

Abstract
Selective covalent modification of a targeted protein is a powerful tool in chemical biology and drug discovery, with applications ranging from identification and characterization of proteins and their functions to the development of targeted covalent inhibitors. Most covalent ligands contain an affinity motif and an electrophilic warhead that reacts with a nucleophilic residue of the targeted protein. Because the electrophilic warhead is prone to react and modify off‐target nucleophiles, its reactivity should be balanced carefully to maximize target selectivity. Arylfluorosulfates have recently emerged as latent electrophiles for selective labeling of context‐specific tyrosine and lysine residues in protein pockets. Here, we review the recent but intense introduction of arylfluorosulfates into the arsenal of available warheads for selective covalent modification of proteins. We highlight the untapped potential of this functional group for use in chemical biology and drug discovery.
Keywords: covalent inhibitors, drug design, electrophilic warheads, medicinal chemistry, protein modifications
1. Introduction
The use of covalent protein ligands for interrogating protein function has proven extremely helpful in chemical biology. However, translation of probes into covalent drugs was abolished from drug discovery programs for a long time, mainly due to concerns regarding their idiosyncratic toxicity. In recent years, however, the development of covalent inhibitors has received a renewed attention, influenced by an exponentially increasing literature of successful examples,1 a better understanding of mechanisms of action,2 refined compound selectivity,3 and several drug approvals (e.g., telaprevir, carfilzomib, ibrutinib, osimertinib, neratinib, and afatinib; Scheme 1).
Scheme 1.
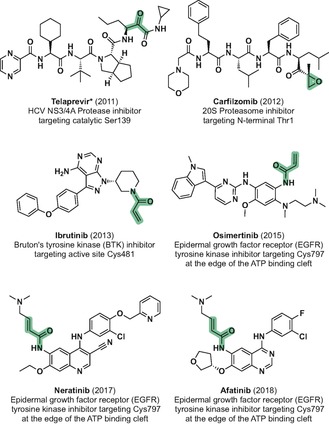
Chemical structures, FDA approval year and targeted residues of recently approved drugs that covalently bind their corresponding targets. The covalently targeted electrophiles are shaded. *Telaprevir forms a reversible hemiketal to the catalytic serine of HCV NS3/4A protease.
Most covalent ligands contain an affinity motif that enables selective binding to a protein active site and, in turn, directs the reactive warhead towards a nucleophilic residue of the target protein. Ideally, this facilitates formation of a covalent, irreversible bond with just the intended functional group among thousands of nucleophiles in the proteome. While a range of covalent ligands has been developed to target a diverse array of proteins, most of them were designed to target an accessible, suitably located nucleophilic cysteine residue in the protein pocket. The unique reactivity of cysteine allows for acrylamides to be the electrophilic functionalities of choice in these cases. These soft electrophiles require proximity to the nucleophilic, targeted cysteine to react, thus minimizing off‐target labeling. It is often the case that the protein of interest lacks a cysteine residue amenable to modification, since cysteines are only found in 1.5 % of protein pockets.4
Exploiting less nucleophilic residues for covalent labeling in protein binding sites has been less common, but there is a rapidly increasing number of examples of compounds capable of selective covalent targeting of tyrosine,5 lysine,6 serine,7 threonine,8 as well as glutamic acid9 residues in the context of activity‐based proteome profiling (ABPP) probes or covalent inhibitors. The development of irreversibly binding probes and inhibitors that rely on covalent modification of Tyr, Lys or Ser residues generally requires compounds bearing considerably reactive electrophilic warheads that can form a covalent bond with the targeted residue. Additionally, the surrounding protein environment enhances the nucleophilicity of the targeted residues by perturbing the pK a values of their side chains. For example, the ϵ‐amino groups of lysine residues display an intrinsic pK a of 10.4, and are most likely positively charged and unavailable for covalent modification under physiological conditions.10 However, an adequate protein environment that effectively buries a particular lysine and/or surrounds it with the appropriate residues can contribute to depress the pK a of its ϵ‐amino group down to five units, making it readily available as a nucleophile.11
Careful selection and optimization of warhead reactivity is essential to maximize target selectivity and minimize the modification of unwanted endogenous nucleophiles. Sulfonyl fluoride‐based warheads have been extensively used for the chemical biology community for covalent modification of a range of proteins, and this subject has been excellently reviewed in detail by Narayanan and Jones.12 Sulfonyl fluorides have been shown to bind covalently to Tyr, Lys and Ser amino acids in protein binding sites, but also to Thr, Cys, and His residues (Scheme 2 a).12, 13 Sulfonyl fluoride‐containing ligands have a balanced reactivity and modest aqueous stability and are incredibly useful tools in chemical biology, particularly explored as ABPP probes. However, the relatively high reactivity and promiscuity of sulfonyl fluoride‐based warheads may jeopardize the efficiency of these probes in whole‐cell investigations or experiments that require extended incubation times, which is an important limitation for the development of covalent inhibitors in drug discovery campaigns.
Scheme 2.
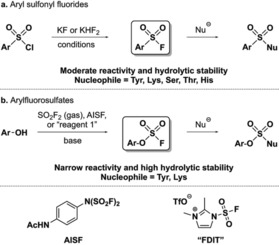
Chemical structure, syntheses and warhead reactivity of a) aryl sulfonyl fluorides and b) arylfluorosulfates. AISF=[4‐(acetylamino)phenyl]‐imidodisulfuryl difluoride. FDIT=1‐(fluorosulfonyl)‐2,3‐dimethyl‐1H‐imidazol‐3‐ium trifluoromethanesulfonate.
A much more stable and closely related sulfur(VI)‐fluoride group, the arylfluorosulfates (Scheme 2 b), has recently emerged as an alternative warhead for covalent modifications of context specific Tyr and Lys, but also Ser residues in protein binding sites that comprise an appropriate protein microenvironment for the sulfur(VI)‐fluoride exchange (SuFEx) reaction to occur.14
In this Minireview we highlight the unique properties of fluorosulfate‐based warheads in the context of other options, provide examples of covalent protein modification by fluorosulfate‐derived probes, and discuss their potential in probe and covalent inhibitor development for drug discovery and general chemical biology investigations.
2. Reactivity of Arylfluorosulfate‐Based Warheads
Sulfonyl fluoride‐derived warheads were broadly popularized in the 1960s as covalent protein modifiers after early reports describing their mechanism of action.15 Irreversible protein binders containing sulfonyl fluorides that take advantage of their relatively low reactivity for selective covalent inhibition of a range of proteins have been continuously developed since then.12 On the other hand, despite arylfluorosulfates being first reported in the 1930s,16 the lack of robust synthetic methods for their preparation resulted in only a few reports entertaining this functional group, and it was not adapted as warhead for covalent modification of proteins until 2015 by Kelly and co‐workers.17 The resurgence of arylfluorosulfates was pioneered by the Sharpless group due to development of a facile method for the synthesis of these compounds (Scheme 2 b).14a Simply incubating phenols with sulfuryl fluoride (gas) and base selectively provides their corresponding arylfluorosulfate derivatives even in the presence of amines, carboxylates, and aliphatic alcohols. Moreover, two bench‐stable “F‐SO2 +” donor reagents have subsequently been reported for arylfluorosulfate syntheses (AISF and FDIT, Scheme 2), thus overcoming the inconvenient handling of sulfuryl fluoride.18 In this regard, another practical transformation of phenols to arylfluorosulfates was also developed by ex situ generation of SO2F2 by means of a two‐chamber reactor.19
Unlike sulfonyl fluorides, arylfluorosulfates have proved to display remarkable stability towards hydrolysis and nucleophiles in general, since they remain intact in phosphate buffer (pH 10) for up to two weeks and in neutral buffers for months. In addition, arylfluorosulfates have been shown to be virtually unreactive in the presence of amino acids or natural products containing an array of different functional groups.14a, 20
In the context of covalent protein modification, this superior stability of arylfluorosulfates compared to sulfonyl fluorides should lead to an even more selective protein labeling and minimized off‐target modification. Granted that the ligand‐binding motif provides sufficient residence time projecting the warhead towards the targeted residue, latent arylfluorosulfate electrophiles have been shown to exclusively react upon activation in particular binding sites. Thus, the surrounding protein environment requires basic residues (Arg, Lys, or His) that decrease the pK a of the targeted nucleophilic residue and/or that facilitate fluoride ion departure (e.g. by hydrogen bonding interactions) to achieve the SuFEx reaction.21
3. Arylfluorosulfate‐Based Targeted Covalent Inhibitors
In the first reported attempt to use fluorosulfates for covalent protein modification, Kelly and co‐workers replaced the sulfonyl fluoride‐based warhead in previously reported fluorogenic transthyretin (TTR) probes22 with an ArOSO2F group (Scheme 3 a).17 Unlike the sulfonyl fluoride‐containing probes 1 and 2, arylfluorosulfates 3 and 4 reacted slowly with the targeted pK a‐perturbed Lys15 ϵ‐amino group of transthyretin and did not reach full conversion, due to their lower reactivity. The additional oxygen atom present in the fluorosulfate‐based warhead could also contribute to the inefficient covalent binding, by decreasing the proximity of the fluorosulfate sulfur atom to the nucleophilic Lys15 side chain or by geometrically compromising the hydrogen bond‐mediated stabilization of the leaving fluorine atom by neighboring residues. In addition, probes 3 and 4 proved to be essentially unreactive towards the whole proteome.
Scheme 3.
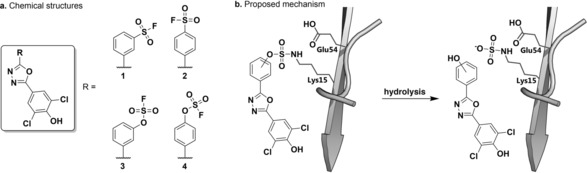
TTR fluorogenic covalent probes. a) Chemical structures of arylfluorosulfate‐ and aryl sulfonyl fluoride‐based TTR probes. b) Proposed mechanism for the observed formal Lys15 sulfamation. TTR=transthyretin.
Interestingly, adducts of TTR with probe 3 or 4 were not detected after more than 8 h incubation time. Only SO3 −‐modified TTR, the corresponding hydrolysis product (Scheme 3 b), was observed by mass spectrometry. This protein‐mediated hydrolysis of the ArOSO2‐covalent adducts appears to be intrinsic to TTR, since hydrolysis of a TTR‐16 (Scheme 5 b) conjugate with formal transfer of SO3 − to Lys15 was also observed over several days.23
Scheme 5.
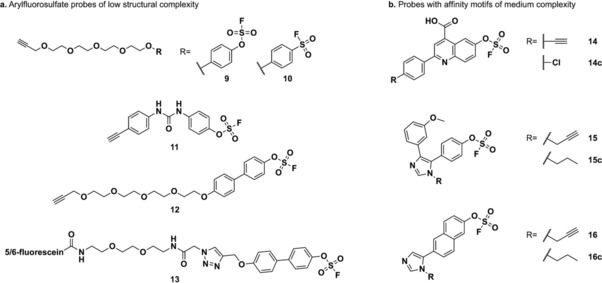
Chemical structures of a) simple alkyne‐functionalized fluorosulfate (9, 11, 12, and 13) and sulfonyl fluoride (10) probes and b) alkyne‐containing arylfluorosulfate probes (14, 15, and 16) and competitors (14 c, 15 c, and 16 c) with intermediate structural complexity.
The propensity and potential applicability of arylfluorosulfates for irreversible protein Lys sulfamation and/or Tyr sulfation remains to be explored in further detail. In a similar approach, attempting to overcome the limited stability of a sulfonyl fluoride‐based covalent ligand of the mRNA decapping scavenger enzyme (DcpS),24 Jones and co‐workers incorporated an arylfluorosulfate electrophile (6) into the previously developed DcpS covalent inhibitor 5 (Scheme 4).25 Remarkably, while the sulfonyl fluoride probe 5 was designed and proved to covalently label Tyr143 in the binding pocket of DcpS (Figure 1 a), fluorosulfate 6 covalently labeled Dcps at the non‐catalytic Ser272 (Figure 1 b). The formation of the covalent adduct was shown by intact mass LC‐MS and native electrospray ionization (ESI) MS analysis, which also revealed the presence of Ser272 dehydrated DcpS, that occurs via β‐elimination of the fluorosulfate‐DcpS conjugate (Scheme 4). Formation of Dha272‐DcpS upon covalent arylfluorosulfate binding takes place rapidly under acidic LC‐MS conditions, but slowly at pH 7.4, as shown by native ESI‐MS. Insights into the general dehydration tendency of Ser‐ and Thr‐modified residues in protein binding sites using ArOSO2F‐based warheads would be highly valuable for the future development of arylfluorosulfate‐based targeted covalent inhibitors.
Scheme 4.
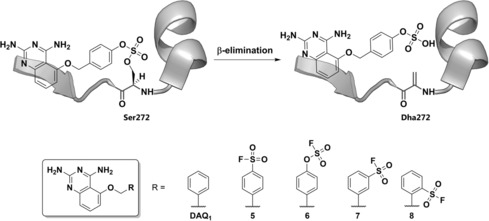
DcpS covalent probes. Top: proposed mechanism for the observed formal Ser272 dehydration. Bottom: chemical structures of non‐covalent probe DAQ1, fluorosulfate‐ and sulfonyl fluoride‐based probes.
Figure 1.
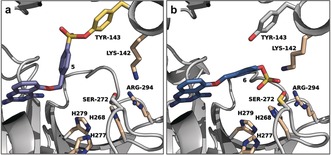
Binding mode of covalent DcpS targeted‐inhibitors. a) Co‐crystal structure of 5 and DcpS (PDB 4QDV). b) Docking pose of 6 in the DcpS binding pocket. Key residues that could catalyze the SuFEx reaction of the arylfluorosulfate with Ser272 are shown as sticks.
While the shorter sulfonyl fluoride warhead of 5 was optimally accommodated towards the Tyr143 side chain (Figure 1 a), the additional bridging oxygen atom of the arylfluorosulfate warhead in compound 6 results in improved positioning of the electrophilic sulfur(IV) atom towards Ser272 (Figure 1 b).
This non‐catalytic serine is neighbored by two basic amino acids (Lys142 and Arg294) and located close to three histidine residues (His268, 277, and 279). This assortment of basic side chains likely contributes to the SuFEx reaction by lowering the pK a of the hydroxyl side chain of Ser272 and/or helping the departure of the fluoride atom. Interestingly, while the para‐isomer 5 labels Tyr143 in the DcpS binding pocket, the ortho‐ and meta‐regioisomers 7 and 8 (Scheme 4) were predicted to locate their electrophilic warheads differently and indeed proved to react with the proximate Tyr113 phenol side chain instead.24 Investigation of the corresponding ortho‐ and meta‐arylfluorosulfate derivatives would provide further insights into their preferential positioning and reactivity. It would also provide valuable information for future attempts to design more stable and selective arylfluorosulfate‐based covalent inhibitors based on known sulfonyl fluoride‐derived compounds. Despite the similarity of both sulfur(VI)‐fluoride groups, the length and geometry of their warheads differ due to the additional oxygen atom present in the fluorosulfate moiety, and we predict that optimization may be required on a case‐by‐case basis. Further characterization of the stability of these compounds revealed that 6 remained intact for up to 24 h in PBS (pH 7.4), while the analogous sulfonyl fluoride 5 was largely hydrolyzed under the same experimental conditions. For a comparable measurement of non‐specific covalent protein labeling, 5 and 6 were incubated with human serum albumin (HSA), and the reactions were followed by LC‐MS. While 5‐HSA and 5(×2)‐HSA covalent adducts formed after 24 h incubation, covalent labeling with 6 was not detected, as a result of the superior kinetic stability and minimal promiscuity of arylfluorosulfate‐based warheads.
In order to sort out the plausible use of arylfluorosulfate‐based warheads in drug discovery, we went a step further and provided metabolic stability and membrane permeability data of both 6 and DAQ1 (Scheme 4), an analogous inhibitor lacking the ‐OSO2F functionality. Interestingly, 6 showed a reduced metabolic turnover rate in human liver microsomes. Arylfluorosulfate 6 was shown to migrate slower than DAQ1, but still displayed acceptable membrane permeability, although the enhanced apparent permeability from the B to A side suggests that efflux is occurring, a behavior not observed for DAQ1. The collection of more ADME data from potential arylflurosulfate‐based covalent inhibitors and their analogs lacking the corresponding electrophile will be important to gain insights into the potential use of fluorosulfate warheads in drug discovery research.
4. Reactivity and Selectivity of Simple Arylfluorosulfate Probes Towards the Human Proteome
Due to their lower reactivity, arylfluorosulfate probes provide a more selective covalent labeling of proteins than the extensively used aryl sulfonyl fluorides in whole human proteome experiments.17 The Kelly and Sharpless groups compared the proteome reactivity of both functional groups and showed that structurally simple arylfluorosulfate‐containing probes 9 and 11 (Scheme 5 a) covalently modified a low number of proteins in HeLa cell culture and lysate, while the analogous aryl sulfonyl fluoride probe 10 (Scheme 5 a) labeled a wide array of proteins under the same conditions.21
SDS‐PAGE separation of proteins from HeLa cells treated with 9 revealed that this probe selectively binds several members of the proteome in the 15 kDa range. Stable isotope labeling by amino acids in cell culture (SILAC)26 experiments were performed to identify these 15 kDa targets to be the fatty acid binding proteins 3 and 5 (FABP3 and FABP5) and the cellular retinoic acid binding protein 2 (CRABP2), which are members of the intracellular lipid binding protein (iLPB) family.27 Complete covalent adduct formation was observed upon incubation of recombinant CRABP2 (2 μm) and 9 (100 μm) after 48 h, and 80 % covalent labeling of recombinant FABP5 was achieved under the same experimental conditions. In addition, recombinant human FABP4, a structurally similar iLBP, formed 90 % covalent adduct with 9 as seen by LC‐MS analysis. However, covalent binding of this probe to FABP3 could not be validated in this or other attempted experiments. In order to accelerate covalent bond formation by enhancing the potency of the affinity motif, biphenyl‐containing probes 12 and 13 (Scheme 5) were synthesized and yielded quantitative covalent adduct formation with recombinant CRABP2 within 1 h. Probes 12 and 13 (100 μm) showed a remarkable and unexpected selectivity for CRABP2, since labeling of FABP3, FABP4, and FABP5 (2 μm) was not detected, even at 24 h incubation times. Selective and nearly complete covalent labeling of overexpressed GFP‐CRABP2 was achieved with the more cell‐permeable probe 12 (20 μm) in living HEK293T cells overexpressing both GFP‐CRABP2 and FABP5‐GFP.
iLBPs contain an arylfluorosulfate‐desirable conserved Arg‐Tyr‐Arg motif in their binding pocket (Figure 2).27 Tandem mass spectrometry experiments28 revealed that arylfluorosulfate probes bind iLBPs via a conserved Tyr residue in their fatty acid‐binding sites. The Arg duo perturb the pK a of the Tyr phenol side chain and/or stabilize the departing fluoride during the sulfur(VI)‐fluoride exchange reaction. Incubation of 13 and recombinant CRABP2 in buffers ranging from pH 4.9 to 10.4 revealed that 13 labels CRABP2 in a pH‐dependent manner (labeling efficiency increases with pH value), and that the phenol side chain of Tyr134 displays an apparent pK a of 7.6, indicating pK a perturbation of the phenol by Arg111 and/or Arg132. To gain insight into the role of the Arg pair in covalent bond formation, CRABP2 mutants Arg111Leu and Arg132Leu were incubated with 13 at different pHs. Both point mutations resulted in a markedly decreased covalent labeling even at pH 10.4, which is higher than the pK a of standard Tyr side chains, suggesting that the neighboring arginines are directly involved in catalyzing the SuFEx reaction.
Figure 2.
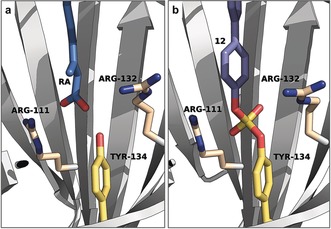
Binding mode of retinoic acid (RA) and 12 to CRABP2. a) Co‐crystal structure of RA and CRABP2 (PDB 2FR3). b) Co‐crystal structure of 12 and CRABP2 (PDB 5HZQ). Key residues are shown as sticks.
A crystal structure resulting from probe 12 in complex with CRABP2 unambiguously proved the formation of a diarylsulfate diester bond between ligand and Tyr134 and showed that the biaryl group engaged in hydrophobic interactions with several residues in the retinoic acid (RA) binding site (Figure 2).29 The crystal structure revealed that the Thr75‐Gly78 β‐turn in CRABP2 provides further space to accommodate the outer ring of 12 than other iLBPs, which explains the significant CRABP2 selectivity of the biphenyl‐based probes.21 In addition, 12‐mediated irreversible inactivation of CRABP2 decreased the transcriptional activation of retinoic acid receptor alpha (RARα), which regulates transcription in a ligand‐dependent manner,30 by inhibiting retinoic acid delivery.
5. Inverse Drug Discovery Using Arylfluorosulfate Probes
With the knowledge that simple arylfluorosulfates display a proteome preference for iLBPs, the Sharpless, Wilson, and Kelly groups ventured into using more structurally complex arylfluorosulfate electrophile probes in an inverse drug discovery approach.23 In this strategy, a small number of arylfluorosulfate clickable probes with intermediate structural complexity was exposed to the human proteome and selectively labeled protein targets were identified and validated by means of mutagenesis experiments, mass spectrometry analysis, and X‐ray crystallography. Three different arylfluorosulfate probes were selected, which contained different aryl motifs, functional groups, as well as H‐bond donors and acceptors. These chemical features endow the probes with sufficient reversible binding energy to preferentially bind particular groups of proteins, and together with the narrow reactivity of the arylfluorosulfate electrophile, should result in a highly selective covalent labeling of different proteome members.
Living HEK293T cells and lysates were treated with alkyne containing probes 14, 15, and 16 alone or in the presence of an excess of competitor (14 c, 15 c, or 16 c, respectively) previous to copper(I)‐catalyzed azide‐alkyne cycloaddition (CuAAC) click reaction31 with either tetramethylrhodamine‐ or diazo biotin‐azide. Labeled proteins were then investigated by fluorescence SDS‐PAGE or tandem mass spectrometry after streptavidin enrichment, trypsin digestion and six‐plex tandem mass tags (TMTs) labeling.32 Disappearance of fluorescence signal (in SDS‐PAGE) or abundance (in MS‐MS) of covalently modified proteins by the alkyne probes in the presence of the corresponding competitors indicate a high extent of covalent protein labeling. Therefore, enabling differentiation between complete irreversible binding of low‐abundance proteins and partial modification of proteins present in higher concentrations.33 Covalently targeted proteome members by each probe were identified by quantitative proteomics and rated on the basis of a high competition ratio (relative abundance of a corresponding protein in an experiment where only alkyne probe vs. alkyne probe plus competitor was applied) and low p‐values. Based on that ranking, 12 different proteins, from which structural information was already available in the Protein Data Bank, were selected for further validation studies. Only in one case, did LC‐ESI‐MS demonstrate that the corresponding recombinant protein did not react upon incubation with the arylfluorosulfate compound, while the recombinant proteins of the remaining 11 selected targets proved to be covalently labeled by their respective probes under the experimental conditions.
Interestingly, a diverse set of proteins was enriched by different probes, marking the predicted importance of the peculiarly distinct binding motifs to achieve selective labeling. A few protein targets were, however, shown to react also with different probes. Glutathione S‐transferase π (GSTP1) was found to be enriched in the volcano plots of both 14 and 15, and tethering experiments corroborated adduct formation in both cases by LC‐ESI‐MS. Mutagenesis experiments of the two potential labeled Tyr residues13c, 34 one at a time (Y7F and Y108F) or both at the same time (Y7F,T108F) revealed that 15 reacts preferentially with Tyr7, but Tyr108 can be labeled as well, since removal of both nucleophiles inside the GSTP1 binding site is necessary to completely abrogate covalent labeling as shown by in‐gel fluorescence. The importance of the neighboring basic Arg13 residue was corroborated by showing that an R13K mutant retained fluorosulfate reactivity, while an R13Q mutation abolished covalent adduct formation.
The multidimensional protein identification technology (MuDPIT) LC‐MS/MS competition ratio experiment identified another glutathione S‐transferase, GSTO1, as target of 14. We corroborated quantitative covalent adduct formation over 48 h with recombinant enzyme and a co‐crystal structure proved sulfate diester bond formation to Tyr229 (Figure 3 b). Mutagenesis experiments highlighted the importance of both proximal Lys57 and Lys59 for labeling (Figure 3), since K57Q and K59Q mutations attenuated covalent adduct formation. However, covalent labeling of these mutants was rescued at pH 10.5, suggesting that the main role of the surrounding Lys residues is to lower the pK a of the Tyr229 phenol in order to promote SuFEx reaction.
Figure 3.
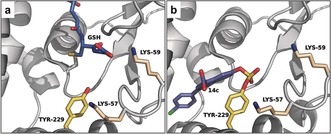
Binding mode of GSH and 14 c to GSTO1. a) Co‐crystal structure of GSH and GSTO1 (PDB 2HVE). b) Co‐crystal structure of 14 c and GSTO1 (PDB 5UI4). Key residues are shown as sticks.
Efforts directed to validate nucleoside diphosphate kinase A (NME1) as a target of 15 led to several interesting findings. LC‐ESI‐MS corroborated complete formation of the NME1‐15 c covalent adduct, which the authors managed to crystallize. The resulting X‐ray crystal structure revealed a sulfamate‐mediated covalent bond to the Lys12 side chain, providing the first example of an X‐ray crystal structure showing a fluorosulfate warhead labeling a lysine residue (Figure 4). This is particularly interesting, because a nearby Tyr52 is also available for covalent bond formation. However, only the side chain of Lys12, which is surrounded by basic Arg105 and His118 residues and predicted to be intrinsically nucleophilic,35 proved to partake in covalent adduct formation, since a MNE1‐K12A mutant failed to react with 15.
Figure 4.
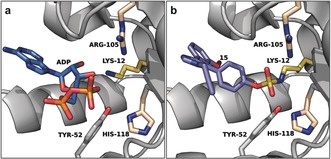
Binding mode of ADP and 15 to NME1. a) Co‐crystal structure of ADP and NME1 (PDB 2HVE). b) Co‐crystal structure of 15 and NME1 (PDB 5UEH). Key residues are shown as sticks.
The remaining eight selected proteins (BLVRA, TIGAR, HSDL2, ALKBH5, TMPT, HMOX2, CRABP2, and TTR) were also validated by measuring covalent adduct formation by LC‐ESI‐MS and, in some cases, the labeling site was assigned by tandem mass spectrometry (MS‐MS) analysis. Interestingly, 15 was found to react with thiopurine S‐methyltransferase (TPMT) by binding to the solvent exposed Lys32 nearby the active site. However, covalent labeling of FTO, which was identified in the competition ratio experiment with probe 14, was not observed and failed to be validated under various experimental conditions. As pointed out, this could be explained by differences between the recombinant and the endogenous protein in a cellular context, as post‐translational modification of FTO or formation of a protein complex could be necessary for covalent modification to occur.23
In vitro functional assays demonstrated a decreased GSTP1 and BLVRA enzymatic activity as a result of treatment with 14 and/or 15 probes. In addition, known inhibitors or endogenous ligands compete with the fluorosulfate‐based probes for the protein binding sites of NME1, CRABP2, and TTR.36 Particularly interesting is the fact that covalent probe 14 is the first reported small molecule probe of hydroxysteroid dehydrogenase‐like protein 2 (HSDL2).37 Several of these proteins possess a high interest for drug discovery and these encouraging results indicate that optimization of the compounds used here could provide potential covalent drug candidates for the treatment of a diverse range of diseases.
6. Summary and Outlook
The recent development of a facile method for the synthesis of arylfluorosulfates has spurred the use of this latent electrophile for covalent protein modification. The low and narrow intrinsic reactivity of fluorosulfate‐based warheads assures minimal covalent labeling of off‐target nucleophiles in a cellular context, making these compounds largely unreactive towards the human proteome unless rigorous conditions are met (e.g. the presence of basic residues that decrease the pK a of the targeted nucleophilic residue and/or facilitate the departure of the fluoride ion).
Similar to the reports discussed herein, several recent examples provide additional encouraging data to start considering the use of arylfluorosulfate‐targeted covalent inhibitors in drug discovery. Thus, proteins containing genetically encoded fluorosulfate‐l‐tyrosine (FSY) have been shown to react with strategically located proximal Tyr, Lys, and His residues on the surfaces of their interacting protein partners in live E. coli, and exhibited no toxicity towards E. coli or mammalian cells at concentrations up to 1 mm.38 These results approved the use of genetically encoded FSY for covalent attachment of protein partners in vivo and suggest the value of arylfluorosulfate‐based compounds for the selective covalent labeling of Tyr, Lys, or His residues on protein surfaces for targeting specific protein‐protein interactions. In addition, reactive covalent docking experiments have already proved useful to predict possible covalent binding sites of arylfluorosulfate probes.23 This suggests that rational targeting of intrinsically reactive Tyr, Lys or even Ser and His residues could be applied in drug discovery for a given protein of interest that displays a fluorosulfate‐desired protein microenvironment, especially with a high‐affinity, reversible ligand in hand that can be modified. Furthermore, a platform for late‐stage compound functionalization has also been developed, that allows for the in situ, selective arylfluorosulfate formation from phenol‐containing compounds in a 96‐well plate format, allowing the generation of arylfluorosulfate compound libraries that can be directly applied in cell‐based or enzymatic assays.39
To date, arylfluorosulfate‐based probes have been found to modify protein pockets preferentially through tyrosine residues (in iLBPs, GSTP1, GSTO1, BLVRA, HMOX2, HSDL2, ALKBH5, and TIGAR proteins) and lysine residues (in TTR, NME1, TMPT, and HMOX2) but also serine in DcpS. We predict that the number of examples of arylfluorosulfate‐derived covalent ligands will substantially increase over the coming years, which should provide further insights into the structure and substituent effects on reactivity, chemical and metabolic stability, as well as covalent target engagement selectivity of these compounds. We anticipate to witness the application of other “SuFEx‐able” warheads (e.g., alkyl or aryliminosulfur oxy(di or mono)fluorides)40 in chemical biology research, which could lead to the development of targeted covalent inhibitors with even lower proteome‐wide reactivity and different amino acid labeling preference.
These are encouraging times for the field of covalent protein modification, and yet the powerful addition of arylfluorosulfates to the catalogue of available warheads promises even more exciting developments in the fields of covalent probes and inhibitors in chemical biology and drug discovery.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Pablo Martín‐Gago is currently an assistant professor at Copenhagen University. He completed his Ph.D. in 2013 at the Institute for Research in Biomedicine (IRB Barcelona), under the supervision of Prof. A. Riera. After a research stay in the group of Prof. G. C. Fu at Caltech, he joined the Max Planck Institute of Molecular Physiology in Dortmund for postdoctoral studies with Prof. H. Waldmann, developing inhibitors of the KRas‐PDEδ interaction.

Biographical Information
Christian A. Olsen received his Ph.D. from the Danish University of Pharmaceutical Sciences. After independently working on the development of novel peptidomimetics, he did his postdoctoral fellowship at The Scripps Research Institute. In 2010 he returned to the Technical University of Denmark and in 2014 he accepted his current position as professor at the University of Copenhagen. He is recipient of the Lundbeck Foundation Fellowship, the EFMC prize for a young medicinal chemist in academia, and an ERC Consolidator grant.

Acknowledgements
We gratefully acknowledge Dr. Andreas S. Madsen for assistance with the Frontispiece graphic. This work was supported by the European Research Council (ERC‐CoG‐725172—SIRFUNCT; C.A.O.).
P. Martín-Gago, C. A. Olsen, Angew. Chem. Int. Ed. 2019, 58, 957.
Contributor Information
Prof. Dr. Pablo Martín‐Gago, Email: pabm@lundbeck.com.
Prof. Dr. Christian A. Olsen, Email: cao@sund.ku.dk.
References
- 1. Singh J., Petter R. C., Baillie T. A., Whitty A., Nat. Rev. Drug Discovery 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Johnson D. S., Weerapana E., Cravatt B. F., Future Med. Chem. 2010, 2, 949–964; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Zhao Z., Bourne P. E., Drug Discovery Today 2018, 23, 727–735. [DOI] [PubMed] [Google Scholar]
- 3. Baillie T. A., Angew. Chem. Int. Ed. 2016, 55, 13408–13421; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13606–13619. [Google Scholar]
- 4. Chen K., Kurgan L., PLoS ONE 2009, 4, e4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Watts A. G., Damager I., Amaya M. L., Buschiazzo A., Alzari P., Frasch A. C., Withers S. G., J. Am. Chem. Soc. 2003, 125, 7532–7533. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Pettinger J., Jones K., Matthew C. D., Angew. Chem. Int. Ed. 2017, 56, 15200–15209; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15398–15408; [Google Scholar]
- 6b. Choi S., Ong D. S. T., Kelly J. W., J. Am. Chem. Soc. 2010, 132, 16043–16051; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Nanna A. R., Li X., Walseng E., Pedzisa L., Goydel R. S., Hymel D., T. R. Burke, Jr. , Roush W. R., Rader C., Nat. Commun. 2017, 8, 1112; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Matos M. J., Oliveira B. L., Martínez-Sáez N., Guerreiro A., Cal P. M. S. D., Bertoldo J., Maneiro M., Perkins E., Howard J., Deery M. J., Chalker J. M., Corzana F., Jiménez-Osés G., Bernardes G. J. L., J. Am. Chem. Soc. 2018, 140, 4004–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Dekker F. J., Rocks O., Vartak N., Menninger S., Hedberg C., Balamurugan R., Wetzel S., Renner S., Gerauer M., Schölermann B., Rusch M., Kramer J. W., Rauh D., Coates G. W., Brunsveld L., Bastiaens P. I. H., Waldmann H., Nat. Chem. Biol. 2010, 6, 449; [DOI] [PubMed] [Google Scholar]
- 7b. Otrubova K., Brown M., McCormick M. S., Han G. W., O'Neal S. T., Cravatt B. F., Stevens R. C., Lichtman A. H., Boger D. L., J. Am. Chem. Soc. 2013, 135, 6289–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bogyo M., McMaster J. S., Gaczynska M., Tortorella D., Goldberg A. L., Ploegh H., Proc. Natl. Acad. Sci. USA 1997, 94, 6629–6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martín-Gago P., Fansa E. K., Winzker M., Murarka S., Janning P., Schultz-Fademrecht C., Baumann M., Wittinghofer A., Waldmann H., Cell Chem. Biol. 2017, 24, 589–597. [DOI] [PubMed] [Google Scholar]
- 10. Platzer G., Okon M., McIntosh L. P., J. Biomol. NMR 2014, 60, 109–129. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Harris T. K., Turner G. J., IUBMB Life 2002, 53, 85–98; [DOI] [PubMed] [Google Scholar]
- 11b. Isom D. G., Castañeda C. A., Cannon B. R., García-Moreno B. E., Proc. Natl. Acad. Sci. USA 2011, 108, 5260–5265; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Yunus A. A., Lima C. D., Nat. Struct. Mol. Biol. 2006, 13, 491; [DOI] [PubMed] [Google Scholar]
- 11d. Hiroshi I., FEBS Lett. 2010, 584, 3464–3468. [DOI] [PubMed] [Google Scholar]
- 12. Narayanan A., Jones L. H., Chem. Sci. 2015, 6, 2650–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Dombrowski K. E., Huang Y. C., Colman R. F., Biochemistry 1992, 31, 3785–3793; [DOI] [PubMed] [Google Scholar]
- 13b. Gushwa N. N., Kang S., Chen J., Taunton J., J. Am. Chem. Soc. 2012, 134, 20214–20217; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Gu C., Shannon D. A., Colby T., Wang Z., Shabab M., Kumari S., Villamor J. G., McLaughlin C. J., Weerapana E., Kaiser M., Cravatt B. F., van der Hoorn R. A. L., Chem. Biol. 2013, 20, 541–548; [DOI] [PubMed] [Google Scholar]
- 13d. Shannon D. A., Weerapana E., Curr. Opin. Chem. Biol. 2015, 24, 18–26; [DOI] [PubMed] [Google Scholar]
- 13e. Fadeyi O., Parikh M. D., Chen M. Z., R. E. Kyne, Jr. , Taylor A. P., O'Doherty I., Kaiser S. E., Barbas S., Niessen S., Shi M., Weinrich S. L., Kath J. C., Jones L. H., Robinson R. P., ChemBioChem 2016, 17, 1925–1930; [DOI] [PubMed] [Google Scholar]
- 13f. Dubiella C., Cui H., Groll M., Angew. Chem. Int. Ed. 2016, 55, 13330–13334; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13524–13528; [DOI] [PubMed] [Google Scholar]
- 13g. Zhao Q., Ouyang X., Wan X., Gajiwala K. S., Kath J. C., Jones L. H., Burlingame A. L., Taunton J., J. Am. Chem. Soc. 2017, 139, 680–685; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13h. Lentz C. S., Sheldon J. R., Crawford L. A., Cooper R., Garland M., Amieva M. R., Weerapana E., Skaar E. P., Bogyo M., Nat. Chem. Biol. 2018, 14, 609–617; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13i. Guardiola S., Prades R., Mendieta L., Brouwer A. J., Streefkerk J., Nevola L., Tarragó T., Liskamp R. M. J., Giralt E., Cell Chem. Biol. 2018, 10.1016/j.chembiol.2018.04.013. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Dong J., Krasnova L., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2014, 53, 9430–9448; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9584–9603; [Google Scholar]
- 14b. Jones L. H., ACS Med. Chem. Lett. 2018, 9, 584–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Baker B. R., Wood W. F., J. Med. Chem. 1968, 11, 650–652; [Google Scholar]
- 15b. Baker B. R., Lourens G. J., J. Med. Chem. 1967, 10, 1113–1122; [DOI] [PubMed] [Google Scholar]
- 15c. Baker B. R., Hurlbut J. A., J. Med. Chem. 1968, 11, 241–245; [DOI] [PubMed] [Google Scholar]
- 15d. Powers J. C., Tanaka T., Harper J. W., Minematsu Y., Barker L., Lincoln D., Crumley K. V., Fraki J. E., Schechter N. M., Lazarus G. G., et al., Biochemistry 1985, 24, 2048–2058; [DOI] [PubMed] [Google Scholar]
- 15e. Deutsch D. G., Lin S., Hill W. A. G., Morse K. L., Salehani D., Arreaza G., Omeir R. L., Makriyannis A., Biochem. Biophys. Res. Commun. 1997, 231, 217–221; [DOI] [PubMed] [Google Scholar]
- 15f. Baker B. R., Ann. N. Y. Acad. Sci. 1971, 186, 214–226. [DOI] [PubMed] [Google Scholar]
- 16. Lange W., Müller E., Ber. Dtsch. Chem. Ges. 1930, 63, 2653–2657. [Google Scholar]
- 17. Baranczak A., Liu Y., Connelly S., Du W.-G. H., Greiner E. R., Genereux J. C., Wiseman R. L., Eisele Y. S., Bradbury N. C., Dong J., Noodleman L., Sharpless K. B., Wilson I. A., Encalada S. E., Kelly J. W., J. Am. Chem. Soc. 2015, 137, 7404–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Zhou H., Mukherjee P., Liu R., Evrard E., Wang D., Humphrey J. M., Butler T. W., Hoth L. R., Sperry J. B., Sakata S. K., Helal C. J., am Ende C. W., Org. Lett. 2018, 20, 812–815; [DOI] [PubMed] [Google Scholar]
- 18b. Guo T., Meng G., Zhan X., Yang Q., Ma T., Xu L., Sharpless K. B., Dong J., Angew. Chem. Int. Ed. 2018, 57, 2605–2610; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2635–2640. [Google Scholar]
- 19. Veryser C., Demaerel J., Bieliūnas V., Gilles P., De Borggraeve W. M., Org. Lett. 2017, 19, 5244–5247. [DOI] [PubMed] [Google Scholar]
- 20. Mukherjee H., Debreczeni J., Breed J., Tentarelli S., Aquila B., Dowling J. E., Whitty A., Grimster N. P., Org. Biomol. Chem. 2017, 15, 9685–9695. [DOI] [PubMed] [Google Scholar]
- 21. Chen W., Dong J., Plate L., Mortenson D. E., Brighty G. J., Li S., Liu Y., Galmozzi A., Lee P. S., Hulce J. J., Cravatt B. F., Saez E., Powers E. T., Wilson I. A., Sharpless K. B., Kelly J. W., J. Am. Chem. Soc. 2016, 138, 7353–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grimster N. P., Connelly S., Baranczak A., Dong J., Krasnova L. B., Sharpless K. B., Powers E. T., Wilson I. A., Kelly J. W., J. Am. Chem. Soc. 2013, 135, 5656–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mortenson D. E., Brighty G. J., Plate L., Bare G., Chen W., Li S., Wang H., Cravatt B. F., Forli S., Powers E. T., Sharpless K. B., Wilson I. A., Kelly J. W., J. Am. Chem. Soc. 2018, 140, 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hett E. C., Xu H., Geoghegan K. F., Gopalsamy A., Kyne R. E., Menard C. A., Narayanan A., Parikh M. D., Liu S., Roberts L., Robinson R. P., Tones M. A., Jones L. H., ACS Chem. Biol. 2015, 10, 1094–1098. [DOI] [PubMed] [Google Scholar]
- 25. Fadeyi O. O., Hoth L. R., Choi C., Feng X., Gopalsamy A., Hett E. C., Kyne R. E., Robinson R. P., Jones L. H., ACS Chem. Biol. 2017, 12, 2015–2020. [DOI] [PubMed] [Google Scholar]
- 26. Ong S.-E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M., Mol. Cell. Proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- 27. Smathers R. L., Petersen D. R., Hum. Genomics 2011, 5, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yates J. R., Eng J. K., McCormack A. L., Schieltz D., Anal. Chem. 1995, 67, 1426–1436. [DOI] [PubMed] [Google Scholar]
- 29. Vaezeslami S., Mathes E., Vasileiou C., Borhan B., Geiger J. H., J. Mol. Biol. 2006, 363, 687–701. [DOI] [PubMed] [Google Scholar]
- 30. Maden M., Nat. Rev. Neurosci. 2007, 8, 755. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Tornøe C. W., Christensen C., Meldal M., J. Org. Chem. 2002, 67, 3057–3064; [DOI] [PubMed] [Google Scholar]
- 31b. Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B., Angew. Chem. Int. Ed. 2002, 41, 2596–2599; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2708–2711; [Google Scholar]
- 31c. Speers A. E., Adam G. C., Cravatt B. F., J. Am. Chem. Soc. 2003, 125, 4686–4687. [DOI] [PubMed] [Google Scholar]
- 32. Dayon L., Hainard A., Licker V., Turck N., Kuhn K., Hochstrasser D. F., Burkhard P. R., Sanchez J.-C., Anal. Chem. 2008, 80, 2921–2931. [DOI] [PubMed] [Google Scholar]
- 33. Wang C., Weerapana E., Blewett M. M., Cravatt B. F., Nat. Methods 2013, 11, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oakley A. J., Bello M. L., Battistoni A., Ricci G., Rossjohn J., Villar H. O., Parker M. W., J. Mol. Biol. 1997, 274, 84–100. [DOI] [PubMed] [Google Scholar]
- 35. Postel E. H., Abramczyk B. M., Levit M. N., Kyin S., Proc. Natl. Acad. Sci. USA 2000, 97, 14194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.
- 36a. Malmquist N. A., Anzinger J. J., Hirzel D., Buxton I. L., Proc. Western Pharmacol. Soc. 2001, 44, 57–59; [PubMed] [Google Scholar]
- 36b. Budhu A. S., Noy N., Mol. Cell. Biol. 2002, 22, 2632–2641; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36c. Bulawa C. E., Connelly S., DeVit M., Wang L., Weigel C., Fleming J. A., Packman J., Powers E. T., Wiseman R. L., Foss T. R., Wilson I. A., Kelly J. W., Labaudinière R., Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ruokun C., Yake X., Fengdong Y., Xinting W., Laijun S., Xianzhi L., Tumor Biol. 2016, 37, 15065–15077. [DOI] [PubMed] [Google Scholar]
- 38. Wang N., Yang B., Fu C., Zhu H., Zheng F., Kobayashi T., Liu J., Li S., Ma C., Wang P. G., Wang Q., Wang L., J. Am. Chem. Soc. 2018, 140, 4995–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu Z., Li J., Li S., Li G., Sharpless K. B., Wu P., J. Am. Chem. Soc. 2018, 140, 2919–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li S., Wu P., Moses E. J., Sharpless K. B., Angew. Chem. Int. Ed. 2017, 56, 2903–2908; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2949–2954. [Google Scholar]