

Abstract
Sialylated glycan structures are known for their immunomodulatory capacities and their contribution to tumor immune evasion. However, the role of aberrant sialylation in colorectal cancer and the consequences of complete tumor desialylation on anti‐tumor immunity remain unstudied. Here, we report that CRISPR/Cas9‐mediated knock out of the CMAS gene, encoding a key enzyme in the sialylation pathway, in the mouse colorectal cancer MC38 cell line completely abrogated cell surface expression of sialic acids (MC38‐Sianull) and, unexpectedly, significantly increased in vivo tumor growth compared to the control MC38‐MOCK cells. This enhanced tumor growth of MC38‐Sianull cells could be attributed to decreased CD8+ T cell frequencies in the tumor microenvironment only, as immune cell frequencies in tumor‐draining lymph nodes remained unaffected. In addition, MC38‐Sianull cells were able to induce CD8+ T cell apoptosis in an antigen‐independent manner. Moreover, low CMAS gene expression correlated with reduced recurrence‐free survival in a human colorectal cancer cohort, supporting the clinical relevance of our work. Together, these results demonstrate for the first time a detrimental effect of complete tumor desialylation on colorectal cancer tumor growth, which greatly impacts the design of novel cancer therapeutics aimed at altering the tumor glycosylation profile.
Keywords: glycosylation, colorectal cancer, sialic acid, CD8+ T cell, apoptosis
Short abstract
What's new?
The current dogma that tumor cells express sialic acids to dampen anti‐tumor immunity has led to the development of novel therapeutic strategies aimed at dismantling sialic acid‐induced tolerance. Yet the effect of a complete loss of tumor sialylation remains to be elucidated. This study is the first to report a detrimental effect of complete tumor desialylation on colorectal cancer tumor growth, which could be attributed to augmented CD8+ T cell apoptosis. The work revisits how tumor‐associated sialic acids influence the anti‐tumor immune response and has implications for the design of novel cancer therapeutics aimed at altering the tumor glycosylation profile.
Abbreviations
- CRC
colorectal cancer
- MAL‐II
Maackia amurensis Lectin II
- MC38‐MOCK
transfection control MC38 cell line
- MC38‐Sianull
CMAS gene knock out MC38 cell line
- MC38‐WT
wild type MC38 cell line
- Neu5Ac
N‐acetylneuraminic acid
- NK
natural killer cell
- NSG
NOD.Cg‐Prkdc scid Il2rg rm1Wjl /SzJ
- OVA
ovalbumin
- Siglecs
Sialic acid‐binding immunoglobulin‐type lectins
- SNA
Sambucus nigra agglutinin
- ST6Gal1
β‐galactoside α2‐6 sialyltransferase 1
- TME
tumor microenvironment
Introduction
A major focus of current cancer research is the elucidation of immune evasion strategies employed by malignant cells within the tumor microenvironment (TME). The TME contains a wide variety of cells, including stromal, endothelial and immune cell subsets. The type, density and localization of immune cells within the TME are defined as the Immunoscore, which can be used to predict clinical outcome.1, 2 As such, cytotoxic CD8+ T cells are capable of eliminating tumor cells and are thus strongly associated with an improved prognosis in many cancer types.3 Nevertheless, as cancers develop, tumor cells acquire the capacity to escape CD8+ T cell‐mediated cytotoxicity through down‐regulation of major histocompatibility complex class I (MHC‐I) molecules and expression of immune checkpoint molecules. The transition of immune surveillance to immune escape has been described as the cancer immunoediting hypothesis.4 Although cancer immunoediting has been widely studied, the contribution of cancer‐specific glycan structures on immune evasion still remains undefined.
Glycosylation is an enzymatic post‐translational process that mediates the attachment of carbohydrate structures to proteins and lipids and functions in a wide range of biological processes including protein folding, cell adhesion and cell signaling.5 Compared to their non‐malignant counterparts, tumor cells generally harbor an aberrant glycosylation profile of N‐glycans, O‐glycans and glycolipids structures that are all known to influence cancer development.6, 7 The cancer‐related glycosylation profile is often characterized by an enhanced expression level of sialylated glycan structures.8 Sialic acids (Sias) are a family of nine‐carbon monosaccharides with N‐acetylneuraminic acid (Neu5Ac) as the most common member. Sialylated glycan structures can be recognized by Sialic acid‐binding immunoglobulin‐type lectins (Siglecs), which are mainly expressed on granulocytes, monocytes, antigen presenting cells and natural killer (NK) cells.9 It is generally accepted that Sias are able to skew immune responses toward immune suppression, since most Siglec receptors contain an immunoreceptor tyrosine‐based inhibitory motif (ITIM) in their cytoplasmic domain. As Sias, although in lower levels, are also expressed on non‐malignant cells, the current dogma states that Sias act as a shield to prevent inappropriate immune reactions against self‐antigens. This dampening of immunity by Sias, therefore, led to the concept that glycans represent a self‐associated molecular pattern (SAMP) recognized by self‐pattern recognition receptors.10
Indeed, numerous studies have evaluated the negative immunoregulatory function of Sias. Targeting of mouse dendritic cells with Sias conjugated to ovalbumin (OVA) augments the differentiation of regulatory T cells both in vitro and in vivo.11 Also, Siglec‐7 present on NK cells inhibits NK cell cytotoxic activity when stimulated with natural sialylated ligands.12, 13 Since tumor cells express enhanced levels of Sias, these cells might exploit the sialylation machinery to suppress anti‐tumor immune responses. Reduced tumor growth as well as enhanced effector T cell responses and diminished regulatory T cell frequencies were found in a mouse model of B16 melanoma wherein the tumor harbored decreased levels of sialylation.14, 15, 16 Moreover, a subset of tumor‐infiltrating T cells upregulates Siglec‐9, which dampens anti‐cancer immunity, thus facilitating immune escape by the tumor.17
The involvement of Sias in suppression of subsequent immune responses, prompted new therapeutic strategies aimed at reducing tumor‐associated Sia expression to enhance anti‐tumor immunity. Xiao et al. developed an anti‐HER2 antibody and sialidase conjugate that simultaneously cleaves Sias from the cell surface and targets tumor cells for antibody‐dependent cell‐mediated cytotoxicity by NK cells.17, 18 More recently, intracellular Sia blockade was shown to be sufficient to enhance CD8+ T cytotoxicity, resulting in reduced tumor burdens.16 While these new therapeutic approaches lower the Sia expression of tumors, the effect of complete tumor desialylation on the anti‐tumor immune response has never been investigated. Moreover, glycosylation patterns differ substantially between and within tumor types,19 urging the need to increase our understanding on how tumor‐associated glycan structures influence the immune system in tumors of different origins.
With our study we are the first to explore the effect of complete tumor desialylation in colorectal cancer (CRC). The adult gastrointestinal tract is a heavily glycosylated area with distinct regional differences, whereby sialic acid levels are higher in the colon compared to the ileum.20, 21, 22 Also, recurrence of CRC tumors is correlated with a distinct glycosylation profile, illustrating the relevance of tumor glycosylation in CRC specifically.23 We assessed tumor growth and the anti‐tumor immune response in vivo using CRISPR/Cas9 glyco‐engineered MC38 CRC cells. Unexpectedly, desialylated MC38 (MC38‐Sianull) cells grew significantly faster in vivo, which we attribute to decreased CD8+ frequencies present within the tumor microenvironment. In addition, we found that MC38‐Sianull cells were capable of inducing CD8+ T cell apoptosis, likely explaining the reduced CD8+ T cells frequencies in vivo. Collectively, these results strongly argue that complete tumor desialylation may have detrimental effects on anti‐tumor immunity, thus greatly impacting the design of novel cancer therapeutics aimed at targeting the tumor sialylation profile.
Materials and Methods
CRISPR/Cas9 constructs
CRISPR/Cas9 constructs were made as previously described.24 The following gRNA strands for murine CMAS were used: CMAS #1: top strand CACCGAATGTGGCCAAACAGTT; bottom strand CTTACACCGGTTTGTCAACAAA; and CMAS #2: top strand CACCGTTTCAGAACTTCTTCGA; bottom strand: CAAAGTCTTGAAGAAGCTCAAA. The gRNA strands were phosphorylated and annealed prior to cloning in the pSpCas9(BB)‐2A‐Puro plasmid, a gift from Feng Zhang (Addgene #62988). Cloning mixtures were treated with PlasmidSafe exonuclease (Epicentre) to digest residual linearized DNA and used for transformation in XL1‐Blue Sublconing‐Grade competent bacteria (Stratagene). The Nucleobond Xtra Midi kit (Macherey‐Nagel) was used to purify the plasmids. Purified constructs were stored at −20 °C until further use.
Generation of the MC38‐Sianull cell line
MC38 cells were cultured in DMEM supplemented with 10% heat inactivated fetal calf serum (FCS, Biowest), 1% penicillin and 1% streptomycin. Cells (70–90% confluency) were transfected with CRISPR/Cas9 constructs (3 μg per 6‐well) targeting the CMAS gene (MC38‐Sianull) or an empty CRISPR/Cas9 construct (MC38‐MOCK). Lipofectamine LTX with PLUS™ reagent (ThermoFisher Scientific) was used for transfection and applied according to the manufacturer's protocol. After 24 h, fresh medium containing 3 μg/mL puromycin was added for selection.
Transfected MC38‐Sianull cells were incubated with 5 μg/mL biotinylated Sia‐specific Maackia amurensis Lectin II (MAL‐II, Vector Laboratories) for 1 h on ice, washed with medium and incubated with streptavidin‐PE (Jackson ImmunoResearch) on ice. After 1 h cells were washed and sorted in bulk on MAL‐IIlow cells.
To obtain supernatant samples, MC38‐MOCK or MC38‐Sianull cells were cultured overnight in 96‐wells plates at a cell density of 3 × 104 cells/200 μL per well. For heat inactivation, supernatant samples were incubated at 65 °C for 5 min.
Surveyor assay
Genomic DNA was isolated with the Quick‐DNA™ kit (Zymo research) according to the manufacturer's protocol. PCR products (732 bp) for the CMAS gene (forward primer: GGCCTGGGATTCAGGAACAT, reverse primer: TGCAGCTGTACCCCAAGCA) were hybridized and treated with Surveyor Nuclease (Surveyor® Mutation Detection Kit, Integrated DNA Technologies) and loaded on a DNA agarose gel to visualize presence or absence of the mutation.
Lectin flow cytometry
The biotinylated plant lectins Sambucus nigra agglutinin (SNA, Vector Laboratories) and Maackia amurensis Lectin II (MAL‐II, Vector Laboratories) were used to detect α2‐6 and α2‐3 Sias, respectively. MC38 cells were incubated with 5 μg/mL of the lectin for 30 min at 37 °C. Cells were washed and incubated with streptavidin‐APC (BD Biosciences) for 30 min at 37 °C. Cells were washed and acquired on the Beckman Coulter Cyan flow cytometer. Data was analyzed with FlowJo v10.
N‐glycan profiling using liquid chromatography mass‐spectrometry (LC–MS)
MC38‐MOCK and MC38‐Sianull cells were washed three times with PBS and resuspended in 100 μL pure water, before homogenization for 45 min in a sonication bath. Samples were centrifuged (500g, 15 min) and 17.5 μL of pure water was added. Five microliters of reaction buffer (250 mM sodium phosphate buffer; pH 7.5) and 1.25 μL of denaturation buffer (2% SDS in 1 M β‐mercaptoethanol) were added. Samples were incubated for 10 min at 100 °C to denature the proteins, afterwards 1.25 μL of Triton X‐100 and 500 units of PNGase F (QABio, California, USA) were added. Samples were vortexed and incubated overnight at 37 °C. The released glycans were then converted to aldoses with 0.1% formic acid, filtered through a protein‐binding membrane and dried.
Released N‐glycans were fluorescently labeled by reductive amination with procainamide as described previously25 using the LudgerTagTM Procainamide Glycan Labelling Kit (LT‐KPROC‐24). Procainamide labeled samples were analyzed by HILIC‐(U)HPLC‐ESI‐MS with fluorescence detection. Samples were injected in 24% pure water/76% acetonitrile onto an ACQUITY UPLC® BEH‐Glycan 1.7 μm, 2.1 × 150 mm column at 40 °C on a Ultimate 3,000 UHPLC instrument (Thermo Scientific, Massachusetts, USA) with a fluoresence detector (λ ex = 310 nm, λ em = 370 nm). A glucose homopolymer ladder labeled with procainamide (Ludger) was used as a standard. ESI‐MS and MS/MS data analysis was performed using Bruker Compass DataAnalysis V4.1 software and GlycoWorkbench software.26
CellTiter‐blue® cell viability assay
The metabolic activity of MC38 cells was analyzed using the CellTiter‐Blue® Cell Viability assay (Promega) and measured on the FLUOstar Galaxy (MTX Lab systems, excitation 560 nm, emission 590 nm).
In vivo tumor experiments
C57BL/6, NOD.Cg‐Prkdc scid Il2rg rm1Wjl /SzJ (NSG, Charles River) and OT‐I mice were used at 8–12 weeks of age and bred at the animal facilities of Amsterdam UMC. For all in vivo experiments, an equal distribution of female and male mice was used. Experiments were performed in accordance with national and international guidelines and regulations.
Tumor cells (2 × 105 per mouse) were injected subcutaneously in the flanks. Tumors were measured three times per week in a double‐blind manner. Total tumor volume was calculated using the formula 4/3 × π × abc (a = width of the tumor/2, b = length/2 and c = the average of a and b). Mice were sacrificed at day 13 after inoculation or when the tumor reached a size of 2000 mm3. Tumor and tumor‐draining lymph nodes were isolated and used for further experiments.
Tumor and lymph node dissociation
Tumors were finely minced and enzymatically digested in RPMI containing 1 mg/mL Collagenase type 4 (Worthington), 30 units/mL DNase I type II (Sigma‐Aldrich) and 100 μg/mL hyaluronidase type V (Sigma‐Aldrich) for 25 min at 37 °C. Lymph nodes were digested with 2 U/mL Liberase TM (Roche) enriched with 30 units/mL DNase I type II for 10 min at 37 °C. All cell suspensions were passed through a 70 μm cell strainer and washed with RPMI supplemented with 10% FCS, 1% penicillin, 1% streptomycin and 1% glutamax. Lymph node digestions were washed twice and directly used for subsequent experiments. Tumor digestion mixtures were loaded on a Ficoll gradient to enrich for lymphocytes and to remove dead cell debris.
Immune cell profiling
Cells were pretreated with 2.4G2 (anti‐CD16/32) for 10 min at RT and cell viability was measured using a fixable viability dye (Zombie NIR, Biolegend), 2 μg/mL DAPI or 7‐AAD (Invitrogen). Cells were stained for 20 min at RT using the following markers: anti‐CD45‐PerCp (30‐F11), anti‐CD31‐FITC (390), anti‐CD3‐BV510 (17A2), anti‐CD4‐A700 (GK1.5), anti‐CD8b‐FITC (YTS156.7.7), anti‐PD‐1‐BV785 (29F.1A12), anti‐NK.1.1‐PE/Dazzle 594 (PK136), anti‐NKp46‐BV421 (29A1.4) (all from Biolegend) and anti‐CD8b‐APC (eBioH35‐17.2, eBioscience). To stain for Foxp3, cells were fixed and permeabilized according to the manufacturer's protocol (Foxp3 Transcription Factor Staining buffer set, eBioscience) and incubated with anti‐Foxp3‐PE antibodies (150D, Biolegend) for 20 min at RT. For tetramer staining, fluorophore‐conjugated MHC‐I peptide complexes were added to the cells prior to cell surface staining. The Dpagt1‐H‐2Kb, Reps1‐H‐2Db and Adpgk‐H‐2Db tetramers were obtained through the NIH Tetramer Core Facility (Emory University, Atlanta) and labeled with PE, APC and BV421, respectively. Samples were acquired on BD LSRFortessa™ X‐20. Data was analyzed with FlowJo v10.
Generation of activated OT‐I CD8+ T cells
To obtain activated CD8+ OT‐I T cells, an OT‐I spleen was minced and meshed through a 70 μm cell strainer. Cells were cultured in RPMI supplemented with 10% heat inactivated FCS, 1% penicillin, 1% streptomycin, 1% glutamax and 50 μM 2‐Mercaptoethanol at a concentration of 3.5 × 106 splenocytes/mL in presence of 0.75 μg/mL OVA257‐264 peptide. After 3 days, 2 mL medium containing 10 U/mL rmIL‐2 was added. At day 5 of culture, CD8+ T cells were purified using Lympholyte separation medium (Cedarlane) and checked for purity by staining for CD8b (>90%).
T cell cytotoxicity assay
MC38‐MOCK and MC38‐Sianull cells were labeled with CFSE (Thermofisher) and CellVue® Claret (Sigma) respectively, according to the manufacturer's protocol. After labeling, cells were pulsed with the OVA257‐264 peptide for 2 h at 37 °C. MC38‐MOCK and MC38‐Sianull cells were plated with or without activated OT‐I CD8+ T cells and after 5 h of incubation, cells were stained with anti‐CD45 and DAPI and acquired on BD LSRFortessa™ X‐20. Data was analyzed with FlowJo v10. Tumor cell viability was calculated by dividing the number of viable MC38 cells incubated with CD8+ T cells by the number of viable MC38 cells cultured without CD8+ T cells x 100%.
CD8+ T cell apoptosis assays
Fresh C57BL/6 splenocytes or activated OT‐I CD8+ T cells were co‐cultured with MC38‐MOCK or MC38‐Sianull cells or supernatants as indicated. PHA‐L (Vector Laboratories) (2 μg/mL) was added to the C57BL/6 splenoctyes for activation. Cells were stained with anti‐CD8b and 7‐AAD and subsequently with Annexin V‐FITC (in Annexin V buffer, BD Bioscience). Non‐apoptotic CD8+ T cells were measured on the BD LSRFortessa™ X‐20 and analyzed with FlowJo v10.
Colorectal cancer cohort
Dataset GSE39582 was obtained from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/gds).27 The median of CMAS gene expression from 566 CRC patients was ranked from low to high and divided into CMAS high (median gene expression >300) and CMAS low (<150) groups. Both groups were used to analyze the recurrence‐free survival.
Statistical analysis
Statistical significance was tested using GraphPad Prism by performing an unpaired nonparametric t‐test. Recurrence free survival was displayed in Kaplan–Meier curves and statistical significance was calculated by Log‐rank Mantel‐Cox test using GraphPad Prism. (*p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant).
Results
Lack of cell surface Sia expression after CMAS gene knockout
We first analyzed the mouse colorectal cancer cell line (MC38 wild type, MC38‐WT) for the presence of sialic acids (Sias). Sias, linked in an α2‐6 or α2‐3 configuration to the underlying galactose residue, can be visualized using the plant lectins Sambucus nigra agglutinin (SNA) and Maackia amurensis lectin II (MAL‐II), respectively. MC38‐WT cells displayed high binding of MAL‐II, while SNA binding was negative (Fig. 1 a), indicating that MC38‐WT cells only expressed α2‐3 linked Sias. In order to reduce Sia expression on the tumor cell surface, we previously targeted the SLC35A1 Sia transporter using shRNA, which resulted in decreased, but still detectable levels of Sia15 (see Fig. 1 b for an overview of the sialylation pathway). However, SLC35A1 was recently shown to be involved in the O‐mannosylation pathway as well.28 Hence, for the present study we mutated the N‐acylneuraminate cytidylyltransferase (CMAS) gene, which catalyzes the activation of Neu5Ac to 5′‐monophosphate N‐acetylneuraminic acid (CMP‐Neu5Ac), the substrate required for the addition of Sia to the growing glycan chain (Fig. 1 b). We designed two different CRISPR/Cas9 guideRNAs, targeting distinct regions in the CMAS gene. Independent of the guideRNA used, both CMAS knockouts (MC38‐Sianull) displayed a complete lack of cell surface Sia (Fig. 1 c). The CMAS gene mutation was confirmed using the Surveyor assay (Supporting Information Fig. 1A) and by mass spectrometric analysis (Fig. 1 d and Supporting Information Table S1). We could only detect one minor sialylated glycan structure on MC38‐Sianull cells, which we assume is a contamination from the MC38 culture medium.
Figure 1.
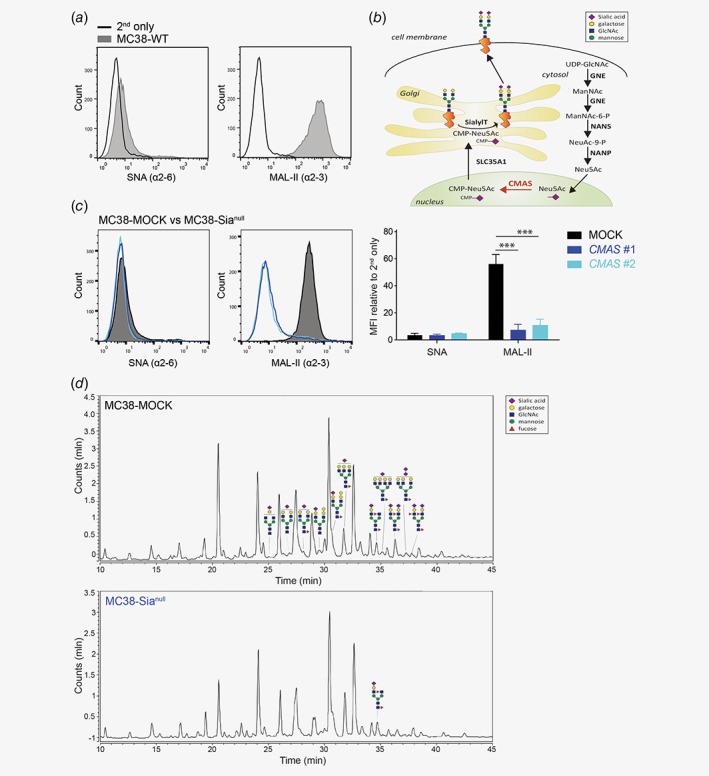
Cell surface sialic acid expression is abrogated in the CMAS gene knockout. (a) The presence of α2‐6 and α2‐3 Sia in MC38‐WT cells was assessed by flow cytometry using the plant lectins SNA and MAL‐II, respectively. (b) Schematic representation of the sialylation pathway. (c) Two CRISPR/Cas9 guideRNAs targeting distinct regions in the CMAS gene both completely abrogate MC38 sialylation (MC38‐Sianull) as measured with plant lectins SNA and MAL‐II. MOCK‐transfected MC38 cells (MC38‐MOCK) were used as a negative control. Relative mean fluorescence intensities (MFI) were calculated relative to MFI of the 2nd antibody only. Data are representative of three independent experiments (a and c). Mean ± SD; ***, p < 0.001. (d) Liquid Chromatography‐Mass spectrometry (LC–MS) analysis of MC38‐MOCK (upper) and MC38‐Sianull (bottom) sialylation. Only representative examples of the identified sialylated glycans structures are depicted. [Color figure can be viewed at wileyonlinelibrary.com]
Manipulation of the glycosylation machinery may influence the proliferation capacity of cells. Nevertheless, MC38‐MOCK and MC38‐Sianull cells displayed identical growth rates under in vitro culture conditions as observed by manual counting and by measuring their metabolic activity (Supporting Information Fig. 1B and C). Thus, we obtained a completely desialyated non‐clonal MC38 cell line through CRISPR/Cas9 genome engineering of the CMAS gene.
MC38‐Sianull cells display enhanced tumor growth in vivo
Next, we investigated the effect of complete tumor desialylation on in vivo tumor growth by injecting MC38‐MOCK and MC38‐Sianull cells into C57Bl/6 mice (Fig. 2 a). Unexpectedly, the MC38‐Sianull tumors grew significantly faster than MC38‐MOCK tumors (Figs. 2 b and 2 c). The tumor mass, weighed directly after isolation, correlated to tumor size (Supporting Information Fig. 2A), confirming the tumor measurements were accurately performed. Moreover, the MC38‐Sianull cells retained their Sia negative phenotype after in vivo passage (Supporting Information Fig. 2B).
Figure 2.
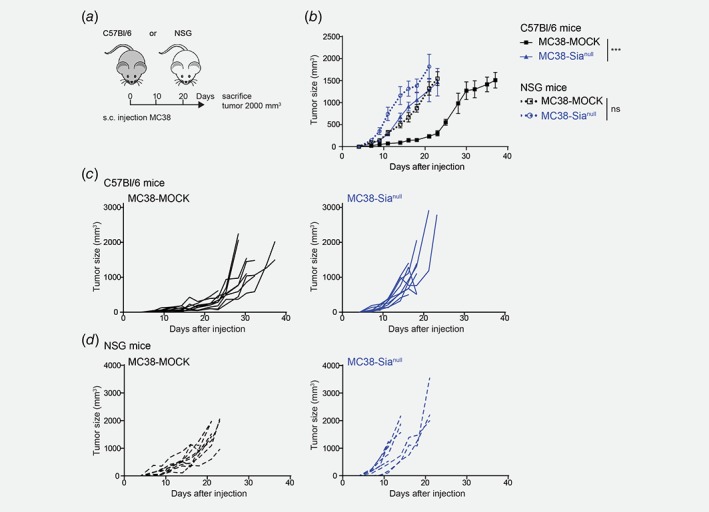
MC38‐Sianull tumors display enhanced tumor growth in vivo. (a) MC38‐MOCK or MC38‐Sianull cells were injected into C57Bl/6 mice (n = 14) and immunodeficient NOD.Cg‐Prkdc scid Il2rg rm1Wjl /SzJ (NSG) mice (n = 9). (b–d) Tumor growth was monitored and depicted as mean tumor size (b) or individual tumor growth curves (c and d). Data are representative of one (d) or three (c) independent experiments. Mean ± SEM; ***, p < 0.001; ns, not significant. [Color figure can be viewed at wileyonlinelibrary.com]
To ascertain that the differences observed in tumor growth were due to alterations in anti‐tumor immunity, we inoculated the MC38‐MOCK and MC38‐Sianull cells into immunodeficient mice (NOD.Cg‐Prkdc scid Il2rg rm1Wjl /SzJ, NSG mice). Compared to immunocompetent mice, MC38‐MOCK tumors grew faster in immunodeficient mice, providing evidence for the existence of immune surveillance toward the MC38‐MOCK tumors (Figs. 2 b and 2 d). In contrast, the MC38‐Sianull only displayed a small increase in tumor growth in the immunodeficient mice compared to the immunocompetent mice (Figs. 2 b and 2 d), suggesting in the case of MC38‐Sianull, the immune system is less able to recognize or can less efficiently combat MC38‐Sianull tumor outgrowth. Together, our data imply that complete CRC desialylation actually augments tumor growth, which might be attributed to a lack of activation or a heightened suppression of anti‐tumor immunity.
MC38‐Sianull tumors harbor less CD8+ T cells
To explore whether tumor desialylation influences anti‐tumor immunity, we analyzed the immune cell composition of the tumor and tumor‐draining lymph nodes at day 13 of tumor development (Fig. 3 a). Compared to MC38‐MOCK, the MC38‐Sianull tumors harbored less cytotoxic CD8+ T cells and NK cells, while CD4+ T cells tended to be lower in MC38‐Sianull tumors (Fig. 3 b). Strikingly, frequencies of Foxp3+CD4+ regulatory T cells were equal in MC38‐MOCK and MC38‐Sianull tumors (Fig. 3 b). Moreover, the reduced levels of CD8+ T cells and NK cells were only observed locally at the tumor site, as the tumor‐draining lymph nodes contained equal frequencies of all subsets investigated (Supporting Information Fig. 3A).
Figure 3.
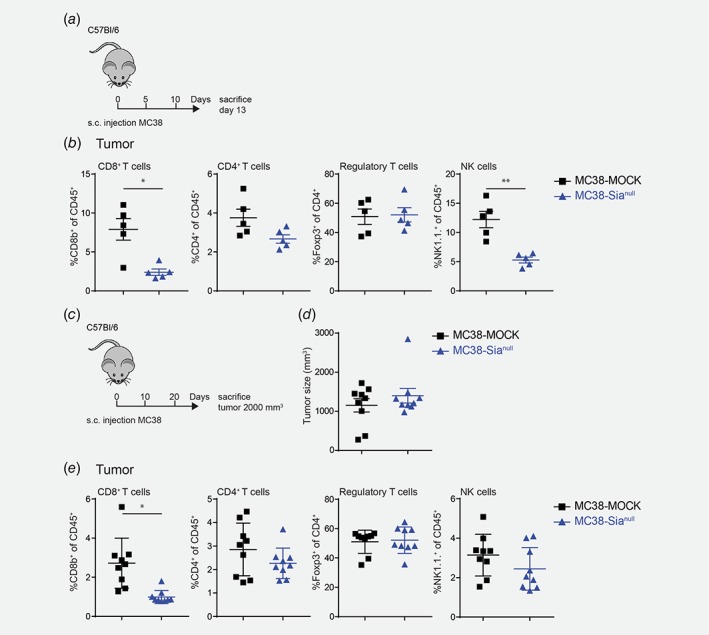
MC38‐Sianull tumors harbor less CD8+ T cells. MC38‐MOCK or MC38‐Sianull cells were injected in C57Bl/6 mice and sacrificed at day 13 of tumor development (a,b, n = 5) or when the tumor reached a size of 2000 mm3 (c–e, n = 9). (b) and (e) Flow cytometric analysis of viable, CD45+ CD8+ T cells (CD3+CD8b+), CD4+ T cells (CD3+CD4+), Regulatory T cells (CD3+CD4+Foxp3+) and NK cells (CD3−NK1.1+) at tumor site after 13 days of tumor development (b) or when the tumor reached a size of 2000 mm3 (e). Data are representative of two independent experiments (a–e). Mean ± SEM; *, p < 0.05; **, p < 0.01. [Color figure can be viewed at wileyonlinelibrary.com]
As MC38 tumors develop, CD8+ T cell frequencies seem to drop.29 Thus, the decreased CD8+ T cell infiltration in MC38‐Sianull tumors might be explained by tumor size, which at day 13 is significantly larger for MC38‐Sianull, and not by the desialylated phenotype (Supporting Information Fig. 4A). Therefore, we also checked the immune composition when tumors had an identical size of 2000 mm3 (Figs. 3 c and 3 d). Interestingly, the reduced CD8+ T cell frequencies in the MC38‐Sianull were maintained in equally sized tumors (Fig. 3 e). In contrast to day 13 of tumor development, tumor‐infiltrated NK cell frequencies were similar in both MC38‐MOCK and MC38‐Sianull tumors (Fig. 3 e). Again, the levels of Foxp3+CD4+ regulatory T cells in the tumor (Fig. 3 e) and the levels of CD8+, CD4+, regulatory T and NK cells in the tumor‐draining lymph nodes remained unaffected (Supporting Information Fig. 3B).
Remarkably, also lower frequencies of PD‐1+CD8+ T cells were found in the MC38‐Sianull tumors compared to the MC38‐MOCK tumors, both at day 13 of tumor development (Fig. 4 a) as well as when the tumors had an equal size (Fig. 4 b). This effect was solely observed at the tumor site and not in the tumor draining lymph nodes (Figs. 4 a and 4 b). PD‐1 is well‐known for its crucial role as a checkpoint molecule in down‐regulating immune responses.30 Nevertheless, PD‐1 expression is primarily induced upon TCR stimulation and lost when T cells fail to receive sustained TCR signaling.31 Therefore, we checked whether the MC38‐Sianull tumors also contained less antigen‐experienced tumor‐specific CD8+ T cells. Yadav et al.29 identified three MC38‐specific neoantigens with a strong immunogenic potential. We tested the CD8+ T cell reactivity toward these neoantigens by generating tetramers specific for these three identified neoantigens. Isolated CD8+ T cells obtained from MC38‐MOCK tumors indeed displayed higher reactivity toward two of the neoantigens, while the response to the third antigen showed a trend toward higher recognition (Fig. 4 c). The specificity of CD8+ T cells in the tumor draining lymph node was indistinguishable between MC38‐MOCK and MC38‐Sianull groups (Supporting Information Fig. 4B). Together, these results indicate that the MC38‐MOCK tumors were infiltrated with more tumor‐specific CD8+ T cells than MC38‐Sianull tumors, thus uncovering a predominant effect of tumor desialylation on the CD8+ T cell compartment exclusively in the tumor microenvironment.
Figure 4.
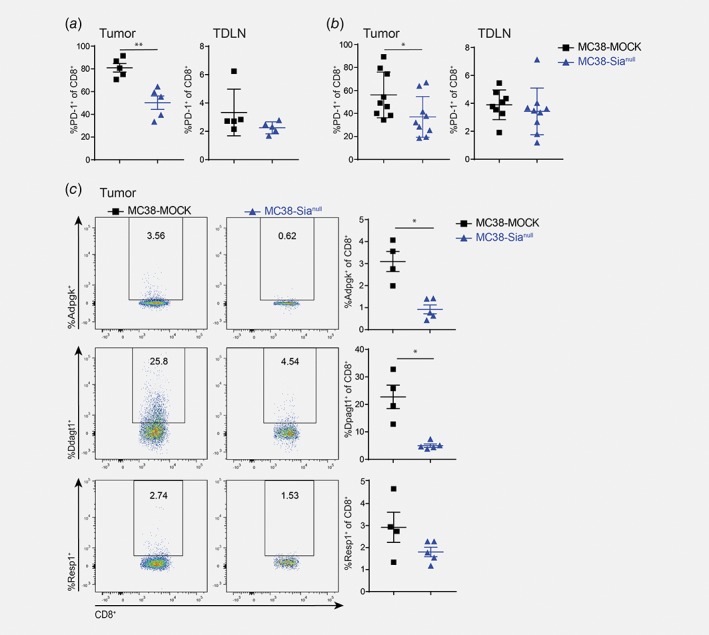
Less PD‐1 positive and MC38 neoantigen‐specifc CD8+ T cells at the MC38‐Sianull tumor site. (a) and (b) Percentage of PD‐1+CD8+ T cells was analyzed at the tumor location or in tumor‐draining lymph nodes with flow cytometry at an early stage of tumor development (a) or when the tumors reached an equal size (b). (c) Tumor infiltrated CD8+ T cells (viable CD45+CD3+CD8b+) were stained with tetramers specific for three different MC38 neoantigens29 and analyzed by flow cytometry. Data are representative of two independent experiments. Mean ± SEM; *, p < 0.05; **, p < 0.01. [Color figure can be viewed at wileyonlinelibrary.com]
MC38‐Sianull tumors induce CD8+ T cell apoptosis
As the presence of CD8+ T cells in the tumor microenvironment correlates with good prognosis,3 we hypothesized that the enhanced growth of the MC38‐Sianull tumors was due to hampered elimination by cytotoxic CD8+ T cells. Moreover, this appeared to be a local effect, as tumor‐specific CD8+ T cell frequencies were unaffected in tumor‐draining lymph nodes. To explore whether MC38‐Sianull cells might be able to suppress the cytotoxic function of CD8+ T‐cells directly, we performed a cytotoxicity assay using OT‐I T cells and OVA‐loaded tumor cells as a model system. Activated OT‐I CD8+ T cells were co‐cultured with OVA257‐264 (SIINFEKL)‐pulsed MC38‐MOCK and MC38‐Sianull cells and assessed for tumor cell eradication. To track the MC38 cells, MC38‐MOCK cells were labeled with CFSE and MC38‐Sianull with CellVue© Claret (Fig. 5 a). After 5 h, OVA257‐264‐pulsed MC38‐Sianull cells were less efficiently killed by the OT‐I CD8+ T cells than the MC38‐MOCK cells (Fig. 5 b), suggesting that upon encounter of desialylated tumor cells, CD8+ T cells are functionally inhibited in their cytotoxic response.
Figure 5.
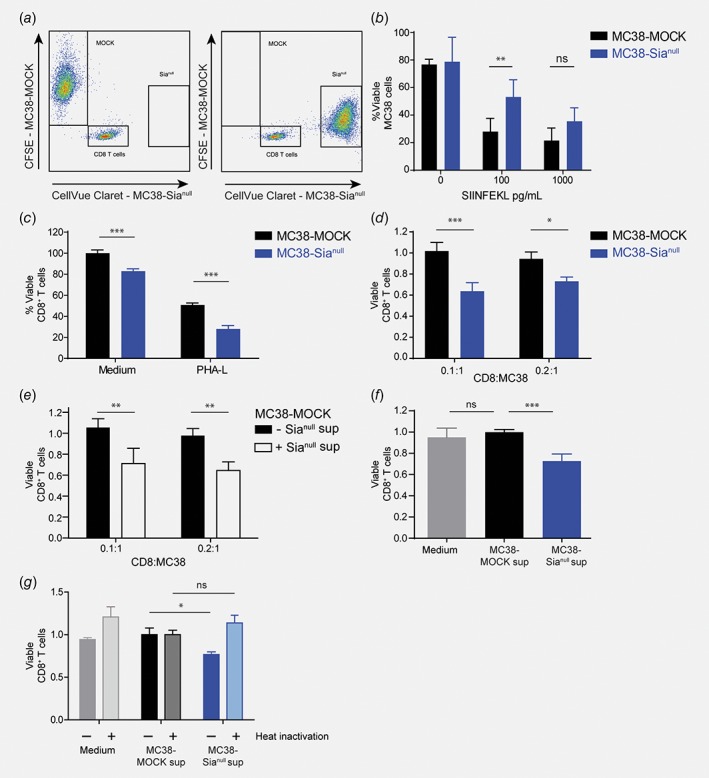
MC38‐Sianull cells induce CD8+ T cell apoptosis. (a) and (b) MC38‐MOCK and MC38‐Sianull cells were pulsed with OVA257‐264 and labeled with CFSE or Cellvue Claret respectively (a). After 5 h of incubation with activated OT‐I CD8+ T cells, viability of MC38 cells was analyzed by flow cytometry and calculated by dividing the number of viable (7‐AAD−Annexin V−) MC38 cells present in the CD8+ T cell co‐culture by the viable MC38 cells cultured without T cells and multiplied by 100% (b). (c–f), Viable CD8+ T cells were analyzed with flow cytometry and gated as CD8b+7‐AAD−Annexin V−. (c) C, C57Bl/6 splenocytes were co‐cultured with MC38‐MOCK or MC38‐Sianull cells in presence of medium or PHA‐L for 48 h. (d) Activated OT‐I CD8+ T cells were co‐cultured with MC38‐MOCK or MC38‐Sianull cells for 24 h. (e) Activated OT‐I CD8+ T cells were co‐cultured with MC38‐MOCK in presence of medium or MC38‐Sianull supernatant for 24 h. (f) and (g) Activated OT‐I CD8+ T cells were cultured with control (f) or heat inactivated (g) medium, MC38‐MOCK or MC38‐Sianull supernatants for 24 h. Data are representative of one (g), two (c, e and f) or three (b and d) independent experiments. Bar, mean ± SD; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. [Color figure can be viewed at wileyonlinelibrary.com]
We next investigated whether MC38‐Sianull cells are able to influence CD8+ T cell function by provoking CD8+ T cell death. Interestingly, fewer viable (7‐AAD−Annexin V−) CD8+ T cells could be recovered from 48 h co‐cultures of wild type C57Bl/6 splenocytes with MC38‐Sianull cells, than from splenocytes and MC38‐MOCK co‐cultures (Fig. 5 c). This effect seemed antigen‐independent, as it occurred both under medium conditions or when T cells were further stimulated with PHA‐L (Fig. 5 c). To mimic activated tumor‐infiltrating CD8+ T cells, we employed antigen‐experienced OT‐I T cells and investigated whether MC38‐Sianull also induced apoptosis of these CD8+ T cells without accompanying antigen‐specific stimulation. Indeed, at low T cell‐tumor ratios, MC38‐Sianull cells induced 40% more CD8+ T cell apoptosis than MC38‐MOCK cells (Fig. 5 d).
We next assessed whether the MC38‐Sianull‐induced apoptosis was mediated by cell‐intrinsic factors or via the secretion of a soluble factor. Therefore, activated OT‐I T cells were co‐cultured with MC38‐MOCK cells in the presence or absence of supernatants derived from MC38‐Sianull cells. Indeed, in the presence of MC38‐Sianull supernatant more CD8+ T cell apoptosis was measured, indicating that MC38‐Sianull cells secrete a soluble pro‐apoptotic mediator (Fig. 5 e). As the apoptosis induction appeared to be antigen‐independent and mediated by a soluble factor in the MC38‐Sianull supernatant, we questioned whether MC38‐Sianull supernatant alone might be sufficient to cause CD8+ T cell apoptosis. To address this, we cultured CD8+ T cells only in the presence of supernatants obtained from MC38‐MOCK or MC38‐Sianull cells or plain medium as a negative control. As expected, CD8+ T cell apoptosis was increased by 30% when cells were cultured in the presence of MC38‐Sianull supernatant instead of plain medium or MC38‐MOCK supernatant (Fig. 5 f). To further characterize the apoptosis inducing factor secreted by MC38‐Sianull cells, supernatants were heat inactivated prior to addition to the CD8+ T cells. Heat inactivation of the MC38‐Sianull supernatant completely abrogated CD8+ T cell apoptosis (Fig. 5 g), suggesting that the MC38‐Sianull‐specific apoptosis mediator is a heat‐sensitive protein or peptide, which acts in an antigen‐independent manner.
Moreover, the augmented CD8+ T cell apoptosis could provide an explanation for both the reduced CD8+ T cell frequencies present in MC38‐Sianull tumors as well as the diminished CD8+ T cell cytotoxicity toward MC38‐Sianull cells in vitro.
Low CMAS gene expression is associated with lower recurrence‐free survival
So far, our results indicate that loss of sialic acids might actually be detrimental in CRC and thereby contradict other studies investigating the role of tumor sialylation on the anti‐tumor immune response.15, 16, 32 To translate our findings to human CRC patients, we analyzed CMAS gene expression in a human colorectal cancer cohort (GSE39582) in relation to disease progression and survival. We stratified the CRC patients according to their CMAS gene expression into two groups, CMAS low (median < 150, n = 74) or CMAS high (median > 300, n = 81) patients, and evaluated their recurrence‐free survival. Interestingly, low CMAS gene expression indeed correlated to a lower recurrence‐free survival in CRC patients (Fig. 6 a), reflecting the results obtained in our in vivo mouse studies. Thus, also in human CRC patients, a low sialylation status of the tumor is correlated to poor prognosis.
Figure 6.
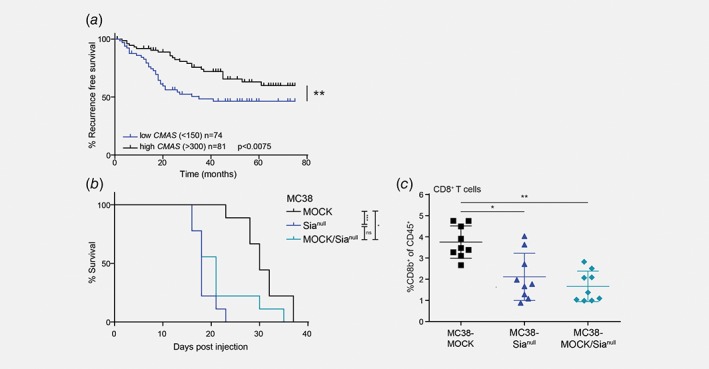
CMAS gene expression levels are correlated to recurrence‐free survival in human colorectal cancer and a partial CMAS knock out is sufficient to drive tumor growth in vivo. (a) CMAS gene expression data was analyzed in the GSE39582 colorectal cancer cohort.27 Patients were stratified in two groups with either low CMAS gene expression (median < 150) or high CMAS gene expression (median > 300) and evaluated for their recurrence‐free survival. (b) MC38‐MOCK, MC38‐Sianull or MC38‐MOCK cells (30%) mixed with MC38‐Sianull cells (70%) were injected into C57Bl/6 mice (n = 9) and sacrificed when the tumor reached a size of 2000 mm3. Tumor growth was measured and survival curves of the mice are displayed. (c) Flow cytometric analysis of viable CD8+ T cells (CD3+CD8b+) at the tumor site when the tumor reached a size of 2000 mm3. Mean ± SD; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. [Color figure can be viewed at wileyonlinelibrary.com]
Partial tumor desialylation is sufficient to drive tumor growth in CRC
Since low CMAS expression correlated to a lower recurrence‐free survival in CRC patients, we tried to mimic this in vivo by investigating whether partial tumor desialylation would be enough to sustain the enhanced tumor growth. Therefore, we inoculated mice with MC38‐MOCK and MC38‐Sianull cells in a 30:70 ratio (MC38‐MOCK/Sianull). Interestingly, the survival curve of the mice injected with the mixed tumors followed the survival curve of mice that received MC38‐Sianull cells only (Fig. 6 b). Moreover, CD8+ T cell frequencies were decreased to an equal level as in the MC38‐Sianull tumors (Fig. 6 c). Together, these data imply that in CRC tumor growth is promoted, even when only a proportion of the malignant cells in the tumor microenvironment is completely desialylated.
Discussion
The upregulation of sialylated glycan structures on the cell surface is a key feature of tumor cells, independent of the tumor type. Sialylated structures are known to regulate immune responses, whereby the current dogma states that tumor cells exploit tumor‐associated Sias to dampen anti‐tumor immunity.14, 15, 16 These findings have led to the development of new therapeutic strategies aimed at reducing tumor‐associated Sia expression to dismantle the Sia‐induced tolerance. However, these novel therapeutic strategies may result in a complete loss of tumor sialylation, yet the effect of a total absence of sialylated structures on tumor growth and anti‐tumor immunity has not been investigated before.
Here, we report for the first time that complete abrogation of Sias on the murine CRC cell line MC38 (MC38‐Sianull) significantly increased tumor growth in vivo. Noteworthy, the MC38 CMAS mutated cells were selected in bulk and sorted based on cell surface expression of Sias. In this way tumor heterogeneity was maintained in our cell lines, allowing us to exclude potential clonal effects. Intestinal mucins are known to be both sialylated and/or sulphated,20 yet in the present study we solely affected the sialylation pathway. Therefore, we cannot exclude that sulphated glycans contribute to tumor development in our model.
The increased tumor growth of MC38‐Sianull cells could be attributed to reduced CD8+ T cell frequencies present within the tumor. Reduced migration of CD8+ T cells toward the tumor core has been correlated with bad prognosis.2, 33, 34 As we did not address the localization of the T cells within the tumor microenvironment, we cannot rule out that CD8+ T cells were hampered in reaching the tumor core. Nevertheless, we did observe that CD8+ T cells displayed diminished cytotoxicity and underwent apoptosis upon encounter of MC38‐Sianull cells. Thus, our study highlights that complete tumor desialylation might be detrimental to the anti‐tumor immune response in CRC, which greatly complicates the design of novel cancer therapeutics aimed at targeting the tumor glycosylation profile.
To abrogate cell surface sialylation we knocked out the CMAS gene, a catalyzing enzyme involved in the sialylation pathway, whereas other studies focused on the CMP‐sialic acid transporter, the SLC35A1 gene. Although both interrupt the sialylation pathway, the consequences of this interruption might significantly differ, since SLC35A1 has been described to be involved in the O‐mannosylation pathway as well.28 E‐cadherin, a protein well known for its role in epithelial‐to‐mesenchymal transition and cancer metastasis,35 is O‐mannosylated, which is also crucial for E‐cadherin‐mediated cell adhesion. However, the choice of the target gene cannot fully explain the observed discrepancies. Teoh et al.32 recently showed decreased lung metastasis upon CMAS gene knock out in a highly metastatic mammary cell line injected in BALB/c mice. We, on the other hand, employed a non‐metastatic CRC model that was inoculated in C57BL/6 mice. The different mouse strains, with their different predisposition toward T helper 2 (Th2) and Th1 responses,36, 37 as well as the different tumor model used, may have impacted the outcome of the respective studies. Discrepancies due to tumor type‐specific effects is further supported by the cohort data presented in both studies32 (Fig. 6 a). Our CRC cohort analysis revealed that CMAS gene expression was positively correlated with prognosis. Strikingly, only a proportion of MC38‐Sianull cells was sufficient to sustain the enhanced tumor growth in vivo. The analysis of sialoglycans in CRC human tissues and their link to CD8+ T cell tumor infiltration are subjects for future studies.
Also in the B16 melanoma model, a reduction in tumor sialylation (B16‐Sialow) had a positive effect on the anti‐tumor immune response, resulting in decreased tumor growth.15, 16 Interestingly, the B16 cell line expresses both α2‐3 and α2‐6‐linked Sias,15 while the MC38 cell line only carries α2‐3‐linked Sias. This raises the possibility that our findings might be related to the unique presence of α2‐3‐linked Sias or conversely the absence of α2‐6‐linked Sias. Indeed, humans generally express higher levels of α2‐6‐linked Sias than mice.38 Unfortunately, the different functions and roles of α2‐3 and α2‐6‐linked Sias and their cognate Siglec receptors in tumor biology are hardly described in literature. In hepatocellular carcinoma, Siglec‐1+ macrophages negatively associate with progression and predict favorable survival.39 Siglec‐1 prefers binding of α2‐3‐linked Sias over α2‐6‐linked Sias40 and lacks the cytoplasmic ITIM domain, common in the majority of Siglec receptors.41 Intriguingly, Siglec‐1+ macrophages are able to scavenge Sia+‐particles from apoptotic tumor cells for subsequent cross‐presentation to tumor‐specific CD8+ T cells.42, 43 Thus, Siglec‐1+ macrophages may actually have the potential to propagate anti‐tumor immunity.
In light of the lack of α2‐6‐linked Sias on our MC38 cells, low and high levels of α2‐6‐linked Sias have indeed been correlated to tumor regression or progression, respectively.44 Especially, high expression of ST6Gal1, the enzyme responsible for α2‐6‐sialylation, has been associated with metastasis and therapeutic failure in colorectal cancer specifically.45, 46 ST6Gal1 modulates tumor differentiation in vivo via an enhanced β1‐integrin function that stimulates cell motility.46 These findings suggest that α2‐6‐linked Sias may be an important instigator of Sia‐mediated immune evasion in CRC. However, conclusive data is lacking to support this hypothesis and should thus be further explored.
One soluble protein known to induce CD8+ T cell apoptosis is Galectin‐1. Produced by tumor cells, Galectin‐1 can bind the Galβ1‐4GlcNAc (LacNAc) epitope,47 which is elevated in our MC38‐Sianull cells after removal of the Sia. Galectin‐1 supports tumor progression through cell–cell and cell‐matrix interactions as well as tumor‐induced angiogenesis and an elevated CD8+ T cell apoptosis.48 MC38‐Sianull cells indeed showed higher mRNA levels of the LGALS1 (Galectin‐1) gene, however, compared to MC38‐MOCK, Galectin‐1 protein concentrations in MC38‐Sianull supernatants were not increased (data not shown). Therefore, more research is required to identify the soluble factor present in the MC38‐Sianull supernatant.
In conclusion, we have demonstrated that complete tumor desialylation of the MC38 mouse CRC cell line augments tumor growth and enhances CD8+ T cell apoptosis. Clearly, discrepancies still exist regarding the expression levels or the linkage type of α2‐3 or α2‐6 of Sias and the cellular origins of the tumor. Therefore, the specific impact of α2‐3 and α2‐6‐linked Sias and their relative amounts on the anti‐tumor immune responses requires further research to fully understand how various tumor types exploit the sialylation machinery to evade anti‐tumor immunity. Nevertheless, we argue that in certain tumor types, for instance CRC, tumor sialylation may actually have a protective effect and we suggest that therapies aimed at abrogating tumor‐associated Sias should be undertaken with caution.
Author Contributions
L.A.M.C designed and performed experiments, analyzed data and wrote paper; A.B performed experiments and analyzed data; J.C.H performed experiments; L.K performed subcutaneous cell injections and mouse experiments; A.Z performed mouse experiments; T.O. performed cell sorting experiments; L.W. performed cell culture experiments; Y.v.K and S.v.V conceived and coordinated the study; S.v.V. designed and supervised the study and wrote paper. All authors contributed to reviewing, and/or revising of the study.
Supporting information
Supplementary Figure 1 Mutating the CMAS gene in the MC38‐Sianull cell line has no effect on cell proliferation rates in vitro. A, The CMAS gene from MC38‐MOCK and MC38‐Sianull cells was amplified with qPCR (742 bp, top arrow) and subsequently tested for the presence of mutations using the Surveyor assay. The Surveyor nuclease enzyme recognizes and cleaves DNA mismatches present in the qPCR product. The CMAS mutation was located after 260 bp of the qPCR product, resulting in a band of 260 bp and 482 bp when cleaved (middle and bottom arrow). B, MC38‐MOCK and MC38‐Sianull were cultured, harvested at day 1, 2, 3 or 4 days and cell numbers were counted manually. C, The metabolic activity of MC38‐MOCK and MC38‐Sianull cells was measured after 4.5 h of culture in a cell concentration‐dependent manner (left) or after the first 24 h (30.000 cells plated, right) using the CellTiter‐Blue® Cell Viability assay. Data are representative of one (A and B) or two (C) independent experiments. Mean ± SD.
Supplementary Figure 2 MC38‐Sianull tumors retain their desialylated phenotype in vivo. A, The manually measured tumor size (mm3) significantly correlates to the tumor mass (mg) weighed directly after tumor resection. B, The sialaylation phenotype of ex vivo MC38‐Sianull tumor cells (CD31−CD45−) was analyzed with the α2‐3‐specific plant lectin MAL‐II. Data are representative of pooled data of two experiments (A) or one experiment (B). Mean ± SEM; *, p < 0.05.
Supplementary Figure 3 Equal immune cell frequencies in the tumor‐draining lymph nodes of MC38‐MOCK and MC38‐Sianull tumors. MC38‐MOCK or MC38‐Sianull cells were injected in C57Bl/6 mice and sacrificed after 13 days of tumor development (A, n = 5) or when the tumor reached a size of 2000 mm3 (B, n = 9). A and B, Flow cytometric analysis of viable, CD45+ CD8+ T cells (CD3+CD8b+), CD4+ T cells (CD3+CD4+), Regulatory T cells (CD3+CD4+Foxp3+) and NK cells (CD3−NK1.1+) in the tumor‐draining lymph node. Data are representative of two independent experiments (A and B). Mean ± SEM.
Supplementary Figure 4 Similar frequencies of neoantigen‐specific CD8+ T cells in tumor‐draining lymph nodes of MC38‐MOCK and MC38‐Sianull tumors. A, MC38‐MOCK or MC38‐Sianull cells were injected in C57Bl/6 mice (n = 5). Tumor growth was monitored and mice were sacrificed 13 days after tumor inoculation. B, Tumor‐draining lymph node cells from MC38‐MOCK and MC38‐Sianull tumors were stained with tetramers specific for three different MC38 neoantigens. The frequencies of tetramer positive CD8+ T cells was indistinguishable between MC38‐MOCK or MC38‐Sianull groups. Data are representative of two independent experiments (A and B). Mean ± SEM; ***, p < 0.01.
Supplementary Table 1 List of possible N‐linked glycans identified in MC38‐MOCK and MC38‐Sianull cells using HILIC‐(U)HPLC‐FLR‐ESI‐MS. Information about the composition, registered or calculated mass [m/z]+ and charge [H+] is provided for the three most abundant N‐glycans detected under each peak of the FLR chromatograms (Fig. 1 d). For each cell line, the possible N‐linked glycans were categorized into four groups, including neutral (non‐fucosylated/non‐sialylated), fucosylated (mono‐, di‐ or tri‐fucosylated), sialylated (mono‐, di‐ or tri‐sialylated) and mixed (fucosylated and sialylated) compositions.
Acknowledgements
We thank our animal facility for caretaking of the animals and Joke den Haan (Amsterdam UMC) for her advice regarding mouse experiments. We thank Daniel Spencer, Rad Kozak and Max Kotsias (Ludger) for their support regarding the mass spectrometry experiments. We thank the NIH Tetramer Core Facility for providing the MHC–peptide complexes.
Conflict of interest: No potential conflicts of interest were disclosed.
The data that support the findings of our study are available from the corresponding author upon reasonable request.
References
- 1. Galon J, Costes A, Sanchez‐Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006;313:1960–4. [DOI] [PubMed] [Google Scholar]
- 2. Galon J, Pages F, Marincola FM, et al. The immune score as a new possible approach for the classification of cancer. J Transl Med 2012;10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fridman WH, Pages F, Sautes‐Fridman C, et al. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012;12:298–306. [DOI] [PubMed] [Google Scholar]
- 4. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007;121:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Varki A, Gagneux P. Biological functions of Glycans In: rd. In: Varki A, Cummings RD, Esko JD, et al., eds Essentials of Glycobiology ed. Cold Spring Harbor, NY, 2015. 77–88. [Google Scholar]
- 6. Brockhausen I. Mucin‐type O‐glycans in human colon and breast cancer: glycodynamics and functions. EMBO Rep 2006;7:599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hakomori S. Cancer‐associated glycosphingolipid antigens: their structure, organization, and function. Acta Anat (Basel) 1998;161:79–90. [DOI] [PubMed] [Google Scholar]
- 8. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 2015;15:540–55. [DOI] [PubMed] [Google Scholar]
- 9. Crocker PR, Varki A. Siglecs in the immune system. Immunology 2001;103:137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Varki A. Since there are PAMPs and DAMPs, there must be SAMPs? Glycan "self‐associated molecular patterns" dampen innate immunity, but pathogens can mimic them. Glycobiology 2011;21:1121–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Perdicchio M, Ilarregui JM, Verstege MI, et al. Sialic acid‐modified antigens impose tolerance via inhibition of T‐cell proliferation and de novo induction of regulatory T cells. Proc Natl Acad Sci USA 2016;113:3329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Falco M, Biassoni R, Bottino C, et al. Identification and molecular cloning of p75/AIRM1, a novel member of the sialoadhesin family that functions as an inhibitory receptor in human natural killer cells. J Exp Med 1999;190:793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nicoll G, Avril T, Lock K, et al. Ganglioside GD3 expression on target cells can modulate NK cell cytotoxicity via siglec‐7‐dependent and ‐independent mechanisms. Eur J Immunol 2003;33:1642–8. [DOI] [PubMed] [Google Scholar]
- 14. Bull C, Boltje TJ, Wassink M, et al. Targeting aberrant sialylation in cancer cells using a fluorinated sialic acid analog impairs adhesion, migration, and in vivo tumor growth. Mol Cancer Ther 2013;12:1935–46. [DOI] [PubMed] [Google Scholar]
- 15. Perdicchio M, Cornelissen LA, Streng‐Ouwehand I, et al. Tumor sialylation impedes T cell mediated anti‐tumor responses while promoting tumor associated‐regulatory T cells. Oncotarget 2016;7:8771–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bull C, Boltje TJ, Balneger N, et al. Sialic acid blockade suppresses tumor growth by enhancing T cell‐mediated tumor immunity. Cancer Res 2018;78:3574–88. [DOI] [PubMed] [Google Scholar]
- 17. Stanczak MA, Siddiqui SS, Trefny MP, et al. Self‐associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J Clin Invest 2018;128:4912–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xiao H, Woods EC, Vukojicic P, et al. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc Natl Acad Sci U S A 2016;113:10304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ashkani J, Naidoo KJ. Glycosyltransferase gene expression profiles classify cancer types and propose prognostic subtypes. Sci Rep 2016;6:26451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robbe C, Capon C, Coddeville B, et al. Structural diversity and specific distribution of O‐glycans in normal human mucins along the intestinal tract. Biochem J 2004;384:307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robbe C, Capon C, Maes E, et al. Evidence of regio‐specific glycosylation in human intestinal mucins: presence of an acidic gradient along the intestinal tract. J Biol Chem 2003;278:46337–48. [DOI] [PubMed] [Google Scholar]
- 22. Robbe‐Masselot C, Maes E, Rousset M, et al. Glycosylation of human fetal mucins: a similar repertoire of O‐glycans along the intestinal tract. Glycoconj J 2009;26:397–413. [DOI] [PubMed] [Google Scholar]
- 23. Mihalache A, Delplanque JF, Ringot‐Destrez B, et al. Structural characterization of Mucin O‐glycosylation may provide important information to help prevent colorectal tumor recurrence. Front Oncol 2015;5:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 2013;8:2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kozak RP, Tortosa CB, Fernandes DL, et al. Comparison of procainamide and 2‐aminobenzamide labeling for profiling and identification of glycans by liquid chromatography with fluorescence detection coupled to electrospray ionization‐mass spectrometry. Anal Biochem 2015;486:38–40. [DOI] [PubMed] [Google Scholar]
- 26. Ceroni A, Maass K, Geyer H, et al. GlycoWorkbench: a tool for the computer‐assisted annotation of mass spectra of glycans. J Proteome Res 2008;7:1650–9. [DOI] [PubMed] [Google Scholar]
- 27. Marisa L, de Reynies A, Duval A, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med 2013;10:e1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Riemersma M, Sandrock J, Boltje TJ, et al. Disease mutations in CMP‐sialic acid transporter SLC35A1 result in abnormal alpha‐dystroglycan O‐mannosylation, independent from sialic acid. Hum Mol Genet 2015;24:2241–6. [DOI] [PubMed] [Google Scholar]
- 29. Yadav M, Jhunjhunwala S, Phung QT, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014;515:572–6. [DOI] [PubMed] [Google Scholar]
- 30. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000;192:1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Simon S, Labarriere N. PD‐1 expression on tumor‐specific T cells: friend or foe for immunotherapy? Oncoimmunology 2017;7:e1364828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Teoh ST, Ogrodzinski MP, Ross C, et al. Sialic acid metabolism: a key player in breast cancer metastasis revealed by metabolomics. Front Oncol 2018;8:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou C, Wu Y, Jiang L, et al. Density and location of CD3(+) and CD8(+) tumor‐infiltrating lymphocytes correlate with prognosis of oral squamous cell carcinoma. J Oral Pathol Med 2018;47:359–67. [DOI] [PubMed] [Google Scholar]
- 34. Melero I, Rouzaut A, Motz GT, et al. T‐cell and NK‐cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov 2014;4:522–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gheldof A, Berx G. Cadherins and epithelial‐to‐mesenchymal transition. Prog Mol Biol Transl Sci 2013;116:317–36. [DOI] [PubMed] [Google Scholar]
- 36. Watanabe H, Numata K, Ito T, et al. Innate immune response in Th1‐ and Th2‐dominant mouse strains. Shock 2004;22:460–6. [DOI] [PubMed] [Google Scholar]
- 37. Hsieh CS, Macatonia SE, O'Garra A. Murphy KM. T cell genetic background determines default T helper phenotype development in vitro. J Exp Med 1995;181:713–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gagneux P, Cheriyan M, Hurtado‐Ziola N, et al. Human‐specific regulation of alpha 2‐6‐linked sialic acids. J Biol Chem 2003;278:48245–50. [DOI] [PubMed] [Google Scholar]
- 39. Zhang Y, Li JQ, Jiang ZZ, et al. CD169 identifies an anti‐tumour macrophage subpopulation in human hepatocellular carcinoma. J Pathol 2016;239:231–41. [DOI] [PubMed] [Google Scholar]
- 40. Hartnell A, Steel J, Turley H, et al. Characterization of human sialoadhesin, a sialic acid binding receptor expressed by resident and inflammatory macrophage populations. Blood 2001;97:288–96. [DOI] [PubMed] [Google Scholar]
- 41. Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol 2007;7:255–66. [DOI] [PubMed] [Google Scholar]
- 42. Asano K, Nabeyama A, Miyake Y, et al. CD169‐positive macrophages dominate antitumor immunity by crosspresenting dead cell‐associated antigens. Immunity 2011;34:85–95. [DOI] [PubMed] [Google Scholar]
- 43. Goswami KK, Ghosh T, Ghosh S, et al. Tumor promoting role of anti‐tumor macrophages in tumor microenvironment. Cell Immunol 2017;316:1–10. [DOI] [PubMed] [Google Scholar]
- 44. Avidan A, Perlmutter M, Tal S, et al. Differences in the sialylation patterns of membrane stress proteins in chemical carcinogen‐induced tumors developed in BALB/c and IL‐1alpha deficient mice. Glycoconj J 2009;26:1181–95. [DOI] [PubMed] [Google Scholar]
- 45. Park JJ, Lee M. Increasing the alpha 2, 6 sialylation of glycoproteins may contribute to metastatic spread and therapeutic resistance in colorectal cancer. Gut Liver 2013;7:629–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hedlund M, Ng E, Varki A, et al. Alpha 2‐6‐linked sialic acids on N‐glycans modulate carcinoma differentiation in vivo. Cancer Res 2008;68:388–94. [DOI] [PubMed] [Google Scholar]
- 47. Cousin JM, Cloninger MJ. The role of Galectin‐1 in cancer progression, and synthetic multivalent Systems for the Study of Galectin‐1. Int J Mol Sci 2016;17:pii: E1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stillman BN, Hsu DK, Pang M, et al. Galectin‐3 and galectin‐1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol 2006;176:778–89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Mutating the CMAS gene in the MC38‐Sianull cell line has no effect on cell proliferation rates in vitro. A, The CMAS gene from MC38‐MOCK and MC38‐Sianull cells was amplified with qPCR (742 bp, top arrow) and subsequently tested for the presence of mutations using the Surveyor assay. The Surveyor nuclease enzyme recognizes and cleaves DNA mismatches present in the qPCR product. The CMAS mutation was located after 260 bp of the qPCR product, resulting in a band of 260 bp and 482 bp when cleaved (middle and bottom arrow). B, MC38‐MOCK and MC38‐Sianull were cultured, harvested at day 1, 2, 3 or 4 days and cell numbers were counted manually. C, The metabolic activity of MC38‐MOCK and MC38‐Sianull cells was measured after 4.5 h of culture in a cell concentration‐dependent manner (left) or after the first 24 h (30.000 cells plated, right) using the CellTiter‐Blue® Cell Viability assay. Data are representative of one (A and B) or two (C) independent experiments. Mean ± SD.
Supplementary Figure 2 MC38‐Sianull tumors retain their desialylated phenotype in vivo. A, The manually measured tumor size (mm3) significantly correlates to the tumor mass (mg) weighed directly after tumor resection. B, The sialaylation phenotype of ex vivo MC38‐Sianull tumor cells (CD31−CD45−) was analyzed with the α2‐3‐specific plant lectin MAL‐II. Data are representative of pooled data of two experiments (A) or one experiment (B). Mean ± SEM; *, p < 0.05.
Supplementary Figure 3 Equal immune cell frequencies in the tumor‐draining lymph nodes of MC38‐MOCK and MC38‐Sianull tumors. MC38‐MOCK or MC38‐Sianull cells were injected in C57Bl/6 mice and sacrificed after 13 days of tumor development (A, n = 5) or when the tumor reached a size of 2000 mm3 (B, n = 9). A and B, Flow cytometric analysis of viable, CD45+ CD8+ T cells (CD3+CD8b+), CD4+ T cells (CD3+CD4+), Regulatory T cells (CD3+CD4+Foxp3+) and NK cells (CD3−NK1.1+) in the tumor‐draining lymph node. Data are representative of two independent experiments (A and B). Mean ± SEM.
Supplementary Figure 4 Similar frequencies of neoantigen‐specific CD8+ T cells in tumor‐draining lymph nodes of MC38‐MOCK and MC38‐Sianull tumors. A, MC38‐MOCK or MC38‐Sianull cells were injected in C57Bl/6 mice (n = 5). Tumor growth was monitored and mice were sacrificed 13 days after tumor inoculation. B, Tumor‐draining lymph node cells from MC38‐MOCK and MC38‐Sianull tumors were stained with tetramers specific for three different MC38 neoantigens. The frequencies of tetramer positive CD8+ T cells was indistinguishable between MC38‐MOCK or MC38‐Sianull groups. Data are representative of two independent experiments (A and B). Mean ± SEM; ***, p < 0.01.
Supplementary Table 1 List of possible N‐linked glycans identified in MC38‐MOCK and MC38‐Sianull cells using HILIC‐(U)HPLC‐FLR‐ESI‐MS. Information about the composition, registered or calculated mass [m/z]+ and charge [H+] is provided for the three most abundant N‐glycans detected under each peak of the FLR chromatograms (Fig. 1 d). For each cell line, the possible N‐linked glycans were categorized into four groups, including neutral (non‐fucosylated/non‐sialylated), fucosylated (mono‐, di‐ or tri‐fucosylated), sialylated (mono‐, di‐ or tri‐sialylated) and mixed (fucosylated and sialylated) compositions.