

Abstract
3-O-Sulfotransferase enzyme (sHS) from Litopenaeus vannamei was cloned and its substrate specificity was investigated against a number of GAG structures, including modified heparin polysaccharides and model oligosaccharides. For the heparin polysaccharides, derived from porcine intestinal mucosa heparin, sulfate groups were incorporated into glucosamine residues containing both N-sulfated and N-acetylated substitution within the regions of the predominant repeating disaccharide, either I-ANS or I-ANAc. However, the resulting polysaccharides did not stabilize antithrombin, which is correlated with anticoagulant activity. It was also shown that the enzyme was able to sulfate disaccharides, I2S-ANS and G-ANAc. The results further illustrate that 3-O-sulfation can be induced outside of the classical heparin-binding pentasaccharide sequence, show that 3-O-sulfation of glucosamine is not a sufficient condition for antithrombin stabilization and suggest that the use of this enzyme during HS biosynthesis may not occur as the final enzymatic step.
1. Introduction
Heparan sulfate (HS) and heparin are sulfated glycosamino-glycans (GAG) composed of repeating disaccharide units of (1 → 4)-linked α-D-glucosamine and uronic acid. Whereas HS disaccharides are predominantly formed by β-D-glucuronate and α-D-glucosamine that can be either N-acetylated or N-sulfated, heparin is more sulfated, composed mainly of α-L-iduronate 2-O-sulfate and α-D-glucosamine N,6-sulfate.1 The average levels of sulfation in HS are close to one per disaccharide, while in heparin, they are around 2.7 per disaccharide.2 These modifications occur in the Golgi apparatus via a series of N-deacetylases/N-sulfotransferases (NDSTs), sulfotransferases and C5-epimerases. HS and heparin biosynthetic processes were first described by Lindahl in 19773 and since then it has been assumed that the enzymatic processing, ultimately resulting in unique substitution patterns, occurs through a hierarchical sequence of enzymatic events3 where NDST is followed by C5-epimerase, 2-O-sulfotransferase, 6-O-sulfotransferase and, lastly, 3-O-sulfotransferase.
The presence of HS biosynthesis enzymes in organisms is related strongly to the emergence of multicellularity and tissue organization, being a characteristic of the eumetazoan lineage.4,5 Moreover, a correlation between the complexity of an organism and the number of HS sulfotransferase isoforms is evident. For instance, the primitive organism C. elegans has just one isoform for each of the five known sulfotransferases, while humans have four NDSTs, one C5-epimerase, one HS2ST, three HS6STs and seven HS3STs.4 Interestingly, rudimentary HS biosynthesis enzymes were found in unicellular and colony-forming organism M. brevicollis, suggesting that GAGs could play a key role in the emergence of multicellularity through extracellular organization and establishment of cell–cell communication.4
HS and heparin are known to regulate a wide range of physiological processes6 and heparin is employed for its pharmacological activity in cardiovascular medicine as an anticoagulant and antithrombotic drug7 since it modulates antithrombin (AT), the principal physiological inhibitor of coagulation in vertebrates. The sequence in heparin which is thought principally responsible for this activity corresponds to a pentasaccharide ANAc/NS,6S–G–ANS,3S,6S–I2S–ANS,6S (AGA*IA), found on average in one-third of the heparin chains.8 This specific sequence was described following fractionation of heparin by affinity chromatography, which revealed that AT high-affinity fractions had 3-O-suIfated glucosamine.9 From then on, this modification has been considered the key for AT-binding,10,11 since pentasaccharide sequences lacking 3-O-suIfated glucosamine have decreased in affinity for AT,11 even though the presence of a 3-O-group in central glucosamine is not essential to activate AT10 since other GAGs and non-GAG based structures are able to activate AT.12,13
Despite being unique and responsible for high-affinity AT-binding, different sequences and compounds, even non-carbohydrates, can exert the same effect.13–15 Furthermore, it is also important to highlight that under normal physiological conditions there is little circulating heparin, indicating that its biological function should be distinguished from its pharma-cological use.1 Furthermore, 3-O-sulfated glucosamine is yet to be found within commonly purified HS.
HS3ST is the largest sulfotransferase family in humans, although the simplest organism in which this enzyme has been reported is M. brevicollis4 suggesting that, evolutionarily, 3-O-suIfated glucosamine could be the most ancient and has played an important role in the emergence of cellular communication. In addition, species that lack AT-mediated coagulation have heparin-like molecules containing high levels of 3-O-sulfated glucosamine with negligible anticoagulation activity.16–19 These data bring to light questions regarding the importance of the 3-O-suIfate group in AT activation and its role regarding the biological functions of HS.
In a previous study, a heparin-like polysaccharide from the shrimp L. vannamei exhibited a higher proportion of 3-O-sulfated glucosamine (A*) than mammalian heparin, however, this compound presented low anticoagulation activity.19 Here, in order to better understand the role of 3-O-sulfotransferase in HS/heparin biosynthesis, the cloning, expression of 3-O-suIfo-transferase from L. vannamei and an investigation into substrate recognition are reported.
2. Materials and methods
2. 1. Materials
[35S]PAPS (3′phosphoadenosine-5′-phosphosuIfate-[35S], 2.3 Ci mmol−1) was purchased from PerkinElmer NEM (Waltham, MA, USA). Hyaluronic acid sodium salt (HA, from Streptococcus sp.) was purchased from Merck Millipore (Kenilworth, NJ, USA). Chondroitin 4-suIfate (C4S, from bovine trachea), chon-droitin 6-sulfate (C6S, from shark cartilage) and dermatan sulfate (DS, from porcine mucosa) were obtained from Sigma-Aldrich Co (St Louis, MO, USA). Heparan sulfate (HS) from bovine pancreas was prepared according to ref. 20. Heparin (Hep) from porcine mucosa was obtained from Bioiberica S.A. (BarceIona, CataIunya, Spain), whiIe chemicaIIy modified heparins were prepared as described in ref. 21. The two oligo-saccharides were synthesized as described in ref. 22.
2.2. Litopenaeus vannamei sulfotransferase (sHS) cloning
To clone the sulfotransferase (sHS) from shrimp L. vannamei, the expressing sequence tag (EST), named Contig 7734-v1, from the Marine Genomics Project database (http://mgnew.clemson.edu/) was used. This EST was used because it showed similarity to human HS3ST1 and HS3ST5 by Blast from NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Thus, a portion of the Contig 7734-v1 was used as a primer to amplify the catalytic domain of the enzyme from L. vannamei using the SMARTer Race 5′/3′ kit. The coding-sequence obtained was performed according to the manufacturer’s instructions. Finally, it was PCR-amplified using a forward primer (5′ GAAGATCTTCCGGAGGCTGCCCCAA 3′) and a reverse primer (5′ GAATTCGAACTTCAGCTGGCCTTAACG 3′). The PCR product was purified by agarose electrophoresis after digestion with BglII and EcoRI endonuclease enzymes, cloned in pRSET A and transformed into E. coli BL21 pLysS competent cells. The coding-sequence from shrimp was confirmed by Sanger sequencing in an Applied Biosystems 3130 Genetic Analyzer.
2.3. Protein expression and purification
After induction with 1 mM IPTG, the protein expression was carried out in LB medium at 37 °C for 1 h and 200 rpm. The bacterial pellet was suspended in 10 mL of Iysis buffer (50 mM NaH2PO4, pH 8.0, 500 mM NaCl, 1 mg mL−1 of Iysozyme), incubated in ice for 30 min and sonicated on ice using six 30 s bursts. Lysed cells were centrifuged (4000g for 20 min, 4 °C) and the supernatant was applied to a HisTrap HP column (GE Healthcare), previousIy equiIibrated with native binding buffer (50 mM NaH2PO4, pH 8.0, 500 mM NaCl, 10 mM imidazole). The column was exhaustively washed with native wash buffer (50 mM NaH2PO4, pH 8.0, 500 mM NaCl, and 40 mM imidazole) and the recombinant protein was eluted with native elution buffer (50 mM NaH2PO4, pH 8.0, 500 mM NaCl, 250 mM imidazole). Fractions were pooled and buffer-exchanged to phosphate-buffered saline using a PD-10 desalting column (GE Healthcare). The recombinant protein was stored at −20 °C.
2.4. In silica analysis
The similarity of HS/Hep sulfotransferases from humans to the cloned-sequence from L. vannamei was analyzed by MUSCLE (MultipIe Sequence Alignment) from EMBL-EBI (http://www.ebi.ac.uk/TooIs/msa/muscIe/). Furthermore, structure protein prediction was performed using a PHYRE2 Protein Fold Recognition Server program (http://www.sbg.bio.ic.ac.uk/phyre2/htmI/page.cgi? id=index).
2.5. Recombinant sHS activity assay
2.5.1. Glycosaminoglycan substrate.
Sulfotransferase activity was determined by incubating 40 μL of purified recombinant enzyme (0.05 μg pL−1) with 100 μg of acceptor substrates and 50 000 cpm [35S]PAPS in 100 μL of reaction buffer (50 mM imidazoIe, pH 7.0, 0.025 μg pL−1 protamine sulfate, 1 mM dithiothreitol). Acceptor substrates were HS, heparin (Hep), modified heparin: O,N-desuIfated-N-reacetylated (HepNAc), O,N-desuIfated-N-resuIfated (HepNSuIfo), N-desuIfated-N-reacetyIated heparin (HepdNSrNAc), C4S, C6S, DS and HA. Reaction mixtures were incubated at 37 °C overnight and the reaction was stopped by heating at 100 °C for 1 min. The 35S-Iabeled products were examined by agarose gel electrophoresis.
2.5.2. Oligosaccharide substrates.
The activity of sHS was also analyzed using two oligosaccharides (G–ANAc and I2S–ANS) as acceptor substrates in order to confirm the specificity of the recombinant enzyme to the heparan sulfate/heparin sequence and whether HS/Hep biosynthesis occurs under the hierarchical model. The reaction mixture contained 50 mM MES, pH 7.0, 10 mM MnCl2, 5 mM MgCl2, 0.5 μg of oligosaccharide, 50 000 cpm [35S]PAPS and 80 μL of recombinant enzyme in a final volume of 200 μL. After incubation at 37 °C overnight, the reaction was stopped by heating at 100 °C for 1 min. In the negative control of each reaction, the enzyme was substituted by water. 35S-labeled products were chromatographed in 500 μL DEAE-Sepharose using an NaCl gradient 0.02–2 M for 22 mL with a flow of 0.250 μL min−1 in an ÄKTA purifier system (GE Healthcare). Fractions (500 μL) were collected and the quantity of 35S-suIfated oligosaccharide was measured by liquid scintillation counting. The values for the blank reaction were subtracted from each run.
2.6. Agarose gel electrophoresis (PDA)
All reaction products were subjected to agarose gel electrophoresis as previously described by Dietrich and Dietrich, 1976.23 Briefly, 25 μg of each acceptor (5 μL) was applied to a 0.55% agarose gel in 0.05 M 1,3-diaminopropane-acetate buffer, pH 9.0 and subjected to electrophoresis at 100 V for 1 h. The gels were fixed with 0.1% cetyltrimethylammonium bromide (CETAVLON) solution for 2 h, dried and stained with toluidine blue solution (0.1% toluidine blue in 1% acetic acid in 50% ethanol), and destained with 1% acetic acid in 50% ethanol solution. Subsequently, the gels were exposed for three days to radiation sensitive films and developed in a Cyclone Storage Phosphor System (Packard Instrument Company Inc., Groningen, Netherlands).
2.7. NMR spectroscopy
Prior to NMR experiments, the 35S-labeIed HepNSulfo was purlified. Briefly, to the sample was added 90% trichloroacetic acid (10% of sample volume) and, after 30 min on ice, the sample was centrifuged and the supernatant was collected. 1 volumes (v/v) of methanol were added and, after 24 h at –20 °C, the precipitate formed was collected by centrifugation (10 000g for 15 min at 4 °C), dried and suspended in 0.5 mL deuterium oxide (D99.99%, Aldrich Chemistry, St Louis, MO, USA) containing 0.006% TSP 3-(trimethylsiIyl)propionic-2,2,3,3-d4 acid. The spectra were obtained with a Superconducting Fourier NMR Spectrometer (AVANCE III 600 MHz, Bruker Corporation) using a Triple Resonance Broadband Inverse (TBI) probe, at Instituito de Química, Unicamp, Campinas, SP, Brazil.
2.8. Differential scanning fluorimetry (DSF)
The capacity of 35S-IabeIed products to stabiIize antithrombin was analyzed as described by Lima et al., 2013.12 Human antithrombin (AT) (1 mg mL−1), previously purified from citrate plasma on a heparin-Sepharose column, was incubated with different substrates in the presence of SyproOrange™ dye (Invitrogen). Firstly, the dye was diluted in water (1 sypro: 50 water (v/v)) and 3.5 μL of this was added to the reaction mixture in PBS buffer. The dye has an excitation wavelength of 300 nm or 470 nm and emits at 570 nm when bound to hydrophobic residues. Different substrates used were 25 μg of unfractionated heparin (UFH) (10 mg mL−1), 62.5 μg of Arixtra pentasaccharide (ANs,6s–G2OH–ANs,6s,3s–I2s–ANs,6s–OMe) (12.5 mg mL−1), 25 μg of each modified heparin and 25 μg of 35S-labeled modified heparins, previously analyzed by PDA. Reaction mixtures were incubated at 31 °C for 2 min, and, then they were subjected to a step-wise temperature gradient from 32 to 85 °C in 0.5 °C steps. Between each temperature step, there was a 5 s incubation period to equilibrate samples. Reactions were developed at 7500 Real Time PCR system (Applied Biosystems) in triplicate. The final curves were generated employing the first derivative of the melting curves.
3. Results
3.1. Phylogenetic analysis of sHS from Litopenaeus vannamei
After cloning and sequencing the sHs from shrimp, the amino acid sequence was predicted using Expasy software: SIB Bioinformatics Resource Portal through the Translate tool (Fig. 1A and S1†). Fig. 1 shows the sHs domains responsible for the carbohydrate and PAPs binding, which are shown in red in the cloned region. Furthermore, comparative analyses were performed, including all HS sulfotransferases from Homo sapiens. These nucleotide and amino acid sequences were obtained from PubMed-NCBI and UniProteinKB databases respectively and aligned using MUSCLE (Fig. 1B and C). The analysis showed that the heparan sulfate 3-O-suIfotransferase family from Homo sapiens (hHs3sT) exhibited homology with sHs, the isoforms 1 and 5 being the closest, and are those thought to be responsible for anticoagulant Hs production. The protein 3D structure was modeled using PHYRE2 Protein Fold Recognition server, shown in Fig. 1D, which highlights the significant structural similarity between sHs and the human heparan sulfate 3-O-suIfotransferase isoform 5.
Fig. 1.
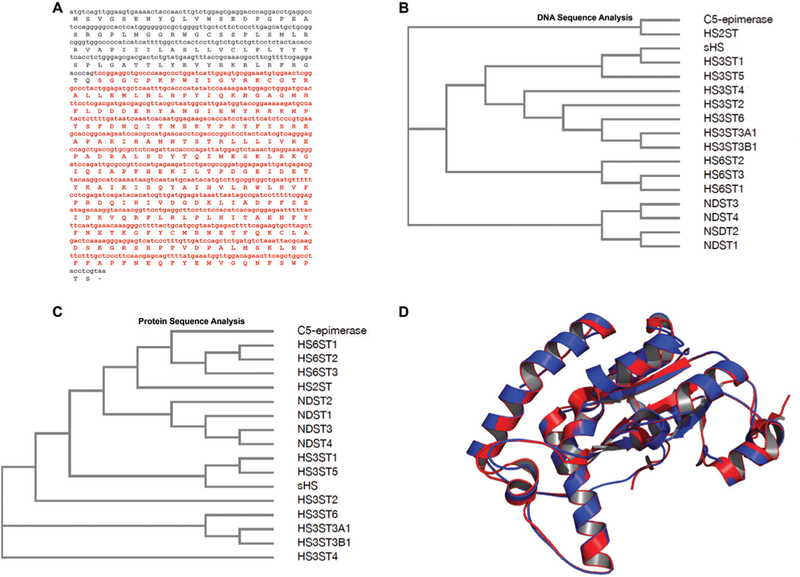
Composite DNA, predicted amino acid sequences and structural model for the shrimp Litopenaeus vannameisulfotransferase. (A) The nucleotide fragment as well as the amino acid sequence in red are related to enzymatic domains responsible for the carbohydrate and PAPS binding. This cDNA sequence (red) was cloned in pRSET A. Amino acid sequence was predicted by using Expasy (http://web.expasy.org/translate/). (B) Cladogram analysis of DNA sequences demonstrates that the isoforms 1 and 5 of 3-O-sulfotransferase (HS3ST) from Homo sapiensdisplay higher identity to sHS. (C) Cladogram analysis of amino acid sequences confirms that the highest similarity is among isoforms 1 and 5 of HS3ST to sHS. (D) Comparison between tertiary structures of sHS from shrimp L. vannamei(red) and HS3ST5 from Homo sapiens(blue). The 3D structure of sHS was modeled based on the human 3-O-sulfotransferase isoform 5 crystal structure (PDB #3BD9) using SWISS-MODEL.37 Structural alignment was performed on The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC. Abbreviations: NDST: N-deacetylase/N-sulfotransferase; HS2ST: 2-O-sulfotransferase; HS3ST: 3-O-sulfotransferase; the isoforms of each enzyme correspond to numbers from 1 to 6.
3.2. Substrate selectivity for sHS
3.2.1. Broad selectivity assays.
Heparan sulfate 3-O-sulfo-transferase transfers sulfate from PAPs (adenosine 3′-phos-phate 5′-phosphosulfate) to the specific 3-OH position of a glucosamine to generate 3-O-sulfated heparan sulfate. The activity assay was based on the transfer of radioactive sulfate from PAP[35S] to selected substrates. First, we tested the substrate selectivity of sHs towards various glycosaminoglycans. Only chemically modified heparins served as substrates for sHs (Fig. 2A) whereas chondroitin 4-sulfate, chondroitin 6-sulfate, dermatan sulfate and hyaluronic acid did not (Fig. S2†), indicating that the sHs modifies only Hs/heparin. Nonetheless, the absence of radioactivity in Hs and heparin may suggest that the 3-OH sites were already modified, resulting in a null sHS action. Furthermore, since N-desulfated-N-reacetylated heparin, which would be the common substrate for NDST, was not modified by sHS, the recombinant enzyme from L. vannamei belongs to the O-sulfotransferase family. Fig. 2A shows the activity assay on modified heparins where both sHS and recombinant human HS3ST5 were able to transfer sulfur to the chemically modified substrates, O,N-desul-fated-N-resulfated and O,N-desulfated-N-reacetylated heparin.
Fig. 2.
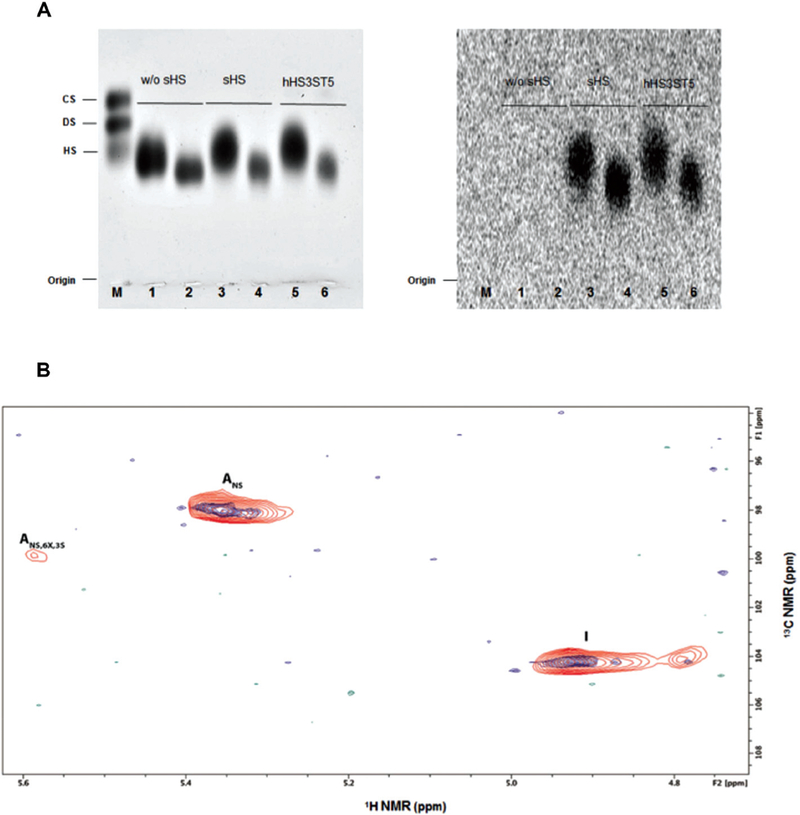
sHS substrate selectivity. (A) Activity assay on modified heparins. 1, 3 and 5: Substrate used was O,N-desulfated-N-resulfated heparin (25 of uronic acid); 2, 4 and 6: using as the substrate O,N-desulfated-N-reacetylated heparin (25 ĝ of uronic acid). In 1 and 2, the reaction mixture did not have the recombinant enzyme in their preparations. Left: PDA gel stained with toluidine blue. Right: PDA gel exposed for three days to radiation sensitive films. Both sHS enzyme as well as recombinant human HS3ST5 were able to transfer [35]S-sulfate to chemically modified heparins. M: Mixture of standard glycosaminoglycans containing chondroitin sulfate (CS), dermatan sulfate (DS) and heparan sulfate (HS) (5 ĝ each). (B) Description of compound HepNSulfo[35]S-sulfate, previously modified by sHS, by two-dimensional heteronuclear single quantum coherence (HSQC) NMR. The HepNSulfo control (blue) and the HepNSulfo[35]S-sulfate (red) spectra displayed similar components, whereas the HepNSulfo[35]S-sulfate showed the presence of 3-O-sulfated glucosamine (ANS,6X,3S), indicating that sHS is indeed a 3-O-sulfotransferase. Abbreviations: I: iduronic acid, Ans: glucosamine N-sulfated and ANS,6X,3S: glucosamine N,3-sulfated, where 6X could be 6OH or 6S.
3.2.2. Determination of sulfation site by NMR.
Proton and carbon NMR chemical shifts of HepNSulfo, used as a control, and HepNSulfo[35]S-sulfate, previously subjected to the sHS action, were assigned by Heteronuclear Single Quantum Coherence (HSQC) spectroscopy experiments. The HepNSulfo[35]S-sulfate exhibited signals similar to those ascribed to the control. Nevertheless, only the HepNSulfo[35]S-sulfate showed a signal at 5.5/99.5 ppm that corresponds to ANS 6X 3S, indicating that sHS was able to transfer the sulfate from PAPS to C3-glucosamine (Fig. 2B).
3.3. 3-O-Sulfotransferase in the HS biosynthesis pathway
3.3.1. Hierarchical vs. non-hierarchical biosynthesis.
We further tested the sHS substrate recognition using two octasaccharides (G-ANAc and I2S-ANS) as substrates for the enzyme. As observed in Fig. 3, both octasaccharides were modified by the sHS and, together with our previous findings, show that sHS does not require either 2-O-, 6-O- or N-sulfate in heparin/HS to modify the polymer and, surprisingly, N-acetylation does not block 3-O-sulfation as anticipated by the hierarchical biosynthetic process where 3-O-sulfation would happen as the final modification step.3 The data show that 3-O-sulfation can occur in distinct biosynthetic steps either being the last HS sulfotransferase in the biosynthesis process or the first one in a non-hierarchical way, according to the oligosaccharides tested.
Fig. 3.
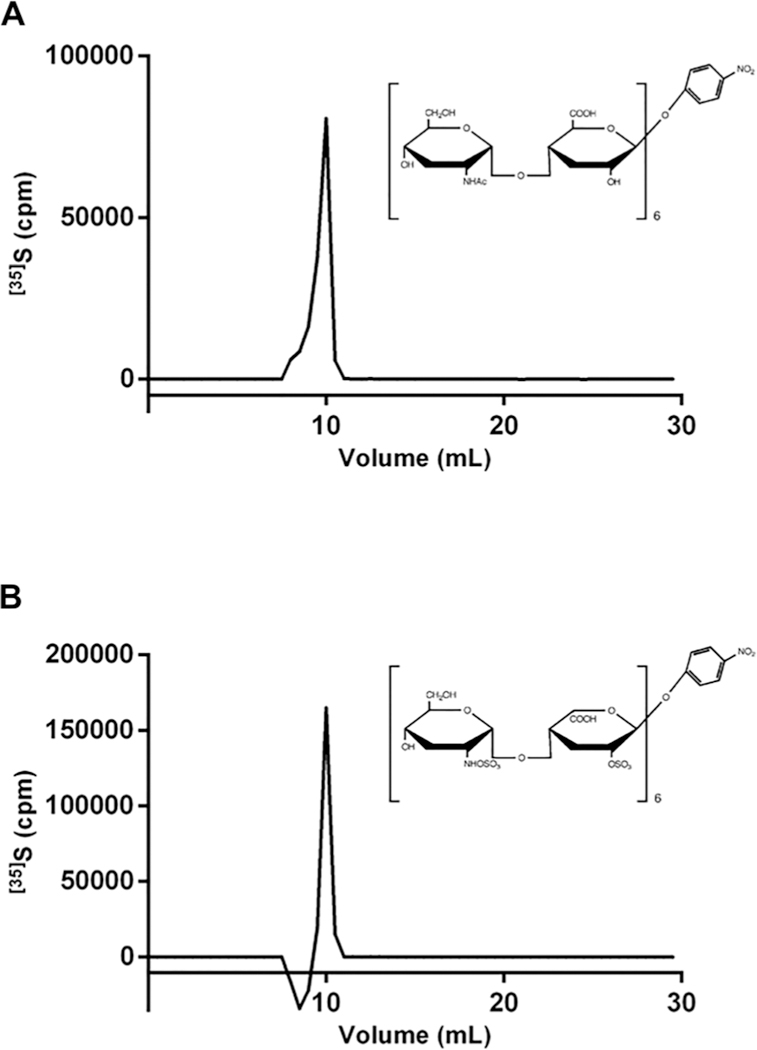
Analysis of 3-O-sulfation oligosaccharides by sHS. [35]S-sulfate products were analyzed using a DEAE-Sepharose column and the 35S radioactivity was measured by liquid scintillation counting. (A) The oligosaccharide Glc-GlcNAc was used as a substrate for the reaction. (B) The oligosaccharide IdoA2S-GlcNS was the acceptor for the sHS activity reaction. The blank run value was subtracted from each test compound.
3.3.2. Tree structure for 3-O-sulfation.
Studies employing the chemoenzymatic approach have revealed that different isoforms of HS3ST could sulfate HS through different pathways in biosynthesis, since HS3ST1 can only work after the 6-O-sulfation step, while HS3ST3 must precede the 6-O-sulfation modification to generate the ANS,3S,6S glucosamine residue.15 In Fig. 4, different 3-O-sulfated heparin structures are shown. The classical HS biosynthetic route is illustrated by the penta-saccharide production, which is synthesized through the hierarchical sequence of enzymatic events (NDST → epimerase → HS2ST → HS6ST → HS3ST) (Fig. 4, sequence 1). Indeed, the hierarchical pathway encapsulates the pentasaccharide biosynthesis yet, and other 3-O-sulfated structures found within mammalian heparin cannot (Fig. 4, sequences 2 and 3).24 Furthermore, sequences from invertebrates of higher proportions of 3-O-sulfation, within “unusual” saccharide sequences and devoid of anticoagulant properties have been found (Fig. 4, sequences 4 and 5).17,19 A tree structure for the biosynthesis of the major heparin and HS disaccharides has been proposed.25 Here, this scheme is expanded and, again, shows that heparin and HS biosynthesis as a whole cannot be described fully using the original description proposed by Lindahl.3
Fig. 4.
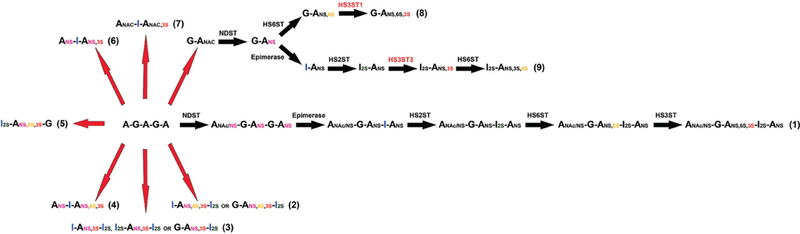
Different 3-O-sulfated heparin sequences found in animals and produced chemoenzymatically. (1) A minimum sequence of heparin (pentasaccharide) involved in AT-binding. It has been proposed that the pentasaccharide is synthesized according to the classical biosynthetic pathway, in which the enzymatic events happen through a hierarchical sequence, as described by Lindahl, 1977.3 (2 and 3) Sequences present in heparin described in Lindahl, 1994.24 (4) Heparin sequence found in clams that does not correlate with affinity for AT.17 (5) Sequence described in shrimp L. vannameithat has negligible anticoagulant activity despite its high affinity for AT and unusually higher proportion of 3-O-sulfated residues.19 (6 and 7) Scheme for 3-O-sulfated heparin sequences described by this study. The starting material for chemical modifications was porcine intestinal mucosa heparin, the schemes show iduronate rather than glucuronate once heparin does have significant higher proportions of iduronate. (8 and 9) Different oligosaccharide substrates required by HS3ST isoforms. While HS3ST3 must precede the 6-O-sulfation to generate the I2S-ANS3S6S, HS3ST1 can only work after the 6-O-sulfation step.15
3.4. Thermostabilizing effects of sHS-treated heparins on AT
As described by Lima et al., 2013,12 the anticoagulant activity of heparin is correlated with the extent to which the complex between the polysaccharide and AT is stabilised, rather than by the secondary structural changes induced in AT by heparin binding. In order to analyze whether the addition of 3-O-sulfate groups induced further AT stabilization, AT was incubated with a range of different substrates and thermostabilization was measured using differential scanning fluorimetry (DSF) (Fig. 5). Only unfractionated heparin (UFH) and the pentasaccharide sequence, classical anticoagulant drugs, stabilized AT (61.75 °C and 65.75 °C respectively) whereas heparins[35]S-sulfate, previously modified by sHS, were unable to do so (Fig. 5). This shows that a 3-O-sulfate group can be introduced into the polysaccharide but, this does not necessarily bestow stabilization on AT, or imply activity.
Fig. 5.
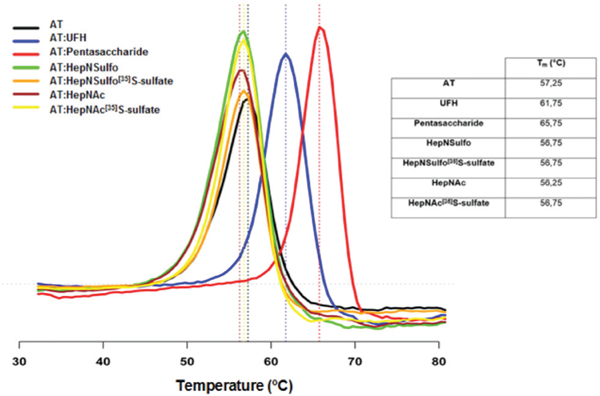
Antithrombin stabilization assay with substrates modified by sHS. UFH (unfractionated heparin) and pentasaccharide (Arixtra™) were used as positive control, since they are already described as anticoagulant drugs. AT was incubated with different substrates and subjected to a step-wise temperature gradient. The melting temperatures for each condition are shown. Only the UFH and the pentasaccharide were able to stabilize AT, whereas none of the compounds (HepNSulfo[35]S-sulfate and HepNAc[35]S-sulfate) modified by the sHS and their counterpart (HepNSulfo and HepNAc) did.
4. Discussion
The first description of 3-O-sulfated glucosamine residues in heparin was suggested by Danishefsky et al., 196926 when, according to methylation analyses, the authors proposed that heparin contained small proportions of 3,6-di-O-sulfoglucos-amine. Owing to the fact that 3-O-sulfation is a rare modification in heparin27 and also in HS28 coupled to the absence of widely available methodologies able to evaluate such a modification, it is challenging to determine the real role and extent of this modification in HS and heparin biosynthetic and biological properties.
Since the discovery of 3-O-sulfate groups in heparin with high-affinity for antithrombin,9 this modification has been considered crucial to the pharmacological role of heparin since the presence of 3-O-sulfation in the central glucosamine of the pentasaccharide (AGA*IA) increases 1000-folds the heparin binding for AT.11 Nonetheless, besides the presence of 3-O-sulfated glucosamine, several other structural features of heparin have been reported to influence the AT activity.11,15,29–31
In this study, we cloned and characterized a sulfotransferase from the shrimp L. vannamei similar to the isoforms 1 and from Homo sapiens. The results suggest that this cloned enzyme is able to transfer sulfate to the C3 position of glucosamine and, since these isoforms are responsible for their production,32,33 also produce anticoagulant polysaccharides. The identity of the 3-O-sulfotransferase was confirmed by radiolabeling activity assays and NMR spectroscopy.
According to the classical HS biosynthesis,34 HS chain modification occurs hierarchically, meaning that 3-O-sulfo-transferase is the last enzyme to modify the HS chains. Nevertheless, both HepNAc or G–ANAc compounds, which show only N-acetylated glucosamine in their structure, were modified by sHS, indicating that the HS sulfotransferases could act on substrates independent of this sequential route,15,25 which emphasizes that 3-O-sulfotransferase works at different steps of the biosynthetic process. It is important to highlight that organisms such as M. brevicollis, which were the first organisms to exhibit HS sulfotransferases, have only HS2ST and HS3ST. Furthermore, it has also been suggested that the epimerase enzyme appears evolutionarily later in a development stage.4 Hence, 3-O-sulfated heparin/HS can be biosynthesized through different pathways.
It has been demonstrated that the high affinity of heparin towards AT requires a pentasaccharide sequence that contains a 3-O-sulfate group in a central glucosamine unit.11,35 However, our results show that the 3-O-sulfated chemically modified heparins were not able to further stabilize AT, compared to their untreated counterparts, suggesting that the presence or abundance of the 3-O-sulfation is not, in itself, sufficient to provide anticoagulant activity.10,19 One may argue that the 3-O-suIfation of the studied polysaccharides is too low to promote AT stabilization but it is important to highlight that these modifications themselves, even within pharmaceutical heparins, are indeed low and, in our work, they were high enough to be detected by NMR, that is, at least 2–3%. Moreover, previous studies have already shown that the presence of 3-O-sulfated groups in heparan is not essential for normal hemostasis, since Hs3t1−/− knockout mice did not exhibit a pro-coagulant phenotype,36 questioning the role of this modification: other heparin features are relevant for such events.19
In summary, our data show that 3-O-suIfate groups are not solely responsible for AT stabilization and that 3-O-sulfotransferases can work in a non-hierarchical fashion during the HS/heparin biosynthetic process.
Supplementary Material
Acknowledegements
The authors wouId Iike to thank CNPq (grant number 442357/20141), CAPES and FAPESP (grant number 2015/0872–3) for financial support.
Abbreviations
- I
α-l-Iduronate
- I2S
Iduronic acid-2-O-suIfate
- G
β-d-Glucuronate
- A*
Glucosamine-3-O-sulfate
- ANAc
N-Acetyl glucosamine
- ANAc,3S
N-Acetyl glucosamine-3-O-sulfate
- ANS
Glucosamine-N-sulfate
- ANS,6S
N-Sulfated glucosamine-6-O-sulfate
- ANS,6S,3S
N-Sulfated glucosamine-6,3-O-sulfate
- NDST
N-Deacetylase/N-sulfotransferase
- HS2ST
Heparan sulfate 2-O-sulfotransferase
- HS6ST
Heparan sulfate 6-O-sulfotransferase
- HS3ST
Heparan sulfate 3-O-sulfotransferase
- HS3ST1
Heparan sulfate 3-O-sulfotransferase 1
- HS3ST5
Heparan sulfate 3-O-sulfotransferase 5
- sHS
Sulfotransferase from shrimp L. vannamei
- hHS3ST
Heparan sulfate 3-O-sulfotransferase from Homo sapiens
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob01533j
Conflicts of interest
There are no confIicts to decIare.
References
- 1.Meneghetti MC, Hughes AJ, Rudd TR, Nader HB, PoweII AK, Yates EA and Lima MA, J. R. Soc., Interface, 2015, 12, 0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Toida T, Yoshida H, Toyoda H, Koshiishi I, Imanari T, HiIeman RE, Fromm JR and Linhardt RJ, Biochem. J, 1997, 322(Pt 2), 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LindahI U, Upsala JMedSci, 1977, 82, 78–79. [DOI] [PubMed] [Google Scholar]
- 4.Ori A, WiIkinson MC and Fernig DG, J. Biol. Chem, 2011, 286, 19892–19904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Medeiros GF, Mendes A, Castro RA, Bau EC, Nader HB and Dietrich CP, Biochim. Biophys. Acta, 2000, 1475, 287–294. [DOI] [PubMed] [Google Scholar]
- 6.Bishop JR, Schuksz M and Esko JD, Nature, 2007, 446, 1030–1037. [DOI] [PubMed] [Google Scholar]
- 7.Jaques LB, Pharmacol. Rev., 1979, 31, 99–166. [PubMed] [Google Scholar]
- 8.LindahI U, Backstrom G, Hook M, Thunberg L, Fransson LA and Linker A, Proc. Natl. Acad. Sci. U. S. A, 1979, 76, 3198–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LindahI U, Backstrom G, Thunberg L and Leder IG, Proc. Natl. Acad. Sci. U. S. A, 1980, 77, 6551–6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richard B, Swanson R and OIson ST, J. Biol. Chem, 2009, 284, 27054–27064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atha DH, Lormeau JC, Petitou M, Rosenberg RD and Choay J, Biochemistry, 1985, 24, 6723–6729. [DOI] [PubMed] [Google Scholar]
- 12.Lima MA, Hughes AJ, Veraldi N, Rudd TR, Hussain R, Brito AS, Chavante SF, Tersariol II, Siligardi G, Nader HB and Yates EA, MedChemComm, 2013, 4, 870–873. [Google Scholar]
- 13.Henry BL, Connell J, Liang A, Krishnasamy C and Desai UR, J. Biol. Chem, 2009, 284, 20897–20908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brito AS, Cavalcante RS, Palhares LC, Hughes AJ, Andrade GP, Yates EA, Nader HB, Lima MA and Chavante SF, Carbohydr. Polym, 2014, 99, 372–378. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Hsieh PH, Xu Y, Thieker D, Chai EJ, Xie S, Cooley B, Woods RJ, Chi L and Liu J, J. Am. Chem. Soc, 2017, 139, 5249–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Straus AH, Sant’anna OA, Nader HB and Dietrich CP, Biochem. J, 1984, 220, 625–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pejler G, Danielsson A, Bjork I, Lindahl U, Nader HB and Dietrich CP, J. Biol. Chem, 1987, 262, 11413–11421. [PubMed] [Google Scholar]
- 18.Dietrich CP, Nader HB, de Paiva JF, Santos EA, Holme KR and Perlin AS, Int. J. Biol. Macromol, 1989, 11, 361–366. [DOI] [PubMed] [Google Scholar]
- 19.Chavante SF, Brito AS, Lima M, Yates E, Nader H, Guerrini M, Torri G and Bisio A, Carbohydr. Res, 2014, 390, 59–66. [DOI] [PubMed] [Google Scholar]
- 20.Dietrich CP and Nader HB, Biochim. Biophys. Acta, 1974, 343, 34–44. [DOI] [PubMed] [Google Scholar]
- 21.Yates EA, Santini F, Guerrini M, Naggi A, Torri G and Casu B, Carbohydr. Res, 1996, 294, 15–27. [DOI] [PubMed] [Google Scholar]
- 22.Liu R, Xu Y, Chen M, Weiwer M, Zhou X, Bridges AS, DeAngelis PL, Zhang Q, Linhardt RJ and Liu J, J. Biol. Chem, 2010, 285, 34240–34249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dietrich CP and Dietrich SM, Anal. Biochem, 1976, 70, 645–647. [DOI] [PubMed] [Google Scholar]
- 24.Lindahl U, Lidholt K, Spillmann D and Kjellen L, Thromb. Res, 1994, 75, 1–32. [DOI] [PubMed] [Google Scholar]
- 25.Rudd TR and Yates EA, Mol. BioSyst, 2012, 8, 1499–1506. [DOI] [PubMed] [Google Scholar]
- 26.Danishefsky I, Steiner H, Bella A and Friedlander A, J. Biol. Chem, 1969, 244, 1741–1745. [PubMed] [Google Scholar]
- 27.Thacker BE, Xu D, Lawrence R and Esko JD, Matrix Biol, 2014, 35, 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tersariol IL, Ferreira TM, Medeiros MG, Porcionatto MA, Moraes CT, Abreu LR, Nader HB and Dietrich CP, Braz. J. Med. Biol. Res, 1994, 27, 2097–2102. [PubMed] [Google Scholar]
- 29.Guerrini M, Guglieri S, Casu B, Torri G, Mourier P, Boudier C and Viskov C, J. Biol. Chem, 2008, 283, 26662–26675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guerrini M, Elli S, Mourier P, Rudd TR, Gaudesi D, Casu B, Boudier C, Torri G and Viskov C, Biochem. J, 2013, 449, 343–351. [DOI] [PubMed] [Google Scholar]
- 31.Guerrini M, Mourier PA, Torri G and Viskov C, GlycoconjugateJ, 2014, 31, 409–416. [DOI] [PubMed] [Google Scholar]
- 32.Datta P, Li G, Yang B, Zhao X, Baik JY, Gemmill TR, Sharfstein ST and Linhardt RJ, J. Biol. Chem, 2013, 288, 37308–37318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duncan MB, Chen J, Krise JP and Liu J, Biochim. Biophys. Acta, 2004, 1671, 34–43. [DOI] [PubMed] [Google Scholar]
- 34.Lindahl U, Kusche M, Lidholt K and Oscarsson LG, Ann. N. Y. Acad. Sci, 1989, 556, 36–50. [DOI] [PubMed] [Google Scholar]
- 35.Petitou M and van Boeckel CA, Angew. Chem., Int. Ed, 2004, 43, 3118–3133. [DOI] [PubMed] [Google Scholar]
- 36.Shworak NW, HajMohammadi S, de Agostini AI and Rosenberg RD, Glycoconjugate J, 2002, 19, 355–361. [DOI] [PubMed] [Google Scholar]
- 37.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L and Schwede T, Nucleic Acids Res, 2014, 42, W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.