

Abstract
A substantial part of biodiversity is thought to have arisen from adaptive radiations in which one lineage rapidly diversified into multiple lineages adapted to many different niches. However, selection and drift reduce genetic variation during adaptation to new niches and may thus prevent or slow down further niche shifts. We tested whether rapid adaptation is still possible from a highly derived ecotype in the adaptive radiation of threespine stickleback on the Haida Gwaii archipelago, Western Canada. In a 19-years selection experiment, we let giant stickleback from a large blackwater lake evolve in a small clearwater pond without vertebrate predators. 56 whole genomes from the experiment and 26 natural populations revealed that adaptive genomic change was rapid in many small genomic regions and encompassed 75% of the adaptive genomic change between 12,000 years old ecotypes. Adaptive genomic change was as fast as phenotypic change in defence and trophic morphology and both were largely parallel between the short-term selection experiment and long-term natural adaptive radiation. Our results show that functionally relevant standing genetic variation can persist in derived adaptive radiation members, allowing adaptive radiations to unfold very rapidly.
The colonization of a new habitat or niche requires rapid adaptation to multiple environmental challenges, i.e. to ‘multifarious’ divergent selection. This is most dramatic in adaptive radiations, where rapid successions of niche and habitat shifts occur within a lineage1-3. However, most adaptive radiations started thousands of generations ago and we don’t know whether major phenotypic and genomic adaptation occurred within the first few generations of colonizing a new habitat, or over longer time scales and thus how ‘rapid’ adaptive radiations unfold. Adaptation may be instantaneous when phenotypic plasticity is involved4,5 or occur over few generations of selection on standing genetic variation6,7 or admixture variation8,9. Alternatively, adaptation may require time for beneficial de novo mutations to arrive, or genomic adaptation may occur slower than phenotypic adaptation if rapid phenotypic plasticity is followed by slower genetic assimilation4,10. Furthermore, each new habitat shift will reduce genetic variation through drift and selection and it is unclear whether further adaptation is hampered or slowed down after a first new niche has been colonized in an adaptive radiation.
Evolution experiments and cases of contemporary evolution, such as in biological invasions, may reveal the speed of phenotypic and genomic adaptation11,12. However, many ‘evolve and re-sequence’ experiments and contemporary evolution studies focussed on single selective agents instead of multifarious fitness landscapes13-21, or phenotypic and genomic adaptation have been studied in isolation22-25. Only few examples of phenotypic and genomic contemporary evolution under multifarious divergent selection have been documented, such as marine threespine stickleback (Gasterosteus aculeatus) colonizing freshwater habitats in artificial and natural selection experiments25-28, showing widespread parallel genomic and phenotypic adaptation compared to thousands of generations older natural populations7,29.
Here, we quantify the speed of genomic adaptation to multifarious divergent selection in a 19 years selection experiment, starting from a phenotypically highly derived adaptive radiation member, and compare rates of phenotypic and genomic change. We expand on a long term investigation of the adaptive radiation of threepine stickleback from the Haida Gwaii archipelago off Western Canada30, where stickleback have colonized multiple watersheds independently and adapted to diverse freshwater habitats including lakes, ponds and streams with vastly divergent biophysical features, predator and parasite communities following glacial retreat ~12,000 years ago31-34. Phenotypic variation in defensive armour35-39 such as dorsal and pelvic spines, pelvic girdle and lateral plates, and in trophic morphology31,39,40 such as in body shape, gape and gill rakers, can be largely explained by three main predictors: predation regime, light spectrum, and lake size30.
A selection experiment along these three axes of selection was in initiated by T.E.R. in 1993: he transplanted 100 adult stickleback from a large, deep, dystrophic, blackwater lake (Mayer Lake) with vertebrate-dominated predation into a small, shallow, eutrophic, previously unoccupied clearwater pond (Roadside Pond) dominated by invertebrate predators41. Mayer Lake contains some of the most derived freshwater stickleback, maximally divergent from the ancestral marine phenotype and occupying the extreme morphospace edge of the Haida Gwaii adaptive radiation30: 8-10 cm long, melanistic ‘giants’ with highly developed predator defence and adaptations to limnetic foraging42,43, low levels of phenotypic variance31 but similar levels of genetic variation as other Haida Gwaii populations34,44. After evolving for 16 years in the new selective regime, six predator defence, four feeding morphology traits and eye size evolved in the expected direction (Fig. 1a), encompassing ~30% of the morphological distance between natural stickleback populations from large lakes and small ponds41 (Fig. 2a). Life history changed from two to one year age of first reproduction and melanism was reduced41,45. Phenotypic evolution was fast with on average 0.15 (0-0.25) haldanes over 11 generations, assuming an average generation time of 1.5 years41. While strong change in the first generation for four traits suggested phenotypic plasticity, other traits showed slower change suggesting genetic change. We use whole genomes from 26 natural populations, including the source Mayer Lake (N = 12), and the transplant population Roadside Pond (N = 11) sampled after evolving for 19 years or 13 generations in the new habitat, to identify the speed and targets of genomic adaptation and the extent of genomic parallelism with the Haida Gwaii adaptive radiation.
Fig. 1. Phenotypic and genomic change in the selection experiment.
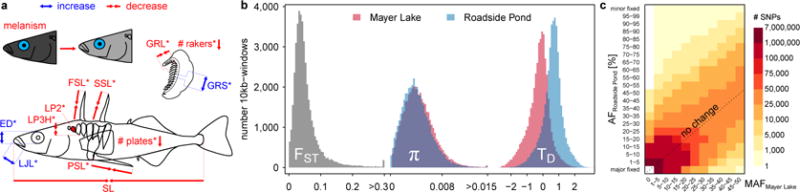
a Summary of the phenotypic change observed in the selection experiment, as reported in Leaver and Reimchen41, with colours indicating trait increase or decrease and asterisks indicating significant change. Phenotypic change in six bony predator defence traits (FSL: first dorsal spine length, SSL: second dorsal spine length, PSL: pelvic spine length, # plates: number of lateral plates, LP3H: lateral plate 3 height, LP2: lateral plate 2 frequency), four feeding morphology traits (LJL: lower jaw length, # rakers: number of gill rakers, GRL: gill raker length, GRS: gill raker spacing) and eye diameter (ED) was in the expected direction, i.e. parallel, given the shift from vertebrate- to invertebrate-dominated predation and zooplankton- to invertebrate-dominated diet and observed phenotypic divergence between large lake and small pond populations in the adaptive radiation on Haida Gwaii41. SL: standard length. b Transplant of 100 adult giant threespine stickleback from Mayer Lake into Roadside Pond and evolution for 13 generations led to moderate genomic differentiation (FST), a minor reduction nucleotide diversity (π) and to a positive shift in the Tajima’s D (TD) distribution. c Even though several rare alleles were fixed, allele frequencies (AF) did not change much over 13 generations. MAF: minor allele frequency.
Fig. 2. Comparison of the extent of phenotypic and genomic evolution in the 19 years selection experiment with the ~12,000 years old adaptive radiation.
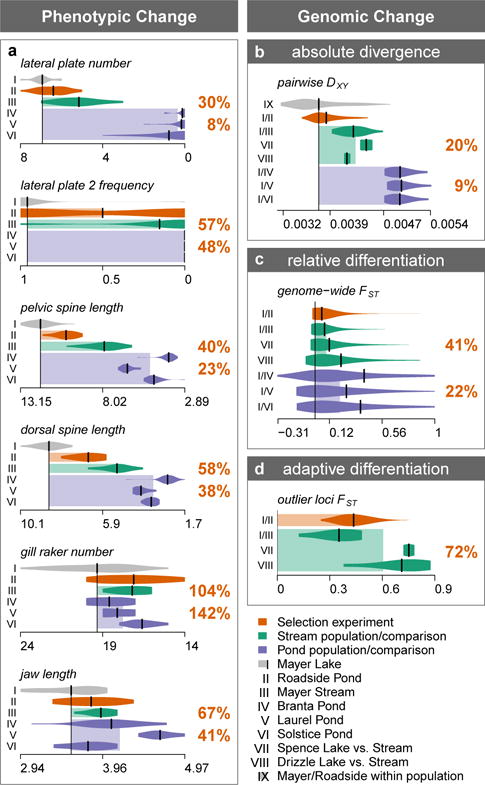
Phenotypic divergence and adaptive genomic differentiation arose rapidly in the selection experiment, comparable in extent to postglacial divergence between large lake and pond or stream ecotypes. a Population means and distributions for six phenotypic traits in Mayer Lake (source population), Roadside Pond (transplant population, orange), postglacial stream (green) and pond (blue) ecotype populations41. b Absolute divergence (DXY), c relative differentiation (FST) and d adaptive differentiation (FST) between postglacial large lake and stream (green) or pond (blue) ecotype populations as well as in the selection experiment (orange). FST estimates for lake-stream and large lake vs. small pond comparisons are based on SNP chip data from a previous study46. Adaptive differentiation SNPs are outlier SNPs in this previous study46 and top 5% FST SNPs in each outlier window for the selection experiment. Numbers are mean percentages of phenotypic or genomic change in the selection experiment compared to postglacial lake vs. stream divergence (upper) and large lake vs. small pond divergence (lower number).
Results
Moderate genome-wide change, but strong change in many small genomic regions
Giant stickleback evolving for 13 generations in a new habitat showed only moderate genome-wide change (Fig. 1b-c), but strong change in many small genomic regions (Fig. 3-4). Allele frequencies (AF) changed on average by 11.4 % (weighted mean |ΔAF|, Fig. 1c), leading to a genomic ‘background’ differentiation between Mayer Lake and Roadside Pond of FST = 0.057 for autosomes and FST = 0.107 for the female sex chromosome (weighted pairwise FST, Fig. 1b). Compared to differentiation observed between natural, postglacial populations, 13 generations of evolution encompassed 41% of the differentiation between lake and stream ecotypes and 22% of the differentiation between stickleback from the large lake Mayer Lake and three independently colonized small ponds on Haida Gwaii (Fig. 2c). Similarly, mean pairwise divergence (DXY), reflecting the sorting of polymorphic, ancient divergent genomic regions between populations on these short time scales, increased marginally (DXY,within populations = 0.0037, DXY,between Mayer-Roadside = 0.0038, t183 = −2.88, P = 0.004) and encompassed 20% of the divergence between naturally occurring lake and stream ecotypes and 9% of the divergence between Mayer Lake and small ponds (Fig. 2b, Supplementary Results).
Fig. 3. Genomic footprints of divergent selection are widespread across the genome.
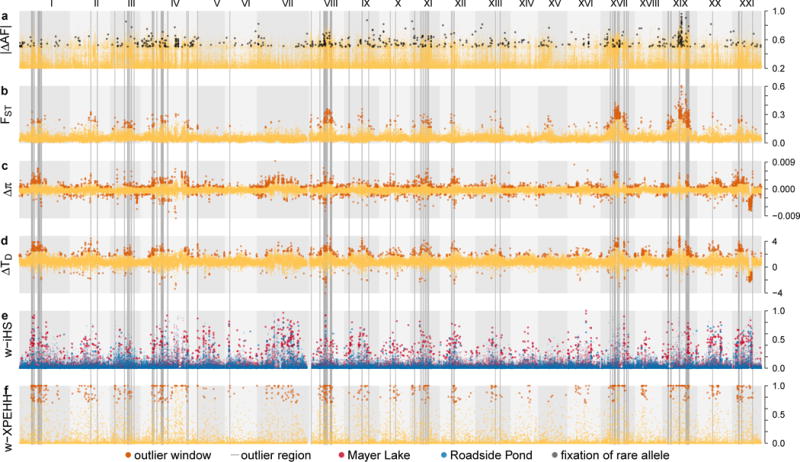
a Absolute allele frequency change (|ΔAF|) at the top 0.1% strongest |ΔAF|-SNPs, with black points highlighting SNPs for which the rarer allele in Mayer Lake went to fixation in Roadside Pond. Grey vertical bars highlight 77 outlier regions across 15 of the 21 stickleback chromosomes (roman numerals) with overlapping top 1% outlier 10kb windows against neutral demographic expectations highlighted with larger, darker points, for the statistics b high differentiation (FST), c change in diversity (Δπ) and d Tajima’s D (ΔTD) and haplotype-based selection statistics e w-iHS and f w-XPEHH (see Methods).
Fig. 4. Local signatures of divergent selection in the genome.
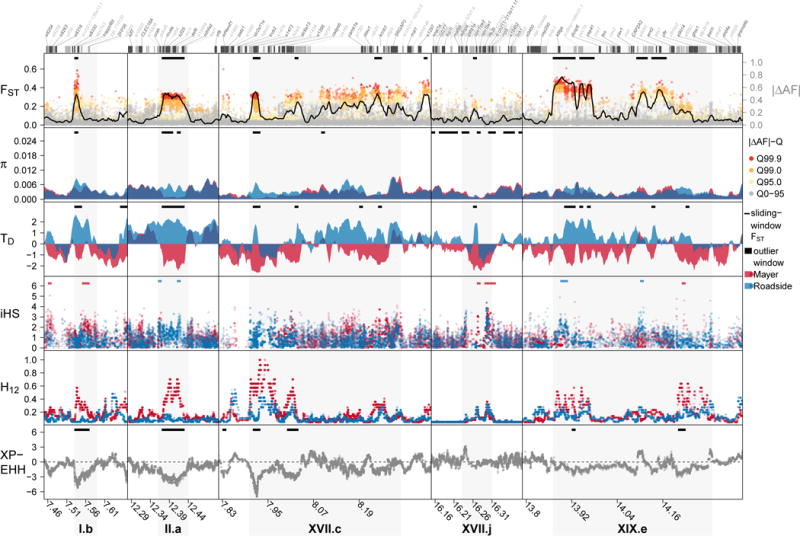
For five of the 77 outlier regions in the genome (grey shading), patterns of differentiation (FST), allele frequency change (|ΔAF|), nucleotide diversity (π), Tajima’s D (TD) and iHS, H12 and XPEHH are shown. Outlier region I.b is centred on a two genes, trappc6bl and bloc1s3, controlling pigmentation in the retina, II.a contains the pigmentation gene mc1r, XVII.j the blue-sensitive colour vision gene opnsw2, likely targets of divergent selection on pigmentation and visual perception. See Supplementary Figs. 5-19 for further outlier regions. The top panel colour code indicates quantiles in the |ΔAF| distribution, black horizontal bars show significant non-overlapping 10kb outlier windows against neutral expectations, gene exons are shown on top (gene names ‘ENSG0000000012345’ shortened to ‘e12345’). Genomic coordinates refer to an improved version of the reference genome80. Lines are sliding-window estimates for 10kb sliding windows with 2.5kb step size, dots are single SNP estimates (|ΔAF|, iHS, XPEHH) or 81-SNP windows (H12).
The transplant of giant Mayer Lake stickleback to Roadside Pond led to a slight loss of genetic diversity and a prominent, genome-wide distortion of the site frequency spectrum (SFS, Fig. 1b, Supplementary Fig. 2a). Mean nucleotide diversity was reduced by 7.4% from πMayer = 0.0047 to πRoadside = 0.0043 (mean 10kb windows, t85940 = 22.56, P < 0.001, Fig. 1b), but local diversity across the genome between both populations remained strongly correlated (Pearson’s r = 0.92, linear regression F2,43444 = 89,485, P < 0.001, Supplementary Fig. 3). The distribution of Tajima’s D was shifted to a positive mean from TD,Mayer = −0.27 to TD,Roadside = 0.60 (mean 10kb windows, t85623 = −197.11, P < 0.001, Fig. 1b), indicating the loss of rare alleles relative to common alleles evident from the observed 1D-SFS (Supplementary Fig. 2a), but Tajima’s D remained correlated across the genome (Pearson’s r = 0.53, linear regression F2,43444 = 17,277, P < 0.001, Supplementary Fig. 3).
Genomic footprints of divergent selection are widespread in the genome
Genome-wide average changes such as diversity loss and a shifted Tajima’s D distribution are likely a product of demographic history, while localized changes in the genome may reflect footprints of selection. To distinguish the two, we reconstructed the demographic history of Mayer Lake and Roadside Pond stickleback (see Methods). The demographic model best fitting the observed 2D-SFS features a bottleneck ~8,200 generations ago, translating to ~12,300 years, in line with the postglacial colonization of Mayer Lake and also recovered the observed population growth of the Roadside Pond population following the transplant41 (Supplementary Fig. 2). We identified signatures of divergent selection between Mayer Lake and Roadside Pond in the genome from outliers for differentiation (FST), change in diversity (Δπ) or Tajima’s D (ΔTD) and haplotype-based selection statistics (iHS, XPEHH) against neutral expectations from demographic history by simulating genomic data under the best-fitting demographic model (Supplementary Fig. 3, see Methods). The simulations reproduced both the observed diversity loss and positive shift in Tajima’s D (Supplementary Fig. 4).
Traces of divergent selection among the habitats are widespread across the genome: we found 77 outlier regions distributed across 15 chromosomes, covering 15.73Mb or 3.6% of the genome (Figs. 3-4, Supplementary Figs. 5-19 and Table 1), exceeding expectations from simulated neutral genomic data (0.16-0.25% of the genome). Outlier regions varied in size between 30kb and 940kb (mean = 204kb, median = 160kb). Three quarters of outlier regions show patterns indicating a near-complete, past selective sweep in Mayer Lake, followed by a quick rise of the previously disfavoured allele to high/intermediate frequency in Roadside Pond: negative Tajima’s D in Mayer Lake, positive Tajima’s D in Roadside Pond, negative XPEHH, significant differentiation and exceptional allele frequency shifts between the populations (Fig. 4, Supplementary Figs. 5-19). The remaining quarter of outlier regions shows an opposite pattern indicating a selective sweep in Roadside Pond but not in Mayer Lake: reduced Tajima’s D and diversity in Roadside Pond, significant H12 patterns for Roadside Pond and a positive XPEHH (Supplementary Figs. 6-19). Both patterns are in agreement with divergent selection between the habitats in the experiment.
We computed linkage disequilibrium (LD) between outlier regions to test whether divergent selection acted on a single region genomic with others hitchhiking, or whether multiple regions responded independently to divergent selection. Significant inter-chromosomal LD was found mainly between outlier regions in the Mayer Lake population (Supplementary Fig. 20), indicating that several regions involved in divergent selection in the experiment are not segregating fully independently in the source population. In the transplant population Roadside Pond however, we found only seven significant inter-chromosomal associations (Supplementary Fig. 20), suggesting that while outlier regions on different chromosomes responded largely independently to divergent selection in the experiment, some uncertainty remains about the exact number of independently selected regions.
Adaptive differentiation, defined as the top 5% single SNP FST estimates from each genomic outlier region, ranged from FST = 0.25 to FST = 0.76 with a mean of FST = 0.44. Allele frequency change at these SNPs ranged from |ΔAF| = 18% to |ΔAF| = 81% with a mean of |ΔAF| = 51%, which under a model of purely selection-driven change would correspond to selection coefficients between s = 0.24 and s = 1 with mean s = 0.62. Compared to naturally evolved, postglacial ecotypes, genomic adaptive differentiation after only 13 generations of evolution in a new ‘ecological theatre’ thus encompassed 72% of the degree of adaptive differentiation found between postglacial lake and stream ecotypes46 and already exceeds adaptive differentiation found between giant Mayer Lake stickleback and its corresponding parapatric stream ecotype (Fig. 2d).
Genomic targets and parallel evolution in experiment and adaptive radiation
We identified potential targets and sources of divergent selection from overlapping genes and QTL47 and from genotype-environment and genotype-phenotype (GE/GP) associations in the Haida Gwaii adaptive radiation. In the latter, we tested whether genomic variation in each outlier region was associated with change in phenotypic and ecological properties in the selection experiment and across one marine and 25 freshwater populations on Haida Gwaii (see Methods, Fig. 5). We found 654 QTL overlapping with outlier regions and 336 candidate genes near the centre of each outlier regions’ selective sweep signature, but no gene ontology term enrichment (Figs. 4 and 5, Supplementary Figs. 5-19 and Tables 2 and 3). 36 outlier regions showed parallel GE/GP associations (Fig. 5).
Fig. 5. Outlier regions and overlapping QTL, candidate genes and genome-phenotype/ecology associations across the adaptive radiation.
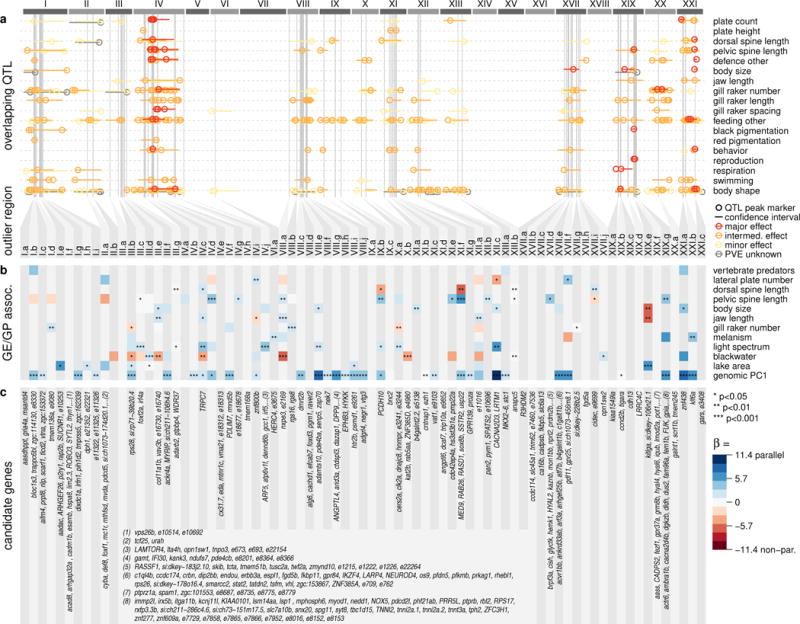
a Distribution of overlapping quantitative trait loci (QTL). Circles indicate QTL peak markers, horizontal bars confidence intervals and colour codes the effect sizes: major (percentage variance explained, PVE > 25%), intermediate (25% > PVE > 5%) and minor effect QTL (PVE < 5%). b Genome vs. environment and phenotype associations (GE/GP assoc.) and directionality of phenotypic and genomic change between selection experiment and Haida Gwaii adaptive radiation. Predictors retained in generalized linear model for each outlier region are shown in coloured squares, with blue boxes representing parallel genomic and phenotypic/ecological change and red boxes non-parallel change (except for genomic PC1, for which directionality cannot be inferred). The colour code shows the relative effect sizes (β). c List of candidate genes centred on divergent selection patterns in outlier regions.
In line with predation landscape being the most important axis of divergent selection in the adaptive radiation30, 96 QTL and one candidate gene controlling predator deference traits overlap with outlier regions. Among these are major effect QTL for lateral plate number, dorsal spine, pelvic spine and pelvic girdle length and many intermediate and minor effect QTL for these traits on additional chromosomes (Fig. 5, Supplementary Table 2). Remarkably, phenotypic variation in pelvic and dorsal spine length across the Haida Gwaii radiation is associated with genomic variation in eleven and six outlier regions, respectively, and the phenotypic and genomic change observed in the selection experiment paralleled the adaptive radiation in ten (pelvic spine) and four (dorsal spine) of these region (Fig. 5). Variation in plate number across the radiation is associated with genomic variation in outlier regions IV.i and XVII.f (parallel) and XII.c (non-parallel). While most annotations for candidate genes did not allow to draw conclusions on defence phenotypes, the eda gene in outlier region IV.e controls lateral plate number48-50 and several associated traits such as lateral line pattern and schooling behavior51,52 (Supplementary Fig. 9). Genetic variation in the eda region may be responsible for the observed reduction of plate number in the selection experiment41, while the lack of an association across the adaptive radiation suggests an involvement of different alleles or genes in other populations.
Variation in light spectrum across the adaptive radiation, the second most important axis of divergent selection30, and the presence of blackwater show many strong, parallel associations with genetic variation in outlier regions also containing multiple candidate genes involved in (colour) vision (Fig. 5). The six outlier regions most strongly associated with light spectrum in parallel between adaptive radiation and the selection experiment contain the photoreceptor opn1sw1 sensitive to UV-light53, TRPC7 involved in eye physiology54, CACNA2D3 associated with night blindness in humans55, the gene atp6v1f involved in retinal pigmentation56,57, the nervous system development genes dennd6b and cers2a expressed in the eye, lens and retina and adamts10 involved in lens development58. In addition, outlier region XVII.j shows parallel association with blackwater habitats and contains the blue-light sensitive photoreceptor opn1sw2, which we demonstrated previously to be under selection between blackwater and clearwater habitats both across adaptive radiation and selection experiment59. Repeated adaptation to light spectrum thus has led to multiple signatures of parallel adaptation on visual perception genes.
Many feeding morphology QTL (n = 277) overlap with outlier regions. Jaw length variation in the adaptive radiation is associated with six outlier regions on five chromosomes, four in parallel and two non-parallel (Fig. 5). QTL for gill raker number overlap with many outlier regions, with two parallel associations across the adaptive radiation on chromosomes I and VIII (Fig. 5, Supplementary Table 2). Similarly, many gill raker length QTL overlap with outlier regions, in particular intermediate effect loci on chromosomes IV and VIII. Also for gill raker spacing, intermediate effect QTL on chromosomes IV and XX and minor effect QTL overlap with outlier regions. The widespread genomic architecture of many feeding morphology related traits thus broadly overlaps with genomic regions under divergent selection. Together with several candidate genes involved in craniofacial development or various metabolic processes (Supplementary Table 3), these overlaps might reflect selection on feeding morphology or diet after the shift from a zooplankton to a benthic invertebrate dominated habitat.
Strikingly, we found several candidate genes (kitlga, mc1r, gpr25, foxd3, bnc2, STYL2, tcta, trappc6bl, bloc1s3, MYRIP, atp6v1f) in outlier regions involved in pigmentation57,60-65. These might be associated with the decreased melanism found in Roadside Pond stickleback, the loss of red nuptial coloration or with sexual selection for blue signals in blackwater Mayer Lake45. The two first genes kitlga and mc1r are well-known targets of divergent selection associated with melanin pigmentation in stickleback, mammals and reptiles61,65,66; and the former overlaps with a large effect QTL for black pigmentation in other wild stickleback populations65. The two genes STYL2 and bloc1s3 overlap with intermediate-effect QTL for red pigmentation67. Outlier regions containing these genes do not show associations with melanism across the Haida Gwaii radiation, nor do melanism-associated regions across the adaptive radiation contain known pigmentation genes (Fig. 5). Finally, body size shows a non-parallel association with outlier region XIX.e containing an intermediate-effect QTL for body size (Supplementary Table 2), while additional, parallel associations are not overlapping with known QTL or gene function. Many outlier regions are associated with several ecological and phenotypic traits, other QTL and with geography (Fig. 5). This might reflect modular trait architecture47,68, selection-driven clustering of adaptive variation or indirect associations with correlated ecological and phenotypic variables across the adaptive radiation.
Discussion
Adaptation of a highly derived threespine stickleback ecotype in the Haida Gwaii adaptive radiation to the opposite multifarious selection regime along the three major axes of selection resulted in rapid and widespread adaptive genomic change over only 13 generations. We previously demonstrated rapid phenotypic change in the selection experiment, encompassing approximately 30% of the phenotypic divergence of 12,000 year old natural populations41 (Fig. 1a, 2a). Here, we found that underlying adaptive genomic variation also responded very fast to divergent selection, with on average 72% of the expected change occurring in the first 13 generations. The evolutionary rate of genomic adaptation is thus very high and comparable in speed to contemporary evolution of Brassica rapa adapting to drought over 7 generations18, showing adaptive differentiation of FST = 0.17-0.44, or to Darwin’s Finches undergoing drought-induced ecological character displacement69, with a selection coefficient of 0.59 on a major effect locus. Genomic adaptation in our selection experiment is faster than in marine stickleback adapting to freshwater habitat over approx. 17 generations in Russia26, where adaptive alleles increased in frequency by 10-50%. Both phenotype, with 0.15 haldanes in 12 generations41, and adaptive genomic variation with a mean FST of 0.46 in 13 generations, thus evolved at a very rapid rate typical of populations colonizing a new adaptive zone in an adaptive radiation70. Rapid adaptation to multifarious divergent selection thus seems to occur similarly fast or faster than adaptation to a single selective force or with a single major locus as in some of these other examples.
Remarkably, the genomic basis of predator defence morphology, colour vision, feeding morphology and pigmentation overlapped with adaptive genomic change in the selection experiment, including some ‘master adaptation genes’ such as eda, opnsw1/2, kitlg and mc1r, frequently involved in repeated divergent adaptation of body armor, colour vision and pigmentation48,49,59,61,65,66,71. Many of these regions showed parallel associations with variation in traits and ecosystem variables across the Haida Gwaii adaptive radiation. This suggests that there was no major gap between phenotypic and genomic change for most of the diverging traits on the contemporary time scale of the selection experiment. Although phenotypic plasticity likely contributed to near-instantaneous phenotypic adaptation for traits such as eye size or gill raker length41, our genomic findings suggest that selection has operated on genetic variation underlying most diverging phenotypic traits. Much of the genetic variation had to be shared with the Haida Gwaii adaptive radiation as standing genetic variation within Mayer Lake and across the archipelago, as outlier regions in the selection experiment evolved in parallel to the radiation in 36 of 77 genomic outlier regions (Fig. 5). Determinism of adaptive evolution thus is not only prevalent in phenotype41, but also in the genome, predicted by the three major axes of natural selection in the radiation30.
Genomic parallelism is somewhat surprising, given that a highly derived phenotype in the adaptive radiation was the source for the selection experiment. Mayer Lake stickleback are vertebrate-predation, blackwater and zooplankton adapted specialists with little phenotypic variance. It is thus conceivable that such a specialist would have lacked the necessary standing genetic variation for rapid adaptation to the opposite extreme ecological theatre. In addition, a bottleneck during the selection experiment reduced genomic variation by 7%. Indeed, some shared alleles may have been lost during 12,000 years of adaptation to a blackwater lake: strong, but non-parallel associations between blackwater habitation and outlier regions on several chromosomes in the selection experiment suggest that different alleles from the adaptive radiation were favoured once the blackwater population had to re-adapt to the clearwater Roadside Pond (Fig. 5). Nevertheless, the Mayer Lake population still retained shared genetic variation at many loci for parallel adaptation in feeding morphology, defence morphology, pigmentation and vision. Genetic variation in Mayer Lake may have been maintained by disruptive or fluctuating selection within Mayer Lake37,38,72, by a large population size in Mayer Lake (Fig. 5) or occasional introgression of adaptive alleles from adjacent stream ecotypes46,73 or nearby lake ecotypes. Linkage disequilibrium between physically unlinked genomic regions containing such variation in Mayer Lake suggests that standing genetic variation is correlated in some individuals (Supplementary Fig. 20), compatible with all three hypotheses. Alternatively, bottlenecks during the colonization of Mayer Lake 12,000 years ago and during the experiment may not have been strong enough to remove adaptive genetic variation, such as in biological invasions where adaptive potential is usually not hampered with reductions in genetic diversity of 15-20%12,74. Drift during habitat shifts may thus rarely hamper sequential and rapid colonization of new niches in an adaptive radiation.
Our results confirm that natural selection generally overrides historical contingency at the genomic level in the adaptive radiation of threespine stickleback on Haida Gwaii, in line with phenotypic patterns30 and previous genomic results for lake-stream and marine-freshwater divergence7,26,27,34,46, and in spite of bottlenecks upon colonization and strong selection acting on new colonizers. Similar selection-driven phenotypic and genomic determinism has been found in other adaptive radiations, based on adaptive introgression or a hybrid swarm origin rather than on standing genetic variation as in the stickleback, e.g. in East African cichlids9,75,76, in Darwin’s Finches69,77 or in Heliconius butterflies78. Except for Darwin’s Finches79 and our experiment however, it remains to be shown whether the colonization of new adaptive zones can occur similarly fast on contemporary time scales. Our findings suggests that multifarious divergent selection acts rapidly on many different genes and regions in the genome and that large steps in both phenotypic and genomic adaptation in adaptive radiations are taken within the first few generations, even when starting from a highly derived adaptive radiation member. Adaptive radiations may thus rapidly advance on contemporary time scales, given enough standing genetic variation in key functional traits and an ecological theatre offering new niche space and imposing multifarious divergent selection.
Methods
Experimental setup, sampling, ethics statement
In May 1993, 100 adult giant threespine stickleback, 50 males and 50 females, were captured in Mayer Lake and transferred to Roadside Pond (aka ‘Mayer Pond’ in Leaver and Reimchen41). In 2004, 12 females were captured in Mayer Lake and in 2012, 11 females were caught in Roadside Pond corresponding to ~13 generations after release, assuming a population-average generation time of 1.5 years. In addition, stickleback from 25 freshwater populations across the Haida Gwaii archipelago representing the range of successfully colonized freshwater habitats, were sampled between 1993 and 2012 (see Table 1 in Marques, et al.59). Stickleback were captured using minnow traps and euthanized with an overdose of tricaine methanesulfonate (MS-222) in agreement with British Columbia’s guidelines for scientific fish collection, under Ministry of Environment permits SM09-51584 and SM10-62059 and University of Victoria Aquatic Unit facility Standard Operating Procedure OA2003. Collections in Naikoon Provincial Park and Drizzle Lake Ecological Reserve were carried out under park use permits: 103171, 103172, 104795 and 104796. Samples were stored in 70% ethanol and the genomes of 58 individuals, including 12 Mayer Lake, 11 Roadside Pond and 1-4 individuals from 25 Haida Gwaii freshwater populations and two mainland British Columbia freshwater populations and one marine population were re-sequenced and are listed in Table 1 in our previous study focussing on the evolution of colour vision59. Alignment, variant and genotype calling and filtering is described in Marques, et al.59. Note that we aligned against an improved ordering of scaffolds of the reference stickleback genome and all genomic coordinates refer to this improved reference80. For the analyses in this study, we used either raw aligned reads with mapping quality ≥ 17 and bases with quality ≥ 17 for statistics computed on genotype likelihoods or one of two subsets from the SNP dataset containing 7,888,602 high-quality SNPs among the 58 sequenced individuals for principal component analysis (unphased SNPs) and haplotype-based statistics (phased and imputed SNPs). The first, ‘selection experiment’ subset contained 4,180,622 SNPs among 12 Mayer Lake and 11 Roadside Pond individuals and the second ‘adaptive radiation’ dataset 6,564,510 SNPs among one marine and 25 natural Haida Gwaii freshwater populations (including Mayer Lake) with one randomly picked individual per population. Read-backed phasing and imputation in both adaptive radiation and selection experiment SNP datasets was performed with SHAPEIT v2.r79081, with phase-informative reads covering on average 7.5% of all heterozygote genotypes and 30.9% of all graph segments.
Population genomic analyses
We described genomic change in the selection experiment with the statistics absolute allele frequency change (|ΔAF|), differentiation (FST), nucleotide diversity (π), and site frequency spectrum (Tajima’s D) computed from genotype likelihoods. We first computed the unfolded two-dimensional site frequency spectrum (2D-SFS) between Mayer Lake and Roadside Pond from aligned autosomal reads with angsd v0.915 and the reference genome as ancestral state. We used the 2D-SFS as prior to estimate FST at single sites as well as FST, Tajima’s D and π in windows of 10kb width, either non-overlapping or sliding with 2kb step size from raw aligned reads in angsd82-84 with the filters as outlined above. Single site FST was calculated from site alphas and betas computed by angsd. We also estimated minor allele frequencies in each population in angsd to calculate absolute allele frequency change (|ΔAF|). We calculated a weighted mean |ΔAF| with the weighted.mean function in R, using each SNP’s ‘starting allele frequency’, the minor allele frequency estimated for the Mayer Lake population, as weights.
We compared the amount of genomic change in the selection experiment to natural populations in the Haida Gwaii radiation for genome-wide differentiation (FST) and absolute divergence (DXY). For absolute divergence between the populations, we computed the unfolded 2D-SFS for pairs of individuals and calculated mean pairwise DXY from the SFS using custom scripts. We computed pairwise DXY within populations Mayer Lake and Roadside Pond to get a baseline of expected pairwise DXY. Then we computed pairwise DXY between populations Mayer Lake vs. Roadside Pond, three lake vs. stream ecotype populations (Mayer Lake vs. Gold Creek, Drizzle Lake vs. inlet and outlet, Spence Lake vs. outlet) and three large lake vs. small pond populations (Mayer Lake vs. Branta, Laurel, Solstice). We also estimated absolute divergence from the proportion of fixed differences among polymorphic sites between pairs of individuals. We annotated SNPs in the ‘adaptive radiation’ dataset using the Ensembl Variant Effect Predictor85 and used the Picard Tool LiftoverVCF v2.7.086 to move the SNPs into the original annotation7. We partitioned SNPs into missense, synonymous, intron, regulatory and intergenic SNPs using SnpSift v4.287 and computed the proportion of fixed differences from the 012 output format of vcftools v0.1.1588. Genome-wide differentiation between populations (FST) was calculated from previously published SNP array data34,46 for the lake vs. stream and large lake vs. small pond comparisons. We ran a locus-by-locus AMOVA for SNPs with at least 3 genotypes per population in arlequin v3.5.2.289 (Supplementary Table 4), resulting in >400 SNPs per comparison that should give an unbiased genome-wide FST estimate90. For lake vs. stream comparisons with multiple stream populations (e.g. Drizzle: inlet and outlet; Mayer: Gold, Woodpile and Spam Creek46), we used hierarchical AMOVAs with each population retained as separate sample but grouped into either lake or stream group (Supplementary Table 4). Alpha and beta estimates from the AMOVA and the FST computation in angsd for Mayer vs. Roadside were pooled to sums of nominators and denominators to get a weighed mean FST estimate91.
We identified likely genomic targets of divergent selection between the source and transplant population with a two-step outlier approach. First, we inferred an optimal, neutral demographic model on the 2D-SFS using fastsimcoal2 v2.692. Second, we simulated neutral genomic data under the best demographic model, against which we identified outlying genomic regions in the observed data. We folded the 2D-SFS using custom scripts, fit 12 different demographic models (Supplementary Fig. 2b and Data 1) to the observed 2D-SFS with fastsimcoal2 and compared their likelihoods using the Akaike information criterion (AIC) following Excoffier, et al.92. We maximized the likelihood of each model from 100 random starting parameter combinations in 10 to max. 50 ECM cycles, with a stopping criterion of 0.001. 100,000 coalescent simulations were used to approximate the expected 2D-SFS. In all simulations, we used a mutation rate of 1.7E-8, following Feulner, et al.93, a founding population size of 2N = 200 individuals for the Roadside Pond population and generated Mayer Lake samples 5 generations prior to Roadside Pond to account for different sampling years (Supplementary Data 1). Likelihood and parameter estimates for each model were obtained from the run with the highest likelihood among the 100 optimizations. For the parameters of the best model, we estimated 95%-confidence intervals as empirical percentiles on parameters from the best of 10 optimization runs on 100 bootstrap replicates of the observed 2D-SFS, with each optimization started from the original parameters of the best model. We computed bootstrap replicates for each autosome separately in angsd and combined 2D-SFS from different autosome with custom scripts. We simulated neutral genomic data under the best demographic model with fastsimcoal2 for four different recombination rates, high = 4-16 cM/Mb, intermediate = 1.5-4 cM/Mb, low = 0.5-1.5 cM/Mb and very low = 0-0.05 cM/Mb. For each recombination range, we generated 1,000 replicate DNA segments of 1Mb length, with a mutation rate of 1.7E-8 and a random recombination rate from that range, assuming a uniform distribution (very low, low recombination rate) or log-uniform distribution (intermediate, high recombination rate) that reflect the frequency of recombination rate variation in the stickleback genome80. We transformed the simulated data into VCF format using custom scripts and computed weighted FST, Tajima’s D and π in non-overlapping 10kb windows using vcftools v0.1.1488.
A selective sweep caused by divergent selection between habitats is expected to lead to excess differentiation (FST) between populations at and around the site under selection, to reduced diversity in the population experiencing the selective sweep and to a shifted SFS, reflected by a strongly negative Tajima’s D upon completion of the sweep. In addition, haplotype-based statistics are able to detect soft and incomplete sweeps within a populations (iHS94 and H1295) or completed sweeps in one of two populations (XPEHH)96. We computed the haplotype-based selection statistics integrated haplotype score (iHS)94, H1295, and cross-population extended haplotype homozygosity (XPEHH)96 for phased and imputed bi-allelic SNPs with minor allele frequency > 5% in the ‘selection experiment’ dataset and for simulated SNP data. We computed iHS and H12 separately for Mayer Lake and Roadside Pond populations, using only SNPs with minor allele frequency >5% the respective population. We calculated the proportion of extreme iHS and XPEHH values (‘w-iHS’, the proportion of |iHS| > 2, following Voight, et al.94 and ‘w-XPEHH’, the proportion of |XPEHH| > 2) in non-overlapping 10kb windows containing more than 10 iHS or XPEHH estimates, respectively, for both observed and simulated datasets. We used selscan v1.1.0b97 with default parameters to compute iHS and XPEHH and the proportion of extreme values in 10kb windows. We also computed H12 for the observed dataset using scripts published alongside the H12 method95, with 81 SNPs bin width, resulting in on average 8.3kb wide windows close to the 10kb windows identified as optimal and robust to various demographic scenarios by Garud, et al.95.
We identified outliers against neutral expectations for six 10kb non-overlapping window statistics: FST, change in nucleotide diversity (Δπ = πRoadside-πMayer), change in Tajima’s D (ΔTD = TD,Roadside - TD,Mayer), w-iHSMayer, w-iHSRoadside and w-XPEHH. Our ability to detect signatures of selective sweeps with window-based statistics depends on local recombination rate, with stronger hitchhiking in low recombination rate regions leading to more prominent signals and a greater variation in such statistics (Supplementary Figs. 3-4). We therefore identified outlier windows separately in genomic regions with high, intermediate, low and very low recombination rate (see above and Supplementary Fig. 4). We assigned 10kb windows to recombination rate bins according to local recombination rates estimated in the middle of each 10kb window as described previously59. For each 10kb window, we computed the empirical quantile of the observed FST, Δπ, ΔTD, w-iHSMayer, w-iHSRoadside and w-XPEHH value against the simulated distribution of the statistic in the respective recombination bin with the function ‘ecdf’ in R v3.3.198. We converted quantiles to two-sided p-values for Δπ and ΔTD and one-sided p-values for the other statistics.
We identified genomic regions likely under divergent selection between Mayer Lake and Roadside Pond (‘outlier regions’) based on overlapping outlier signatures in these six selection statistics. To capture the shared signal, we applied Fisher’s combined probability test to the four to six p-values in each 10kb window, as implemented in the R-package ‘metap’. P-values from Fisher’s combined probability test were corrected for multiple testing using the false discovery rate method99 implemented in ‘p.adjust’, converted to q-values (= 1 – padj) and z-transformed using the R function ‘qnorm’. We used a Hidden Markov model (HMM) approach to group adjacent 10bk windows into outlier regions. The z-transformed q-values were used as input to HMMs with two or three normally distributed states. We optimized parameters of both HMMs from 1,000 random starting parameters using the Baum-Welch algorithm implemented in the R-package ‘HiddenMarkov’. The three-state HMM better fit the data according to the Akaike information criterion and was thus used to assign all 42,996 10kb-windows to the three states using the Viterbi algorithm. Preliminary outlier regions were obtained from joining adjacent windows assigned to the state capturing highly significant Fisher’s combined probability test p-values. Then, only outlier regions that contained significant outliers with p < 0.01 for each of the statistics (a) FST, (b) Δπ or ΔTD and (c) w-iHSMayer, w-iHSRoadside or w-XPEHH as well as outlier regions with strongly aligned signatures for these statistics plus H12Mayer or H12Roadside were retained in the final set of outlier regions reflecting divergent selection between Mayer Lake and Roadside Pond. We did not further analyse signatures of e.g. shared directional or background selection, which should result in reduced diversity and Tajima’s D or significant haplotype-based statistics in both populations, but not in differentiation between the populations.
We quantified adaptive differentiation between the source and transplant population by computing single SNP FST and retaining the top 5% FST SNPs in each outlier region, thereby containing likely the few SNPs under selection and many more linked, hitchhiking SNPs in each region affected by divergent selection. We compared this distribution of adaptive differentiation to adaptive differentiation among three postglacial pairs of lake and stream ecotypes on Haida Gwaii, using FST estimates from only those SNPs previously identified to be under selection in the respective ecotype comparison46, which also likely reflect SNPs hitchhiking and to a lesser degree direct targets of selection. For the top 5% FST SNPs in each outlier region, we computed the expected selection coefficient under a pure selection model, based on the allele frequency changes at these SNPs over 12.7 generations and assuming incomplete dominance h = 0.5 following equation 3.2 in Gillespie100. These calculations likely overestimate selection coefficients due to unaccounted contributions of drift and should thus be interpreted with caution.
We computed linkage disequilibrium (LD) as r2 between the most divergent 15 SNPs polymorphic in both Mayer Lake and Roadside Pond for each outlier region using vcftools, both within and between chromosomes. We assessed whether LD between outlier regions on different chromosomes exceeded neutral expectations of no linkage disequilibrium. We derived the neutral distribution of LD with the observed sample sizes by randomly choosing SNPs outside outlier regions from each chromosome with at least 500kb distance between SNPs on the same chromosome (N = 617) and computing inter-chromosomal LD between these SNPs. Then, we determined whether the mean observed LD between two outlier regions is greater than the 95%-quantile of the neutral observed inter-chromosomal LD distribution.
We associated the genomic signatures of divergent selection with potential sources of selection in the experiment and the adaptive radiation by studying their gene content and the gene’s functional annotations, from their overlap with previously described stickleback QTL that have been mapped in genetic studies of specific phenotypes47, and from genotype-phenotype/ecology associations across the Haida Gwaii radiation. First, we identified candidate genes by inspecting the patterns of 10kb sliding window statistics FST, πMayer, πRoadside, Tajima’s D and single locus statistics iHSMayer, iHSRoadside, H12Mayer, H12Roadside, XPEHH and |ΔAF| visually (Fig. 4, Supplementary Figs. 5-19) and retained a list of genes centred on or adjacent to selective sweep signatures (Supplementary Table 3). Then, we tested this list of candidate genes for enrichment of gene ontology terms using the String database v10101 and retrieved functional and expression information from zebrafish58 and related mouse, rat and human databases102,103. In addition, we identified overlap between outlier regions and QTL previously identified in other stickleback populations in the Northern Hemisphere using the list of Peichel and Marques47 and the peak marker location and confidence intervals reported there. QTL were grouped into major, intermediate or minor effect size classes respectively, when they explained > 25%, between 5% and 25%, or < 5%, respectively, of the phenotypic variation in the experiments47.
Finally, we determined whether outlier regions in the selection experiment evolved in predictable directions given the environmental contrast. In the absence of replicate experimental ponds, we used genotype-phenotype and genotype-environment associations across the larger Haida Gwaii stickleback adaptive radiation with many natural replicates to infer whether the same genomic regions evolved in parallel direction. For each outlier region, we identified the SNPs with the strongest allele frequency change between Mayer Lake and Roadside Pond (top 1% |ΔAF|), assigned the alleles as Mayer Lake-like or Roadside Pond-like based on in which population they are more frequent, extracted the genotypes for those SNPs from the adaptive radiation SNP dataset containing single genomes of one marine and 25 freshwater populations including Mayer Lake, recoded the alleles as 0 (Mayer-like) or 1 (Roadside-like), combined them into multi-dimensional scaling (MDS) factors and polarized the MDS factors for Mayer Lake to be represented by low and Roadside Pond by high values in R. Next, we used the genomic MDS factor of each outlier region as response variable in a generalized linear model with 12 phenotypic and ecological properties of the 26 Haida Gwaii populations as predictors (see below). For each outlier region’s generalized linear model, we performed variable selection by iteratively removing non-significant predictors (χ2 tests, P > 0.1). Parallelism was inferred if the MDS factor of an outlier region was positively associated with the predictors, no parallelism if the association was negative.
We used the presence of blackwater (transmission at 400nm < 74%), of vertebrate predators in a population, and whether the population consists of predominantly melanistic phenotypes as binary predictors. Continuous predictors were lake area (log-transformed), light spectrum, mean body size (standard length), mean lateral plate number (excluding fully plated individuals), mean dorsal spine length, mean pelvic spine length, mean jaw length and mean gill raker number, the linear measurements size-corrected as described in Reimchen, et al.30 and all scaled and standardized to mean zero and standard deviation one. We polarized all predictors so that the change from Mayer Lake to Roadside Pond would represent a positive shift, i.e. a shift from blackwater to clearwater, from melanism to reduced melanism, a decrease in lake area, body size, lateral plate number, pelvic spine length and gill raker number, and an increase in jaw length41. As last predictor, we included geographic structuring by using the first principal components axis from genomic variation in the adaptive radiation SNP dataset. We used genotype likelihoods from the adaptive radiation SNP dataset, computed the site allele frequency spectrum to get a covariance matrix as implemented in angsd and ngsCovar84 and performed the eigenvalue decomposition in R to get the first principal component. We visualized phenotypic change in the selection experiment41 and phenotypic divergence between Mayer Lake, it’s stream ecotype (Mayer Stream = Gold Creek46), Laurel, Branta and Solstice39 from the data of this earlier work by using the size-correction of Leaver and Reimchen41 within each population for all datasets combined. Analyses were performed on Compute Canada’s WestGrid computer cluster infrastructure (www.westgrid.ca).
Supplementary Material
Acknowledgments
We thank Bruce Deagle, Stephen D. Leaver, Craig B. Lowe, Shannon D. Brady, Jason Turner, Kerstin Lindblad-Toh and the Broad Genomics Platforms for help with sequences, samples and morphometric analysis and Belaid Moa for bioinformatics support. This work was funded by the National Research Council Canada grant NRC2354 to T.E.R. and the National Institute of Health grants 3P50HG002568-09S1 ARRA and 3P50HG002568 to D.M.K.
Footnotes
Data availability. Aligned sequences can be accessed under accession SRP100209 on the NCBI short read archive (www.ncbi.nlm.nih.gov/sra).
Code availability. Custom scripts to compute DXY from the SFS, to fold 2D-SFS and to fit HMM are available on https://github.com/marqueda.
Author contributions
T.E.R. conceived the study, ran the experiment, collected fish and ecological data in the field and acquired morphological data; D.M.K., F.C.J. and F.D.P. generated sequencing data and genotype calls, D.A.M. designed and performed all subsequent analyses and wrote the manuscript with contributions from all co-authors.
Competing interests
The authors declare no competing financial interests.
Requests for materials should be addressed to T.E.R.
References
- 1.Schluter D. The ecology of adaptive radiation. Oxford University Press; 2000. [Google Scholar]
- 2.Grant PR. Speciation and the Adaptive Radiation of Darwin Finches. American Scientist. 1981;69:653–663. [Google Scholar]
- 3.Losos JB, Jackman TR, Larson A, Queiroz K, Rodriguez-Schettino L. Contingency and determinism in replicated adaptive radiations of island lizards. Science. 1998;279:2115–2118. doi: 10.1126/science.279.5359.2115. [DOI] [PubMed] [Google Scholar]
- 4.West-Eberhard MJ. Developmental plasticity and evolution. Oxford University Press; 2003. [Google Scholar]
- 5.Muschick M, Barluenga M, Salzburger W, Meyer A. Adaptive phenotypic plasticity in the Midas cichlid fish pharyngeal jaw and its relevance in adaptive radiation. BMC Evol Biol. 2011;11:116. doi: 10.1186/1471-2148-11-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrett RD, Schluter D. Adaptation from standing genetic variation. Trends Ecol Evol. 2008;23:38–44. doi: 10.1016/j.tree.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Jones FC, et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature. 2012;484:55–61. doi: 10.1038/nature10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seehausen O. Hybridization and adaptive radiation. Trends Ecol Evol. 2004;19:198–207. doi: 10.1016/j.tree.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Meier JI, et al. Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat Commun. 2017;8:14363. doi: 10.1038/ncomms14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Price TD, Qvarnstrom A, Irwin DE. The role of phenotypic plasticity in driving genetic evolution. Proc Biol Sci. 2003;270:1433–1440. doi: 10.1098/rspb.2003.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlotterer C, Kofler R, Versace E, Tobler R, Franssen SU. Combining experimental evolution with next-generation sequencing: a powerful tool to study adaptation from standing genetic variation. Heredity (Edinb) 2016;116:248. doi: 10.1038/hdy.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrett SCH, I CR, Dlugosch KM, Rieseberg LH. Invasion Genetics: The Baker and Stebbins Legacy. Wiley-Blackwell; 2016. [Google Scholar]
- 13.Burke MK, et al. Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature. 2010;467:587–590. doi: 10.1038/nature09352. [DOI] [PubMed] [Google Scholar]
- 14.Fritz ML, et al. Contemporary evolution of a Lepidopteran species, Heliothis virescens, in response to modern agricultural practices. Mol Ecol. 2018;27:167–181. doi: 10.1111/mec.14430. [DOI] [PubMed] [Google Scholar]
- 15.Tobler R, et al. Massive habitat-specific genomic response in D. melanogaster populations during experimental evolution in hot and cold environments. Mol Biol Evol. 2014;31:364–375. doi: 10.1093/molbev/mst205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graves JL, Jr, et al. Genomics of parallel experimental evolution in Drosophila. Mol Biol Evol. 2017 doi: 10.1093/molbev/msw282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang Y, Wright SI, Agrawal AF. Genome-wide patterns of genetic variation within and among alternative selective regimes. PLoS Genet. 2014;10:e1004527. doi: 10.1371/journal.pgen.1004527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franks SJ, Kane NC, O’Hara NB, Tittes S, Rest JS. Rapid genome-wide evolution in Brassica rapa populations following drought revealed by sequencing of ancestral and descendant gene pools. Mol Ecol. 2016;25:3622–3631. doi: 10.1111/mec.13615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van’t Hof AE, Edmonds N, Dalikova M, Marec F, Saccheri IJ. Industrial melanism in British peppered moths has a singular and recent mutational origin. Science. 2011;332:958–960. doi: 10.1126/science.1203043. [DOI] [PubMed] [Google Scholar]
- 20.Reid NM, et al. The genomic landscape of rapid repeated evolutionary adaptation to toxic pollution in wild fish. Science. 2016;354:1305–1308. doi: 10.1126/science.aah4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fraser BA, Kunstner A, Reznick DN, Dreyer C, Weigel D. Population genomics of natural and experimental populations of guppies (Poecilia reticulata) Mol Ecol. 2015;24:389–408. doi: 10.1111/mec.13022. [DOI] [PubMed] [Google Scholar]
- 22.Hendry AP, Kinnison MT. Perspective: The pace of modern life: Measuring rates of contemporary microevolution. Evolution. 1999;53:1637–1653. doi: 10.1111/j.1558-5646.1999.tb04550.x. [DOI] [PubMed] [Google Scholar]
- 23.Reznick DN, Ghalambor CK. The population ecology of contemporary adaptations: what empirical studies reveal about the conditions that promote adaptive evolution. Genetica. 2001;112–113:183–198. [PubMed] [Google Scholar]
- 24.Stockwell CA, Hendry AP, Kinnison MT. Contemporary evolution meets conservation biology. Trends Ecol Evol. 2003;18:94–101. [Google Scholar]
- 25.Bell MA, Aguirre WE, Buck NJ. Twelve years of contemporary armor evolution in a threespine stickleback population. Evolution. 2004;58:814–824. doi: 10.1111/j.0014-3820.2004.tb00414.x. [DOI] [PubMed] [Google Scholar]
- 26.Terekhanova NV, et al. Fast evolution from precast bricks: genomics of young freshwater populations of threespine stickleback Gasterosteus aculeatus. PLoS Genet. 2014;10:e1004696. doi: 10.1371/journal.pgen.1004696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lescak EA, et al. Evolution of stickleback in 50 years on earthquake-uplifted islands. Proc Natl Acad Sci U S A. 2015;112:E7204–7212. doi: 10.1073/pnas.1512020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aguirre WE, Bell MA. Twenty years of body shape evolution in a threespine stickleback population adapting to a lake environment. Biol J Linn Soc. 2012;105:817–831. [Google Scholar]
- 29.Hohenlohe PA, et al. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 2010;6:e1000862. doi: 10.1371/journal.pgen.1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reimchen TE, Bergstrom C, Nosil P. Natural selection and the adaptive radiation of Haida Gwaii stickleback. Evol Ecol Res. 2013;15:241–269. [Google Scholar]
- 31.Moodie GEE, Reimchen TE. Phenetic variation and habitat differences in Gasterosteus populations of Queen Charlotte Islands. Syst Zool. 1976;25:49–61. [Google Scholar]
- 32.Reimchen TE, Douglas S. Differential contribution of the sexes to prefledged young in red-throated loons. Auk. 1985;102:198–201. [Google Scholar]
- 33.Bergstrom CA, Reimchen TE. Habitat dependent associations between parasitism and fluctuating asymmetry among endemic stickleback populations. J Evol Biol. 2005;18:939–948. doi: 10.1111/j.1420-9101.2005.00930.x. [DOI] [PubMed] [Google Scholar]
- 34.Deagle BE, Jones FC, Absher DM, Kingsley DM, Reimchen TE. Phylogeography and adaptation genetics of stickleback from the Haida Gwaii archipelago revealed using genome-wide single nucleotide polymorphism genotyping. Mol Ecol. 2013;22:1917–1932. doi: 10.1111/mec.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reimchen TE. Predator handling failures of lateral plate morphs in Gasterosteus aculeatus: Functional implications for the ancestral plate condition. Behaviour. 2000;137:1081–1096. [Google Scholar]
- 36.Reimchen TE. Spine deficiency and polymorphism in a population of Gasterosteus aculeatus - an adaptation to predators. Can J Zool. 1980;58:1232–1244. [Google Scholar]
- 37.Reimchen TE, Nosil P. Temporal variation in divergent selection on spine number in threespine stickleback. Evolution. 2002;56:2472–2483. doi: 10.1111/j.0014-3820.2002.tb00172.x. [DOI] [PubMed] [Google Scholar]
- 38.Reimchen TE, Nosil P. Variable predation regimes predict the evolution of sexual dimorphism in a population of threespine stickleback. Evolution. 2004;58:1274–1281. doi: 10.1111/j.0014-3820.2004.tb01706.x. [DOI] [PubMed] [Google Scholar]
- 39.Reimchen TE, Stinson EM, Nelson JS. Multivariate differentiation of parapatric and allopatric populations of threespine stickleback in the Sangan river watershed, Queen-Charlotte islands. Can J Zool. 1985;63:2944–2951. [Google Scholar]
- 40.Spoljaric MA, Reimchen TE. 10 000 years later: evolution of body shape in Haida Gwaii three-spined stickleback. J Fish Biol. 2007;70:1484–1503. [Google Scholar]
- 41.Leaver SD, Reimchen TE. Abrupt changes in defence and trophic morphology of the giant threespine stickleback (Gasterosteus sp.) following colonization of a vacant habitat. Biol J Linn Soc. 2012;107:494–509. [Google Scholar]
- 42.Moodie GEE. Morphology, life-History, and ecology of an unusual stickleback (Gasterosteus aculeatus) in Queen-Charlotte Islands, Canada. Can J Zool. 1972;50:721–732. [Google Scholar]
- 43.Moodie GEE. Predation, natural selection and adaptation in an unusual threespine stickleback. Heredity. 1972;28:155–167. [Google Scholar]
- 44.Oreilly P, Reimchen TE, Beech R, Strobeck C. Mitochondrial-DNA in Gasterosteus and pleistocene glacial refugium on the Queen-Charlotte-Islands, British-Columbia. Evolution. 1993;47:678–684. doi: 10.1111/j.1558-5646.1993.tb02122.x. [DOI] [PubMed] [Google Scholar]
- 45.Flamarique IN, Bergstrom C, Cheng CL, Reimchen TE. Role of the iridescent eye in stickleback female mate choice. J Exp Biol. 2013;216:2806–2812. doi: 10.1242/jeb.084889. [DOI] [PubMed] [Google Scholar]
- 46.Deagle BE, et al. Population genomics of parallel phenotypic evolution in stickleback across stream-lake ecological transitions. Proc Biol Sci. 2012;279:1277–1286. doi: 10.1098/rspb.2011.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peichel CL, Marques DA. The genetic and molecular architecture of phenotypic diversity in sticklebacks. Philos Trans R Soc Lond B Biol Sci. 2017;372 doi: 10.1098/rstb.2015.0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peichel CL, et al. The genetic architecture of divergence between threespine stickleback species. Nature. 2001;414:901–905. doi: 10.1038/414901a. [DOI] [PubMed] [Google Scholar]
- 49.Colosimo PF, et al. Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science. 2005;307:1928–1933. doi: 10.1126/science.1107239. [DOI] [PubMed] [Google Scholar]
- 50.Colosimo PF, et al. The genetic architecture of parallel armor plate reduction in threespine sticklebacks. PLoS Biol. 2004;2:E109. doi: 10.1371/journal.pbio.0020109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wark AR, et al. Genetic architecture of variation in the lateral line sensory system of threespine sticklebacks. G3. 2012;2:1047–1056. doi: 10.1534/g3.112.003079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greenwood AK, Wark AR, Yoshida K, Peichel CL. Genetic and neural modularity underlie the evolution of schooling behavior in threespine sticklebacks. Curr Biol. 2013;23:1884–1888. doi: 10.1016/j.cub.2013.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rennison DJ, Owens GL, Heckman N, Schluter D, Veen T. Rapid adaptive evolution of colour vision in the threespine stickleback radiation. Proc Biol Sci. 2016;283:20160242. doi: 10.1098/rspb.2016.0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perez-Leighton CE, Schmidt TM, Abramowitz J, Birnbaumer L, Kofuji P. Intrinsic phototransduction persists in melanopsin-expressing ganglion cells lacking diacylglycerol-sensitive TRPC subunits. Eur J Neurosci. 2011;33:856–867. doi: 10.1111/j.1460-9568.2010.07583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakajima Y, Moriyama M, Hattori M, Minato N, Nakanishi S. Isolation of ON bipolar cell genes via hrGFP-coupled cell enrichment using the mGluR6 promoter. J Biochem. 2009;145:811–818. doi: 10.1093/jb/mvp038. [DOI] [PubMed] [Google Scholar]
- 56.Amsterdam A, et al. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci U S A. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nuckels RJ, Ng A, Darland T, Gross JM. The vacuolar-ATPase complex regulates retinoblast proliferation and survival, photoreceptor morphogenesis, and pigmentation in the zebrafish eye. Investigative ophthalmology & visual science. 2009;50:893–905. doi: 10.1167/iovs.08-2743. [DOI] [PubMed] [Google Scholar]
- 58.Howe DG, et al. ZFIN, the zebrafish model organism database: increased support for mutants and transgenics. Nucleic acids research. 2013;41:D854–860. doi: 10.1093/nar/gks938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marques DA, et al. Convergent evolution of SWS2 opsin facilitates adaptive radiation of threespine stickleback into different light environments. PLoS Biol. 2017;15:e2001627. doi: 10.1371/journal.pbio.2001627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gwynn B, Smith RS, Rowe LB, Taylor BA, Peters LL. A mouse TRAPP-related protein is involved in pigmentation. Genomics. 2006;88:196–203. doi: 10.1016/j.ygeno.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 61.Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA, Crossland JP. A single amino acid mutation contributes to adaptive beach mouse color pattern. Science. 2006;313:101–104. doi: 10.1126/science.1126121. [DOI] [PubMed] [Google Scholar]
- 62.Dickinson ME, et al. High-throughput discovery of novel developmental phenotypes. Nature. 2016;537:508–514. doi: 10.1038/nature19356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ignatius MS, Moose HE, El-Hodiri HM, Henion PD. colgate/hdac1 repression of foxd3 expression is required to permit mitfa-dependent melanogenesis. Dev Biol. 2008;313:568–583. doi: 10.1016/j.ydbio.2007.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patterson LB, Parichy DM. Interactions with iridophores and the tissue environment required for patterning melanophores and xanthophores during zebrafish adult pigment stripe formation. PLoS Genet. 2013;9:e1003561. doi: 10.1371/journal.pgen.1003561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller CT, et al. cis-Regulatory changes in Kit ligand expression and parallel evolution of pigmentation in sticklebacks and humans. Cell. 2007;131:1179–1189. doi: 10.1016/j.cell.2007.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosenblum EB, Hoekstra HE, Nachman MW. Adaptive reptile color variation and the evolution of the Mc1r gene. Evolution. 2004;58:1794–1808. doi: 10.1111/j.0014-3820.2004.tb00462.x. [DOI] [PubMed] [Google Scholar]
- 67.Malek TB, Boughman JW, Dworkin I, Peichel CL. Admixture mapping of male nuptial colour and body shape in a recently formed hybrid population of threespine stickleback. Mol Ecol. 2012;21:5265–5279. doi: 10.1111/j.1365-294X.2012.05660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miller CT, et al. Modular skeletal evolution in sticklebacks is controlled by additive and clustered quantitative trait Loci. Genetics. 2014;197:405–420. doi: 10.1534/genetics.114.162420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lamichhaney S, et al. A beak size locus in Darwin’s finches facilitated character displacement during a drought. Science. 2016;352:470–474. doi: 10.1126/science.aad8786. [DOI] [PubMed] [Google Scholar]
- 70.Gingerich PD. Rates of evolution: effects of time and temporal scaling. Science. 1983;222:159–161. doi: 10.1126/science.222.4620.159. [DOI] [PubMed] [Google Scholar]
- 71.Rennison DJ, Owens GL, Taylor JS. Opsin gene duplication and divergence in ray-finned fish. Mol Phylogenet Evol. 2012;62:986–1008. doi: 10.1016/j.ympev.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 72.Reimchen TE. Predator-induced cyclical changes in lateral plate frequencies of Gasterosteus. Behaviour. 1995;132:1079–1094. [Google Scholar]
- 73.Stinson EM. MSc thesis. University of Alberta; 1983. Threespine sticklebacks (Gasterosteus aculeatus) in Drizzle lake and its inlet, Queen Charlotte islands: ecological and behavioural relationships and their relevance to reproductive isolation. [Google Scholar]
- 74.Dlugosch KM, Parker IM. Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol. 2008;17:431–449. doi: 10.1111/j.1365-294X.2007.03538.x. [DOI] [PubMed] [Google Scholar]
- 75.Keller I, et al. Population genomic signatures of divergent adaptation, gene flow and hybrid speciation in the rapid radiation of Lake Victoria cichlid fishes. Mol Ecol. 2013;22:2848–2863. doi: 10.1111/mec.12083. [DOI] [PubMed] [Google Scholar]
- 76.McGee MD, Neches RY, Seehausen O. Evaluating genomic divergence and parallelism in replicate ecomorphs from young and old cichlid adaptive radiations. Mol Ecol. 2016;25:260–268. doi: 10.1111/mec.13463. [DOI] [PubMed] [Google Scholar]
- 77.Lamichhaney S, et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature. 2015;518:371–375. doi: 10.1038/nature14181. [DOI] [PubMed] [Google Scholar]
- 78.Dasmahapatra KK, et al. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012;487:94–98. doi: 10.1038/nature11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grant PR, Grant BR. Unpredictable evolution in a 30-year study of Darwin’s finches. Science. 2002;296:707–711. doi: 10.1126/science.1070315. [DOI] [PubMed] [Google Scholar]
- 80.Glazer AM, Killingbeck EE, Mitros T, Rokhsar DS, Miller CT. Genome assembly improvement and mapping convergently evolved skeletal traits in sticklebacks with genotyping-by-sequencing. G3. 2015;5:1463–1472. doi: 10.1534/g3.115.017905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Delaneau O, Howie B, Cox AJ, Zagury JF, Marchini J. Haplotype estimation using sequencing reads. Am J Hum Genet. 2013;93:687–696. doi: 10.1016/j.ajhg.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Korneliussen TS, Albrechtsen A, Nielsen R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinformatics. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nielsen R, Korneliussen T, Albrechtsen A, Li Y, Wang J. SNP calling, genotype calling, and sample allele frequency estimation from New-Generation Sequencing data. PLoS One. 2012;7:e37558. doi: 10.1371/journal.pone.0037558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fumagalli M, et al. Quantifying population genetic differentiation from next-generation sequencing data. Genetics. 2013;195:979–992. doi: 10.1534/genetics.113.154740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McLaren W, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Broad Institute. Picard Tools. http://broadinstitute.github.io/picard (accessed 30 January 2017)
- 87.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 90.Willing EM, Dreyer C, van Oosterhout C. Estimates of genetic differentiation measured by F(ST) do not necessarily require large sample sizes when using many SNP markers. PLoS One. 2012;7:e42649. doi: 10.1371/journal.pone.0042649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bhatia G, Patterson N, Sankararaman S, Price AL. Estimating and interpreting FST: The impact of rare variants. Genome Res. 2013;23:1514–1521. doi: 10.1101/gr.154831.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Excoffier L, Dupanloup I, Huerta-Sanchez E, Sousa VC, Foll M. Robust demographic inference from genomic and SNP data. PLoS Genet. 2013;9:e1003905. doi: 10.1371/journal.pgen.1003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Feulner PG, et al. Genomics of divergence along a continuum of parapatric population differentiation. PLoS Genet. 2015;11:e1004966. doi: 10.1371/journal.pgen.1004966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garud NR, Messer PW, Buzbas EO, Petrov DA. Recent selective sweeps in North American Drosophila melanogaster show signatures of soft sweeps. PLoS Genet. 2015;11:e1005004. doi: 10.1371/journal.pgen.1005004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sabeti PC, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–U912. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Szpiech ZA, Hernandez RD. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 2014;31:2824–2827. doi: 10.1093/molbev/msu211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.R Development Core Team. R: A language and environment for statistical computing. http://www.r-project.org/ (accessed 20 April 2016)
- 99.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57:289–300. [Google Scholar]
- 100.Gillespie JH. Population genetics: a concise guide. 2nd. Johns Hopkins University Press; 2004. [Google Scholar]
- 101.Szklarczyk D, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Blake JA, et al. Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic acids research. 2017;45:D723–D729. doi: 10.1093/nar/gkw1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shimoyama M, et al. The Rat Genome Database 2015: genomic, phenotypic and environmental variations and disease. Nucleic acids research. 2015;43:D743–750. doi: 10.1093/nar/gku1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.