

Abstract
The phytonutrient ursolic acid (UA), present in apples, rosemary, and other plant sources, has anti‐cancer properties in a number of systems, including skin cancers. However, few reports have examined upstream mechanisms by which UA may prevent or treat cancer. Recent reports have indicated UA induces death of cancer cell lines via AMP‐activated protein kinase (AMPK), an energy‐sensing kinase which possesses both pro‐metabolic and anti‐cancer effects. Other studies have shown UA activates peroxisome proliferator activated receptor α (PPARα) and the glucocorticoid receptor (GR). Here, we found the cytotoxic effect of UA in skin carcinoma cells required AMPK activation. In addition, two inhibitors of PPARα partially reversed the cytotoxic effects of UA, suggesting its effects are at least partially mediated through this receptor. Finally, inhibition of the GR did not reverse the effects of UA nor did this compound bind the GR under the conditions of experiments performed. Overall, studies elucidating the anti‐cancer effects of UA may allow for the development of more potent analogues utilizing similar mechanisms. These studies may also reveal the mediators of any possible side effects or resistance mechanisms to UA therapy.
Keywords: AMP‐activated protein kinases, neoplasms, phytochemicals
Abbreviations
- AMPK
AMP‐activated protein kinase
- GR
glucocorticoid receptor
- LDH
lactate dehydrogenase
- MTT reagent
thiazolyl blue tetrazolium bromide
- PPAR
peroxisome proliferator activated receptor α
- UA
ursolic acid
1. INTRODUCTION
The natural phytonutrient ursolic acid (UA) has been shown to have both anti‐cancer1, 2, 3, 4, 5 and anti‐diabetic6, 7 effects in a variety of in vitro and in vivo systems. A number of studies indicated UA activates the energy sensor AMP‐activated protein kinase (AMPK),6, 8, 9 which has both anti‐cancer and anti‐diabetic properties. AMPK is activated by both exercise10, 11 and calorie restriction.12, 13 These lifestyle factors induce a negative energy balance, and the resulting increases in the AMP/ATP ratio allosterically interact with and allow activation of AMPK via phosphorylation at threonine 172 (Thr172) by upstream kinases such as LKB1 and CAMKKβ.14, 15 Activated AMPK stimulates catabolic processes such as fat oxidation and glucose uptake and inhibits anabolic processes such as fatty acid synthesis and gluconeogenesis to restore ATP levels.16, 17
AMPK also has anti‐tumor properties. In this regard, AMPK loss leads to increased tumor formation in lymphoma‐susceptible mice.18 LKB1, the upstream kinase of AMPK, also functions as a tumor suppressor.19 In addition, recent studies have shown the cytotoxic effects of UA against cancer cells are mediated by AMPK. The AMPK inhibitor Compound C decreased UA‐mediated pro‐apoptotic signaling in liver cancer cells.8 Another report found UA‐induced apoptosis of bladder cancer cells was suppressed by knockdown of AMPK.9 In addition, inhibitory phosphorylation of AMPK was shown to increase during progression to human SCC,20 suggesting a potential role of AMPK as a suppressor of skin carcinoma formation or viability. These studies further suggest that the effects of UA against skin tumor growth in vivo3 may be mediated, at least in part, by AMPK activation.
Despite many reports demonstrating the anti‐cancer effects of UA, very few studies have attempted to determine which upstream proximal receptor(s) contribute to these effects. The peroxisome proliferator activated receptor α (PPARα) is primarily expressed in the liver, kidney, skeletal muscle, and heart and is important for lipid homeostasis and may have anti‐inflammatory properties.21 Ligands for PPARα have been shown to inhibit skin tumorigenesis in vivo22 and activate AMPK.23 Also, synthesized derivatives of a UA isomer bound different PPARs24 and resulted in downstream anti‐inflammatory and anti‐skin tumor effects.25 In addition, UA enhanced PPARα promoter‐binding and overall transcriptional activity in HepG2 liver cancer cells. However, UA did not directly bind PPARα.26 Overall, these studies suggest that the anti‐cancer effect of UA may also involve activation of PPARα through a mechanism independent of ligand binding.
Another potential candidate receptor for UA is the glucocorticoid receptor (GR), which has anti‐inflammatory properties both through transrepressive interactions with pro‐inflammatory transcription factors and through transactivation of promoters leading to synthesis of anti‐inflammatory proteins.27 The structure of UA strongly resembles that of typical glucocorticoids, which also inhibit skin tumor promotion.28, 29 UA also enhanced GR nuclear translocation,30, 31 and UA‐mediated decreases in MMP‐9 expression are reversed by GR antagonist RU486 in fibrosarcoma cells.30 Finally, UA has been shown to slightly bind the GR in breast cancer cells, even though UA did not affect GR‐dependent transcription.31 Although these studies indicate UA does not impact the transcriptional activity of the GR, UA could still exert beneficial effects by mediating transrepressive protein‐protein interactions between the GR and oncogenic transcription factors,32, 33 which are suggested to mediate the anti‐skin cancer effects of GR.34
In the current studies, we used pharmacological inhibitors of AMPK, PPARα, and GR to determine if these pathways contribute to the cytotoxic effects of UA in skin cancer cells. An accurate characterization of the anti‐cancer effects of UA will assist with future clinical aspects, such as explanation of potential side effects, identification of resistance mechanisms, and the possibility of encountering target polymorphisms among non‐melanoma skin cancer patients.
2. MATERIALS AND METHODS
2.1. Reagents
GW6471 and MK 886 were purchased from Tocris Bioscience (Bristol, UK). UA, Compound C, RU486, dexamethasone, cortisol, corticosterone, and thiazolyl blue tetrazolium bromide (MTT reagent) were obtained from Sigma (St. Louis, MO). A kit for assaying lactate dehydrogenase (LDH) was purchased from Roche (Indianapolis, IN). [3H]dexamethasone was obtained from American Radiolabeled Chemicals (St. Louis, MO). Human recombinant GR was purchased from Invitrogen (Grand Island, NY). Primary antibodies against phosphorylated AMPK (Thr172) and AMPK were obtained from Cell Signaling Technology (Danvers, MA), while primary antibody against β‐actin was purchased from Abcam (Cambridge, MA).
2.2. Cell culture
A mouse squamous cell carcinoma cell line (Ca3/7) and a mouse skin papilloma cell line (MT1/2) were used for the current studies. The cells were maintained in Joklik MEM supplemented with 8% FBS, 50 U/mL penicillin, 50 ng/mL streptomycin, 10 μg/mL transferrin, 50 μg/mL gentamicin sulfate, 5 μg/mL insulin, 5 ng/mL EGF, 10 μM o‐phosphorylethanolamine, and 10 μM 2‐aminoethanol. The immortalized human keratinocyte cell line, HaCaT,35 were maintained in high glucose DMEM + GlutaMAX supplemented with 10% fetal bovine serum, 50 U/mL penicillin, and 50 ng/mL streptomycin. All cells were grown in an incubator at 5% CO2 and 37°C. The murine papilloma cell line MT1/2 was generated from papilloma from the MNNG/TPA skin carcinogenesis protocol. The squamous cell carcinoma cell line Ca3/7 was created with carcinoma from mice treated with the DMBA/TPA skin carcinogenesis protocol.36, 37, 38 MT1/2 and Ca3/7 cells were were obtained from the University of Texas M.D. Anderson Cancer Center, Science Park‐Research Division, Smithville, TX. Ca3/7 and HaCaT cells were verified free of mycoplasma the year before experiments were performed. The cell culture working environment was demonstrated to be mycoplasma‐free after concluding all experiments. Each vial of cells was used within 2 months of thawing.
2.3. Western blotting
Ca3/7 cells were incubated with 10 μM AMPK inhibitor Compound C or 0.1% DMSO vehicle for 4 h followed by incubation with UA or 0.1% DMSO vehicle for 6 h. Cells were rinsed twice with PBS and lysed in buffer containing 1% Triton X‐100, 0.5% IGEPAL, 0.05 M TrisHCl and 0.1 M NaCl as well as protease/phosphatase inhibitors and 5 mM EDTA. Proteins were extracted by centrifugation and quantified by Bradford. Proteins were denatured in XT Sample Buffer containing XT reducing agent. Proteins were separated on 4‐12% gradient gels and transferred onto PVDF membranes, which were blocked in 5% bovine serum albumin or 5% milk in TBST for 1 h and incubated overnight with primary antibody. Blots were rinsed, incubated with secondary antibody for 1 h and developed with Pierce ECL2 Western blotting substrate.
2.4. MTT assay for cell viability
Ca3/7 cells were pretreated with indicated doses of Compound C or 0.1% DMSO vehicle (4 h) or GW6471, MK 886, or 0.1% DMSO vehicle (1 h) followed by UA or 0.1% DMSO for 24 h. In the figures, “vehicle” refers to groups treated with UA but without the AMPK inhibitor Compound C, the PPARα inhibitors GW6471 and MK 866, or the GR inhibitor RU486. MTT reagent (0.5 mg/mL) was added, and cells were incubated for an additional 3 h. Plates were centrifuged at 100g for 5 min, media was removed, and formazan crystals were solubilized with 100 μL DMSO. Plates were measured at 570 nm with a background subtraction at 650 nm, using a Biotek Synergy HT spectrophotometer (Biotek, Winooski, VT).
2.5. LDH assay for cell death
Ca3/7 cells were pretreated with 10 μM Compound C or 0.1% DMSO vehicle for 4 h followed by indicated doses of UA or 0.1% DMSO vehicle for 24 h. For positive control wells, 2% Triton X‐100 was added. Cell plates were centrifuged at 100g for 5 min, and 50 μL supernatant/well was transferred to a 96 well plates containing the LDH reagent prepared according to the manufacturer's instructions. Plates were incubated for 20 min in the dark, then read at 490 nm with background subtraction at 650 nm using a Biotek Synergy HT spectrophotometer.
2.6. Radioligand binding assays
HaCaT cells were plated at 4.0 × 105 cells per well in 12‐well plates in high glucose DMEM with 10% Chelex‐treated FBS. Cells were treated with up to 25 μM UA or control glucocorticoids dexamethasone, cortisol, or corticosterone for 24 h. DMSO was used as the vehicle control for UA and ethanol was used for all other compounds. Cells were incubated in media containing the same concentrations of compounds as well as 10 nM [3H]dexamethasone for 2 h. Cells were rinsed three times with PBS and checked under the microscope to verify no cell loss. Cells were lysed for 1 h at 4°C with 0.5 mL 1 M NaOH. [3H] radioactivity in lysates was counted with a Multi‐Purpose Scintillation Counter from Beckman Coulter. Background radioactivity from similarly‐manipulated cell‐free wells was subtracted from each reading.
In order to confirm the binding assay results obtained in HaCaT cells, a cell‐free system utilizing human recombinant GR was employed. The reaction was conducted in a binding buffer consisting of 10 mM potassium phosphate, 0.1 mM EDTA, 10 mM sodium molybdate, and 5 mM DTT. A 30 nM [3H]dexamethasone, 30 nM human recombinant GR, and indicated doses of UA or control glucocorticoids were combined in binding buffer and incubated for 2 h at 4°C. A 5% dextran‐coated charcoal was added to each tube to chelate unbound ligands for removal. Samples were centrifuged and [3H] in each clear supernatant was counted with a Multi‐Purpose Scintillation Counter from Beckman Coulter.
2.7. siRNA transient transfection
Ca3/7 cells (1 × 106) cells were transiently transfected with mouse AMPK siRNA with the use of Amaxa Nucleofector Technology according to the manufacturer's protocol (Lonza Cologne GmbH, 50829 Köln, Germany). AMPK siRNA (sc‐45312), purchased from Santa Cruz Biotechnology, Inc., (Dallas, TX), consisted of pools of three to five target‐specific 19‐25 nt siRNAs designed to knockdown gene expression. The doses of AMPK siRNA were 100 and 200 nM. The scrambled siRNA was used at 200 nM. The effect of AMPK siRNA on cell proliferation was tested in transfected cells by MTT assay at a cell density of 3000 cells/well. Knockdown of AMPK protein expression was confirmed by Western blot analysis.
2.8. Statistical analyses
For siRNA knockdown experiments, differences between the two groups were determined by two‐tailed Student's t‐test. Statistically significant differences between treatment groups in other experiments were determined by using ANOVA. IC50 and LD50 values were calculated using GraphPad Prism.
3. RESULTS
3.1. UA decreased carcinoma cell viability via AMPK activation
In these experiments, we first used the AMPK inhibitor, Compound C, to determine whether UA toxicity in Ca3/7 skin carcinoma cells is mediated by AMPK activity. Western blotting experiments revealed UA enhanced AMPK activity at cytotoxic doses, and AMPK phosphorylation was significantly prevented by Compound C (Figure 1). Cell viability (MTT assay) and cell death (LDH assay) analyses showed Compound C significantly prevented the effects of UA in Ca3/7 skin carcinoma cells (Figure 2). Finally, siRNA knockdown of AMPK led to a reversal of UA‐mediated decreases in Ca3/7 cell viability (Figure 3). These results reveal AMPK activation plays a role in the ability of UA to inhibit the viability of Ca3/7 skin carcinoma cells.
Figure 1.
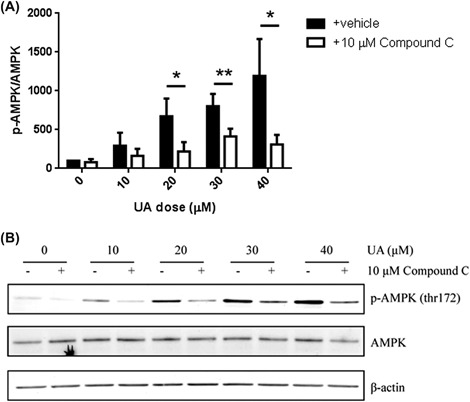
UA‐mediated AMPK activation was reversed with Compound C in Ca3/7 cells. A, Compound C reversed UA‐mediated AMPK activation in Ca3/7 cells (average of four independent experiments, *P < 0.05, **P < 0.01). B, Representative Western blot shown
Figure 2.
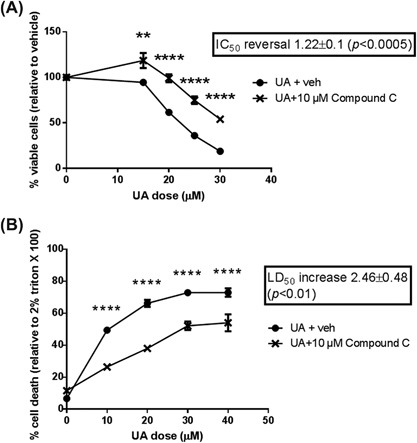
The cytotoxic effect of UA on Ca3/7 cells was reversed by AMPK inhibition. A, UA‐mediated decrease in Ca3/7 viability is reversed by AMPK inhibitor Compound C (IC50 reversal calculated from average of six independent experiments, n = 3/exp, representative experiment shown). B, UA‐mediated increase in LDH release in Ca3/7 is reversed by Compound C (LD50 increase calculated from average of three independent experiments, n = 5/exp, representative experiment shown). For all experiments, **P < 0.01, ****P < 0.001
Figure 3.
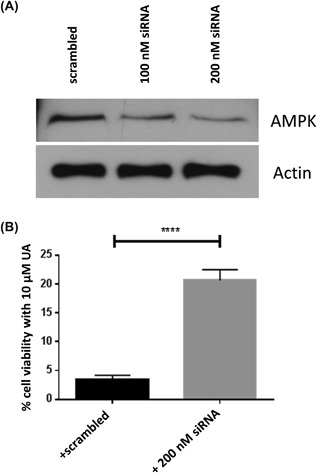
The cytotoxic effect of UA on Ca3/7 cells was reversed by siRNA knockdown of AMPK. A, Western blot showing knockdown of AMPK. B, siRNA knockdown of AMPK makes Ca3/7 cells resistant to cytotoxicity from 10 μM UA. (n = 6, ****P < 0.001)
3.2. PPARα partially mediated the cytotoxic effects of UA
As noted in the Introduction, a number of studies have shown UA can activate PPARα, which is also activated by other anti‐skin cancer compounds.22 We performed experiments to determine whether PPARα contributes to UA‐mediated death of skin cancer cells. Mouse skin papilloma cells (MT1/2) or Ca3/7 cells were pretreated with the PPARα inhibitors GW6471 or MK 886 for 1 h prior to adding UA at increasing doses. Cells were haversted 24 h later. GW6471 slightly but significantly increased the IC50 of UA on MT1/2 (Figure 4A) and Ca3/7 (Figure 4B) cell viability. In order to confirm these findings, we repeated these experiments with another PPARα inhibitor, MK 886, which is less potent than GW6471. Similar effects were observed, as MK 886 significantly increased the IC50 of UA in both MT1/2 (Figure 4C) and Ca3/7 (Figure 4D) cell lines. Overall, these results indicate that UA‐mediated cytotoxicity in skin cancer cells is at least partially mediated by PPARα.
Figure 4.
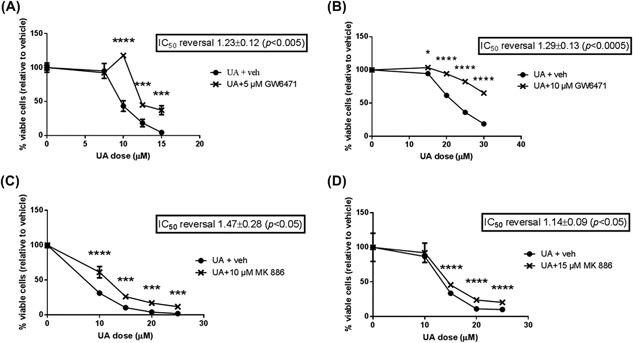
UA‐mediated decreases in MT1/2 and Ca3/7 viability were blunted by PPARα inhibitors. A, UA‐mediated decreases in MT1/2 viability were partially reversed by PPARα inhibitor GW6471 (IC50 reversal calculated from average of five independent experiments n = 3/exp, representative experiment shown). B, UA‐mediated decreases in Ca3/7 viability were partially reversed by PPARα inhibitor GW6471 (IC50 reversal calculated from average of six independent experiments n = 3/exp, representative experiment shown). C, UA‐mediated decreases in MT1/2 viability were partially reversed by PPARα inhibitor MK 886 (IC50 reversal calculated from average of four independent experiments, n = 3/exp, representative experiment shown). D, UA‐mediated decreases in Ca3/7 viability were partially reversed by PPARα inhibitor MK886 (IC50 reversal calculated from average of four independent experiments, n = 3/exp, representative experiment shown). For all experiments, *P < 0.05, ***P < 0.005, ****P < 0.001
3.3. GR did not mediate the cytotoxic effects of UA
A number of studies have shown UA may modulate the GR, which mediates the effects of skin cancer‐suppressing glucocorticoids.28, 29, 30, 34 We found the GR inhibitor RU486, which inhibits both the transactivation and transrepression properties of GR,39 did not suppress UA‐mediated decreases in Ca3/7 cell viability (Figure 5A). On the contrary, RU486 significantly enhanced decreases in Ca3/7 viability in response to UA. This indicates that GR activity may mediate a cytoprotective resistance to UA treatment, or RU486 may enhance the effects of UA through a GR activity‐independent mechanism.
Figure 5.
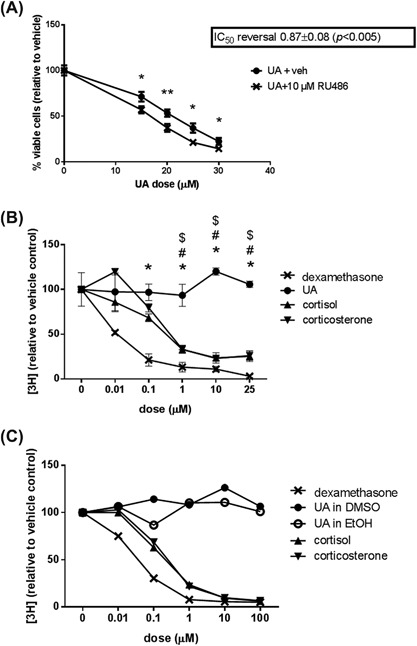
The anti‐cancer effects of UA are not mediated by the GR. A, GR inhibitor RU486 enhanced the anti‐proliferative effect of UA in Ca3/7 cells (IC50 values calculated from average of six independent experiments, n = 3/exp, representative experiment shown, *P < 0.05, **P < 0.01). B, UA did not bind the ligand‐binding site on GR in HaCaT cells (n = 2, * indicates P < 0.05 between UA and dexamethasone, # indicates P < 0.05 between UA and cortisol, $ indicates P < 0.05 between UA and corticosterone). C, UA did not bind human recombinant GR in a cell‐free system (n = 1)
UA has been demonstrated to have GR‐activating properties, and the structure of UA resembles that of typical glucocorticoids. In order to determine if UA functions as a typical glucocorticoid, we performed radioligand binding assays. These assays determine if UA can displace radiolabeled dexamethasone, which should be bound to the traditional ligand‐binding site. In initial experiments, we employed the human keratinocyte cell line HaCaT, in which UA is less cytotoxic. Non‐toxic doses of UA up to 25 μM did not displace [3H]dexamethasone, while control glucocorticoids dexamethasone, cortisol, and corticosterone all competed with [3H]dexamethasone for binding (Figure 5B). In other experiments we incubated UA with [3H]dexamethasone over a series of time points (1‐8 h), all of which showed no decrease in [3H]dexamethasone in cell lysates (data not shown). These results also suggest UA does not interact with GR as a traditional glucocorticoid in cell culture. In order to confirm the lack of UA binding to GR in HaCaT cells was not due to cellular manipulations such as efflux, metabolism, or sequestration of UA, we employed a simple cell‐free system using human recombinant GR. In this study, we also used EtOH as the vehicle for UA, to confirm the use of different vehicles did not complicate the findings. Similar to the cell culture studies, UA did not displace [3H]dexamethasone from human recombinant GR, while dexamethasone, cortisol, and corticosterone all competed for binding (Figure 5C).
4. DISCUSSION
The current results indicate that AMPK activity at least partially mediates the effects of UA on viability of Ca3/7 skin carcinoma cells. In other studies, the anti‐cancer effects of UA have also been shown to depend on AMPK in liver cancer cells and bladder cancer cells.8, 9 In addition, studies using PPARα inhibitors GW6471 and MK 886 showed that the UA‐mediated decrease in the viability of MT1/2 and Ca3/7 cells was also at least partially dependent on PPARα activity. A number of interactions between PPARα and AMPK have been demonstrated. AMPK contributes to phosphorylation and transcriptional activity of PPARα40 as well as to PPARα expression.41 Also, PPARα ligands induce activation of AMPK, and AMPK is required for the downstream effects of PPARα ligands.42 A positive feedback loop between AMPK and PPARα could explain how the ability of UA to induce death of skin cancer cells is reversed by both AMPK and PPARα inhibitors as seen in the current experiments.
Experiments exploring the possible role of the GR showed that RU486 unexpectedly enhanced the anti‐cancer effects of UA in the skin carcinoma cell line Ca3/7. Considering the GR‐activating effects of UA in other systems and the anti‐cancer effects of different glucocorticoids, we expected the anti‐cancer effects of UA to be at least partially prevented with the GR antagonist. Interestingly, dexamethasone has been shown to enhance expression of p‐glycoprotein, which was reversed with RU486.43, 44 P‐glycoprotein is a drug‐effluxing pump which contributes to chemoresistance to a wide variety of xenobiotics, including anti‐cancer compounds.45 In addition, our recent studies show that the cytotoxic effects of UA are enhanced with inhibition of p‐glycoprotein.5 In our system, RU486 may decrease the basal expression of p‐glycoprotein resulting in increased intracellular UA and enhanced cytotoxicity.46 Regardless, our GR binding assays indicate that despite the steroid‐like structure of UA and similar downstream effects to other glucocorticoids, UA is not a direct ligand for the GR, at least not at the characteristic binding site for glucocorticoids. UA may interact with and impact the GR through an allosteric interaction, although future experiments are needed to confirm this in our system.
Recent studies from our laboratories using an in vivo model of skin tumor promotion have shown that UA and related triterpenes given topically can activate AMPK in the epidermis of mice treated with TPA.4, 47 AMPK activation correlated with increased phosphorylation of ULK1 (Ser555) suggesting that AMPK activation by UA and related triterpenes may lead to induction of autophagic cell death. Recent studies have also shown that PPARα activation can regulate autophagy.48 Autophagic cell death may play an important role in the reduced survival seen in skin cancer cells observed with UA. Our future studies will explore the possible interactions between UA and AMPK‐ and PPARα‐mediated autophagic death pathways in both premalignant and malignant skin cancer cells.
In summary, both the AMPK and PPARα pathways contribute significantly to the cytotoxic effects of UA in skin cancer cells. Furthermore, the effects of UA on cell survival were not dependent on the GR. In addition, UA did not function as a typical GR ligand in the cell lines used. Future studies utilizing in vivo models of skin cancer may further confirm the mechanism of action of UA. This would involve the generation of mouse models lacking AMPK or PPARα in the skin, perhaps through crossing skin‐specific Cre recombinase mice with mice with floxed AMPK or PPARα. These mice could then be treated with the DMBA/TPA skin carcinogenesis protocol and UA to determine if AMPK or PPARα mediate the ability of UA to reduce skin cancer formation in vivo. These results delineating the mechanism of action of UA may allow for the development of related anti‐skin cancer compounds which operate through similar means. Full characterization of UA may also provide molecular explanations in case any side effects or resistance mechanisms arise during UA therapy, especially in non‐melanoma skin cancer patients with certain polymorphisms in the targets for this compound.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
ACKNOWLEDGMENTS
Radioactive ligand binding experiments were performed using a Multi‐Purpose Scintillation Counter from Beckman Coulter provided by the Department of Pharmacology at The University of Texas Health Science Center at San Antonio. Financial Support was provided by the National Institutes of Health (R01 CA164159, P30 CA54174), a Clinical and Translational Science Award (UL1RR025767), the American Cancer Research Center and Foundation, an Oppenheimer Multi‐Investigator Research Award, and a pre‐doctoral fellowship from the Pharmaceutical Research and Manufacturers of America Foundation.
Junco JJ, Cho J, Mancha A, et al. Role of AMPK and PPARα in the anti‐skin cancer effects of ursolic acid. Molecular Carcinogenesis. 2018;57:1698–1706. 10.1002/mc.22890
Statement of implication: The anti‐skin cancer effects of ursolic acid are partially mediated through AMPK and PPARα⋅
REFERENCES
- 1. Shanmugam MK, Dai X, Kumar AP, Tan BK, Sethi G, Bishayee A. Ursolic acid in cancer prevention and treatment: molecular targets, pharmacokinetics and clinical studies. Biochem Pharmacol. 2013;85:1579–1587. [DOI] [PubMed] [Google Scholar]
- 2. De Angel RE, Smith SM, Glickman RD, Perkins SN, Hursting SD. Antitumor effects of ursolic acid in a mouse model of postmenopausal breast cancer. Nutr Cancer. 2010;62:1074–1086. [DOI] [PubMed] [Google Scholar]
- 3. Huang MT, Ho CT, Wang ZY, et al. Inhibition of skin tumorigenesis by rosemary and its constituents carnosol and ursolic acid. Cancer Res. 1994;54:701–708. [PubMed] [Google Scholar]
- 4. Cho J, Rho O, Junco J, et al. Effect of combined treatment with ursolic acid and resveratrol on skin tumor promotion by 12‐O‐tetradecanoylphorbol‐13‐Acetate. Cancer Prev Res (Phila). 2015;8:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Junco JJ, Mancha A, Malik G, et al. Resveratrol and P‐glycoprotein inhibitors enhance the anti‐skin cancer effects of ursolic acid. Mol Cancer Res. 2013;11:1521–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ha do T, Tuan DT, Thu NB, et al. Palbinone and triterpenes from Moutan Cortex (Paeonia suffruticosa, Paeoniaceae) stimulate glucose uptake and glycogen synthesis via activation of AMPK in insulin‐resistant human HepG2 Cells. Bioorg Med Chem Lett. 2009;19:5556–5559. [DOI] [PubMed] [Google Scholar]
- 7. Kunkel SD, Elmore CJ, Bongers KS, et al. Ursolic acid increases skeletal muscle and brown fat and decreases diet‐induced obesity, glucose intolerance and fatty liver disease. PLoS ONE. 2012;7:e39332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Son HS, Kwon HY, Sohn EJ, et al. Activation of AMP‐activated protein kinase and phosphorylation of glycogen synthase kinase3 beta mediate ursolic acid induced apoptosis in HepG2 liver cancer cells. Phytother Res. 2013;27:1714–1722. [DOI] [PubMed] [Google Scholar]
- 9. Zheng QY, Jin FS, Yao C, Zhang T, Zhang GH, Ai X. Ursolic acid‐induced AMP‐activated protein kinase (AMPK) activation contributes to growth inhibition and apoptosis in human bladder cancer T24 cells. Biochem Biophys Res Commun. 2012;419:741–747. [DOI] [PubMed] [Google Scholar]
- 10. Birk JB, Wojtaszewski JF. Predominant alpha2/beta2/gamma3 AMPK activation during exercise in human skeletal muscle. J Physiol. 2006;577:1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoene M, Lehmann R, Hennige AM, et al. Acute regulation of metabolic genes and insulin receptor substrates in the liver of mice by one single bout of treadmill exercise. J Physiol. 2009;587:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Canto C, Auwerx J. Calorie restriction: is AMPK a key sensor and effector? Physiology (Bethesda). 2011;26:214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang P, Zhang RY, Song J, et al. Loss of AMP‐activated protein kinase‐alpha2 impairs the insulin‐sensitizing effect of calorie restriction in skeletal muscle. Diabetes. 2012;61:1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salminen A, Hyttinen JM, Kaarniranta K. AMP‐activated protein kinase inhibits NF‐kappaB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl). 2011;89:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5'‐AMP for allosteric stimulation, activation, and deactivation of AMP‐activated protein kinase. J Biol Chem. 2006;281:32207–32216. [DOI] [PubMed] [Google Scholar]
- 16. Viollet B, Lantier L, Devin‐Leclerc J, et al. Targeting the AMPK pathway for the treatment of Type 2 diabetes. Front Biosci. 2009;14:3380–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hardie DG, Ross FA, Hawley SA. AMP‐activated protein kinase: a target for drugs both ancient and modern. Chem Biol. 2012;19:1222–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Faubert B, Boily G, Izreig S, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang X, Wullschleger S, Shpiro N, et al. Important role of the LKB1‐AMPK pathway in suppressing tumorigenesis in PTEN‐deficient mice. Biochem J. 2008;412:211–221. [DOI] [PubMed] [Google Scholar]
- 20. Einspahr JG, Calvert V, Alberts DS, et al. Functional protein pathway activation mapping of the progression of normal skin to squamous cell carcinoma. Cancer Prev Res (Phila). 2012;5:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non‐alcoholic fatty liver disease. J Hepatol. 2015;62:720–733. [DOI] [PubMed] [Google Scholar]
- 22. Thuillier P, Anchiraico GJ, Nickel KP, et al. Activators of peroxisome proliferator‐activated receptor‐alpha partially inhibit mouse skin tumor promotion. Mol Carcinog. 2000;29:134–142. [DOI] [PubMed] [Google Scholar]
- 23. Xiao X, Su G, Brown SN, Chen L, Ren J, Zhao P. Peroxisome proliferator‐activated receptors gamma and alpha agonists stimulate cardiac glucose uptake via activation of AMP‐activated protein kinase. J Nutr Biochem. 2010;21:621–626. [DOI] [PubMed] [Google Scholar]
- 24. Wang Y, Porter WW, Suh N, et al. A synthetic triterpenoid, 2‐cyano‐3,12‐dioxooleana‐1,9‐dien‐28‐oic acid (CDDO), is a ligand for the peroxisome proliferator‐activated receptor gamma. Mol Endocrinol. 2000;14:1550–1556. [DOI] [PubMed] [Google Scholar]
- 25. Place AE, Suh N, Williams CR, et al. The novel synthetic triterpenoid, CDDO‐imidazolide, inhibits inflammatory response and tumor growth in vivo. Clin Cancer Res. 2003;9:2798–2806. [PubMed] [Google Scholar]
- 26. Jia Y, Bhuiyan MJ, Jun HJ, et al. Ursolic acid is a PPAR‐alpha agonist that regulates hepatic lipid metabolism. Bioorg Med Chem Lett. 2011;21:5876–5880. [DOI] [PubMed] [Google Scholar]
- 27. Strehl C, Buttgereit F. Optimized glucocorticoid therapy: teaching old drugs new tricks. Mol Cell Endocrinol. 2013;380:32–40. [DOI] [PubMed] [Google Scholar]
- 28. Slaga TJ, Klein‐Szanto AJ, Fischer SM, Weeks CE, Nelson K, Major S. Studies on mechanism of action of anti‐tumor‐promoting agents: their specificity in two‐stage promotion. Proc Natl Acad Sci USA. 1980;77:2251–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Strawhecker JM, Pelling JC. Inhibition of mouse skin tumorigenesis by dexamethasone occurs through a Ha‐ras‐independent mechanism. Carcinogenesis. 1992;13:2075–2080. [DOI] [PubMed] [Google Scholar]
- 30. Cha HJ, Park MT, Chung HY, et al. Ursolic acid‐induced down‐regulation of MMP‐9 gene is mediated through the nuclear translocation of glucocorticoid receptor in HT1080 human fibrosarcoma cells. Oncogene. 1998;16:771–778. [DOI] [PubMed] [Google Scholar]
- 31. Kassi E, Sourlingas TG, Spiliotaki M, et al. Ursolic acid triggers apoptosis and Bcl‐2 downregulation in MCF‐7 breast cancer cells. Cancer Invest. 2009;27:723–733. [DOI] [PubMed] [Google Scholar]
- 32. Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF‐kappa B and the glucocorticoid receptor. Proc Natl Acad Sci USA. 1994;91:752–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang‐Yen HF, Chambard JC, Sun YL, et al. Transcriptional interference between c‐Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein‐protein interaction. Cell. 1990;62:1205–1215. [DOI] [PubMed] [Google Scholar]
- 34. Chebotaev D, Yemelyanov A, Budunova I. The mechanisms of tumor suppressor effect of glucocorticoid receptor in skin. Mol Carcinog. 2007;46:732–740. [DOI] [PubMed] [Google Scholar]
- 35. Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Conti CJ, Fries JW, Viaje A, Miller DR, Morris R, Slaga TJ. In vivo behavior of murine epidermal cell lines derived from initiated and noninitiated skin. Cancer Res. 1988;48:435–439. [PubMed] [Google Scholar]
- 37. Klann RC, Fitzgerald DJ, Piccoli C, Slaga TJ, Yamasaki H. Gap‐junctional intercellular communication in epidermal cell lines from selected stages of SENCAR mouse skin carcinogenesis. Cancer Res. 1989;49:699–705. [PubMed] [Google Scholar]
- 38. Miller DR, Viaje A, Rotstein J, Aldaz CM, Conti CJ, Slaga TJ. Induction of terminal differentiation‐resistant epidermal cells in mouse skin and in papillomas by different initiators during two‐stage carcinogenesis. Cancer Res. 1989;49:410–414. [PubMed] [Google Scholar]
- 39. Honer C, Nam K, Fink C, et al. Glucocorticoid receptor antagonism by cyproterone acetate and RU486. Mol Pharmacol. 2003;63:1012–1020. [DOI] [PubMed] [Google Scholar]
- 40. Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta. 2007;1771:952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee SK, Lee JO, Kim JH, et al. Coenzyme Q10 increases the fatty acid oxidation through AMPK‐mediated PPARalpha induction in 3T3‐L1 preadipocytes. Cell Signal. 2012;24:2329–2336. [DOI] [PubMed] [Google Scholar]
- 42. Okayasu T, Tomizawa A, Suzuki K, Manaka K, Hattori Y. PPARalpha activators upregulate eNOS activity and inhibit cytokine‐induced NF‐kappaB activation through AMP‐activated protein kinase activation. Life Sci. 2008;82:884–891. [DOI] [PubMed] [Google Scholar]
- 43. Pavek P, Cerveny L, Svecova L, et al. Examination of Glucocorticoid receptor alpha‐mediated transcriptional regulation of P‐glycoprotein, CYP3A4, and CYP2C9 genes in placental trophoblast cell lines. Placenta. 2007;28:1004–1011. [DOI] [PubMed] [Google Scholar]
- 44. Narang VS, Fraga C, Kumar N, et al. Dexamethasone increases expression and activity of multidrug resistance transporters at the rat blood‐brain barrier. Am J Physiol Cell Physiol. 2008;295:C440–C450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marquez B, Van Bambeke F. ABC multidrug transporters: target for modulation of drug pharmacokinetics and drug‐drug interactions. Curr Drug Targets. 2011;12:600–620. [DOI] [PubMed] [Google Scholar]
- 46. Jurado R, Lopez‐Flores A, Alvarez A, Garcia‐Lopez P. Cisplatin cytotoxicity is increased by mifepristone in cervical carcinoma: an in vitro and in vivo study. Oncol Rep. 2009;22:1237–1245. [DOI] [PubMed] [Google Scholar]
- 47. Cho J, Tremmel L, Rho O, et al. Evaluation of pentacyclic triterpenes found in Perilla frutescens for inhibition of skin tumor promotion by 12‐O‐tetradecanoylphorbol‐13‐acetate. Oncotarget. 2015;6:39292–39306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee JM, Wagner M, Xiao R, et al. Nutrient‐sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]