

Abstract
The ontogeny of acute myeloid leukemia is a multistep process. It is driven both by features of the malignant clone itself as well as by environmental pressures, making it a unique process in each individual. The technological advancements of recent years has increased our understanding about the different steps that take place at the genomic level. It is now clear that malignant clones evolve, expand and change even during what seem to be clinically healthy or “cured” periods. This opens a wide window for new therapeutic and monitoring opportunities. Moreover, prediction and even early prevention have become possible goals to be pursued. The aim of this review is to shed light upon recent observations in leukemia evolution and their clinical implications. We present a critical view of these concepts in order to assist clinicians when interpreting results of the ever growing myriad of genomic diagnostic tests. We wish to help clinicians incorporate genetic tests into their clinical assessment and enable them to provide genetic counseling to their patients.
Introduction
Acute myeloid leukemia (AML) is a clonal disorder that originates in leukemic stem and progenitor cells, termed blasts.1 AML clinically manifests with the accumulation of these immature cells that exhibit uncontrolled growth and decreased apoptosis, and that lack normal differentiation. AML is clinically defined when blasts make up 20% or more of the cellular component of the bone marrow (BM). These cells inhibit normal hematopoiesis resulting in BM failure.2 Although most of the leukemic cells can be eradicated during the first course of therapy, most patients succumb to disease relapse within the first two years.3
Acute myeloid leukemia is characterized by a relatively small (compared to solid tumors), recurrent set of somatic mutations, designated leukemia driver mutations.4 The various mutations can be used to classify AML subtypes. Genomic analysis of 1540 AML patients identified distinct AML subgroups according to their mutational background. Some mutations (such as in the DNMT3A gene) are shared by few subgroups, some of the mutations co-occur (e.g. DNMT3A, NPM1 and FLT3-ITD), while others are not usually found in the same clone (TP53 and NPM1).5
Most somatic mutations in AML occur stochastically across the genome without any foci of localized hypermutation.6 Theoretically, if the effective size of the stem cell population had been large enough, and it had been given enough replication cycles, it would have been reasonable to assume that almost every possible mutation can be found at the single cell level. Yet, not all mutations are shared by all AML subtypes. Moreover, a specific mutational signature with an elevated rate of C>T transitions was found in AML. This mutational signature was related to spontaneous deamination of 5-methyl-cytosine and was correlated with age.6 This implies that the aging BM niche exerts a selective pressure on the leukemic stem cells and shapes their mutational profile.
Clones become fitter as they accumulate mutations and evolve. However, studies in other malignancies suggest that most mutations that are being accumulated during cancer evolution are deleterious to tumor fitness,7 and are called “passenger mutations”. Therefore, it might seem paradoxical that, in some cases, an increased number of somatic mutations predict a worse prognosis and a more rapid evolution.8 It is, therefore, important to stress that it is not only the number of mutations, but also the identity of the specific mutation acquired, that determines progression.9 Only these true “driver” mutations confer an advantageous phenotype.
Over recent years, studies aimed at depicting the clonal structure of AML have been published. These studies performed deep sequencing of primary AML samples taken from patients, patient-derived xenografts, and from in vitro cultures, and used the various somatic variant allele frequencies (VAF) as measures of clonal sizes [assuming no copy number variability (CNV) or loss of heterozygosities (LOH)]. These studies shed light on the selection and expansion that these clones undergo during their evolution and following therapy. Combining this information with clinical data from different time points improves our ability to predict treatment outcomes, enabling us to personalize therapy. Moreover, this approach raises the hope that healthy individuals can be screened for AML, and maybe even preventing the disease; something that was once considered unachievable.
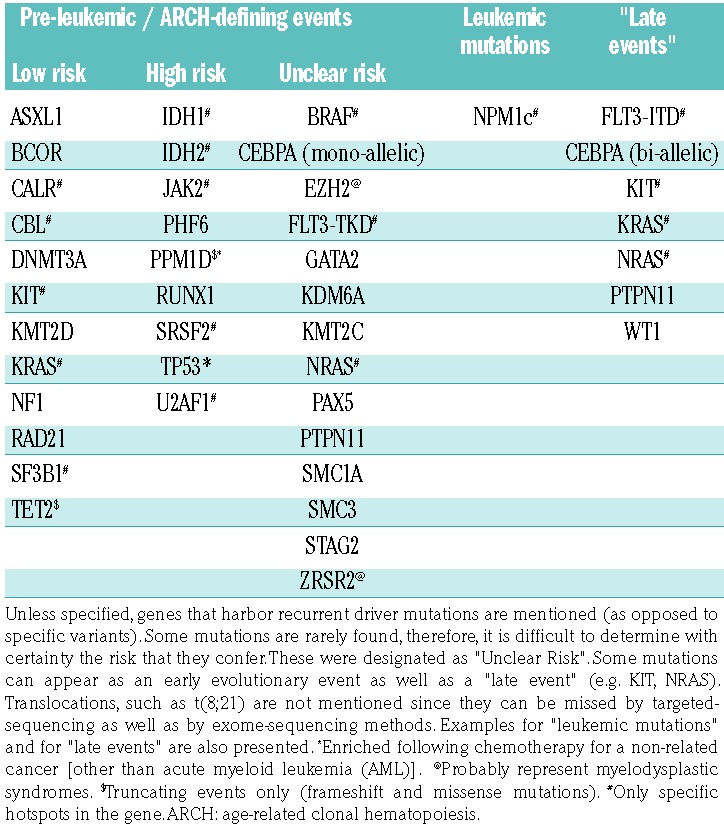
The clonal evolution of most AML subtypes can be viewed as a multistep process. It is now well accepted that this process can be schematically divided into three stages according to the clinical presentation. Each evolutionary stage has a different time frame and is characterized by typical somatic mutations that are, therefore, categorized into three groups according to the timing of their appearance: pre-leukemic, leukemic, and late events (Table 1). Although acute promyelocytic peukemia (APL), AML with KDM2A translocations, and the core-binding factor (CBF) leukemias do not have a clear pre-leukemic stage, they too develop over time and acquire late events, as discussed below.
Table 1.
Age-related clonal hematopoiesis defining events and the risk that they confer for acute myeloid leukemia progression.9,22
This review summarizes our current knowledge regarding somatic mutations in AML, their contribution to clonal fitness under different selective environmental conditions, and their correlation with patients’ clinical outcomes.
Pre-leukemic stage
Pre-leukemic mutations are somatic mutations that are found in leukemic blasts as well as in hematopoietic progenitors and mature cells from different lineages that share a common ancestral stem cell. This stem cell, which is still capable of differentiation, is defined as a pre-leukemic hematopoietic stem and progenitor cell (preL-HSPC).10,11 Such preL-HSPCs were isolated from AML patients at diagnosis, remission and relapse.10–12 By definition, the term pre-leukemic can only be inferred retrospectively, after the diagnosis of AML has been made.
Following these findings, somatic mutations were identified in the hematopoietic system of healthy individuals in various allele frequencies increasing with age, a phenomenon that was termed age-related clonal hematopoiesis (ARCH).13,14 In fact, deep sequencing techniques revealed the presence of DNMT3A and TET2 mutations in nearly all individuals; however, most of them were at lower VAF, in comparison to the original reports (median VAF 0.0024), and remained stable over time.15 Such a ubiquitous phenomenon probably represents the general structure of the aging human hematopoietic system, as the same findings could not be replicated among young individuals (aged 20-29 years).16 The large number of human hematopoietic stem cells (HSC) (estimated to be within the range of 50,000-200,000)17 and the number of somatic mutations in each adult single HSC (approx. 1000 mutations) suggest an estimated HSC pool mutation burden of 108,17 and explain why somatic mutations can be present at low VAF in every individual.14 However, with age, an exponential increase in the prevalence of ARCH13,14,16 occurs. In addition, this correlation with age is specific to mutations found in DNMT3A, TET2 and a few additional candidate driver genes,14 suggesting that mutations in these genes confer a selective advantage to HSPCs. The selective advantage that mutations in DNMT3A and TET2 confer is probably introduced during the third or fourth decade of life. A second selective advantage is introduced later, during the fifth decade and onward, reflecting the aging BM selecting for HSC-carrying spliceosome machinery mutations (SRSF2, U2AF1, SF3B1, ZRSR2, DDX41, EZH2, ASXL1, etc.).18,19 The fact that different individuals can carry clones of different sizes at the same age suggests differences in risk factors for clonal expansion.
The presence of such a mutated clone in allele frequency of more than 2% is termed clonal hematopoiesis of indeterminate potential (CHIP).20 It was found to be associated with development of hematologic cancers, cardiovascular morbidity, chronic obstructive pulmonary disease, and with an increase in all-cause mortality.13,14,21 While these clones predict a somewhat dismal prognosis, only a small number of individuals that harbor such a clone will eventually develop AML (approx. 0.1-3%).13,14
Nevertheless, since some of these clones do evolve into full-blown leukemia, these observations can be exploited to build a model for early detection of AML, at its pre-clinical, pre-leukemic phase. In fact, two recently published papers studied large prospective cohorts of healthy individuals and compared benign clonal hematopoiesis (one that did not evolve into AML) with malignant, pre-AML, clonal hematopoiesis. These studies used deep targeted sequencing methods to search for mutations in driver genes in a total of 307 cases (individuals that subsequently developed AML) and 626 age- and gender-matched controls (individuals that did not develop AML) in blood samples obtained 6-9 years before AML diagnosis. These studies found discriminative characteristics between the two cohorts.9,22
Clone size
A pre-leukemic state is manifested by an increased incidence of clonal hematopoiesis. Setting a 10% threshold for VAF value significantly discriminated pre-AML from controls. Thirty-nine percent of pre-AML individuals have clones of this size, as opposed to 4% of control individuals. Although statistically significant, there is a large overlap between VAF values of driver mutations found in benign and in malignant ARCH (especially for DNMT3A and TET2 mutations) that precludes it from being a single predictor of a rare disease such as AML.
Number of accumulated mutations
Pre-AML individuals have significantly more mutations in driver genes (including a few variants in the same gene, e.g. in DNMT3A22) per individual (not necessarily in the same clone) when compared with controls. This is especially evident among older individuals (>60-65 years of age) underscoring the time frame required for mutations to accumulate. Nevertheless, it is important to note that a substantial number of patients (20-46%) do develop AML without having a mutation in any driver gene prior to the diagnosis. This decreases the negative predictive value of these models.
When measured at a certain time point, these two characteristics indirectly reflect increased clonal fitness, as manifested by increased expansion and increased number of replications with the accumulation of mutations over time. These characteristics were also found to be predictive of progression to myeloid neoplasms when found during the evaluation of unexplained cytopenias.8
Specific high-risk mutations
Progression to AML was found to be preceded by accumulation of specific high-risk mutations. These mutations were more prevalent among pre-leukemic clonal hematopoiesis when compared to benign ARCH. Specifically, the presence of spliceosome-machinery mutations in SRSF2 P95R and U2AF1 Q157P as well as in TP53, in IDH1 R132, in IDH2 R140 and in RUNX1 (even at VAF values <10%) confer the highest risk for subsequent AML development in healthy individuals.9,22 When these clones appear at a relatively young age (>50 years) they tend to evolve into AML. The simple explanation for this could be that there is more time for AML transformation to take place. Another explanation could be that the environment that positively selected this clone continues to exert its selective pressure, eventually leading to AML. Table 1 summarizes the various ARCH-defining events and the risk that each of them confers for AML progression.
Temporal progression
While some low-risk clones can remain stable over a period of 3-10 years,15 clones that are characterized by high-risk mutations show a more rapid increase in their size, as manifested by an increase in their VAF values over time9,22 (Figure 1). Additional prospective cohorts might better define which clone develops to other hematologic malignancies [e.g. myelodysplastic syndromes (MDS) or myeloproliferative neoplasms (MPN)].
Figure 1.

High-risk and low-risk pre-leukemic somatic mutations. X: an acquired somatic mutation. Clone size [as manifested by variant allele frequency (VAF) value] is represented by the size of the oval shape. Clonal expansion is represented by the rising curve. (A) Low-risk age-related clonal hematopoiesis (ARCH) mutations, such as DNMT3A or TET2 mutations, are acquired at a relatively young age (marked in white). Most of these clones will not progress to acute myeloid leukemia (AML). (B) Pre-leukemic clones, characterized by similar low-risk mutations have an increased fitness (as manifested by an increased VAF). They acquire additional pre-leukemic mutations (marked in yellow and red), not necessarily in the same clone. These cells are hematopoietic stem and progenitor cells (HSPCs), still capable of differentiation and sustain hematopoiesis. Once a clone acquires a leukemic, transforming, mutation (NPM1, for instance, marked in green) it will progress rapidly to an overt AML with loss of differentiation capacity and uncontrolled proliferation. Retrospectively, its preceding clones are referred to as pre-leukemic. Leukemic mutations are shared by all the leukemic blasts, hence they have a high VAF (50%) in the leukemic clone. Late events (e.g. FLT3-ITD, marked in purple) appear later along the AML evolutionary trajectory, are shared by subclones, and represent the clonal heterogeneity of the leukemia; they have VAFs ≤50%. The exact timing of AML diagnosis can vary. Therefore, late events are usually already present when the actual diagnosis is made. Single cells or sub-populations have to be sequenced in order to accurately determine the order of acquisition of the mutations. (C) Spliceosomal machinery, and other high-risk mutations (such as SRSF2, U2AF1, IDH1, IDH2 and TP53 that are marked in pink) are usually acquired at a more advanced age. These clones expand more rapidly (as manifested by the rate of increase of their VAF value) and most will lead to AML.
Progression to AML depends on the identity of the initiating mutation and on the identity of additional mutations that are subsequently accumulated. It is conceivable that many factors influence the timing of the appearance of the mutation and the positive selection of such a clone, among which are probably the underlying specific germline background.
In addition, specific environmental pressures confer a selective advantage to HSCs carrying specific mutations; clear examples are TP53 and PPM1D mutations that are enriched following exposure to chemotherapy and radiotherapy. Chemotherapy does not increase the number of somatic single nucleotide variants or the percentage of chemotherapy-related transversions. Rather, it positively selects for pre-existing TP53 and PPM1D mutated clones.23–29
Moreover, a third-generation, single-molecule real-time sequencing assay with long-read length of AML and MDS samples exposed different TP53 variants residing on different alleles in each sample.30 This emphasizes the importance of the environmental conditions that select a certain phenotype, thus enabling the evolution of a few clones in parallel, all sharing similar driver mechanisms (TP53 mutations). It is important to note that chemotherapy exerts a selective pressure regardless of the specific mutation that characterizes the pre-leukemic clone. Following chemotherapy, BM is enriched with pre-leukemic clones and their prevalence increases by 10% or even 30% among younger and elderly individuals, respectively, when compared to their prevalence in the general age-matched population.24,13 The clones that were selected can neither be categorized according to a certain mutation, nor according to a certain chemotherapy (with the exception of topoisomerase II inhibitors, for which see below). However, they can be divided into three groups according to patient age groups, with younger individuals enriched with DNMT3A mutated clones. This holds true also for AML patients in remission that were found to have residual pre-leukemic clones.19,31 This implies that most pre-leukemic clones have an inherent chemoresistance (Figure 2), a phenomenon that was also shown in both in vivo and in vitro models.32
Figure 2.

Pre-leukemic clones have inherent chemoresistance. Pre-leukemic clones (blue) undergo positive selection by chemotherapy administered for a non-related cancer [other than acute myeloid leukemia (AML)]. They expand and evolve into t-AML (green). Pre-leukemic hematopoietic stem and progenitor cells (HSPCs) have inherent chemoresistance, thus they also survive following AML induction chemotherapy and reconstitute clonal hematopoiesis. Most relapses occur within the first 2 years and originate from residual leukemic clones that can be identified at diagnosis and that were not eradicated by AML therapy. Rare events of second AML (red) stem from (mostly, the same) pre-leukemic clones that evolved again into AML following a more prolonged latency.
The time frame of evolution from pre-leukemia to AML depends both on the context (extrinsic factors) and the driver mutations (intrinsic factors). Pre-leukemia in healthy individuals usually progresses slowly with a latency period that can sometimes be as long as 20 years. Presence of specific mutations was correlated with a shorter timeframe, as in the case of RUNX1 mutations (associated with a rapid progression to AML of <2 years) and of TP53 mutations.22 Specifically, following chemotherapy, TP53 mutated clones, as well as PPM1D mutated clones, evolve to hematologic malignancies within as few as 6 months to up to 10 years.23,24 Another example is MLL-rearranged AML; this usually develops within 6-18 months following exposure to topoisomerase II inhibitors. In contrast to therapy-related AML with somatic mutations in TP53 or PPM1D, where chemotherapy selects pre-existing mutated clones, MLL-rearrangement is assumed to be induced directly by topoisomerase II inhibitors.19,33
Although IDH mutations can be viewed as high-risk mutations, their presence was not found to be correlated with a shortened AML latency.22 This can be explained by the fact that some of the high-risk mutations occur early along the evolutionary trajectory of the clone; they can be considered as the initiating event (U2AF2, SRSF2, TP53 and RUNX1). Other high-risk mutations (IDH1 and IDH2) tend to appear later and require additional, co-operating, driver mutations in order to progress into AML. Indeed, certain combinations of mutations (when found in the same individual) can shorten the time to AML diagnosis, such as when DNMT3A is found with spliceosomal machinery mutations.22
Importantly, the size of the clone9 and the number of mutations identified9,22 mean a shortened interval for AML progression. Clones with increased fitness, as manifested by their size (VAF) and replication rate (number of mutations), are prone to acquire additional mutations until transforming, ‘leukemic’, mutations occur.
Leukemic stage
Molecular analysis of AML reveals a set of somatic mutations that are only present in the leukemic blasts but are absent in normal hematopoietic progenitors, in non-myeloid, and in mature cells. These mutations were not found in healthy individuals.9,13,14,22
These mutations can be further divided into leukemic mutations, which are shared by all the leukemic blasts, and late events. While the former represent the leukemic transformation, the latter represent subclones, as can be inferred from their lower VAF value (Figure 1).
Since there are already subclones at the time of diagnosis, determining the order of acquisition of the mutations is limited by the sensitivity of the sequencing method used to detect rare clones. It can be achieved by reconstructing the phylogenetic tree of the leukemia with single cell or sub-population sequencing.34 However, there is a general consensus that one mutation can be considered to be leukemic; this is a small insertion/duplication in the terminal exon of NPM1 that causes the mutant protein to be aberrantly localized in the cytoplasm (hence, designated NPM1c). Detection of this mutation accompanies a dramatic change in the phenotype of the clone with a rapid proliferation and acquisition of additional mutations. When individuals were found to harbor an NPM1c mutated clone, they were subsequently diagnosed with NPM1c AML within up to 3 months.22,35 This mutation was most clearly shown to be a marker for leukemic blasts when risk for relapse was found to be correlated with its presence in blood samples of AML patients in remission.36
Targeting cells harboring transforming, leukemic, mutations seems plausible not only for monitoring purposes, but also for therapy.37 In fact, genomic editing of this mutation in cell lines as well as in primary human AML samples using the CRISPR/Cas9 system disrupted the mutant allele and led to nuclear re-localization of the protein. This reverted the leukemic phenotype, resulting in differentiation and a reduced proliferation rate. A nuclear export inhibitor had a similar effect on NPM1c, both at the molecular level as well as at the cellular level, when tested on a cell line and on primary AML samples. It also resulted in prolonged survival of NPM1c-mutated leukemic mice.38
Late events
Late events are mutations that appear later on during leukemic evolution, represent clonal selection and heterogeneity, and are not shared by all leukemic blasts. Examples for such variants are activating mutations in tyrosine kinase receptors (such as FLT3-ITD, KIT, RAS), emphasizing their role in increasing clonal proliferation capacity.39 Other examples are WT1,39 transcription factor CEBPA bi-allelic mutations,19 and del(7q) in TP53-mutated AML.23 Interestingly, there are some exceptions to these “rules”: some mutations that were detected in healthy individuals, for example, IDH2, were also described as late events in AML.34 In addition, the FLT3 D835 mutation, usually considered as a late event, was found to be pre-leukemic.9
Some late events co-occur with certain leukemic events, for example, the FLT3-ITD and NPM1 mutations.4 This strong link between leukemic mutations and late events, rather than between pre-leukemic mutations and late events, was recently demonstrated by Höllein et al.40 They described patients with NPM1-mutant AMLs that developed a NPM1 wild-type AML after therapy. Both types of leukemia evolved on a similar pre-leukemic background. Interestingly, FLT3-ITD was significantly more frequent among NPM1-mutant AML.40 Late events, such as FLT3-ITD or KIT mutations, can be found in unique AML subtypes, such as APL and CBF-AML, respectively, as described below.
Targeting cells according to late events can prove beneficial and was employed using tyrosine kinase inhibitors.41 Nevertheless targeting a few subclones can result in a positive selection of other subclones with a different genomic landscape.42 Monitoring residual clones using late events as clonal markers should be done with great care, since this approach might miss subclones lacking these markers.43,44
Unique acute myeloid leukemia subtypes
A few AML subtypes, such as CBF-AML and APL, are diagnosed by the presence of their typical chromosomal aberrations, regardless of actual blast count.45,46 These chromosomal abnormalities, which result in novel fusion RNA transcripts, are being used for monitoring minimal residual disease following therapy and dictate pre-emptive treatment upon detection.47,48 While this implies that these chromosomal aberrations should be regarded as leukemic events, some of the chromosomal abnormalities of CBF-AML were retrospectively detected in blood samples taken more than 10 years before the patients were diagnosed with AML,49 as well as up to 7 years following allogeneic transplantation in patients without evidence of disease.50 In fact, when stem cells and cells from various lineages were sorted from BM of CBF-AML patients in remission, t(8;21) translocations were identified in B cells as well as in myeloid cells. This suggests that this event occurred at the stem cell level, still capable of differentiation.51
A debate continues over the cell of origin of APL. It was shown that APL blasts have a gene expression profile with T-cell lymphoid features52 and that they can engraft immune-deficient mice.53 This raises the possibility that APL blasts (especially blasts of the hypogranular variant) originate in early multipotent progenitors prior to lineage commitment.54 Nevertheless, t(15;17) translocation was identified only in CD34 positive (CD34+) precursor cells of the myeloid lineage and not in B or T lymphocytes.55 Thus, it is generally accepted that the APL transforming event occurs at a more committed progenitor cell.56 In contrast to the aforementioned pre-leukemic HSPCs (e.g. DNMT3A mutated), these cells do not maintain multilineage hematopoiesis.
Leukemic cells that harbor APL and CBF-AML translocations acquire additional mutations as they evolve. Similarly to other AML subtypes, additional somatic mutations that were identified in these clones involved tyrosine kinase receptor genes (e.g. RAS, FLT3 and KIT).
Clinical implications and future challenges
Pre-leukemic stage: prediction and screening
A reliable screening strategy for a rare disease, such as AML with an estimated incidence of 4:100,000,57 should have a high positive predictive value. In order to improve current models, specific variants that were described in hematologic malignancies, rather than specific genes, should be used for screening. In addition, a better understanding of the selective pressures and the germline background, under which pre-AML clones evolve, is required. While such an understanding still remains elusive, the term “fitness” encompasses both clonal intrinsic factors and environmental selective pressures. Thus, exposing a detrimental, malignant, clonal evolution requires a dynamic, longitudinal follow up of healthy individuals, rather than relying on a single blood test that depicts a static picture of the hematopoietic clonal structure (as suggested by the term CHIP). Therefore, AML screening programs should use a 2% VAF threshold as well as a documentation of clonal temporal evolution (by having at least two assessments of the clonal mutational profile 6-12 months apart). This should be incorporated into a refined definition of ARCH.58 Documenting ARCH-defining events in two consecutive tests will allow a better characterization of the clones and increase the confidence in their status, thus facilitating patient risk stratification.
Screening for therapy-related AML can be performed by detecting pre-leukemic clones at the time of initial chemotherapy treatment (administered for a non-AML tumor), and patients should be monitored at least once again to define ARCH.59
Additional improvement in the positive predictive value of such a model might also require inclusion of yet undescribed non-genomic, evolutionary events that manifest in the epigenome or in post-transcriptional or post-translational landscapes of the pre-malignant clone. It is still not known what influences these events: whether there are cause-and-effect relations between specific mutations and these non-genomic events, or whether they are influenced by the environment itself. As an example of the latter, p53 (wild-type at the genomic level) was shown to acquire a mutant-like post-translational conformation following stimulation by growth factors in AML cells.60
Incorporating clinical data into prediction models is expected to improve their accuracy and enable risk stratification for individuals carrying ARCH.9 However, pre-AML individuals were shown to have only subtle abnormalities in their blood count measures, often within the normal limits, with a large overlap with values documented in controls.9,13,21
Pre-leukemic stage: prevention
Once prediction tools become more reliable, prospective, intervening clinical trials can be initiated. When planning such clinical trials two major challenges arise: 1) the low incidence of AML in the general population; and 2) its prolonged latency. The former can be mitigated by registering a large cohort of participants and by patient selection based on risk stratified according to clonal temporal progression (using the refined ARCH definition) and on their clinical data.8,9,13 Overcoming its prolonged latency requires a long follow up of the participants and might require a prolonged or indefinite treatment period to suppress pre-leukemic clones.
Targeting pre-leukemic clones as a means to prevent AML needs to be performed with caution. This might cause aplasia when the entire hematopoietic system is comprised by this clone (e.g. as is the case with a high VAF TET2 clone). This can also give a selective advantage to a different pre-leukemic clone that resides in the shared microenvironment. An effective intervention must target both the clone with its driver genes and the environment that enabled it to flourish.
Leukemic stage
Targeting leukemic mutations might be an effective way to eliminate the malignant clone,38 thus preventing relapse. This is particularly true in AML because most relapses arise during the first 2 years and stem from the original clone detected at diagnosis.34,61,62
Can pre-leukemic mutations serve as a target for therapy as well? On the one hand, all leukemic cells share these mutations. Targeting IDH1 using a specific inhibitor resulted in a 30% complete remission rate among 125 IDH1-mutated relapsed/refractory AML patients. These responses lasted a median of 8 months. Remission was accompanied by a decrease in IDH1 VAF values.63
Similar results were obtained when using an IDH2 specific inhibitor, with an overall response rate of 40.3% and a median response duration of 5.8 months.64 These results of phase I trials, using IDH1/2-targeted monotherapy, need to be repeated in larger cohorts of treatment-naïve patients before conclusions can be drawn. On the other hand, the use of early, pre-leukemic, mutations as markers of the malignant clone should be handled with caution since following chemotherapy, the presence of most residual pre-leukemic clones (carrying DNMT3A, TET2 or ASXL1 mutations) do not increase risk for early (4-year) relapse unless accompanied by other mutations.19,31
In addition, one should bear in mind that, in contrast to the pre-leukemic stage, once overt leukemia evolves, targeting pre-leukemic somatic mutations might not prove effective. This can be inferred from the fact that some patients develop relapsed NPM1-mutated leukemia, lacking their DNMT3A pre-leukemic mutations.65 This can represent a biological phenomenon in which the relative contribution of these early driver mutations to clonal fitness diminishes as the clone evolves to overt leukemia. Indeed, IDH1/2 inhibition induced differentiation of the malignant clone in only 5-7% of the patients and clearance of IDH1 mutated clone was noted only in 21% of clinically responding patients.63 In patients treated with an IDH2 inhibitor, differentiation of the blasts without elimination of the malignant clone was documented,64 in line with previous reports about IDH2 being acquired following AML transformation (as a late event).34 Improving efficacy might be achieved by combining drugs that target both leukemic and late events.
Relapse
Most AML patients experience relapse originating in leukemic stem cells (LSC) that belong to the leukemic clone and that can already be identified at the time of diagnosis. It is, therefore, imperative to identify and target these cells. Two main subtypes of AML were identified. The first AML subtype contains rare stem cells that have a stem/progenitor-like immunophenotype. In the other subtype, relapse originates from the major CD33+ blast population and is more dependent on growth factors when studied in vivo.34,61 Studying gene expression profiles of these subtypes revealed that this division is correlated with French-American-British (FAB) classification. The first subtype is enriched for FAB M4/M5 subtypes and the other is enriched for the less differentiated AML subtypes (M0/M1/M2). Nevertheless, relapse-initiating LSC in both groups had similar gene expression profiles and, as expected, the relapsing clone was found to be characterized by an increased number of LSC.34 Such a “leukemic stemness” transcriptional signature can be used to predict prognosis and to monitor patients in remission.66 Targeting LSC has been studied extensively in xenograft models but less so in clinical trials. A recent study suggests that the combination of Azacitidine and Venetoclax target LSCs, as identified both immune-phenotypically and by their transcriptomics signature.67 The number of participants in this trial was small, and, although this therapy does not induce remission in all patients (only in approx. 67%68), and some relapse while on therapy, this is an important step towards LSC-targeted therapy.
Survivorship
Following the elimination of the leukemic clone, chemotherapy-resistant clones, which can sometimes be detected in low VAF values at the time of AML diagnosis,29 expand and re-populate the BM.29,40 Some of these clones harbor ARCH mutations,29,31 and some of these clones are truly pre-malignant since they go on and evolve into MDS69 or a second AML, albeit following a more prolonged latency than the relapse of the original leukemia (median 33.7-43 months vs. 8.6-14 months, respectively). Second AML should be diagnosed as a distinct entity whenever leukemic mutations that characterize the primary (diagnosis) AML are not identified at relapse (Table 1). Second AML was described to occur in 10-14% of the patients experiencing relapse.40,65 However, second AML should not be considered as a relapse. Sometimes, second AML does not share its pre-leukemic mutations with the primary AML (Table 1). This is underlined by the identification of pre-leukemic clones of second AML that lack the primary AML pre-leukemic DNMT3A, TET2, SRSF2 or RUNX1 mutations.40,70 Nevertheless, most of these second-AML-initiating clones share the same early, pre-leukemic mutations as the primary AML clone,40,65 and some even evolve similarly to the primary clone and acquire a different mutation in the same gene,70,71 emphasizing the role of an environmental selective pressure (Figure 2).
In this regard, environmental influence is best demonstrated when patients that undergo allogeneic stem cell transplantation develop an AML that originates in the donor hematopoietic cells. Two main reasons can lead to this very rare outcome (estimated to occur following 0.08% of transplants72): 1) a pre-existing pre-leukemic clone in the stem cell donation; and 2) evolution of a new leukemic clone in the recipient following the transplantation. Although the stem cell source (BM vs. peripheral blood) did not influence the risk for donor cell leukemia, environmental factors seem to be crucial in promoting the malignant clone. Multivariate analysis revealed three risk factors associated with development of donor cell leukemia: 1) the use of growth factors; 2) in vivo T-cell depletion; and 3) having a previous allograft. These risk factors imply that a reduced immune surveillance and increased replication signals create a more permissive environment that allows the development of the malignant clone. As an emphasis, two different trajectory leukemic evolutions were described following a DNMT3A-mutated pre-leukemic clone donation. While the donor developed NPM1-mutated, FLT3-ITD AML, the recipient developed NPM1 SMC1A-mutated AML.73 Therefore, when monitoring AML patients in remission, predicting a rare, second AML becomes somewhat analogous to predicting transformation from pre-leukemia to AML. Here, too, some residual or newly evolving pre-leukemic clones confer increased risk for second AML development, heralded by clonal expansion. The exact risk stratification still needs to be validated by appropriately designed studies. These studies need to use broad sequencing panels instead of a panel dictated only by the mutations found at diagnosis.
Conclusions
Recently published studies reveal that the evolutionary trajectory of AML begins many years before the patient is actually diagnosed. It is a multistep process characterized by Darwinian evolution with clonal selection and expansion. Much is still unknown regarding the various factors that influence the path that clones in the hematopoietic system follow. They consist of both clon al-intrinsic as well as environmental factors. Both factors are influenced by each patient’s germline background. We can improve our understanding firstly by depicting the exact route that pre-leukemic clones take on their way to becoming AML. It is important to remember that the trajectory of these clones does not end when patients achieve a complete (even molecular) remission. Residual, as well as new pre-leukemic clones, continue to evolve along the same path. Familiarizing ourselves with the ontogeny of AML and incorporating it into clinical prac-tice will expand our therapeutic opportunities and grant us additional time points for intervention. Most importantly, a truly holistic treatment must also consider the environmental pressures under which AML evolved and address them. This will give a comprehensive meaning to the term “cure”.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/104/5/872
References
- 1.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. [DOI] [PubMed] [Google Scholar]
- 2.Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152. [DOI] [PubMed] [Google Scholar]
- 3.Estey E. Acute myeloid leukemia: 2016 update on risk-stratification and management. Am J Hematol. 2016;91(8):824–846. [DOI] [PubMed] [Google Scholar]
- 4.The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(9):2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McFarland CD, Korolev KS, Kryukov GV, Sunyaev SR, Mirny LA. Impact of deleterious passenger mutations on cancer progression. Proc Natl Acad Sci U S A. 2013;110(8):2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malcovati L, Gallì A, Travaglino E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129(25):3371–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ableson S, Collord G, Ng SWK, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559(7714):400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A. 2014;111(7):2548–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lal R, Lind K, Heitzer E, et al. Somatic TP53 mutations characterize preleukemic stem cells in acute myeloid leukemia. Blood. 2017;129(18):2587–2591. [DOI] [PubMed] [Google Scholar]
- 13.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7: 12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Acuna-Hidalgo R, Sengul H, Steehouwer M, et al. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am J Hum Genet. 2017;101(1):50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee-Six H, Øbro NF, Shepherd MS, et al. Population dynamics of normal human blood inferred from somatic mutations. Nature. 2018;561(7724):473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mckerrell T, Park N, Moreno T, et al. Leukemia associated somatic mutations drive distinct patterns of age related clonal hemopoiesis. Cell Rep. 2015;10(8):1239–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindsey RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Busque L, Buscarlet M, Mollica L, Levine RL. Concise review: age-related clonal hematopoiesis: stem cells tempting the devil. Stem Cells. 2018;36(9):1287–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buscarlet M, Provost S, Zada YF, et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. 2017;130(6):753–762. [DOI] [PubMed] [Google Scholar]
- 22.Desai P, Mencia-Trinchant N, Savenkov O, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. 2018;24(7):1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gibson CJ, Lindsley RC, Tchekmedyian V, et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol. 2017;35(14):1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coombs CC, Zehir A, Devlin SM, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. 2017;21(3): 374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu JI, Dayaram T, Tovy A, et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell stem Cell. 2018;23(5):700–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kahn JD, Miller PG, Silver AJ, et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood. 2018;132(11):1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindsley RC, Saber W, Mar BG, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med. 2017;376(6):536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong TN, Miller CA, Klco JM, et al. Rapid expansion of preexisting nonleukemic hematopoietic clones frequently follows induction therapy for de novo AML. Blood. 2016;127(7):893–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lodé L, Ameur A, Coste T, et al. Single- molecule DNA sequencing of acute myeloid leukemia and myelodysplastic syndromes with multiple TP53 alterations. Haematologica. 2018;103(1):e13–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378(13):1189–1199. [DOI] [PubMed] [Google Scholar]
- 32.Guryanova OA, Shank K, Spitzer B, et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med. 2016;22(12):1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broeker PL, Super HG, Thirman MJ, et al. Distribution of 11q23 breakpoints within the MLL breakpoint cluster region in de novo acute leukemia and in treatment-related acute myeloid leukemia: correlation with scaffold attachment regions and topoisomerase II consensus binding sites. Blood. 1996;87(5):1912–1922. [PubMed] [Google Scholar]
- 34.Shlush LI, Mitchell A, Heisler L, et al. Tracing the origins of relapse in acute myeloid leukemia to stem cells. Nature. 2017;547(7661):104–108. [DOI] [PubMed] [Google Scholar]
- 35.Grove CS, Bolli N, Manes N, et al. Rapid parallel acquisition of somatic mutations after NPM1 in acute myeloid leukemia evolution. Br J Haematol. 2017;176(5):825–829. [DOI] [PubMed] [Google Scholar]
- 36.Ivy A, Hills RK, Simpson MA, et al. Assessment of minimal residual disease in standard-risk AML. N Engl J Med. 2016; 374(5):422–433. [DOI] [PubMed] [Google Scholar]
- 37.Yap TA, Gerlinger M, Futreal PA, Pusztai L, Swanton C. Intratumor heterogeneity: seeing the wood for the trees. Sci Transl Med. 2012;4(127):127ps10. [DOI] [PubMed] [Google Scholar]
- 38.Brunetti L, Gundry MC, Sorcini D, et al. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell. 2018;34(3):499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Höllein A, Meggendorfer M, Dicker F, et al. NPM1 mutated AML can relapse with wild-type NPM1: persistent clonal hematopoiesis can drive relapse. Blood Adv. 2018;2(22):3118–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pemovska T, Kontro M, Yadav B, et al. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 2013;3(12):1416–1429. [DOI] [PubMed] [Google Scholar]
- 43.Shouval R, Shlush LI, Yehudai-Resheff S, et al. Single cell analysis exposes intra-tumor heterogeneity and suggests that FLT3-ITD is a late event in leukemogenesis. Exp Hematol. 2014;42(6):457–463. [DOI] [PubMed] [Google Scholar]
- 44.Abdelhamid E, Preudhomme C, Helevaut N, et al. Minimal residual disease monitoring based on FLT3 internal tandem duplication in adult acute myeloid leukemia. Leuk Res. 2012;36(3):316–323. [DOI] [PubMed] [Google Scholar]
- 45.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. [DOI] [PubMed] [Google Scholar]
- 46.Swerdlow SH, Campo E, Harris NL, et al., editors. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008. [Google Scholar]
- 47.Grimwade D, Jovanovic JV, Hills RK, et al. Prospective minimal residual disease monitoring to predict relapse of acute promyelocytic leukemia and to direct pre-emptive arsenic trioxide therapy. J Clin Oncol. 2009;27(22):3650–3658. [DOI] [PubMed] [Google Scholar]
- 48.Jourdan E, Boissel N, Chevret S, et al. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood. 2013;121(12):2213–2223. [DOI] [PubMed] [Google Scholar]
- 49.Wiemels JL, Xiao Z, Buffler PA, et al. In utero origin of t(8;21) AML1-ETO translocations in childhood acute myeloid leukemia. Blood. 2002;99(10):3801–3805. [DOI] [PubMed] [Google Scholar]
- 50.Jurlander J, Caligiuri MA, Ruutu T, et al. Persistence of the AML1/ETO fusion transcript in patients treated with allogeneic bone marrow transplantation for t(8;21) leukemia. Blood. 1996;88(6):2183–2191. [PubMed] [Google Scholar]
- 51.Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci U S A. 2000;97(13):7521–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chapiro E, Delabesse E, Asnafi V, et al. Expression of T-lineage-affiliated transcripts and TCR rearrangements in acute promyelocytic leukemia: implications for the cellular target of t(15;17). Blood. 2006;108(10): 3484–3493. [DOI] [PubMed] [Google Scholar]
- 53.Ailles LE, Gerhard B, Kawagoe H, Hogge DE. Growth characteristics of acute myelogenous leukemia progenitors that initiate malignant hematopoiesis in nonobese diabetic/severe combined immunodeficient mice. Blood. 1999;94(5):1761–1772. [PubMed] [Google Scholar]
- 54.Grimwade D, Enver T. Acute promyelocytic leukemia: where does it stem from? Leukemia. 2004;18(3):375–384. [DOI] [PubMed] [Google Scholar]
- 55.Haferlach T, Löffler H, Nickenig C, et al. Cell lineage specific involvement in acute promyelocytic leukaemia (APL) using a combination of May-Grunwald-Giemsa staining and fluorescence in situ hybridization techniques for the detection of the translocation t(15;17)(q22;q12). Br J Haematol. 1998;103(1):93–99. [DOI] [PubMed] [Google Scholar]
- 56.Turhan AG, Lemoine FM, Debert C, et al. Highly purified primitive hematopoietic stem cells are PML-RARA negative and generate nonclonal progenitors in acute promyelocytic leukemia. Blood. 1995;85(8): 2154–2161. [PubMed] [Google Scholar]
- 57.SEER program [Internet]. National Cancer Institute; 2018. - [cited 2019 January 10]. Available from: https://seer.cancer.gov/ [Google Scholar]
- 58.Shlush LI. Age related clonal hematopoiesis. Blood. 2018;131(5):496–504. [DOI] [PubMed] [Google Scholar]
- 59.Berger G, Kroeze LI, Koorenhof-Scheele TN, et al. Early detection and evolution of preleukemic clones in therapy-related myeloid neoplasms following autologous SCT. Blood. 2018;131(16):1846–1857. [DOI] [PubMed] [Google Scholar]
- 60.Zhang W, Deisseroth AB. Conformational change of p53 protein in growth factor-stimulated human myelogenous leukemia cells. Leuk Lymphoma. 1994;14(3-4):251–255. [DOI] [PubMed] [Google Scholar]
- 61.Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parkin B, Ouillette P, Li Y, et al. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood. 2013;121(2):369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Di Nardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386–2398. [DOI] [PubMed] [Google Scholar]
- 64.Stein EM, Di Nardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krönke J, Bullinger L, Teleanu V, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122(1):100–108. [DOI] [PubMed] [Google Scholar]
- 66.Ng SW, Mitchell A, Kennedy JA, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540(7633):433–437. [DOI] [PubMed] [Google Scholar]
- 67.Pollyea DA, Stevens BM, Jones CL, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24(12):1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Di Nardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Herold S, Sockel K, Sayehli C, et al. Evolution of NPM1-negative therapy-related myelodysplastic syndromes following curative treatment of NPM1- mutant AML. Leukemia. 2017;31(10):2247–2251. [DOI] [PubMed] [Google Scholar]
- 70.Kohlmann A, Nadarajah N, Alpermann T, et al. Monitoring of residual disease by next-generation deep-sequencing of RUNX1 mutations can identify acute myeloid leukemia patients with resistant disease. Leukemia. 2014;28(1):129–137. [DOI] [PubMed] [Google Scholar]
- 71.Webersinke G, Kranewitter W, Deutschbauer S, et al. Switch of the mutation type of the NPM1 gene in acute myeloid leukemia (AML): relapse or secondary AML? Blood Cancer J. 2014;4(6):e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Engel N, Rovo A, Badoglio M, et al. European experience and risk factor analysis of donor cell-derived leukaemias/MDS following haematopoietic cell transplantation. Leukemia. 2019;33(2):508–517. [DOI] [PubMed] [Google Scholar]
- 73.Hahn CN, Ross DM, Feng J, et al. A tale of two siblings: two cases of AML arising from a single pre-leukemic DNMT3A mutant clone. Leukemia. 2015;29(10):2101–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.