

Abstract
The Ras/mitogen‐activated protein kinase (MAPK) pathway controls fundamental cellular processes such as proliferation, differentiation, and apoptosis. The dual‐specificity phosphatase 6 (DUSP6) regulates cytoplasmic MAPK signaling by dephosphorylating and inactivating extracellular signal‐regulated kinase (ERK1/2) MAPK. To determine the role of DUSP6 in the maintenance of intestinal homeostasis, we characterized the intestinal epithelial phenotype of Dusp6 knockout (KO) mice under normal, oncogenic, and proinflammatory conditions. Our results show that loss of Dusp6 increased crypt depth and epithelial cell proliferation without altering colonic architecture. Crypt regeneration capacity was also enhanced, as revealed by ex vivo Dusp6 KO organoid cultures. Additionally, loss of Dusp6 induced goblet cell expansion without affecting enteroendocrine and absorptive cell differentiation. Our data also demonstrate that Dusp6 KO mice were protected from acute dextran sulfate sodium‐induced colitis, as opposed to wild‐type mice. In addition, Dusp6 gene deletion markedly enhanced tumor load in Apc Min/+ mice. Decreased DUSP6 expression by RNA interference in HT29 colorectal cancer cells enhanced ERK1/2 activation levels and promoted both anchorage‐independent growth in soft agar as well as invasion through Matrigel. Finally, DUSP6 mRNA expression in human colorectal tumors was decreased in advanced stage tumors compared with paired normal tissues. These results demonstrate that DUSP6 phosphatase, by controlling ERK1/2 activation, regulates colonic inflammatory responses, and protects the intestinal epithelium against oncogenic stress.
Keywords: colitis, colorectal cancer, Dusp6, ERK, intestinal epithelium, proliferation
Abbreviations
- APC
adenomatous polyposis coli
- CRC
colorectal cancer
- DSS
dextran sulfate sodium
- DUSP
dual‐specificity phosphatase
- EdU
5‐ethynyl‐2‐deoxyuridine
- EGF
epidermal growth factor
- ERK
extracellular signal‐regulated kinase
- FBS
fetal bovine serum
- KRAS
Kirsten rat sarcoma viral oncogene homolog
- IEC
intestinal epithelial cells
- MAPK
mitogen‐activated protein kinase
- MEK
MAPK/Erk kinase
- MKP
mitogen‐activated protein kinase phosphatase
- Min
multiple intestinal neoplasia
- MSI
microsatellite instability
- MSS
microsatellite stable
- PCNA
proliferating cell nuclear antigen
- PI3K
phosphatidylinositol 3‐kinase
- shRNA
short hairpin RNA
- TGF
transforming growth factor
- Wnt
wingless
1. INTRODUCTION
The intestinal epithelium is constantly renewed and repaired throughout life by the action of multipotent stem cells that give rise to a population of proliferative and undifferentiated cells. These progenitor cells, also called transit‐amplifying (T/A) cells, proliferate rapidly while migrating along the crypt axis. In the small intestine, the progenitor cells cease their proliferation after two to three divisions and differentiate in absorptive enterocytes, mucus‐producing goblet cells, hormone‐secreting enteroendocrine cells, or Paneth cells. While the first three cell types differentiate during an upward migration from the crypt to the adjacent villus, Paneth cells differentiate during a downward movement to the crypt base. In contrast to the small intestine, the colonic epithelium displays epithelial caps instead of villi and deeper crypts devoid of Paneth cells (Barker, 2014; Crosnier, Stamataki, & Lewis, 2006).
A central role for wingless (Wnt), bone morphogenetic proteins/transforming growth factor β (TGFβ), Notch, and epidermal growth factor (EGF) signal transduction pathways in the maintenance, proliferation, and differentiation of intestinal stem and progenitor cells has been demonstrated in several murine models (Andreu et al., 2005; Carragher et al., 2010; van Es et al., 2005Feng et al., 2011; Fre et al., 2005; Haigis et al., 2008; Haramis et al., 2004; He et al., 2004; Janssen et al., 2002; Jensen et al., 2000; Korinek et al., 1998; Kuhnert et al., 2004; Milano et al., 2004; Pinto, Gregorieff, Begthel, & Clevers, 2003; Sansom et al., 2004; Yang, Bermingham, Finegold, & Zoghbi, 2001; Zecchini, Domaschenz, Winton, & Jones, 2005). EGF and its orthologs epiregulin and TGFα, control epithelial cell proliferation by activating the Ras/Raf/MEK/ERK (extracellular signal‐regulated kinase) mitogen‐activated protein kinase pathway (MAPK) and phosphatidylinositol 3‐kinase (PI3K)/Akt signaling pathways (Riese & Cullum, 2014; Wee & Wang, 2017). More specifically, we and others have observed a close correlation between the activation of MAPK/Erk kinase (MEK)/ERK signaling and proliferation on cultured intestinal epithelial cells (IEC; Aliaga, Deschênes, Beaulieu, Calvo, & Rivard, 1999; Boucher, Jean, Vézina, & Rivard, 2004; Dionne et al., 1998; Paquin, Cagnol, Carrier, Leblanc, & Rivard, 2013; Rhoads et al., 1997; Rivard, Boucher, Asselin, & L’Allemain, 1999; Sheng, Bernabe, Guo, & Warner, 2006). Indeed, pharmacological inhibition of MEK activity blocks IEC cell cycle progression in G1 phase (Aliaga et al., 1999; Balmanno, Chell, Gillings, Hayat, & Cook, 2009; Paquin et al., 2013; Rivard et al., 1999). Accordingly, high ERK activity is detected in progenitor cells of the crypt T/A zone in human intestinal epithelium where ERK1/2 may direct the proliferation to differentiation switch (Aliaga et al., 1999). Indeed, we and others have reported that ERK1/2 are selectively inactivated during absorptive cell differentiation (Aliaga et al., 1999; Ding, Wang, & Evers, 2001; Lemieux et al., 2011; Taupin & Podolsky, 1999). These data support the hypothesis that ERK1/2 kinase activity must be shut down to initiate the differentiation process in the intestine, at least in enterocytes.
ERK1/2 activation depends on phosphorylation of threonine and tyrosine amino acids in a specific TEY motif. Dephosphorylation of these residues by specific dual‐specificity phosphatases (DUSP), also known as mitogen‐activated protein kinase phosphatases (MKP), mediates the end of the signal. There are two groups of ERK1/2‐targeting DUSP depending on their subcellular localization, namely, DUSP1/MKP‐1, DUSP2/PAC‐1, DUSP4/MKP‐2, and DUSP5 localized in the nucleus, or DUSP6/MKP‐3, DUSP7/MKP‐X, and DUSP9/MKP‐4 localized in the cytoplasm (Kidger & Keyse, 2016; Owens & Keyse, 2007). DUSPs are characterized by their variable N‐terminal MAPK‐binding region including the kinase interaction motif (KIM) that governs substrate specificity and stability of interaction (Owens & Keyse, 2007; Tanoue, Adachi, Moriguchi, & Nishida, 2000). Very few studies have analyzed the expression or function of these enzymes in the intestine. Interestingly, the elevated DUSP1 expression is detected in differentiated villus cells but not in proliferating crypt cells in the adult intestine (Noguchi et al., 1993). In the mouse intestine, Dusp6 is predominantly detected in the differentiated epithelium (Ruan et al., 2016). In line with these results, expression of both DUSP1 and DUSP6 markedly increases during enterocyte differentiation of human Caco‐2/15 cells, hence correlating with dramatic inhibition of ERK1/2 activities (Aliaga et al., 1999; Taupin & Podolsky, 1999). These observations suggest that ERK activity is tightly controlled by DUSP, both in its duration and intensity, during IEC differentiation.
In the current study, we have focused on the role of DUSP6, the first MKP to be characterized with absolute substrate specificity for ERK as opposed to either JNK or p38 (Groom, Sneddon, Alessi, Dowd, & Keyse, 1996; Muda et al., 1996). More specifically, this phosphatase recognizes and binds the biphosphorylated ERK (pT183pY185) via its conserved KIM domain. Binding to ERK1/2 allosterically increases the catalytic activity of DUSP6 which then dephosphorylates ERK1/2 (Camps et al., 1998).
To determine the role of DUSP6 in intestinal homeostasis, we have analyzed the intestinal epithelial phenotype of Dusp6 knockout (KO) mice under normal, oncogenic, and proinflammatory conditions. Our results demonstrate that DUSP6, by controlling ERK1/2 activation, regulates colonic inflammatory responses and protects the intestinal epithelium against oncogenic stress.
2. MATERIALS AND METHODS
2.1. Antibodies and reagents
Primary antibodies were obtained from the following sources: DUSP6 (ab76310) from Abcam (Toronto, Canada), β‐actin (MAB1501R) from Millipore (Oakville, Canada), ERK2 (sc‐154) and MEK2 (sc‐524) from Santa Cruz Biotechnology (Santa Cruz, CA), phosphorylated ERK1/2 (M8159) from Sigma‐Aldrich (Oakville, Canada), phosphorylated MEK1/2 (9121) from Cell Signaling Technology (Danvers, MA, USA), chromogranin A (20085) from ImmunoStar (Hudson, WI), proliferating cell nuclear antigen (PCNA; 18197) from Abcam and Ki67 (GTX16667) from Genetex (Irvine, CA). Horseradish peroxidase antibodies were obtained from GE Healthcare Life Sciences (Mississauga, Canada) and alkaline phosphatase‐conjugated antibodies from Promega (Madison, WI). For immunofluorescence, Alexa Fluor 488 conjugated antibodies were obtained from Molecular Probes (Waltham, MA). Other materials were purchased from Sigma‐Aldrich unless stated otherwise.
2.2. Animals
Dusp6 +/− mice were obtained from Jefferey D. Molkentin (Cincinnati, OH; Maillet et al., 2008) and C57BL6/J‐Apc Min/+ mice were purchased from Jackson Laboratory. Dusp6 +/−mice were bred to generate Dusp6 −/− experimental mice and Dusp6 +/+ control littermates. For tumor initiation experiments, Apc Min/+ mice were crossed with Dusp6 +/− mice to obtain double heterozygous Dusp6 +/− ;Apc Min/+ mice. Dusp6 +/− ;Apc Min/+ mice were then bred with Dusp6 +/− mice to obtain experimental Dusp6 −/− ;Apc Min/+ mice and Dusp6 +/+ ;Apc Min/+ control littermates. For genotyping, genomic DNA was extracted with a 25 mM NaOH/0.2 mM ethylenediaminetetraacetic acid (EDTA) solution heated at 95°C for 1 hr, followed by addition of an equal volume of 40 mM Tris‐HCl (pH 5.5). Primer sequences and polymearse chain reaction (PCR) conditions are available upon request. All experiments were approved by the Animal Research Ethics Committee of the Faculty of Medicine and Health Sciences of the Université de Sherbrooke (Protocols: 348‐13B and 348‐18B).
2.3. Macroadenomas count, histological staining, immunofluorescence, and immunohistochemistry
Methylene blue‐stained polyps were visualized under an SZ51 stereomicroscope (Olympus, Richmond Hill, Canada). Polyp sizes were measured with a digital caliper (Thermo Fisher Scientific, Waltham, MA) and polyp numbers were counted from the duodenum to the rectum as previously described (Perreault, Sackett, Katz, Furth, & Kaestner, 2005). Tissues were fixed, paraffin embedded, sectioned, and stained as described before (Leblanc et al., 2017). Immunohistochemistry staining was performed with the Dako EnVision+System Kit (Dako, Santa Clara, CA) according to the manufacturer’s recommendations. Immunofluorescence against chromogranin A was performed as described previously (C. S. Lee, Perreault, Brestelli, & Kaestner, 2002). Mucus secretion was visualized by Alcian blue staining performed on distal colon tissues fixed with Carnoy’s solution (10% glacial acetic acid, 30% chloroform, and 60% ethanol). For immunofluorescence, images were taken with a Leica DLMB2 microscope equipped with a DFC300FX camera and Leica FireCAM 3.4.1 Software (Leica, Concord, Canada). Otherwise, slides were visualized with a NanoZoomer slide scanner and NDP.view2 software (Hamamatsu, Boston, MA). All cell counts were performed on well‐oriented crypts in a double‐blind manner.
2.4. Colonoid culture
Twelve‐week‐old murine colons were cut into 5‐mm pieces and thoroughly washed in phosphate‐buffered saline (PBS). Colon fragments were incubated for 1 hr in 5 mM EDTA/PBS solution. EDTA was replaced with PBS and colonic pieces were shaken vigorously until crypt dissociation. Crypts were then centrifuged at 350g for 3 min, and the pellet was washed twice with PBS before pellet resuspension in Matrigel (BD Corning, Corning, NY) and plating in a 48‐well plate (Corning Costar; Corning). Colonoids were cultured in Advanced DMEM/F‐12 culture medium (Gibco, Waltham, MA) supplemented with 1 mM N‐acetylcysteine, 50 ng/ml murine EGF (Life Technologies, Waltham, MA), B27 supplement 1× (Life Technologies), N2 supplement 1× (Life Technologies), 150 ng/ml recombinant Wnt3a (Abcam) in addition to 10% R‐spondin 1% and 5% Noggin conditioned media. Noggin‐Fc and R‐spondin 1‐Fc conditioned supernatants were recovered from HEK293T cell lines stably expressing R‐spondin 1‐Fc (Dr. C. Kuo, Stanford University, Stanford, CA) or Noggin‐Fc (Dr. G. R. van den Brink, Hubrecht Institute, Utrecht, The Netherlands). Culture medium was changed every 2 days. Photos were taken at Days 5, 7, and 9 with an inversed Zeiss Axiovert 200M microscope (Zeiss, Toronto, Canada). Relative organoid areas were measured by encircling the periphery of each organoid in Image J software (NIH). Colonoid proliferation was evaluated by 5‐ethynyl‐29‐deoxyuridine (EdU) incorporation for 1 hr, followed by 4% paraformaldehyde fixation and EdU detection with the Click‐it EdU Alexa Fluor 555 Imaging Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Organoids were visualized by confocal microscopy with a Zeiss LSM 880 laser scan microscope using Zen Black software (Zeiss).
2.5. Dextran sulfate sodium (DSS) treatment
Eleven‐week‐old mice were administered 2% DSS (36–50 kDa; MP Biomedicals) in drinking water ad libitum for 7 days. Disease activity index (DAI) was measured at sacrifice according to Cooper, Murthy, Shah, and Sedergran (1993) criteria: stool consistency (0–4), rectal bleeding (0–4), colon hardening (0–4), and occult bleeding (0–4). Histological score was based on the presence or absence of mucosal architecture destruction, immune cell infiltration, muscle thickening, Goblet cell depletion, and crypt abscesses, as previously described (Coulombe et al., 2016).
2.6. Cell culture
HT29 and HCT116 colon carcinoma cell lines (ATCC, Manassas, VA) were grown in McCoy’s medium containing 10% fetal bovine serum (FBS). The retroviral shDUSP6 pSUPERretro expression vector was obtained from Transat (Saint‐Priest, France). Retroviruses were produced in HEK293T cells and were used to infect HT29 and HCT116 cells. Infected cells were selected with 2,5 µg/ml puromycin. For all experiments, at least three different cell populations originating from three different infections were analyzed.
2.7. Soft agarose and invasion assays
Soft agarose assays were performed with 30,000 cells per well for HT29 cells and 15,000 cells per well for HCT116 cells as previously described (Bian et al., 2016). After 14 days, colonies were stained with MTT ([3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) for 5 hr at 37°C. Images were acquired using an Infinity VX2 1100/26MX Imaging System (Vilber Lourmat, Marne‐la‐Vallée, France) and colonies were counted with the Image J software (NIH). Invasion assays were performed with 30,000 cells per insert, for both HT29 and HCT116 cells, in 24‐well plate Corning® BioCoat™ Matrigel® invasion chambers with 8‐µm polycarbonate membranes according to the manufacturer’s procedure. Briefly, cells were seeded in media without serum and media containing 20% FBS was used as chemoattractant. After a 72 hr incubation for HT29 cells and 48 hr incubation for HCT116 cells, noninvasive cells were removed with two cotton swabs. Invading cells were fixed with methanol 100% and stained with crystal violet 1%. Invading cells were observed with a Leica DLMB2 microscope and the total number of cells per insert was manually counted.
2.8. Human colorectal tissues
Samples from colorectal tumors and paired normal tissues, at least 10 cm from a tumor, were resected, processed, classified, and graded from patients who did not receive neoadjuvant therapy as previously described (Bian et al., 2016). Tissues were collected after obtaining the patient’s written consent, according to the protocol approved by the Institutional Human Subject Review Board of the Centre Hospitalier Universitaire de Sherbrooke. Total RNA was extracted using the Totally RNA Kit (Invitrogen, Waltham, MA) and processed according to the manufacturer’s instructions. Genomic DNA was extracted from formalin‐fixed paraffin‐embedded tissue using an FFPE DNA Isolation Kit for Cells and Tissues (Qiagen, Quebec City, Canada) and amplified by PCR. Presence of mutations in tumor samples was detected by direct sequencing by the Plateforme de Séquençage et de Génotypage des Génomes du Centre de Recherche du Centre Hospitalier de l’Université Laval, Quebec City, QC, Canada). Molecular characterization included microsatellite instability status (MSI panel markers: BAT25, BAT26, D5S346, D2S123, D17S250), oncogenic Kirsten rat sarcoma viral oncogene homolog (KRAS; codons 12, 13, and 61) and BRAF (V600E) mutations. Pathological and clinical data were obtained from medical records and are provided in Supporting Information Tables S1 and S2.
2.9. Western blot analysis
Proteins were isolated from scraped colonic mucosa enrichments of 12‐week‐old mice in chilled radioimmunoprecipitation assay buffer. For HT29 and HCT116 cell cultures, protein samples were prepared in Laemmli buffer as previously described (Bian et al., 2016). Western blot analyses were performed as described previously (Bian et al., 2016).
2.10. RNA extraction and quantitative real‐time PCR (qPCR)
Mouse colonic RNA was extracted with the Qiagen RNeasy Kit according to the manufacturer’s protocol, including on‐column DNase digestion. Reverse transcription was performed with the Transcriptor reverse transcriptase (Roche, Mississauga, Canada) according to the manufacturer’s instructions. Total RNA from human colorectal tissues was extracted with the Totally RNA Kit and reverse transcription was performed using AMV‐RT (Roche) according to the manufacturer’s protocol. Quantitative PCRs were performed by the RNomics Platform of the Université de Sherbrooke. Target expression was quantified relatively to Pum1, Psmc4, and Tbp expression for murine tissues. DUSP6 expression was normalized to MRLP19, PUM1, and RPL13A expression levels in human colorectal cancer (CRC) tissue samples. We have selected reference genes according to the library of about 10 genes (human and mouse) used by the RNomics Platform of the Université de Sherbrooke. The platform tested several reference genes and chose the three most stable ones with geNorm (https://genorm.cmgg.be/). Primer sequences and PCR conditions are available upon request.
2.11. Statistical analyses
Statistics were calculated using Student’s two‐tailed t tests if not stated otherwise. Data are expressed as mean ± SEM if not stated otherwise. Results were considered statistically significant at *p ≤ 0.05 and **p ≤ 0.01. Graphs and statistics were generated with GraphPad Prism (GraphPad Software Inc., La Jolla, CA). Densitometric analyses were performed using Image J software (NIH).
3. RESULTS
3.1. Dusp6 deletion promotes colonic epithelial cell proliferation
To characterize the role of Dusp6 in the regulation of intestinal homeostasis, we compared wild‐type Dusp6 +/+ mice to Dusp6 −/− KO mice. Dusp6 −/− mice have been previously characterized and are viable and fertile (Maillet et al., 2008). Western blot analyses of colonic mucosal protein enrichments confirmed the loss of Dusp6 expression in Dusp6 −/− mice, resulting in increased ERK1/2 phosphorylation levels, supporting ERK1/2 pathway excessive activation in the Dusp6‐deficient colonic epithelium (Figure 1a). Histological analysis of Dusp6 −/− mouse colon sections did not reveal apparent alterations in colonic gland architecture except a significant increase in crypt depth (Figure 1b). In line with these observations, mutant mouse colons displayed increased proliferative cell numbers in comparison to control littermates, as assessed by Ki67 staining (Figure 1c). Of importance, increased proliferation was not a result of stem cell compartment expansion since expression of intestinal stem cell markers, namely, Lgr5 and Ascl2, was not significantly modulated in Dusp6‐deficient colons, as determined by qPCR analysis (Figure 1d).
Figure 1.
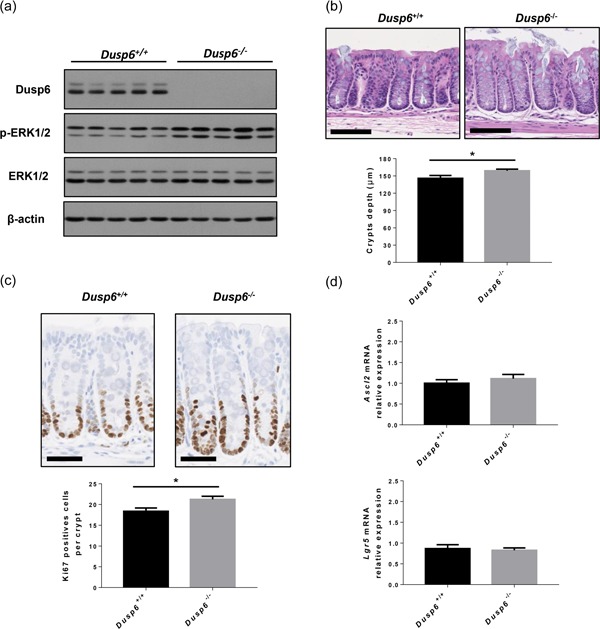
Dusp6 deletion promotes colonic epithelial cell proliferation. (a) ERK1/2 phosphorylation and Dusp6 expression were analyzed by western blot on colonic mucosal enrichments from 12‐week‐old Dusp6 knockout and control mice (n = 5). β‐Actin and ERK1/2 expression served as loading controls. Representative immunoblots are shown. (b) Colon tissue architecture was visualized with H&E staining and crypt depth was measured (n = 11). Scale bars=100 µm. (c) Immunohistochemistry against Ki67 was performed to measure proliferation. The number of positive cells per crypt was counted (n = 12). Scale bars=50 µm. (d) Relative Ascl2 and Lgr5 mRNA expression were quantified by qPCR with RNAs isolated from 12‐week‐old murine total colonic extracts (n = 11). Relative expression was normalized to housekeeping genes Pum1, Tbp, and Psmc4 expression. Data are expressed as mean ± SEM. Student’s t test; *p ≤ 0.05. DUSP6: dual‐specificity phosphatase 6; ERK: extracellular signal‐regulated kinase; H&E: hematoxylin and eosin; mRNA: messenger RNA; qPCR: quantitaive polymerase chain reaction
To assess the intrinsic effect of Dusp6 deficiency on intestinal epithelial cell regeneration, we used a crypt colonoid culture system. As shown in Figure 2a, loss of Dusp6 significantly promoted colonic organoid development. Indeed, Dusp6 ‐/‐ colonoid sizes were larger after 7 and 9 days in culture compared to control colonoids (Figure 2b). Furthermore, Dusp6‐deficient colonoid proliferation was increased in comparison to wild‐type colonoids, as determined by EdU incorporation (Figure 2c). These data indicate that Dusp6 deletion leads to increased IEC proliferation.
Figure 2.
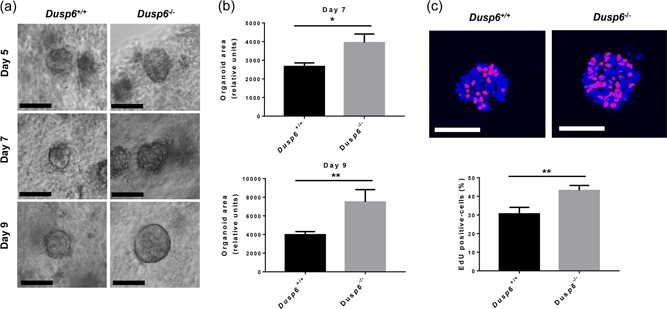
Dusp6 deletion stimulates colonoid development. (a) Phase‐contrast images of Dusp6 +/+ and Dusp6 ‐/‐ colon organoids after 5, 7, and 9 days of culture. Scale bars: 100 µm. (b) Relative organoid areas after 7 and 9 days of culture (n ≥ 33). (c) EdU incorporation (red) and DAPI staining (blue) were used to evaluate the ratio of proliferative cells per organoid (n = 16). Scale bars: 50 µm. Graphs are representative of at least three independent experiments conducted with different mice. Data are expressed as mean ± SEM. Student‘s t test; *p ≤ 0.05, **p ≤ 0.01. DAPI: 4′,6‐diamidino‐2‐phenylindole; DUSP6: dual‐specificity phosphatase 6; EdU: 5‐ethynyl‐2‐deoxyuridine
3.2. Dusp6 deletion induces goblet cell expansion without affecting enteroendocrine and absorptive cell differentiation
We next verified whether loss of Dusp6 modulates IEC differentiation in the colon. Dusp6 deletion did not alter enteroendocrine cell differentiation. Indeed, the number of chromogranin‐positive cells was not increased in mutant mouse colons in comparison to control mice, as assessed by immunofluorescence (Figure 3a). Additionally, expression of the enteroendocrine cell marker Neurogenin‐3 was not significantly modified without Dusp6 (Figure 3b). Dusp6 deletion also did not alter absorptive cell differentiation as both control and mutant mouse colons exhibited similar expression level of the absorptive cell marker Slc26a3, as determined by qPCR (Figure 3b). Hence, Dusp6 deletion does not affect enteroendocrine and absorptive cell differentiation.
Figure 3.
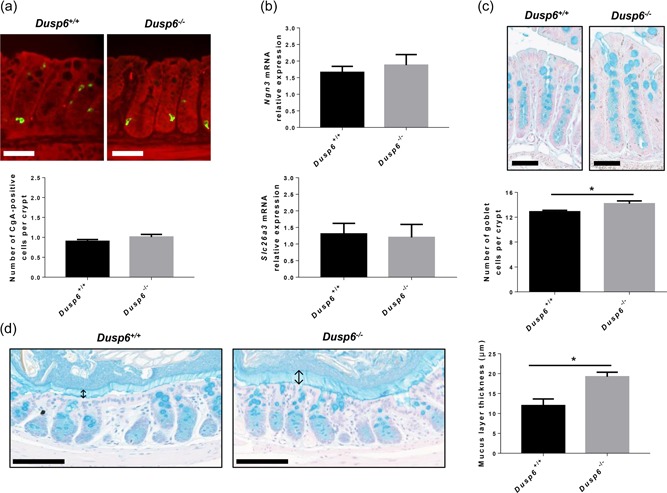
Dusp6 deletion induces goblet cell expansion without affecting enteroendocrine and absorptive cell differentiation. (a) Chromogranin A immunofluorescence with Evan’s blue counterstaining was performed on 12‐week‐old Dusp6 +/+ and Dusp6 −/− murine colon sections to evaluate the number of enteroendocrine cells per crypt. Scale bars=50 µm (n = 7). (b) Relative Ngn3 and Slc26a3 mRNA expression was quantified by qPCR with RNAs isolated from 12‐week‐old murine total colonic extracts (n = 7). Relative expression was normalized to housekeeping genes Pum1, Tbp, and Psmc4 expression. (c) Alcian blue staining was used to visualize Goblet cells in Dusp6 +/+ and Dusp6 ‐/‐ colon sections. Scale bars: 50 µm. The number of Alcian blue positive cells per crypt was counted (n = 7). (d) Carnoy’s fixation followed by Alcian blue staining was performed on 12‐week‐old Dusp6 +/+ and Dusp6 −/− mouse distal colon sections to measure the mucus layer thickness (n ≥ 3). ↕Mucus layer. Scale bars=100 µm. Data are expressed as mean ± SEM. Student’s t test; *p ≤ 0.05. DUSP6: dual‐specificity phosphatase 6; mRNA: messenger RNA; qPCR: quantitaive polymerase chain reaction
In contrast, the number of goblet cells was significantly increased in Dusp6 −/− mouse colons in comparison to controls, as determined by Alcian blue staining which detects acidic mucins (Figure 3c). Notably, a thicker inner mucus layer was observed in Dusp6‐deficient mouse colon lumen (Figure 3d), as evidenced by Carnoy’s fixation. This indicates that increased colonic ERK‐dependent signaling after Dusp6 deletion selectively promotes goblet cell expansion and secretory function.
3.3. Dusp6 deletion protects against DSS‐induced colitis in mice
We next assessed the susceptibility of Dusp6 +/+ and Dusp6 ‐/‐ mice to colonic injury induced by DSS added in drinking water (Cooper et al., 1993). After 7 days, control mice had lost nearly 20% of their body weight (Figure 4a) and exhibited signs of diarrhea and rectal bleeding, resulting in high DAI (Figure 4b). In contrast, Dusp6 ‐/‐ mice treated with DSS exhibited less weight loss (Figure 4a), a decreased DAI associated with milder diarrhea and bloody stool symptoms (Figure 4b) and decreased histological alterations at the microscopic level (Figure 4c). These findings demonstrate that Dusp6 inhibition protects the colon from DSS‐induced acute injury.
Figure 4.
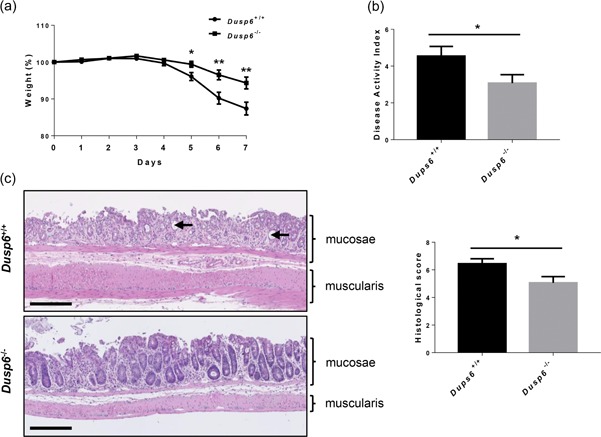
Dusp6 deletion protects against DSS‐induced colitis in mice. Eleven‐week‐old Dusp6 +/+ and Dusp6 ‐/‐ mice were treated with 2% DSS in drinking water for 7 days. (a) Body weight was measured every day during treatment (n = 21). (b) Disease activity index of Dusp6 knockout mice and control littermates was calculated by scoring stool softness, occult fecal blood, rectal bleeding, and colon rigidity at sacrifice (n ≥ 21). (c) Hematoxylin and eosin staining was performed on Dusp6 +/+ and Dusp6 −/− colon tissue to score the damage according to the extent of destruction of normal mucosal architecture (0, normal; 1, 2, and 3, respectively, mild, moderate, and extensive damage), presence and degree of cellular infiltration (0, normal; 1, 2, and 3, mild, moderate, and transmural infiltration, respectively), extent of muscle thickening (0, normal; 1, 2, and 3, respectively mild, moderate, and extensive thickening), presence or absence of crypt abscesses (0, absent; 1, present; see arrowheads) and the presence or absence of goblet cell depletion (0, absent; 1, present). (n = 18). Scale bars=250 µm. Data are expressed as mean ± SEM. Student’s t test; *p ≤ 0.05, **p ≤ 0.01. DUSP6: dual‐specificity phosphatase 6; DSS: dextran sulfate sodium
3.4. Dusp6 deletion increases intestinal tumorigenesis in Apc Min/+ mice
Our data show that loss of Dusp6 in IEC results in ERK signaling activation. We and others have shown previously that ERK is important for intestinal tumor development (Carragher et al., 2010; C. S. Lee, Perreault, et al., 2002; H.‐W. Lee, Ahn, et al., 2002; Lemieux et al., 2009; Wang et al., 2013). To determine whether Dusp6 deletion could affect intestinal tumorigenesis, we crossed Dusp6 −/− mice with Apc Min/+ mice. These mice, heterozygous for an Apc truncated mutation frequently found in human sporadic CRC, spontaneously develop intestinal adenomas (Moser, Pitot, & Dove, 1990; Su et al., 1992). As shown in Figure 5a, Dusp6 deficiency promoted intestinal tumor load in Apc Min/+ mice both in the small intestine and colon. Small intestinal tumors were also larger in Dusp6 −/−; Apc Min/+ mice as opposed to control Dusp6 +/+ ;Apc Min/+ mice (Figures 5b,c). In addition, expression of the proliferation marker PCNA was more elevated in double mutant Dusp6 −/−;Apc Min/+ polyps (Figures 5d,e). We have also determined phospho‐ERK1/2 expression to assess MAPK signaling. As shown in Figure 5d, polyps from Apc Min/+ mice displayed consistently much higher phosphorylation levels of ERK1/2 in comparison to the corresponding benign epithelium (margin). This is consistent with the observation that ERK signaling activation drives intestinal tumorigenesis in Apc Min/+ mice (S. H. Lee et al., 2010). However, upon deletion of Dusp6 in Apc Min/+ mice, phospho‐ERK levels were barely increased in the intestinal margins of some Dusp6 KO mice (lanes 7 and 9 vs. lanes 1, 3, and 5) but not in polyps (lanes 8, 10, and 12 vs. 2, 4, and 6). Taken together, these results suggest that Dusp6 deficiency promotes intestinal tumor initiation and proliferation, in part by promoting ERK1/2 activation.
Figure 5.
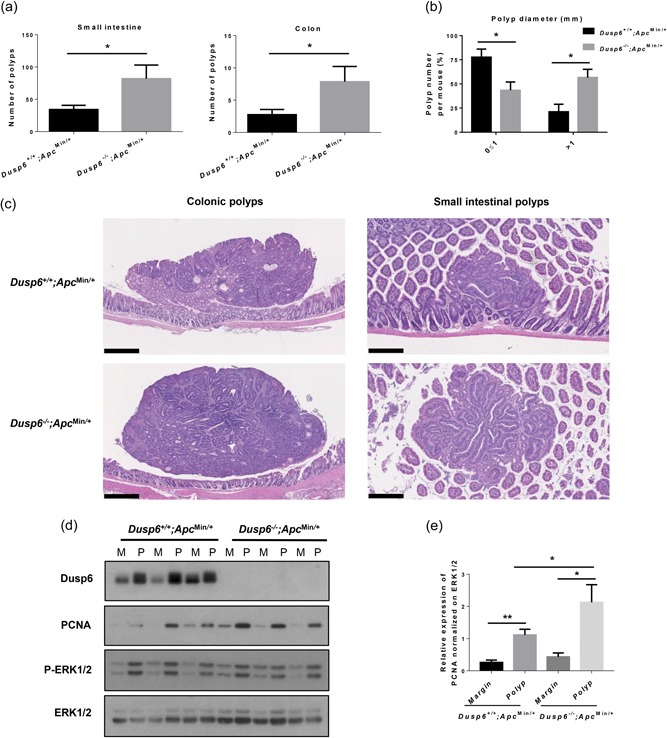
Dusp6 deletion increases intestinal tumorigenesis in Apc Min/+ mice. (a) Polyp numbers were counted in 13‐week‐old Dusp6 +/+ ; Apc Min/+ and Dusp6 ‐/‐; Apc Min/+ small intestine and colon (n ≥ 7). (b) Small intestinal polyp size (diameter) was compared between Dusp6 +/+ ; Apc Min/+ and Dusp6 ‐/‐;Apc Min/+ mice (n = 5). (c) Dusp6 +/+; Apc Min/+ and Dusp6 ‐/‐; Apc Min/+ polyp histology was visualized with hematoxylin and eosin staining in the small intestine (scale bars: 250 µm) and colon (scale bars: 500 µm; representative of n ≥ 4). (d) Dusp6, PCNA, and phosphorylated ERK1/2 levels were analyzed by western blot of polyp (P) and healthy marge (M) protein extracts (n = 3). ERK1/2 expression was used as loading control. Representative immunoblots are shown. (e) Densitometric analysis of PCNA expression relative to ERK1/2 (n = 7). Data are expressed as mean ± SEM. Student’s t test; *p < 0.05, **p < 0.01. DUSP6: dual‐specificity phosphatase 6; ERK: extracellular signal‐regulated kinase; PCNA: proliferating cell nuclear antigen
3.5. Decreased DUSP6 expression is associated with human CRC progression
To determine DUSP6 contribution in human CRC cells, we reduced DUSP6 protein expression in established human CRC HT29 and HCT116 cell lines, by a specific anti‐DUSP6 short hairpin RNA. DUSP6 suppression led to increased ERK1/2 phosphorylation levels in HT29 cells (Figure 6a). As expected, no difference was seen in the levels of phosphorylated MEK1/2. This increased ERK1/2 activity in DUSP6‐depleted HT29 cells was associated with an enhanced capacity to grow in soft agar and to invade Matrigel (Figures 6b,c). In contrast, DUSP6‐depleted HCT116 cells did not display increased MEK1/2 nor ERK1/2 activation or enhanced soft agar growth and invasion (Figures 6b,c). We also determined DUSP6 mRNA expression levels in 66 human paired CRC specimens consisting of resection margins and primary tumors from our biobank. As shown in Figure 6d, relative amounts of DUSP6 transcripts were significantly reduced in CRC tumors compared to normal specimens, mostly in advanced stage tumor samples (Figure 6e). Of note, decreased DUSP6 gene expression was more prominent in microsatellite stable (MSS) tumors (Figure 6f). By contrast, there did not appear to be a significant difference relative to KRAS or BRAF mutations (Figure 6g). These data suggest that DUSP6 inactivation may favor colorectal oncogenesis.
Figure 6.
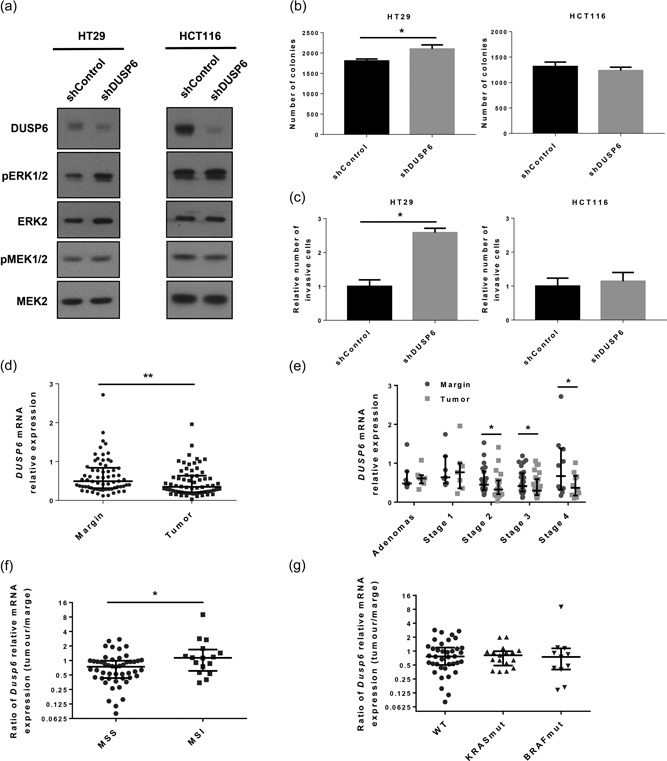
Decreased DUSP6 expression is associated with human colorectal cancer progression. HT29 and HCT116 colorectal carcinoma cells were stably infected with lentiviruses encoding for a control shRNA or a shRNA against DUSP6. (a) ERK phosphorylation, MEK phosphorylation, and DUSP6 expression were analyzed by western blot. ERK2 and MEK2 expression were used as loading control. Representative immunoblots are shown. (b) HT29 and HCT116 cells were cultured in soft agarose for 14 days before MTT staining. Graphs are representative of three independent experiments. Data are expressed as mean ± SEM. Student’s t test; *p ≤ 0.05. (c) Invasion of shControl‐ and shDUSP6‐expressing HT29 and HCT116 cells was evaluated in Matrigel‐coated transwells. After 72 hr for HT29 cells and 48 hr for HCT116 cells, invading cells were fixed, stained with 1% crystal violet and counted (n = 3 independent experiments for HT29 cells and n = 5 for HCT116 cells). Data are expressed as mean ± SEM. Student’s t test; *p ≤ 0.05. (d) Relative DUSP6 mRNA expression was analyzed by qPCR with RNAs isolated from human colorectal tumor specimens compared to paired adjacent healthy tissue. Relative expression was normalized to housekeeping genes MRPL19, SDHA, and YWHAZ expression (n = 66). Data are expressed as a median ± interquartile range. The Wilcoxon signed‐rank test; **p ≤ 0.01 (e) Relative DUSP6 mRNA expression was assessed by qPCR with RNAs isolated from colorectal tumors and paired healthy adjacent tissue separated by stage (n = 7 for adenomas and Stage 1, n = 22 for Stage 2, n = 27 for Stage 3 and n = 10 for Stage 4). Relative expression was normalized to housekeeping genes MRPL19, SDHA, and YWHAZ expression. Data are expressed as a median ± interquartile range. The Wilcoxon signed‐rank test; *p ≤ 0.05. (f) Ratio of DUSP6 mRNA expression (tumor/paired adjacent healthy tissue) was compared between MSS (n = 50) and MSI (n = 16) patients. Relative expression was normalized to housekeeping genes MRPL19, SDHA, and YWHAZ expression. Data are expressed as a median ± interquartile range. Mann–Whitney U test; *p ≤ 0.05. (g) Ratio of DUSP6 mRNA expression (colorectal tumor/paired adjacent healthy tissue) was compared between wild‐type (n = 38), KRAS‐mutated (n = 17), and BRAF‐mutated (n = 11) patients. Relative expression was normalized to housekeeping genes MRPL19, SDHA, and YWHAZ expression. Data are expressed as a median ± interquartile range. Mann–Whitney U test. DUSP6: dual‐specificity phosphatase 6; ERK: extracellular signal‐regulated kinase; MEK: mitogen‐activated protein kinase/Erk kinase; mRNA: messenger RNA; qPCR: quantitaive polymerase chain reaction; shRNA: short hairpin RNA
4. DISCUSSION
We have shown here an important role of DUSP6, a cytoplasmic phosphatase that negatively regulates ERK1/2, in the maintenance of homeostasis in the colon. Indeed, Dusp6 gene deletion in mice is sufficient to consistently increase, albeit modestly, endogenous ERK1/2 phosphorylation in the colonic mucosa. This increased ERK1/2 activity correlates with a stimulation of colonic crypt cell proliferation in Dusp6 KO mice. By culturing colonic organoids, we further show that an increase in crypt cell proliferation in Dusp6 −/− mice is intrinsic to epithelial cells. These findings are in agreement with previous studies demonstrating the central role of the KRas/ERK MAPK pathway in the control of epithelial proliferation in both the small and large intestines (Aliaga et al., 1999; Feng et al., 2011; Haigis et al., 2008; Paquin et al., 2013; Rivard et al., 1999; Voisin et al., 2008). Notably, phosphorylated and activated forms of ERK1/2 have been mostly found in undifferentiated proliferative crypt cells in human intestine (Aliaga et al., 1999), hence supporting their function in epithelial cell proliferation. Interestingly, the expression of stem cell markers Ascl2 and Lgr5 is not affected in Dusp6 KO colons, suggesting that crypt‐based columnar stem cells were not affected by the Dusp6 deficiency. Therefore, the increase number of proliferative cells observed in Dusp6−/− mice may mostly result from an increased number of transit‐amplifying cells, cells which are undifferentiated and in transition between stem cells and differentiated cells. These results are reminiscent of those of Feng et al. (2011) showing that KRas activation in the colonic epithelium while stimulating proliferation, does not expand the crypt stem cell compartment.
ERK signaling has been previously shown to negatively regulate enterocyte and enteroendocrine cell differentiation in the intestine (Basak et al., 2017; Ding et al., 2001; Lemieux et al., 2011). However, our data show that the absence of Dusp6 expression does not affect absorptive nor enteroendocrine cell differentiation in the colon. This might be explained by the modest increase of endogenous ERK1/2 activity observed in Dusp6 −/− colonic epithelium. Such modulation of ERK activity might not be sufficient to significantly perturb absorptive and endocrine cell differentiation pathways. However, Dusp6 deletion is sufficient to deregulate colonic goblet cell number and function, suggesting that goblet cell differentiation is positively affected by ERK signaling and may be more sensitive to ERK1/2 activity changes. Accordingly, Feng et al. (2011) as well as Yamashita et al. (2014) previously observed goblet cell expansion in mouse intestines expressing oncogenic KRas or MEK1. Furthermore, activation of ERK signaling in airway epithelial and CRC cells promotes MUC2 gene transcription and mucus production (Dilly et al., 2015; Hatayama, Iwashita, Kuwajima, & Abe, 2007; Kim et al., 2002; Kuracha, Thomas, Loggie, & Govindarajan, 2017; C. S. Lee, Perreault, et al., 2002; H.‐W. Lee, Ahn, et al., 2002; Li et al., 1998; Perrais, Pigny, Copin, Aubert, & Van Seuningen, 2002; Suzuki, Takeuchi, Ishinaga, Basbaum, & Majima, 2008). Overall, these observations suggest a role for the Ras/ERK pathway in goblet cell fate and differentiation. However, the exact molecular mechanisms involved remain to be elucidated.
These modulations in crypt cell proliferation and differentiation observed in Dusp6 KO intestine prompted us to determine whether Dusp6 play a role in the intestinal response against injury. We, therefore, investigate a model of ulcerative colitis in the large intestine. Treatment of wild‐type mice with DSS induces acute colitis. Interestingly, Dusp6 KO mice exhibited less weight loss, diarrhea, and bloody stools after DSS treatment as well as milder mucosal alterations at the microscopic level. Thus, Dusp6 deficiency in IEC likely protects the colonic mucosae against injury‐induced inflammation. Increased IEC proliferation, enhanced goblet cell numbers as well as mucus production may be all implicated in colitis protection. Indeed, proliferation is important for wound healing after epithelial damage in the intestine and the mucus layer is protective against adhesion and invasion by microbes and antigens (Sturm & Dignass, 2008). For instance, mice knockout for Muc2 spontaneously develops chronic inflammation in the colon and rectum (Van der Sluis et al., 2006). Additionally, Ruan et al. (2016) have recently measured the in vivo gut permeability in Dusp6 −/− mice using fluorescein isothiocyanate dextran molecule and found reduced paracellular permeability in comparison to wild‐type mice. Therefore, a tightening of the epithelial barrier could also contribute to the protective effect against colitis observed in Dusp6 ‐/‐ mice. Together, these data indicate that Dusp6 maintains epithelial barrier function in colonic mucosae.
By contrast, another study (Bertin et al., 2015) showed that Dusp6 deletion accelerated and exacerbated inflammation in interleukin‐10 deficient (Il10 −/−) mouse model of colitis. The discrepancy between our data and the study of Bertin et al. may be explained by the different experimental models used for colitis induction. Herein, we used DSS which triggers inflammation by causing chemical injury directly to the intestinal epithelium, resulting in exposure of the lamina propria and submucosa to luminal antigens and bacteria (Low, Nguyen, & Mizoguchi, 2013). Thus, by stimulating goblet cell function as well as crypt proliferation and regeneration, Dusp6 deletion may protect the colonic epithelium against erosion induced by DSS. By contrast, the enterocolitis in Il10 −/− mice is largely attributed to dysfunctional CD4+ T‐cell activation (Roers et al., 2004). In their study, Bertin et al. (2015) clearly demonstrated that Dusp6‐deficient CD4+ T cells have increased ERK1/2 activation and production of interferon γ upon TCR stimulation. They also showed that Dusp6 −/− naïve CD4+ T cells exhibited a greater propensity to differentiate along the Th1 axis in vitro. Therefore, it is not surprising that Dusp6 deletion exacerbated spontaneous colitis in the Il10 deficient mice.
Aberrant activation of ERK signaling is commonly linked to intestinal tumorigenesis and CRC. Notably, mutations in genes encoding KRAS and BRAF, the upstream activators of ERK1/2 kinases. occur in approximately 40% and 10%, respectively, of all CRCs (Andreyev, Norman, Clarke, Cunningham, & Oates, 1998; Davies et al., 2002; Rajagopalan et al., 2002; Samowitz et al., 2000). Furthermore, several studies have demonstrated that inhibition of MEK/ERK signaling prevents CRC cell growth in cell culture and mouse xenografts (Balmanno et al., 2009; Kress, Raabe, & Feller, 2010; Lemieux et al., 2009; Sebolt‐Leopold et al., 1999; Solit et al., 2006; Voisin et al., 2008; Wang et al., 2013). However, DUSP functions in the oncogenicity of KRAS/ERK pathway in CRC are not known. Herein, we demonstrated that Dusp6 deletion promotes intestinal tumor load and size in Apc Min/+ mice, supporting the requirement for ERK1/2 activity in Apc Min/+‐dependent murine intestinal tumorigenesis (S. H. Lee et al., 2010). Indeed, polyps from Apc Min/+ mice displayed much higher phosphorylation levels of ERK1/2 in comparison to the corresponding benign epithelium (margin). Surprisingly, we did not detect a further increase in phospho‐ERK levels in Dusp6‐deficient polyps. The reason why this was not observed in polyps is not clear but is consistent with other reports (Maillet et al., 2008). It is possible that ERK1/2 phosphorylation is induced earlier during the tumorigenic process of Apc Min/+ mice in the absence of Dusp6. Herein, the mice were killed at 3 months of age to obtain a significant number of polyps (Moser et al., 1990). To better analyze the regulation of ERK activity during polyposis, it would have been necessary to also sacrifice mice earlier, at the age of 1 or 2 months. That being said, the possibility that Dusp6 dephosphorylates other targets than ERK1/2 cannot be totally excluded. For instance, Dusp6 has been shown to interact with other kinases such as CK2 (Hagan, Knutson, & Lange, 2013) and ERK5 (Razmara, Eger, Rorsman, Heldin, & Lennartsson, 2012; Sarközi et al., 2007). In this regard, ERK5 is particularly interesting since this MAPK can rescue intestinal epithelial turnover and tumor cell proliferation upon ERK1/2 inhibition (de Jong et al., 2016).
Notably, we found that DUSP6 gene expression decreases in patient‐derived colorectal tumors, mostly in advanced stage tumors. While we did not detect a significant association between DUSP6 expression and KRAS or BRAF mutations in the small number of adenocarcinomas analyzed, a correlation between low DUSP6 transcript levels and MSS status of tumors was observed. Interestingly, MSS tumors are generally more aggressive than MSI tumors and are more often associated with lymph‐node metastases and distant spread (Popat, Hubner, & Houlston, 2005). Therefore, one could speculate that the low DUSP6 expression in MSS tumor cells would contribute to their invasive and metastatic properties by enhancing ERK activity. Accordingly, in HT29 human colon carcinoma cells, silencing of DUSP6 expression resulted in enhanced activation of ERK1/2 and this correlates with increased anchorage‐independent growth ability and invasion capacity. Together, these findings provide evidence that DUSP6 expression limits intestinal tumor growth and progression.
Surprisingly, DUSP6 silencing did not alter the oncogenic properties of HCT116 cells nor ERK activation levels. The fact that HT29 cells are BRAF mutated while HCT116 cells bear a KRAS mutation (Davies et al., 2002) is a plausible explanation for this discrepancy between these two cell lines. Indeed, there is a correlation between ERK1/2 activation and BRAF, but not KRAS, mutation status in CRC cells and tumors (Yeh et al., 2009). In addition, we cannot exclude that HCT116 cells may express higher levels of other DUSP, which in turn can also negatively regulate ERK phosphorylation. For instance, we previously observed higher DUSP4 expression levels in HCT116 cells in comparison to HT29 cells (Cagnol & Rivard, 2013). Regardless of of the reason that explains this difference in ERK phosphorylation modulation between the two cell lines, our results suggest that DUSP6 silencing must result in increased ERK1/2 phosphorylation to stimulate anchorage‐independent growth ability and invasion capacity in CRC cells.
A tumor suppressive role for DUSP6 has also been suggested in several cancers (Chan et al., 2008; Furukawa, Sunamura, Motoi, Matsuno, & Horii, 2003; Okudela et al., 2009; Xu, Furukawa, Kanai, Sunamura, & Horii, 2005). Accordingly, expression levels of DUSP6 are indeed reduced in pancreatic, ovarian and lung cancers (Chan et al., 2008; Furukawa et al., 2003; Okudela et al., 2009; Xu et al., 2005) and DUSP6 overexpression suppresses cancer cell growth (Chan et al., 2008; Furukawa et al., 2003; Okudela et al., 2009). However, other studies have hinted at an oncogenic function for DUSP6. Indeed, DUSP6 is upregulated in various cancer cell lines including those from glioblastomas, papillary thyroid carcinoma and gastric cancers and its enforced expression in these cancer cell lines clearly increases their growth, migration and/or tumorigenic potential in vivo (Degl’Innocenti et al., 2013; Messina et al., 2011; Wu et al., 2018). The mechanisms explaining why DUSP6 can act as a tumor suppressor in some cancers and as an oncogene in others still remain to be determined.
In conclusion, our results demonstrate that the DUSP6 phosphatase, by limiting the activation of ERK signaling, regulates colonic inflammatory response and protects the colonic epithelium against oncogenic stress.
AUTHOR’S CONTRIBUTIONS
K. B., M.‐J. L., A. M., and S. C. performed research. J. C. C. provided the tumor specimens. N. R., K. B., M.‐J. L. and S. C. analyzed data. M.‐J. L., N. R., and K. B. wrote the paper. N. R. designed research. All authors read and approved the final manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors thank Pr Claude Asselin (Université de Sherbrooke) for critical reading of the manuscript and Gérald Bernatchez (Université de Sherbrooke) for technical assistance. The authors also thank the RNomics and Electron Microscopy and Histology Platforms of the Faculté de Médecine et des Sciences de la Santé of the Université de Sherbrooke. Special thanks to Dr. Jefferey D. Molkentin (University of Cincinnati, OH) for providing the Dusp6 +/− mice, Dr. C. Kuo (Stanford University, CA) for providing the R‐spondin 1 producing cell line and Dr. G.R. van den Brink (Hubrecht Institute, The Netherlands) for providing the Noggin producing cell line. The biobank of CRC specimens was supported by a team grant on digestive epithelium from the Canadian Institutes of Health Research (CTP‐82942). K. B. is the recipient of a Centre de Recherche Médicale de l’Université de Sherbrooke (CRMUS) fellowship. N. R. and J. C. C. are members of the Fonds de la Recherche en Santé du Québec—funded Centre de Recherche du Centre Hospitalier Universitaire de Sherbrooke. N. R. is a recipient of a Canada Research Chair in Colorectal Cancer and Inflammatory Cell Signaling (227765). This researchstudy was supported by a Canadian Institutes of Health Research grant to N. R. (MOP‐119593).
Beaudry K, Langlois MJ, Montagne A, et al. Dual‐specificity phosphatase 6 deletion protects the colonic epithelium against inflammation and promotes both proliferation and tumorigenesis. J Cell Physiol. 2019;234:6731–6745. 10.1002/jcp.27420
References
REFERENCES
- Aliaga, J. C. , Deschênes, C. , Beaulieu, J. F. , Calvo, E. L. , & Rivard, N. (1999). Requirement of the MAP kinase cascade for cell cycle progression and differentiation of human intestinal cells. The American Journal of Physiology, 277(3 Pt 1), G631–G641. [DOI] [PubMed] [Google Scholar]
- Andreu, P. , Colnot, S. , Godard, C. , Gad, S. , Chafey, P. , Niwa‐Kawakita, M. , & Romagnolo, B. (2005). Crypt‐restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development, 132(6), 1443–1451. 10.1242/dev.01700 [DOI] [PubMed] [Google Scholar]
- Andreyev, H. J. N. , Norman, A. R. , Clarke, P. A. , Cunningham, D. , & Oates, J. R. (1998). Kirsten ras mutations in patients with colorectal cancer: The Multicenter “RASCAL” Study. Journal of the National Cancer Institute, 90(9), 675–684. 10.1093/jnci/90.9.675 [DOI] [PubMed] [Google Scholar]
- Balmanno, K. , Chell, S. D. , Gillings, A. S. , Hayat, S. , & Cook, S. J. (2009). Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY‐142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. International Journal of Cancer, 125(10), 2332–2341. 10.1002/ijc.24604 [DOI] [PubMed] [Google Scholar]
- Barker, N. (2014). Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nature Reviews Molecular Cell Biology, 15(1), 19–33. 10.1038/nrm3721 [DOI] [PubMed] [Google Scholar]
- Basak, O. , Beumer, J. , Wiebrands, K. , Seno, H. , van Oudenaarden, A. , & Clevers, H. (2017). Induced quiescence of Lgr5 + stem cells in intestinal organoids enables differentiation of hormone‐producing enteroendocrine cells. Cell Stem Cell, 20(2), 177–190. e4. 10.1016/j.stem.2016.11.001 [DOI] [PubMed] [Google Scholar]
- Bertin, S. , Lozano‐Ruiz, B. , Bachiller, V. , García‐Martínez, I. , Herdman, S. , Zapater, P. , … González‐Navajas, J. M. (2015). Dual Specificity Phosphatase 6 (DUSP6) regulates CD4 + T cell functions and restrains the spontaneous colitis in IL‐10 deficient mice. Mucosal Immunology, 8(3), 505–515. 10.1038/mi.2014.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian, B. , Mongrain, S. , Cagnol, S. , Langlois, M. J. , Boulanger, J. , Bernatchez, G. , … Rivard, N. (2016). Cathepsin B promotes colorectal tumorigenesis, cell invasion, and metastasis. Molecular Carcinogenesis, 55(5), 671–687. 10.1002/mc.22312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher, M.‐J. , Jean, D. , Vézina, A. , & Rivard, N. (2004). Dual role of MEK/ERK signaling in senescence and transformation of intestinal epithelial cells. American Journal of Physiology, 286(5), G736–G746. 10.1152/ajpgi.00453.2003 [DOI] [PubMed] [Google Scholar]
- Cagnol, S. , & Rivard, N. (2013). Oncogenic KRAS and BRAF activation of the MEK/ERK signaling pathway promotes expression of dual‐specificity phosphatase 4 (DUSP4/MKP2) resulting in nuclear ERK1/2 inhibition. Oncogene, 32(5), 564–576. 10.1038/onc.2012.88 [DOI] [PubMed] [Google Scholar]
- Camps, M. , Nichols, A. , Gillieron, C. , Antonsson, B. , Muda, M. , Chabert, C. , & Arkinstall, S. (1998). Catalytic activation of the phosphatase MKP‐3 by ERK2 mitogen‐activated protein kinase. Science, 280(5367), 1262–1265. [DOI] [PubMed] [Google Scholar]
- Carragher, L. A. S. , Snell, K. R. , Giblett, S. M. , Aldridge, V. S. S. , Patel, B. , Cook, S. J. , … Pritchard, C. A. (2010). V600EBraf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16INK4a. EMBO Molecular Medicine, 2(11), 458–471. 10.1002/emmm.201000099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, D. W. , Liu, V. W. S. , Tsao, G. S. W. , Yao, K. ‐M. , Furukawa, T. , Chan, K. K. L. , & Ngan, H. Y. S. (2008). Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis, 29(9), 1742–1750. 10.1093/carcin/bgn167 [DOI] [PubMed] [Google Scholar]
- Cooper, H. S. , Murthy, S. N. , Shah, R. S. , & Sedergran, D. J. (1993). Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Laboratory Investigation; a Journal of Technical Methods and Pathology, 69(2), 238–249. [PubMed] [Google Scholar]
- Coulombe, G. , Langlois, A. , De Palma, G. , Langlois, M.‐J. , McCarville, J. L. , Gagné‐Sanfaçon, J. , … Rivard, N. (2016). SHP‐2 phosphatase prevents colonic inflammation by controlling secretory cell differentiation and maintaining host‐microbiota homeostasis. Journal of Cellular Physiology, 231(11), 2529–2540. 10.1002/jcp.25407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosnier, C. , Stamataki, D. , & Lewis, J. (2006). Organizing cell renewal in the intestine: Stem cells, signals and combinatorial control. Nature Reviews Genetics, 7(5), 349–359. 10.1038/nrg1840 [DOI] [PubMed] [Google Scholar]
- Davies, H. , Bignell, G. R. , Cox, C. , Stephens, P. , Edkins, S. , Clegg, S. , … Futreal, P. A. (2002). Mutations of the BRAF gene in human cancer. Nature, 417(6892), 949–954. 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- Degl’innocenti, D. , Romeo, P. , Tarantino, E. , Sensi, M. , Cassinelli, G. , Catalano, V. , … Borrello, M. G. (2013). DUSP6/MKP3 is overexpressed in papillary and poorly differentiated thyroid carcinoma and contributes to neoplastic properties of thyroid cancer cells. Endocrine‐Related Cancer, 20(1), 23–37. 10.1530/ERC-12-0078 [DOI] [PubMed] [Google Scholar]
- Dilly, A. K. , Song, X. , Zeh, H. J. , Guo, Z. S. , Lee, Y. J. , Bartlett, D. L. , & Choudry, H. A. (2015). Mitogen‐activated protein kinase inhibition reduces mucin 2 production and mucinous tumor growth. Translational Research, 166(4), 344–354. 10.1016/j.trsl.2015.03.004 [DOI] [PubMed] [Google Scholar]
- Ding, Q. , Wang, Q. , & Evers, B. M. (2001). Alterations of MAPK activities associated with intestinal cell differentiation. Biochemical and Biophysical Research Communications, 284(2), 282–288. 10.1006/bbrc.2001.4969 [DOI] [PubMed] [Google Scholar]
- Dionne, S. , D’agata, I. D. , Ruemmele, F. M. , Levy, E. , St‐Louis, J. , Srivastava, A. K. , … Seidman, E. G. (1998). Tyrosine kinase and MAPK inhibition of TNF‐α‐ and EGF‐stimulated IEC‐6 cell growth. Biochemical and Biophysical Research Communications, 242(1), 146–150. 10.1006/bbrc.1997.7922 [DOI] [PubMed] [Google Scholar]
- van Es, J. H. , Van gijn, M. E. , Riccio, O. , Van den born, M. , Vooijs, M. , Begthel, H. , … Clevers, H. (2005). Notch/γ‐secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature, 435(7044), 959–963. 10.1038/nature03659 [DOI] [PubMed] [Google Scholar]
- Feng, Y. , Bommer, G. T. , Zhao, J. , Green, M. , Sands, E. , Zhai, Y. , … Fearon, E. R. (2011). Mutant KRAS promotes hyperplasia and alters differentiation in the colon epithelium but does not expand the presumptive stem cell pool. Gastroenterology, 141(3), 1003–1013. e10. 10.1053/j.gastro.2011.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fre, S. , Huyghe, M. , Mourikis, P. , Robine, S. , Louvard, D. , & Artavanis‐Tsakonas, S. (2005). Notch signals control the fate of immature progenitor cells in the intestine. Nature, 435(7044), 964–968. 10.1038/nature03589 [DOI] [PubMed] [Google Scholar]
- Furukawa, T. , Sunamura, M. , Motoi, F. , Matsuno, S. , & Horii, A. (2003). Potential tumor suppressive pathway involving DUSP6/MKP‐3 in pancreatic cancer. The American Journal of Pathology, 162(6), 1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom, L. A. , Sneddon, A. A. , Alessi, D. R. , Dowd, S. , & Keyse, S. M. (1996). Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual‐specificity phosphatase. The EMBO Journal, 15(14), 3621–3632. [PMC free article] [PubMed] [Google Scholar]
- Hagan, C. R. , Knutson, T. P. , & Lange, C. A. (2013). A common docking domain in progesterone receptor‐B links DUSP6 and CK2 signaling to proliferative transcriptional programs in breast cancer cells. Nucleic Acids Research, 41(19), 8926–8942. 10.1093/nar/gkt706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis, K. M. , Kendall, K. R. , Wang, Y. , Cheung, A. , Haigis, M. C. , Glickman, J. N. , … Jacks, T. (2008). Differential effects of oncogenic K‐Ras and N‐Ras on proliferation, differentiation and tumor progression in the colon. Nature Genetics, 40(5), 600–608. 10.1038/ng [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haramis, A.‐P. G. , Begthel, H. , Born, M. , , van den Es, J. , van Jonkheer, S. , Offerhaus, G. J. A. , & Clevers, H. (2004). De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science, 303(5664), 1684–1686. 10.1126/science.1093587 [DOI] [PubMed] [Google Scholar]
- Hatayama, H. , Iwashita, J. , Kuwajima, A. , & Abe, T. (2007). The short chain fatty acid, butyrate, stimulates MUC2 mucin production in the human colon cancer cell line, LS174T. Biochemical and Biophysical Research Communications, 356(3), 599–603. 10.1016/j.bbrc.2007.03.025 [DOI] [PubMed] [Google Scholar]
- He, X. C. , Zhang, J. , Tong, W.‐G. , Tawfik, O. , Ross, J. , Scoville, D. H. , … Li, L. (2004). BMP signaling inhibits intestinal stem cell self‐renewal through suppression of Wnt–β‐catenin signaling. Nature Genetics, 36(10), 1117–1121. 10.1038/ng1430 [DOI] [PubMed] [Google Scholar]
- Janssen, K.‐P. , Marjou, F. E. , Pinto, D. , Sastre, X. , Rouillard, D. , Fouquet, C. , … Robine, S. (2002). Targeted expression of oncogenic K‐ras in intestinal epithelium causes spontaneous tumorigenesis in mice. Gastroenterology, 123(2), 492–504. [DOI] [PubMed] [Google Scholar]
- Jensen, J. , Pedersen, E. E. , Galante, P. , Hald, J. , Heller, R. S. , Ishibashi, M. , … Madsen, O. D. (2000). Control of endodermal endocrine development by Hes‐1. Nature Genetics, 24(1), 36–44. 10.1038/71657 [DOI] [PubMed] [Google Scholar]
- de Jong, P. R. , Taniguchi, K. , Harris, A. R. , Bertin, S. , Takahashi, N. , Duong, J. , … Raz, E. (2016). ERK5 signalling rescues intestinal epithelial turnover and tumour cell proliferation upon ERK1/2 abrogation. Nature Communications, 7, 11551 10.1038/ncomms11551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidger, A. M. , & Keyse, S. M. (2016). The regulation of oncogenic Ras/ERK signalling by dual‐specificity mitogen activated protein kinase phosphatases (MKPs). Seminars in Cell & Developmental Biology, 50, 125–132. 10.1016/j.semcdb.2016.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y.‐D. , Jeon, J.‐Y. , Woo, H. J. , Lee, J. C. , Chung, J. H. , Song, S. Y. , … Baek, S. H. (2002). Interleukin‐1beta induces MUC2 gene expression and mucin secretion via activation of PKC‐MEK/ERK, and PI3K in human airway epithelial cells. Journal of Korean Medical Science, 17(6), 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek, V. , Barker, N. , Moerer, P. , Van donselaar, E. , Huls, G. , Peters, P. J. , & Clevers, H. (1998). Depletion of epithelial stem‐cell compartments in the small intestine of mice lacking Tcf‐4. Nature Genetics, 19(4), 379–383. 10.1038/1270 [DOI] [PubMed] [Google Scholar]
- Kress, T. R. , Raabe, T. , & Feller, S. M. (2010). High Erk activity suppresses expression of the cell cycle inhibitor p27Kip1 in colorectal cancer cells. Cell Communication and Signaling: CCS, 8(1), 1 10.1186/1478-811X-8-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnert, F. , Davis, C. R. , Wang, H. ‐T. , Chu, P. , Lee, M. , Yuan, J. , … Kuo, C. J. (2004). Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf‐1. Proceedings of the National Academy of Sciences, 101(1), 266–271. 10.1073/pnas.2536800100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuracha, M. R. , Thomas, P. , Loggie, B. W. , & Govindarajan, V. (2017). Bilateral blockade of MEK‐ and PI3K‐mediated pathways downstream of mutant KRAS as a treatment approach for peritoneal mucinous malignancies. PLOS One, 12(6), e0179510 10.1371/journal.pone.0179510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblanc, C. , Langlois, M. ‐J. , Coulombe, G. , Vaillancourt‐Lavigueur, V. , Jones, C. , Carrier, J. C. , … Rivard, N. (2017). Epithelial Src homology region 2 domain‐containing phosphatase‐1 restrains intestinal growth, secretory cell differentiation, and tumorigenesis. FASEB Journal, 31(8), 3512–3526. 10.1096/fj.201601378R [DOI] [PubMed] [Google Scholar]
- Lee, C. S. , Perreault, N. , Brestelli, J. E. , & Kaestner, K. H. (2002). Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes & Development, 16(12), 1488–1497. 10.1101/gad.985002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. ‐W. , Ahn, D. ‐H. , Crawley, S. C. , Li, J. ‐D. , Gum, J. R. , Basbaum, C. B. , & Kim, Y. S. (2002). Phorbol 12‐myristate 13‐acetate up‐regulates the transcription of MUC2 intestinal mucin via Ras, ERK, and NF‐κB. Journal of Biological Chemistry, 277(36), 32624–32631. 10.1074/jbc.M200353200 [DOI] [PubMed] [Google Scholar]
- Lee, S. H. , Hu, L. ‐L. , Gonzalez‐Navajas, J. , Seo, G. S. , Shen, C. , Brick, J. , … Raz, E. (2010). ERK activation drives intestinal tumorigenesis in Apcmin/+mice. Nature Medicine, 16(6), 665–670. 10.1038/nm.2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux, E. , Boucher, M. ‐J. , Mongrain, S. , Boudreau, F. , Asselin, C. , & Rivard, N. (2011). Constitutive activation of the MEK/ERK pathway inhibits intestinal epithelial cell differentiation. American Journal of Physiology Gastrointestinal and Liver Physiology, 301(4), G719–G730. 10.1152/ajpgi.00508.2010 [DOI] [PubMed] [Google Scholar]
- Lemieux, É. , Bergeron, S. , Durand, V. , Asselin, C. , Saucier, C. , & Rivard, N. (2009). Constitutively active MEK1 is sufficient to induce epithelial‐to‐mesenchymal transition in intestinal epithelial cells and to promote tumor invasion and metastasis. International Journal of Cancer, 125(7), 1575–1586. 10.1002/ijc.24485 [DOI] [PubMed] [Google Scholar]
- Li, J.‐D. , Feng, W. , Gallup, M. , Kim, J.‐H. , Gum, J. , Kim, Y. , & Basbaum, C. (1998). Activation of NF‐κB via a Src‐dependent Ras‐MAPK‐pp90rsk pathway is required for Pseudomonas aeruginosa‐induced mucin overproduction in epithelial cells. Proceedings of the National Academy of Sciences of the United States of America, 95(10), 5718–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low, D. , Nguyen, D. D. , & Mizoguchi, E. (2013). Animal models of ulcerative colitis and their application in drug research. Drug Design, Development and Therapy, 7, 1341–1357. 10.2147/DDDT.S40107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillet, M. , Purcell, N. H. , Sargent, M. A. , York, A. J. , Bueno, O. F. , & Molkentin, J. D. (2008). DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. The Journal of Biological Chemistry, 283(45), 31246–31255. 10.1074/jbc.M806085200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina, S. , Frati, L. , Leonetti, C. , Zuchegna, C. , Di Zazzo, E. , Calogero, A. , & Porcellini, A. (2011). Dual‐specificity phosphatase DUSP6 has tumor‐promoting properties in human glioblastomas. Oncogene, 30(35), 3813–3820. 10.1038/onc.2011.99 [DOI] [PubMed] [Google Scholar]
- Milano, J. , McKay, J. , Dagenais, C. , Foster‐Brown, L. , Pognan, F. , Gadient, R. , … Ciaccio, P. J. (2004). Modulation of notch processing by γ‐secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicological Sciences, 82(1), 341–358. 10.1093/toxsci/kfh254 [DOI] [PubMed] [Google Scholar]
- Moser, A. , Pitot, H. , & Dove, W. (1990). A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science, 247(4940), 322–324. [DOI] [PubMed] [Google Scholar]
- Muda, M. , Theodosiou, A. , Rodrigues, N. , Boschert, U. , Camps, M. , Gillieron, C. , … Arkinstall, S. (1996). The dual specificity phosphatases M3/6 and MKP‐3 are highly selective for inactivation of distinct mitogen‐activated protein kinases. The Journal of Biological Chemistry, 271(44), 27205–27208. [DOI] [PubMed] [Google Scholar]
- Noguchi, T. , Metz, R. , Chen, L. , Mattéi, M. G. , Carrasco, D. , & Bravo, R. (1993). Structure, mapping, and expression of erp, a growth factor‐inducible gene encoding a nontransmembrane protein tyrosine phosphatase, and effect of ERP on cell growth. Molecular and Cellular Biology, 13(9), 5195–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okudela, K. , Yazawa, T. , Woo, T. , Sakaeda, M. , Ishii, J. , Mitsui, H. , … Kitamura, H. (2009). Down‐regulation of DUSP6 expression in lung cancer —Its mechanism and potential role in carcinogenesis. The American Journal of Pathology, 175(2), 867–881. 10.2353/ajpath.2009.080489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens, D. M. , & Keyse, S. M. (2007). Differential regulation of MAP kinase signalling by dual‐specificity protein phosphatases. Oncogene, 26(22), 3203–3213. 10.1038/sj.onc.1210412 [DOI] [PubMed] [Google Scholar]
- Paquin, M.‐C. , Cagnol, S. , Carrier, J. C. , Leblanc, C. , & Rivard, N. (2013). ERK‐associated changes in E2F4 phosphorylation, localization and transcriptional activity during mitogenic stimulation in human intestinal epithelial crypt cells. BMC Cell Biology, 14, 33 10.1186/1471-2121-14-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrais, M. , Pigny, P. , Copin, M.‐C. , Aubert, J.‐P. , & Van Seuningen, I. (2002). Induction of MUC2 and MUC5AC mucins by factors of the epidermal growth factor (EGF) family is mediated by EGF Receptor/Ras/Raf/extracellular signal‐regulated kinase cascade and Sp1* . Journal of Biological Chemistry, 277(35), 32258–32267. 10.1074/jbc.M204862200 [DOI] [PubMed] [Google Scholar]
- Perreault, N. , Sackett, S. D. , Katz, J. P. , Furth, E. E. , & Kaestner, K. H. (2005). Foxl1 is a mesenchymal modifier of Min in carcinogenesis of stomach and colon. Genes & Development, 19(3), 311–315. 10.1101/gad.1260605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto, D. , Gregorieff, A. , Begthel, H. , & Clevers, H. (2003). Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes & Development, 17(14), 1709–1713. 10.1101/gad.267103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popat, S. , Hubner, R. , & Houlston, R. S. (2005). Systematic review of microsatellite instability and colorectal cancer prognosis. Journal of Clinical Oncology, 23(3), 609–618. 10.1200/JCO.2005.01.086 [DOI] [PubMed] [Google Scholar]
- Rajagopalan, H. , Bardelli, A. , Lengauer, C. , Kinzler, K. W. , Vogelstein, B. , & Velculescu, V. E. (2002). Tumorigenesis: RAF/RAS oncogenes and mismatch‐repair status. Nature, 418(6901), 934 10.1038/418934a [DOI] [PubMed] [Google Scholar]
- Razmara, M. , Eger, G. , Rorsman, C. , Heldin, C.‐H. , & Lennartsson, J. (2012). MKP3 negatively modulates PDGF‐induced Akt and Erk5 phosphorylation as well as chemotaxis. Cellular Signalling, 24(3), 635–640. 10.1016/j.cellsig.2011.11.001 [DOI] [PubMed] [Google Scholar]
- Rhoads, J. M. , Argenzio, R. A. , Chen, W. , Rippe, R. A. , Westwick, J. K. , Cox, A. D. , … Brenner, D. A. (1997). L‐glutamine stimulates intestinal cell proliferation and activates mitogen‐activated protein kinases. American Journal of Physiology–Gastrointestinal and Liver Physiology, 272(5), G943–G953. 10.1152/ajpgi.1997.272.5.G943 [DOI] [PubMed] [Google Scholar]
- Riese, D. J. , & Cullum, R. L. (2014). Epiregulin: Roles in normal physiology and cancer. Seminars in Cell & Developmental Biology, 0, 49–56. 10.1016/j.semcdb.2014.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivard, N. , Boucher, M. J. , Asselin, C. , & L’Allemain, G. (1999). MAP kinase cascade is required for p27 downregulation and S phase entry in fibroblasts and epithelial cells. The American Journal of Physiology, 277(4 Pt 1), C652–C664. [DOI] [PubMed] [Google Scholar]
- Roers, A. , Siewe, L. , Strittmatter, E. , Deckert, M. , Schlüter, D. , Stenzel, W. , … Müller, W. (2004). T cell‐specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. The Journal of Experimental Medicine, 200(10), 1289–1297. 10.1084/jem.20041789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan, J.‐W. , Statt, S. , Huang, C.‐T. , Tsai, Y.‐T. , Kuo, C.‐C. , Chan, H.‐L. , … Kao, C. Y. (2016). Dual‐specificity phosphatase 6 deficiency regulates gut microbiome and transcriptome response against diet‐induced obesity in mice. Nature Microbiology, 2, 16220 10.1038/nmicrobiol.2016.220 [DOI] [PubMed] [Google Scholar]
- Samowitz, W. S. , Curtin, K. , Schaffer, D. , Robertson, M. , Leppert, M. , & Slattery, M. L. (2000). Relationship of Ki‐ras mutations in colon cancers to tumor location, stage, and survival: A population‐based study. Cancer Epidemiology, Biomarkers & Prevention, 9(11), 1193–1197. [PubMed] [Google Scholar]
- Sansom, O. J. , Reed, K. R. , Hayes, A. J. , Ireland, H. , Brinkmann, H. , Newton, I. P. , … Winton, D. J. (2004). Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes & Development, 18(12), 1385–1390. 10.1101/gad.287404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarközi, R. , Miller, B. , Pollack, V. , Feifel, E. , Mayer, G. , Sorokin, A. , & Schramek, H. (2007). ERK1/2‐driven and MKP‐mediated inhibition of EGF‐induced ERK5 signaling in human proximal tubular cells. Journal of Cellular Physiology, 211(1), 88–100. 10.1002/jcp.20909 [DOI] [PubMed] [Google Scholar]
- Sebolt‐Leopold, J. S. , Dudley, D. T. , Herrera, R. , Becelaere, K. V. , Wiland, A. , Gowan, R. C. , … Saltiel, A. R. (1999). Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nature Medicine, 5(7), 810–816. 10.1038/10533 [DOI] [PubMed] [Google Scholar]
- Sheng, G. , Bernabe, K. Q. , Guo, J. , & Warner, B. W. (2006). Epidermal growth factor receptor–mediated proliferation of enterocytes requires p21waf1/cip1 expression. Gastroenterology, 131(1), 153–164. 10.1053/j.gastro.2006.05.007 [DOI] [PubMed] [Google Scholar]
- Van der Sluis, M. , De Koning, B. A. E. , De Bruijn, A. C. J. M. , Velcich, A. , Meijerink, J. P. P. , Van Goudoever, J. B. , … Einerhand, A. W. C. (2006). Muc2‐deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology, 131(1), 117–129. 10.1053/j.gastro.2006.04.020 [DOI] [PubMed] [Google Scholar]
- Solit, D. B. , Garraway, L. A. , Pratilas, C. A. , Sawai, A. , Getz, G. , Basso, A. , … Rosen, N. (2006). BRAF mutation predicts sensitivity to MEK inhibition. Nature, 439(7074), 358–362. 10.1038/nature04304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm, A. , & Dignass, A. U. (2008). Epithelial restitution and wound healing in inflammatory bowel disease. World Journal of Gastroenterology, 14(3), 348–353. 10.3748/wjg.14.348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, L. , Kinzler, K. , Vogelstein, B. , Preisinger, A. , Moser, A. , Luongo, C. , … Dove, W. (1992). Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science, 256(5057), 668–670. [DOI] [PubMed] [Google Scholar]
- Suzuki, S. , Takeuchi, K. , Ishinaga, H. , Basbaum, C. , & Majima, Y. (2008). Leukotriene D4 upregulates MUC2 gene transcription in human epithelial cells. Pharmacology, 81(3), 221–228. 10.1159/000112866 [DOI] [PubMed] [Google Scholar]
- Tanoue, T. , Adachi, M. , Moriguchi, T. , & Nishida, E. (2000). A conserved docking motif in MAP kinases common to substrates, activatorsand regulators. Nature Cell Biology, 2(2), 110–116. 10.1038/35000065 [DOI] [PubMed] [Google Scholar]
- Taupin, D. , & Podolsky, D. K. (1999). Mitogen‐activated protein kinase activation regulates intestinal epithelial differentiation. Gastroenterology, 116(5), 1072–1080. [DOI] [PubMed] [Google Scholar]
- Voisin, L. , Julien, C. , Duhamel, S. , Gopalbhai, K. , Claveau, I. , Saba‐El‐Leil, M. K. , … Meloche, S. (2008). Activation of MEK1 or MEK2 isoform is sufficient to fully transform intestinal epithelial cells and induce the formation of metastatic tumors. BMC Cancer, 8, 337 10.1186/1471-2407-8-337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Velho, S. , Vakiani, E. , Peng, S. , Bass, A. J. , Chu, G. C. , … Haigis, K. M. (2013). Mutant N‐Ras protects colorectal cancer cells from stress‐induced apoptosis and contributes to cancer development and progression. Cancer Discovery, 3(3), 294–307. 10.1158/2159-8290.CD-12-0198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee, P. , & Wang, Z. (2017). Epidermal growth factor receptor cell proliferation signaling pathways. Cancers, 9(5), 1–45. 10.3390/cancers9050052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Q.‐N. , Liao, Y.‐F. , Lu, Y.‐X. , Wang, Y. , Lu, J.‐H. , Zeng, Z.‐L. , … Xu, R. H. (2018). Pharmacological inhibition of DUSP6 suppresses gastric cancer growth and metastasis and overcomes cisplatin resistance. Cancer Letters, 412, 243–255. 10.1016/j.canlet.2017.10.007 [DOI] [PubMed] [Google Scholar]
- Xu, S. , Furukawa, T. , Kanai, N. , Sunamura, M. , & Horii, A. (2005). Abrogation of DUSP6 by hypermethylation in human pancreatic cancer. Journal of Human Genetics, 50(4), 159–167. 10.1007/s10038-005-0235-y [DOI] [PubMed] [Google Scholar]
- Yamashita, H. , Kotani, T. , Park, J. , Murata, Y. , Okazawa, H. , Ohnishi, H. , … Matozaki, T. (2014). Role of the protein tyrosine phosphatase Shp2 in homeostasis of the intestinal epithelium. PLoS One, 9(3), e92904 10.1371/journal.pone.0092904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Q. , Bermingham, N. A. , Finegold, M. J. , & Zoghbi, H. Y. (2001). Requirement of math1 for secretory cell lineage commitment in the mouse intestine. Science, 294(5549), 2155–2158. 10.1126/science.1065718 [DOI] [PubMed] [Google Scholar]
- Yeh, J. J. , Routh, E. D. , Rubinas, T. , Peacock, J. , Martin, T. D. , Shen, X. J. , … Der, C. J. (2009). KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer. Molecular Cancer Therapeutics, 8(4), 834–843. 10.1158/1535-7163.MCT-08-0972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecchini, V. , Domaschenz, R. , Winton, D. , & Jones, P. (2005). Notch signaling regulates the differentiation of post‐mitotic intestinal epithelial cells. Genes & Development, 19(14), 1686–1691. 10.1101/gad.341705 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information