

Abstract
Sickle cell disease is an autosomal recessive genetic red cell disorder with a worldwide distribution. Growing evidence suggests a possible involvement of complement activation in the severity of clinical complications of sickle cell disease. In this study we found activation of the alternative complement pathway with microvascular deposition of C5b-9 on skin biopsies from patients with sickle cell disease. There was also deposition of C3b on sickle red cell membranes, which is promoted locally by the exposure of phosphatidylserine. In addition, we showed for the first time a peculiar “stop-and-go” motion of sickle cell red blood cells on tumor factor-α–activated vascular endothelial surfaces. Using the C3b/iC3b binding plasma protein factor Has an inhibitor of C3b cell-cell interactions, we found that factor H and its domains 19-20 prevent the adhesion of sickle red cells to the endothelium, normalizing speed transition times of red cells. We documented that factor H acts by preventing the adhesion of sickle red cells to P-selectin and/or the Mac-1 receptor (CD11b/CD18), supporting the activation of the alternative pathway of complement as an additional mechanism in the pathogenesis of acute sickle cell related vaso-occlusive crises. Our data provide a rationale for further investigation of the potential contribution of factor H and other modulators of the alternative complement pathway with potential implications for the treatment of sickle cell disease.
Introduction
Sickle cell disease (SCD; OMIM # 603903) is an autosomal recessive genetic red blood cell (RBC) disorder with a worldwide distribution. SCD results from a point mutation (βS, 6V) in codon 6 of the β-globin gene where the insertion of valine in place of glutamic acid leads to the production of a defective form of hemoglobin, termed hemoglobin S (HbS).1–3 Pathophysiological studies have shown that intravascular sickling in capillaries and small vessels leads to vaso-occlusion and impaired blood flow. Vaso-occlusive events in the microcirculation result from a complex and only partially understood scenario involving interactions between different cell types. These cells include dense, dehydrated sickle cells, reticulocytes, abnormally activated endothelial cells, leukocytes and platelets.1–4 Plasma factors such as coagulation system cytokines and oxidized pro-inflammatory lipids may also be involved. In addition, cyclic polymerization-depolymerization promotes RBC membrane oxidation and reduces RBC survival in the peripheral circulation.1,5,6 The resulting increase in free hemoglobin and free heme, a consequence of the saturation of the physiological system and local reduction of nitric oxide bioavailability, leads to a pro-coagulant state with increased risk of thrombotic events.2,3,7–10 All this evidence indicates that sickle cell vasculopathy is a crucial player in RBC adhesion and in the development of acute vaso-occlusion in SCD patients.
Although progress has been made in recent decades in understanding the pathogenesis of SCD, the molecular events involved in these processes are still only partially delineated. Whereas a key role for complement activation has been highlighted in chronic inflammatory processes characterized by hemolysis and inflammatory vasculopathy such as atypical hemolytic uremic syndromes and paroxysmal nocturnal hemoglobinuria11–14 the involvement of complement in SCD has been less extensively explored. Previous studies have revealed: (i) an activation of the alternative complement pathway (AP) in SCD patients; (ii) a reduction in the activating proteases factors B and D, modulating complement activation; (iii) a decrease in the plasma levels of factor H (FH), the major soluble regulator of AP activation; and (iv) increased deposition of the complement opsonin C3b on RBC exposing phosphatidylserine.15–22 Preliminary data from a mouse model of SCD suggest a possible role for complement activation in the generation of vaso-occlusive crises, as an additional disease mechanism contributing to the severity of acute clinical manifestations related to SCD.23,24
Because of its potential detrimental effects on host cells, the AP is finely regulated by membrane-bound and soluble regulators. Circulating FH plays a particularly important role, since this regulator not only binds to C3b and prevents the formation of C3b convertases, but it is also able to recognize self-associated molecular patterns such as sialic acid and glycosaminoglycans present on the membranes of most healthy cells.25–27 Any interference with this recognition process, resulting from either polymorphisms or blocking antibodies against FH, may have severe pathological consequences as described for atypical hemolytic uremic syndromes and other complement-mediated disorders.28
Here, we found that sickle RBC are characterized by membrane deposition of C3b, which acts as a marker for the activation of the AP on sickle RBC. We sought to determine whether C3b deposition on RBC might possibly stimulate vaso-occlusive crises by favoring cell-cell interactions. Indeed, we now demonstrate for the first time a peculiar ex vivo motion profile (“stop-and-go” behavior) of SCD red cells during their transit on vascular endothelial surfaces, a motion that prolongs their transit on the vascular endothelial surface and promotes the adhesion of sickle RBC. We show that FH and its 19-20 domain,29,30 which primarily targets C3b, prevent the adhesion of sickle RBC to the endothelium. We further document that FH acts by preventing the adhesion of sickle RBC to P-selectin and/or the receptor Mac-1 (CD11b/CD18). Our data provide a rationale for further investigation of FH and other modulators of the AP as novel disease-modifying molecules with potential implications for the treatment of the clinical manifestations of SCD.
Methods
Study design
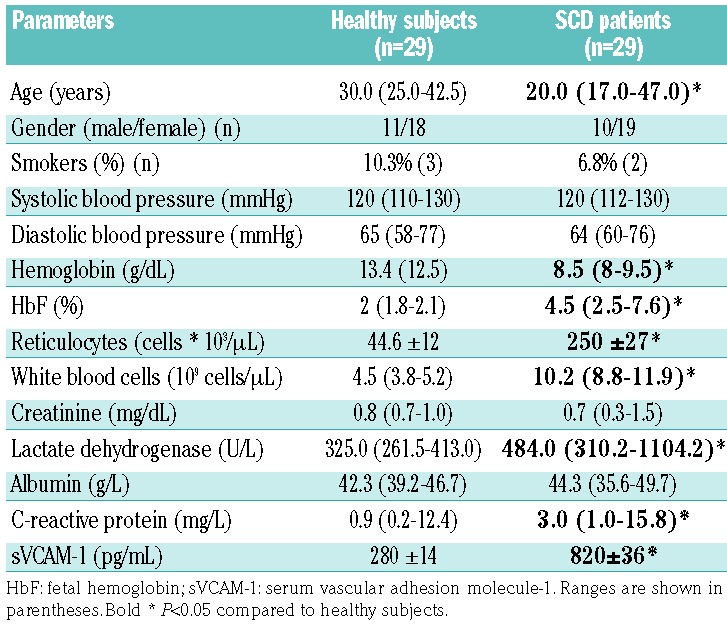
We studied SCD subjects (n=29; 26 SS and 3 Sβ0) referred to the Department of Medicine, University of Verona and Azienda Ospedaliera Integrata of Verona (Italy) between January 2012 and January 2017. SCD patients were evaluated at steady state, and none of them had been on either hydroxyurea or a transfusion regimen during the 6 months immediately prior to our analysis. Healthy controls were matched by age, sex and ethnic background. The study was approved by the Ethical Committee of the Azienda Ospedaliera Integrata of Verona (Italy) and informed consent was obtained from patients and healthy controls (ethical approval FGRF13IT). Table 1 shows the demographic characteristics of both the healthy subjects and SCD patients studied. Biochemical and hematologic parameters as well as plasma levels of C3 and C4 were determined according to clinical and laboratory standards at the Laboratory of Medicine, University of Verona and Azienda Ospedaliera Integrata of Verona (Italy). Plasma C5a (EIA Quidel Corp., San Diego, CA, USA), plasma vascular cell adhesion molecule-1 (Invitrogen, Carlsbad, CA, USA) and serum FH (Hycult Biotech, Uden, the Netherlands) were determined by enzyme-linked immunosorbent assays, according to the manufacturers’ protocols.31
Table 1.
Demographic, hematologic and biochemical data for healthy subjects and patients with sickle cell disease.
Evaluation of C5b-9 complement deposition on fixed skin biopsies
Skin punch biopsies were carried out on the volar surface of the left arm on apparently normal skin. The samples were paraffin-embedded and stained with hematoxylin and eosin before examination; they were also used to estimate the presence of C5b-9 complement deposition within the microvasculature.32 Details on the immunofluorescent and immunohistochemical staining assays are reported in the Online Supplementary Methods.32–34
Measurements of phosphatidylserine-positive and C3d-positive red blood cells
Phosphatidylserine-positive cells were detected as previously reported.35–37 Details are given in the Online Supplementary Methods.
Red blood cell adhesion assay
The real-time adhesion of RBC collected from healthy and SCD subjects on inactive or activated endothelium [in the absence or presence of tumor necrosis factor-alpha (TNF-α), respectively] was determined as previously reported38–40 with or without FH or the FH19-20 or FH68 domains.41 Details are provided in the Online Supplementary Methods.
Development of an algorithm to determine red blood cell transit and flux trajectory
The algorithm to determine RBC transit and flux trajectory is described in the Online Supplementary Methods.
P-selectin and Mac-1 expression in vitro in vascular endothelial cells
Details on immunoblotting42 and the cytofluorimetric analysis to determine P-selectin and Mac-1 expression are given in the Online Supplementary Methods.
Statistical analysis
All calculations were performed using the IBM SPSS 20.0 statistical package (IBM Inc., Armonk, NY, USA). The level of aggregation was expressed as median values with the minimum-maximum range and shown by means of either dot or box plots. Data were analyzed with non-parametric tests, the Mann-Whitney U test for unpaired samples and the Wilcoxon signed-rank test for paired samples. A P value <0.05 was considered statistically significant.
Results
Patients with sickle cell disease show activation of the alternative complement pathway and increased C3d-positive red blood cells
Untreated SCD subjects at steady state were studied. As shown in Table 1, reduced hemoglobin, increased reticulocyte counts and raised levels of plasma lactate dehydrogenase, all signs of chronic hemolytic anemia, were observed. In SCD patients, we also found significant increases of the levels of plasma C-reactive protein and surface vascular cell adhesion molecule-1, indicating the presence of a chronic inflammatory vasculopathy in agreement with previous reports.2,3,9,43 Serum C3 and C4 levels were similar in healthy and SCD subjects (data not shown); whereas levels of the complement activation fragment C5a were significantly elevated in SCD patients when compared to the levels in healthy individuals (Figure 1A); statistical analyses were performed to exclude the possible contribution of confounding factors such as gender or smoking status. Our finding is consistent with previous reports, and confirms the substantial complement activation in SCD patients, likely via the AP.15–22
Figure 1.

The adhesion of C3d-positive sickle red blood cells is prevented by factor H. (A) Plasma samples from healthy controls (AA) and patients with sickle cell disease (SCD) were tested by enzyme-linked immunosorbent assay (see Methods section); *P<0.05 AA versus SCD; n=10 in each group. (B) Deposition of C5b-9 (orange fluorescence) assessed by immunofluorescent staining involving the abluminal aspect of the microvasculature in apparently normal skin of a patient with SCD (direct immunofluorescence; original magnification x100). Inset. Detail of the vessels showing intense granular deposition of C5b-9; direct immunofluorescence; original magnification: ×400. Nuclei were stained with Prolong Gold antifade reagent with DAPI (blue fluorescence). The image shown is one representative image of 16 others with similar results. Lower panel. The percentage of vessels positive (+) for C5b9 granular deposition in skin biopsies from healthy subjects (AA) and sickle cell patients (SCD). Data are shown as means ± SD **P<0.002 AA versus SCD. (C) Left panel. Representative immunohistochemical image of a normal skin biopsy from a SCD patient showing a small vessel in the superficial dermis (arrow) characterized by co-expression of C5b9 (brown) and CD31 (red); for comparison a C5b9-negative vessel (circle) only decorated with CD31 staining (red) is highlighted. One representative image is shown; all 16 gave similar results (n=16). Right panel. Quantification of C5b9-positive vessels in skin biopsies from healthy subjects (see Online Supplementary Figure S2) and SCD patients. Data are shown as means ± SD; **P<0.01 AA versus SCD. (D) Percentages of C3d+ red blood cells (RBC) in healthy donors (AA) and in sickle cell subjects (n=16 AA; n=16 SCD). The dashed line indicates the threshold of normality, corresponding to C3d+ <0.5% of RBC; *P<0.05 AA versus SCD; **P<0.01 AA versus SCD. (E) Percentages of phosphatidylserine-positive (PS+) RBC in healthy donors (AA) and in sickle cell subjects (n=20 AA; n=32 SCD). The dashed line indicates the threshold of normality, corresponding to PS+ <0.5 % of RBC; **P<0.01 AA versus SCD.
Since studies in atypical hemolytic uremic syndromes have shown that skin biopsy might be a feasible method for documenting AP activation by C5b-9 vascular deposition,32 we obtained skin biopsies from SCD patients at steady state. We considered skin to be an interesting window of observation in SCD, since (i) it might be involved in the clinical manifestations of SCD, such as leg ulcers; and (ii) it has been widely used in SCD mouse models to functionally characterize the microvasculature.44–47 As shown in Figure 1B, we observed microvascular intense, focal granular deposition of C5b-9 in small vessels throughout the dermis of SCD patients. This pattern is similar to that reported in skin biopsies from patients with atypical hemolytic uremic syndrome, which is a thrombotic microangiopathy related to complement dysfunction.32 No deposition of C5b9 was observed in skin from healthy controls (Online Supplementary Figure S1A,B). Co-localization of C59b deposits and CD31+ skin vessels was confirmed by immunohistochemical staining only in the skin biopsies from SCD patients (Figure 1C, Online Supplementary Figure S2A,B).
Taken together, our data indicate an activation of the AP in SCD, with possible involvement of complement in SCD vasculopathy and in related cellular adhesion events.
To understand whether AP activation occurs directly on sickle RBC, we measured the amounts of circulating RBC carrying C3-derived opsonins by detecting the presence of the common C3d fragment.11,12 As shown in Figure 1D, higher numbers of C3d+ RBC were found in SCD patients in the steady state than in healthy subjects (representative scatter-plots are shown in Online Supplementary Figure S3). This fraction further increased in a subgroup of SCD patients during acute pain crises (SS steady state 2.5±0.8% versus SS pain crisis: 6.1±0.7% P<0.02; n=10), in agreement with an observation by Mold et al.18 Since a previous report linked activation of the AP with deposition of C3 opsonins to the exposure of phosphatidylserine on SCD RBC surfaces, we evaluated the percentage of phosphatidylserine-positive RBC in our SCD patients17 and did indeed find higher percentages of phosphatidylserine-positive RBC in SCD patients than in healthy subjects in agreement with previous studies35,36 (Figure 1E). In contrast to paroxysmal nocturnal hemoglobinuria, in which opsonization leads to rapid lysis of affected RBC.11,12 the presence of functional membrane regulators on SCD erythrocytes allowed a low level of C3-fragment deposition, which might also be considered as a marker of complement activation on sickle RBC. We, therefore, reasoned that the presence of C3b, iC3b or C3dg may function as a site of adhesion of SCD RBC to activated vascular endothelial surfaces, which may carry ligands for C3 fragments, such as Mac-1 and P-selectin.48,49 To study this possibility, we used the C3b and iC3b binding plasma protein FH as an inhibitor25–27 and saw no significant differences in FH serum levels between healthy subjects and SCD patients [AA median: 841 (range 616–1528) μg/mL versus SCD median: 980 (741–1301) μg/mL; P=NS].
Factor H prevents the adhesion of sickle red blood cells to tumor necrosis factor-α-activated vascular endothelium
We performed an ex vivo adhesion assay using TNF-α-activated vascular endothelial cells and monitored, in real time, the adhesion of RBC collected from healthy and SCD subjects to inactive (no TNF-α) or activated endothelium (plus TNF-α) as previously described.38–40 Consistent with the literature, we observed significantly increased adhesion of SCD RBC to TNF-α-activated endothelium when compared to normal RBC (Figure 2A, Online Supplementary Video S1). We next investigated the effect of FH on the adhesion of healthy or SCD RBC to activated vascular endothelium. The dose-response curve with FH showed a significant reduction in RBC adhesion at concentrations ≥9 nM (Figure 2B). This was more pronounced for SCD RBC than for healthy RBC (Figure 2B). We, therefore, expanded the tests to include a larger number of SCD patients using FH concentrations of 9 or 18 nM; as expected, reduced adhesion of SCD RBC to the vascular endothelium was observed in the presence of either 9 nM or 18 nM FH (Figure 3A). Previous studies have identified two major regions on FH (i.e., domains 6-8 and 19-20) as the putative sites of interaction with cell surface constituents such as glycosaminoglycans; furthermore, domains 19-20 are also responsible for binding to sialic acids or C3d-containing deposits.29 We, therefore, evaluated whether any of these segments had activity similar to that of full-length FH.
Figure 2.

Adhesion of red blood cells to endothelium activated or not by tumor necrosis factor-α. (A) Adhesion of healthy (AA) or sickle cell disease (SCD) red blood cells (RBC) on immortalized endothelium (EA926.hy) treated or not with tumor necrosis factor-α (TNF-α) under flow conditions (data are expressed as cells/mm2). The data were obtained from six separate comparable experiments. All calculations were performed using the IBM SPSS 20.0 statistical package (IBM Inc., Armonk, NY, USA). The results of the adhesion tests are expressed as median values with the minimum-maximum range and are illustrated by box plots. The data were analyzed with non-parametric tests, the Mann-Whitney U test for unpaired samples and the Wilcoxon signed-rank test for paired samples. A value of P<0.05 is considered statistically significant. (B) Dose-response curve for factor H (FH) in adhesion assays for healthy (AA) or sickle (SCD) RBC. Data were obtained at 6 min flux on endothelium treated with either vehicle or TNF-α. The curves are representative of six separate and independent experiments with similar results.
Figure 3.

Factor H and its 19-20 segment normalized the transit of sickle red blood cells on the tumor necrosis factor-α-activated vascular endothelial surface. (A) Sickle cell adhesion after 6 min of perfusion on activated or non-activated endothelium (±TNF-α) in the presence of FH 9 nM or 18 nM final concentration. The data shown are representative of six other independent assays with similar results. Wilcoxon test: *indicates the corresponding significance. A P value <0.05 is considered statistically significant. Statistical analysis as in Figure 2A. (B) Sickle cell adhesion after 6 min of perfusion on activated or non-activated endothelium (± TNF-α) in the presence of FH and its fragments 19-20 and 6-8 (18 nM final concentration). Data shown are representative of six other independent assays with similar results. A P value <0.05 is considered statistically significant. Statistical analysis as in Figure 2A.
Perfusion experiments performed on TNF-α-activated endothelium demonstrated that, like intact FH, FH19-20 (18 nM) strongly prevented the adhesion of sickle RBC to the surface of TNF-α-activated vascular endothelium. Conversely, FH6-8 showed a trend, which did not reach statistical significance, toward a reduction in RBC adhesion (Figure 3B). Our data indicate that FH prevents sickle cell adhesion to the activated endothelium through its interaction with cell-surface sialic acids and C3b/iC3b found on the surface of pathological RBC (Figure 3B).
Factor H and its 19-20 domain normalize the sickle red blood cell trajectory and transverse velocity on tumor necrosis factor-α-activated vascular endothelium
We then developed a new algorithm for RBC to analyze their trajectory and transverse velocity, the velocity component perpendicular to the direction of flow of the RBC during their transit in the flow-chamber. The trajectory of each sickle RBC appeared irregular in space and was not uniform in time, especially when compared to that of healthy RBC (Figure 4A,B). This observation was based on the high transverse displacement and the presence of frequent stop-and-go motion that characterized the sickle RBC transit on the activated vascular endothelium. The transverse velocity is a parameter that is correlated with the disturbed movement of a particle. The path of healthy RBC appeared to be more regular, uniform, and parallel to the flow (Figure 4A, Online Supplementary Video S2). Thus, healthy RBC crossed the field of view more rapidly than did sickle RBC (0.8–1.2 s versus 1.7–2 s, respectively; P<0.05). In the presence of FH, the trajectory of sickle RBC was regularized (Figure 4C), reducing their transverse velocity to 7.71 ± 6.7 μm/s (average decrease of 81.13% versus vehicle-treated sickle RBC). The same behavior appeared in sickle RBC treated only with the FH 19-20 domain (Figure 4D), with the transverse speed being 6.56 ± 5.6 μm/s (an average decrease of 83.94% versus vehicle-treated sickle RBC). When the particle kinematics were quantified (Figure 3E), we found that the absolute value of the instantaneous transverse speed of sickle RBC was 40.86 ± 27.6 μm/s, whereas that of healthy cells was only 5.64 ± 4.9 μm/s (a difference of 86.20%). When healthy cells were compared to either FH- or FH19-20-treated sickle RBC, the absolute values for instantaneous transverse speed decreased significantly, reaching values similar to those observed for healthy RBC.
Figure 4.

Factor H and its 19-20 fragment normalized the “stop-and-go” motion of sickle red blood cells. (A) Trajectory of three representative healthy (AA) red blood cells (RBC) in the field of view: each coordinate indicates their centroid at every consecutive frame (flow direction: x axis). (B) Trajectory of three representative sickle (SCD) RBC, showing the “stop-and-go” motion. (C) Trajectory of three representative sickle RBC treated with fator H (FH) (18 nM). (D) Trajectory of three representative sickle RBC treated with FH 19-20 segment (18 nM). (E) Absolute values for instantaneous transverse speed expressed as mean ± standard deviation (**P<0.01 versus healthy RBC).
Anti-P-selectin and anti-Mac-1 antibodies prevent adhesion of sickle red blood cells to tumor necrosis factor-α-activated vascular endothelial surface
To better understand the binding proteins that may be important for adhesion of C3b/iC3b+ SCD red cells, we then evaluated the effects of anti-P-selectin or anti-Mac-1 antibodies on the adhesion of SCD to TNF-α-activated vascular endothelium. We chose these two molecules for the following reasons: (i) they are both modulated in endothelial cells:2,50–55 (ii) P-selectin has been reported to bind C3b present on the membranes of circulating cells such as platelets;56 and (iii) Mac-1 is a well-known receptor for iC3b, and potentially C3dg associated with the cell membrane.25 In addition, P-selectin has been reported to bind RBC, targeting red cell plasma-membrane sialic acid, whereas the presence of a Mac-1 binding site on RBC membranes is still under investigation.29,48,51,57 In our model, the expression of P-selectin and Mac-1 was significantly increased on TNF-α-activated vascular endothelium when compared to the expression on vehicle-treated cells (Online Supplementary Figure S4A). Both anti-P-selectin and anti-Mac1 antibodies prevented the adhesion of sickle RBC to the activated vascular endothelium (Figure 5A). Whereas the effect of the anti-P-selectin antibody on RBC adhesion may be mediated through interference with two different targets (i.e., iC3b and/or sialic acid), the interplay between Mac-1 and iC3b deposited on sickle RBC is considered to be more selective.25,58 The anti-adhesive effect of the anti-Mac-1 antibody was more pronounced than that of the anti-P-selectin antibody, but the difference was not statistically significant. No effect on the adhesion of sickle RBC was documented in the presence of a control antibody (Online Supplementary Figure S4B). Collectively, these findings indicate that C3b/iC3b is deposited on sickle RBC membranes as a new ligand, bridging sickle RBC to the activated vascular endothelial surface through binding to the pro-adhesive molecules P-selectin and/or Mac-1.
Figure 5.

The anti-adhesive effect of factor H involves P-selectin and Mac-1 pro-adhesive molecules. (A) Adhesion of sickle cell disease red blood cells (SCD RBC) after 6 min of perfusion on activated or non-activated endothelium (± TNF-α) pre-coated with either anti-P-selectin antibody or anti-Mac1 (CD11b/CD18) antibody. Data shown are representative of six other independent assays with similar results. Wilcoxon test: *indicates the corresponding significance. Adhesion is expressed as median values with a minimum-maximum range and illustrated by box plots. A value of P<0.05 is considered statistically significant. Statistical analysis as in Figure 2A. (B) Schematic model of the beneficial action of factor H in reducing adhesion of C3-derived opsonins on sickle RBC to the TNF-α-activated vascular endothelium. C3 split-fragments on erythrocytes might favor cell-cell interactions through P-selectin and Mac-1. P-selectin might bind RBC through two different targets, iC3b and/or sialic acid; in contrast, Mac-1 targets only iC3b deposits on sickle RBC as a more selective interaction. FH and FH19-20 segment normalized the transit of sickle RBC across the TNF-α-activated vascular endothelial surface, abolishing the “stop-and-go” behavior of the sickle RBC. This effect positively affected (shortened) the transit time of sickle RBC, thereby reducing the likelihood of the RBC sickling during their transit through the microcirculation AP: alternative complement pathway; SCD: sickle cell disease; PS: phosphatidylserine; FH: factor H.
Discussion
In this study, we confirmed activation of the AP in SCD and demonstrated, for the first time, cutaneous vascular deposition of C5b-9, supporting the involvement of complement in the microvascular injury associated with SCD.
We then showed that activation of the AP results in C3 split-fragments being bound to the sickle RBC surface, thereby contributing to the adhesion of RBC to the inflammatory activated vascular endothelial surface. Notably, decoration of erythrocytes with C3 opsonins was more evident in patients undergoing vaso-occlusive crises, further supporting the involvement of complement in microvascular injury in SCD. Since sickle RBC, in contrast to paroxysmal nocturnal hemoglobinuria RBC, still contain the functional complement regulators CD55 and CD59, phosphatidylserine-mediated complement activation may allow for a certain degree of opsonization without inducing hemolysis. This situation would lead to the observed accumulation of C3d-containing opsonins (i.e., C3b, iC3b, C3dg), which have all been associated with cell interactions and signaling functions.59 Thus, we propose that C3 split-fragments on RBC might favor cell-cell interactions, supporting previous observations of reduced RBC-cell interactions in mice genetically lacking the C3 complement fraction.51,60
We also demonstrated here that FH prevents the adhesion of sickle RBC and normalizes their trajectory and transverse velocity on TNF-α-activated vascular endothelial surfaces through a mechanism involving P-selectin and/or Mac-1 as pro-adhesion molecule(s). FH binds to C3b/iC3b molecules present on cell surfaces and inhibits the AP. The algorithm developed in the present study allowed us to go further in describing RBC adhesion to the endothelial surface. Indeed, we showed that SCD RBC adhere to endothelial cells with a peculiar dynamic behavior not shown by healthy RBC. The stop-and-go motion of sickle RBC contributes to the irregular trajectory of these cells during their transit on the vascular endothelial surface. This slow and irregular movement clearly affects the speed transition time of sickle RBC when compared to healthy control RBC, possibly contributing to the reduction in blood flow in the microcirculation that generally characterizes the early phase of an acute vaso-occlusive crisis.1,5,61–63 It is thus conceivable that our flow-based methodology could have a positive impact on monitoring therapy for SCD. Both FH and its FH19-20 segment normalized the transit of sickle RBC across the TNF-α-activated vascular endothelial surface, abolishing the “stop-and-go” behavior of sickle RBC. This effect is of great importance because the transition time of sickle RBC in the microcirculation is critically related to the HbS polymerization time and the generation of dense, dehydrated RBC, which contribute to the development of the acute clinical manifestations of SCD.1,10 It is important to note that FH appears to exert a non-canonical function in preventing cell adhesion. This complement regulator typically inhibits complement activation via the AP by accelerating the decay of C3 convertases and by mediating the degradation of C3b to iC3b and C3dg via the plasma protease factor I. Since our experiments were performed using purified cells in the absence of plasma or serum, factor I and the components involved in convertase formation cannot have been involved in the observed effects. Rather, FH appears to interfere directly with cell-cell interaction events between sickle RBC and endothelial cells.
To further elucidate the mechanism of complement opsonin-mediated adhesion and the role of FH as an anti-adhesive molecule for SCD RBC, we pre-coated vascular endothelial cells with either anti-P-selectin or anti-Mac-1 antibodies. Both surface molecules are expressed on activated vascular endothelial cells and have been associated with opsonin interactions. Mac-1 is well-established as a functionally important receptor for iC3b which contributes to phagocytosis and cell activation. Whereas, the interplay of complement with P-selectin is less well described, several studies have shown interactions between C3b and P-selectin.56,64,65 We found that the adhesion of sickle RBC could be prevented by either anti-P-selectin or anti-Mac-1 antibody, indicating that both molecules contribute to the adhesion of sickle RBC to the vascular endothelium. In our studies, blocking Mac-1 had a more pronounced effect on adhesion than did impairing P-selectin activity. The beneficial impact of interfering with Mac-1 in SCD has been supported by the reduction of RBC-neutrophil interactions in SCD mice treated with anti-Mac-1 antibody51 or in Mac-1-deficient SCD mice.57 Thus, our data indicate that complement is involved in the interaction between sickle RBC and the endothelium, pointing to a new additional mechanism contributing to the biocomplexity of acute events in SCD.
Our study therefore has potential implications for the clinical management of SCD. Current treatments in development are focused on the role of selectins in the pathogenesis of SCD. For example, an anti-P-selectin antibody (crizanlizumab) that has been used to treat SCD has been reported to affect the period of time between acute pain crises in SCD patients.52,66,67 Similarly, the small molecule-based pan-selectin inhibitor, rivipansel, currently undergoing phase III clinical trials, is able to reduce the time required to resolve vaso-occlusive crises with a reduction in opioid treatment.68 Our findings suggest that targeting complement opsonization and/or opsonin-mediated cell adhesion could provide an alternative strategy. Whereas the use of exogenous full-length FH as a therapeutic tool is associated with some challenges, several smaller variants of the regulator have shown promise in preclinical trials for complement-mediated diseases such as paroxysmal nocturnal hemoglobinuria. Given the importance of FH domains 19-20 in interfering with RBC adhesion, mini-FH constructs containing this domain pair may be considered, since they may affect both AP activity and the adhesive function of existing opsonins.69,70 Alternatively, blocking opsonization itself at the level of C3 activation is also expected to impair complement-mediated adhesion.
In conclusion, we have first shown that complement activation on sickle RBC participates in the adhesion of sickle erythrocytes to the TNF-α-activated vascular endothelium (Figure 5B). We then further demonstrated that the FH19-20 segment is as efficient as FH in preventing the adhesion of sickle RBC, and results in normalization of sickle RBC transit across the vascular endothelial surface. We suggest that chronic hemolysis may require high levels of FH to prevent RBC adhesion and entrapment in the microcirculation. Finally, our data indicate that FH might act as a multimodal molecule, preventing the opsonization of sickle RBC with C3 opsonins and targeting the interaction of sickle RBC with the endothelium through the adhesion molecules P-selectin and Mac-1 (Figure 5B). Our findings provide a rationale for considering FH-based inhibitors and other modulators of the AP as potential new therapeutic options in SCD.
Supplementary Material
Acknowledgments
This work was supported in part by grants FUR 2016-2017 to LDF and by NIH grant AI068730 to JDL We thank Dr. Sara Ugolini, Dr. Monica Battiston and Dr. Francesco Agostini for technical support, Ing. Leoardo Buscemi for LB software writing and Ing. Vincenzo Insalaca for video editing. We also thank Dr. Letizia Delmonte and Dr. Elisa Vencato for their contribution in preliminary experiments and Dr. Deborah McClellan for editing the manuscript.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/104/5/919
References
- 1.De Franceschi L, Cappellini MD, Olivieri O. Thrombosis and sickle cell disease. Semin Thromb Hemost. 2011;37(3):226–236. [DOI] [PubMed] [Google Scholar]
- 2.Kato GJ, Hebbel RP, Steinberg MH, Gladwin MT. Vasculopathy in sickle cell disease: biolo gy, pathophysiology, genetics, translational medicine, and new research directions. Am J Hematol. 2009;84(9):618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hebbel RP. The systems biology-based argu ment for taking a bold step in chemoprophylaxis of sickle vasculopathy. Am J Hematol. 2009;84(9):543–545. [DOI] [PubMed] [Google Scholar]
- 4.Parise LV, Telen MJ. Erythrocyte adhesion in sickle cell disease. Curr Hematol Rep. 2003;2(2):102–108. [PubMed] [Google Scholar]
- 5.De Franceschi L, Corrocher R. Established and experimental treatments for sickle cell disease. Haematologica. 2004;89(3):348–356. [PubMed] [Google Scholar]
- 6.Sabaa N, de Franceschi L, Bonnin P, et al. Endothelin receptor antagonism prevents hypoxia-induced mortality and morbidity in a mouse model of sickle-cell disease. J Clin Invest. 2008;118(5):1924–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vinchi F, De Franceschi L, Ghigo A, et al. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation. 2013;127(12):1317–1329. [DOI] [PubMed] [Google Scholar]
- 8.Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. 2013;121(8): 1276–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hebbel RP. Adhesion of sickle red cells to endothelium: myths and future directions. Transfus Clin Biol. 2008;15(1-2):14–18. [DOI] [PubMed] [Google Scholar]
- 10.Telen MJ. Beyond hydroxyurea: new and old drugs in the pipeline for sickle cell disease. Blood. 2016;127(7):810–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Risitano AM, Notaro R, Marando L, et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009;113(17):4094–4100. [DOI] [PubMed] [Google Scholar]
- 12.Risitano AM, Notaro R, Pascariello C, et al. The complement receptor 2/factor H fusion protein TT 30 protects paroxysmal nocturnal hemoglobinuria erythrocytes from complement-mediated hemolysis and C3 fragment. Blood. 2012;119(26):6307–6316. [DOI] [PubMed] [Google Scholar]
- 13.Frimat M, Tabarin F, Dimitrov JD, et al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood. 2013;122(2):282–292. [DOI] [PubMed] [Google Scholar]
- 14.Roumenina LT, Loirat C, Dragon-Durey MA, et al. Alternative complement pathway assessment in patients with atypical HUS. J Immunol Methods. 2011;365(1-2):8–26. [DOI] [PubMed] [Google Scholar]
- 15.Test ST, Woolworth VS. Defective regulation of complement by the sickle erythrocyte: evidence for a defect in control of membrane attack complex formation. Blood. 1994;83(3):842–852. [PubMed] [Google Scholar]
- 16.Chudwin DS, Papierniak C, Lint TF, Korenblit AD. Activation of the alternative complement pathway by red blood cells from patients with sickle cell disease. Clin Immunol Immunopathol. 1994;71(2):199–202. [DOI] [PubMed] [Google Scholar]
- 17.Wang RH, Phillips G, Jr, Medof ME, Mold C. Activation of the alternative complement pathway by exposure of phosphatidylethanolamine and phosphatidylserine on erythrocytes from sickle cell disease patients. J Clin Invest. 1993;92(3):1326–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mold C, Tamerius JD, Phillips G., Jr Complement activation during painful crisis in sickle cell anemia. Clin Immunol Immunopathol. 1995;76(3 Pt 1):314–320. [DOI] [PubMed] [Google Scholar]
- 19.Gavriilaki E, Mainou M, Christodoulou I, et al. In vitro evidence of complement activation in patients with sickle cell disease. Haematologica. 2017;102(12):e481–e482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koethe SM, Casper JT, Rodey GE. Alternative complement pathway activity in sera from patients with sickle cell disease. Clin Exp Immunol. 1976;23(1):56–60. [PMC free article] [PubMed] [Google Scholar]
- 21.Strauss RG, Asbrock T, Forristal J, West CD. Alternative pathway of complement in sickle cell disease. Pediatr Res. 1977;11(4):285–289. [DOI] [PubMed] [Google Scholar]
- 22.de Ciutiis A, Polley MJ, Metakis LJ, Peterson CM. Immunologic defect of the alternate pathway-of-complement activation post-splenectomy: a possible relation between splenectomy and infection. J Natl Med Assoc. 1978;70(9):667–670. [PMC free article] [PubMed] [Google Scholar]
- 23.Schaid TR, Nguyen J, Chen C, et al. Complement activation in a murine model of sickle cell disease: inhibition of vaso-occlusion by blocking C5 activation. Blood. 2016;128(22):158. [Google Scholar]
- 24.Merle NS, Grunenwald A, Rajaratnam H, et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight. 2018;3(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009; 9(10):729–740. [DOI] [PubMed] [Google Scholar]
- 26.de Cordoba SR, de Jorge EG. Translational mini-review series on complement factor H: genetics and disease associations of human complement factor H. Clin Exp Immunol. 2008;151(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kajander T, Lehtinen MJ, Hyvarinen S, et al. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc Natl Acad Sci U S A. 2011;108(7):2897–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. 2016;12(7):383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu J, Wu YQ, Ricklin D, et al. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat Immunol. 2009;10(7): 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgan HP, Schmidt CQ, Guariento M, et al. Structural basis for engagement by complement factor H of C3b on a self surface. Nat Struct Mol Biol. 2011;18(4):463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dworkis DA, Klings ES, Solovieff N, et al. Severe sickle cell anemia is associated with increased plasma levels of TNF-R1 and VCAM-1. Am J Hematol. 2011;86(2):220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magro CM, Momtahen S, Mulvey JJ, et al. Role of the skin biopsy in the diagnosis of atypical hemolytic uremic syndrome. Am J Dermatopathol. 2015;37(5):349–356; quiz 357-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scambi C, Ugolini S, Jokiranta TS, et al. The local complement activation on vascular bed of patients with systemic sclerosis: a hypothesis-generating study. PLoS One. 2015;10(2):e0114856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vianello A, Vencato E, Cantini M, et al. Improvement of maternal and fetal outcomes in women with sickle cell disease treated with early prophylactic erythrocytapheresis. Transfusion. 2018;58(9):2192–2201. [DOI] [PubMed] [Google Scholar]
- 35.de Jong K, Emerson RK, Butler J, et al. Short survival of phosphatidylserine-exposing red blood cells in murine sickle cell anemia. Blood. 2001;98(5):1577–1584. [DOI] [PubMed] [Google Scholar]
- 36.de Jong K, Larkin SK, Styles LA, Bookchin RM, Kuypers FA. Characterization of the phosphatidylserine-exposing subpopulation of sickle cells. Blood. 2001;98(3):860–867. [DOI] [PubMed] [Google Scholar]
- 37.de Franceschi L, Turrini F, Honczarenko M, et al. In vivo reduction of erythrocyte oxidant stress in a murine model of beta-thalassemia. Haematologica. 2004;89(11):1287–1298. [PubMed] [Google Scholar]
- 38.Hebbel RP. Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest. 1997;100(11 Suppl):S83–86. [PubMed] [Google Scholar]
- 39.Montes RA, Eckman JR, Hsu LL, Wick TM. Sickle erythrocyte adherence to endothelium at low shear: role of shear stress in propagation of vaso-occlusion. Am J Hematol. 2002;70(3):216–227. [DOI] [PubMed] [Google Scholar]
- 40.Barabino GA, McIntire LV, Eskin SG, Sears DA, Udden M. Endothelial cell interactions with sickle cell, sickle trait, mechanically injured, and normal erythrocytes under controlled flow. Blood. 1987;70(1):152–157. [PubMed] [Google Scholar]
- 41.Jokiranta TS, Hellwage J, Koistinen V, Zipfel PF, Meri S. Each of the three binding sites on complement factor H interacts with a distinct site on C3b. J Biol Chem. 2000;275(36):27657–27662. [DOI] [PubMed] [Google Scholar]
- 42.Matte A, De Falco L, Federti E, et al. Peroxiredoxin-2: a novel regulator of iron homeostasis in ineffective erythropoiesis. Antioxid Redox Signal. 2018;28(1):1–14. [DOI] [PubMed] [Google Scholar]
- 43.Elmariah H, Garrett ME, De Castro LM, et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am J Hematol. 2014;89(5):530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kalambur VS, Mahaseth H, Bischof JC, et al. Microvascular blood flow and stasis in transgenic sickle mice: utility of a dorsal skin fold chamber for intravital microscopy. Am J Hematol. 2004;77(2):117–125. [DOI] [PubMed] [Google Scholar]
- 45.Vercellotti GM, Zhang P, Nguyen J, et al. Hepatic overexpression of hemopexin inhibits inflammation and vascular stasis in murine models of sickle cell disease. Mol Med. 2016;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Serjeant GR. Leg ulceration in sickle cell anemia. Arch Intern Med. 1974;133(4):690–694. [PubMed] [Google Scholar]
- 47.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033–1048. [DOI] [PubMed] [Google Scholar]
- 48.Matsui NM, Borsig L, Rosen SD, et al. P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood. 2001;98(6):1955–1962. [DOI] [PubMed] [Google Scholar]
- 49.Manwani D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood. 2013;122(24):3892–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wood K, Russell J, Hebbel RP, Granger DN. Differential expression of E- and P-selectin in the microvasculature of sickle cell transgenic mice. Microcirculation. 2004;11(4): 377–385. [DOI] [PubMed] [Google Scholar]
- 51.Hidalgo A, Chang J, Jang JE, et al. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med. 2009;15(4):384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ataga KI, Kutlar A, Kanter J. Crizanlizumab in sickle cell disease. N Engl J Med. 2017;376(18):1796. [DOI] [PubMed] [Google Scholar]
- 53.Cherqui S, Kurian SM, Schussler O, et al. Isolation and angiogenesis by endothelial progenitors in the fetal liver. Stem Cells. 2006;24(1):44–54. [DOI] [PubMed] [Google Scholar]
- 54.Aranguren XL, Agirre X, Beerens M, et al. Unraveling a novel transcription factor code determining the human arterial-specific endothelial cell signature. Blood. 2013;122(24):3982–3992. [DOI] [PubMed] [Google Scholar]
- 55.Lotzer K, Dopping S, Connert S, et al. Mouse aorta smooth muscle cells differentiate into lymphoid tissue organizer-like cells on combined tumor necrosis factor receptor-1/lymphotoxin beta-receptor NF-kappaB signaling. Arterioscler Thromb Vasc Biol. 2010;30(3):395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201(6):871–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen G, Chang J, Zhang D, et al. Targeting Mac-1-mediated leukocyte-RBC interactions uncouples the benefits for acute vaso-occlusion and chronic organ damage. Exp Hematol. 2016;44(10):940–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kassebaum NJ, Jasrasaria R, Naghavi M, et al. A systematic analysis of global anemia burden from 1990 to 2010. Blood. 2014;123(5):615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 -the “Swiss Army Knife” of innate immunity and host defense. Immunol Rev. 2016;274(1):33–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beller DI, Springer TA, Schreiber RD. Anti-Mac-1 selectively inhibits the mouse and human type three complement receptor. J Exp Med. 1982;156(4):1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Franceschi L, Franco RS, Bertoldi M, et al. Pharmacological inhibition of calpain-1 prevents red cell dehydration and reduces Gardos channel activity in a mouse model of sickle cell disease. FASEB J. 2013;27(2):750–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stocker JW, De Franceschi L, McNaughton-Smith GA, et al. ICA-17043, a novel Gardos channel blocker, prevents sickled red blood cell dehydration in vitro and in vivo in SAD mice. Blood. 2003;101(6):2412–2418. [DOI] [PubMed] [Google Scholar]
- 63.de Franceschi L, Baron A, Scarpa A, et al. Inhaled nitric oxide protects transgenic SAD mice from sickle cell disease-specific lung injury induced by hypoxia/reoxygenation. Blood. 2003;102(3):1087–1096. [DOI] [PubMed] [Google Scholar]
- 64.Saggu G, Cortes C, Emch HN, et al. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and C3(H2O). J Immunol. 2013;190(12):6457–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Atkinson C, Zhu H, Qiao F, et al. Complement-dependent P-selectin expression and injury following ischemic stroke. J Immunol. 2006;177(10):7266–7274. [DOI] [PubMed] [Google Scholar]
- 66.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376(5):429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Slomski A. Crizanlizumab prevents sickle cell pain crises. JAMA. 2017;317(8):798. [DOI] [PubMed] [Google Scholar]
- 68.Telen MJ, Wun T, McCavit TL, et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood. 2015;125(17):2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harder MJ, Anliker M, Hochsmann B, et al. Comparative analysis of novel complement-targetedinhibitors, miniFH, and the natural regulators factor H and factor H-like protein 1 reveal functional determinants of complement regulation. J Immunol. 2016;196(2): 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nichols EM, Barbour TD, Pappworth IY, et al. An extended mini-complement factor H molecule ameliorates experimental C3 glomerulopathy. Kidney Int. 2015;88(6): 1314–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.