

ABSTRACT
Studies on the mechanism of integrin inside-out activation have been focused on the role of β-integrin cytoplasmic tails, which are relatively conserved and bear binding sites for the intracellular activators including talin and kindlin. Cytoplasmic tails for α-integrins share a conserved GFFKR motif at the membrane-proximal region and this forms a specific interface with the β-integrin membrane-proximal region to keep the integrin inactive. The α-integrin membrane-distal regions, after the GFFKR motif, are diverse both in length and sequence and their roles in integrin activation have not been well-defined. In this study, we report that the α-integrin cytoplasmic membrane-distal region contributes to maintaining integrin in the resting state and to integrin inside-out activation. Complete deletion of the α-integrin membrane-distal region diminished talin- and kindlin-mediated integrin ligand binding and conformational change. A proper length and correct amino acids in α-integrin membrane-distal region was found to be important for integrin inside-out activation. Our data establish an essential role for the α-integrin cytoplasmic membrane-distal region in integrin activation and provide new insights into how talin and kindlin induce the high-affinity integrin conformation that is required for fully functional integrins.
KEY WORDS: Integrin cytoplasmic domain, Inside-out activation, Talin, Kindlin, Platelet, Leukocyte
INTRODUCTION
Integrins are cell surface molecules composed of α and β subunits, each containing a large extracellular domain, a short transmembrane (TM) domain, and usually a short cytoplasmic tail (CT). The combination of 18 α- integrin and eight β-integrin subunits results in 24 human integrins, and these play important roles in diverse biological processes including cell migration, morphogenesis, hemostasis and the immune response (Hynes, 2002). Integrins transmit signals between extracellular ligands and the intracellular cytoskeleton through bidirectional signaling across cell membrane (Kim et al., 2011b; Springer and Dustin, 2012). Intracellular stimulations impinging on the integrin CT induce conformational changes that transform resting integrin into a high-affinity ligand-binding-competent state (known as integrin activation or inside-out signaling). Ligand binding to the extracellular domain induces integrin conformational rearrangement and transmits signals to the cytoplasmic side (known as integrin outside-in signaling). The integrin TM and CT domains play pivotal roles in the bidirectional signaling process.
The large-scale integrin conformational rearrangements, including the changing from a bent to an extended, and from a closed to an open headpiece conformation, have been directly visualized by electron microscopy (EM) (Takagi et al., 2002; Nishida et al., 2006; Ye et al., 2010; Eng et al., 2011; Zhu et al., 2012), crystallography (Xiao et al., 2004; Zhu et al., 2013), and small angle X-ray scattering (Mould et al., 2003; Eng et al., 2011), and this process in now known to involve the exposure of epitopes (known as ligand-induced-binding sites, LIBS) that are masked in the inactive state (Frelinger et al., 1991; Sims et al., 1991; Humphries, 2004; Byron et al., 2009). It has been demonstrated that both integrin extension and headpiece opening are required for high-affinity ligand binding (Chen et al., 2010; Springer and Dustin, 2012), which is crucial for platelet aggregation mediated by integrin αIIbβ3 at the bleeding sites and neutrophil arrest mediated by integrin αLβ2 at the inflamed tissues (Lefort et al., 2012).
Integrin inside-out activation is regulated through the CT. Sequence alignment shows that β-integrin CTs are relatively conserved at both the membrane-proximal (MP) and membrane-distal (MD) regions, which have common binding sites for regulatory proteins like talin and kindlin (supplementary material Fig. S1) (Anthis and Campbell, 2011; Kim et al., 2011b; Margadant et al., 2011; Morse et al., 2014). In contrast, α-integrin CTs are only highly conserved at the MP regions, which have a Gly-Phe-Phe-Lys-Arg (GFFKR) motif, whereas the MD regions vary substantially both in sequence and length (supplementary material Fig. S1). It is known that the α- integrin CT MP region is required for keeping the integrin inactive (O'Toole et al., 1991; O'Toole et al., 1994; Hughes et al., 1995; Hughes et al., 1996; Lu and Springer, 1997; Zhu et al., 2009), but the role of α-integrin CT MD region in integrin activation is not well characterized.
Mutational studies have suggested that the specific associations of α-integrin and β-integrin TM and CT domains maintain integrin in the resting state (O'Toole et al., 1994; Hughes et al., 1995; Luo et al., 2005; Partridge et al., 2005; Berger et al., 2010). This was confirmed by an analysis of the structure of the αIIbβ3 TM and CT domains after disulfide crosslinking using intact integrin on the cell surface (Zhu et al., 2009) (Fig. 1A) and by NMR using the isolated peptides either in lipid bicelles (Lau et al., 2009) (Fig. 1B) or in organic solvent (Yang et al., 2009) (Fig. 1C). The three structures share a similar interface at the TM domain, but differ at the CT (Fig. 1A–C). Although the disulfide-based and the NMR structure in lipid bicelles have a similar conformation at the CT MP regions (Fig. 1A,B), there are different interpretations of how the conserved residues, such as β3-K716, contribute to integrin activation (Zhu et al., 2009; Kim et al., 2011a; Kurtz et al., 2012). The NMR structure in organic solvent has a very different conformation at the MP region as compared with the other structures (Fig. 1C). It is not known whether such structural diversity is due to the different environments used for structure determination or whether it represents a dynamic conformational change. The structure of αIIb CT MD region is not well-defined and varies among all the structures (Fig. 1A–C). It is not known whether this region contributes to maintaining the resting integrin conformation.
Fig. 1.

Effect of cytoplasmic truncation on αIIbβ3 integrin activation. (A–C) The transmembrane (TM) and cytoplasmic structures of αIIbβ3 determined by (A) disulfide crosslinking and Membrane Rosetta (xxx reference xxx), (B) by NMR in lipid bicelles (PDB code: 2K9J; xxx reference xxx), (C) by NMR in organic solvent (PDB code: 2KNC; xxx reference xxx). TM regions are red. The cytoplasmic membrane-proximal (MP) and membrane-distal (MD) regions are cyan and gray, respectively. Interfacial residues are shown as sticks with red oxygens and blue nitrogens or as Cα spheres (for glycine), and marked with stars in the αIIbβ3 sequences shown. The three structures were superimposed based on the TM region and shown in horizontal separation on the page. (D) PAC-1 binding of the αIIb CT truncations (Tr, shown schematically above the bars); (E) PAC-1 binding of the β3 CT truncations. 293FT cells were co-transfected with αIIb truncations and wild-type β3 (WT), or with β3 truncations and αIIb WT. PAC-1 binding was measured by flow cytometry and presented as the mean fluorescence intensity (MFI) normalized to integrin expression. Data are presented as mean±s.e.m. (n=3).
In the inside-out integrin activation, the cytosolic protein talin (which has two isoforms, talin-1 and -2) binds to both the MD and MP regions of the β-integrin CT through its head domain, which is composed of F0, F1, F2 and F3 subdomains, resulting in conformational changes in the integrin that are propagated to the extracellular domains across cell membrane (Kim et al., 2011b). Kindlin (which has three isoforms, kindlin-1, -2 and -3) has been found to cooperate with talin during integrin activation through direct binding to the β-integrin CT MD region (Ma et al., 2008; Moser et al., 2008; Harburger et al., 2009; Malinin et al., 2009; Moser et al., 2009b; Svensson et al., 2009; Bledzka et al., 2012). Studies on how talin changes integrin conformation have been focused on the β-integrin TM and CT domains, and have been described in many elegant reviews (Moser et al., 2009a; Shattil et al., 2010; Anthis and Campbell, 2011; Kim et al., 2011b; Calderwood et al., 2013; Das et al., 2014). Recent structural and functional studies suggest that talin-1 binding to the β3 CT changes the tilt angel of β3 TM domain in the cell membrane, thus disturbing the α-integrin–β-integrin interfaces at the TM and MP regions (Kalli et al., 2011; Kim et al., 2011a; Kim et al., 2012). A crystal structure of the talin-2 F2-F3 domain bound to β1D-CT suggests that there is an ionic interaction between a lysine residue in the talin-2 F3 domain and the conserved aspartic acid residue of the β1D integrin CT MP region (Anthis et al., 2009). This interaction was proposed to be important in integrin activation by disrupting the putative salt-bridge (between αIIb-R995 and β3-D723) at the α-integrin–β-integrin MP interface that has been suggested to maintain the resting integrin state (Hughes et al., 1996). However, it is not known if or how the α-integrin CT, and especially the MD region, participates in talin- and kindlin-induced integrin activation.
In this study, we examined the role of αIIb, αV and αL integrin CT MD regions in the regulation of integrin activation. We report that the α-integrin CT MD region helps maintain integrin in the resting state and is indispensible in talin- and kindlin-induced integrin conformational change and ligand binding. The proper length and correct amino acids are important for the α-integrin CT MD region to exert its effect on integrin activation. During the preparation of our manuscript, Li et al. (Li et al., 2014) reported that the αIIb CT MD is required for αIIbβ3 inside-out activation and proposed a model of ‘steric clashes’ between the talin-1 head and αIIb CT MD region that is involved in integrin activation. Our data of αIIbβ3 are consistent with their results, but our comprehensive analysis on multiple integrins suggests a more complicated mechanism by which α-integrin CT MD region regulates integrin activation.
RESULTS
The MP CT region is the minimum structural requirement for maintaining αIIbβ3 integrin in the resting state
To determine the minimum structure requirement of αIIbβ3 CT for maintaining the resting state, we performed a serial truncation mutagenesis. αIIb CT truncations before F993 greatly reduced integrin cell surface expression, whereas other truncations had a minor effect (supplementary material Fig. S2A). Similarly, β3 CT truncations before but not after I719 greatly reduced integrin expression (supplementary material Fig. S2B). The ligand-mimetic monoclonal antibody (mAb) PAC-1 was used to assess αIIbβ3 activation. PAC-1 binding to all the αIIb CT truncations after R995 was comparable to that for wild-type (WT) integrin (Fig. 1D). Truncation after αIIb-K994 only slightly increased PAC-1 binding, whereas truncations after αIIb V990, G991, F992 and F993 greatly enhanced PAC-1 binding (Fig. 1D, truncations are denoted by the suffix Tr). For β3 integrin, there was no detectable activation for all the β3 CT truncations after I721 (Fig. 1E). β3-W715Tr, β3-K716Tr and β3-L717Tr strongly induced PAC-1 binding, and truncation after β3-L719 weakly induced PAC-1 binding (Fig. 1E). The combination of the αIIb-K994Tr and β3-I721Tr had comparable PAC-1 binding to WT αIIbβ3 (data not shown). These results demonstrate that the MP sequence, KVGFFK or KVGFFKR in the αIIb CT, and the MP sequence of KLLITI or KLLIT in the β3 CT are the minimum requirement for maintaining αIIbβ3 integrin in the resting state.
MD αIIb CT truncations amplify the activating effect of β3 mutations
Truncations after the αIIb CT MP region might decrease the threshold for integrin activation because of structure incompleteness. To test this possibility, we performed a PAC-1 binding assay for the αIIb CT truncations co-expressed with the constitutively active β3 mutants, β3-G708L or β3-K716A. These β3 mutations are known to activate αIIbβ3 by disturbing the TM interaction (Luo et al., 2005; Zhu et al., 2009; Kim et al., 2011a). Compared with WT αIIb, truncation after αIIb-F993, K994, R995, N996, R997 or P998 strongly enhanced PAC-1 binding to both β3-G708L and β3-K716A mutants (Fig. 2A,B). This is in contrast with the very subtle or undetectable activating effect of these αIIb truncations when paired with WT β3 (Fig. 1D). The truncation-amplified activating effect was decreased with the increase of the length of αIIb CT MD region (Fig. 2A,B). More strikingly, the β3-G708A mutation very weakly induced PAC-1 binding when paired with WT αIIb, but its activating effect was greatly enhanced upon deletion of the αIIb CT MD region (Fig. 2C). In addition, the enhanced activating effect of αIIb CT MD deletion on the β3-G708L mutation was close to the maximal level, given that Mn2+ did not further increase PAC-1 binding (Fig. 2C). Similar results were obtained with the active β3-G135A mutant, which facilitates formation of the active conformation of β3-I domain (Zhang et al., 2013). Complete deletion of the αIIb CT MD region enhanced the activation of β3-G135A (data not shown). These data demonstrate that deletion of the αIIb CT MD region renders the integrin hyper-reactive to β3-activating mutations. Therefore, the αIIb CT MD region contributes to maintaining integrin in the resting state.
Fig. 2.

αIIb CT truncations synergize or compensate the activating effect of the transmembrane and cytoplasmic mutations. PAC-1 binding of 293FT cells co-transfected with the indicated αIIb constructs (shown schematically above the bars in A and E) and (A) β3-G708L, (B) β3-K716A, (C) β3-G708A and β3-G708L, (D) β3-D723R or (E) β3 WT. 293FT transfectants of αIIbβ3 WT and αIIb-F993Tr plus β3 WT are shown as controls in A, B and D. PAC-1 binding was measured by flow cytometry in the buffer containing 1♣mM Ca2+ and 1♣mM Mg2+ (Ca/Mg) (for A–E) or 0.2♣mM Ca2+ plus 2♣mM Mn2+ (Ca/Mn) (for C) and presented as the MFI normalized to integrin expression. Data are presented as mean±s.e.m. (n=3).
MD αIIb CT truncation abolishes the activating effect of αIIb-R995 and β3-D723 mutations
Integrin activation induced by the αIIb-R995 and β3-D723 mutations has been shown to depend on talin-1 binding to the β3 CT, which is distinct from the activation mechanism promoted by the activating mutations at the TM domain (Wegener et al., 2007). We asked whether the αIIb CT MD truncations exert different effects on αIIb-R995 and β3-D723 mutations. The β3-D723R mutant constitutively bound PAC-1 when paired with αIIb WT (Fig. 2D). However, PAC-1 binding was reverted to a WT level when β3-D723R was paired with the αIIb truncations F993Tr, K994Tr, R995Tr or N996Tr, in which all or most of the MD region is deleted (Fig. 2D). Strikingly, although both αIIb-F993Tr and β3-D723R were constitutively active when paired with their WT partners, their combination abolished the activating effect (Fig. 2D). This is in sharp contrast with the amplified activation mediated by the αIIb truncations for the β3-G708L and β3-K716A mutants (Fig. 2A,B). Retaining two or more residues of the αIIb MD region restored or increased integrin activation induced by the β3-D723R mutation (Fig. 2D).
We further verified our results with the αIIb-R995A and αIIb-R995D mutation. Both of these mutations render αIIbβ3 active to bind PAC-1 in the context of full-length αIIb (Fig. 2E). Their activating effects were abolished when the αIIb CT MD region was completely deleted (Fig. 2E). By contrast, the activation by αIIb-F992A or αIIb-F993A mutation was enhanced upon the αIIb CT MD deletion (Fig. 2E). These data suggest that the αIIb CT MD region plays a positive role in integrin inside-out activation.
The α-integrin CT MD region is required for talin-1-induced integrin activation
The deactivating effect of αIIb CT MD truncation on αIIb-R995 and β3-D723 mutations raises the intriguing possibility that the αIIb CT MD region might participate in talin-mediated integrin activation. We performed a well-established integrin activation assay by overexpressing of an EGFP-tagged talin-1 head domain (EGFP–TH) (Bouaouina et al., 2012). As expected, overexpression of EGFP–TH significantly activated WT αIIbβ3 as determined by the amount of PAC-1 binding (Fig. 3A). The EGFP–TH-induced activation was much more substantial for integrin bearing αIIb-R995A or β3-D723A mutation, indicating a synergetic effect (Fig. 3A). However, EGFP-TH failed to induce PAC-1 binding to the αIIb-R995Tr mutant, i.e. the αIIb CT MD region was deleted (Fig. 3A). Remarkably, the strong activating effect of EGFP–TH on αIIb-R995A or β3-D723A was also completely lost in the absence of the αIIb CT MD region (Fig. 3A). The expression levels of EGFP–TH or integrins were comparable among all the transfectants (supplementary material Fig. S2C). These results clearly demonstrate the requirement of αIIb CT MD region in talin-1-head-induced αIIbβ3 activation.
Fig. 3.

The α-integrin CT MD region is required for talin- and kindlin-induced integrin activation. (A–C) Complete deletion of the α-integrin CT MD region abolished talin-1-head-induced activation of (A) αIIbβ3, (B) αVβ3, and (C) αLβ2 integrins. Ligand binding was measured by flow cytometry with 293FT cells co-transfected with the indicated integrin constructs (also shown schematically above the bars) and EGFP or EGFP–TH. The EGFP and integrin double-positive cells were analyzed. (D–F) The α-integrin CT MD region is required for kindlin-induced activation of (D) αIIbβ3, (E) αVβ3, and (F) αLβ2 integrins. Ligand binding was measured by flow cytometry with 293FT cells co-transfected with the indicated integrin constructs and EGFP–TH plus mCherry (mC), mC-tagged kindlin-2 (mC–K2), or mC-tagged kindlin-3 (mC–K3). The integrin, EGFP, and mCherry triple-positive cells were analyzed. Data are presented as the MFI of the ligand normalized to integrin expression. Data are presented as mean±s.e.m. (n≥3). *P<0.05; **P<0.01; ns, not significant (unpaired two-tailed Student's t-tests).
We next examined whether the CT MD region is also important for talin-induced activation of other α-integrins. We found that overexpression of EGFP–TH significantly enhanced the binding of fibronectin type III domain 9–10 (Fn9-10) and intercellular adhesion molecule 1 (ICAM-1) to αVβ3 and αLβ2, respectively (Fig. 3B,C). In contrast, when the CT MD region was completely deleted for αV or αL integrin (αV-R993Tr or αL-R1094Tr), the talin-1 head failed to augment ligand binding for these integrins (Fig. 3B,C). The expression levels of EGFP–TH or integrin are comparable among the transfectants (supplementary material Fig. S2D,E). Thus, the requirement for an α-integrin CT MD region for talin-mediated integrin activation could be a generalized property of integrins.
The α-integrin CT MD region is required for kindlin-induced integrin activation
Having demonstrated the indispensable role of the α-integrin CT MD region in talin-induced integrin activation, we next asked whether the α-integrin CT MD region is also required for kindlin-mediated integrin activation. Given that integrin activation by kindlin requires the presence of talin (Ma et al., 2008), we examined the effect of deletion of the α-integrin CT MD domain on kindlin-mediated integrin activation with co-expression of the talin-1 head. Overexpression of mCherry–kindin-2 (mC–K2) significantly enhanced PAC-1 binding to WT αIIbβ3 (Fig. 3D). We also detected a slight increase of PAC-1 binding induced by mCherry–kindlin-3 (mC–K3), but it was not statistically significant compared with the mCherry control (Fig. 3D). In the absence of αIIb CT MD region (αIIb-R995Tr), mC-K2-mediated PAC-1 binding was greatly and significantly (P<0.01) reduced compared with the WT (Fig. 3D). As a control, the talin-1 head and kindlins failed to induce PAC-1 binding to αIIb–β3-R724Tr owing to the deletion of binding sites for talin-1 and kindlin (Fig. 3D). The expression levels of EGFP–TH, mC–K2 or mC–K3 were comparable among all the transfectants (supplementary material Fig. S3A,B). These data demonstrate that the αIIb CT MD region is required for kindlin-mediated αIIbβ3 activation.
Similar results were obtained with αVβ3 and αLβ2 integrins. In the presence of the talin-1 head, kindlin-2 but not kindlin-3 markedly enhanced fibronectin (Fn) binding to WT αVβ3 (Fig. 3E). Both kindlin-2 (P<0.01) and kindlin-3 (P<0.05) significantly increased ICAM-1 binding to WT αLβ2, but the level of increase by kindlin-3 was significantly (P<0.05) lower than by kindlin-2 (Fig. 3F). Deletion of the CT MD region of αV (αV-R993Tr) or αL (αL-R1094Tr) significantly (P<0.01) and greatly reduced the kindlin-2-mediated ligand binding (Fig. 3E,F). The kindlin-3-induced ICAM-1 binding was also greatly reduced for the αL-R1094Tr–β2 mutant (Fig. 3F). The expression levels of EGFP–TH, and kindlin-2 or kindlin-3 were comparable among the transfectants of αVβ3 (supplementary material Fig. S3C,D) and αLβ2 (material Fig. S3E,F). These data strongly demonstrate that the α-integrin CT MD region is also required for kindlin-mediated integrin activation.
The kindlin-2-mediated ligand binding was not completely abolished in the absence of the α-integrin CT MD region. The low level increase of ligand binding upon addition of kindlin-2 was statistically significant for αIIb-R995Tr–β3 (P<0.05) and αL-R1094Tr–β2 (P<0.01) (Fig. 3D,F), but not for αV-R993Tr–β3 (Fig. 3E). In addition, we could detect a moderate increase (P=0.05) of kindlin-3-mediated ICAM-1 binding to the αL-R1094Tr–β2 mutant (Fig. 3F). Given that PAC-1, Fn, and ICAM-1 are all multivalent ligands, the low level of ligand binding was probably due to the integrin clustering effect of kindlin as suggested by a recent study (Ye et al., 2013).
The α-integrin CT MD region is required for PMA-stimulated integrin activation in K562 cells
We further validated our data with by expressing the αIIbβ3 integrin in human K562 cells. PMA was used to stimulate integrin activation, which mimics the physiological integrin inside-out activation cascade (Banno and Ginsberg, 2008). We did not detect PMA-induced PAC-1 binding to WT αIIbβ3 (data not shown). However, PMA induced PAC-1 binding to αIIb–β3-D723A in a concentration-dependent manner (Fig. 4A), indicating that the β3-D723A mutation enhanced integrin sensitivity to PMA stimulation. Consistent with the results obtained upon overexpression of talin-1 head and kindlins, complete deletion of αIIb CT MD region abolished PMA-induced PAC-1 binding (Fig. 4A,B).
Fig. 4.

The αIIb CT MD region is required for PMA-stimulated integrin activation in K562 cells. (A) Dose–response curve for PMA-induced PAC-1 binding to αIIbβ3 integrin expressed in K562 cells. Transfected K562 cells were incubated with PMA and then with PAC-1. Flow cytometry was used to measure PAC-1 binding. Data are presented as the MFI of the ligand normalized to integrin expression. Data are presented as mean±s.e.m (n=4). (B) Representative flow cytometry plots of PMA-induced (at 1♣∀M) PAC-1 binding to K562 cells expressing the indicated αIIbβ3 integrins.
Retaining two residues of α-integrin CT MD region partially restores talin-1-induced activation of αIIbβ3, but not αVβ3 and αLβ2 integrins
We next asked how the α-integrin CT MD region participates in talin-mediated integrin activation. Structural superposition of talin bound to the β-CT and αIIbβ3 TM-CT complexes suggests that there can be steric clashes between the talin head and αIIb MD residues that are close to β3 and immediately follow the GFFKR motif (i.e. residues N996–R997) (supplementary material Fig. S4A,B). To test whether the potential steric clash plays a role in talin-mediated integrin activation, we made truncated αIIb constructs that had the two native residues, NR997Tr, two small residues, GG997Tr and AA997Tr, or two bulky residues, YY997Tr and WW997Tr, at the MD region (Fig. 5A). If the steric clashes are important, we would expect to see that the mutations with bulky residues would have a greater activating effect on talin-mediated integrin activation than the mutations with small residues. All the mutants had comparable levels of PAC-1 binding to that shown by WT αIIb when paired with β3 WT, except that the WW997Tr mutant was constitutively active (Fig. 5A). The Talin-1 head augmented PAC-1 binding to all the truncated αIIb mutants co-expressed with WT β3 (Fig. 5B). The WW997Tr mutant had a much higher level of PAC-1 binding than other constructs owing to its constitutive activation (Fig. 5B). We consistently observed that a lower level of PAC-1 binding was induced by the talin-1 head for the YY997Tr mutant than for the other constructs (Fig. 5B). We then examined the same αIIb mutants in the context of β3-D723A mutation, which is expected to enhance the sensitivity of the assay. The Talin-1 head induced similar levels of PAC-1 binding to αIIb-NR997Tr, αIIb-GG997Tr, and αIIb-AA997Tr mutants co-expressed with β3-D723A, but the binding was significantly (P<0.001) lower than for αIIb-WT co-expressed with β3-D723A (Fig. 5C). Strikingly, talin-1-head-induced PAC-1 binding to αIIb-YY997Tr mutant was significantly (P<0.01) lower than with αIIb-NR997Tr and the other constructs (Fig. 5C). When the NR997 was replaced by YY997 in the context of full-length αIIb, the αIIb-YY997 mutation also significantly reduced talin-1-head-mediated activation of β3-D723A (Fig. 5C). We next extended the C-termini of αIIb CT with a 29-residue tag of V5 epitope plus hexahistidine, which would be expected to provide more clashes with talin-1 head. We found that the response to talin-1-head-induced activation for V5–His-tagged αIIb was indistinguishable from WT αIIb (data not shown). This result, in accordance with the YY997 mutation data, suggests that it is not plausible to assign a simple ‘steric clash’ model to the mechanism by which αIIb CT MD region participates in integrin activation.
Fig. 5.

Retaining two residues at the α CT MD region partially restores talin-1 head induced activation of αIIbβ3, but not αVβ3 and αLβ2 integrins. (A) PAC-1 binding of 293FT cells transfected with β3 WT and the indicated αIIb constructs constructs (also shown schematically above the bars). (B) EGFP–TH-induced PAC-1 binding of the 293FT cells transfected with β3 WT and the indicated αIIb constructs. (C) EGFP–TH-induced PAC-1 binding of 293FT cells transfected with β3-D723A and the indicated αIIb constructs. (D) EGFP–TH induced Fn9-10 binding of 293FT cells transfected with αVβ3 constructs. (E) EGFP–TH induced ICAM-1 binding of 293FT cells transfected with αLβ2 constructs. EGFP and integrin double-positive cells were analyzed by flow cytometry. Ligand binding was presented as the MFI of the ligand normalized to integrin expression. Data are presented as mean±s.e.m (n≥3). *P<0.05; **P<0.01; ***P<0.001; ns, not significant (unpaired two-tailed Student's t-tests).
We extended our study to αVβ3 and αLβ2 integrins. Consistent with αIIbβ3, the presence of β3-D723A or the equivalent β2-D709A mutation significantly enhanced talin-1-head-induced binding of Fn9-10 and ICAM-1 to αVβ3 and αLβ2, respectively (Fig. 5D,E). Complete deletion of the αV and αL CT MD region (αV-R993Tr and αL-R1094Tr) totally abolished the activating effect of the talin-1 head (Fig. 5D,E). However, retaining two residues in the αV and αL CT MD region (αV-R995Tr and αL-R1096Tr) did not restore talin-1-head-induced integrin activation (Fig. 5D,E). This is in contrast to the result that showing that retaining two residues at the αIIb CT MD region partially restores talin-1-induced αIIbβ3 activation. In addition, retaining eight residues for the αL CT MD region (αL-A1102Tr) was still not sufficient to restore talin-1-head-induced integrin activation to the WT αL level (Fig. 5E). Thus, different α integrins might have different requirements for the CT MD region in talin-1-mediated activation. The proper amino acids and correct length of α-integrin CT MD region are required for sufficient integrin activation.
The α-integrin CT MD region is required for talin- and kindin-induced integrin conformational change
It is generally accepted that talin activates integrin through inducing conformational change (Anthis and Campbell, 2011; Kim et al., 2011b). By using conformation-dependent LIBS mAbs, we examined the talin-1-head-induced conformational change of integrin ectodomains. Consistent with our previous results (Zhang et al., 2013), overexpression of the talin-1 head significantly increased the binding of the β3 LIBS mAb 319.4, but not the αIIb LIBS mAb 370.3 to WT αIIbβ3 (Fig. 6A,B). The β3-D723A or αIIb-R995A mutation markedly enhanced the talin-1-head-induced binding of both mAbs to full-length αIIbβ3 (Fig. 6A,B). However, deletion of αIIb CT MD region significantly reduced talin-1-head-induced mAb binding (Fig. 6A,B). Similarly, the talin-1 head significantly increased the binding of the β2 LIBS mAbs KIM127 and M24 to WT αLβ2, but not to αL-R1094Tr–β2 (Fig. 6C,D). The expression levels of talin-1 head were comparable among the transfectants (data not shown). Thus, the α-integrin CT MD region is required for talin-induced integrin conformational change.
Fig. 6.

Effect of α-integrin CT truncation on talin-1 head induced integrin conformational change. (A,B) Talin-1-head-induced αIIbβ3 epitope exposure for LIBS mAbs 319.4 and 370.3. (C,D) Talin-1-head-induced αLβ2 epitope exposure for mAbs KIM127 and M24. 293FT cells were transfected with the indicated integrin constructs (shown under B and D) and EGFP or EGFP–TH. Cells were first incubated with the biotinylated LIBS mAbs and then stained with PE-labeled streptavidin and Alexa-Fluor-647-labeled mAb AP3 (for αIIbβ3), or stained with Alexa-Fluor-647-labeled streptavidin and PE-labeled TS2/4 (for αLβ2). The EGFP and integrin double-positive cells were analyzed by flow cytometry. The binding of LIBS mAb is presented as the MFI normalized to integrin expression. Data are presented as mean±s.e.m (n≥3). *P<0.05; **P<0.01; ***P<0.001; ns, not significant (unpaired two-tailed Student's t-tests).
We next asked whether the α-integrin CT MD region is also important for kindlin-mediated integrin conformational change. When co-expressed with the talin-1 head, kindlin-2, but not kindlin-3 significantly increased the binding of the mAbs 319.4 and 370.3 to WT αIIbβ3 (Fig. 7A,B). This is consistent with the data that kindlin-2 but not kindlin-3 enhanced ligand binding to WT αIIbβ3 (Fig. 3D). Kindlin-2 also significantly enhanced the binding of KIM127 and M24 to WT αLβ2 (Fig. 7C,D). We also detected kindlin-3-induced binding of M24 to WT αLβ2 (P<0.05) (Fig. 7D). The binding of KIM127 was also increased by kindlin-3, but it is not statistically significant (Fig. 7C). By contrast, both kindlin-2 and kindlin-3 failed to induce the binding of mAbs to the truncated αIIb-R995Tr–β3 and αL-R1094Tr–β2 integrins (Fig. 7). The expression levels of the talin-1 head and kindlin-2 or kindlin-3 were comparable among the integrin transfectants (data not shown). These results demonstrate that the α-integrin CT MD region is also required for kindlin-mediated integrin conformational change.
Fig. 7.
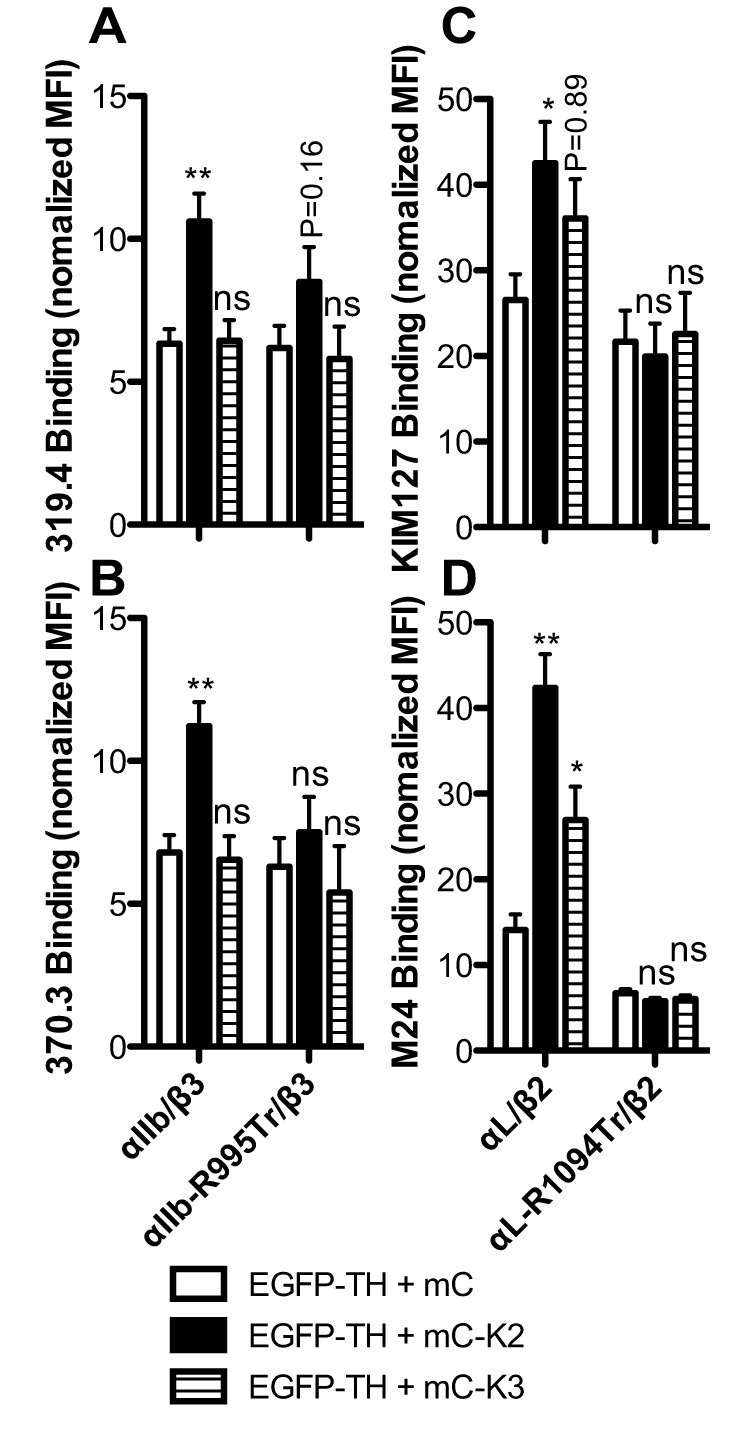
Effect of α-integrin CT truncation on kindlin-induced integrin conformational change. (A,B) Binding of LIBS mAb 319.4 and 370.3 to αIIbβ3 induced by the overexpression of EGFP–TH plus mCherry (mC)-tagged kindlin-2 (mC–K2) or kindlin-3 (mC–K3). (C,D) Binding of LIBS mAb KIM127 and M24 to αLβ2 induced by the overexpression of EGFP–TH plus mC–K2 or mC–K3. 293FT cells were co-transfected with the integrin constructs (shown under B and D) and EGFP–TH plus mC, mC–K2, or mC–K3. The integrin, EGFP, and mCherry triple-positive cells were analyzed by flow cytometry. The binding of LIBS mAb was presented as the MFI normalized to integrin expression. Data are presented as mean±s.e.m (n≥3). *P<0.05; **P<0.01; ***P<0.001; ns, not significant (unpaired two-tailed t-tests were used for the comparison with mC control group).
Deletion of the αIIb integrin CT MD region reduced αIIbβ3 transmembrane heterodimerization but did not affect the binding of the talin-1 head
To have direct evidence that the αIIb CT MD region contributes to αIIbβ3 TM association, we performed a disulfide crosslinking assay with integrins on the cell surface. Consistent with our previous data (Zhu et al., 2009), disulfide bonds were formed at high efficiency between the cysteine mutations of interfacial residues αIIb-G972C and β3-V700C at the TM domains (Fig. 8A), indicating the close TM association of αIIbβ3 (Fig. 1A). However, the disulfide bond formation was significantly reduced upon the deletion of αIIb CT MD region (Fig. 8A), suggesting a disassociation of TM domains. As a control, deletion of both the MP and the MD regions of αIIb CT (αIIb-G972C-V990Tr) further reduced disulfide bond formation (Fig. 8A). This is consistent with the ligand-binding assay. Deletion of αIIb CT MD region greatly enhanced PAC-1 binding to the αIIb-G972C mutant but not the β3-V700C mutant, whereas deletion of the αIIb CT MP region further enhanced PAC-1 binding due to the increased dissociation of αIIbβ3 transmembrane domains (Fig. 8A,B). This data clearly demonstrate that the αIIb CT MD region contributes to αIIbβ3 transmembrane association, which helps to keep integrin inactive. We next asked whether deletion of the αIIb CT MD affects the binding of talin-1 head to αIIbβ3 integrin. As shown in Fig. 8C, the EGFP–talin-1-head was co-immunoprecipitated with both WT αIIbβ3 and αIIb-R995Tr–β3 mutant, suggesting that the loss of talin-mediated integrin activation in the absence of αIIb CT MD region is not due to the effect on talin-1 head binding.
Fig. 8.

Deletion of αIIb CT MD region reduced αIIb and β3 transmembrane domain association but did not affect the binding of the talin-1 head. (A) Disulfide crosslinking of indicated αIIbβ3 constructs expressed in 293FT cells. Immunoprecipitates of [35S]-labeled integrins were subjected to non-reducing SDS-PAGE and autoradiography (upper panel). The quantification of crosslinking efficiency is shown in the lower panel. Data are presented as mean±s.e.m (n≥3). (B) PAC-1 binding of 293FT cells transfected with indicated αIIbβ3 constructs in the buffer containing 1♣mM Ca2+ and 1♣mM Mg2+ (Ca/Mg) or 0.2♣mM Ca2+ plus 2♣mM Mn2+ (Ca/Mn). (C) Co-immunoprecipitation of the talin-1 head with αIIbβ3 integrin. 293FT cells were co-transfected with αIIbβ3 constructs and EGFP or EGFP–TH. Integrins were immunoprecipitated (IP) with mAb 10E5 and subjected to western blotting (WB) with anti-EGFP and anti-β3 antibodies.
DISCUSSION
We present evidence to support the previously unappreciated structural roles of the α-integrin CT MD region in integrin activation. In addition to the well-conserved MP region, the α-integrin CT MD region also contributes to maintaining integrin in the resting state. Deletion of the αIIb CT MD region lowers the threshold of mutation-induced integrin activation. This is due to the reduced association of αIIbβ3 TM domains in the absence of the αIIb CT MD region as demonstrated by a disulfide crosslinking assay. By contrast, we demonstrate that the α-integrin CT MD region is required for talin- and kindlin-induced inside-out activation of αIIb, αV, and αL integrins, which is indicative of an essential and conserved mechanism of integrin family activation. The α-integrin CT MD region is dispensable for talin binding but is required for talin- and kindlin-induced integrin conformational change. Our data explain to a certain extent the previous observations that deletion of the α-integrin CT MD region diminished cell adhesion mediated by α1, α2, α4, αV and α6 integrins (Kassner and Hemler, 1993; Kawaguchi and Hemler, 1993; Shaw and Mercurio, 1993; Filardo and Cheresh, 1994; Kassner et al., 1994; Kawaguchi et al., 1994; Yauch et al., 1997; Abair et al., 2008) and that αL CT MD deletion reduced the sensitivity of αLβ2 integrin to PMA stimulation (Lu and Springer, 1997).
It is not readily known how the α-integrin CT MD region contributes to maintaining the resting state because current structural information shows diverse structures for integrin cytoplasmic tails (supplementary material Fig. S4). Several models could be proposed according to the data available (supplementary material Fig. S4C). First, our data demonstrate that in addition to the MP regions, the αIIb MD region contributes to integrin TM association on the cell surface. A recent report has shown that complete deletion of the αIIb CT MD region reduced the interaction of isolated αIIb and β3 TM-CT peptides co-expressed on the cell surface (Li et al., 2014). Our disulfide crosslinking experiments (Zhu et al., 2009) and a direct interaction assay (Ginsberg et al., 2001) indicate a close association of αIIb MD region with β3 cytoplasmic tail. Thus, the αIIb MD region might contribute to the structural stability of an αIIbβ3 TM-CT heterodimer through direct interaction with the β3 cytoplasmic tail. This interaction might be sequence specific given that replacing the αIIb CT with an α5 CT rendered αIIbβ3 constitutively active despite αIIb and α5 having a very similar CT MP region (O'Toole et al., 1991). Furthermore, replacing the αIIb CT MD region with a penta-alanine sequence reduces the interaction of isolated αIIb and β3 TM-CT peptides (Li et al., 2014). Second, the C-terminal αIIb CT MD region is unique with seven acidic amino acids (supplementary material Fig. S1) that have been shown to interact with the N-terminal αIIb CT MP region in a NMR structure of membrane-anchored αIIb CT peptide (Vinogradova et al., 2000). This interaction might be important in keeping the resting integrin state, but the same interaction was not found in the NMR structure of the αIIbβ3 TM-CT heterodimer determined in organic solvent (Yang et al., 2009) (Fig. 1C). Another possible mechanism of α-integrin CT MD region contributing to the inactive state is through its interaction with the negative integrin regulators. Most of the negative regulators identified so far bind to the conserved α-integrin CT MP region (Pouwels et al., 2012; Bouvard et al., 2013; Morse et al., 2014), but the MD region might also contribute to the interaction to some extent, which helps maintain the resting integrin state.
It has been shown that membrane-permeant peptides containing the αIIb CT MD sequences specifically blocked αIIbβ3 activation in platelets (Ginsberg et al., 2001; Koloka et al., 2008). A recent study has suggested that this is because the exogenous αIIb CT MD peptides inhibit the association of talin with αIIbβ3 in thrombin-stimulated platelets (Gkourogianni et al., 2013). However, this is unlikely owing to competition with or steric hindrance of the talin-binding sites of β3 CT given that deletion of the αIIb CT MD region did not increase the binding of talin-1 head to β3 CT (Fig. 8C and (Li et al., 2014). Talin activates integrin by disrupting the α-integrin–β-integrin TM–CT association, whereas the αIIb CT MD region negatively regulates this process by enhancing the structural stability of the αIIbβ3 TM-CT heterodimer.
In addition to the negative regulation of α CT MD region in integrin activation, our data demonstrate a positive role for the α-integrin CT MD region in talin-induced integrin activation. Displacement of α-integrin CT MD region by the β CT-bound talin-1 head domain might destabilize the α-integrin–β-integrin TM–CT dimerization, leading to integrin activation. The potential steric clashes between the talin-1 head and αIIb CT MD residues based on structure superposition have been proposed to play a role in this process (Yang et al., 2009; Zhu et al., 2009; Ye et al., 2011). This hypothesis was tested recently by replacing the αIIb CT MD region with a penta-alanine peptide or the CT MD sequence of α5 or αL integrin (Li et al., 2014). It has been shown that the αIIb chimeras remain responsive to talin-1-head-induced activation. Based on this result, it has been concluded that the αIIb CT MD region participates in integrin activation through steric clashing with talin-1 head without the requirement of a specific sequence. We examined the potential role of steric clashes using the truncated αIIb constructs that retain the first two residues of αIIb CT MD region (i.e. NR997Tr) given that the structural superposition showed major steric clashes between these residues and the talin-1 head (supplementary material Fig. S4B). The steric clash model is not consistent with the results showing that the bulky YY997 mutations significantly reduced, rather than enhancing, the talin-mediated αIIbβ3 activation. It should be noted that both the length and sequence of αIIb CT MD region are important to maintain the resting integrin state. Indeed, truncating the αIIb CT or swapping the αIIb CT MD region with other amino acids renders αIIbβ3 more active than WT integrin as shown in our study and by Li et al. (Li et al., 2014). In addition, the conformational flexibility of both the αIIb and β3 CT MD region (Zhu et al., 2009; Metcalf et al., 2010) would eliminate clashes in a talin–αIIbβ3 complex. Thus, the steric clash model is not the sole mechanism through which the αIIb CT MD region participates in talin-induced integrin activation.
An interaction between αIIb CT and talin has been reported (Knezevic et al., 1996; Yuan et al., 2006; Gingras et al., 2009; Raab et al., 2010). Although it might not have a major contribution to αIIbβ3 and talin interaction, a transient and weak interaction between αIIb CT and talin might stabilize the active conformation of αIIb CT during integrin activation. Recent biochemical and molecular dynamic simulation studies indicate that the αIIb CT increases its membrane embedding upon integrin inside-out activation (Kurtz et al., 2012; Provasi et al., 2014). The αIIb-YY997 mutation might affect the membrane embedding of αIIb TM-CT and reduces integrin activation since tyrosine residues tend to locate at the membrane boundary (Killian and von Heijne, 2000). A cluster of negatively charged residues at αIIb CT MD region may disfavor the negatively charged cell membrane and inhibit the membrane embedding of αIIb TM-CT, while an interaction between talin-head and αIIb CT MD acidic residues as indicated by a molecular dynamic simulation study (Provasi et al., 2014) might facilitate the membrane embedding of αIIb TM-CT to activate integrin. In addition, the capability of talin to activate integrin depends on its binding to both β-integrin CT and cell membrane (Wegener et al., 2007; Anthis et al., 2009; Moore et al., 2012; Song et al., 2012; Kalli et al., 2013). The interaction between α CT and talin might help talin maintain a proper orientation for the favorable interaction with β-integrin CT and cell membrane in order to stabilize the active conformation of TM-CT (supplementary material Fig. S4C). Considering the significant diversity of the α-integrin CT MD region, it is tempting to propose that this diversity might regulate integrin inside-out activation in an α-integrin-specific manner as it has been suggested that the α-integrin CT MD regions determine the specificity of integrin outside-in signaling (Chan et al., 1992; Shaw et al., 1995; Sastry et al., 1999; Na et al., 2003; Goel et al., 2014).
Another potential mechanism of regulating integrin activation by α-integrin CT is through its interacting proteins. Interestingly, most α-integrin-CT-binding proteins identified so far interact with the conserved membrane-proximal GFFKR motif and function as inhibitors of integrin activation (Pouwels et al., 2012; Bouvard et al., 2013; Morse et al., 2014), but the mechanism of their negative regulation is not well-defined. A Rap1 effector, RapL, has been shown to activate αLβ2 integrin through binding to the αL CT MD sequence proximal to the GFFKR motif (Katagiri et al., 2003). It is not known how RapL cooperates with talin and/or kindlin to activate αLβ2 integrin. The requirement of α-integrin CT MD region for integrin activation suggests that as-yet undiscovered regulatory proteins that interact with α-integrin CT MD region are involved in integrin activation, probably in an integrin-specific manner.
It has been shown that deletion of the αIIb CT MD region did not affect αIIbβ3-mediated cell adhesion and spreading, but led to aberrant recruitment of αIIbβ3 into focal adhesions formed by other integrins (Ylänne et al., 1993). The underlined mechanism is not known. The same phenomenon was observed with α2 integrin (Kawaguchi et al., 1994). However, deletion of the α CT MD region of α1, α2, α4, αV or α6 integrin diminished cell adhesion (Kassner and Hemler, 1993; Kawaguchi and Hemler, 1993; Shaw and Mercurio, 1993; Filardo and Cheresh, 1994; Kassner et al., 1994; Kawaguchi et al., 1994; Yauch et al., 1997; Abair et al., 2008), indicating that there are different regulatory roles of α CT MD regions among different integrins.
Our data on the β3-K716A mutation have important implications for the current model of integrin inside-out activation. It has been suggested that the β3-K716 residue helps to maintain a proper tilt angle for the β3 TM domain in the resting state by placing its basic side chain near negatively charged phospholipid head groups (Kim et al., 2011a). It has been proposed that the β3-K716A mutation or talin binding to β3 CT induces integrin activation by changing the tilt angle of β3 TM domain to disturb the αIIbβ3 TM-CT interaction. It has also been shown that the tilt angle change of β3 TM domain can occur upon adding the talin-1 head to the isolated β3 TM-CT peptide embedded in lipid nanodiscs in the absence of αIIb TM-CT peptide (Kim et al., 2012). We found that complete deletion of αIIb CT MD region greatly enhanced the activation of β3-K716A. If the talin-1 head could induce integrin activation by changing the tilt angle of the β3 TM domain as β3-K716A does, which seems to be independent of αIIb CT, we would expect to see an enhanced activation mediated by the talin-1 head in the absence of the αIIb CT MD region. However, an opposite result was obtained. The talin-1 head failed to activate integrin when the α-integrin CT MD region was completely deleted. By using a cysteine-scanning accessibility method with intact integrin on the cell surface, another group has suggested that β3-K716 is embedded in the lipid bilayer both in the inactive and active conformation. They detected conformational change of the αIIb TM-CT but not the β3 TM-CT domain in activating integrin mutants (Kurtz et al., 2012). Our structural study revealed that interactions between the β3-K716 side chain and the αIIb F992 and K994 backbone carbonyl oxygens are important in maintaining the resting integrin state (Zhu et al., 2009). These interactions were observed in a molecular dynamic simulation study starting with the NMR structure of αIIbβ3 TM-CT determined in lipid bicelles (Provasi et al., 2014). The direct interaction between β3-K716 and αIIb CT was also observed in a NMR study (Metcalf et al., 2010). Clearly, further investigations are required to reconcile these discrepancies.
It has been proposed that the conserved αIIb-R995 and β3-D723 form a salt bridge that restrains αIIbβ3 in the resting state (Hughes et al., 1996). One of the current models suggests that talin-1 head binding to the β3 CT breaks the putative salt-bridge to induce integrin activation (Anthis et al., 2009; Anthis and Campbell, 2011). Despite the very disruptive effect of αIIb-R995 and β3-D723 mutations on αIIbβ3 TM dimerization observed using isolated TM-CT peptides (Kim et al., 2009; Lau et al., 2009), their mutations only moderately activated full-length integrins (Peyruchaud et al., 1998; Ma et al., 2006; Ghevaert et al., 2008; Zhu et al., 2009). Remarkably, we found that the αIIb-K993Tr and αIIb-K994Tr mutation, in which the αIIb-R995 was deleted, abolished rather than synergized integrin activation induced by β3-D723 mutations and/or by the talin-1 head. In sharp contrast, it was only in the presence of αIIb CT MD region that the αIIb-R995 or β3-D723 mutations greatly synergized αIIbβ3 activation and the conformational change induced by the talin-1 head. Thus, although αIIb-R995 and β3-D723 are involved in integrin activation, they might not be central in structurally maintaining integrin in the resting state. Mutagenesis data has suggested that the extracellular domains are also involved in stabilizing the αIIbβ3 TM dimer as well as their membrane embedding (Lau et al., 2009; Kurtz et al., 2012). Caution should be taken when interpreting data obtained using isolated TM-CT peptides.
Natural mutations of αIIb-R995 and β3-D723 (αIIb-R995Q or αIIb-R995W and β3-D723H) have been identified in patients with thrombocytopenia (a relative decrease of platelets in blood) (Peyruchaud et al., 1998; Ghevaert et al., 2008; Kunishima et al., 2011). Although it was not studied in these patients, our data here suggest that αIIbβ3 integrin with αIIb-R995 or β3-D723 mutations might be more responsive to agonist stimulation leading to more platelet aggregation than in wild type, which might simultaneously cause a reduction in the amount of platelets in blood. An equivalent Arg to Ala mutation of α4 integrin CT has been found to aberrantly activate α4 integrin in genetic knock-in mice (Imai et al., 2008). We also found that the equivalent β2-D709A mutation renders αLβ2 integrin hyper-responsive to talin-1-head-induced activation. Interestingly, an equivalent Asp to Ala mutation in β1 integrin CT did not result in obvious defect in vivo (Czuchra et al., 2006), despite the fact that the β1 subunit forms heterodimers with 12 α-integrin subunits. Thus, the conserved α-Arg and β-Asp of the integrin CT might play different roles in regulating integrin activation among different integrins.
Among the caveats of our study is that the effect of αIIb CT MD mutations on talin-1-head-mediated integrin activation was much more dramatic with the β3-D723A mutant compared with WT β3. It should be noted that overexpression of talin-1 head only moderately activates WT integrin. Increasing the DNA amount for transfection did not further increase integrin activation (data not shown). It is possible that overexpression of talin-1 head alone might not be sufficient to induce maximal integrin activation owing to the lack of the recruitment process of talin-1 head to the integrin tail. In the physiological situation, the active full-length talin might apply traction force to its bound β-integrin CT through the coupled actin cytoskeleton (Zhu et al., 2008; Schürpf and Springer, 2011), and thus exert a more disruptive effect on the integrin TM-CT interaction than that mediated by the talin head alone. The β3-D723A mutation or other activating mutations like αIIb-R995A and β3-G135A might facilitate talin-head-induced integrin activation by decreasing the energy barrier. It significantly increases talin-1-head-induced integrin activation by more than 20-fold compared with WT. Moreover, PMA stimulates a significant increase in soluble ligand binding to the αIIb–β3-D723A mutant but not to the WT in K562 cells in suspension. Consistent with the soluble ligand-binding assay, the β3-D723A or αIIb-R995A mutation significantly enhanced talin-1-head-induced integrin conformational change. Thus, the combination of β3-D723A and αIIb CT MD mutations greatly improves the sensitivity of our assay. Similarly, the combination of the β3 activating mutations and αIIb CT MD truncations enabled us to reveal the contribution of αIIb CT MD region to maintaining the resting state.
Talin-1-head-induced integrin extension, but not headpiece opening, has been directly visualized by EM using the purified intact αIIbβ3 embedded in the lipid nanodiscs (Ye et al., 2010). We have recently shown that talin-1-head-induced integrin conformational change needs to be propagated to the ligand-binding site, probably through headpiece opening, in order to activate integrin (Zhang et al., 2013). In this study, by using the conformation-dependent mAbs that report integrin extension (319.4 for β3 and 370.3 for αIIb; KIM127 for β2) and headpiece opening (M24 for β2), we further demonstrated that the talin-1 head induced extension and headpiece opening of integrin. In particular, we detected enhanced integrin conformation change (extension and headpiece opening) when the talin-1 head was co-expressed with kindlins. Kindlin-2 or -3 exerted more effect on M24 binding than did KIM127 binding to αLβ2. This is consistent with a recent study showing that both talin-1 and kindlin-3 were required for inducing the extended open headpiece conformation of αLβ2 (Lefort et al., 2012). Remarkably, the talin-1-head- and kindlin-induced integrin extension and headpiece opening require the presence of an α-integrin CT MD region. However, because how kindlins induce integrin activation remains unknown, the requirement of an α-integrin CT MD region for kindlin-mediated integrin activation can only be interpreted as a secondary effect due to the loss of effectiveness of talin according to the current data.
We found that the talin-1-head- and kindlin-induced binding of β3 LIBS mAb 319.4 is not completely abolished in the absence of the αIIb CT MD region. This is in contrast with the binding of αIIb LIBS mAb 370.3. This indicates that the talin-1 head might exert some extent of conformational change on the β3 subunit even in the absence αIIb CT MD region, but that a fully active integrin conformation induced by talin and kindlin requires the involvement of the αIIb CT MD region. In summary, our study provides new insights into integrin inside-out activation and suggests that further structural studies are required to understand the precise mechanism by which the α-integrin CT MD region is involved in talin- and kindlin-mediated integrin activation.
MATERIALS AND METHODS
DNA constructs
DNA constructs of human αIIbβ3, αVβ3, αLβ2, and the EGFP-tagged mouse talin-1 head (EGFP–TH) were as described previously (Zhu et al., 2007; Bouaouina et al., 2008; Zhang et al., 2013). Human kindlin-2 or kindlin-3 was cloned into the pmCherry-C1 vector (Clontech). Mutations were introduced by site-directed mutagenesis with the QuikChange kit (Agilent Technologies).
Antibodies and ligands
PAC-1 (BD Bioscience) is a ligand-mimetic mAb (IgM) specific for activated αIIbβ3 integrin (Shattil et al., 1985). AP3 is a conformation-independent anti-β3 mAb (Kouns et al., 1991). 319.4 is a LIBS mAb that binds to β3 I-EGF domains (Zhang et al., 2013). 370.3 is a LIBS mAb that binds to the αIIb calf-1 domain (Zhang et al., 2013). PE-labeled TS2/4 (BioLegend) is a non-functional anti-αL mAb (Sanchez-Madrid et al., 1982). KIM127 (which binds to I-EGF-2 domain) and mAb 24 (M24, which binds to βI domain) are anti-β2 LIBS mAbs (Dransfield and Hogg, 1989; Robinson et al., 1992; Nishida et al., 2006; Chen et al., 2010). Human Fn9-10 was as described previously (Takagi et al., 2001). Human fibronectin and ICAM-1 (with a C-terminal human IgG1 Fc tag, ICAM-1-Fc) were purchased from Sigma-Aldrich and Sino-Biological, respectively.
Soluble ligand binding assay by flow cytometry with 293FT transfectants
PAC-1 binding of 293FT (Life Technologies) cells transfected with αIIbβ3 only were as described previously (Zhang et al., 2013). For talin-1-head-induced ligand binding, 293FT cells were co-transfected with integrin constructs and EGFP or EGFP–TH for at least 24♣hours. For kindlin-induced ligand binding, 293FT cells were co-transfected with integrin constructs plus EGFP–TH and mCherry or mCherry–kindlin for at least 24♣hours. Ligand binding was performed in HBSGB buffer (20♣mM HEPES pH♣7.4, 150♣mM NaCl, 5.5♣mM glucose, and 1% BSA) plus 5♣mM EDTA or 1♣mM Ca2+ and 1♣mM Mg2+ (Ca/Mg) at 25°C for 30♣min with 5♣∀g/ml PAC-1 for αIIbβ3, 20♣∀g/ml each of ICAM-1-Fc and biotin-labeled mouse anti-human IgG1 Fc for αLβ2, or 50♣∀g/ml Alexa-Fluor-647-labeled Fn9-10 or Fn for αVβ3. Cells were then washed and incubated with Ca/Mg on ice with the detecting reagents: 10♣∀g/ml each of biotinylated AP3, PE-labeled streptavidin and Alexa-Fluor-647-labeled goat anti-mouse IgM for αIIbβ3, biotinylated AP3 and PE-labeled streptavidin for αVβ3, PE-labeled TS2/4, and Alexa Fluor 647-labeled streptavidin for αLβ2. Integrin and EGFP double-, or integrin, EGFP, and mCherry triple-positive cells were acquired for calculating mean fluorescence intensity (MFI) by flow cytometry. Ligand binding was presented as the normalized MFI, that is ligand MFI (after subtracting the ligand MFI in EDTA) as a percentage of integrin MFI.
PMA-induced integrin ligand binding in K562 cells
Human K562 cells were transfected with αIIbβ3 constructs by Lipofectamine 2000 (Life Technologies). Cells were incubated in RPMI-1640 medium plus 1% BSA with PMA for 10♣minutes followed by a 10-minute incubation with PAC-1. Cells were then washed and incubated with Alexa-Fluor-488-labeled AP3 and Alexa-Fluor-647-labeled goat anti-mouse IgM on ice for 30♣minutes. Integrin-positive cells were acquired for calculating MFI by flow cytometry. Ligand binding was presented as MFI normalized by integrin expression.
LIBS epitope exposure
Talin-1-head- and kindlin-induced LIBS epitope exposure was as described previously (Zhang et al., 2013). In brief, 293FT transfectants were first incubated with or without (as background) the biotinylated LIBS mAb in Ca/Mg at 25°C for 30♣mins, and then washed and incubated with the detecting reagents: Alexa-Fluor-647-labeled AP3 and PE-labeled streptavidin for αIIbβ3, PE-labeled TS2/4 and Alexa-Fluor-647-labeled streptavidin for αLβ2. Integrin and EGFP double-, or integrin, EGFP and mCherry triple-positive cells were analyzed for calculating the MFI. LIBS mAb binding is presented as MFI normalized to the integrin expression, that is LIBS mAb MFI (after subtracting the background MFI) as a percentage of integrin MFI.
Disulfide crosslinking and immunoprecipitation
Metabolic labeling with [35S]cysteine/methionine and disulfide crosslinking using intact cells of 293FT transfectants were as described previously (Zhu et al., 2009). Formation of disulfide bonds was induced by treating the cells with CuSO4 and o-phenanthroline on ice. Integrins were immunoprecipitated with anti-αIIb mAb 10E5 from the cell lysate and subjected to nonreducing SDS-PAGE and autoradiography. For the talin-1-head-binding assay, αIIbβ3 integrins were immunoprecipitated with mAb 10E5 from cells co-transfected with EGFP or EGFP–TH. Binding of the talin-1 head was detected by western blotting with rabbit anti-EGFP polyclonal antibody (Origene). β3 integrin was detected with rabbit anti-β3 H-96 (Santa Cruz Biotechnology).
Acknowledgements
We thank Drs. Daniel Bougie, Richard Aster, Barry Coller, Chafen Lu, and Timothy Springer for providing antibodies; David Calderwood for providing the DNA construct of EGFP-tagged mouse talin-1-head domain; Peter Newman for critical reading of the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
J.L., Z.W., A.M.M.T. and J.Z. performed the experiments and analyzed data. Y-Q.M. made the kindlin constructs. J.Z. designed the study, prepared the figures and wrote the manuscript.
Funding
This work was supported by a Scientist Development Grant from the American Heart Association [grant number 12SDG12070059 to J.Z.]; and an ASH Scholar Award for junior faculty from the American Society of Hematology [to J.Z.].
References
- Abair T. D., Bulus N., Borza C., Sundaramoorthy M., Zent R. and Pozzi A. (2008). Functional analysis of the cytoplasmic domain of the integrin α1 subunit in endothelial cells. Blood 112, 3242-3254. doi:10.1182/blood-2007-12-126433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthis N. J. and Campbell I. D. (2011). The tail of integrin activation. Trends Biochem. Sci. 36, 191-198. doi:10.1016/j.tibs.2010.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthis N. J., Wegener K. L., Ye F., Kim C., Goult B. T., Lowe E. D., Vakonakis I., Bate N., Critchley D. R., Ginsberg M. H. et al. (2009). The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 28, 3623-3632. doi:10.1038/emboj.2009.287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banno A. and Ginsberg M. H. (2008). Integrin activation. Biochem. Soc. Trans. 36, 229-234. doi:10.1042/BST0360229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger B. W., Kulp D. W., Span L. M., DeGrado J. L., Billings P. C., Senes A., Bennett J. S. and DeGrado W. F. (2010). Consensus motif for integrin transmembrane helix association. Proc. Natl. Acad. Sci. USA 107, 703-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bledzka K., Liu J., Xu Z., Perera H. D., Yadav S. P., Bialkowska K., Qin J., Ma Y. Q. and Plow E. F. (2012). Spatial coordination of kindlin-2 with talin head domain in interaction with integrin β cytoplasmic tails. J. Biol. Chem. 287, 24585-24594. doi:10.1074/jbc.M111.336743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaouina M., Lad Y. and Calderwood D. A. (2008). The N-terminal domains of talin cooperate with the phosphotyrosine binding-like domain to activate β1 and β3 integrins. J. Biol. Chem. 283, 6118-6125. doi:10.1074/jbc.M709527200 [DOI] [PubMed] [Google Scholar]
- Bouaouina M., Harburger D. S. and Calderwood D. A. (2012). Talin and signaling through integrins. Methods Mol. Biol. 757, 325-347. doi:10.1007/978-1-61779-166-6_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvard D., Pouwels J., De Franceschi N. and Ivaska J. (2013). Integrin inactivators: balancing cellular functions in vitro and in vivo. Nat. Rev. Mol. Cell Biol. 14, 430-442. doi:10.1038/nrm3599 [DOI] [PubMed] [Google Scholar]
- Byron A., Humphries J. D., Askari J. A., Craig S. E., Mould A. P. and Humphries M. J. (2009). Anti-integrin monoclonal antibodies. J. Cell Sci. 122, 4009-4011. doi:10.1242/jcs.056770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood D. A., Campbell I. D. and Critchley D. R. (2013). Talins and kindlins: partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 14, 503-517. doi:10.1038/nrm3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan B. M. C., Kassner P. D., Schiro J. A., Byers H. R., Kupper T. S. and Hemler M. E. (1992). Distinct cellular functions mediated by different VLA integrin α subunit cytoplasmic domains. Cell 68, 1051-1060. doi:10.1016/0092-8674(92)90077-P [DOI] [PubMed] [Google Scholar]
- Chen X., Xie C., Nishida N., Li Z., Walz T. and Springer T. A. (2010). Requirement of open headpiece conformation for activation of leukocyte integrin alphaXbeta2. Proc. Natl. Acad. Sci. USA 107, 14727-14732. doi:10.1073/pnas.1008663107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czuchra A., Meyer H., Legate K. R., Brakebusch C. and Fässler R. (2006). Genetic analysis of β1 integrin “activation motifs” in mice. J. Cell Biol. 174, 889-899. doi:10.1083/jcb.200604060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M., Subbayya Ithychanda S., Qin J. and Plow E. F. (2014). Mechanisms of talin-dependent integrin signaling and crosstalk. Biochim. Biophys. Acta 1838, 579-588. doi:10.1016/j.bbamem.2013.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dransfield I. and Hogg N. (1989). Regulated expression of Mg2+ binding epitope on leukocyte integrin α subunits. EMBO J. 8, 3759-3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng E. T., Smagghe B. J., Walz T. and Springer T. A. (2011). Intact αIIbβ3 extends after activation measured by solution X-ray scattering and electron microscopy. J. Biol. Chem. 286, 35218-35226. doi:10.1074/jbc.M111.275107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filardo E. J. and Cheresh D. A. (1994). A beta turn in the cytoplasmic tail of the integrin alpha v subunit influences conformation and ligand binding of αvβ3. J. Biol. Chem. 269, 4641-4647. [PubMed] [Google Scholar]
- Frelinger A. L. III, Du X. P., Plow E. F. and Ginsberg M. H. (1991). Monoclonal antibodies to ligand-occupied conformers of integrin α IIb β 3 (glycoprotein IIb-IIIa) alter receptor affinity, specificity, and function. J. Biol. Chem. 266, 17106-17111. [PubMed] [Google Scholar]
- Ghevaert C., Salsmann A., Watkins N. A., Schaffner-Reckinger E., Rankin A., Garner S. F., Stephens J., Smith G. A., Debili N., Vainchenker W. et al. (2008). A nonsynonymous SNP in the ITGB3 gene disrupts the conserved membrane-proximal cytoplasmic salt bridge in the alphaIIbbeta3 integrin and cosegregates dominantly with abnormal proplatelet formation and macrothrombocytopenia. Blood 111, 3407-3414. doi:10.1182/blood-2007-09-112615 [DOI] [PubMed] [Google Scholar]
- Gingras A. R., Ziegler W. H., Bobkov A. A., Joyce M. G., Fasci D., Himmel M., Rothemund S., Ritter A., Grossmann J. G., Patel B. et al. (2009). Structural determinants of integrin binding to the talin rod. J. Biol. Chem. 284, 8866-8876. doi:10.1074/jbc.M805937200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg M. H., Yaspan B., Forsyth J., Ulmer T. S., Campbell I. D. and Slepak M. (2001). A membrane-distal segment of the integrin α IIb cytoplasmic domain regulates integrin activation. J. Biol. Chem. 276, 22514-22521. doi:10.1074/jbc.M101915200 [DOI] [PubMed] [Google Scholar]
- Gkourogianni A., Egot M., Koloka V., Moussis V., Tsikaris V., Panou-Pomonis E., Sakarellos-Daitsiotis M., Bachelot-Loza C. and Tsoukatos D. C. (2013). Palmitoylated peptide, being derived from the carboxyl-terminal sequence of the integrin α cytoplasmic domain, inhibits talin binding to αIIbβ3. Platelets 25, 619-627. [DOI] [PubMed] [Google Scholar]
- Goel H. L., Gritsko T., Pursell B., Chang C., Shultz L. D., Greiner D. L., Norum J. H., Toftgard R., Shaw L. M. and Mercurio A. M. (2014). Regulated splicing of the α6 integrin cytoplasmic domain determines the fate of breast cancer stem cells. Cell Reports 7, 747-761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harburger D. S., Bouaouina M. and Calderwood D. A. (2009). Kindlin-1 and -2 directly bind the C-terminal region of β integrin cytoplasmic tails and exert integrin-specific activation effects. J. Biol. Chem. 284, 11485-11497. doi:10.1074/jbc.M809233200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes P. E., O'Toole T. E., Ylänne J., Shattil S. J. and Ginsberg M. H. (1995). The conserved membrane-proximal region of an integrin cytoplasmic domain specifies ligand binding affinity. J. Biol. Chem. 270, 12411-12417. doi:10.1074/jbc.270.21.12411 [DOI] [PubMed] [Google Scholar]
- Hughes P. E., Diaz-Gonzalez F., Leong L., Wu C., McDonald J. A., Shattil S. J. and Ginsberg M. H. (1996). Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J. Biol. Chem. 271, 6571-6574. [DOI] [PubMed] [Google Scholar]
- Humphries M. J. (2004). Monoclonal antibodies as probes of integrin priming and activation. Biochem. Soc. Trans. 32, 407-411. doi:10.1042/BST0320407 [DOI] [PubMed] [Google Scholar]
- Hynes R. O. (2002). Integrins: bidirectional, allosteric signaling machines. Cell 110, 673-687. doi:10.1016/S0092-8674(02)00971-6 [DOI] [PubMed] [Google Scholar]
- Imai Y., Park E. J., Peer D., Peixoto A., Cheng G., von Andrian U. H., Carman C. V. and Shimaoka M. (2008). Genetic perturbation of the putative cytoplasmic membrane-proximal salt bridge aberrantly activates α(4) integrins. Blood 112, 5007-5015. doi:10.1182/blood-2008-03-144543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A. C., Campbell I. D. and Sansom M. S. (2011). Multiscale simulations suggest a mechanism for integrin inside-out activation. Proc. Natl. Acad. Sci. USA 108, 11890-11895. doi:10.1073/pnas.1104505108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A. C., Campbell I. D. and Sansom M. S. (2013). Conformational changes in talin on binding to anionic phospholipid membranes facilitate signaling by integrin transmembrane helices. PLOS Comput. Biol. 9, e1003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassner P. D. and Hemler M. E. (1993). Interchangeable α chain cytoplasmic domains play a positive role in control of cell adhesion mediated by VLA-4, a β 1 integrin. J. Exp. Med. 178, 649-660. doi:10.1084/jem.178.2.649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassner P. D., Kawaguchi S. and Hemler M. E. (1994). Minimum α chain cytoplasmic tail sequence needed to support integrin-mediated adhesion. J. Biol. Chem. 269, 19859-19867. [PubMed] [Google Scholar]
- Katagiri K., Maeda A., Shimonaka M. and Kinashi T. (2003). RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat. Immunol. 4, 741-748. doi:10.1038/ni950 [DOI] [PubMed] [Google Scholar]
- Kawaguchi S. and Hemler M. E. (1993). Role of the α subunit cytoplasmic domain in regulation of adhesive activity mediated by the integrin VLA-2. J. Biol. Chem. 268, 16279-16285. [PubMed] [Google Scholar]
- Kawaguchi S., Bergelson J. M., Finberg R. W. and Hemler M. E. (1994). Integrin α 2 cytoplasmic domain deletion effects: loss of adhesive activity parallels ligand-independent recruitment into focal adhesions. Mol. Biol. Cell 5, 977-988. doi:10.1091/mbc.5.9.977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killian J. A. and von Heijne G. (2000). How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 25, 429-434. doi:10.1016/S0968-0004(00)01626-1 [DOI] [PubMed] [Google Scholar]
- Kim C., Lau T. L., Ulmer T. S. and Ginsberg M. H. (2009). Interactions of platelet integrin alphaIIb and β3 transmembrane domains in mammalian cell membranes and their role in integrin activation. Blood 113, 4747-4753. doi:10.1182/blood-2008-10-186551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C., Schmidt T., Cho E. G., Ye F., Ulmer T. S. and Ginsberg M. H. (2011a). Basic amino-acid side chains regulate transmembrane integrin signalling. Nature 481, 209-213. doi:10.1038/nature10697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C., Ye F. and Ginsberg M. H. (2011b). Regulation of integrin activation. Annu. Rev. Cell Dev. Biol. 27, 321-345. doi:10.1146/annurev-cellbio-100109-104104 [DOI] [PubMed] [Google Scholar]
- Kim C., Ye F., Hu X. and Ginsberg M. H. (2012). Talin activates integrins by altering the topology of the β transmembrane domain. J. Cell Biol. 197, 605-611. doi:10.1083/jcb.201112141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knezevic I., Leisner T. M. and Lam S. C. (1996). Direct binding of the platelet integrin alphaIIbbeta3 (GPIIb-IIIa) to talin. Evidence that interaction is mediated through the cytoplasmic domains of both alphaIIb and β3. J. Biol. Chem. 271, 16416-16421. doi:10.1074/jbc.271.27.16416 [DOI] [PubMed] [Google Scholar]
- Koloka V., Christofidou E. D., Vaxevanelis S., Dimitriou A. A., Tsikaris V., Tselepis A. D., Panou-Pomonis E., Sakarellos-Daitsiotis M. and Tsoukatos D. C. (2008). A palmitoylated peptide, derived from the acidic carboxyl-terminal segment of the integrin alphaIIb cytoplasmic domain, inhibits platelet activation. Platelets 19, 502-511. doi:10.1080/09537100802266875 [DOI] [PubMed] [Google Scholar]
- Kouns W. C., Newman P. J., Puckett K. J., Miller A. A., Wall C. D., Fox C. F., Seyer J. M. and Jennings L. K. (1991). Further characterization of the loop structure of platelet glycoprotein IIIa: partial mapping of functionally significant glycoprotein IIIa epitopes. Blood 78, 3215-3223. [PubMed] [Google Scholar]
- Kunishima S., Kashiwagi H., Otsu M., Takayama N., Eto K., Onodera M., Miyajima Y., Takamatsu Y., Suzumiya J., Matsubara K. et al. (2011). Heterozygous ITGA2B R995W mutation inducing constitutive activation of the αIIbβ3 receptor affects proplatelet formation and causes congenital macrothrombocytopenia. Blood 117, 5479-5484. doi:10.1182/blood-2010-12-323691 [DOI] [PubMed] [Google Scholar]
- Kurtz L., Kao L., Newman D., Kurtz I. and Zhu Q. (2012). Integrin αIIbβ3 inside-out activation: an in situ conformational analysis reveals a new mechanism. J. Biol. Chem. 287, 23255-23265. doi:10.1074/jbc.M112.360966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau T. L., Kim C., Ginsberg M. H. and Ulmer T. S. (2009). The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J. 28, 1351-1361. doi:10.1038/emboj.2009.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort C. T., Rossaint J., Moser M., Petrich B. G., Zarbock A., Monkley S. J., Critchley D. R., Ginsberg M. H., Fässler R. and Ley K. (2012). Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood 119, 4275-4282. doi:10.1182/blood-2011-08-373118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A., Guo Q., Kim C., Hu W. and Ye F. (2014). Integrin αIIb tail distal of GFFKR participates in inside-out αIIbβ3 activation. Journal of Thrombosis and Haemostasis 12, 1145-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C. F. and Springer T. A. (1997). The α subunit cytoplasmic domain regulates the assembly and adhesiveness of integrin lymphocyte function-associated antigen-1. J. Immunol. 159, 268-278. [PubMed] [Google Scholar]
- Luo B.-H., Carman C. V., Takagi J. and Springer T. A. (2005). Disrupting integrin transmembrane domain heterodimerization increases ligand binding affinity, not valency or clustering. Proc. Natl. Acad. Sci. USA 102, 3679-3684. doi:10.1073/pnas.0409440102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y. Q., Yang J., Pesho M. M., Vinogradova O., Qin J. and Plow E. F. (2006). Regulation of integrin alphaIIbbeta3 activation by distinct regions of its cytoplasmic tails. Biochemistry 45, 6656-6662. doi:10.1021/bi060279h [DOI] [PubMed] [Google Scholar]
- Ma Y. Q., Qin J., Wu C. and Plow E. F. (2008). Kindlin-2 (Mig-2): a co-activator of β3 integrins. J. Cell Biol. 181, 439-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinin N. L., Zhang L., Choi J., Ciocea A., Razorenova O., Ma Y. Q., Podrez E. A., Tosi M., Lennon D. P., Caplan A. I. et al. (2009). A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat. Med. 15, 313-318. doi:10.1038/nm.1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margadant C., Monsuur H. N., Norman J. C. and Sonnenberg A. (2011). Mechanisms of integrin activation and trafficking. Curr. Opin. Cell Biol. 23, 607-614. doi:10.1016/j.ceb.2011.08.005 [DOI] [PubMed] [Google Scholar]
- Metcalf D. G., Moore D. T., Wu Y., Kielec J. M., Molnar K., Valentine K. G., Wand A. J., Bennett J. S. and DeGrado W. F. (2010). NMR analysis of the alphaIIb β3 cytoplasmic interaction suggests a mechanism for integrin regulation. Proc. Natl. Acad. Sci. USA 107, 22481-22486. doi:10.1073/pnas.1015545107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore D. T., Nygren P., Jo H., Boesze-Battaglia K., Bennett J. S. and DeGrado W. F. (2012). Affinity of talin-1 for the β3-integrin cytosolic domain is modulated by its phospholipid bilayer environment. Proc. Natl. Acad. Sci. USA 109, 793-798. doi:10.1073/pnas.1117220108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse E. M., Brahme N. N. and Calderwood D. A. (2014). Integrin cytoplasmic tail interactions. Biochemistry 53, 810-820. doi:10.1021/bi401596q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser M., Nieswandt B., Ussar S., Pozgajova M. and Fässler R. (2008). Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 14, 325-330. doi:10.1038/nm1722 [DOI] [PubMed] [Google Scholar]
- Moser M., Legate K. R., Zent R. and Fässler R. (2009a). The tail of integrins, talin, and kindlins. Science 324, 895-899. doi:10.1126/science.1163865 [DOI] [PubMed] [Google Scholar]
- Moser M., Bauer M., Schmid S., Ruppert R., Schmidt S., Sixt M., Wang H. V., Sperandio M. and Fässler R. (2009b). Kindlin-3 is required for β2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 15, 300-305. doi:10.1038/nm.1921 [DOI] [PubMed] [Google Scholar]
- Mould A. P., Symonds E. J., Buckley P. A., Grossmann J. G., McEwan P. A., Barton S. J., Askari J. A., Craig S. E., Bella J. and Humphries M. J. (2003). Structure of an integrin-ligand complex deduced from solution x-ray scattering and site-directed mutagenesis. J. Biol. Chem. 278, 39993-39999. doi:10.1074/jbc.M304627200 [DOI] [PubMed] [Google Scholar]
- Na J., Marsden M. and DeSimone D. W. (2003). Differential regulation of cell adhesive functions by integrin α subunit cytoplasmic tails in vivo. J. Cell Sci. 116, 2333-2343. doi:10.1242/jcs.00445 [DOI] [PubMed] [Google Scholar]
- Nishida N., Xie C., Shimaoka M., Cheng Y., Walz T. and Springer T. A. (2006). Activation of leukocyte β2 integrins by conversion from bent to extended conformations. Immunity 25, 583-594. doi:10.1016/j.immuni.2006.07.016 [DOI] [PubMed] [Google Scholar]
- O'Toole T. E., Mandelman D., Forsyth J., Shattil S. J., Plow E. F. and Ginsberg M. H. (1991). Modulation of the affinity of integrin α IIb β 3 (GPIIb-IIIa) by the cytoplasmic domain of α IIb. Science 254, 845-847. doi:10.1126/science.1948065 [DOI] [PubMed] [Google Scholar]
- O'Toole T. E., Katagiri Y., Faull R. J., Peter K., Tamura R., Quaranta V., Loftus J. C., Shattil S. J. and Ginsberg M. H. (1994). Integrin cytoplasmic domains mediate inside-out signal transduction. J. Cell Biol. 124, 1047-1059. doi:10.1083/jcb.124.6.1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge A. W., Liu S., Kim S., Bowie J. U. and Ginsberg M. H. (2005). Transmembrane domain helix packing stabilizes integrin alphaIIbbeta3 in the low affinity state. J. Biol. Chem. 280, 7294-7300. doi:10.1074/jbc.M412701200 [DOI] [PubMed] [Google Scholar]
- Peyruchaud O., Nurden A. T., Milet S., Macchi L., Pannochia A., Bray P. F., Kieffer N. and Bourre F. (1998). R to Q amino acid substitution in the GFFKR sequence of the cytoplasmic domain of the integrin IIb subunit in a patient with a Glanzmann's thrombasthenia-like syndrome. Blood 92, 4178-4187. [PubMed] [Google Scholar]
- Pouwels J., Nevo J., Pellinen T., Ylänne J. and Ivaska J. (2012). Negative regulators of integrin activity. J. Cell Sci. 125, 3271-3280. doi:10.1242/jcs.093641 [DOI] [PubMed] [Google Scholar]
- Provasi D., Negri A., Coller B. S. and Filizola M. (2014). Talin-driven inside-out activation mechanism of platelet αIIbβ3 integrin probed by multimicrosecond, all-atom molecular dynamics simulations. Proteins 82, 3231-3240. doi:10.1002/prot.24540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M., Daxecker H., Edwards R. J., Treumann A., Murphy D. and Moran N. (2010). Protein interactions with the platelet integrin α(IIb) regulatory motif. Proteomics 10, 2790-2800. doi:10.1002/pmic.200900621 [DOI] [PubMed] [Google Scholar]
- Robinson M. K., Andrew D., Rosen H., Brown D., Ortlepp S., Stephens P. and Butcher E. C. (1992). Antibody against the Leu-CAM β-chain (CD18) promotes both LFA-1- and CR3-dependent adhesion events. J. Immunol. 148, 1080-1085. [PubMed] [Google Scholar]
- Sanchez-Madrid F., Krensky A. M., Ware C. F., Robbins E., Strominger J. L., Burakoff S. J. and Springer T. A. (1982). Three distinct antigens associated with human T-lymphocyte-mediated cytolysis: LFA-1, LFA-2, and LFA-3. Proc. Natl. Acad. Sci. USA 79, 7489-7493. doi:10.1073/pnas.79.23.7489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry S. K., Lakonishok M., Wu S., Truong T. Q., Huttenlocher A., Turner C. E. and Horwitz A. F. (1999). Quantitative changes in integrin and focal adhesion signaling regulate myoblast cell cycle withdrawal. J. Cell Biol. 144, 1295-1309. doi:10.1083/jcb.144.6.1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schürpf T. and Springer T. A. (2011). Regulation of integrin affinity on cell surfaces. EMBO J. 30, 4712-4727. doi:10.1038/emboj.2011.333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil S. J., Hoxie J. A., Cunningham M. and Brass L. F. (1985). Changes in the platelet membrane glycoprotein IIb.IIIa complex during platelet activation. J. Biol. Chem. 260, 11107-11114. [PubMed] [Google Scholar]
- Shattil S. J., Kim C. and Ginsberg M. H. (2010). The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 11, 288-300. doi:10.1038/nrm2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw L. M. and Mercurio A. M. (1993). Regulation of α 6 β 1 integrin laminin receptor function by the cytoplasmic domain of the α 6 subunit. J. Cell Biol. 123, 1017-1025. doi:10.1083/jcb.123.4.1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw L. M., Turner C. E. and Mercurio A. M. (1995). The α 6A β 1 and α 6B β 1 integrin variants signal differences in the tyrosine phosphorylation of paxillin and other proteins. J. Biol. Chem. 270, 23648-23652. doi:10.1074/jbc.270.40.23648 [DOI] [PubMed] [Google Scholar]
- Sims P. J., Ginsberg M. H., Plow E. F. and Shattil S. J. (1991). Effect of platelet activation on the conformation of the plasma membrane glycoprotein IIb-IIIa complex. J. Biol. Chem. 266, 7345-7352. [PubMed] [Google Scholar]
- Song X., Yang J., Hirbawi J., Ye S., Perera H. D., Goksoy E., Dwivedi P., Plow E. F., Zhang R. and Qin J. (2012). A novel membrane-dependent on/off switch mechanism of talin FERM domain at sites of cell adhesion. Cell Res. 22, 1533-1545. doi:10.1038/cr.2012.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer T. A. and Dustin M. L. (2012). Integrin inside-out signaling and the immunological synapse. Curr. Opin. Cell Biol. 24, 107-115. doi:10.1016/j.ceb.2011.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson L., Howarth K., McDowall A., Patzak I., Evans R., Ussar S., Moser M., Metin A., Fried M., Tomlinson I. et al. (2009). Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 15, 306-312. doi:10.1038/nm.1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi J., Erickson H. P. and Springer T. A. (2001). C-terminal opening mimics ‘inside-out’ activation of integrin α5β1. Nat. Struct. Biol. 8, 412-416. doi:10.1038/87569 [DOI] [PubMed] [Google Scholar]
- Takagi J., Petre B. M., Walz T. and Springer T. A. (2002). Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110, 599-11. doi:10.1016/S0092-8674(02)00935-2 [DOI] [PubMed] [Google Scholar]
- Vinogradova O., Haas T., Plow E. F. and Qin J. (2000). A structural basis for integrin activation by the cytoplasmic tail of the α IIb-subunit. Proc. Natl. Acad. Sci. USA 97, 1450-1455. doi:10.1073/pnas.040548197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener K. L., Partridge A. W., Han J., Pickford A. R., Liddington R. C., Ginsberg M. H. and Campbell I. D. (2007). Structural basis of integrin activation by talin. Cell 128, 171-182. doi:10.1016/j.cell.2006.10.048 [DOI] [PubMed] [Google Scholar]
- Xiao T., Takagi J., Coller B. S., Wang J. H. and Springer T. A. (2004). Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 432, 59-67. doi:10.1038/nature02976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Ma Y. Q., Page R. C., Misra S., Plow E. F. and Qin J. (2009). Structure of an integrin alphaIIb β3 transmembrane-cytoplasmic heterocomplex provides insight into integrin activation. Proc. Natl. Acad. Sci. USA 106, 17729-17734. doi:10.1073/pnas.0909589106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauch R. L., Felsenfeld D. P., Kraeft S.-K., Chen L. B., Sheetz M. P. and Hemler M. E. (1997). Mutational evidence for control of cell adhesion through integrin diffusion/clustering, independent of ligand binding. J. Exp. Med. 186, 1347-1355. doi:10.1084/jem.186.8.1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F., Hu G., Taylor D., Ratnikov B., Bobkov A. A., McLean M. A., Sligar S. G., Taylor K. A. and Ginsberg M. H. (2010). Recreation of the terminal events in physiological integrin activation. J. Cell Biol. 188, 157-173. doi:10.1083/jcb.200908045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F., Kim C. and Ginsberg M. H. (2011). Molecular mechanism of inside-out integrin regulation. Journal of Thrombosis and Haemostasis 9 Suppl. 1, 20-25. doi:10.1111/j.1538-7836.2011.04355.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F., Petrich B. G., Anekal P., Lefort C. T., Kasirer-Friede A., Shattil S. J., Ruppert R., Moser M., Fässler R. and Ginsberg M. H. (2013). The mechanism of kindlin-mediated activation of integrin αIIbβ3. Curr. Biol. 23, 2288-2295. doi:10.1016/j.cub.2013.09.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylänne J., Chen Y., O'Toole T. E., Loftus J. C., Takada Y. and Ginsberg M. H. (1993). Distinct functions of integrin α and β subunit cytoplasmic domains in cell spreading and formation of focal adhesions. J. Cell Biol. 122, 223-233. doi:10.1083/jcb.122.1.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W., Leisner T. M., McFadden A. W., Wang Z., Larson M. K., Clark S., Boudignon-Proudhon C., Lam S. C. and Parise L. V. (2006). CIB1 is an endogenous inhibitor of agonist-induced integrin alphaIIbbeta3 activation. J. Cell Biol. 172, 169-175. doi:10.1083/jcb.200505131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Liu J., Jiang X., Haydar N., Zhang C., Shan H. and Zhu J. (2013). Modulation of integrin activation and signaling by α1/α1′-helix unbending at the junction. J. Cell Sci. 126, 5735-5747. doi:10.1242/jcs.137828 [DOI] [PubMed] [Google Scholar]
- Zhu J., Boylan B., Luo B.-H., Newman P. J. and Springer T. A. (2007). Tests of the extension and deadbolt models of integrin activation. J. Biol. Chem. 282, 11914-11920. doi:10.1074/jbc.M700249200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Luo B. H., Xiao T., Zhang C., Nishida N. and Springer T. A. (2008). Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol. Cell 32, 849-861. doi:10.1016/j.molcel.2008.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Luo B. H., Barth P., Schonbrun J., Baker D. and Springer T. A. (2009). The structure of a receptor with two associating transmembrane domains on the cell surface: integrin alphaIIbbeta3. Mol. Cell 34, 234-249. doi:10.1016/j.molcel.2009.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Choi W.-S., McCoy J. G., Negri A., Zhu J., Naini S., Li J., Shen M., Huang W., Bougie D. et al. (2012). Structure-guided design of a high-affinity platelet integrin αIIbβ3 receptor antagonist that disrupts Mg²⁺ binding to the MIDAS. Sci. Transl. Med. 4, 125ra32. doi:10.1126/scitranslmed.3003576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Zhu J. and Springer T. A. (2013). Complete integrin headpiece opening in eight steps. J. Cell Biol. 201, 1053-1068. doi:10.1083/jcb.201212037 [DOI] [PMC free article] [PubMed] [Google Scholar]