

Abstract
Ruxolitinib is the front-line non-palliative treatment for myelofibrosis (MF). However, a significant number of patients lose or present suboptimal response, are resistant or have unacceptable toxicity. In an attempt to improve response and avoid the adverse effects of this drug, we evaluated the combination of 17 drugs with ruxolitinib in ex vivo models of peripheral blood mononuclear cells from MF patients and cell lines. We found that the combination ruxolitinib and nilotinib had a synergistic effect against MF cells (ΔEC50 nilotinib, −21.6%). Moreover, the addition of prednisone to combined ruxolitinib/nilotinib improved the synergistic effect in all MF samples studied. We evaluated the molecular mechanisms of combined ruxolitinib/nilotinib/prednisone and observed inhibition of JAK/STAT (STAT5, 69.2+11.8% inhibition) and MAPK (ERK, 29.4+4.5% inhibition) signaling pathways. Furthermore, we found that the triple therapy combination inhibited collagen protein and COL1A1 gene expression in human bone marrow mesenchymal cells. Taken together, we provide evidence that combined ruxolitinib/nilotinib/prednisone is a potential therapy for MF, possibly through the anti-fibrotic effect of nilotinib, the immunomodulatory effect of ruxolitinib and prednisone, and the anti-proliferative effect of ruxolitinib. This combination will be further investigated in a phase Ib/II clinical trial in MF.
Introduction
Myelofibrosis (MF) is a Philadelphia chromosome-negative chronic myeloproliferative neoplasm (MPN) clinically characterized by stem cell-derived clonal myeloproliferation and a reactive cytokine-driven increase in bone marrow (BM) fibrosis.1,2 Patients with MF have a poor prognosis and a median survival of 5.8 years.1
Dysregulation of JAK/STAT signaling is the main cause of MPN and, accordingly, inhibitors of the JAK/STAT signal transduction pathway are currently the best clinical approach to treat this disease. Discovered in 2005,3–7 a mutation in the JAK2 gene resulting in a substitution of valine for phenylalanine (V617F) was found in approximately 90% of patients with polycythemia vera (PV), and in 50-60% of patients with essential thrombocythemia (ET) and primary myelofibrosis (PMF).8 In addition, mutations in the MPL gene, which encodes the thrombopoietin receptor, were found in approximately 1% of patients with MPN,9 and 12% of patients with MPN (35-50% of MF) have mutations in calreticulin (CALR).10–12 Interestingly, the mutated forms of CALR acquire the ability to activate the thrombopoietin receptor and, therefore, constitutively activate the JAK/STAT pathway.13,14
Ruxolitinib, a JAK1/JAK2 inhibitor, is the first and only drug approved by the European Medicines Agency for the treatment of PMF, post-PV MF, and post-ET MF15 and is the first-line treatment for MF. Results of the COMFORT-I and II clinical trials showed that ruxolitinib produced a reduction in spleen volume, improved MF-related symptoms, and was associated with prolonged overall survival of patients compared with controls.15
Despite the beneficial effect of ruxolitinib, a high percentage of patients lose their response at some point during treatment, and others are refractory or present a suboptimal response. Because of this, the use of drug combinations might increase the effectiveness of the treatment and response time, and overcome treatment resistance. Indeed, numerous studies have used this premise, and a number of combinations have been tested in clinical trials with varying success. For instance, whereas the combination of ruxolitinib with simtuzumab (clinicaltrials.gov identifier: 01369498) produced no clinical benefit,16 and the combination with lenalidomide (clinicaltrials.gov identifier: 01375140) had to be terminated early because the efficacy objectives were not achieved,17 the combination with danazol (clinicaltrials.gov identifier: 01732445) achieved a hematologic stabilization but did not increase the response to ruxolitinib.18 Other combinations including ruxolitinib with buparlisib (clinicaltrials.gov identifier: 01730248) or with panobinostat (clinicaltrials.gov identifiers: 01693601 and 01433445) are currently under evaluation in clinical trials. In this scenario, the objective of the present study was to develop a drug combination that enhances the effect of ruxolitinib in the treatment of MF.
The proposed combination in this work, ruxolitinib, nilotinib and prednisone, is the result of testing 17 drugs with ruxolitinib to evaluate the best therapy for MF. We hypothesized that this combination would be synergic through a decrease in the proinflammatory status by ruxolitinib and prednisone19 and the known antifibrogenic effect of nilotinib,20 and would result in a better histological response.
Methods
Primary samples and cell lines
Peripheral blood (PB) samples were collected from MF patients and from healthy donors after obtaining informed consent in accordance with the guidelines of the 12 Octubre Hospital ethics committee and the Declaration of Helsinki. The diagnosis of MF was based on 2016 World Health Organization criteria.21 PB mononuclear cells (PBMCs) were isolated from 6-10 mL of PB by density gradient centrifugation (Ficoll-Paque™ PLUS, GE Healthcare, Little Chalfont, UK) and were cultured (0.4×106 cells/mL) in Methocult TM GF_H4535 supplemented with 20 ng/mL IL-3 and 50 ng/mL Stem Cell Factor (both from StemCell Technologies, Vancouver, Canada) at 37°C in a humidified atmosphere containing 5% CO2. For the drug screening study, samples from 9 patients were used; age range was 49-83 years, there were 5 males and 4 females, and 6 of them harbored the JAK2-V617F mutation (Online Supplementary Table S1). For synergy studies, all patients (aged 66-83 years) had the JAK2-V617F mutation (3 males and 2 females). No patient had been treated previously (Online Supplementary Table S2).
The BA/F3 wild-type (BA/F3 WT), BA/F3 with JAK2-V617F mutation (BA/F3 V617F JAK2), and WEHI cell lines were kindly provided by Dr Quintás-Cardama (MD Anderson Cancer Center, Houston, TX, USA). The WEHI cell line, which produces IL-3, was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Biowest, Nuaillé, France) with 10% heat-inactivated fetal bovine serum (FBS). BA/F3 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Biowest, Nuaillé, Francia) with 10% FBS plus 10% conditioned medium from WEHI cells. The SET2 cell line (DSMZ, Braunschweig, Germany), which harbors the JAK2-V617F mutation, was cultured in RPMI 1640 with 20% FBS. The HS27a human BM mesenchymal cell line (DSMZ) was cultured in DMEM with 10% FBS.
Dose response and synergy analysis
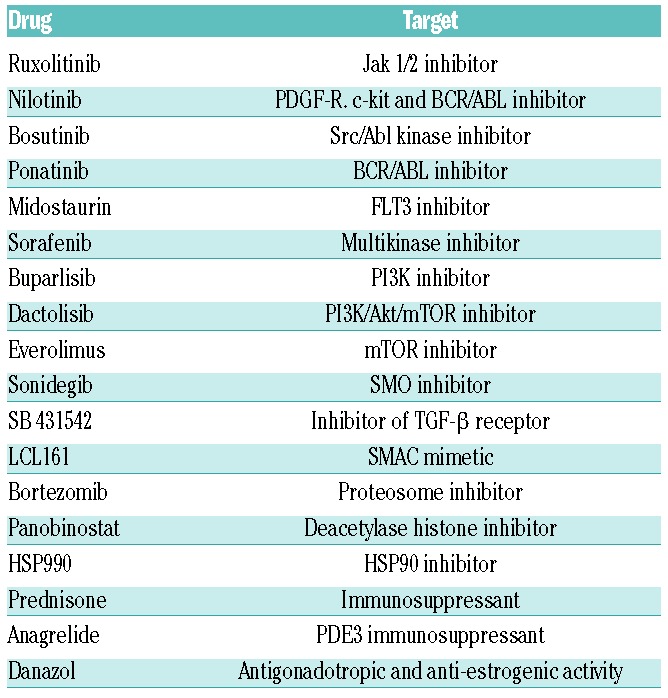
A total of 10,000-20,000 cells of the different cell lines were seeded per well in 96-well plates in the presence of the drugs alone (Table 1 and Online Supplementary Table S3) or in combination with ruxolitinib. Dimethyl sulfoxide (DMSO) was used as vehicle. After 48-72 hours (h), cell viability was measured by flow cytometry with Annexin V-phycoerythrin (Biolegend, San Diego, CA, USA) or the metabolic WST-8 assay (Cell Counting Kit - 8 BioChemika; Sigma-Aldrich). Drugs were purchased from Sigma-Aldrich (St. Louis, MO, USA), Tocris (Bristol, UK) or kindly donated by Novartis (Basel, Switzerland).
Table 1.
Drugs used in the screening to search for the best combination with ruxolitinib.
Peripheral blood mononuclear cells were treated as follows: a) directly delivering the drugs to methylcellulose solid culture at the outset of the experiment (ex vivo, model A); or b) after 14 days of methylcellulose culture (ex vivo, model B). In model B, colony-forming cells were collected, washed with phosphate buffer saline (PBS) and cultured in RPMI with 10% FBS at 15,000 cells per well in 96-well plates in the presence of the drugs alone (Table 1) or in combination with ruxolitinib, for 72 h. DMSO was used as vehicle at a maximum concentration of 0.5%. Flow cytometry to measure myeloid cell viability was performed with monoclonal antibodies against CD45-allophycocyanin-Cy7, CD13-allophycocyanin and Annexin V-phycoerythrin (all from Biolegend, San Diego, CA, USA) using the ExviTech automated flow cytometry plataform.
Collagen I expression study
Hs27a cells were treated with 100 nM ruxolitinib, 1 μM nilotinib or 1 μM prednisone or their combinations for 1 h. Subsequently, 2 ng/mL of TGF-β (R&D Systems, Minneapolis, MN, USA) was added and cells were incubated for a further 24 h. Immunocytochemistry and quantitative polymerase chain reaction (qPCR) analysis was used to measure collagen I expression.
Protein array and western blotting
The effects of 32 nM ruxolitinib, 1.6 μM nilotinib, 0.8 μM prednisone and their combinations, on protein phosphorylation were analyzed using the Human Phospho-kinase Array (Proteome ProfilerTM, R&D Systems) and by western blotting. Antibodies against phosphorylated or non-phosphorylated STAT5 and ERK 1/2 proteins (Cell Signaling Technology, Danvers, MA, USA) were used in western blotting analysis. Tubulin was used as a loading control and was purchased from Abcam (Cambridge Science Park, Cambridge, UK). Proteins were visualized with the ChemiDoc MP imaging system (BioRad laboratories, Hercules, CA, USA), quantified, corrected for housekeeping expression, and normalized to control samples using the ImageLab software program (v.5.1, BioRad).
Quantitative polymerase chain reaction
Total RNA was prepared with the AllPrepTM DNA/RNA Micro Kit (Qiagen, Hilden, Germany). Reverse transcription reaction was carried out using the High Capacity cDNA Reverse Transcription Kit system (Life Technologies, Carlsbad, MA, USA). Real-time PCR was performed with Taqman Gene Expression Master Mix and gene-specific Taqman probe COL1A1 (Hs00164004_m1) using the 7900HT Fast Real-Time PCR Systems platform (all from Life Technologies). Normalized gene expression levels were calculated using GAPDH mRNA expression as a housekeeping gene.
Immunocytochemistry
HS27a cells were fixed with 4% paraformaldehyde (Merck Millipore, Billerica, MA, USA) permeabilized and blocked with 0.25% Triton X-100 plus 1% BSA in PBS for 30 minutes (min). Slides were incubated with antibodies against collagen I (Abcam) for one hour, followed by a 5-min incubation with 3% H2O2 to inactivate endogenous peroxidase. After incubation with a peroxidase-conjugated secondary antibody for 1 h, signals were revealed with 3,3 diaminobenzidine (Abcam). Counterstaining was performed with Carazzi’s hematoxylin (AppliChem Panreac, Darmstadt, Germany). Images were visualized on the Eclipse 80i (Nikon) microscope equipped with a DS-Fi1 camera (Nikon, Minato, Tokyo, Japan). Stained areas were calculated with ImageJ (Rasband, W.S., ImageJ, NIH, Bethesda, MD, USA).
Statistical analysis
The analysis of drug dose-response was performed using the non-linear regression model (Equation 1):
where C is the drug concentration; E is the drug effect; Emax, the maximum drug efficacy in terms of survival; E0, survival when only DMSO is applied; EC50, drug concentration in which 50% of the total drug action is achieved; and γ the slope of the curve. The area under curve (AUC) of dose response curves was also calculated.
The study of the behavior of drugs in combination was performed using ΔEC50, the percentage of difference between EC50 of each drug in combination with ruxolitinib minus their EC50 in monotherapy.
Synergy analysis was performed using Calcusyn v.2.0 (Biosoft, Ferguson, MO, USA). The calculations performed by the program are based on the median-effect equation formulated by Chou.22 The combination index (CI) is the parameter by which the synergy or antagonism of two drugs were quantified (Equation 2):
where CD is the concentration of each drug, CR is the ruxolitinib concentration, and is the concentration of a drug in the presence of another drug that causes a certain effect. A CI <0.8 indicates synergism.
Shapiro-Wilk and Levene’s robust test statistic were applied to evaluate normality and homoscedasticity, respectively. Linear regression was performed for the correlation of time of response with ex vivo activity of ruxolitinib. For the statistical analysis of phospho-kinase array, an ANOVA test was performed. A t-test was used to assess whether the CI of each combination was significantly synergistic. For collagen expression assays and phosphoproteomics studies, Student t-test was used when the populations were normal and the non-parametric Wilcocox t-test when they were not. P<0.05 was considered statistically significant. Statistical analyses were performed with GraphPad Prism v.6.00 for Windows (GraphPad Software, La Jolla, CA, USA) or STATA v.13 (StataCorp., College Station, TX, USA).
Results
Ruxolitinib activity in cell lines and patients’ samples
We first evaluated the activity of ruxolitinib in JAK2-mutated cell lines. Ruxolitinib efficiently inhibited the viability of BA/F3 and SET2 V617F JAK2 cells with an EC50 of 35 nM and 25 nM, respectively (Online Supplementary Figure S1A). The EC50 for ruxolitinib in BA/F3 WT cells was 212 nM, indicating the importance of the JAK2-V617F mutation for the activity of ruxolitinib. Nonetheless, when we compared the activity of ruxolitinib in patients’ PBMCs with or without a JAK2 mutation, using ex vivo model A, we found that its activity was not significantly different, with an EC50 of 55 nM. For this reason, subgroups based on the mutation in JAK2 were not studied further.
To determine the best cell model to screen drugs in combination with ruxolititinb, its activity was tested in the two different ex vivo culture models. The only method that provided a sufficient number of cells for screening was model B, although the EC50 for ruxolititinb using this model was 0.747 μM. Greater ruxolitinib activity was found when PBMCs were seeded in methylcellulose in the presence of ruxolitinib (model A: EC50 = 43 nM) (Online Supplementary Table S4 and Online Supplementary Figure S1B). Moreover, if ex vivo activity of ruxolitinib was compared with the time of response to ruxolitinib of each patient sample, it was found that both models A and B distinguished patients’ samples with responses >6 months (Online Supplementary Figure S1C).
BCR/ABL or ABL kinase inhibitors and PDGFR and TGFβ R inhibitors are effective combinations with ruxolitinib in cell lines and patients’ samples
To examine the best combination with ruxolitinib, dose-response curves of all tested drugs in monotherapy or in combination with ruxolitinib were first analyzed in BA/F3 V617F JAK2 cells using an automated flow cytometry platform. Drugs exhibiting the best behavior in the presence of ruxolitinib were then selected to perform the same assay using PBMCs of MF patients in ex vivo model B. Drugs with more activity in the screening were also included in dose-response assays in monotherapy with patients’ samples.
Results showed that the BCR/ABL or SRC/ABL tyrosine kinase inhibitors (TKI) nilotinib and bosutinib, respectively, together with danazol, a synthetic androgen reported to reverse anemia,23 and SB432542, an inhibitor of the TGF-β receptor related to the fibrogenic processes, were among the four best combinations in BA/F3 JAK2 V617F cells (Figure 1A). Accordingly, they presented the lowest increments between their EC50 in the presence or absence of ruxolitinib (ΔEC50 nilotinib = −92.4%; ΔEC50 bosutinib = −87.7%; ΔEC50 danazol = −80.1%; ΔEC50 SB432542 = −77.1%). When tested in patients’ samples, of the two BCR/ABL inhibitors, only nilotinib showed a lower EC50 in the presence of ruxolitinib than in its absence (ΔEC50 nilotinib = −21.6%), together with SB432542 (ΔEC50 = −11.7%) (Figure 1B).
Figure 1.

Effect of each drug in combination with 100 nM ruxolitinib in BA/F3 JAK2-V617F3 cell line (A) or in patients’ peripheral blood mononuclear cells (B). y-axis: the increment between the EC50 for each drug in the presence of ruxolitinib minus its EC50 in monotherapy. Results are expressed as the mean±Standard Deviation (SD) of 2 independent experiments in cell lines (A) and median and interquartile range in patients’ samples (B).
Online Supplementary Table S5 shows the drugs listed by their Emax and EC50. The most active drugs in BA/F3 JAK2 V617F cells were the histone deacetylase HDAC6 inhibitor panobinostat (EC50 = 0.041μM), the proteasome inhibitor bortezomib (EC50 = 0.041 μM), and the immunomodulatory HSP90 inhibitor HSP990 (EC50 = 0.045 μM). Interestingly, these inhibitors were also the most active drugs in patients’ samples (Table 2), showing an EC50 of 0.008 μM, 0.033 μM and 0.041 μM, respectively. Furthermore, prednisone, an immunosuppressant used to control symptoms of MF by decreasing the levels of cytokines and growth factors such as TGF-β,24 showed an EC50 of 0.144 μM. Other active drugs in the low micromolar range included the signaling pathway inhibitors BKM120 (EC50 = 3.331 μM) and ponatinib (EC50 = 5.530 μM) (Online Supplementary Table S5). Patients’ PBMCs were even more sensitive to these drugs: EC50 BKM120 = 0.893 μM, EC50 ponatinib = 1.716 μM (Table 2).
Table 2.
Results of the dose-response curves of drugs in monotherapy in patients’ samples after 72 hours of incubation with drugs: median. First (Q1) and third (Q3) quartiles.

Curiously, drugs used in treatment of MF such as danazol or prednisone did not have any effect on BA/F3 JAK2 V617F viability (Online Supplementary Table S5), but prednisone was effective in combination with ruxolitinib (ΔEC50 = −13.9%) (Figure 1A). Patients’ samples showed the same response (ΔEC50 = −18.0%) (Figure 1B).
Ruxolitinib/nilotinb/prednisone combination has a synergistic effect on myeloid cell lines and patient peripheral blood mononuclear cells
Given the synergistic effect of nilotinib with ruxolitinib in BA/F3 JAK2 V617F cells, we next studied the effect of adding prednisone to this combination in myeloid cell lines and patients’ PBMCs. As well as being a potent BCR/ABL TKI, nilotinib has been reported to inhibit the PDGF receptor, which is involved in fibrogenesis.20
A synergistic effect with all combinations of the three drugs tested was found in BA/F3 JAK2 V617F and SET2 cells (Table 3). By contrast, only four of the eight doses tested were synergistic in BA/F3 wild-type (WT) cells, again suggesting an important role for the V617F mutation in the activity of the drug combination. We repeated this assay using monotherapy or combination regimens in model A cultures (PBMCs) to test the effect of the combinations in myeloid progenitors. Myelofibrosis patients’ samples were sensitive to nilotinib and prednisone with an EC50 of 6.6 μM and 13.1 μM, respectively. Moreover, all combinations tested (ruxolitinib/nilotinib and ruxolitinib/niloitnib/prednisone) exhibited synergic behavior in at least two of the five patients’ samples tested. In addition, the combination of 160 nM ruxolitinib, 8 μM nilotinib and 0.8 μM prednisone was synergistic in all the patients’ samples tested (Table 4).
Table 3.
Combination Index of ruxolitinib (R), nilotinib (N) and prednisone (P) in samples of cell lines. SET2: BA/F3 JAK2 wt or V617F cell lines were incubated with R, N, P or their combination for 48 hours and then Wst8 was performed.

Table 4.
Combination Index (CI) of ruxolitinib (R), nilotinib (N) and prednisone (P) in samples of myelofibrosis (MF) patients and healthy donors. Mononuclear cells from peripheral blood of MF patients and healthy donors were seeded at 200,000-500,000 cell/mL in Metocult supplemented with SCF and IL3 in presence of drugs for two weeks. Then flow cytometry analysis was performed.

Ruxolitinib/nilotinb/prednisone combination blocks JAK/STAT and MAPK signaling
The signaling pathways affected by the ruxolitinib/nilotinib/prednisone combination or monotherapy were next characterized in SET2 cells using the Proteome ProfilerTM phosphoprotein array after treatment for 30 min. Results showed that, among others, phosphorylation of STAT5 was significantly inhibited 88.94%, TOR 76.55%, ERK 87.55%, and SRC 76.88% (Figure 2) by drug combination (ANOVA ***P<0.005). Western blotting confirmed that the phosphorylation of STAT5 and ERK was inhibited by ruxolitinib in monotherapy by 69.8±8.1% and 87.7±2.3%, respectively. In combination with nilotinib and prednisone, STAT5 and ERK were inhibited by 69.2±11.8% and 29.4±4.5%, respectively (Figure 3A and B). By contrast, phosphorylation of AKT was not inhibited in any case (data not shown).
Figure 2.

Screening for signaling proteins affected by 32 nM ruxolitinib (R), 1.6 μM nilotinib (N), 0.8 μM prednisone (P), and their combinations, using a phospho-protein array. C: Control. The SET2 cell line was incubated with R, N, P or their combination for 48 hours and phosphoprotein analysis was performed.
Figure 3.

Effect of 32 nM ruxolitinib (R), 1.6 μM nilotinib (N), 0.8 μM prednisone (P), and their combinations, on the phosphorylation of ERK1/2 and STAT5 in cell lines. Results are expressed as the mean±Standard Error of Mean. Data are representative of at least 2 separate experiments. *P<0.05; **P<0.01; ***P<0.001. C: control.
Ruxolitinib/nilotinib/prednisone combination decreases the synthesis of collagen I in bone marrow mesenchymal cells
Myelofibrosis is characterized by an increase in collagen deposition, among other fibrillar proteins, in BM, which prevents its proper functioning. To study the effect of ruxolitinib, nilotinib and prednisone (in monotherapy and in combination), on collagen expression, we utilized the HS27a mesenchymal cell line, together with TGF-β as an inductor of collagen expression. TGF-β increased the expression of COL1A1 200.9% over untreated cells (Figure 4A). Nilotinib decreased the mRNA expression of COL1A1 in HS27a cells by 60.2±0.9% in monotherapy, by 51.9±2.9% in combination with ruxolitinib and 62.2±1.9% in combination with ruxolitinib and prednisone. As a complementary test, we measured collagen expression by immunocytochemistry, finding that collagen synthesis was reduced, especially in monotherapy (79.13±20.5%) and in combination with ruxolitinib (79.20±2.3%) (Figure 4B and Online Supplementary Figure S2).
Figure 4.

Effect of ruxolitinib (R), nilotinib (N), prednisone (P), and their combination, on collagen I mRNA expression using quantitative polymerase chain reaction or protein expression by immunocytochemistry. HS27a cell line was incubated with R, N, P or their combination for 1 hour (h) and then for 24 h with 2 ng/mL TGF-β. Results were expressed as the mean±Standard Error of Mean. Data are representative of at least 2 independent experiments. *P<0.05; **P<0.01.
Discussion
The management of MF remains challenging, even in the era of TKIs and personalized medicine. The discovery of the V617F mutation in JAK2 as a physiopathogenic mechanism of MPN3-7 prompted the development of JAK2 inhibitors and represented a revolution in the treatment of MF. Currently, the only approved JAK2 inhibitor for the treatment of MF and PV in the second-line is ruxolitinib,25 which has been shown to be effective in reducing hepatosplenomegaly, resolving disease-related symptoms, and producing a significant increase in overall survival when compared with conventional therapies.26 Nevertheless, there are some limitations to the use of ruxolitinib, including hematologic toxicity (anemia and thrombocytopenia) and a failure to achieve histopathological and molecular complete responses.15 Accordingly, the unmet clinical need to increase the effectiveness of the treatment and decrease its toxicity guides the search for combination treatments with ruxolitinib.
The main objective of this work was to evaluate the best combination of drugs for the treatment of MF, maintaining ruxolitinib as a therapeutic base and reducing its toxicity while maintaining its efficacy. Since MF is not characterized by large amounts of pathological cells, as in acute myeloid leukemia,27 it is challenging to develop an ex vivo model to screen 17 combinations. Consequently, we elected to utilize myeloid cells obtained from 14-day old cultures of PBMCs in methylcellulose (ex vivo model B), which produce a sufficient supply of cells for screening. Interestingly, the most active drugs in monotherapy coincided with the most active drugs in preclinical trials, including bortezomib,28 panobinostat29 and HSP990.30 Therefore, the search for combination treatments with ruxolitinib is in response to the need to increase the effectiveness of the treatment and decrease its toxicity.
We found that the best combinations were those with BCR/ABL inhibitors, nilotinib and bosutinib, used for the treatment of chronic myeloid leukemia (CML).31 Nevertheless, TKIs such as perifosine or BKM120, corticosteroids such as prednisone, androgens such as danazol, and the TGF-β receptor inhibitor SB431542, also decreased the EC50 in combination with ruxolitinib. It is interesting to note that some of these combinations have already been tested in clinical trials, such as ruxolitinib plus danazol (clinicaltrials.gov identifier: 01732445);18 this study showed that while there was no improvement in hematologic response, stabilization of the patients was achieved. Regarding ruxolitinib plus BKM120 (clinicaltrials.gov identifier: 01730248), although no results have yet been reported, preliminary analyses (ASH 2015) are not encouraging.
Interestingly, it has previously been described that the combination nilotinib plus ruxolitinib can eliminate CD34+ leukemic progenitors in CML32 and Philadelphia chromosome positive acute lymphoblastic leukemia;33 however, it was not known whether this also holds for MF. We show here synergistic behavior of nilotinib plus ruxolitinib, which blocks colony formation in clonogenic assays with patients’ PMBCs, and inhibits the phosphorylation of both STAT5 and ERK 1/2. Furthermore, the inclusion of prednisone is key to achieve synergy in the survival assays of cell lines and increases the power of synergy in patients’ samples. In addition, this combination inhibited the synthesis of collagen in BM mesenchymal cells, which is important to achieve histopathological response. The antifibrogenic effect of nilotinib has been previously described in skin cells,20,34 liver35 and muscle,36 via its ability to inhibit the PDGF receptor, which is directly involved in the induction of collagen synthesis. In addition, ruxolitinib is able to stabilize, or even ameliorate, fibrosis through its ability to reduce the proinflamatory state, which is typical in MF.37 Similar results are seen with corticosteroids like prednisone, which decreases the levels of cytokines and proinflammatory growth factors including TGF-β.24
Our results indicate that the combination ruxolitinib, nilotinib and prednisone would eliminate more efficiently pathological cells, stopping and/or reverting MF. It must be remembered, however, that this evidence was obtained in vitro and the impact on the clinical situation still needs to be proved. With this is mind, and given that all the drugs are approved for clinical practice, we have recently initiated a clinical trial (the RuNic Trial; clinicaltrials.gov identifier: 02973711) which does not require an in vivo analysis in animal models.
In summary, MF is a complex disease in which alterations in tyrosine kinase-related signaling participates in the amplification of hematopoietic clones and in the increased production of cytokines and growth factors by pathological cell clones, which stimulates MF.38 It is often advantageous to use combinations of drugs that target different pathways involved in the pathophysiology of a disease, shown here with the objective of decreasing fibrosis of the BM, and not only eradicating the tumor clone, as previously attempted.15,30,39,40 Our results lead us to hypothesize that the combination therapy ruxolitinib and prednisone might provide a dampened proinflammatory environment and nilotinib would block fibrosis. Accordingly, the combination ruxolitinib/nilotinib/prednisone is configured as a therapeutic strategy against MF that aims to enhance the effect of ruxolitinib, and promote the reduction of fibrosis in the BM and reduce inflammation. As mentioned above, this combination will be studied in a phase Ib/II clinical trial in MF (the RuNic Trial; clinicaltrials.gov identifier: 02973711).
Further information is available in the Online Supplementary Appendix.
Supplementary Material
Acknowledgments
The authors would like to thank to Carmen Delgado, from H12O Hematology Department for the support with the samples, Kenneth McCreath for language support, and to all patients who participated in the study.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/104/5/937
Funding
This study was supported by the Subdirección General de Investigación Sanitaria (Instituto de Salud Carlos III, Spain) grants PI13/02387 and PI16/01530, and the CRIS against Cancer foundation grant 2014/0120. M.L. holds a postdoctoral fellowship of the Spanish Ministry of Economy and Competitiveness (FPDI- 2013-16409).
References
- 1.Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: The 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22(1):14–22. [DOI] [PubMed] [Google Scholar]
- 2.Tefferi A. How I treat myelofibrosis. Blood. 2011;117(13):3494–3504. [DOI] [PubMed] [Google Scholar]
- 3.James C, Ugo V, Le Couédic J-P, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005; 434(7037):1144–1148. [DOI] [PubMed] [Google Scholar]
- 4.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005; 7(4):387–397. [DOI] [PubMed] [Google Scholar]
- 5.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. [DOI] [PubMed] [Google Scholar]
- 6.Zhao R, Xing S, Li Z, et al. Identification of an acquired JAK2 mutation in poly-cythemia vera. J Biol Chem. 2005;280(24):22788–22792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. [DOI] [PubMed] [Google Scholar]
- 8.Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129(6):667–679. [DOI] [PubMed] [Google Scholar]
- 9.Ma W, Zhang X, Wang X, et al. MPL mutation profile in JAK2 mutation-negative patients with myeloproliferative disorders. Diagn Mol Pathol. 2011;20(1):34–39. [DOI] [PubMed] [Google Scholar]
- 10.Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. [DOI] [PubMed] [Google Scholar]
- 11.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ha J-S, Kim Y-K. Calreticulin exon 9 mutations in myeloproliferative neoplasms. Ann Lab Med. 2015;35(1):22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marty C, Pecquet C, Nivarthi H, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood. 2016;127(10):1317–1324. [DOI] [PubMed] [Google Scholar]
- 14.Chachoua I, Pecquet C, El-Khoury M, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. 2016; 127(10):1325–1335. [DOI] [PubMed] [Google Scholar]
- 15.Verstovsek S, Gotlib J, Mesa RA, et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J Hematol Oncol. 2017;10(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verstovsek S, Savona MR, Mesa RA, et al. A phase 2 study of simtuzumab in patients with primary, post-polycythaemia vera or post-essential thrombocythaemia myelofibrosis. Br J Haematol. 2017;176(6):939–949. [DOI] [PubMed] [Google Scholar]
- 17.Daver N, Cortes J, Newberry K, et al. Ruxolitinib in combination with Lenalidomide as therapy for patients with myelofibrosis. Haematologica. 2015; 100(8):1058–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gowin K, Kosiorek H, Dueck A, et al. Multicenter phase 2 study of combination therapy with ruxolitinib and danazol in patients with myelofibrosis. Leuk Res. 2017;6031–6035. [DOI] [PubMed] [Google Scholar]
- 19.Mascarenhas J, Hoffman R. A comprehensive review and analysis of the effect of ruxolitinib therapy on the survival of patients with myelofibrosis. Blood. 2013; 121(24):4832–4837. [DOI] [PubMed] [Google Scholar]
- 20.Akhmetshina A, Dees C, Pileckyte M, et al. Dual inhibition of c-abl and PDGF receptor signaling by dasatinib and nilotinib for the treatment of dermal fibrosis. FASEB J. 2008;22(7):2214–2222. [DOI] [PubMed] [Google Scholar]
- 21.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127(20):2391–2405. [DOI] [PubMed] [Google Scholar]
- 22.Chou T-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol Rev. 2006;58(3):621–681. [DOI] [PubMed] [Google Scholar]
- 23.Cervantes F, Isola IM, Alvarez-Larrán A, Hernández-Boluda J-C, Correa J-G, Pereira A. Danazol therapy for the anemia of myelofibrosis: assessment of efficacy with current criteria of response and long-term results. Ann Hematol. 2015;94(11):1791–1796. [DOI] [PubMed] [Google Scholar]
- 24.Yu W, Guo F, Song X. Effects and mechanisms of pirfenidone, prednisone and acetylcysteine on pulmonary fibrosis in rat idiopathic pulmonary fibrosis models. Pharm Biol. 2017;55(1):450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vainchenker W, Leroy E, Gilles L, Marty C, Plo I, Constantinescu SN. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Res. 2018;7:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vannucchi AM, Kantarjian HM, Kiladjian J-J, et al. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 ran-domized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica. 2015;100(9):1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennett TA, Montesinos P, Moscardo F, et al. Pharmacological Profiles of Acute Myeloid Leukemia Treatments in Patient Samples by Automated Flow Cytometry: A Bridge to Individualized Medicine. Clin Lymphoma Myeloma Leuk. 2014; 14(4):305–318. [DOI] [PubMed] [Google Scholar]
- 28.Wagner-Ballon O, Pisani DF, Gastinne T, et al. Proteasome inhibitor bortezomib impairs both myelofibrosis and osteosclerosis induced by high thrombopoietin levels in mice. Blood. 2007;110(1):345–353. [DOI] [PubMed] [Google Scholar]
- 29.Evrot E, Ebel N, Romanet V, et al. JAK1/2 and Pan-deacetylase inhibitor combination therapy yields improved efficacy in preclinical mouse models of JAK2V617F-driven disease. Clin Cancer Res. 2013; 19(22):6230–6241. [DOI] [PubMed] [Google Scholar]
- 30.Marubayashi S, Koppikar P, Taldone T, et al. HSP90 is a therapeutic target in JAK2- dependent myeloproliferative neoplasms in mice and humans. J Clin Invest. 2010; 120(10):3578–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green MR, Newton MD, Fancher KM. Off-Target Effects of BCR-ABL and JAK2 Inhibitors. Am J Clin Oncol. 2016;39(1): 76–84. [DOI] [PubMed] [Google Scholar]
- 32.Gallipoli P, Cook A, Rhodes S, et al. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood. 2014;124(9):1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kong Y, Wu Y-L, Song Y, et al. Ruxolitinib/nilotinib cotreatment inhibits leukemia-propagating cells in Philadelphia chromosome-positive ALL. J Transl Med. 2017;15(1):184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luchetti MM, Moroncini G, Jose Escamez M, et al. Induction of Scleroderma Fibrosis in Skin-Humanized Mice by Administration of Anti-Platelet-Derived Growth Factor Receptor Agonistic Autoantibodies. Arthritis Rheumatol. 2016; 68(9):2263–2273. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y, Wang Z, Kwong SQ, et al. Inhibition of PDGF, TGF-β, and Abl signaling and reduction of liver fibrosis by the small molecule Bcr-Abl tyrosine kinase antagonist Nilotinib. J Hepatol. 2011;55(3):612–625. [DOI] [PubMed] [Google Scholar]
- 36.Lemos DR, Babaeijandaghi F, Low M, et al. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat Med. 2015;21(7):786–794. [DOI] [PubMed] [Google Scholar]
- 37.Massaro F, Molica M, Breccia M. How ruxolitinib modified the outcome in myelofi- brosis: focus on overall survival, allele burden reduction and fibrosis changes. Expert Rev Hematol. 2017;10(2):155–159. [DOI] [PubMed] [Google Scholar]
- 38.Lataillade J-J, Pierre-Louis O, Hasselbalch HC, et al. Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood. 2008;112(8): 3026–3035. [DOI] [PubMed] [Google Scholar]
- 39.Barosi G, Gattoni E, Guglielmelli P, et al. Phase I/II study of single-agent bortezomib for the treatment of patients with myelofibrosis. Clinical and biological effects of proteasome inhibition. Am J Hematol. 2010;85(8):616–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tefferi A, Lasho TL, Begna KH, et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N Engl J Med. 2015;373(10):908–919. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.