

Abstract
The ability to measure the levels of diagnostically relevant proteins, such as antibodies, directly at the point of care could significantly impact healthcare. Thus motivated, we explore here the E-DNA “scaffold” sensing platform, a rapid, convenient, single-step means to this end. These sensors comprise a rigid nucleic acid “scaffold” attached via a flexible linker to an electrode and modified on its distal end with a redox reporter and a protein binding “recognition element”. The binding of a targeted protein reduces the efficiency with which the redox reporter approaches the electrode, resulting in an easily measured signal change when the sensor is interrogated voltammetrically. Previously we have demonstrated scaffold sensors employing a range of low molecular weight haptens and linear peptides as their recognition elements. Expanding on this here we have characterized sensors employing much larger recognition elements (up to and including full length proteins) in order to (1) define the range of recognition elements suitable for use in the platform; (2) better characterize the platform’s signaling mechanism to aid its design and optimization; and (3) demonstrate the analytical performance of sensors employing full-length proteins as recognition elements. In doing so we have enlarged the range of molecular targets amenable to this rapid and convenient sensing platform.
Keywords: electrochemical biosensor, DNA, protein, protein detection, antibody detection
Graphical Abstract
There remains a pressing need for improved methods for measuring the levels of diagnostically relevant protein biomarkers directly at the point of care. For example, while lateral flow immunoassays1 have seen significant implementation even for home use,2–5 they provide at best semi-quantitative results regarding the presence or absence of a target. Fluorescence polarization assays,6 which have also seen some deployment at the point-of-care, are, in contrast, quantitative. This approach, which is predicated on using fluorescence anisotropy to measure binding-induced changes in the tumbling time of a fluorescently labeled, target-binding receptor, requires careful background subtraction and large sample volumes, rendering it rather slow and cumbersome for point of care applications. It is thus only used for the highest value targets, such as markers of heart attack in emergency rooms.7,8
Motivated by the lack of rapid, convenient, and quantitative point-of-care molecular diagnostics, numerous approaches to this end have been proposed, including surface plasmon resonance-based devices,9 quartz crystal microbalances,10 surface acoustic wave sensors,11 and time-resolved surface fluorescence chips.12,13 To date, however, these have failed to achieve clinical relevance due to their inability to discriminate between authentic target binding and the nonspecific binding of the many other proteins found in realistically complex clinical samples. In short, the ability to rapidly and conveniently measure the levels of specific proteins in crude clinical samples at the point of care remains an important, unmet medical need.
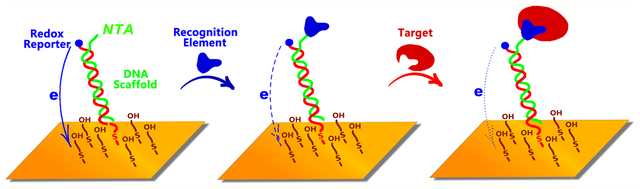
In response to the need for improved point-of-care molecular diagnostics we have developed the E-DNA “scaffold” platform, an electrochemical analog of fluorescence polarization that is convenient and rapid enough to deploy at the point of care and selective enough to work in relatively crude, unprocessed clinical samples.14,15 The approach employs a nucleic acid duplex “scaffold” via an attached flexible linker to the surface of an interrogating electrode. Its other, distal end is modified with a protein binding “recognition element”, which is typically a short peptide (e.g., a linear epitope) or small molecule (e.g., a hapten), and a redox reporter (methylene blue) that generates an easily measurable electrochemical signal (Figure 1). The binding of a target protein, such as an antibody, to the recognition element reduces the efficiency with which the redox reporter approaches the electrode, producing a change in electron transfer that is easily measured via square wave voltammetry. To date, such E-DNA sensors have been described for the achieving of rapid (few minute), quantitative measurements of a number of antibodies and non-antibody protein targets in only minimally diluted blood serum or cell extracts.14–17
Figure 1.

E-DNA scaffold sensors are composed of a redox-reporter-modified, double stranded nucleic acid “scaffold” attached via a flexible linker to self-assembled monolayer deposited on a gold electrode. Their distal end is modified with a redox-reporter (here methylene blue) and a target-binding recognition element. Here we have attached these recognition elements using a nitrilotriacetic acid (NTA) on the scaffold that, in the presence of copper, binds to a His-tag on the recognition element. (a) In the absence of a recognition element the reporter readily approaches the electrode, (b) producing a large redox current. (c) Upon addition of a recognition element (during sensor fabrication) this current is (d) reduced, presumably due to the steric bulk of the recognition element. (e) The binding of a macromolecular target, such as an antibody, (f) reduces this current still further (Figure S1), producing an easily measurable signal indicative of the presence of the target. Here we explore how large a recognition element can be employed before the reduction in current it causes is so great that target binding no longer produces a sufficient additional signal change to support target detection.
To date, we have only employed small molecules (e.g., digoxin) or short peptides (e.g., epitopes of 16 or fewer amino acids) as recognition elements in the E-DNA scaffold platform, with the rationale being that, as the recognition element becomes larger the additional signal change produced by target binding will ultimately fall to zero. Given this, there should be a maximum recognition element size that the platform can accommodate before the baseline change in current (change upon addition of the recognition element to the scaffold) is so great that no additional signal change is observed upon target binding. Here, we have addressed this question by characterizing how the size of the sensor’s recognition element affects signaling. In doing so we demonstrate sensors in this class that, for the first time, employ full-length proteins as their recognition elements.
As a first exploration of the extent to which the size of the recognition element alters the scaffold sensor’s baseline current we compared the behavior of sensors fabricated using either the 7 kDa P3 domain18 of CheA or the full length, 71 kDa monomeric protein19 as the recognition element (Figure 2). To do so we expressed the two recognition elements with carboxy-terminal hexa-his-tags, taking advantage of the ability of this sequence to bind copper nitrilotriacetic acid to facilely attach each to an NTA-modified scaffold13,20,21 (Figure 2a). Characterizing these sensors, we see that, as expected, the size of the recognition-element matters: while the addition of the 7 kDa P3 domain to the scaffold reduces baseline peak current by ~15% (relative to that seen for the unmodified scaffold), the addition of the full-length, 71 kDa protein reduces it by ~35% (Figure 2b).
Figure 2.

Addition of recognition elements to the scaffold reduces the current observed in the absence of target in a size-dependent manner. (a) To show this we attached either the 7 kDa P3 domain of CheA or the full length, 71 kDa CheA to the scaffold. (b) Upon the addition of the P3 domain the signaling current is reduced by only ~15% relative to that seen for the unmodified scaffold (the dark bars represent the signal change seen for control sensors lacking NTA). Addition of the full-length protein, in contrast, causes the signaling current to decrease by 35%. Throughout, the error bars reflect the standard deviations of replicates performed using multiple independently fabricated sensors.
To further explore the relationship between signaling and recognition element size we next fabricated sensors employing his-tagged polypeptides and proteins spanning a much wider range of molecular weights: CheY (14 kDa),22 CdiACTMHI813(17 kDa),23 p24 (24 kDa),24 green fluorescent protein (GFP) (29 kDa), HER2 (69 kDa),25 and GlnRS (88 kDa).26 We also included in this set the proteins anti-Dig IgG (150 kDa), which we bound to a scaffold to which Dig had been covalently added15 and streptavidin (211 kDa), which was attached to a biotin-modified scaffold. The addition of these “recognition elements” to the scaffold monotonically reduces baseline signaling current until the protein reaches ~70 kDa, after which the current reduction plateaus at ~35% signal change relative to the unmodified scaffold (Figure 3). Of note, this molecular weight corresponds to a protein of ~7 nm in diameter (Figure S2); i.e., at this point the radius of the recognition element approaches the width of the double-helical scaffold (2 nm) plus the length of the methylene blue and its linker (~1 nm) and thus the receptor is large enough to physically prevent the methylene blue from contacting the electrode when the side of the scaffold to which it is attached approaches the surface.
Figure 3.

As the recognition element becomes larger, the signaling current remaining after it is attached to the scaffold is reduced. (a) Shown is the signal reduction observed upon addition of various recognition elements to the scaffold as a function of their molecular weight. This change in current increases monotonically until the recognition element reaches ~70 kDa, after which the signal change plateaus at ~35% for these his-tag-linked polypeptides and proteins.
Simulations of the scaffold sensor support our argument that the dependence of the baseline peak current on molecular weight stems from simple geometric exclusion. To see this, we simulated a model of the scaffold that employs three adjustable parameters: the flexibility of the linker connecting the scaffold to the surface (θ), the flexibility of the linker connecting the recognition element to the scaffold (φ), and the size of the recognition element (r) (Figure 4a). We find that, while the simulated rate of electron transfer is largely independent of the first parameter (Figure S3), it is a strong function of the size of the recognition element and the flexibility of the linker connecting it to the scaffold (Figure 4b). Specifically, plots of (relative) estimated transfer rate versus recognition element molecular weight trace out shapes similar to those seen in our experimental results, with the curves plateauing when, as is true in the experimental case, the radius of the recognition element approaches the sum of the diameter of the scaffold and the length of the methylene blue and its linker. The current suppression seen at the plateau, however, is a function of the flexibility of the linker connecting the recognition element to the scaffold (Figure 4b). This occurs because more flexible recognition elements can move to avoid colliding with the surface, an effect that presumably explains why the sensors we have characterized here, which use a highly flexible his-tag linker, plateau at lower signal suppression (~35% versus upward of 50%) than the signal suppression we have previously seen in sensors employing shorter, less flexible linkers.15,16
Figure 4.

(a) To better understand the origins of E-DNA scaffold signaling, here we have simulated a simple modeled sensor that employs only three parameters: the flexibility of the linker connecting the scaffold to the surface (θ), the flexibility of the linker connecting the recognition element to the scaffold (φ), and the size of the recognition element (r). (b) Estimated transfer efficiency is a strong function of the size of the recognition element and the flexibility of the linker connecting it to the scaffold (the width of the distribution that ϕ adopts), with plots of estimated transfer rate (normalized against the transfer rate of the unmodified scaffold) versus the molecular weight of the recognition element tracing out curves similar to those seen in our experimental results (Figure 3). The value at which the signal change plateaus, however, is a strong function of the flexibility of the recognition-element-to-scaffold linker (the standard deviation of the Gaussian-distribution of the population of angles, ϕ, employed in each simulation is denoted).
Further simulations are also consistent with our model of the scaffold sensor mechanism. These suggest, for example, that signaling is independent of the flexibility of the linker connecting the redox reporter to the scaffold because such flexibility alters transfer from both the modified and unmodified scaffolds equivalently (Figure S4). Conversely, our simulations suggest that, if we place the redox reporter in the middle of the scaffold (at base pair 11, rather than on the distal terminus), the sensor’s signal is a less strong function of recognition element size (which should enable the use of larger recognition elements at the cost of a reduced signal change upon target binding). Consistent with our model, however, under these conditions signaling is also independent of the flexibility of the linker connecting the recognition element to the scaffold (Figure S5).
The above results suggest that, while the signal change induced by target binding will fall as the recognition element increases in size, there is nevertheless “room” to exploit larger recognition elements than the largest we have previously employed (a 16-residue, 2 kDa linear epitope from HIV16). To test this, we designed sensors employing full-length protein antigens (thus including conformational epitopes, and not just linear epitopes) for the detection of specific antibodies (Figure 5). As the first test-case example of this we employed the 24 kDa HIV p24 antigen as the recognition element. The resultant sensor easily measures anti-HIV antibodies with a detection limit of a few nanomolar (Figure 5a). This is 1 to 2 orders of magnitude poorer than an ELISA (due to the enzyme-driven signal amplification that ELISAs employ), but ELISAs only achieve this improved detection limit at the cost of being complex, multistep, and far slower than E-DNA sensors. As our second example we employed the 27 kDa green fluorescent protein (GFP) to detect anti-GFP antibodies (Figure 5c). Using polyclonal anti-GFP antisera as our target we observed a 12% signal change upon antibody binding (Figure S6a) and observed the expected Langmuir isotherm binding with a dissociation constant of 6.3 ng/mL (Figure S6b).
Figure 5.

(a) The ability to apply full-length proteins in the E-DNA scaffold platform expands the range of targets it can detect to, for example, antibodies that bind conformational epitopes. Using green fluorescent protein (GFP)−NTA complex as the recognition element, we easily detect anti-GFP antibodies with a total signal change of about ~12% (Figure S6a), a value easily comparable to the signal change seen in typical fluorescence polarization assays. (b) Using the HIV protein p24 as the recognition element we can similarly detect antibodies specific for this virus. The error bars reflect the standard deviations of replicates performed using multiple independently fabricated sensors. (c) The smaller the recognition element, the larger the signal change seen upon target binding. To show this here we have employed the following recognition elements: Dig (0.5 kDa), p24 (24 kDa), GFP (27 kDa), and HER2 (69 kDa). The error bars reflect standard deviations of replicates performed using multiple independently fabricated sensors.
As expected, the target response of antibody-detecting EDNA scaffold sensors are also dependent on the size of the recognition element. To see this, we characterized sensors employing Dig (0.5 kDa; which was attached covalently to the scaffold rather than via a His-tag), p24 (24 kDa), GFP (27 kDa), and HER2 (69 kDa) as recognition elements (Figure 5c; Figure S7). Upon binding its antibody target the sensor employing the Dig recognition element exhibits a 35% change in signal. As expected, this is similar to the change in current we would expect in transitioning from an unmodified scaffold (Dig is only 0.5 kDa, we presume it does not significantly alter the current seen from the unmodified scaffold) to a scaffold presenting a 150 kDa antibody. The intermediate-sized recognition elements p24 and GFP, in contrast, produce signal changes of 21% and 25%, respectively, upon addition of the recognition element to the scaffold. They correspondingly exhibit only 12% and 13% signal changes upon target binding. Finally, addition of the much higher molecular weight HER2 recognition element to the scaffold leads to a ~30% reduction in signaling current, and thus target binding produces only minimal additional signal change.
We have determined the extent to which the signaling of EDNA scaffold sensors depends on the size of their recognition element, finding that as the recognition element becomes larger its steric bulk reduces electron transfer and thus reduces the signal change observed upon target binding. This effect “saturates” when the recognition reaches ~70 kDa (Figure 3), eliminating any additional signal change associated with target binding (Figure 5c). Perhaps not coincidentally, the radius of a protein of this molecular weight is similar to the width of the scaffold plus the length of the methylene blue and its linker, suggesting that these observations arise due to simple steric blocking. Recognition elements of under ~25 kDa, however, still produce adequate signal change to support target detection. At ~17%, for example, the signal change seen when a 25 kDa recognition element binds its target antibody is larger than the typical signal change seen in fluorescence polarization assays,7,8 a technique that, as noted above, has seen significant deployment at the point of care despite its far slower and more cumbersome nature than the E-DNA platform.
The observation that sensors utilizing recognition elements of up to ~25 kDa remain responsive to the binding of their target provides opportunities to expand this sensor platform. To this end, here we have demonstrated E-DNA sensors that employ full-length antigens as their recognition element, improving the potential impact of the approach. Specifically, the use of full-length antigens expands the approach to the detection of antibodies that recognize conformational epitopes comprising two or more sequence-distant elements of the antigen.27 The use of full-length antigens also provides a ready means of simultaneously monitoring antibodies against different linear epitopes of the same antigen, which, given that different patients sometimes produce antibodies against different epitopes in the same antigen,28,29 should improve clinical sensitivity.
While the above analysis focused on the detection of antibodies, we have previously shown the platform can be used to detect non-antibody targets recognized by short polypep-tides.17 The ability to utilize recognition elements of up to 25 kDa should significantly expand upon this ability. For example, the results presented here suggest that the platform will be amenable to the incorporation of single domain antibodies, such as Camelid30 or other single chain variable fragments antibodies,31 which are under 25 kDa and can be used to recognize a wide range of nonantibody protein targets.32
Supplementary Material
■. ACKNOWLEDGMENTS
This work was supported by the NIH through grant R01GM118560 (K.W.P) and R01GM59544 (F.W.D). C.P. thanks the Generalitat de Catalunya for the Beatriu de Pinoś fellowship (2014 BP_A 00068) and the Lindros Award for supporting his research.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssensors.8b00311.
Methods and further data supporting the conclusions described here (PDF)
The authors declare no competing financial interest.
■ REFERENCES
- (1).Yalow RS; Berson SA Immunoassay of endogenous plasma insulin in man. J. Clin. Invest 1960, 39, 1157–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Jameson DM; Ross JA Fluorescence polarization/anisotropy in diagnostics and imaging. Chem. Rev 2010, 110, 2685–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lea WA; Simeonov A Fluorescence polarization assays in small molecule screening. Expert Opin. Drug Discovery 2011, 6, 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Brangel P; Sobarzo A; Parolo C; Miller BS; Howes PD; Gelkop S; Lutwama JJ; Dye JM; McKendry RA; Lobel L; Stevens MM A Serological Point-of-Care Test for the Detection of IgG Antibodies against Ebola Virus in Human Survivors. ACS Nano 2018, 12, 63–73. [DOI] [PubMed] [Google Scholar]
- (5).Parolo C; Merkoci A Paper-based nanobiosensors for diagnostics. Chem. Soc. Rev 2013, 42, 450–457. [DOI] [PubMed] [Google Scholar]
- (6).Dandliker WB; Kelly RJ; Dandliker J; Farquhar J; Levin J Fluorescence polarization immunoassay. Theory and experimental method. Immunochemistry 1973, 10, 219–227. [DOI] [PubMed] [Google Scholar]
- (7).Nielsen K; Gall D Fluorescence polarization assay for the diagnosis of brucellosis: a review. J. Immunoassay Immunochem 2001, 22, 183–201. [DOI] [PubMed] [Google Scholar]
- (8).Rossi AM; Taylor CW Analysis of protein-ligand interactions by fluorescence polarization. Nat. Protoc 2011, 6, 365–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Blow N Proteins and proteomics: life on the surface. Nat. Methods 2009, 6, 389–393. [Google Scholar]
- (10).Cheng CI; Chang YP; Chu YH Biomolecular interactions and tools for their recognition: focus on the quartz crystal microbalance and its diverse surface chemistries and applications. Chem. Soc. Rev 2012, 41, 1947–1971. [DOI] [PubMed] [Google Scholar]
- (11).Länge K; Rapp BE; Rapp M Surface acoustic wave biosensors: a review. Anal. Bioanal. Chem 2008, 391, 1509–1519. [DOI] [PubMed] [Google Scholar]
- (12).Rant U; Arinaga K; Fujita S; Yokoyama N; Abstreiter G; Tornow M Dynamic Electrical Switching of DNA Layers on a Metal Surface. Nano Lett. 2004, 4, 2441–2445. [Google Scholar]
- (13).Langer A; Hampel PA; Kaiser W; Knezevic J; Welte T; Villa V; Maruyama M; Svejda M; Jahner S; Fischer F; Strasser R; Rant U Protein analysis by time-resolved measurements with an electro-switchable DNA chip. Nat. Commun 2013, 4, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cash KJ; Ricci F; Plaxco KW A general electrochemical method for label-free screening of protein-small molecule interactions. Chem. Commun 2009, 41, 6222–6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cash KJ; Ricci F; Plaxco KW An electrochemical sensor for the detection of protein-small molecule interactions directly in serum and other complex matrices. J. Am. Chem. Soc 2009, 131, 6955–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).White RJ; Kallewaard HM; Hsieh W; Patterson AS; Kasehagen JB; Cash KJ; Uzawa T; Soh HT; Plaxco KW Wash-free, electrochemical platform for the quantitative, multiplexed detection of specific antibodies. Anal. Chem 2012, 84, 1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bonham AJ; Paden NG; Ricci F; Plaxco KW Detection of IP-10 protein marker in undiluted blood serum via an electro-chemical E-DNA scaffold sensor. Analyst 2013, 138, 5580–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang X; Vallurupalli P; Vu A; Lee K; Sun S; Bai W; Wu C; Zhou H; Shea JE; Kay LE; Dahlquist FW The linker between the dimerization and catalytic domains of the CheA histidine kinase propagates changes in structure and dynamics that are important for enzymatic activity. Biochemistry 2014, 53, 855–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Stock A; Chen T; Welsh D; Stock J CheA protein, a central regulator of bacterial chemotaxis, belongs to a family of proteins that control gene expression in response to changing environmental conditions. Proc. Natl. Acad. Sci. U. S. A 1988, 85, 1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sigal GB; Bamdad C; Barberis A; Strominger J; Whitesides GM A self-assembled monolayer for the binding and study of histidine-tagged proteins by surface plasmon resonance. Anal. Chem 1996, 68, 490–497. [DOI] [PubMed] [Google Scholar]
- (21).Kang D; Sun S; Kurnik M; Morales D; Dahlquist FW; Plaxco KW New Architecture for Reagentless, Protein-Based Electrochemical Biosensors. J. Am. Chem. Soc 2017, 139, 12113–12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mo G; Zhou H; Kawamura T; Dahlquist FW Solution structure of a complex of the histidine autokinase CheA with its substrate CheY. Biochemistry 2012, 51, 3786–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Willett JLE; Gucinski GC; Fatherree JP; Low DA; Hayes CS Contact-dependent growth inhibition toxins exploit multiple independent cell-entry pathways. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 11341–11346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Deshmukh L; Schwieters CD; Grishaev A; Ghirlando R; Baber JL; Clore GM Structure and dynamics of full-length HIV-1 capsid protein in solution. J. Am. Chem. Soc 2013, 135, 16133–16147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cho H; Mason K; Ramyar KX; Stanley AM; Gabelli SB; Denney DW Jr.; Leahy DJ Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [DOI] [PubMed] [Google Scholar]
- (26).Rodriguez-Hernandez A; Bhaskaran H; Hadd A; Perona JJ Synthesis of Glu-tRNA(Gln) by engineered and natural aminoacyltRNA synthetases. Biochemistry 2010, 49, 6727–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Luzzago A; Felici F; Tramontano A; Pessi A; Cortese R Mimicking of discontinuous epitopes by phage-displayed peptides, I. Epitope mapping of human H ferritin using a phage library of constrained peptides. Gene 1993, 128, 51–57. [DOI] [PubMed] [Google Scholar]
- (28).Langedijk JP; Schalken JJ; Tersmette M; Huisman JG; Meloen RH Location of epitopes on the major core protein p24 of human immunodeficiency virus. J. Gen. Virol 1990, 71, 2609–2614. [DOI] [PubMed] [Google Scholar]
- (29).Tang S; Zhao J; Wang A; Viswanath R; Harma H; Little RF; Yarchoan R; Stramer SL; Nyambi PN; Lee S; Wood O; Wong EY; Wang X; Hewlett IK Characterization of immune responses to capsid protein p24 of human immunodeficiency virus type 1 and implications for detection. Clin. Vaccine Immunol 2010, 17, 1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Muyldermans S Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem 2013, 82, 775–797. [DOI] [PubMed] [Google Scholar]
- (31).Holliger P; Hudson PJ Engineered antibody fragments and the rise of single domains. Nat. Biotechnol 2005, 23, 1126–1136. [DOI] [PubMed] [Google Scholar]
- (32).Harmsen MM; De Haard HJ Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol 2007, 77, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.