

Abstract
Background
Beta cell function in type 1 diabetes is commonly assessed as the average plasma C-peptide concentration (CPAVE) following a mixed meal. Monitoring of disease progression and response to disease-modifying therapy would benefit from a simpler, more convenient and less costly measure. Therefore, we determined if CPAVE could be reliably estimated from routine clinical parameters.
Method
Clinical and fasting biochemical data from eight randomised therapy trials involving participants with recently-diagnosed type 1 diabetes were used to develop and validate linear models to estimate CPAVE and to test their accuracy in estimating loss of beta cell function and response to immune therapy.
Results
A model based on disease duration, body mass index, insulin dose, HbA1c, fasting plasma C-peptide and fasting plasma glucose most accurately estimated loss of beta cell function (area under ROC 0.89; 95% CI 0.87, 0.92) and was superior to the commonly used insulin dose-adjusted HbA1c (IDAA1C) measure (area under ROC 0.72; 95% CI 0.68, 0.76). Model-estimated CPAVE (CPEST) reliably identified treatment effects in randomised trials. CPEST, compared to CPAVE, required only a modest (up to 17%) increase in sample size for equivalent statistical power.
Conclusion
CPEST, approximated from six parameters at a single time-point, accurately identifies loss of beta cell function in type 1 diabetes and is comparable to CPAVE for identifying treatment effects. CPEST could serve as a convenient and economical measure of beta cell function in the clinic and as a primary outcome measure in trials of disease-modifying therapy in type 1 diabetes.
Keywords: Adult, Beta cell function, Children, Clinical trial, Linear model, Immune therapy, Immune Tolerance Network, TrialNet, Type 1 diabetes
Introduction
Therapies targeting pancreatic islet autoimmunity are being tested for their ability to preserve insulin-secreting beta cells and modify the natural history of type 1 diabetes after diagnosis [1]. The widely accepted measure of their efficacy is the average plasma C-peptide concentration during the first two hours of a mixed meal test (CPAVE) [2]. However, the measurement of CPAVE requires ingestion of a liquid meal and at least seven venous blood samples. A more convenient measure would streamline the assessment of beta cell function, particularly when disease-modifying therapies enter routine clinical practice.
In clinical trials, the biologic agents rituximab, teplizumab and abatacept have been shown to improve beta cell function for at least one year in people with recently-diagnosed type 1 diabetes [3–5]. Improved CPAVE in these trials was associated with a decrease in insulin requirement and in HbA1c, suggesting these routine clinical measures may be useful surrogates of beta cell function. Indeed, insulin dosage and HbA1c are used to calculate ‘insulin dose-adjusted HbA1c’ (IDAA1C), which identifies type 1 diabetes children with residual beta cell function [6, 7]. Other studies in children and adults at high risk of developing type 1 diabetes have shown that HbA1c, age and body mass index (BMI) correlate with the C-peptide response to oral glucose [8–10], again suggesting that these routine measures could also serve as useful surrogates of beta cell function in the clinic.
We aimed to develop a simple and reliable model that could accurately estimate CPAVE, based on a combination of routine clinical measures and fasting plasma C-peptide (FCP). Data from eight trials involving people with recently-diagnosed type 1 diabetes [3, 4, 11–16] were used to build predictive models to approximate CPAVE and derive estimates of variability for use in future trial design.
Methods
Study participants gave informed consent if adult and assent if aged under 18 years. All studies were approved by the responsible ethics committee and were carried out in accordance with the Declaration of Helsinki as revised in 2008. Clinical and biochemical data from the TrialNet (TN)-02, −05, −08, −09 and −14 clinical trials (Table 1) [3, 11–13] were extracted from the TrialNet data repository in April 2014. In all of these trials, predominantly white participants were assessed at 0, 3, 6 and 12 months after enrolment and, for TN-08 and TN-14, also at 9 months. Additional data from the Immune Tolerance Network (ITN)-27, −28 and −45 trials [14–16] were extracted in February 2016 and comprised clinical and biochemical measures obtained at the 0-, 6- and 12-month time points. Data from Australian adults with recently-diagnosed type 1 diabetes participating in an ongoing clinical trial of empagliflozin in recently-diagnosed type 1 diabetes (ACTRN12617000016336) were obtained April 2018. Plasma C-peptide concentrations in TrialNet and ITN trials were determined to sensitivities of 0.017 and 0.05nmol/L with TOSOH 2000 and TOSOH 1800 autoanalysers (TOSOH, South San Francisco, CA), respectively. In Australia, C-peptide and HbA1c were measured by Melbourne Health Pathology (Parkville, Australia) using ARCHITECT (Abbott, Wiesbaden, Germany) and Ultra2 (Primus Diagnostics, Kansas City, MO) kits respectively.
Table 1.
Baseline characteristics of participants aged less than 21 years according to trial and treatment group
| Trial | TN-02 | TN-05 | TN-08 | TN-09 | TN-14 | ITN-27 | ITN-28 | ITN-45 |
|---|---|---|---|---|---|---|---|---|
| Registration number | NCT00100178 | NCT00279305 | NCT00529399 | NCT00505375 | NCT00947427 | NCT00129259 | NCT00515099 | NCT00965458 |
| Treatment(s) | Mycophenolate and daclizumab | Rituxumab | GAD s.c. immunisation | Abatacept | Cankinumab | Teplizumab | Anti-thymocyte globulin | Alefacept |
| Main Trial Outcome | No preservation of CPAVE | Preserved CPAVE at 12 months | No preservation of CPAVE | Preserved CPAVE at 12 months | No preservation of CPAVE | Preserved CPAVE at 12 months | No preservation of CPAVE | No preservation of CPAVE |
| N | 75 | 58 | 105 | 95 | 66 | 73 | 38 | 29 |
| Percent males | 60 | 64 | 52 | 57 | 53 | 59 | 63 | 59 |
| Percent Hispanic or Latino | 4 | 5 | 8 | 6 | 6 | 3 | 6 | 4 |
| Age (years) | 13.5±3.0 | 13.8±2.7 | 12.7±4.3 | 12.6±3.7 | 11.8±3.6 | 12.1±2.7 | 15.9±2.5 | 16.1±2.1 |
| Body mass index (kg/m2) | 20.6±3.4 | 21.4±4.2 | 20.0±3.5 | 20.5±4.4 | 20.3±5.0 | 19.5±4.0 | 22.4±2.9 | 22.5±5.7 |
| Diabetes duration (days) | 54±21 | 63±22 | 63±18 | 60±18 | 49±21 | 49±7 | 50±20 | 51±23 |
| HbA1c (mmol/mol) | 61±15 | 56±12 | 49±11 | 48±9 | 53±12 | 58±12 | 51±12 | 55±16 |
| HbA1c (percentage units) | 7.7±1.4 | 7.3±1.1 | 6.6±1.0 | 6.5±0.8 | 7.0±1.1 | 7.5±1.1 | 6.8±1.1 | 7.2±1.5 |
| Insulin dose (U/kg) | 0.27±0.26 | 0.42±0.21 | 0.40±0.24 | 0.40±0.26 | 0.38±0.25 | 0.42±0.27 | 0.41±0.26 | 0.38±0.19 |
| CPAVE (nmol/L) | 0.68±0.29 | 0.75±0.39 | 0.71±0.30 | 0.75±0.40 | 0.64±0.33 | 0.70±0.31 | 0.98±0.45 | 0.83±0.41 |
Continuous data are mean±SD
After receipt of the archived data, missing weight, height, insulin dose and HbA1c values were imputed where possible by filling backward or forward from the nearest time point (if within 1 month) or by averaging values either side of the missing value. Undetectable C-peptide concentrations observed in TrialNet and ITN datasets were assigned values of half of the lower limit of detection. Because daily insulin requirements are ~20% lower with insulin pump compared to injection therapy [17], the daily insulin dose of TrialNet participants who reported using insulin pumps was multiplied by 1.25.
Correlation and receiver-operator curve (ROC) analyses were performed using Prism software (v6.0g for Mac; GraphPad, CA). Data modelling was performed using R software v3.3.2 (www.r-project.org). Half of the participants aged<21 years at baseline were randomly assigned to train the Linear Mixed Models to determine the estimated CPAVE (CPEST) and a Validation Dataset, comprising data from the remaining participants aged<21 years at baseline, was used to identify the best models. CPAVE was log-transformed after adding 1 [18] and eight covariates were chosen for inclusion in the prediction model: age, sex, body mass index (BMI), diabetes duration, insulin dose per kilogram body weight, fasting plasma C-peptide (FCP), fasting plasma glucose (FG) and HbA1c. Participant ID was added as a random effect to account for the repeated measurements from the same individual. The ‘dredge’ function in the MuMIn library (v1.15.6) was used to construct 256 models from all possible combinations of variables and these models were ranked by Akaike Information Criterion (AIC), corrected for a finite sample size. To validate the rankings of the models, the lmer function in the lme4 library (v1.1–13) was used to rebuild the models in the Validation Dataset based on the relevant inputs, thereby enabling their AIC values to be determined. To compare treatment arms of clinical trials, mixed models were fitted using lmer with a random intercept per participant and adjusted for sex, age and baseline loge(CPAVE+1) or loge(CPEST+1). The lmer-Test package was used to calculate p values based on F statistics for treatment comparisons.
Power calculations for the comparison of two groups with equal variance were performed using placebo-group data from the Validation Dataset and Stata (v14.2) software (StataCorp LLC, TX). They were based on the mean and standard deviation (SD) of the loge(CPAVE+1) values and a conservative approximation of the SD of loge(CPEST+1) values, calculated by combining the variance of loge(CPAVE+1) values with an estimated variance of the difference between the loge(CPAVE+1) and loge(CPEST+1) values according to the formula:
A standard trial design that assumed a treatment effect of 50% increase in loge(CPAVE+1) at 12 months, two-tailed α= 0.05, power = 0.8 and 2:1 (active:placebo) randomisation was used to estimate the required number of participants.
Results
Developing and validating equations to interpolate beta cell function
The baseline characteristics of participants whose data were used to develop the models are presented according to clinical trial and treatment assignment in Table 1. Initially, we used data from participants aged less than 21 years to fit and test linear models for three reasons: i) this age group accounts for over 75% of classic type 1 diabetes presentations [19]; ii) beta cell function declines more slowly in older people [20, 21]; and iii) preservation of beta cell function is more characteristic of younger participants in trials of biologic agents [22]. Half of the participants were randomly assigned to train linear models to estimate CPAVE using one or more of the eight input variables of age, sex, body mass index (BMI), diabetes duration, insulin dose per kilogram, fasting C-peptide (FCP), fasting glucose (FG) and HbA1c. Based on one to eight predictor variables, the Akaike Information Criterion (AIC) was used to identify the most accurate models, hereafter referred to as M1 to M8. The coefficients and associated standard errors of the variables included in the eight models are provided in ESM Table 1. Data from the remaining half of the participants were used to validate the models. Model 6 (M6), which is based on BMI, diabetes duration, insulin dose per kilogram, FCP, FG and HbA1c, was chosen for subsequent testing because its AIC was lowest in the Validation Dataset (Figure 1). Within the Validation Dataset, M6-modelled CPAVE (hereafter called CPEST) and observed CPAVE were strongly correlated (r2=0.816, p<0.001). The equation for M6 is loge(CPEST +1) = 0.317 + 0.00956×BMI(kg/m2) - 0.000159×duration(days) + 0.710×FCP(nmol/l) - 0.0117×FG(mmol/l) - 0.0186×HbA1c(%) - 0.0665×insulin(U/kg) (ESM Method file).
Figure 1. Performance characteristics of eight models to estimate loge(CPAVE+1) from single-time point data.
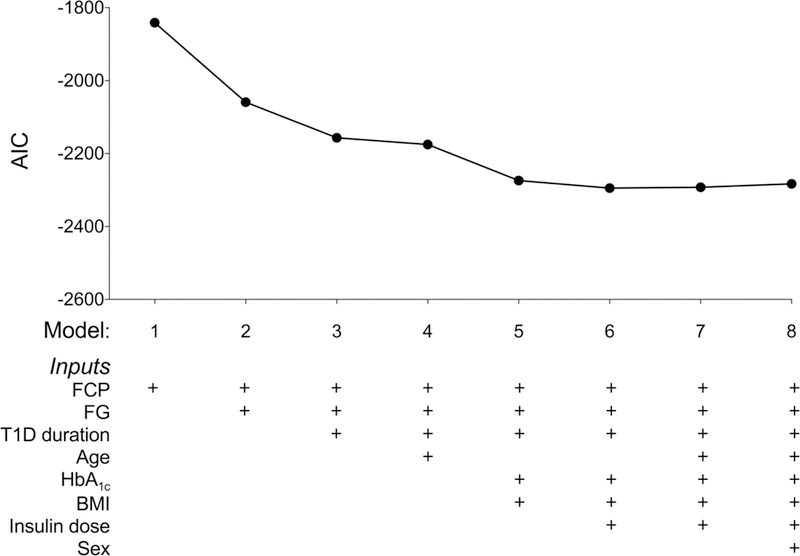
The components of each model are indicated below the graph of the Akaike Information Criterion (AIC) against the number of model variables in the context of the Validation Dataset. Model 6 was used to calculate CPEST values. FCP: fasting C-peptide; FG: fasting glucose; BMI: body mass index.
Because M6 did not require age as an input, we determined if it might also be accurate in the 150 trial participants aged over 21 years whose data were not included in either the Training or Validation datasets (baseline characteristics presented in ESM Table 2). Correlation analysis of data from 554 meal tests performed during the first trial year again demonstrated a strong correlation between CPAVE and CPEST (r2=0.729, p<0.001). Strong agreement between CPAVE and CPEST (r2=0.869, p<0.001) was also observed when M6 was applied to data from 31 meal tests from 10 participants (3 females, 7 males, aged 18 to 37 years at diagnosis; ESM Table 3) in an ongoing Australian trial of empagliflozin in recently-diagnosed type 1 diabetes.
Applying CPEST to clinical practice
Receiver-operator curve (ROC) analysis of the Validation Dataset was performed to determine how accurately CPEST identified significant loss of beta cell function at 3, 6 and 12 months after clinical trial entry, defined as a decrease of 7.5% or more of the baseline CPAVE [20, 23]. The ROC curves (Figure 2) show areas under the curve ranging from 0.86 (95% CI 0.81, 0.91) to 0.91 (95% CI 0.87, 0.95). When tested for the ability to identify significant loss of beta cell function at 3, 6 and 12 months compared to baseline, CPEST furnished an area under the ROC (AUROC) of 0.89 (95% CI 0.87, 0.92). The corresponding AUROC for trial participants aged over 21 years was 0.88 (95% CI 0.84, 0.91). We also determined how accurately insulin dose-adjusted HbA1c (IDAA1C), an extant clinical measure of beta cell function [6], identified trial participants who had lost significant beta cell function. The AUROC of the ratio of baseline to 3-, 6- and 12-month IDAA1C was markedly lower at 0.72 (95% CI 0.68, 0.76).
Figure 2. CPEST accuracy.
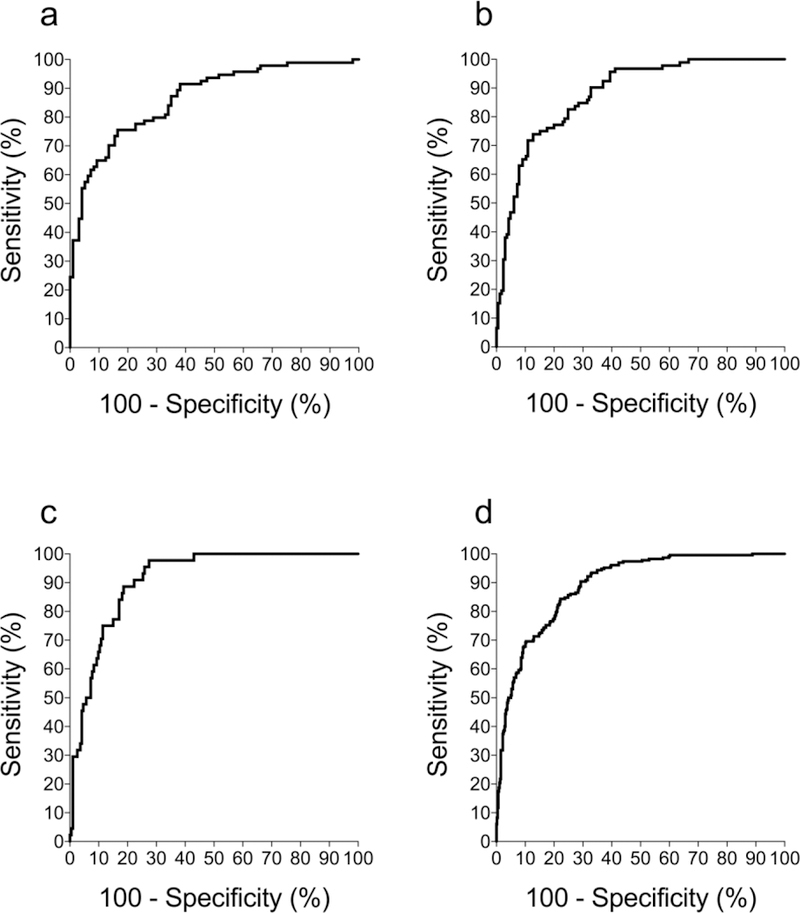
ROC analysis to determine how accurately CPEST identified participants whose CPAVE decreased by more than 7.5% of the baseline value at 3 (a), 6 (b), 12 (c) months after clinical trial entry. The ROC analysis for 7.5% decrease of CPAVE at 3, 6 or 12 months is shown at d. The respective AUROCs (95% CIs) for a-d were 0.86 (0.81, 0.91), 0.88 (0.84, 0.92), 0.91 (0.87, 0.95) and 0.89 (0.87, 0.92). These analyses used the Validation Dataset, which was derived from half of the participant population and was fully independent of the dataset used to develop the CPEST model.
Implications for clinical trial design
The potential suitability of CPEST as an alternative primary outcome measure for clinical trials was then assessed. All available data from participants (children and adults) in the TN-05 rituximab [4], TN-09 abatacept [3] and ITN-27 teplizumab [15] trials were analysed. The major conclusion from each trial, that the active therapy preserved beta cell function over the first year after diagnosis, held regardless of whether CPAVE or CPEST was used to compare treatment groups (Figure 3). We also applied CPEST to data from the other five negative trials and observed similar treatment effects (ESM Figure).
Figure 3. Outcomes of TN-05 (rituximab), TN-09 (abatacept) and ITN-27 (teplizumab) trials according to CPAVE and CPEST.
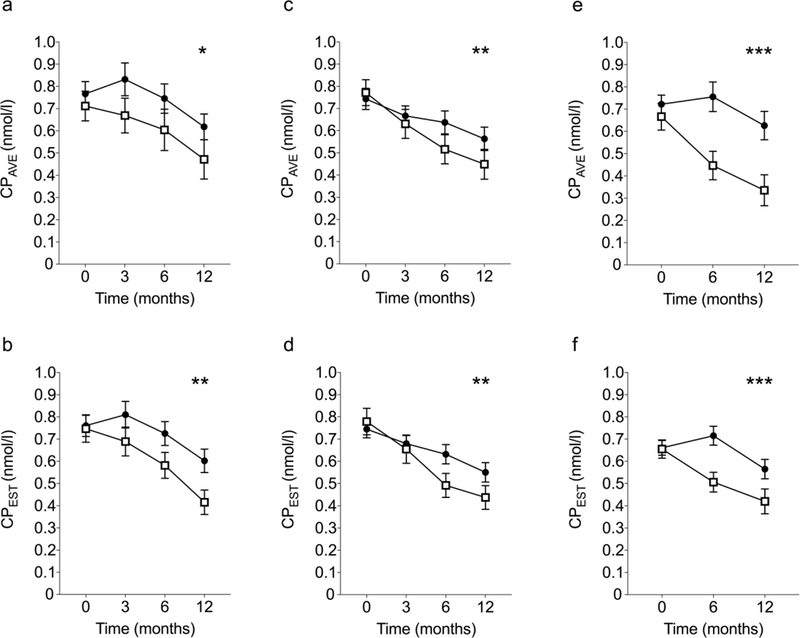
Outcomes for active (filled circles) and placebo (open squares) participants in TN-05 (a,b; 51 active and 29 placebo participants), TN-09 (c,d; 74 active and 31 placebo participants) and ITN-27 (e,f; 54 active and 25 placebo participants) are shown as mean ± SEM. CPAVE measured by meal test is presented in the top panels (a,c,e) and CPEST measured from single time point measures is presented in the bottom panels (b,d,f). Differences between treatment groups were determined using a mixed model that corrects for baseline CPAVE (or CPEST), age and sex, with significance between treatment groups indicated as *, ** and *** for p<0.05, <0.01 and <0.001 respectively.
To examine implications for clinical trial design, the standard deviation (SD) of loge(CPEST+1) values was conservatively estimated by combining the variance of loge(CPAVE+1) values with the variance of the difference between the loge(CPAVE+1) and loge(CPEST+1) values, as outlined in Methods. Using 12-month placebo-group data from the Validation Dataset from participants aged<21 years, the mean±SDs of loge(CPAVE+1) and loge(CPEST+1) were 0.320±0.218 and 0.331±0.166, respectively. The variance of the difference between these values was 0.0087, resulting in an estimated SD for loge(CPEST+1) of 0.237. When the loge(CPAVE+1) mean±SD and the estimated SD for loge(CPEST+1) were applied to a standard trial design that assumed a treatment effect of 50% increase in loge(CPAVE+1) at 12 months (i.e. Δ=0.160), two-tailed α=0.05 and 2:1 (active:placebo) randomisation, the number of participants required to achieve 80% power was 69 for loge(CPAVE+1) and 81, i.e. 17% higher, for loge(CPEST+1). When the validation data were combined with placebo-group data from adult participants aged over 21 years (Combined Dataset), the mean±SD for loge(CPAVE+1) and loge(CPEST+1) increased to 0.370±0.227 and 0.377±0.174, respectively, and the estimated SD for loge(CPEST+1) to 0.247, yielding Δ=0.185 and a requirement for 57 participants if loge(CPAVE+1) was the primary outcome measure, and 66, i.e. 16% higher, if loge(CPEST+1) was used. If geometric means for loge(CPAVE+1) were instead used as the basis for power calculations, the use of loge(CPEST+1) as the primary outcome measure required 17% and 13% more participants, respectively, in the context of the Validation Dataset and Combined Dataset.
Discussion
Using six, single time point measures, we describe a model (CPEST) for estimating CPAVE that reliably identifies loss of beta cell function in children and adults with recently-diagnosed type 1 diabetes. The accuracy of CPEST was comparable to that of CPAVE and was superior to that of IDAA1C. When applied to data from the active and placebo arms of three trials of immune modulators that preserved beta cell function, CPEST identified differences in beta cell function over the first year that were similar to those identified using CPAVE. These findings reinforce the strong correlation between FCP and CPAVE in people with recently-diagnosed type 1 diabetes [8, 20] and suggest that the relatively simple biochemical measurement of HbA1c, FCP and FG combined with BMI, insulin dose and disease duration may be sufficient to assess an individual’s response to disease-modifying therapy.
CPEST did not require age as an input despite the known strong association of age with beta cell function and with its rate of decline following diagnosis [20, 24]. Whereas age was an input for Model 4, it was not used in the optimal models that incorporated 5 or 6 inputs, which instead used HbA1c, BMI and insulin dose. Clearly these other clinical measures accounted for the effect of age on beta cell function. During model development with the Training Dataset, using age as an input did not always increase accuracy. For example, of the eight models based on four inputs that were more accurate than Model 3, only two (including Model 4) included age as an input. Similarly, of the six models based on five inputs that were more accurate than Model 4, only three used age.
Power calculations, based on a conservative estimate of the SD of loge(CPEST+1), indicated that sample size would need to increase by up to 17% if CPEST was used as a primary outcome measure. However, because the SD of loge(CPEST+1) was lower than the SD of loge(CPAVE+1), it is possible that modelled values are inherently less variable and therefore more accurate measures of beta cell function. This may be explained by the fact that a single fasting test eliminates variation attributable to meal ingestion and multiple sampling. Alternatively, incorporation of fasting glucose in the model may account for day-to-day variation in insulin sensitivity [25], which in turn could alter beta cell function [26] and increase CPAVE variability between meal tests. It will be important to establish the power of CPEST relative to CPAVE in future trials because CPEST is simpler and much more convenient. Even if subsequent testing shows that using CPEST would require a modest increase in sample size, this would need to be balanced against its potential to improve participant recruitment and satisfaction. CPEST also enables more frequent assessment of beta cell function during a trial and obviates the need to admit participants to a clinical trials unit for a meal test, thereby reducing trial costs.
In the clinical setting, the ability of CPEST to identify individuals who lose beta cell function commends it for routine use in monitoring an individual’s beta cell function over time and determine their response to disease-modifying therapy. CPEST is also likely to be useful for larger Phase 3 and 4 trials, and for studies of type 1 diabetes cohorts that aim to identify factors associated with disease progression and the relationship between C-peptide preservation and long-term complications such as hypoglycaemia unawareness and rates of micro- and macro-vascular disease.
IDAA1C is a measure of beta cell function that has gained acceptance in clinical practice because it reliably identifies children with type 1 diabetes who have substantial beta cell reserve, defined as a peak plasma C-peptide response to a mixed meal of greater than 0.3nmol/l (0.9ng/ml) [6, 7]. However, our analysis shows that IDAA1C has relatively poor accuracy for diagnosing significant loss of beta cell function, in accord with an earlier study that showed IDAA1C was not a reliable surrogate of CPAVE during the first 4 years following the diagnosis of type 1 diabetes [21]. Therefore, compared to modelled CPEST, IDAA1C is not suitable for assessing disease-modifying therapy.
Lastly, several caveats are in order. Our cohort comprised participants who were mostly of European descent and had type 1 diabetes for no more than 100 days when CPAVE was first measured. Therefore, the accuracy of our model in other ethnic groups or those with longer-standing type 1 diabetes is uncertain. In addition, despite the model’s accuracy in the two adult populations tested, caution should be exercised in applying it to other adult populations until its accuracy is further confirmed. Finally, because FCP and HbA1c were measured at only three laboratories, the generalisability of CPEST should be determined in the context of other laboratories and assay platforms.
In summary, CPEST modelled from six routine clinical and biochemical parameters is an accurate measure of beta cell function in children and young adults with recently-diagnosed type 1 diabetes. The simplicity and convenience of CPEST combined with its superior accuracy when compared to IDAA1C argues for its implementation and further validation in assessing beta cell function in clinical trials and during the course of routine clinical care.
Supplementary Material
Research in context.
What is already known about this subject?
Measuring average C-peptide after a mixed meal, the gold standard measure of beta cell function in type 1 diabetes, is laborious and inconvenient.
Insulin dose-adjusted HbA1c (IDAA1C), based on HbA1c and insulin dose, is widely used as a simple measure of beta cell function in routine care but this measure is not accurate and is not ideal for assessing responses to disease-modifying therapy.
What is the key question?
Can a more accurate measure of beta cell function in type 1 diabetes be developed from routine clinical measures?
What are the new findings?
Estimated C-peptide (CPEST), based on six routine measures, accurately identifies significant loss of beta cell function and reliably identifies treatment effects in randomised trials of immune therapy for type 1 diabetes.
CPEST is more accurate than IDAA1C
How might this impact on clinical practice in the foreseeable future?
CPEST could serve as a simple measure of beta cell function in routine practice and as a more economical and acceptable primary outcome measure in future trials of disease-modifying therapy.
Acknowledgements
We are grateful to the trial participants and to M Ritchie (Molecular Medicine Division, Walter and Eliza Hall Institute, Australia) and A Gorelik (Epicentre, Royal Melbourne Hospital, Australia) for statistical advice.
We also thank the following ITN and TrialNet investigators who contributed to original data:
ITN027AI AbATE
Department of Immunobiology and Internal Medicine, Yale University, New Haven, CT (Prof K C Herold MD); Immune Tolerance Network, San Francisco, CA (M R Ehlers MBChB, PhD, Peter H Sayre MD PhD); National Institutes of Allergy and Infectious Diseases, Bethesda, MD (J McNamara MD); Immune Tolerance Network, Bethesda, MD (S Aggarwal PhD, D Phippard PhD); Rho Federal Systems Division, Chapel Hill, NC (K D Boyle MS, L Keyes-Elstein DrPH); University of California San Francisco, San Francisco, CA (Prof S E Gitelman MD, Prof J A Bluestone PhD); Barbara Davis Center, University of Colorado, Aurora, CO (Prof P A Gottlieb MD); Benaroya Research Institute, Seattle, WA (C J Greenbaum MD); and Pacific Northwest Diabetes Research Institute, Seattle, WA (W Hagopian MD PhD)
ITN028AI START
University of California San Francisco, San Francisco, CA (S E Gitelman, J A Bluestone), Barbara Davis Center, University of Colorado, Aurora, CO (P A Gottlieb), Emory University, Atlanta, GA (E I Felner), Children’s Hospital of Philadelphia, Philadelphia, PA (S M Willi), Children’s Hospital of Los Angeles, Los Angeles, CA (L K Fisher), University of Minnesota, Minneapolis, MN (A Moran), University of California San Diego, San Diego, CA (M Gottschalk), Children’s Mercy Hospital, Kansas City, MO (W V Moore), Rho Federal Systems Division, Chapel Hill, NC (A Pickney, L Keyes-Elstein), Immune Tolerance Network, Bethesda, MD (K M Harris, S Kanaparthi, D Phippard) and San Francisco, CA (M R Ehlers), National Institute of Allergy and Infectious Diseases, Bethesda, MD (L Ding)
ITN045AI T1DAL
Immune Tolerance Network, Bethesda, MD (K M Harris, N Lim, S Kanaparthi, D Phippard), Rho Federal Systems Division, Chapel Hill, NC (A Pinckney, L Keyes-Elstein), Creighton Diabetes Center, Omaha, NE (M S Rendell), Emory University, Atlanta, GA (EI Felner), University of North Carolina, Durham, NC (J M Dostou), University of California San Francisco, San Francisco, CA (S E Gitelman), University of Arizona, Tucson, AZ (K J Griffin), University of Iowa, Iowa City, Iowa (E Tsalikian), Barbara Davis Center, University of Colorado, Aurora, CO (P A Gottlieb), Benaroya Research Institute, Seattle, WA (C J Greenbaum, A Long, G T Nepom), Massachusetts General Hospital, Boston, MA (N A Sherry), Children’s Mercy Hospital, Kansas City, MO (W V Moore), Children’s Hospital Los Angeles, Los Angeles, CA (R Monzavi), Children’s Hospital of Philadelphia, Philadelphia, PA (S M Willi), The University of Texas, Southwestern Medical Center, Dallas, TX (P Raskin), Immune Tolerance Network, San Francisco, CA (C L Soppe, M R Ehlers), National Institutes of Allergy and Infectious Diseases, Bethesda, MD (M L Fitzgibbon, J McNamara), Immune Tolerance Network, Bethesda, MD (K M Harris, N Lim, S Kanaparthi, D Phippard), Rho Federal Systems Division, Chapel Hill, NC (A Pinckney, L Keyes-Elstein)
Type 1 Diabetes TrialNet Study Group
Steering Committee: C. J. Greenbaum, Chair (Benaroya Research Institute), M. Atkinson (University of Florida), D. Baidal (University of Miami), M. Battaglia (San Raffaele Diabetes Research Institute), D. Becker (University of Pittsburgh), P. Bingley (University of Bristol), E. Bosi (San Raffaele University), J. Buckner (Benaroya Research Institute), M. Clements (Children’s Mercy Hospital), P. Colman (Walter & Eliza Hall Institute of Medical Research), L. DiMeglio (Indiana University), S. Gitelman, (University of California, San Francisco), R. Goland (Columbia University), P. Gottlieb (University of Colorado Barbara Davis Center for Childhood Diabetes), K. Herold (Yale University), M. Knip (University of Helsinki), J. Krischer (University of South Florida), A. Lernmark (Skane University), W. Moore (Children’s Mercy Hospital), A. Moran (University of Minnesota), A. Muir (Emory University), J. Palmer (University of Washington), M. Peakman (King’s College), L. Philipson (University of Chicago), P. Raskin (University of Texas Southwestern), M. Redondo (Baylor University), H. Rodriguez (University of South Florida), W. Russell (Vanderbilt University), L. Spain (National Institute of Diabetes and Digestive and Kidney Diseases [NIDDK]), D.A. Schatz (University of Florida), J. Sosenko (University of Miami), J. Wentworth (Walter & Eliza Hall Institute of Medical Research), D. Wherrett (University of Toronto), D. Wilson (Stanford University), W. Winter (University of Florida), A. Ziegler (Technical University Munich).Past Members: M. Anderson (University of California, San Francisco), P. Antinozzi (Wake Forest University), C. Benoist (Joslin Diabetes Center), J. Blum (Indiana University), K. Bourcier, P. Chase (University of Colorado Barbara Davis Center for Childhood Diabetes), M. Clare-Salzler (University of Florida), R. Clynes (Columbia University), G. Eisenbarth (University of Colorado Barbara Davis Center for Childhood Diabetes), C. G. Fathman (Stanford University), G. Grave (National Institute of Child Health and Human Development), B. Hering (University of Minnesota), R. Insel (Juvenile Diabetes Research Foundation), F. Kaufman (Children’s Hospital Los Angeles), T. Kay (St Vincent’s Institute of Medical Research), E. Leschek (NIDDK), J. Mahon (University of Western Ontario), J.B. Marks (University of Miami), K. Nanto-Salonen (University of Turku), G. Nepom (Benaroya Research Institute), T. Orban (Joslin Diabetes Center), R. Parkman (Children’s Hospital Los Angeles), M. Pescovitz (Indiana University), J. Peyman (National Institute of Allergy and Infectious Disease), A. Pugliese (University of Miami), B. Roep (Leiden University Medical Center), M. Roncarolo (San Raffaele University), P. Savage (NIDDK), O. Simell (University of Turku), R. Sherwin (Yale University), M. Siegelman (University of Texas Southwestern), J.S. Skyler (University of Miami), A. Steck (University of Colorado Barbara Davis Center for Childhood Diabetes), J. Thomas (Vanderbilt University), M. Trucco (University of Pittsburgh), J. Wagner (University of Minnesota).
TrialNet Executive Committee
Carla J. Greenbaum, Jeffrey P. Krischer, Ellen Leschek, Lisa Rafkin, Lisa Spain.Past Members: Katarzyna Bourcier, Catherine Cowie, Mary Foulkes, Richard Insel, Heidi Krause-Steinrauf, John M. Lachin, Saul Malozowski, John Peyman, John Ridge, Peter Savage, Jay S. Skyler, Stephanie J. Zafonte.
Chair’s Office
Carla J. Greenbaum, Lisa Rafkin, Jay M. Sosenko.Past Member: Jay S. Skyler, Norma S. Kenyon, Irene Santiago.
TrialNet Coordinating Center (University of South Florida)
Jeffrey P. Krischer, Brian Bundy, Michael Abbondondolo, Timothy Adams, Ilma Asif, Matthew Boonstra, Brian Bundy, David Cuthbertson, Christopher Eberhard, Steve Fiske, Julie Ford, Jennifer Garmeson, Heather Guillette, Susan Geyer, Brian Hays, Courtney Henderson, Martha Henry, Kathleen Heyman, Belinda Hsiao, Kaleena Dezsi, Christina Karges, Amanda Kinderman, Lindsay Lane, Ashley Leinbach, Shu Liu, Jennifer Lloyd, Jamie Malloy, Kristin Maddox, Julie Martin, Jessica Miller, Eric Milliot, Margaret Moore, Sarah Muller, Thuy Nguyen, Jodie Nunez, Ryan O’Donnell, Melissa Parker, MJ Pereyra, Nichole Reed, Tina Stavros, Roy Tamura, Keith Wood, Rebecca Wood, Ping Xu, Kenneth Young. Past Staff Members: Persida Alies, Darlene Amado, Franz Badias, Aaron Baker, Monica Bassi, Craig Beam, David Boulware, London Bounmananh, Susan Bream, Cristina Burroughs, Mary Deemer, Doug Freeman, Jessica Gough, Jinin Ginem, Moriah Granger, Mary Holloway Michelle Kieffer, Page Lane, Pat Law, Cristin Linton, Lavanya Nallamshetty, Vanessa Oduah, Yazandra Parrimon, Kate Paulus, Jennifer Pilger, Joy Ramiro, AQesha Luvon Ritzie, Amy Roberts, Kelly Sadler, Archana Sharma, Audrey Shor, Xiaohong Song, Amanda Terry, Jeanne Weinberger, Margaret Wootten.
Previous Coordinating Center (George Washington University)
John M. Lachin, Mary Foulkes, Pamela Harding, Heidi Krause-Steinrauf, Susan McDonough, Paula F. McGee, Kimberly Owens Hess, Donna Phoebus, Scott Quinlan, Erica Raiden.
TrialNet Clinical Network HUB (Benaroya Research Institute):
Carla J. Greenbaum, Emily Batts, Chris Buddy, Julie Hunt, Kristin Kirpatrick, Mary Ramey, Ann Shultz, Chris WebbPast Member: Melita Romasco.
NIDDK Staff
Judith Fradkin, Ellen Leschek, Lisa Spain.Past Member: Peter Savage.
Data Safety and Monitoring Board
Sean Aas (Georgetown University), Emily Blumberg (University of Pennsylvania), Chair, Gerald Beck (Cleveland Clinic), Rose Gubitosi-Klug (Case Western Reserve), Lori Laffel (Joslin Diabetes Center), Sean Aas (Georgetown University), Robert Vigersky (Medtronic), Dennis Wallace (Research Triangle Institute).Past Members: Jonathan Braun (University of California Los Angeles), David Brillon (Cornell University), Ake Lernmark (Lund University), Bernard Lo (University of California San Francisco), Herman Mitchell (Rho Inc.), Ali Naji (University of Pennsylvania), Jorn Nerup (University of Copenhagen), Trevor Orchard (University of Pittsburgh), Michael Steffes (University of Minnesota), Anastasios Tsiatis (North Carolina State University), Robert Veatch (Georgetown University), Bernard Zinman (University of Toronto).
Infectious Disease Safety Committee
Brett Loechelt (Children’s National Medical Center) (Medical Monitor), Lindsey Baden (Harvard University), Michael Green (University of Pittsburgh), Adriana Weinberg (University of Colorado).
Laboratory Directors
Santica Marcovina (University of Washington), Jerry P. Palmer, Adriana Weinberg, Liping Yu (University of Colorado Barbara Davis Center for Childhood Diabetes), Sunanda Babu (University of Colorado Barbara Davis Center for Childhood Diabetes) William Winter (University of Florida).Past Member: George S. Eisenbarth (late).
Personnel at Sites Participating in these Trials (TN02 TN05 TN09 TN08 TN14)
Barbara Davis Center for Childhood Diabetes-P. Gottlieb, J. Barker, S. Barry, M. Belzer, L Briggs, H. P. Chase, A. Conley, D. DiDomenico, W. Kastelic, D. Lehr, J. Lungaro, L. Meyers, A. Michels, R. Ohman, A. Proto, A. Wallace, L. Weiner; Benaroya Research Institute-C. Greenbaum, L. Allen, E. Batts, J. Bollyky, J. Buckner, A. Dove, D. Hefty, N. Hilderman, J. Klein, K. Kuhns, M. McCulloch-Olson, C. Murphy, M. Ramey, S. Sanda, D. Tridgell, H. Vendettuoli, C. Webber; Children’s Hospital Los Angeles-M. Geffner, R. Monzavi, M. Bock, L. Fisher, M. Gonzalez, M. Halvorson, D. Jeandron, F. Kaufman, J. Wood; Columbia University-R. Goland, M. Chan, R. Clynes, S. Cook, M. Gallagher, R. Gandica, E. Greenberg, A. Kurland, S. Pollak, K. Smith, J. Trast, A. Wolk; Indiana University-L. DiMeglio, J. Blum, L. Christner, E. Eugster, C. Evans-Molina, L. Ford, J. Fuqua, N. Haddad, T. Hannon, R. Hufferd, B. Jagielo, C. Kruse, E. Melvin, R. Mirmira, T. Nebesio, M. Nicholson, V. Patrick, M. Pescovitz, M. Rigby, E. Sims, M. Spall, K. Swinney, J. Terrell, E.Walvoord, S. Woerner; Joslin Diabetes Center-J. Gaglia, C. Alleyn, D. Conboy, S. Fay, R. Jackson, H. Jalahej, T. Orban, A. Ricker, S. Szubowicz, J. Wolfsdorf, H. Zhang; San Raffaele Hospital-E. Bosi, L. Falqui, P. Grogan, L. Molteni, M. Pastore; Stanford University-D. Wilson, T. Aye, B. Baker, B. Berry, B. Buckingham, T. Esrey, J. Liu, J. Perry, A. Rigby, A. Soto; Technical University Munich-A. Ziegler, T. Kaupper, M. Walter; The Hospital for Sick Children-D. Wherrett, B. Ahenkorah, J. Cevallos, L. Eisel, R. Kovalakovska, M. Mehan, M. Ricci, A. Roode, M. Sriskandarajah, R. Steger, F. Sultan; University of California San Francisco-S. Gitelman, S. Adi, M. Anderson, A. Berhel, K. Breen, J. Buchanan, K. Fraser, A. Gerard-Gonzalez, L. Hamid, C. Hamilton, L. Hawkins, A. Jain, P. Jossan, K. Ko, R. Lustig, S. Moassesfar, A. Mugg, D. Ng, C. O’Brien, S. Phelps, P. Prahalod, T. Rodriguez, S. Rosenthal, L. Stiehl, J. Tarkoff, L. Taylor, C. Torok, M. Wertz, R. Wesch; University of Florida-D. Schatz, A. Abraham, M. Atkinson, M. Clare‐Salzler, G. Cole, R. Cook, J. Ferguson, M. Haller, E. Hicks, D. Mancini; University of Maryland Hospital-D. Counts, M. Burr; University of Miami-J. Marks, C. Blaschke, D. Matheson, B. Acosta, L. Arazo, R. Arce, M. Cisneros, A. Pugliese; University of Minnesota-A. Moran, T. Albright-Fischer, M. Boes, C. Gibson, B. Hering, C. Kwong, J. Leschyshyn, B. Nathan, S. Peterson-Eck, J. Smith, A. Street, J. Wagner; University of Pittsburgh-D. Becker, B. Copemen, K. Delallo, A. Diaz, B. Elnyczky, D. Groscost, D. Gwynn, I. Libman, B. Pasek, K. Riley, F. Toledo, M. Trucco, G. Wohlers; University of Texas Southwestern-P. Raskin, M. Alford, J. Arthur, R. Davis, S. Fernandez, T. Harden, M. Hutchins, L. Pruneda, J. Richard, M. Siegelman, N. Torres; Vanderbilt University-W. Russell, M. Black, F. Brendle, A. Brown, D. Moore, E. Pittel, J. Thomas; Walter and Eliza Hall Institute of Medical Research-M. Bjorasen, J. French; Yale University-K. Herold, L. Feldman, R. Sherwin.
Funding
This work was supported by the Juvenile Diabetes Research Foundation (JDRFA Clinical Practitioner Fellowship to J.M.W. and JDRF Strategic Research Agreement to C.E-M.) and the Australian National Health and Medical Research Council (NHMRC) (Program Grant 1037321 to L.C.H. and CRE 1078106 Fellowship to J.M.W.). L.C.H. is a Senior Principal Research Fellow of the NHMRC. This work was made possible through Victorian State Government Operational Infrastructure Support and Australian National Health and Medical Research Council Research Institute Infrastructure Support Scheme. This manuscript includes clinical and biochemical data provided by the TrialNet data repository from clinical trials TN-02, TN-05, TN-08, TN-09 and TN-14. TrialNet is currently funded by NIH grants U01 DK061010, U01 DK061034, U01 DK061042, U01 DK061058, U01 DK085461, U01 DK085465, U01 DK085466, U01 DK085476, U01 DK085499, U01 DK085509, U01 DK103180, U01 DK103153, U01 DK103266, U01 DK103282, U01 DK106984, U01 DK106994, U01 DK107013, U01 DK107014, UC4 DK106993, and the Juvenile Diabetes Research Foundation International (JDRF). Research reported in this publication was also performed as a project of the Immune Tolerance Network and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health under Award Number UM1AI109565. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funding sources had no role in the writing of the manuscript or the decision to submit it for publication.
Abbreviations
- AIC
Akaike information criterion
- AUROC
area under the receiver operator curve
- BMI
body mass index
- CI
confidence interval
- CPAVE
average C-peptide over two hours following mixed meal challenge
- CPEST
estimated CPAVE
- FG
fasting glucose
- FCP
fasting C-peptide
- IDAA1C
insulin dose-adjusted HbA1c
- ITN
Immune Tolerance Network
- M6
model 6
- ROC
receiver operator curve
- SD
standard deviation
Footnotes
Data availability
Data used for this study can be accessed by application through the TrialNet (www.trialnet.org) and Immune Tolerance Network (www.immunetolerance.org) websites.
Duality of interest
Dr Gitelman received funding from the Immune Tolerance Network (in turn funded by NIAID) for his role as principal investigator of the START trial (ITN-28). Dr Geyer received a grant from NIDDK for unrelated work. No other author has a duality of interest associated with this manuscript.
Contribution statement
JMW devised the study, and analysed the data and prepared the manuscript with NGB, LCG and LCH. All named authors contributed to collection, collation, analysis and interpretation of the data and helped revise the manuscript and approved it for publication. Authors listed in Acknowledgements contributed by performing the TrialNet and ITN clinical trials.
Guarantor statement
JMW takes full responsibility for the work as a whole, including the study design, access to data, and the decision to submit and publish the manuscript.
Suggested Tweet
Measure beta cell function in type 1 diabetes without a meal test. A simpler method was published today @WEHI_research@TheRMH
References
- [1].Skyler JS (2013) Primary and secondary prevention of Type 1 diabetes. Diabetic medicine : a journal of the British Diabetic Association 30: 161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Greenbaum CJ, Mandrup-Poulsen T, McGee PF, et al. (2008) Mixed-meal tolerance test versus glucagon stimulation test for the assessment of beta-cell function in therapeutic trials in type 1 diabetes. Diabetes care 31: 1966–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Orban T, Bundy B, Becker DJ, et al. (2011) Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet 378: 412–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. (2009) Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. The New England journal of medicine 361: 2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sherry N, Hagopian W, Ludvigsson J, et al. (2011) Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet 378: 487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mortensen HB, Hougaard P, Swift P, et al. (2009) New definition for the partial remission period in children and adolescents with type 1 diabetes. Diabetes care 32: 1384–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Max Andersen ML, Hougaard P, Porksen S, et al. (2014) Partial remission definition: validation based on the insulin dose-adjusted HbA1c (IDAA1C) in 129 Danish children with new-onset type 1 diabetes. Pediatr Diabetes 15: 469–476 [DOI] [PubMed] [Google Scholar]
- [8].Sosenko JM, Krischer JP, Palmer JP, et al. (2008) A risk score for type 1 diabetes derived from autoantibody-positive participants in the diabetes prevention trial-type 1. Diabetes care 31: 528–533 [DOI] [PubMed] [Google Scholar]
- [9].Sosenko JM, Skyler JS, Palmer JP, et al. (2013) The prediction of type 1 diabetes by multiple autoantibody levels and their incorporation into an autoantibody risk score in relatives of type 1 diabetic patients. Diabetes care 36: 2615–2620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sosenko JM, Geyer S, Skyler JS, et al. (2017) The influence of body mass index and age on C-peptide at the diagnosis of type 1 diabetes in children who participated in the diabetes prevention trial-type 1. Pediatr Diabetes [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gottlieb PA, Quinlan S, Krause-Steinrauf H, et al. (2010) Failure to preserve beta-cell function with mycophenolate mofetil and daclizumab combined therapy in patients with new- onset type 1 diabetes. Diabetes care 33: 826–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wherrett DK, Bundy B, Becker DJ, et al. (2011) Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet 378: 319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Moran A, Bundy B, Becker DJ, et al. (2013) Interleukin-1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double-blind, placebo-controlled trials. Lancet 381: 1905–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gitelman SE, Gottlieb PA, Rigby MR, et al. (2013) Antithymocyte globulin treatment for patients with recent-onset type 1 diabetes: 12-month results of a randomised, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol 1: 306–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Herold KC, Gitelman SE, Ehlers MR, et al. (2013) Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 62: 3766–3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rigby MR, DiMeglio LA, Rendell MS, et al. (2013) Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol 1: 284–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pickup JC (2012) Insulin-pump therapy for type 1 diabetes mellitus. The New England journal of medicine 366: 1616–1624 [DOI] [PubMed] [Google Scholar]
- [18].Bundy BN, Krischer JP, Type 1 Diabetes TrialNet Study G (2016) A model-based approach to sample size estimation in recent onset type 1 diabetes. Diabetes Metab Res Rev 32: 827–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Beck RW, Tamborlane WV, Bergenstal RM, et al. (2012) The T1D Exchange clinic registry. J Clin Endocrinol Metab 97: 4383–4389 [DOI] [PubMed] [Google Scholar]
- [20].Greenbaum CJ, Beam CA, Boulware D, et al. (2012) Fall in C-peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite Type 1 Diabetes TrialNet data. Diabetes 61: 2066–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hao W, Gitelman S, DiMeglio LA, Boulware D, Greenbaum CJ, Type 1 Diabetes TrialNet Study G (2016) Fall in C-Peptide During First 4 Years From Diagnosis of Type 1 Diabetes: Variable Relation to Age, HbA1c, and Insulin Dose. Diabetes care 39: 1664–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wherrett DK, Chiang JL, Delamater AM, et al. (2015) Defining pathways for development of disease-modifying therapies in children with type 1 diabetes: a consensus report. Diabetes care 38: 1975–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Herold KC, Gitelman SE, Masharani U, et al. (2005) A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes 54: 1763–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Barker A, Lauria A, Schloot N, et al. (2014) Age-dependent decline of beta-cell function in type 1 diabetes after diagnosis: a multi-centre longitudinal study. Diabetes Obes Metab 16: 262–267 [DOI] [PubMed] [Google Scholar]
- [25].Moberg E, Kollind M, Lins PE, Adamson U (1995) Day-to-day variation of insulin sensitivity in patients with type 1 diabetes: role of gender and menstrual cycle. Diabetic medicine : a journal of the British Diabetic Association 12: 224–228 [DOI] [PubMed] [Google Scholar]
- [26].Bergman RN, Phillips LS, Cobelli C (1981) Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest 68: 1456–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.