2004; Yang et al., 2011). WRM-1 does not activate POP-1/TCF transcription but, instead, facilitates POP-1 phosphorylation by Nemo-like kinase and subsequent POP-1 nuclear export in signaled daughters (Korswagen et al., 2000). In the Wnt-independent daughter cell, nuclear levels of WRM-1 are lowered by APC-dependent microtubule-mediated nuclear export, leading to decreased nuclear WRM-1 levels compared with the Wnt-dependent daughter cell. Wnt signaling inhibits APC and microtubule stability in the Wnt-dependent daughter, leading to elevated nuclear WRM-1 and decreased POP-1 (Sugioka et al., 2011). The resulting reciprocal asymmetry of SYS-1 and POP-1 gives rise to one nucleus with a low SYS-1∶POP-1 ratio that results in target gene repression (the Wnt-independent daughter) and another nucleus with a high SYS-1∶POP-1 ratio that results in Wnt target gene activation (the Wnt-dependent daughter; Fig. 1; Jackson and Eisenmann, 2012). Whether and to what extent the WβA branches that control POP-1 and SYS-1 asymmetries intersect remains uncertain, primarily because SYS-1 regulation is poorly understood compared with POP-1 regulation.
Fig. 1.
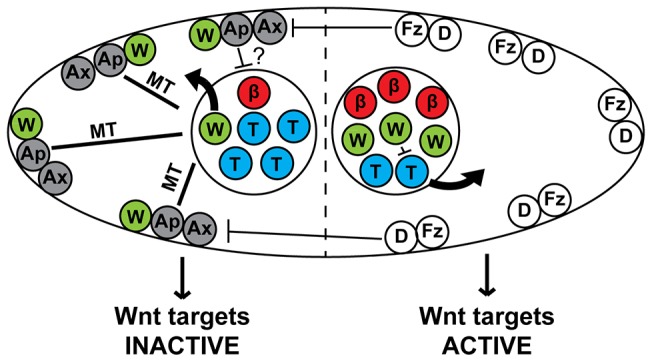
A model of the WβA pathway in the epithelial seam stem cells. Negative regulators of the TCF-export pathway (PRY-1/Axin, APR-1/APC) are localized to the anterior cortex by positive regulators of Wnt signaling on the posterior cortex (Frizzled, Disheveled). APR-1/APC stabilizes microtubules (MT) in the anterior daughter to increase WRM-1/β-catenin nuclear export and cortical localization. Higher nuclear levels of WRM-1/β-catenin in the posterior daughter cell facilitate POP-1/TCF nuclear export. How SYS-1/β-catenin asymmetry is generated is presently unclear, although previous data indicate that APR-1/APC might be involved (?). β, SYS-1/β-catenin (red); W, WRM-1/β-catenin (green); T, POP-1/TCF (blue); Fz, Frizzled (white); D, Disheveled (white); Ax, PRY-1/Axin (gray); Ap, APR-1/APC (gray).
The C. elegans epithelial seam cells are especially amenable to the analysis of Wnt-mediated control of stem cell fate and β-catenin regulation. These visually accessible cells repeatedly divide asymmetrically in a stem-cell-like pattern during larval development, and cell fate determination for each of these divisions is dependent on the WβA pathway (supplementary material Fig. S1; Mizumoto and Sawa, 2007; Kanamori et al., 2008; Huang et al., 2009; Banerjee et al., 2010; Gleason and Eisenmann, 2010; Ren and Zhang, 2010; Harterink et al., 2011; Yamamoto et al., 2011; Gorrepati et al., 2013; Hughes et al., 2013). Thus, the lineage maintains a constant number of seam cells (posterior WβA-signaled daughters) while increasing the number of hypodermal nuclei (anterior Wnt-independent daughters). Activation or repression of WβA results in increased or decreased seam cell numbers, respectively. At least one direct target of SYS-1 and POP-1, EGL-18, is also differentially expressed in the seam daughters; EGL-18 expression is elevated in the posterior cell with a high SYS-1∶POP-1 ratio and is reduced or absent in the anterior daughter with a low SYS-1∶POP-1 ratio (Gorrepati et al., 2013). Other recent studies have analyzed the genetics of Wnt signaling components in C. elegans and the effects these components have on seam cell number, providing several potential regulators of SYS-1 asymmetry (Huang et al., 2009; Banerjee et al., 2010; Gleason and Eisenmann, 2010; Ren and Zhang, 2010).
Vertebrate β-catenin negative regulation requires the APC scaffold, which represents a candidate SYS-1 regulator. Loss of APC function results in inappropriate β-catenin stabilization and tumorigenesis in mammals (Clevers and Nusse, 2012). Loss of the C. elegans APC homolog, APR-1, also results in the inappropriate expansion of a stem cell population, in this case the worm seam cells. APR-1 regulates the asymmetric nuclear localization of WRM-1 during ACD; however, as discussed above, studies in the embryo indicate that APR-1 accomplishes this not by binding directly to WRM-1 but, instead, through asymmetric stabilization of astral microtubules that then control WRM-1 nuclear export (Sugioka et al., 2011; Mizumoto and Sawa, 2007). In order to affect asymmetric microtubule stability, APR-1 displays Wnt-dependent asymmetric localization to the anterior cortex of seam cells (Fig. 1; Sugioka et al., 2011; Mizumoto and Sawa, 2007). Finally, knocking down APR-1 expression by using RNAi [apr-1(RNAi)] results in symmetrical SYS-1 localization in the nuclei of asymmetrically dividing embryonic cells, raising the possibility that APR-1 might also regulate SYS-1 asymmetry in the seam cells (Fig. 1; Huang et al., 2007).
Because APC regulation of β-catenin in the canonical Wnt pathway requires β-catenin phosphorylation by CKIα, the worm homolog of CKIα, KIN-19, is also a candidate SYS-1 negative regulator. In mammals, CKIα phosphorylates β-catenin and APC, priming both proteins for additional phosphorylation by GSK3β, after which β-catenin is bound by APC and degraded (Clevers and Nusse, 2012; Price, 2006). kin-19(RNAi) results in more than double the average number of seam cells found in a wild-type worm, indicating that KIN-19 acts as a negative regulator of seam cell fate (Banerjee et al., 2010; Gleason and Eisenmann, 2010). Interestingly, loss of the worm homolog of GSK3β, GSK-3, alone or in combination with other WβA pathway members, does not cause defects in seam cell number, indicating that KIN-19, unlike mammalian CKIα, regulates cell fate independently of GSK3β (Banerjee et al., 2010; Gleason and Eisenmann, 2010). However, the identity of the KIN-19 targets during WβA signaling is unclear because there are two β-catenins, SYS-1 and WRM-1, that cooperate to properly specify seam cell fate – the kin-19(RNAi) seam cell hyperplasia phenotype is dependent on the function of both β-catenins. Therefore, the mechanism by which KIN-19 negatively regulates seam cell fate and how KIN-19 relates to APR-1 function is unknown.
Axin is another major scaffold for β-catenin destruction complex proteins, such as GSK3β and CKIα (Clevers and Nusse, 2012). The C. elegans Axin homolog, PRY-1, negatively regulates seam cell fate and canonical Wnt signaling in C. elegans (Korswagen et al., 2002; Gleason and Eisenmann, 2010). PRY-1 localizes to the anterior cortex of dividing seam cells, indicating that its function in the seam cells might be involved in the WβA pathway (Mizumoto and Sawa, 2007). Unlike apr-1 loss of function, pry-1 mutants display normal nuclear localization of WRM-1 in the nuclei of early larval seam cell daughters, although the establishment of cortical WRM-1 asymmetry prior to division can be delayed (Mizumoto and Sawa, 2007). PRY-1 physically interacts with APR-1, and pry-1 mutants display defects in the cortical asymmetry of APR-1, resulting in a symmetric distribution of APR-1 across the cortex of the mother cell (Korswagen et al., 2002; Mizumoto and Sawa, 2007). However, because WRM-1 nuclear asymmetry defects are not seen in pry-1 mutants in the V5.p seam cell, the significance of PRY-1 regulation of APR-1 cortical asymmetry and the mechanism underlying PRY-1-mediated negative regulation of seam cell fate are unclear (Mizumoto and Sawa, 2007; Gleason and Eisenmann, 2010).
Here, we examine the role of APC and other members of the β-catenin destruction complex in SYS-1 regulation during asymmetric division of C. elegans seam cells. We show that KIN-19 regulates seam cell fate by negative regulation of SYS-1 levels in the anterior seam cell daughter. Additionally, KIN-19 acts as a positive regulator of posterior WRM-1 localization upstream of APR-1 localization. We find that loss of APR-1 function causes a similar defect in SYS-1 negative regulation to loss of KIN-19 function. Interestingly, PRY-1/Axin is not required for the negative regulation of SYS-1 or WRM-1 levels as would be suggested from previous Wnt–β-catenin studies. Rather, we demonstrate a novel Axin function in mother cell polarity, because pry-1 mutants show randomized SYS-1 and WRM-1 localization in seam cell daughters, likely due to mislocalized APR-1. These results establish that APR-1 has two major functions in the seam cells: (1) negatively regulating SYS-1 levels in the anterior daughter in conjunction with KIN-19 and (2) promoting WRM-1 nuclear export in the anterior daughter (Sugioka et al., 2011). kin-19(RNAi) divisions show APR-1 mislocalization to the posterior daughter, which results in inappropriate WRM-1 nuclear export but not SYS-1 depletion. Therefore, we propose a model whereby the interaction of APR-1/APC and PRY-1/Axin with KIN-19/CKIα distinguishes between microtubule (and thus WRM-1) and SYS-1 regulatory roles. Taken together, these data reveal multiple functions of APC that are dictated by Wnt signaling during in vivo asymmetric stem cell division.
RESULTS
SYS-1/β-catenin localization is dynamic during seam cell division
In order to investigate how the components of the WβA pathway direct ACD in worms, we established a system to examine ACD in vivo by visualizing the distribution of SYS-1, the β-catenin that controls TCF target gene activation, in seam cells. We chose to test this hypothesis in seam cells because many WβA components have been genetically determined to positively and negatively regulate seam cell fate, and the subcellular localization of several pathway members has already been examined. However, as the localization pattern of SYS-1/β-catenin had not yet been determined for seam cell divisions, we first characterized SYS-1 localization during the seam cell cycle. The seam cells divide asymmetrically several times during C. elegans development; however, the final asymmetric division during late larval development is the most strongly affected by RNAi for Wnt components and was thus the focus of our studies (Banerjee et al., 2010; Gleason and Eisenmann, 2010). In order to analyze the localization of SYS-1 in the seam cells, we examined the expression pattern of a rescuing Psys-1::YFP::SYS-1 transgene (Fig. 2A–D; Phillips et al., 2007). Between divisions, SYS-1 localized weakly to the cortex and cytoplasm. Prior to metaphase, SYS-1 localized to the nucleus and cytoplasm (Fig. 2A). During metaphase and anaphase, SYS-1 localized to bright puncta that behaved similarly to centrosomes (Fig. 2B). Centrosomal localization was confirmed by the colocalization of mCherry::SYS-1 with GFP-tagged β-tubulin (supplementary material Fig. S2), consistent with SYS-1 centrosomal localization observed in other tissues (Phillips et al., 2007; Huang et al., 2007). Asymmetric localization of SYS-1 was established during early telophase, during which SYS-1 levels were rapidly lowered in the anterior daughter while SYS-1 levels increased in the nuclear region of the posterior daughter (Fig. 2C). SYS-1 asymmetry was maintained after cytokinesis into late telophase when it localized primarily in the nucleus of the posterior daughter (Fig. 2D). Within 1 hour after division, nuclear levels of SYS-1 decreased and localization at the cortex and cytoplasm increased, but SYS-1 eventually became undetectable by 2 hours after division (Fig. 2E). Mutation and/or knockdown of Wnt pathway components has been found to alter fate in all asymmetrically dividing seam cell lineages, further demonstrating that the WβA pathway functions in each seam cell lineage (Banerjee et al., 2010; Gleason and Eisenmann, 2010). Consistent with this, SYS-1 localization followed the pattern reported here during all observed seam cell divisions, thus establishing a baseline system to study the function of the WβA pathway during ACD. We therefore treated the V seam cells in their final (L4) division as a single unit with regards to SYS-1 function and regulation.
Fig. 2.

KIN-19 and APR-1 are required for asymmetric localization of SYS-1 during seam cell division. (A–E) YFP::SYS-1 localization in wild-type/empty feeding vector (EV) seam cells. (F–J) YFP::SYS-1 localization in kin-19(RNAi) cells. (K–O) YFP::SYS-1 localization in apr-1(RNAi) cells. In A–O, the upper images display YFP::SYS-1 and the lower images display the corresponding DIC images. Blue dotted lines, cell boundaries; white dashed circles, nuclear boundaries. Scale bars: 5 µm. (A,F,K) Prior to division, SYS-1 localizes to the cortex and nucleus. (B,G,L) During metaphase, SYS-1 localizes to centrosomes in all cells and to the cortex in kin-19(RNAi) cells (arrowheads). (C,H,M) Early telophase. In empty-vector controls, SYS-1 localizes asymmetrically to the nuclear regions of the dividing cell. In kin-19(RNAi) and apr-1(RNAi) cells, SYS-1 localizes symmetrically to the nuclear regions. Arrowheads, cortical SYS-1 localization. (D,I,N) Late telophase. SYS-1 levels in empty-vector control nuclei remain asymmetric. SYS-1 levels in the nuclei of kin-19(RNAi) cells remain symmetric. In apr-1(RNAi) cells, nuclear levels of SYS-1 become asymmetric, although nuclear levels of SYS-1 in anterior daughters remain higher than those of empty-vector controls. (E,J,O) 1 hour post-division. In controls, the anterior daughter has fused to hyp7. In kin-19(RNAi) and apr-1(RNAi) cells, both daughters have adopted the seam fate. (P,Q) The average nuclear (P) and whole-cell (Q) YFP::SYS-1 fluorescence intensity after telophase. White bars, anterior daughters; gray bars, posterior daughters. The n values are as follows: EV, n = 41; kin-19(RNAi), n = 28; apr-1(RNAi), n = 32; apr-1+kin-19(RNAi), n = 22. Data show the mean±s.e.m.; *P<0.05; ***P<0.001; n.s.P>0.05. Asterisks inside columns are a comparison to anterior empty-vector control.
KIN-19/CKIα and APR-1/APC negatively regulate SYS-1 in the anterior daughter cell
Because KIN-19 and APR-1 negatively regulate seam cell fate and because the identity of SYS-1 negative regulators is a major unanswered question in this pathway, we examined SYS-1 localization after KIN-19 and APR-1 RNAi. Consistent with the published cell fate analyses (Banerjee et al., 2010; Gleason and Eisenmann, 2010), we found that APR-1 and KIN-19 knockdown resulted in symmetric divisions that produced two elongated seam cells, rather than the wild-type pattern of one elongated seam cell and one spherical cell that fuses to the hypodermal syncytium (Fig. 2E,J,O). We observed that SYS-1 levels were higher in the anterior daughters of these symmetric seam cell divisions after kin-19 and apr-1 RNAi treatment (Fig. 2I,N). Both kin-19(RNAi) and apr-1(RNAi) seam cells followed the wild-type localization pattern until metaphase, at which point we observed a noticeable increase in cortical SYS-1 in kin-19(RNAi) (Fig. 2G, arrowheads). At telophase in kin-19(RNAi) and apr-1(RNAi) cells, SYS-1 was inappropriately maintained in the nuclear region of both the anterior and posterior daughters (Fig. 2H,M). Additionally, unlike wild-type seam cells, kin-19(RNAi) and apr-1(RNAi) seam cells maintained cortical SYS-1 during division (Fig. 2H,M, arrowheads). Because SYS-1 functions in the nucleus, we quantified SYS-1 fluorescence intensity in anterior and posterior nuclei in wild-type, kin-19(RNAi) and apr-1(RNAi) worms during late telophase (analogous to Fig. 2D,I,N). YFP::SYS-1 levels were significantly higher in the nuclei of anterior seam cell daughters in kin-19(RNAi) and apr-1(RNAi) versus control larvae fed empty RNAi vector (EV; Fig. 2P). To account for the observed increases in cortical SYS-1 localization, we also quantified SYS-1 localization over the entire cells and, again, we observed significant increases in SYS-1 levels in both kin-19(RNAi) and apr-1(RNAi) cells (Fig. 2Q). Interestingly, after apr-1(RNAi), we saw persistent elevation of SYS-1 throughout the entire cell, even though the nuclear elevation was transient (Fig. 2N), which suggests that APR-1 has a heretofore unknown function in SYS-1 nuclear retention or distribution. Taken together, these data indicate that APR-1 and KIN-19 are major negative regulators of SYS-1 in the anterior seam cell daughters.
If APR-1 acts as a scaffold for the formation of a complex of SYS-1 and KIN-19, then loss of either APR-1 or KIN-19 would be expected to result in identical phenotypes. Alternatively, APR-1 and KIN-19 might represent two distinct mechanisms of SYS-1 negative regulation. To distinguish between these possibilities, we performed a double knockdown of APR-1 and KIN-19 [apr-1+kin-19(double RNAi)]. We found that SYS-1 localization in apr-1+kin-19(double RNAi) seam cells was similar to that of apr-1(RNAi) seam cells (supplementary material Fig. S3A–D), and quantification showed that SYS-1 fluorescence in apr-1+kin-19(double RNAi) seam cells and nuclei was not significantly higher than in apr-1(RNAi) or kin-19(RNAi) cells or nuclei (P>0.05, Fig. 2P,Q). Although these animals are likely not null for KIN-19 or APR-1 function, the lack of enhancement in the double knockdown organisms compared with either single knockdown suggests that APR-1 and KIN-19 are both essential negative regulators of SYS-1 that act in a single pathway in the anterior seam cell daughter; however, the possibility remains that they regulate SYS-1 levels by separate mechanisms.
PRY-1/Axin loss randomizes SYS-1 localization
Similar to scaffolding by APC, Axin acts as a scaffold in the conserved β-catenin destruction complex alongside CKIα. Because the C. elegans homologs of APC and CKIα regulate SYS-1/β-catenin levels (above), and because PRY-1/Axin also acts as a negative regulator of seam cell fate (Gleason and Eisenmann, 2010), we investigated PRY-1 as a potential regulator of SYS-1 localization that cooperates with APC during seam cell division. Defects in SYS-1 localization in the seam cells of worms carrying the mutant pry-1 allele mu38 [pry-1(mu38)] become apparent during telophase (Fig. 3A; supplementary material Fig. S3E–H). Average YFP::SYS-1 fluorescence was significantly lower in the nuclei of both anterior and posterior daughter cells in pry-1(mu38) versus wild-type worms (Fig. 3C). However, we observed extensive variability in the degree and polarity of SYS-1 asymmetry between the two pry-1(mu38) daughters, so we calculated the difference in fluorescence between posterior and anterior daughter nuclei to estimate the ‘polarity’ of SYS-1 localization. We found that nuclear SYS-1 levels in pry-1(mu38) daughter pairs ranged from a complete reversal of wild-type asymmetry, to symmetry, to an essentially wild-type pattern of localization (Fig. 3A,D). Average SYS-1 levels in pry-1 mutants were decreased because the level of SYS-1 in the posterior nucleus was occasionally about as high as the average in wild-type, but was never higher and was typically lower. SYS-1 levels in reversed or symmetrical divisions were never as high as those in wild-type posterior daughters. From these data, we conclude that PRY-1 regulates the polarity of SYS-1 localization, but that PRY-1 is not essential for the reduction of SYS-1 levels in the anterior daughter, indicating that PRY-1 is important for establishing the site of SYS-1 negative regulation but not for regulating SYS-1 protein levels directly.
Fig. 3.

PRY-1 controls the polarity of SYS-1 asymmetry. (A) Representative images of YFP::SYS-1 localization in pry-1(mu38) seam cell daughters during late telophase. (B) YFP::SYS-1 localization in pry-1(mu38);apr-1(RNAi) cells. Blue dotted lines, cell boundaries; white dashed circles, nuclear boundaries. Scale bars: 5 µm. (C) Average nuclear YFP::SYS-1 fluorescence intensity after telophase. White bars, anterior daughters; gray bars, posterior daughters. Data show the mean±s.e.m.; *P<0.05; **P<0.01; ***P<0.001; n.s.P>0.05. Asterisks inside columns are a comparison to anterior empty-vector control (EV). Data for the empty-vector control are the same as those shown in Fig. 2, as all experiments were conducted as part of the same cohort. (D) Graph showing the posterior-anterior (P-A) difference in YFP::SYS-1 nuclear fluorescence. The x-axis indicates the difference in posterior and anterior YFP::SYS-1 fluorescence intensity in individual pairs of seam cell daughters, the y-axis indicates the number of daughter pairs with a difference in SYS-1 intensity in a given range. The n values are as follows: EV, n = 41; pry-1(mu38), n = 20; apr-1(RNAi)+pry-1(mu38), n = 32.
PRY-1 controls the asymmetric cortical localization of APR-1 during an earlier seam cell division (V5.p; Mizumoto and Sawa, 2007), and APR-1 is required for the negative regulation of SYS-1 in later seam cell ACDs (Fig. 2). We therefore tested the hypothesis that the negative regulation of SYS-1 levels in seam cells remains functional in pry-1 mutants by examining whether the SYS-1 polarity defects observed in pry-1(mu38) daughter cells are dependent on APR-1. SYS-1 levels in both daughter nuclei in pry-1(mu38);apr-1(RNAi) worms are significantly increased, thus making the divisions more symmetrical compared with those of wild-type or pry-1(mu38) organisms, although SYS-1 levels in the posterior daughter are still significantly reduced in pry-1(mu38);apr-1(RNAi) compared with wild-type worms (Fig. 3B–D). Above, we have described a ‘nuclear retention’ phenotype that is defective in apr-1(RNAi) seam cells, where SYS-1 levels were elevated in the anterior daughter but distributed more to the cytoplasm and cortex than the nucleus (Fig. 2N). The cortical pattern of SYS-1 localization in pry-1(mu38);apr-1(RNAi) cells resembles that of apr-1(RNAi) cells, with the addition of a polarity defect whereby the daughter with higher cortical levels of SYS-1 can be anterior or posterior (Fig. 3B). Because APR-1-dependent SYS-1 negative regulation still occurs in pry-1 mutants, these data indicate that PRY-1 polarizes the site of SYS-1 negative regulation, likely through APR-1, and that PRY-1 also polarizes the nuclear retention mechanism that becomes apparent after apr-1(RNAi).
KIN-19, APR-1 and PRY-1 differentially regulate WRM-1 localization
Another potential cause of ACD defects is through the regulation of the WRM-1 branch of the WβA pathway and, indeed, the kin-19 and apr-1 seam cell hyperplasia phenotypes are dependent upon WRM-1 function (Banerjee et al., 2010; Gleason and Eisenmann, 2010). This could be due to either KIN-19-mediated control of WRM-1 localization, or the fact that the loss of WRM-1 decreases the SYS-1∶POP-1 ratio to such an extent that an increase in SYS-1 levels after KIN-19 loss remains insufficient to activate target gene expression. To determine whether KIN-19 and APR-1 regulate the WRM-1–POP-1 branch of the WβA pathway, we observed WRM-1 localization in the nuclei of V seam cells after terminal division in kin-19(RNAi) and apr-1(RNAi) larvae. Levels of WRM-1::YFP fluorescence were significantly lower in the posterior (signaled) daughter nuclei of kin-19(RNAi) seam cells compared with those of controls (Fig. 4A,B). Additionally, a visible increase in the level of cortical WRM-1 was observed in the posterior daughter of kin-19(RNAi) divisions, as would be expected from the lower nuclear levels of WRM-1 (Fig. 4B, arrowheads). Conversely, we found that WRM-1 levels were significantly higher in the anterior daughters of apr-1(RNAi) seam cells compared with those of controls, similar to the reported phenotype in the T seam cell division (Fig. 4A,C; Mizumoto and Sawa, 2007). Although KIN-19 and APR-1 have similar roles in SYS-1 regulation, it appears that their roles in regulating nuclear WRM-1 levels are distinct.
Fig. 4.

KIN-19 positively regulates posterior WRM-1 nuclear localization. (A) WRM-1::YFP localization during late telophase in (A) empty-vector control cells (EV), (B) kin-19(RNAi) and (C) apr-1(RNAi) seam cell daughters. Blue dotted lines, cell boundaries; white dashed circles, nuclear boundaries; arrowheads, posterior cortical WRM-1 puncta. (D) Average nuclear WRM-1::YFP fluorescence intensity after telophase. White bars, anterior daughters; gray bars, posterior daughters. The n values are as follows: EV, n = 43; kin-19(RNAi), n = 33; apr-19(RNAi), n = 17. Data show the mean±s.e.m.; *P<0.05; n.s.P>0.05. Asterisks inside columns are a comparison to anterior empty-vector control. (E,F) GFP::WRM-1 nuclear localization in wild-type (w.t.)/WM75 (E) and pry-1(mu38) cells (F). White bars indicate daughter pairs. Scale bars: 5 µm.
Because APR-1 and KIN-19 regulate WRM-1 (Fig. 4B–D), we also investigated whether PRY-1 has a role in the WRM-1 branch of the WβA pathway. pry-1(mu38) larvae have been previously shown to experience a delay in the establishment of GFP::WRM-1 cortical asymmetry during the earlier division of the V5.p seam cell, but this is eventually resolved, and the two daughter cells display proper asymmetric nuclear localization of WRM-1 (Mizumoto and Sawa, 2007). To determine whether this is the case in later seam cell divisions, we observed GFP::WRM-1 nuclear localization in pry-1(mu38) cells during late telophase of terminal V seam cell divisions. Pwrm-1::GFP::WRM-1 was used, as Pscm::WRM-1::YFP expression was severely reduced in pry-1(mu38) worms (data not shown), likely because of transgene promoter differences. Pwrm-1::GFP::WRM-1 is a rescuing transgene that asymmetrically localizes to daughter cell nuclei; however, its lower expression levels compared with those of Pscm::WRM-1::YFP generally make its use less desirable (Nakamura et al., 2005; Mizumoto and Sawa, 2007). In control worms, GFP::WRM-1 localized to seam cell daughter nuclei in the expected wild-type pattern in all observed divisions (n = 30) (Fig. 4E). However, in pry-1(mu38) worms, seam cell daughters displayed a reversed pattern of GFP::WRM-1 localization in 30% of observed divisions (n = 20; Fig. 4F). We conclude from this that PRY-1 controls the polarity of WRM-1 nuclear localization during terminal seam cell division, but that PRY-1 is unnecessary for WRM-1 nuclear export. Taken together with our SYS-1 data, these data indicate that PRY-1 controls the site of regulation of both SYS-1 and WRM-1, but that PRY-1 is dispensable for their respective destabilization or nuclear export.
Changes in APR-1 localization do not necessarily correspond to disruption of SYS-1 nuclear asymmetry
KIN-19 regulates WRM-1 (Fig. 4), and, in the C. elegans embryo, nuclear levels of WRM-1 are regulated by microtubules in an APR-1-dependent manner (Sugioka et al., 2011). However, it is unknown whether KIN-19 regulates WRM-1 through APR-1, so we examined APR-1 localization after kin-19 RNAi treatment. In order to identify defective ACDs and to establish baseline timepoints for APR-1 comparison, we examined the colocalization of APR-1::YFP with mCherry::SYS-1. In control worms, expression of the rescuing transgenes osIs13 (Papr-1::APR-1::YFP) and uiwIs4 (Psys-1::mCherry::SYS-1) led to significantly enriched APR-1 localization on the anterior cortex compared with the posterior cortex, and these animals also displayed the wild-type pattern of SYS-1 localization as described above (n = 23; Fig. 5; Fig. 6A,C). The timing of APR-1 and SYS-1 colocalization shows that SYS-1 asymmetry occurs after APR-1 asymmetry, consistent with APR-1-mediated regulation of SYS-1 (Fig. 5). When osIs13; uiwIs4 worms were exposed to kin-19(RNAi), a significant increase in cortical localization of APR-1::YFP was observed in both the anterior and posterior daughter cells (n = 25; Fig. 6B,F). For quantification of cortical levels of APR-1, the cortical domains of each daughter were delineated by hand based on fluorescence images, and it is likely that these domains included some cytoplasm. These values therefore likely underestimate the differences in cortical intensity. Therefore, in addition, we examined the average anterior-posterior difference in cortical APR-1 intensity, to highlight changes in asymmetry between daughters and treatments (Fig. 6G). From these data, we conclude that: (1) the lower posterior nuclear levels of WRM-1 seen in kin-19(RNAi) can be attributed to the mislocalization of APR-1 to the posterior daughter during seam cell division, and (2) APR-1 that is mislocalized to the posterior cortex in the absence of KIN-19 cannot negatively regulate SYS-1 levels.
Fig. 5.

Reciprocal asymmetry of APR-1 and SYS-1 is achieved during telophase. Timecourse of APR-1 and SYS-1 colocalization during seam cell division. (A) Prior to division. (B) Early metaphase. (C) Late metaphase. (D) Anaphase. (E) Early telophase. The timecourse indicates that APR-1 asymmetry precedes SYS-1 asymmetry and that APR-1 and SYS-1 asymmetries are best observed simultaneously during telophase. Top row, APR-1::YFP (green in merge). Middle row, SYS-1::mCherry (red in merge). Bottom row, merge. Time is indicated as the time since first exposure (+0 min) in minutes. Scale bars: 5 µm.
Fig. 6.

KIN-19 controls APR-1 and SYS-1 localization, whereas WRM-1 controls only APR-1. All images show osIs13(APR-1::YFP); uIwIs4(mCherry::SYS-1). (A) Wild-type control. Cortical APR-1::YFP (left, arrowheads; green in merge) and nuclear SYS-1::mCherry (center, white dashed circles; red in merge) display proper asymmetric localizations (merge on right). (B) kin-19(RNAi). (C) Wild-type seam cell held at 26.5°C for 3 hours prior to division. (D) wrm-1(ne1982) at the permissive temperature of 20°C. (E) wrm-1(ne1982) at the restrictive temperature of 26.5°C for 3 hours prior to division. Reduced cortical levels of APR-1::YFP were observed but no changes in SYS-1 nuclear asymmetry were observed. Arrowheads, cortical APR-1 localization; white dashed circles, nuclei. Scale bars: 5 µm. (F) Quantification of cortical APR-1::YFP fluorescence above backgrounds. White bars, anterior daughters; gray bars, posterior daughters. (G) Average anterior-posterior (A-P) difference in cortical APR-1::YFP fluorescence. ts, temperature sensitive. The n values are as follows: EV at 20°C, n = 23; kin-19(RNAi) at 20°C, n = 24; wild-type at 26.5°C, n = 25; wrm-1(ne1982) at 20°C, n = 26; wrm-1(ne1982) at 26.5°C, n = 28. Data show the mean±s.e.m.; *P<0.05; ***P<0.001.
Loss of function of wrm-1 results in lower levels of APR-1 on the anterior cortex of the seam cells during division (Mizumoto and Sawa, 2007), potentially providing an additional background against which the effects of APR-1 dysregulation during seam cell division can be investigated. To determine whether APR-1 mislocalization in this case affects SYS-1 localization, we examined the localization of APR-1::YFP and mCherry::SYS-1 in a temperature-sensitive wrm-1 mutant, ne1982ts. At the permissive temperature of 20°C, we found that cortical levels of APR-1 were significantly higher in the anterior daughter compared with the posterior daughter, while wild-type SYS-1 asymmetry was observed in all divisions (n = 26; Fig. 6D,F,G). When wrm-1(ne1982) larvae were held at 26.5°C for 3 hours prior to the final seam cell division, we found that anterior cortical levels of APR-1 were significantly reduced compared with those of anterior daughters of larvae held at the permissive temperature (P<0.05, Mann-Whitney test), whereas wild-type SYS-1 asymmetry was still observed in all divisions (n = 28; Fig. 6E–G). Wild-type worms held at 26.5°C for 3 hours prior to division displayed no defects in cortical APR-1 asymmetry (n = 25; Fig. 6C,F,G). We conclude from these data that mislocalization of APR-1 as a result of the loss of wrm-1 function does not translate into defects in SYS-1 localization.
To determine whether the SYS-1 polarity defect observed in pry-1(mu38) seam cells was correlated with APR-1 mislocalization, we examined APR-1 and SYS-1 localization in pry-1(mu38) animals. pry-1(mu38); osIs13; uiwIs4 worms displayed enriched APR-1 on the posterior cortex in 29% of observed divisions – a reversal of the wild-type pattern (n = 28; Fig. 7A,B). This abnormal pattern of APR-1::YFP localization was established during metaphase and was maintained after division was completed (supplementary material Fig. S4). In pry-1(mu38) seam cell divisions that had abnormally localized APR-1, SYS-1 was mislocalized in 88% of observed divisions (n = 8), whereas 100% of the divisions in wild-type worms displayed wild-type patterns of APR-1 and SYS-1 localization (n = 23) (P<0.0001, Fisher's exact test). Thus, in contrast to the loss of WRM-1 and KIN-19, defects in APR-1 localization in pry-1 mutants correspond to defects in SYS-1 levels, and we conclude that the randomization of SYS-1 asymmetry seen in pry-1 mutants is due to defects in APR-1 localization. Additionally, as we observed an instance in which cortical APR-1 was essentially symmetrical between both daughters in addition to examples of reversals of polarity (Fig. 7B), the lower average levels of SYS-1 in pry-1(mu38) nuclei (Fig. 3C) might be related to more variable average cortical levels of APR-1 in both daughter cells.
Fig. 7.
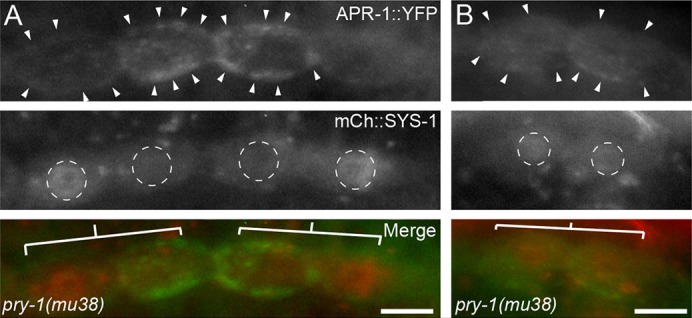
Defective APR-1 localization in pry-1(mu38) correlates with defective SYS-1 localization. (A) Representative images of APR-1::YFP (green in merge) and SYS-1::mCherry (red in merge) localization in pry-1(mu38) seam cell daughters during late telophase. The leftmost pair of cells represents a reversal of both APR-1 and SYS-1 asymmetry. The rightmost pair represents a wild-type localization pattern for APR-1 and SYS-1. (B) Seam cell division in pry-1(mu38) in which cortical APR-1 and nuclear SYS-1 are symmetric between daughters. Arrowheads, enriched cortical APR-1; white dashed circles, nuclei; white bars, daughter pairs. Scale bars: 5 µm.
DISCUSSION
Here, we have identified an additional role of APR-1/APC during the asymmetric division of C. elegans seam cells, beyond its role in regulating WRM-1/β-catenin nuclear export; in a conserved fashion across metazoans, APR-1 controls the localization of SYS-1/β-catenin during asymmetric divisions of the epithelial stem cells of C. elegans. We have shown that the CKIα homolog KIN-19 cooperates with APR-1/APC to negatively regulate seam cell fate through SYS-1, but that KIN-19 also positively regulates WRM-1 levels, likely through regulation of APR-1/APC. These two functions of APR-1/APC operate in the same asymmetrically dividing cell and are differentially regulated by KIN-19. We show that PRY-1/Axin is dispensable for the negative regulation of the nuclear levels of SYS-1 and WRM-1, but that PRY-1 is necessary for the proper localization of the complexes regulating SYS-1 and those regulating WRM-1 during ACD. Taken together, these results (summarized in Fig. 8A) provide the major framework for understanding APR-1/APC-mediated regulation of the nuclear localization of both SYS-1 and WRM-1 in WβA-dependent cell divisions. In addition, these results provide new perspectives on the functions of conserved members of the β-catenin destruction complex and how the function of these proteins might be regulated during ACD.
Fig. 8.

Proposed model for SYS-1 destruction during seam cell division. (A) Table summarizing main findings. Columns indicate the loss-of-function treatment, rows indicate the protein localization affected. (B) Model for β-catenin regulation in the seam cells of a wild-type worm. Two distinct pools of APR-1 are present at the anterior cortex, a microtubule (MT)-associated WRM-1-regulating pool (shown in green) and a KIN-19- and PRY-1-associated SYS-1-regulating pool (shown in red). KIN-19 inhibits posterior cortical localization of APR-1, thereby removing it from the posterior daughter (dashed outlines). β, SYS-1/β-catenin (red); W, WRM-1/β-catenin (green); Ax, SYS-1-regulating PRY-1/Axin (red); Ax, WRM-1-regulating PRY-1/Axin (green); Ap, SYS-1-regulating APR-1/APC (red); Ap, WRM-1-regulating APR-1/APC (green); K, KIN-19/CKIα (red); Fz, Frizzled (white); D, Disheveled (white).
It remains unclear how APC localization is regulated during Wnt signaling in general and during Wnt-induced ACD specifically. Instead of a complex that is co-regulated by upstream Wnt pathway factors, the novel pry-1 mutant phenotype demonstrates that PRY-1/Axin controls the localization of at least one other destruction complex member, APR-1/APC. Although Axin is generally thought of as necessary for β-catenin degradation, PRY-1 is not required for reductions in SYS-1 levels. Instead, PRY-1 regulates the polarity of SYS-1 asymmetry during terminal seam cell division, through its control of APR-1. It is still unclear how Wnt signaling polarizes PRY-1 itself, although repressive interactions with asymmetrically localized Dishevelled proteins (Mizumoto and Sawa, 2007) seem a likely candidate. The observation that SYS-1 is, on average, positively regulated by PRY-1 suggests that APR-1 localization or activity is expanded in pry-1 mutants. Previous work has shown that cortical APR-1 is symmetric in pry-1(mu38) cells prior to division of the V5.p seam cell, and that PRY-1 does not regulate the nuclear localization of WRM-1 during this same division (Mizumoto and Sawa, 2007). However, our data show that, in later divisions (equivalent to Vn.pppp), loss of pry-1 function results in reversals of APR-1 cortical asymmetry, in addition to a reduced frequency of cortical symmetry, and that PRY-1 regulates the polarity of both SYS-1 and WRM-1 nuclear localization. This difference could be explained by differences in the lineages. The V5.p division occurs at the same time as the other Vn.p divisions; however, the anterior daughter of V5.p takes on a unique lineage that results in the birth of the PDE and PVD neurons, whereas the other Vn.pa cells continue the stereotyped seam cell lineage. Interestingly, although the Vn.p divisions (with the exception of V5.p) result in two seam cell daughters, it has been reported that APR-1, POP-1 and WRM-1 are all asymmetrically localized during this apparently cell-fate-symmetric division (Wildwater et al., 2011; Hughes et al., 2013). How the function and regulation of WβA signaling is different between the Vn.p division and the later seam cell divisions represents an interesting area of further study to determine how the function of Wnt signaling might change within a single stem cell lineage over time. Because pry-1(mu38) is a nonsense mutation that is present in each seam cell nucleus through each of their several divisions, it is also of interest that some mu38 seam cells still divide with a wild-type polarity of Wnt signaling components, consistent with our conclusion that PRY-1 loss randomizes the polarity of the mother cell.
The finding that KIN-19 regulates both SYS-1 and WRM-1 asymmetry was unexpected. Our KIN-19 phenotypic analysis shows that KIN-19 has a positive role in WRM-1 nuclear localization and a negative role in regulating SYS-1 levels, in addition to its previously known negative role in fate determination of seam cells (Banerjee et al., 2010; Gleason and Eisenmann, 2010). The observation that KIN-19 regulates SYS-1, WRM-1 and APR-1 begs the question of whether any of these proteins are directly phosphorylated by KIN-19. We identified several CKI consensus phosphorylation sites in SYS-1, WRM-1 and APR-1 but, given the relative commonality of this sequence, more work is required to determine the significance of these sites. Our discovery that KIN-19 regulates cortical APR-1 polarity provides a starting point for future research questions, such as whether the APR-1 that is localized by KIN-19 is functionally distinct from the APR-1 that is localized by WRM-1. How does Wnt signaling distinguish between the functions of APR-1 in such a situation? pry-1(mu38) seam cells display markedly different SYS-1 and WRM-1 localization phenotypes compared with those of kin-19(RNAi) cells, despite the fact that pry-1 and kin-19 both display APR-1 localization defects. If PRY-1 and KIN-19 both regulate cortical asymmetry of APR-1, why are the resultant phenotypes in SYS-1 and WRM-1 localization so dissimilar?
We propose a reconciling model in which KIN-19/CKIα and WRM-1/β-catenin subdivide APR-1/APC into two distinct pools: a WRM-1-regulating pool and a SYS-1-regulating pool. In the first pool, APR-1 functions in WRM-1 regulation through association with microtubules at the cortex (Sugioka et al., 2011). In a second pool, entry into which appears to be dependent on KIN-19, APR-1 reduces SYS-1 levels. This model predicts that kin-19(RNAi) results in changes in SYS-1 and WRM-1 localization because KIN-19 loss results in a single WRM-1-regulating pool of APR-1. Loss of KIN-19 also increases the level of WRM-1-regulating APR-1 at the posterior cortex, so we hypothesize that KIN-19 normally restricts this pool of APR-1 to the cytoplasm. wrm-1(ne1982) also reduces the amount of anterior cortical APR-1 but does not affect SYS-1 nuclear asymmetry, further demonstrating that the WRM-1- and SYS-1-regulating functions of APR-1 are regulated separately. Previous experiments in mammalian tissue culture have indicated that CKI-dependent phosphorylation of APC greatly increases its affinity for β-catenin (Rubinfeld et al., 2001; Ha et al., 2004), suggesting that KIN-19-dependent phosphorylation of APR-1 might act as a switch between APR-1-mediated regulation of WRM-1 (through microtubules) or SYS-1 (through direct interaction). A graphical representation of this model is shown in Fig. 8B.
One caveat to our current model is that it is still unclear exactly where APR-1 localizes when it is engaged in the negative regulation of SYS-1. wrm-1(ts) mutants show wild-type SYS-1 asymmetry and loss of much, but not all, of the APR-1 from the cortex, suggesting that SYS-1 could be regulated by cytoplasmic APR-1 that is somehow qualitatively different from the cytoplasmic APR-1 in the posterior daughter. However, we see no difference in APR-1 cytoplasmic levels in the two wild-type daughter cells that display SYS-1 asymmetry or in pry-1 mutants that have defective cortical APR-1 asymmetry and corresponding defective SYS-1 regulation. We therefore prefer our simpler model of cortical APR-1 having a potent effect on SYS-1 negative regulation. However, if cytoplasmic APR-1 does regulate SYS-1, this further reinforces our overall conclusion that WRM-1 and SYS-1 are regulated by different pools of APR-1; if APR-1 is removed from the cortex and can still regulate SYS-1 levels in the anterior daughter cell, then the localization and function of SYS-1-regulating APR-1 must be distinct from those of WRM-1-regulating APR-1 that localizes to the cortex.
The ability of APR-1 to promote nuclear retention of SYS-1 in the anterior daughter cell is intriguing, given the negative nuclear role for mammalian APC in the expression of Wnt target genes (Neufeld, 2009). Vertebrate APC competes with TCF for binding to nuclear β-catenin as a mechanism to restrict the aberrant activation of Wnt target genes (Neufeld et al., 2000), suggesting that the apparent nuclear anchoring of SYS-1 by APR-1 might be an effect of competition for SYS-1 binding between nuclear APR-1 and POP-1. Alternatively, APR-1 might inhibit the interaction of SYS-1 with an as-yet-unknown protein that functions to export SYS-1 from the nucleus of the anterior seam cell daughter. Our results from the pry-1(mu38);apr-1(RNAi) double loss-of-function experiments indicate that this mechanism is also polarized by the function of PRY-1. Future work will examine the role of nuclear APC in C. elegans and determine whether nuclear APC localization is required for nuclear β-catenin retention in mammals.
The role of SYS-1 in the transcriptional activation of TCF target genes and its regulation at the level of protein stability make it a useful model for the study of mammalian β-catenin, in part because SYS-1 lacks a potentially confounding adhesive function. Although the function of WRM-1 is not conserved in mammalian β-catenin, the fact that the localization of the former is controlled by APC means that WRM-1 localization can serve as a proxy for APC function, the regulation of which is complex in both mammals and C. elegans. A recent in vitro study has shown that asymmetric presentation of Wnt protein results in asymmetric localization of APC and β-catenin, as well as asymmetric cell fate specification in mouse embryonic stem cells (Habib et al., 2013). A similar mechanism for Wnt-induced asymmetric division and cell fate acquisition has been proposed to occur in the mammalian intestinal crypt (Quyn et al., 2010; Bellis et al., 2012), although ACD in this tissue is difficult to observe in vivo. Studying the regulation of SYS-1, WRM-1 and APR-1 in the C. elegans seam cells thus provides an in vivo opportunity for understanding the regulation of β-catenin and APC in Wnt signaling during ACD that is currently lacking in other systems. To our knowledge, this is the first in vivo evidence indicating that Wnt signaling components can distinguish between multiple functions of APC simultaneously and in the same cell. We believe that the C. elegans seam cells provide a powerful system that can be used to address the multiple functions of APC and how they are regulated in vivo.
MATERIALS AND METHODS
Strains
The C. elegans strains used in this study were: (1) BTP1 [qIs95 (Psys-1::VENUS::SYS-1) III], (2) BTP117 [pry-1(mu38) I; qIs95 III], (3) BTP118 [unc-76(e911) V; osIs13(Papr-1::APR-1::VENUS; unc-76 (+)), uiwIs4 (Psys-1::mCherry::SYS-1)], (4) BTP119 [pry-1(mu38) I; unc-76(e911); osIs13; uiwIs4], (5) BTP126 [wrm-1(ne1982) III; osIs13; uiwIs4], (6) BTP128 [uiwIs4; Pscm::GFP::β-tubulin], (7) BTP132 [pry-1(mu38) I; neIs2 (pwrm-1::GFP::WRM-1; pRF4(rol-6)) IV], (8) BTP142 [wrm-1(ne1982) III; wIs51(Pscm::GFP)], (9) HS1325 [unc-76(e911) V; osEx229(Ppry-1::pry-1::GFP+unc-76(+))], (10) HS1417 [osIs5 (Pscm::WRM-1::VENUS) II], (11) JR667 [unc-119(e2498::Tc1) III; wIs51(Pscm::GFP) V] and (12) WM75 [wrm-1(tm514) III; neIs2 IV].
Larvae analysis
RNAi was performed using standard techniques using the pL4440 feeding vector (Timmons et al., 2001). apr-1+kin-19(RNAi) was performed by inserting sequences homologous to both apr-1 and kin-19 RNA into a single RNAi vector, as described previously (Min et al., 2010). Synchronized L1 worms were grown at 20°C until late L3, when the terminal seam cell division occurs. L3 worms were then immobilized using 10 µM muscimol on 6% agarose pads for live imaging. Cells and their corresponding nuclei were identified by differential interference contrast (DIC) microscopy prior to fluorescence image capture (see Fig. 2). The n values refer to the number of daughter pairs imaged and compared.
RNAi efficacy was quantified by semi-quantitative RT-PCR, with mRNA decreases ranging from 73–80%. RNAi was further validated by feeding RNAi to worms expressing the seam cell marker Pscm::GFP. We observed increases in seam cell number similar to those reported previously for RNAi-mediated knockdown of kin-19 and apr-1 (Banerjee et al., 2010, Gleason and Eisenmann, 2010). RNAi-mediated knockdown of apr-1 was additionally validated by performing knockdown in worms expressing APR-1::YFP. The expression of APR-1::YFP was visibly eliminated in these larvae.
In the wrm-1(ne1982) temperature-shift experiment, temperature-shifted worms were held at 20°C until 3 hours prior to division, when they were shifted to the restrictive temperature of 26.5°C (Gleason and Eisenmann, 2010). The average number of seam cells in wrm-1(ne1982) mutants was 15.7±0.12 (n = 20, ±s.e.m.) at 20°C and was significantly reduced to 12.2±0.57 (n = 20) in worms that were shifted to 26.5°C 3 hours before their final division, indicating that our temperature-shift regimen was sufficient to affect cell fate (P<0.0001, Mann-Whitney test).
Fluorescence quantification
Images were obtained using a Zeiss Axioplan compound fluorescent microscope and Zeiss Axiovision software. Raw YFP image data was exported into TIFF format and analyzed using ImageJ software. Nuclei, bodies and cortices of seam cells were identified in DIC images and then the mean protein fluorescence in fluorescence images was quantified using ImageJ. Background fluorescence was normalized for each experiment by using the same channels and exposures to image seam cells in N2 wild-type worms (n = 14 for each exposure setting). All statistical comparisons were performed with either Fisher's Exact test or the Mann-Whitney test and VassarStats software (Lowry, 2010).
Acknowledgments
We thank the following colleagues from the University of Iowa, IA: Lori Adams, Diane Slusarski, Doug Houston and Sarit Smolikove for helpful comments. We also thank Hitoshi Sawa (National Institute of Genetics, Mishima, Shizuoka, Japan) and Sander van den Heuvel (Utrecht University, Utrecht, The Netherlands) for the osIs13 transgene and Mike Herman (Kansas State University, Manhattan, KS) for Pscm::GFP::β-tubulin. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health Office of Research Infrastructure Programs [grant number P40 OD010440].
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
A.T.B. designed experiments, performed experiments and prepared the manuscript. B.T.P. designed experiments and prepared the manuscript.
Funding
This work was supported by research grants from the March of Dimes Foundation [grant number 5-FY11-109]; and from the American Cancer Society [grant number RSG-11-140-01-DDC].
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.146514/-/DC1
References
- Banerjee D., Chen X., Lin S. Y., Slack F. J. (2010). kin-19/casein kinase Iα has dual functions in regulating asymmetric division and terminal differentiation in C. elegans epidermal stem cells. Cell Cycle 9, 4748–4765. 10.4161/cc.9.23.14092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth A. I. M., Caro-Gonzalez H. Y., Nelson W. J. (2008). Role of adenomatous polyposis coli (APC) and microtubules in directional cell migration and neuronal polarization. Semin. Cell Dev. Biol. 19, 245–251. 10.1016/j.semcdb.2008.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellis J., Duluc I., Romagnolo B., Perret C., Faux M. C., Dujardin D., Formstone C., Lightowler S., Ramsay R. G., Freund J-N. et al. (2012). The tumor suppressor Apc controls planar cell polarities central to gut homeostasis. J. Cell Biol. 198, 331–341. 10.1083/jcb.201204086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocardo M., Henderson B. R. (2008). APC shuttling to the membrane, nucleus and beyond. Trends Cell Biol. 18, 587–596. 10.1016/j.tcb.2008.09.002 [DOI] [PubMed] [Google Scholar]
- Cicalese A., Bonizzi G., Pasi C. E., Faretta M., Ronzoni S., Giulini B., Brisken C., Minucci S., Di Fiore P. P., Pelicci P. G. (2009). The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 138, 1083–1095. 10.1016/j.cell.2009.06.048 [DOI] [PubMed] [Google Scholar]
- Clevers H., Nusse R. (2012). Wnt/β-catenin signaling and disease. Cell 149, 1192–1205. 10.1016/j.cell.2012.05.012 [DOI] [PubMed] [Google Scholar]
- Gleason J. E., Eisenmann D. M. (2010). Wnt signaling controls the stem cell-like asymmetric division of the epithelial seam cells during C. elegans larval development. Dev. Biol. 348, 58–66. 10.1016/j.ydbio.2010.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrepati L., Thompson K. W., Eisenmann D. M. (2013). C. elegans GATA factors EGL-18 and ELT-6 function downstream of Wnt signaling to maintain the progenitor fate during larval asymmetric divisions of the seam cells. Development 140, 2093–2102. 10.1242/dev.091124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R. A., Kaplan K. B. (2003). Chromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by a dominant mutation in APC. J. Cell Biol. 163, 949–961. 10.1083/jcb.200307070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha N-C., Tonozuka T., Stamos J. L., Choi H-J., Weis W. I. (2004). Mechanism of phosphorylation-dependent binding of APC to beta-catenin and its role in beta-catenin degradation. Mol. Cell 15, 511–521. 10.1016/j.molcel.2004.08.010 [DOI] [PubMed] [Google Scholar]
- Habib S. J., Chen B-C., Tsai F-C., Anastassiadis K., Meyer T., Betzig E., Nusse R. (2013). A localized Wnt signal orients asymmetric stem cell division in vitro. Science 339, 1445–1448. 10.1126/science.1231077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harterink M., Kim D. H., Middelkoop T. C., Doan T. D., van Oudenaarden A., Korswagen H. C. (2011). Neuroblast migration along the anteroposterior axis of C. elegans is controlled by opposing gradients of Wnts and a secreted Frizzled-related protein. Development 138, 2915–2924. 10.1242/dev.064733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvitz H. R., Herskowitz I. (1992). Mechanisms of asymmetric cell division: two Bs or not two Bs, that is the question. Cell 68, 237–255. 10.1016/0092--8674(92)90468--R [DOI] [PubMed] [Google Scholar]
- Huang S., Shetty P., Robertson S. M., Lin R. (2007). Binary cell fate specification during C. elegans embryogenesis driven by reiterated reciprocal asymmetry of TCF POP-1 and its coactivator beta-catenin SYS-1. Development 134, 2685–2695. 10.1242/dev.008268 [DOI] [PubMed] [Google Scholar]
- Huang X., Tian E., Xu Y., Zhang H. (2009). The C. elegans engrailed homolog ceh-16 regulates the self-renewal expansion division of stem cell-like seam cells. Dev. Biol. 333, 337–347. 10.1016/j.ydbio.2009.07.005 [DOI] [PubMed] [Google Scholar]
- Hughes S., Brabin C., Appleford P. J., Woollard A. (2013). CEH-20/Pbx and UNC-62/Meis function upstream of rnt-1/Runx to regulate asymmetric divisions of the C. elegans stem-like seam cells. Biology Open 6, 20134549 10.1242/bio.20134549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson B. M., Eisenmann D. M. (2012). β-catenin-dependent Wnt signaling in C. elegans: teaching an old dog a new trick. Cold Spring Harb. Perspect. Biol. 4, a007948 10.1101/cshperspect.a007948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori T., Inoue T., Sakamoto T., Gengyo-Ando K., Tsujimoto M., Mitani S., Sawa H., Aoki J., Arai H. (2008). Beta-catenin asymmetry is regulated by PLA1 and retrograde traffic in C. elegans stem cell divisions. EMBO J. 27, 1647–1657. 10.1038/emboj.2008.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khramtsov A. I., Khramtsova G. F., Tretiakova M., Huo D., Olopade O. I., Goss K. H. (2010). Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am. J. Pathol. 176, 2911–2920. 10.2353/ajpath.2010.091125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd A. R., III, Miskowski J. A., Siegfried K. R., Sawa H., Kimble J. (2005). A beta-catenin identified by functional rather than sequence criteria and its role in Wnt/MAPK signaling. Cell 121, 761–772. 10.1016/j.cell.2005.03.029 [DOI] [PubMed] [Google Scholar]
- Knoblich J. A. (2008). Mechanisms of asymmetric stem cell division. Cell 132, 583–597. 10.1016/j.cell.2008.02.007 [DOI] [PubMed] [Google Scholar]
- Korswagen H. C., Herman M. A., Clevers H. C. (2000). Distinct beta-catenins mediate adhesion and signalling functions in C. elegans. Nature 406, 527–532. 10.1038/35020099 [DOI] [PubMed] [Google Scholar]
- Korswagen H. C., Coudreuse D. Y. M., Betist M. C., van de Water S., Zivkovic D., Clevers H. C. (2002). The Axin-like protein PRY-1 is a negative regulator of a canonical Wnt pathway in C. elegans. Genes Dev. 16, 1291–1302. 10.1101/gad.981802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M-C., Gay F., Odom R., Shi Y., Lin R. (2004). Phosphorylation by the beta-catenin/MAPK complex promotes 14-3-3-mediated nuclear export of TCF/POP-1 in signal-responsive cells in C. elegans. Cell 117, 95–106. 10.1016/S0092--8674(04)00203--X [DOI] [PubMed] [Google Scholar]
- Lowry R. (2010). VassarStats Available at: http://vassarstats.net/index.html [Google Scholar]
- MacDonald B. T., Tamai K., He X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26. 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min K., Kang J., Lee J. (2010). A modified feeding RNAi method for simultaneous knock-down of more than one gene in Caenorhabditis elegans. Biotechniques 48, 229–232. 10.2144/000113365 [DOI] [PubMed] [Google Scholar]
- Mizumoto K., Sawa H. (2007). Cortical beta-catenin and APC regulate asymmetric nuclear beta-catenin localization during asymmetric cell division in C. elegans. Dev. Cell 12, 287–299. 10.1016/j.devcel.2007.01.004 [DOI] [PubMed] [Google Scholar]
- Morrison S. J., Kimble J. (2006). Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 441, 1068–1074. 10.1038/nature04956 [DOI] [PubMed] [Google Scholar]
- Nakamura M., Zhou X. Z., Lu K. P. (2001). Critical role for the EB1 and APC interaction in the regulation of microtubule polymerization. Curr. Biol. 11, 1062–1067. 10.1016/S0960--9822(01)00297--4 [DOI] [PubMed] [Google Scholar]
- Nakamura K., Kim S., Ishidate T., Bei Y., Pang K., Shirayama M., Trzepacz C., Brownell D. R., Mello C. C. (2005). Wnt signaling drives WRM-1/beta-catenin asymmetries in early C. elegans embryos. Genes Dev. 19, 1749–1754. 10.1101/gad.1323705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld K. L. (2009). Nuclear APC. Adv. Exp. Med. Biol. 656, 13–29. 10.1007/978--1--4419--1145--2_2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld K. L., Nix D. A., Bogerd H., Kang Y., Beckerle M. C., Cullen B. R., White R. L. (2000). Adenomatous polyposis coli protein contains two nuclear export signals and shuttles between the nucleus and cytoplasm. Proc. Natl. Acad. Sci. USA 97, 12085–12090. 10.1073/pnas.220401797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumüller R. A., Knoblich J. A. (2009). Dividing cellular asymmetry: asymmetric cell division and its implications for stem cells and cancer. Genes Dev. 23, 2675–2699. 10.1101/gad.1850809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips B. T., Kimble J. (2009). A new look at TCF and beta-catenin through the lens of a divergent C. elegans Wnt pathway. Dev. Cell 17, 27–34. 10.1016/j.devcel.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips B. T., Kidd A. R., 3rd, King R., Hardin J., Kimble J. (2007). Reciprocal asymmetry of SYS-1/beta-catenin and POP-1/TCF controls asymmetric divisions in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 104, 3231–3236. 10.1073/pnas.0611507104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell A. E., Shung C-Y., Saylor K. W., Müllendorff K. A., Weiss J. B., Wong M. H. (2010). Lessons from development: A role for asymmetric stem cell division in cancer. Stem Cell Res. 4, 3–9. 10.1016/j.scr.2009.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. A. (2006). CKI, there's more than one: casein kinase I family members in Wnt and Hedgehog signaling. Genes Dev. 20, 399–410. 10.1101/gad.1394306 [DOI] [PubMed] [Google Scholar]
- Quyn A. J., Appleton P. L., Carey F. A., Steele R. J. C., Barker N., Clevers H., Ridgway R. A., Sansom O. J., Näthke I. S. (2010). Spindle orientation bias in gut epithelial stem cell compartments is lost in precancerous tissue. Cell Stem Cell 6, 175–181. 10.1016/j.stem.2009.12.007 [DOI] [PubMed] [Google Scholar]
- Ren H., Zhang H. (2010). Wnt signaling controls temporal identities of seam cells in Caenorhabditis elegans. Dev. Biol. 345, 144–155. 10.1016/j.ydbio.2010.07.002 [DOI] [PubMed] [Google Scholar]
- Reya T., Clevers H. (2005). Wnt signalling in stem cells and cancer. Nature 434, 843–850. 10.1038/nature03319 [DOI] [PubMed] [Google Scholar]
- Rocheleau C. E., Yasuda J., Shin T. H., Lin R., Sawa H., Okano H., Priess J. R., Davis R. J., Mello C. C. (1999). WRM-1 activates the LIT-1 protein kinase to transduce anterior/posterior polarity signals in C. elegans. Cell 97, 717–726. 10.1016/S0092--8674(00)80784--9 [DOI] [PubMed] [Google Scholar]
- Rubinfeld B., Tice D. A., Polakis P. (2001). Axin-dependent phosphorylation of the adenomatous polyposis coli protein mediated by casein kinase 1ε. J. Biol. Chem. 276, 39037–39045. 10.1074/jbc.M105148200 [DOI] [PubMed] [Google Scholar]
- Sawa H. (2012). Control of cell polarity and asymmetric division in C. elegans. Curr. Top. Dev. Biol. 101, 55–76. 10.1016/B978--0--12--394592--1.00003--X [DOI] [PubMed] [Google Scholar]
- Sugioka K., Mizumoto K., Sawa H. (2011). Wnt regulates spindle asymmetry to generate asymmetric nuclear β-catenin in C. elegans. Cell 146, 942–954. 10.1016/j.cell.2011.07.043 [DOI] [PubMed] [Google Scholar]
- Timmons L., Court D. L., Fire A. (2001). Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263, 103–112. 10.1016/S0378--1119(00)00579--5 [DOI] [PubMed] [Google Scholar]
- Wildwater M., Sander N., de Vreede G., van den Heuvel S. (2011). Cell shape and Wnt signaling redundantly control the division axis of C. elegans epithelial stem cells. Development 138, 4375–4385. 10.1242/dev.066431 [DOI] [PubMed] [Google Scholar]
- Yamamoto Y., Takeshita H., Sawa H. (2011). Multiple Wnts redundantly control polarity orientation in Caenorhabditis elegans epithelial stem cells. PLoS Genet. 7, e1002308 10.1371/journal.pgen.1002308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X-D., Huang S., Lo M-C., Mizumoto K., Sawa H., Xu W., Robertson S., Lin R. (2011). Distinct and mutually inhibitory binding by two divergent {beta}-catenins coordinates TCF levels and activity in C. elegans. Development 138, 4255–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumbrunn J., Kinoshita K., Hyman A. A., Näthke I. S. (2001). Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 β phosphorylation. Curr. Biol. 11, 44–49. 10.1016/S0960--9822(01)00002--1 [DOI] [PubMed] [Google Scholar]