

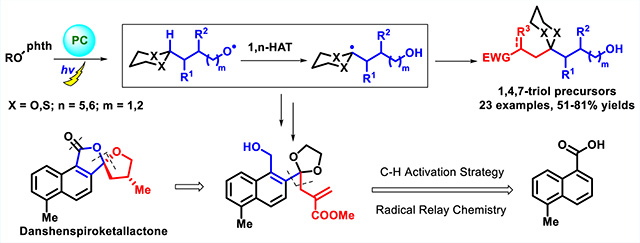
Abstract
Visible-light-induced generation of dithianyl and dioxolanyl radicals via selective hydrogen atom transfer (HAT) has been achieved. This radical relay tactic enables remote C(sp3)—H functionalization to permit rapid access to polyol and spiroketal segments, and in turn has been exploited as a key synthetic construct in the total synthesis of the danshenspiroketallactones. The conformational stability of the danshenspiroketallactones has also been defined via experiments and DFT calculations.
Graphical Abstract
Anion Relay Chemistry (ARC) comprises a one-flask multicomponent union tactic employing solvent-controlled Brook rearrangements.1 Central to the ARC tactic are dithiane-containing linchpins, in which the dithiane moiety serves as an acyl anion equivalent for the construction of carbon—carbon bonds.2 The significance of this synthetic tactic comprises the efficient rapid assembly of natural products with diverse architectural features, ranging from polyols, spiroketals, and polyene macrolides to polypropionate scaffolds, and as such has been a recent theme in our laboratory.3
The multicomponent ARC union protocol relies on [1,4]-Brook rearrangements4 wherein an initially generated alkoxy anion can be triggered to undergo in situ 1,4-silyl group migration to afford a new carbanion (Scheme 1a), which in turn can be readily trapped by diverse electrophiles. The resulting coupled products provide ample bandwidth for downstream functionalization to furnish various 1,3,5-polyol motifs. Brook rearrangements involving 1,5-negative charge migration, although recently studied by us and others,5 are for the most part synthetically not viable, primarily due to the harsh conditions required to initiate the silyl group migration.
Scheme 1.

Anion Relay Chemistry and Proposed Radical Relay Chemistry
Recently, radical relay chemistry has been developed to functionalize remote C(sp3)—H bonds via 1,5-hydrogen atom transfer (1,5-HAT).6 For example, initially generated alkoxyl6a–c or aminyl radicals6d–f are known to undergo in situ 1,5-or 1,6-HAT to furnish carbon radicals via a photoredox catalysis cycle. As such, this reactivity migration, although a challenge via remote Brook anionic rearrangement, would comprise an important extension and complement to the ARC tactic (Scheme 1b). We therefore explored 1,5- and 1,6-HAT radical relay chemistry to access various 1,4,7-polyol or 1,4,8-polyol fragments via visible-light photoredox catalysis.
Spiroketals possessing [5.5]- or [5.6]-oxygen architectures are abundant in a wide array of natural products possessing diverse biological activities (Figure 1).7 Examples include: cephalosporolide I (1),8 isolated from a marine-derived fungus that inhibits both 3α-hydroxysteroid dehydrogenase and xanthine-oxidase; berkelic acid (2),9 a fungal metabolite that exhibits activity against ovarian cancer; aquilarinoside A (3),10 the principal component of Aquilaria sinensis, a highly valuable agarwood; and danshenspiroketallactone (4)11 and the epimer (5),12 both isolated from Salvia miltiorrhiza, known as Danshen, which is employed in traditional Chinese medicine as a remedy for renal failure, stroke and heart disease. Most synthetic strategies to construct the core architecture of such [5.5]- or [5.6]-spiroketals rely on acid-catalyzed cyclization of 1,4,7- or 1,4,8-polyol precursors (or their equivalents). However, compared to 1,3,5-polyols, synthetic methods to access 1,4,7-polyol precursors remain limited, usually requiring multiple oxidation states or protecting group manipulations.7 An effective catalytic method to rapidly access such precursors, especially via direct C(sp3)-H functionalization, would thus be of considerable interest.
Figure 1.

Natural products containing [5.5]- or [5.6]-spiroketal skeleton.
Toward this end, the reaction sequence based on Chen’s cleavage–translocation–allylation sequence was proposed as outlined in Scheme 2. We envisioned that, under visible light, the photocatalyst fac-Ir(ppy)3 could be excited and reduced by the Hantzsch ester, known to furnish a reduced photocatalyst, which would reduce N-alkoxyphthalimides (6) to yield alkoxyl radicals (Scheme 2) via the Hantzsch ester radical cation.6a,13 The resulting alkoxyl radical A could then undergo a 1,5-HAT reaction to generate either a dithianyl or dioxolanyl radical B, which comprise carbonyl or methylene radical equivalents, that in turn could engage with an acceptor (7a) to yield coupled adducts. Although dithianes and ketals are extensively employed in various reactions, dithianyl and dioxolanyl radicals generated via photoredox catalysis are much less studied.14
Scheme 2.

Proposed Reaction Sequence
To initiate this study, we selected 2-[3-(1,3-dioxolan-2-yl)propoxy]-isoindoline-1,3-dione 6a (Table 1) as a model substrate and 2 equiv of methyl 2-[(phenylsulfonyl)methyl]-acrylate (7a) as the radical acceptor. After extensive screening, we found that employing fac-Ir(ppy)3 as the photoredox catalyst in THF led to coupled product 8a in 45% yield (entry 1), whereas other photocatalysts gave only lower yields (entries 2–3). Interestingly, the choice of solvent proved crucial to achieve efficiency for the 1,5-HAT process. Less polar ether solvents, such as 1,4-dioxane or methyl tert-butyl ether (MTBE), led to higher yields, along with less of the quenched byproduct 8aa′ (entries 4–5). Further investigation of the reaction solvent revealed that a combination of MTBE and dioxane (9:1) provided the best results, with an isolated yield of 8a of 73% (entry 6), employing 2 equiv of the acceptor 7a. Pleasingly, the loading of the acceptor (7a) could be decreased to 1.5 equiv and still resulted in a 68% yield (entry 7). Control experiments demonstrated the essential role of photoredox catalyst and visible light for this reaction (entries 8–9).
Table 1.
Optimization of Reaction Conditions
 | |||
|---|---|---|---|
| entry | photocatalyst | solvent | yielda (%) of 8a |
| 1 | fac-Ir(ppy)3 | THF | 45 |
| 2 | Ru(bpy)3(PF6)2 | THF | 38 |
| 3 | Ir(ppy)2(dtbbpy)PF6 | THF | 34 |
| 4 | fac-Ir(ppy)3 | dioxane | 61 |
| 5 | fac-Ir(ppy)3 | MTBE | 65 |
| 6 | fac-Ir(ppy)3 | MTBE/dioxane = 9:1 | 76 (73) |
| 7b | fac-Ir(ppy)3 | MTBE/dioxane = 9:1 | 68 |
| 8 | – | MTBE/dioxane = 9:1 | trace |
| 9c | fac-Ir(ppy)3 | MTBE/dioxane = 9:1 | trace |
Reaction conditions: 6a (0.1 mmol), 7a (0.2 mmol), photocatalyst (1 mol %), Hantzsch ester (0.15 mmol), 1 mL solvent, rt, blue LED, 5 h. The yield of 8a was determined by 1H NMR analysis. Isolated products are obtained by flash chromatography with yields given in parentheses.
7a (0.15 mmol).
No light.
With these conditions established, we examined the scope of the reaction with respect to the structural diversity of the N-alkoxyphthalimides (Table 2). Dioxolanyl, dioxanyl, dithiolanyl, and dithianyl radicals in turn can be successfully generated under the above conditions, leading to allylated products 8a–8d in moderate to good yields (61–73%). Importantly, steric hindrance at the α- or β-position did not influence the reaction. Instead, the Thorpe—Ingold effect appears to increase the efficiency of 1,5-HAT to provide higher yields of 8e (80%) and 8f (72%). Also noteworthy, the generated dioxolanyl radical was tolerated with a stereogenic center at the α-position (6g), without erosion of the enantiomeric excess (97% ee). We were also pleased to discover that with 6h the dioxolanyl radical could be generated by exploiting 1,6-HAT, leading to the C—H allylated product 8h in 59% yield. This result demonstrates the potential of this method for the construction of various [5.6]-spiroketal scaffolds. In addition to aliphatic substituents, aromatic substituents are also tolerated in this HAT reaction. With a phenyl group at the α-position (6i), a small decrease in the yield of 8i was obtained. We further tested whether the radical translocation could proceed at an ortho benzylic carbon. However, when 6j was treated via our standard protocol with 2 equiv of the radical acceptor, only a trace amount of desired product 8j was observed. The major result was a pair of diastereomers 8l in a 1:1 ratio (Table 2b). To elucidate the structure, oxidation with Dess-Martin periodinane afforded the bisalkylated ketone 8j′ in 56% overall yield, the structure of which was assigned by extensive NMR/HRMS analysis. The interesting structure of 8j′ revealed that a tandem 1,5-HAT/1,6-HAT sequence had occurred in the reaction (Scheme 3). This unique radical relay reaction, different from the radical process with aliphatic substrates (6a–6i), is presumably favored by both the conformational restraints of intermediate C (Scheme 3) and the resultant more stable benzyl radical D. Decreasing the acceptor loading to 1 equiv significantly increased the yield of monoallylated products, leading to benzyl C—H functionalized products (8j–8k) in 51–54% yield. This result is important because it provides a novel tactic to construct benzannulated spiroketal moieties, which will be demonstrated as the key step in the total synthesis of danshenspiroketallactone 4 and epi-danshenspiroketallactone 5 (vide infra).
Table 2.
Substrates Scope for the N-Alkoxyphthalimides
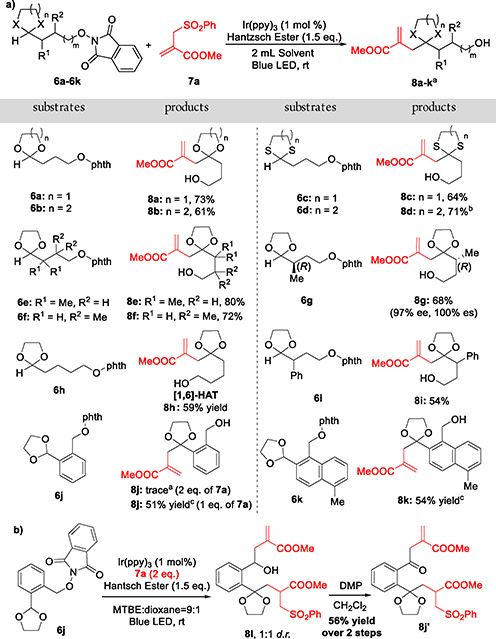 |
aReaction conditions: 6 (0.2 mmol), 7a (0.4 mmol), Ir(ppy)3 (1 mol %), Hantzsch ester (0.3 mmol), 2 mL of MTBE/dioxane = 9:1, rt, blue LED, 5 h. Yields of isolated products are given. b2 mL of THF. c6j or 6k (0.2 mmol), 7a (0.2 mmol), Hantzsch ester (0.24 mmol), 2 mL of dioxane.
Scheme 3.

Possible Mechanism for the Generation of 8j′
We next explored various radical acceptors including allyl sulfones and Michael acceptors (Scheme 4). Here we discovered that allyl sulfones with ethyl ester or cyanide as the electron-withdrawing group react smoothly with the dioxolanyl and dithianyl radicals to furnish C(sp3)—H allylation adducts 9a–9d in good yield (70–78%). Best results were obtained when 2 equiv of the acceptors were employed. However, with phenyl as the electron-withdrawing group, the radical addition of the dioxolanyl radical to allyl sulfones was not successful (9e); only quenched byproduct 8aa′ (Table 1) was observed. Acceptors with remote functional groups, such as alkynes and dienes, were also tested; 9f and 9g were obtained in 56% and 75% yield respectively, thus providing a scaffold amenable to additional synthetic applications. Interestingly, Michael acceptors (7g–7k) readily reacted with the dioxolanyl radical to yield alkylated products (9h–9l), although a slightly more polar solvent was required (MTBE/dioxane = 1:1). In these reactions, Hantzsch ester served as the reductant to reduce the ensuing radicals. For example, aryl acrylates with different functional groups on the aromatic ring (7g–7j) were explored; all were well tolerated to deliver the desired products 9h–9k in 53–63% yield. The dioxolanyl radical also underwent radical addition to 2-methylenemalonate 7k to furnish product 9l in 81% yield.
Scheme 4.

Substrate Scope for the Radical Acceptors
aReaction conditions: 6a or 6d (0.2 mmol), 7 (0.4 mmol), Ir(ppy)3 (1 mol %), Hantzsch ester (0.3 mmol), MTBE/dioxane = 9:1, rt, blue LED, 5 h. Yields of isolated products are given. bMTBE/dioxane = 1:1.
To demonstrate the utility of this radical relay chemistry, we devised a synthesis of the danshenspiroketallactones 4 and 5 (Scheme 5). Both danshenspiroketallactones 4 and 5 comprise monobenzannulated [5.5]-spiroketals, a rare spirocyclic unit in natural products.15 The first total synthesis of the danshenspiroketallactones 4 and 5, reported by Brimble and co-workers in 2012,16a employed directed metalation and a late-stage oxidative spiroketalization.16b In both the original isolation paper12 and the Brimble synthesis, a mixture of 4 and 5 was obtained without further separation. Here, we record the total synthesis of the danshenspiroketallactones 4 and 5 utilizing [1,5]-radical relay chemistry, wherein the generation and functionalization of the dioxolanyl radical plays a key role in the construction of the 1,4,7-polyol precursor (8k, Scheme 5). Noteworthy here, the pure format of 4 and 5 were obtained with high performance liquid chromatography (HPLC).
Scheme 5.

Total Synthesis of the Danshenspiroketallactones
Our synthesis began with commercially available 5-methyl-1-naphthoic acid 10 (Scheme 5). Palladium(II)-catalyzed ortho C—H activation17 followed by a base workup furnished an ortho-hydroxymethyl-naphthoic acid, which in turn was elaborated to 11 by a two-step sequence involving oxidation and methylation. The ethylene acetal was then installed in 12, followed by reduction and a Mitsunobu reaction with N-hydroxyphthalimide to yield 6k that sets the stage for the envisaged [l,5]-radical relay reaction sequence using photoredox catalysis.
Pleasingly, alkoxyl radical generation followed by 1,5-HAT under the above conditions using photoredox catalysis converted 6k to 8k in a 54% yield, which in turn was subjected to hydrogenation using Wilkinson’s catalyst to deliver 13. Upon exposure to LiAlH4 and acidification, the ester group in 13 was reduced and the resulting diol underwent cyclization to furnish two diastereomers, 14 and 15, in 2:1 d.r. as an inseparable mixture. Formation of the minor diastereomer is presumed to result from the weak dependence of [5.5]-spiroketals on the anomeric effect.7 Completion of the total synthesis of danshenspiroketallactones 4 and 5 was then achieved via benzylic oxidation of the mixture of 14 and 15. Notably, by employing peroxide 16,18 the α-alkoxy benzyl carbon could be chemoselectively oxidized in 80% yield, thus providing a mixture of danshenspiroketallactones 4 and 5. Separation of 4 and 5 via silica gel chromatography proved not possible, because epi-danshenspiroketallactone 5 was found to undergo rapid isomerization to 4 on silica gel.12 Pleasingly, employing HPLC, we were able to separate successfully the mixture and obtained pure danshenspiroketallactone 4 and the epimer 5, with 1H and 13C NMR data identical in all respects with the published data.11,12,16a Noteworthy, this is the first time that pure synthetic danshenspiroketallactones (4 and 5) have been obtained. The total synthesis was achieved in 10 steps with an 11% overall yield.
Not surprisingly, treatment of the synthetic sample of either 4 or the mixture of 4 and 5 (1:1) with TFA in CH2Cl2 led to a mixture of 4 and 5 in a ratio of 1:0.4. To evaluate the thermodynamic stability of 4 and 5, DFT calculations were conducted.19 The optimized structures are shown in Figure 2. Isomer 4 is 0.8 kcal/mol more stable than 5, in agreement with the observed experimental ratio. Analysis of the optimized structures (Figure 2; 4 and ent-5) reveals that the methyl substituent (C17) achieves an equatorial conformation in both diastereomers. In the case of 4, this can be achieved in such a way that the conformation of the tetrahydrofuran ring has mostly staggered arrangements about the CC bonds (see Newman projection below 4), whereas 5 has eclipsing interactions that destabilize this conformation.20
Figure 2.

Conformational structures of 4 and 5 (ent-5 is Shown). Newman projections along the C13–C16 bond are shown.
In summary, we have designed and validated a new visible-light-induced protocol to generate dithianyl and dioxolanyl radicals via 1,5- or 1,6-HAT. The resulting radicals engage in conjugate additions to achieve formal allylation and alkylation. This radical relay reaction provides an efficient method to construct various 1,4,7-polyols or spiroketals and, in turn, was utilized as the key synthetic tactic in the total synthesis of danshenspiroketallactones 4 and 5.
Supplementary Material
ACKNOWLEDGMENTS
Financial support was provided by the NIH through Grants CA-19033 and GM-29028. We thank Dr. Charles W. Ross III for providing mass spectral data.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.9b00271.
Compound preparation and characterization, computational studies, and copies of 1H and 13C NMR spectra (PDF)
REFERENCES
- (1).Smith AB III; Xian M Anion Relay Chemistry: An Effective Tactic for Diversity Oriented Synthesis. J. Am. Chem. Soc 2006, 128, 66–67. [DOI] [PubMed] [Google Scholar]
- (2).Smith AB III; Adams CM Evolution of Dithiane-Based Strategies for the Construction of Architecturally Complex Natural Products. Acc. Chem. Res 2004, 37, 365–377. [DOI] [PubMed] [Google Scholar]
- (3).(a) Ai Y; Kozytska MV; Zou Y; Khartulyari AS; Smith AB III Total Synthesis of (−)-Enigmazole A. J. Am. Chem. Soc 2015, 137, 15426–15429. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nguyen MH; Imanishi M; Kurogi T; Smith AB III Total Synthesis of (−)-Mandelalide A Exploiting Anion Relay Chemistry (ARC): Identification of a Type II ARC/CuCN Cross-Coupling Protocol. J. Am. Chem. Soc 2016, 138, 3675–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Q; Deng Y; Smith AB III Total Synthesis of (−)-Nahuoic Acid Ci (Bii). J. Am. Chem. Soc 2017, 139, 13668–13671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Brook AG Molecular Rearrangement of Organosilicon Compounds. Acc. Chem. Res 1974, 7, 77–84. [Google Scholar]; (b) Moser WH The Brook Rearrangement in Tandem Bond Formation Strategies. Tetrahedron 2001, 57, 2065–2084. [Google Scholar]
- (5).(a) Smith AB III; Xian M; Kim W-S; Kim D-S The [1,5]-Brook Rearrangement: An Initial Application in Anion Relay Chemistry. J. Am. Chem. Soc 2006, 128, 12368–12369. [DOI] [PubMed] [Google Scholar]; (b) Gao L; Lu J; Song Z; Lin X; Xu Y; Yin Z [1,5]-Brook Rearrangement: An Overlooked but Valuable Silyl Migration to Synthesize Configurationally Defined Vinylsilane. The Unique Steric and Electronic Effects of Geminal Bis(silane). Chem. Commun. 2013, 49, 8961–8963. [DOI] [PubMed] [Google Scholar]; (c) Liu Q; Chen Y; Zhang X; Houk KN; Liang Y; Smith AB III Type II Anion Relay Chemistry: Conformational Constraints to Achieve Effective [1,5]-Vinyl Brook Rearrangement. J. Am. Chem. Soc 2017, 139, 8710–8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Zhang J; Li Y; Zhang F; Hu C; Chen Y Generation of Alkoxyl Radicals by Photoredox Catalysis Enables Selective C(sp3)-H Functionalization under Mild Reaction Conditions. Angew. Chem., Int. Ed 2016, 55, 1872–1875; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 1904–1907. [Google Scholar]; (b) Wang C; Harms K; Meggers E Catalytic Asymmetric C(sp3)-H Functionalization under Photoredox Conditions by Radical Translocation and Stereocontrolled Alkene Addition. Angew. Chem., Int. Ed. 2016, 55, 13495–13498; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 13693–13696. [Google Scholar]; (c) Hu A; Guo J; Pan H; Tang H; Gao Z; Zuo Z δ-Selective Functionalization of Alkanols Enabled by Visible-Light-Induced Ligand-to-Metal Charge Transfer. J. Am. Chem. Soc 2018, 140, 1612–1616. [DOI] [PubMed] [Google Scholar]; (d) Martínez C; Muniz K An Iodine-Catalyzed Hofmann-Luffler Reaction. Angew. Chem., Int. Ed 2015, 54, 8287–8291; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2015, 127, 8405–8409. [Google Scholar]; (e) Chu JCK; Rovis T Amide-Directed Photoredox-Catalyzed C-C Bond Formation at Unactivated sp3 C-H bonds. Nature 2016, 539, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Catalytic Alkylation of Remote C-H bonds Enables by Proton-Coupled Electron Transfer. Nature 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Perron F; Albizati KF Chemistry of Spiroketals. Chem. Rev 1989, 89, 1617–1661. [Google Scholar]
- (8).Li X; Yao Y; Zheng Y; Sattler I; Lin W Cephalosporolides H and I, Two Novel Lactones from a Marine-Derived Fungus, Penicillium sp. Arch. Pharmacal Res 2007, 30, 812–815. [DOI] [PubMed] [Google Scholar]
- (9).Stierle AA; Stierle DB; Kelly K Berkelic Acid, A Novel Spiroketal with Selective Anticancer Activity from an Acid Mine Waste Fungal Extremophile. J. Org. Chem 2006, 71, 5357–5360. [DOI] [PubMed] [Google Scholar]
- (10).Qi J; Lu J; Liu J; Yu B Flavonoid and a Rare Benzophenone Glycoside from the Leave of Aquilaria Sinensis. Chem. Pharm. Bull 2009, 57, 134–137. [DOI] [PubMed] [Google Scholar]
- (11).Kong D; Lu X; Teng M; Rao Z The Structure of Danshenspiroketallactone of Dan-Shen (Salvia Miltiorrhiza Bunge). Acta. Pharm. Sin 1985, 20, 747–751. [PubMed] [Google Scholar]
- (12).Hou WL; Shaoxing C; Lee JN; Snyder JK Epi-Danshenspiroketallactone from Salvia Miltiorrhize. Phytochemistry 1988, 27, 290–292. [Google Scholar]
- (13).(a) Kim S; Lee TA; Song Y Facile Generation of Alkoxy Radicals from N-Alkoxyphthalimides. Synlett 1998, 1998, 471–472. [Google Scholar]; (b) Zlotorzynska M; Sammis GM Photoinduced Electron-Transfer-Promoted Redox Fragmentation of N-Alkoxyphthalimides. Org. Lett 2011, 13, 6264–6267. [DOI] [PubMed] [Google Scholar]; (c) Zhu H; Leung JCT; Sammis GM Strategies to Control Alkoxy Radical-Initiated Relay Cyclizations for the Synthesis of Oxygenated Tetrahydrofuran Motifs. J. Org. Chem 2015, 80, 965–979. [DOI] [PubMed] [Google Scholar]
- (14).(a) Mosca R; Fagnoni M; Mella M; Albini A Synthesis of Monoprotected 1,4-Diketones by Photoinduced Alkylation of Enones with 2-Substituted-1,3-Dioxolanes. Tetrahedron 2001, 57, 10319–10328. [Google Scholar]; (b) Gualandi A; Matteucci E; Monti F; Baschieri A; Armaroli N; Sambri L; Cozzi PG Photoredox Radical Conjugate Addition of Dithiane-2-Carboxylate Promoted by an Iridium (III) Phenyl-Tetrazole Complex: a Formal Radical Methylation of Michael Acceptors. Chem. Sci 2017, 8, 1613–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nielsen MK; Shields BJ; Liu J; Williams MJ; Zacuto MJ; Doyle AG Mild, Redox-Neutral Formylation of Aryl Chlorides through the Photocatalytic Generation of Chlorine Radicals. Angew. Chem., Int. Ed 2017, 56, 7191–7194; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 7297–7300. [Google Scholar]
- (15).Sperry J; Wilson ZE; Rathwell DCK; Brimble MA Isolation, Biological Activity and Synthesis of Benzannulated Spiroketal Natural Products. Nat. Prod. Rep 2010, 27, 1117–1137. [DOI] [PubMed] [Google Scholar]
- (16).(a) Chorley DF; Chen JL; Furkert DP; Sperry J; Brimble MA Total Synthesis of Danshenspiroketallactone. Synlett 2012, 2012, 128–130. [Google Scholar]; (b) Cryptoacetalides, analogues of danshenspiroketal-lactones, were synthesized employing the same reactions: Zou, Deiters Y, A. Total Synthesis of Cryptoacetalide. J. Org. Chem. 2010, 75, 5355–5358. [DOI] [PubMed] [Google Scholar]
- (17).Zhang Y; Shi B; Yu J Palladium(II)-Catalyzed ortho Alkylation of Benzoic Acids with Alkyl Halides. Angew. Chem., Int. Ed 2009, 48, 6097–6100; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2009, 121, 6213–6216. [Google Scholar]
- (18).Ochiai M; Ito T; Takahashi H; Nakanishi A; Toyonari M; Sueda T; Goto S; Shiro M Hypervalent (tert-Butylperoxy)iodanes Generate Iodine-Centered Radicals at Room Temperature in Solution: Oxidation and Deprotection of Benzyl and Allyl Ethers, and Evidence for Generation of α-Oxy Carbon Radicals. J. Am. Chem. Soc 1996, 118, 7716–7730. [Google Scholar]
- (19).All calculations were carried out at the M062X/6–31G(d)/SMD(CH2Cl2)//M062X/6–311++G(2d, p)/SMD(CH2Cl2) level using Gaussian 09, revision A.02 (for full citation and computational details, see Supporting Information).
- (20).Boese AD; Boese R Tetrahydrothiophene and Tetrahydrofuran, Computational and X-ray Studies in the Crystalline Phase. Cryst. Growth Des 2015, 15, 1073–1081. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.