

Abstract
Over the past decade, there has been a renewed interest in the use of transition metal polypyridyl complexes as photoredox catalysts for a variety of innovative synthetic applications. Many derivatives of these complexes are known, and the effect of ligand modifications on their efficacy as photoredox catalysts has been the subject of extensive, systematic investigation. However, the influence of the photocatalyst counteranion has received little attention, despite the fact that these complexes are generally cationic in nature. Herein, we demonstrate that counteranion effects exert a surprising, dramatic impact on the rate of a representative photocatalytic radical cation Diels–Alder reaction. A detailed analysis reveals that counteranion identity impacts multiple aspects of the reaction mechanism. Most notably, photocatalysts with more non-coordinating counteranions yield a more powerful triplet excited state oxidant and longer radical cation chain length. It is proposed that this counteranion effect arises from Coulombic ion-pair interactions between the counteranion and both the cationic photoredox catalyst and the radical cation intermediate, respectively. The comparatively slower rate of reaction with coordinating counteranions can be rescued by using hydrogen-bonding anion binders that attenuate deleterious ion-pairing interactions. These results demonstrate the importance of counteranion identity as a variable in the design and optimization of photoredox transformations and suggest a novel strategy for the optimization of organic reactions using this class of transition metal photocatalysts.
Graphical Abstract
Introduction
Ruthenium(II) polypyridyl complexes have been among the most widely studied molecular photocatalysts for a variety of applications. The photophysical, electrochemical, and physical properties of this class of luminescent transition metal complexes have been extensively characterized.1 They generally exhibit strong absorbance in the visible spectrum, feature high intersystem crossing efficiency, and can participate in a diverse range of photoinduced electron- and energy-transfer processes. Because of these attractive features, Ru(II) photocatalysts were instrumental in the early development of solar fuels technologies;2 in addition, some of the best light-harvesting sensitizers for dye-sensitized solar cells belong to this family of complexes.3 Over the past decade, the recognition that Ru(bpy)32+ and its analogues are also useful photocatalysts for organic transformations has stimulated a renewal of interest in photochemical synthesis.4 Because of the exceptional utility of Ru(bpy)32+ in so many diverse applications, numerous structurally varied Ru(II) polypyridyl photocatalysts have been prepared, and the effects of ligand modifications on catalyst properties are well-understood (Figure 1).5
Figure 1.

Structurally varied Ru(II) photocatalysts.
The effect of counteranion structure on the photoactivity of these cationic complexes, on the other hand, has not been subject to similar systematic study. In this paper, we document the discovery of the unexpected impact counteranion identity plays on the efficiency of a radical cation Diels–Alder cycloaddition, a representative photoredox transformation. We rationalize the observed rate increase as the consequence of (1) a change in the photocatalyst ground-state electrochemical properties, (2) a significant shift in its triplet-state energy, and (3) an increase in the efficiency of radical cation chain propagation. The results reported herein suggest that this counterion effect may be an unappreciated but important phenomenon in many photoredox reactions. Understanding the impact of this experimental variable, therefore, should benefit the growing community of scholars interested in the use of these complexes as photoredox catalysts in organic chemistry.
Results and Discussion
Counterion Effects in Radical Cation Cycloadditions
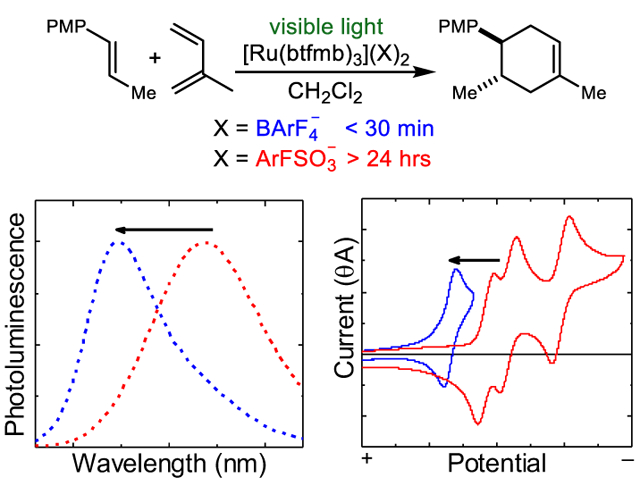
Several years ago, we reported that visible light photoredox catalysis offered an efficient means to conduct radical cation Diels–Alder cycloadditions between a wide range of electron-rich styrenes and diverse dienes.6 The highly electron-deficient [Ru(bpz)3](BArF4)2 complex7 proved to be a potent photocatalyst for this transformation, providing excellent rates and yields at ambient temperatures with as little as 0.5 mol% of photocatalyst.8 Our proposal for the mechanism of this reaction is briefly summarized in Scheme 1. Photoexcitation of Ru(bpz)32+ with visible light results in the efficient formation of a long-lived redox-active triplet state. The electron-deficient bpz ligands render the photoexcited catalyst a substantially stronger oxidant (+1.4 V vs SCE) than the parent Ru(bpy)32+ catalyst (+0.89 V vs SCE), enabling the one-electron photooxidation of anethole (4, +1.1 V vs SCE). The resulting alkene radical cation undergoes rapid [4+2] cycloaddition with diene 5 to afford product radical cation 6•+.9 Formation of the neutral product can occur by one of two mechanisms: either radical chain-propagating oxidation of another equivalent of alkene 4 or by chain-terminating oxidation of the reduced Ru(bpz)3+ catalyst.10 The latter process regenerates the photoactive Ru(II) state of the catalyst and closes the catalytic cycle.
Scheme 1.

Proposed Photocatalytic Radical Cation Diels–Alder Cycloaddition Mechanism
Despite the efficiency and broad scope of this reaction, the Ru(bpz)32+ chromophore suffers from limited solubility in non-polar organic solvents. Empirical screening indicated that the reaction proceeds more rapidly in these solvents and resulted in the use of CH2Cl2 in optimized reaction conditions. Nevertheless, the solubility of the bpz complex in CH2Cl2 is modest, and reactions conducted at moderate catalyst loadings are often visibly heterogeneous. These constraints limited our ability to systematically investigate catalyst structure on the rate of photoredox reactions. We thus became interested in the use of strongly oxidizing photocatalysts with greater lipophilicity that might be freely soluble in low-dielectric solvents.
As a starting point for these studies, we prepared a series of photocatalysts based upon the Ru(btfmb)32+ chromophore (Figure 1, 3; btfmb = 4,4’-bis(trifluoromethyl)-2,2’-bipyridyl). The photophysical and electrochemical properties of the homoleptic [Ru(btfmb)3](PF6)2 complex were previously investigated in acetonitrile by Furue and Kamachi.11 Given the oxidizing potential reported for its excited state (+1.3 V vs SCE), we hypothesized that Ru(btfmb)32+ would be an effective photooxidative catalyst for electron-rich styrenes such as 4. Moreover, we hoped that the lipophilic CF3 substituents would improve the solubility of the photocatalyst in non-polar organic solvents compared to Ru(bpz)32+. In order to maximize the organic solubility of this chromophore, we prepared a series of salts bearing a variety of lipophilic counteranions (3a–f) and assessed their activities in a model photoreaction.
Figure 3.

(A) Effect of anion-binding co-catalyst 7 on photoluminescence of 3d. (B) Stern–Volmer plot in the absence and presence of ion binder 7.
The results of this initial catalyst screen for the radical cation Diels–Alder cycloaddition between anethole (4) and isoprene (5) are summarized in Table 1. In general, these various photocatalysts each promoted the reaction, but surprisingly, the rates of reaction varied dramatically depending on counteranion identity. While the cycloaddition of 4 and 5 is complete in 20 min using 1 mol% of BArF4− catalyst 3a, the reaction proceeds to only 55% yield after 24 h with the analogous triflate catalyst 3b, 14% yield with the 3,5-bis(trifluoromethyl)benzenesulfonate (ArFSO3−) catalyst 3d, and only 2% yield with the tosylate catalyst 3e. No conversion was observed in this timeframe using the carboxylate complex 3f. Thus, there appears to be a correlation between the rate of product formation and the non-coordinating nature of the catalyst counteranions.12 Unaware of any previous report of similarly dramatic counterion effects on the rate of organic photoredox transformations, we elected to investigate the origins of this phenomenon.
Table 1.
Counteranion Effect on the Rate of Radical Cation Diels–Alder Cycloaddition.a
 | ||||
|---|---|---|---|---|
| entry | catalyst | time | yield 6b | unreacted 4b |
| 1 | 3a (BArF4−) | 20min | 98% | 0% |
| 2 | 3b (TfO−) | 24 h | 55% | 37% |
| 3 | 3d (ArFSO3−) | 24 h | 14% | 80% |
| 4 | 3e (TsO−) | 24 h | 2% | 95% |
| 5 | 3f (ArFCO2−) | 24 h | 0% | 100% |
General conditions: anethole (0.06 mmol), isoprene (0.18 mmol), CH2Cl2 (0.08 M), [Ru] (1 mol%). Each reaction was subjected to 3 freeze-pump-thaw cycles prior to irradiation from a 23 W CFL bulb.
1H NMR yields referenced to trimethyl(phenyl)silane internal standard.
To begin to understand this effect, we performed Stern–Volmer analyses of the relationship between the concentration of anethole (4) in CH2Cl2 and the photoluminescence intensity of the BArF4–, PF6–, and ArFSO3– complexes of Ru(btfmb)32+. This investigation demonstrated that the degree of excited-state quenching between the photoexcited catalyst and the organic substrate (i.e., the Stern–Volmer constant, Ksv) decreased by two orders of magnitude from the least coordinating counteranion, BArF4–, to the most coordinating counteranion in this study, ArFSO3− (Figure 2A). This is consistent with the markedly superior reactivity of the BArF4− complex. The value of Ksv is dependent both upon the excited-state lifetime of the photocatalyst (τ) and the bimolecular electron-transfer rate constant (kq); Ksv = τkq. To deconvolute whether the large change in the Stern–Volmer constant arises primarily from a change in catalyst triplet lifetime or in the electron-transfer rate constant, we measured t for each Ru(btfmb)32+ complex (Table 2). These results show that while the counteranion does have an influence on triplet lifetime, the effect is relatively small — approximately two-fold over the range of counteranions investigated. The impact on kq, therefore, is much larger, spanning two orders of magnitude from ArFSO3− 3d to BArF4– 3a. Thus, the unanticipated conclusion from these preliminary studies is that the identity of the photocatalyst counteranion can impact a photooxidative reaction by dramatically altering the intrinsic rate constant of bimolecular electron transfer to the photocatalyst excited state.
Figure 2.

A. Stern–Volmer plots for excited-state quenching of catalysts 3a–c in CH2Cl2. B/C. Effect of counteranion identity on excitedstate properties of [Ru(btfmb)3](X)2, X is indicated in the legend. (B) Absorption (solid line) and photoluminescence (dashed line) spectra in MeCN. (C) Absorption (solid line) and photoluminescence (dashed line) spectra in CH2Cl2.
Table 2.
Spectroscopic Properties and Stern–Volmer Quenching Constantsa
| entry | catalyst | λabs (nm) |
λem (nm) |
KSV (x 102 M−1) |
τ (ns) |
kq (x 108 M−1 s−1) |
|---|---|---|---|---|---|---|
| 1 | 3a | 453 | 573 | 17 | 520 | 33 |
| 2 | 3c | 458 | 605 | 4.2 | 860 | 4.9 |
| 3 | 3d | 458 | 618 | 0.61 | 950 | 0.64 |
Data acquired in CH2Cl2.
This finding was surprising. While the importance of the bipyridyl ligand structure in the design and optimization of photocatalytic reactions is well appreciated,13 the effect of the catalyst counterion on photocatalytic reaction rates has received significantly less attention. Meyer and coworkers have examined ion-pairing effects on the photophysics of Ru(II) polypyridyl chromophores.14 These investigations show that addition of Cl– to solutions of [Ru(bpy)2(deeb)](PF6)2 in CH2Cl2 results in the formation of tight ion pairs and a concomitant decrease in triplet excited-state energy and lifetime.15 However, neither the impact of structurally complex organic counteranions on the photochemical properties of Ru(II) complexes, nor the effects of ion pairing on the rate of synthetic photocatalytic applications, have been systematically explored.16
Spectroscopic, Electrochemical, and Quantum Yield Studies
We have collected absorption, emission, and electrochemical data for a representative series of Ru(btfmb) 2+ complexes bearing BArF4–, PF6–, ArFSO3–, and TsO– counteranions. These data are depicted in Figure 2B and C. First, we obtained spectral data for these complexes in acetonitrile (Figure 2B). Both the absorption and emission spectra are superimposable in MeCN, consistent with the attenuated impact of ion pairing in high-dielectric solvents. On the other hand, Coulombic effects are more significant in non-polar solvents, which are often ideal for applications in organic synthesis. Figure 2C shows absorption and emission spectra for the same series of catalysts in CH2Cl2. The impact of counteranion identity on the absorption spectrum in this relatively non-polar solvent is modest, suggesting that if differences in Coulombic interactions exert any influence on the ground-state properties of the photocatalyst or on the singlet excited state, it is a small effect. In contrast, the photoluminescence spectra of the various complexes differ markedly. Most notably, the λmax of photoluminescence varies by 52 nm from the least coordinating (BArF4–, 573 nm) to the most coordinating (TsO–, 625 nm) counteranion, corresponding to a substantial energy difference of 4.2 kcal/mol (0.18 eV).
Thus, altering the identity of the counteranion produces an unexpectedly large change in the energy of the emissive triplet state of Ru*(btfmb)32+. One would expect these changes to be reflected in excited-state redox potentials. To quantify this effect, we measured the one-electron reduction potential E(Ru2+/+) of the Ru(btfmb)32+ complexes in CH2Cl2. Each measurement was made using a matching n-Bu4N+X– salt as a supporting electrolyte in order to avoid complications arising from counteranion exchange. Counteranions of lower Lewis basicity resulted in significant anodic shifts in the ground-state potentials, with the largest and most significant effect observed for the BArF4– counteranion (Table 3). This effect can also be rationalized as a consequence of ion pairing where the one-electron reduction of the least electrostatically stabilized BAF4− complex is more energetically favorable than the tightly ion-paired tosylate complex. To calculate the excited-state redox potential, we made the commonly utilized assumption17,18 that the Gibbs free energy change for the S0 to T1 transition is represented by the energy of the corresponding photoluminescence maximum (ΔGES). The catalytically relevant first triplet excited-state reduction potential E(Ru2+*/+) can then be approximated from the sum of ΔGES and E(Ru2+/+). As the data in Table 3 show, these potentials span a range of 480 mV (11 kcal/mol), with the BArF4–complex having the most positive reduction potential of +1.52 V vs SCE. The conclusion from these studies, therefore, is that the degree of ion pairing has a synergistic effect on both the excited-state triplet energy and on the ground-state electrochemical potential, leading to a large net dependence of photooxidant strength on the identity of the catalyst counteranion. These results are consistent with the experimentally observed effect of counteranion identity on the radical cation Diels–Alder reaction described above. The most non-coordinating counteranion (BArF4–) results in the largest driving force for photoinduced electron transfer, consistent with a faster rate of photoinitiation and a shorter reaction time.
Table 3.
Ground- and Excited-State Redox Potentials for Ru(btfmb)32+ in CH2 Cl2a
| entry | catalyst | ΔGES | E(Ru2+/+) | E(Ru2+*/+) |
|---|---|---|---|---|
| 1 | 3a | 2.17 eV | −0.65 V | +1.52 V |
| 2 | 3c | 2.05 eV | −0.89 V | +1.16 V |
| 3 | 3d | 2.01 eV | −0.93 V | +1.08 V |
| 4 | 3e | 1.99 eV | −0.95 V | +1.04 V |
Electrochemical potentials were measured through cyclic voltammetry in a standard three-electrode set-up, scan rate = 100 mV/s. A 100 mM solution of a n-Bu4N+X– salt that matched the photocatalyst counteranion was used as a supporting electrolyte. Potentials were corrected to SCE through an external ferrocene reference.
In order to rationalize the impact of counteranion identity on the triplet excited-state energy of the Ru(btfmb)32+ chromophore, we propose an explanation based upon an empirical physical model for charge redistribution between the ground and electronically excited states of this canonical class of transition metal photocatalysts (Scheme 2). The ground state of the Ru(btfmb)32+ chromophore has D3 symmetry and consequently cannot support a permanent dipole moment. On the other hand, the emissive states of Ru(II)* tris(bipyridyl) complexes are understood to be metal-to-ligand charge-transfer (MLCT) triplets, and considerable experimental evidence supports the contention that the transferred electron is localized to a single ligand without significant delocalization across the other two ligands.19 Thus, electronically excited Ru*(btfmb)32+ is best conceptualized as a C2-symmetric, charge-separated state with an oxidized Ru(III) core and a single reduced btfmb•– ligand (Scheme 2). This lower-symmetry MLCT state would therefore be expected to have a very large dipole moment. Meyer has estimated the dipole moment of the triplet Ru*(bpy)32+ state to be approximately 14 D.20 If the photocatalyst exists largely in an ion-paired state in non-polar solvents, stabilizing charge–dipole interactions should have a larger effect on the triplet excited state than they do on the ground state. One would further expect that more strongly coordinating anions, which produce tighter ion pairs, would better stabilize the triplet excited state. Finally, a strong solvent dependence would be consistent with this model, as charge–dipole interactions are attenuated by increasing solvent dielectric.
Scheme 2.

Representation of the ground state (no dipole) and triplet excited state (significant dipole) for [Ru(btfmb)3](X)2.
In the radical cation Diels–Alder reaction, the radical cation intermediates (4•+ and 6•+) would also be expected to exist as ion pairs, and the most reasonable counteranion would be that introduced by the photocatalyst.16 We wondered if this ion-pairing interaction might also affect the dynamics of the product-forming cycloaddition and chain propagation steps as well as the photoinitiation step. To investigate this question, we utilized the same protocol we previously described for estimating the chain length in radical cation cycloadditions.10 First, we measured the reaction quantum yield with the BArF4– and ArFSO3 catalysts through chemical actinometry (see Supporting Information). The quantum yield using the BArF4– catalyst 3a (1 mol%) was measured to be Φ = 26, comparable to the value we determined for the corresponding [Ru(bpz)3](BArF4)2 catalyst in previous studies.10 However, when ArFSO3– complex 3d was utilized as the photocatalyst, we measured a significantly decreased quantum yield of Φ = 0.35. To correct for the differing efficiency of photoinitiation by photocatalysts with different excited-state oxidation potentials, we divided the measured quantum yield values by the quenching fraction. The quenching of 3a by anethole was highly efficient (Q > 0.99), and thus the estimated average chain length is the same as the quantum yield (CL = 26). The quenching fraction for 3d was lower (Q = 0.83), but the resulting average chain length was still calculated to be quite low (CL = 0.42). Thus, in addition to influencing the ground and excited states of the photocatalyst, the counteranion impacts the efficiency of the subsequent radical chain reaction.
These results have several significant implications. First, counteranion identity is a previously underappreciated variable in the optimization of organic photoredox reactions that has the potential to dramatically impact the success and efficiency of synthetically useful organic reactions. Second, counteranion effects impact multiple aspects of the photocatalytic mechanism, including the energy of the reactive triplet excited state, the rate of the electron-transfer photoinitiation event, and the dynamics of nonphotochemical product-forming radical chain propagation events. Finally, because the strength of ion-pairing interactions is sensitive to solvent dielectric, these counterion effects are expected to be most important in the relatively non-polar organic solvents that are often optimal for synthetic applications. Such effects may be important in a much wider range of organic photoredox reactions than previously appreciated.
Hydrogen-Bonding Anion Binders as Co-Catalysts
The model proposed above suggests that the rate of photocatalytic Diels–Alder cycloaddition is strongly influenced by Coulombic interactions between the counteranion and both the cationic photocatalyst and the radical cation intermediates. As a further test of this model, we hypothesized that other strategies for disrupting ion pairing might be used to exert a similar effect. In particular, we drew inspiration from a concept pioneered by Jacobsen: hydrogen-bonding organocatalysts can accelerate reactions involving various cationic reactive intermediates by binding their associated counteranions.21,22
We hypothesized that the tight ion pairing between a Lewis basic counteranion and Ru*(btfmb)32+ could be disrupted by addition of an appropriate hydrogen-bonding anion binder, recapitulating the rate increases we observed using weakly coordinating counteranions.
Our investigations focused on the use of sulfonate complex 3d as a photoredox catalyst for the radical cation Diels–Alder reaction. As described previously, 3d is markedly less reactive than the optimal BArF4– complex 3a. Several recent reports have shown that C3v-symmetric thiophosphotriamide 7 is an effective hydrogen-bond donor for binding sulfonate anions,23 and we imagined that the sequestration of the ArFSO3– counteranion by 7 might attenuate its propensity to participate in tight ion-pairing interactions. To test this hypothesis, we conducted Diels–Alder cycloadditions using 1 mol% of 3d in the presence and absence of 7 (Table 4). These experiments showed a large rate increase for the Diels–Alder cycloaddition upon addition of just 20 mol% of 7. Under these conditions, the reaction is complete within 2 h, while only 5% yield of 6 is formed at the same timepoint in the absence of the anion binder. Notably, there is no observable formation of cycloadduct upon irradiation in the presence of 7 without photocatalyst 3d. This demonstrates that the thiophosphotriamide is not photocatalytically active, and the improvement in photoredox activity thus arises from a synergistic cocatalytic effect.
Table 4.
Effect of Ion-binder Co-catalyst 7 on Radical Cation Diels–Alder reaction.
 | ||||
|---|---|---|---|---|
| entry | [Ru] cat 3d | H-bond cat 7 | time | yield 6 |
| 1 | 1 mol% | 20 mol% | 2 h | 99% |
| 2 | 1 mol% | none | 2 h | 5% |
| 3 | none | 20 mol% | 2 h | 0% |
We also investigated whether co-catalyst 7 had an influence on the photophysical properties of ArFSO3– photocatalyst 3d consistent with our proposed model (Figure 3A). The addition of 7 to 3d induced a large hypsochromic shift in the photoluminescence maximum. This shift was in the direction of the emission maximum of BArF4– catalyst 3a, consistent with the expectation that added 7 would decrease the extent of ion pairing. In contrast, the addition of 7 to the BArF4– complex 3a yielded no change in the emission maximum, even at 50-fold excess of 7 relative to 3a. This experiment supports the contention that the effect arises from a specific interaction between the thiophosphotriamide and the sulfate counteranion, rather than an interaction with some other component of the reaction mixture or a general medium effect. To further support this contention, we investigated interaction in CD2Cl2 using 1H NMR spectroscopy. Titration of thiophosphotriamide 7 with n-Bu4N+ ArFSO3– resulted in a significant shift of the aromatic C–H resonances of 7. A fit of these data to a 1:1 binding model provided an association constant of 1.3 × 106.
The influence of added thiophosphotriamide 7 replicated the effect of weakly coordinating anions in other regards as well. First, Stern–Volmer analysis indicates that the rate of quenching of 3d by anethole is significantly faster upon the addition of the thiophosphotriamide (Figure 3B). This is consistent with the observed increase in triplet excited-state energy with greater concentrations of 7. Second, we observed a substantial effect on the radical cation chain length (see Supporting Information). The addition of 20 mol% of 7 yielded a 20-fold increase in both the quantum yield and apparent radical chain length of the reaction, suggesting that anion binder 7 can influence the dynamics of the chain process by disrupting ion pairing.
Thus, we have been able to recapitulate the observed effect of non-coordinating anions on the photocatalytic activity of ArFSO3– catalyst 3d using hydrogen-bonding co-catalyst 7. The thiophosphotriamide disrupts ion pairing by binding the sulfonate counteranion, which results in significant increases to the photocatalyst’s triplet excited-state energy, the rate of photoinduced electron transfer, the length of the radical cation chain process, and the overall efficiency of the photocatalytic Diels–Alder process. These results support the contention that Coulombic effects can be more significant in photoreactions than previously appreciated. Moreover, these results suggest that the use of anion-binding organocatalysts could be a conceptually orthogonal strategy for optimization of the growing class of synthetically useful photoredox transformations.
Conclusion
The studies summarized above suggest several important implications for the design, understanding, and optimization of photocatalytic processes. First, counterion effects can exert a significant impact on the observed rate of radical cation reactions initiated by photoredox catalysis. The degree of ion pairing between the counteranion and both the Ru(II) photoredox catalyst and the oxidized radical cation intermediate can influence the efficiency of multiple steps in the mechanism of these reactions. Thus, these studies indicate that modulating the degree of ion pairing is an important unexplored variable in the optimization of this class of transformations, and we have described two complementary approaches that can successfully increase the overall rate of a radical cation cycloaddition by several orders of magnitude. Second, the Coulombic interactions that are the putative origin of these effects are most significant in relatively non-polar solvents such as those that are often optimal for synthetic applications. This could indicate that counteranion identity is a particularly important variable for organic photoredox reactions compared to better-established applications of Ru(II) photoredox catalysts in solar energy conversion and biology, where the use of water or other high-dielectric solvents might mask the impact of ion pairing. As interest in the use of this class of transition metal photoredox catalyst for synthetic applications continues to grow, a deeper understanding of the impact of ion-pairing effects will be critical to developing a complete, detailed understanding of the mechanisms in this class of synthetically useful transformations.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (GM095666). The NMR and mass spectroscopy facilities at UW–Madison are funded in part by NIH (S10OD020022–1), NSF (CHE-1048642), the University of Wisconsin, and a generous gift from Paul J. and Margaret M. Bender.
Footnotes
(Supporting Information. General experimental preparation, synthesis of Ru(II) polypyridyl catalysts, reaction conditions, UV-Vis and photoluminescence spectroscopy, cyclic voltammetry, and NMR characterization. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- 1.(a) Kalyanasundaram K “Photophysics, Photochemistry and Solar Energy Conversion with Tris(bipyridyl)ruthenium(II) and Its Analogues,” Coord. Chem. Rev 1982, 46, 159–244. [Google Scholar]; (b) Roundhill DM “Photochemistry, Photophysics, and Photoredox Reactions of Ru(bpy)32+ and Related Complexes,” in Modern Inorganic Chemistry: Photochemistry and Photophysics of Metal Complexes, Springer: New York, 1994, pp. 165–215. [Google Scholar]
- 2.Lehn JM; Sauvage JP “Chemical Storage of Light Energy. Catalytic Generation of Hydrogen by Visible Light or Sunlight. Irradiation of Neutral Aqueous Solutions,” Nouv. J. Chim 1977, 1, 449–451. [Google Scholar]
- 3.O’Regan B; Grätzel M “A Low-Cost, High-Efficiency Solar Cell Based on Dye-Sensitized Colloidal TiO2 Films,” Nature 1991, 353, 737–740. [Google Scholar]
- 4.For reviews, see:; a) Narayanam JMR; Stephenson CRJ “Visible Light Photoredox Catalysis: Applications in Organic Synthesis,” Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]; b) Prier CK; Rankic DA; MacMillan DWC “Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis,”Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Romero NA; Nicewicz DA “Organic Photoredox Catalysis,” Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]; d) Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC “The Merger of Transition Metal and Photocatalysis,” Nature Rev. Chem 2017, 1, 0052. [Google Scholar]; e) Zou YQ; Hörmann FM; Bach T “Iminium and Enamine Catalysis in Enantioselective Photo-chemical Reactions,” Chem. Soc. Rev 2018, 47, 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Silvi M; Melchiorre P “Enhancing the Potential of Enantioselective Organocatalysis with Light,” Nature 2018, 554, 41–49. [DOI] [PubMed] [Google Scholar]
- 5.Juris A; Balzani V; Barigelletti F; Campagna S; Belser P; von Zelewsky A “Ru(II) Polypyridine Complexes: Photophysics, Photochemistry, Electrochemistry, and Chemiluminescence,” Coord. Chem. Rev 1988, 84, 85–277. [Google Scholar]
- 6.(a) Lin S; Ischay MA; Fry CG; Yoon TP “Radical Cation Diels-Alder Cycloadditions by Visible Light Photocatalysis,” J. Am. Chem. Soc 2011, 133, 19350–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lin S; Padilla CE; Ischay MA; Yoon TP “Visible Light Photocatalysis of Intramolecular Radical Cation Diels–Alder Cycloadditions,” Tetrahedron Lett. 2012, 53, 3073–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lin SS; Lies SD; Gravatt CS; Yoon TP “Radical Cation Cycloadditions Using Cleavable Redox Auxiliaries,” Org. Lett 2017, 19, 368–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Crutchley RJ; Lever ABP “Ruthenium(II) Tris(bipyrazyl) Dication – A New Photocatalyst,” J. Am. Chem. Soc 1980, 102, 7128–7129. [Google Scholar]; (b) Rillema DP; Allen G; Meyer TJ; Conrad D “Redox Properties of Ruthenium(II) Tris Chelate Complexes Containing the Ligands 2,2’-bipyrazine, 2,2’-bipyridine, and 2,2’-bipyrimidine,” Inorg. Chem 1983, 22, 1617–1622. [Google Scholar]
- 8.Several other photocatalytic radical cation Diels–Alder reactions have been reported using alternate photocatalysts. For leading references, see:; (a) Stevenson SM; Shores MP; Ferreira EM “Photooxidizing Chromium Catalysts for Promoting Radical Cation Cycloadditions,” Angew. Chem. Int. Ed 2015, 54, 6506–6510. [DOI] [PubMed] [Google Scholar]; (b) Pitre SP; Scaiano JC; Yoon TP “Photocatalytic Indole Diels–Alder Cycloadditions Mediated by Heterogenous Platinum-Modified Titanium Dioxide,” ACS Catal. 2017, 7, 6440–6444. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gieseler A; Steckhan E; Wiest O; Knoch F “Photochemically Induced Radical Cation Diels–Alder Reaction of Indole and Electron-Rich Dienes,” J. Org. Chem 1991, 56, 1405–1411. [Google Scholar]
- 9.Bauld NL “Cation Radical Cycloadditions and Related Sigmatropic Reactions,” Tetrahedron 1989, 45, 5307–5363. [Google Scholar]
- 10.Cismesia MA; Yoon TP “Characterizing Chain Processes in Visible Light Photoredox Catalysis,” Chem. Sci 2015, 6, 5426–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furue M; Maruyama K; Oguni T; Naiki M; Kamachi M “Trifluoromethyl-substituted 2,2’-bipyridine Ligands. Synthetic Control of ExcitedState Properties of Ruthenium(II) Tris-Chelate Complexes,” Inorg. Chem 1992, 31, 3792–3795. [Google Scholar]
- 12.Strauss SH “The Search for Larger and More Weakly Coordinating Anions” Chem. Rev 1993, 93, 927. [Google Scholar]
- 13.(a) Ischay MA; Ament MS; Yoon TP “Crossed Intermolecular [2+2] Cycloaddition of Styrenes by Visible Light Photocatalysis,” Chem. Sci 2012, 2046–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Douglas JJ; Nguyen JD; Cole KP; Stephenson CRJ “Enabling Novel Photoredox Reactivity via Photocatalyst Selection,” Aldrichim. Acta 2014, 47, 15–25. [Google Scholar]
- 14.(a) Ward WM; Farnum BH; Siegler M; Meyer GJ “Chloride Ion-Pairing with Ru(II) Polypyridyl Compounds in Dichloromethane,” J. Phys. Chem. A 2013, 117, 8883–8894. [DOI] [PubMed] [Google Scholar]; (b) Wehlin SAM; Troian-Gautier L; Li G; Meyer GJ “Chloride Oxidation by Ruthenium Excited-States in Solution,” J. Am. Chem. Soc, 2017, 139, 12903–12906. [DOI] [PubMed] [Google Scholar]; (c) Troian-Gautier L; Wehlin SAM; Meyer GJ “Photophysical Properties of Tetracationic Ruthenium Complexes and Their Ter-Ionic Assemblies with Chloride,” Inorg. Chem, 2018, 57, 12232–12244. [DOI] [PubMed] [Google Scholar]
- 15.The chemical shifts associated with the ligands in the 1H NMR of the photocatalysts vary as a function of counteranion identity and are consistent with observations made by Meyer. These data are summarized in the Supporting Information. See:; Li G; Swords WB; Meyer GJ “Bromide Photo-oxidation Sensitized to Visible light in Consecutive Ion Pairs,” J. Am. Chem. Soc 2017, 139, 14983–14991. [DOI] [PubMed] [Google Scholar]
- 16.Four recent reports have described the influence of chiral counteranions on the enantioselectivity of photocatalytic radical cation reactions:; (a) Morse PD; Nguyen TM; Cruz CL; Nicewicz DA “Enantioselective Counter-Anions in Photoredox Catalysis: The Asymmetric Cation Radical Diels–Alder Reaction,” Tetrahedron 2018, 74, 3266–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang Z; Li H; Li S; Zhang M–T; Luo S “A Chiral Ion-Pair Photoredox Organocatalyst: Enantioselective Anti-Markovnikov Hydroetherification of Alkenols,” Org. Chem. Front, 2017, 4, 1037–1041. [Google Scholar]; (c) Wang H; Ren Y; Wang K; Man Y; Xiang Y; Li N; Tang B “Visible Light-Induced Cyclization Reactions for the Synthesis of 1,2,4–Triazolines and 1,2,4–Triazoles,” Chem. Commun, 2017, 53, 9644–9647. [DOI] [PubMed] [Google Scholar]; (d) Gentry EC; Rono LJ; Hale ME; Matsuura R; Knowles RR “Enantioselective Synthesis of Pyrroloindolines via Noncovalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products,” J. Am. Chem. Soc, 2018, 140, 3394–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adamson QW; Namnath J; Shastry VJ; Slawson V “Thermodynamic Inefficiency of Conversion of Solar Energy to Work,” J. Chem. Ed 1984, 61, 221. [Google Scholar]
- 18.Flamigni L; Barbieri A; Sabatini C; Ventura B; Barigelletii F “Photochemistry and Photophysics of Coordination Compounds: Iridium,” Top. Curr. Chem 2007, 281, 143–203. [Google Scholar]
- 19.(a) Forster M; Hester RE “Resonance Raman Investigation of Electronically Excited Ru(bipyridine)32+ Using a CW Laser,” Chem. Phys. Lett 1981, 81, 42–47. [Google Scholar]; (b) Bradley PG; Kress N; Hornberger BA; Dallinger RF; Woodruff WH “Vibrational Spectroscopy of the Electronically Excited State. 5. Time-Resolved Resonance Raman Study of Tris(bipyridine) Ruthenium(II) and Related Complexes. Definitive Evidence for the Localized MLCT State,” J. Am. Chem. Soc 1981, 103, 7441–7446. [Google Scholar]; (c) Ceulemans A; Vanquickenborne LG “On the Charge-Transfer Spectra of Iron(II)-and Ruthenium(II)-Tris(2,2’-bipyridyl) Complexes,” J. Am. Chem. Soc 1981, 103, 2238–2241. [Google Scholar]; (d) Ohsawa Y; DeArmond MK; Hanck KW; Morris DE; Whitten DG; Neveux PE “Spatially Isolated Redox Orbitals: Evidence from Low-Temperature Voltammetry,” J. Am. Chem. Soc 1983, 105, 6522–6524. [Google Scholar]
- 20.Kober EM; Sullivan BP; Meyer TJ “Solvent Dependence of Metal-to-Ligand Charge-Transfer Transitions. Evidence for Initial Electron Localization in MLCT Excited States of 2,2’-Bipyridine Complexes of Ruthenium(II) and Osmium(II),” Inorg. Chem 1984, 23, 2098–2104. [Google Scholar]
- 21.(a) Reisman SE; Doyle AG; Jacobsen EN “Enantioselective Thiourea-Catalyzed Additions to Oxocarbenium Ions,” J. Am. Chem. Soc 2008, 130, 7198–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Birrell JA; Desrosiers J-N; Jacobsen EN “Enantioselective Acylation of Silyl Ketene Acetals through Fluoride Anion-Binding Catalysis,” J. Am. Chem. Soc 2011, 133, 13872–13875. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wasa M; Liu RY; Roche SP; Jacobsen EN “Asymmetric Mannich Synthesis of α-Amino Esters by Anion-Binding Catalysis,” J. Am. Chem. Soc 2014, 136, 12872–12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gansäuer has recently reported the use of hydrogen bond-donating cocatalysts to influence the rate of titanocene-mediated electron transfer reactions. See:; Liedtke T; Spannring P; Riccardi L; Gansäuer A “Mechanism-Based Condition Screening for Sustainable Catalysis in Single-Electron Steps by Cyclic Voltammetry,” Angew. Chem. Int. Ed 2018, 57, 5006–5010. [DOI] [PubMed] [Google Scholar]
- 23.(a) Cranwell PB; Hiscock R; Haynes CJE; Light ME; Wells NJ; Gale PA “Anion Recognition and Transport Properties of Sulfamide-, Phosphoric Triamide- and Thiophosphoric Triamide-Based Receptors,” Chem. Commun 2013, 49, 874–876. [DOI] [PubMed] [Google Scholar]; (b) Borovika A; Tang PI; Klapman S; Nagorny P “Thiophosphoramide-Based Cooperative Catalysts for Brønsted Acid Promoted Ionic Diels–Alder Reactions,” Angew. Chem. Int. Ed 2013, 52, 13424–13428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.