

Abstract
Photocaging facilitates non‐invasive and precise spatio‐temporal control over the release of biologically relevant small‐ and macro‐molecules using light. However, sub‐cellular organelles are dispersed in cells in a manner that renders selective light‐irradiation of a complete organelle impractical. Organelle‐specific photocages could provide a powerful method for releasing bioactive molecules in sub‐cellular locations. Herein, we report a general post‐synthetic method for the chemical functionalization and further conjugation of meso‐methyl BODIPY photocages and the synthesis of endoplasmic reticulum (ER)‐, lysosome‐, and mitochondria‐targeted derivatives. We also demonstrate that 2,4‐dinitrophenol, a mitochondrial uncoupler, and puromycin, a protein biosynthesis inhibitor, can be selectively photoreleased in mitochondria and ER, respectively, in live cells by using visible light. Additionally, photocaging is shown to lead to higher efficacy of the released molecules, probably owing to a localized and abrupt release.
Keywords: BODIPY, mitochondria, organelles, photocages, photo-release
The interior of cells is a highly organized environment. Cellular tasks such as energy production, protein synthesis, and many others, are functionally compartmentalized in organelles, along with most of the biomolecules required for their execution. Studying localized processes, including those taking place inside organelles, often makes use of small‐molecules to manipulate their progress.1 Unfortunately, most small‐molecules lack an innate specificity for cellular locations; they tend to disperse randomly in cells, not necessarily arriving at the desired place owing to unsuitable physical‐chemical properties or conversely, ending up in too many locations in an unspecific manner.
Photocaging2 is an effective light‐mediated controlled‐release strategy that enables activation of small‐ or macro‐bioactive molecules with high spatio‐temporal resolution.3 This strategy is widely utilized to achieve localized control over the activation of bioactive molecules in vitro and in vivo.4 However, organelles are scattered or dispersed in cells in a manner that renders selective irradiation of a complete organelle (e.g., mitochondria or golgi apparatus) impractical, undermining the utility of the strategy in this context. Over the years, several chemical motifs have been determined to passively accumulate in specific sub‐cellular compartments owing to their chemical and/or physical properties (e.g., pH, charge, and hydrophobicity). Such motifs include, for example, triphenylphosphonium5 (TPP), phenyl sulfonamide,6 and tertiary/secondary amines7 that tend to accumulate in mitochondria, endoplasmic reticulum (ER), and lysosomes/endosomes, respectively. Their coupling to small‐ and even macro‐molecules was found to effectively impart sub‐cellular specificity to the entire conjugate.
The combination of sub‐cellular targeting with photocaging, providing a means of preserving the advantages of the two strategies while overcoming their distinct limitations, has been effectively demonstrated using 2‐nitrobenzyl derivatives caging groups.8 However, the use of UV‐excitable photocages often leads to inherent hurdles, including sample overheating and phototoxicity. More recent work has effectively improved the utility of targeted photocages by using coumarins, excitable at around 400 nm, as caging groups.9
Herein, we establish a range of organelle‐targeted photocages based on the recently introduced, visible‐light excitable (>500 nm) meso‐methyl BODIPY photocage10 (Figure 1). To this end, we also developed a straightforward, post‐synthetic method for the functionalization of BODIPYs that streamlines their synthesis.
Figure 1.
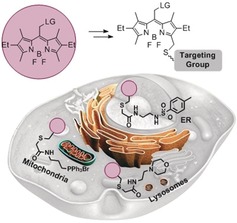
Schematic of ER‐, lysosome‐, and mitochondria‐targeted BODIPY photocages.
The photoreaction efficiency in BODIPY photocages,10d as well as in many other photocaging groups,11 is highly sensitive to changes in electronic and steric properties of the core. We therefore first sought to establish a robust synthetic approach for chemical functionalization of BODIPY photocages without affecting their photorelease ability.
The synthetic procedure is outlined in Figure 2 A. Using a one‐pot, two‐step protocol, that is, bromination with N‐bromosuccinimide (NBS) followed by nucleophilic substitution, compounds 1–5 were synthesized. This is a modified approach to an earlier report,12 which leverages the nucleophilic character of BODIPY α‐methyls to introduce bromine as a leaving group. In this case, the superior nucleophilicity of thiols is employed to introduce less nucleophilic yet reactive functional groups through appropriate bifunctional molecules. Thus, various unprotected functional groups including amine, thiol, chloro, carboxylic acid, and azide groups, all key building blocks in the construction of more elaborate molecules, could be simply and directly introduced. We verified that the reactive functionalities installed can be further conjugated in the context of BODIPY photocages by reacting each of them in a compatible manner (i.e., amidation, Michael addition to N‐ethyl maleimide, and copper‐mediated click reaction) (compounds 6–10, Figure S1).
Figure 2.
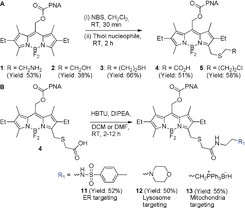
A) Synthetic scheme of compounds 1–5. B) Synthetic scheme of organelle‐targeting compounds 11–13.
The photophysical and photoreaction properties of all synthesized compounds are summarized in Tables S1. Compared to previously reported BODIPY photocages,10a,10b,10d all new derivatives (1–10) present a 5–7 nm red‐shift in both absorbance and fluorescence λ max, with molar extinction coefficient values in the typical BODIPY range (ca. 35 000–70 000 m −1 cm−1). Photoreaction properties were determined by UV/Vis spectroscopy, monitoring p‐nitroaniline (PNA) release (see Supporting Information). Compounds 5–8 and 10 are stable in the absence of light but release PNA in response to irradiation with visible light (545/30 nm, 42 mW cm−2), presenting comparable photoreaction properties (quantum yield (Φ r), half‐life (t 1/2), and chemical yield) to previously reported BODIPY photocages (Table S1, Figure S2). These results confirm that the above‐described post‐synthetic methodology can be straightforwardly applied to conjugate BODIPY photocages through diverse chemical functionalities while retaining their spectroscopic and photoreaction properties.
We next applied the synthetic approach to generate a panel of organelle‐targeted BODIPY photocages. A direct HBTU‐mediated coupling of compound 4 with different amine‐bearing organelle‐directing groups such as phenyl sulfonamide (ER targeting), morpholine (lysosome targeting), and TPP (mitochondria targeting), afforded targeted BODIPY photocages 11–13, respectively (Figure 2 B). Compounds 11–13 are stable in the absence of light and show controlled release of PNA upon irradiation with visible light (Table S1 and Figure S3). Their cellular uptake and sub‐cellular localization were evaluated in HeLa cells by live‐cell imaging using commercial stains for ER, lysosomes, and mitochondria (Figures 3 and Figures S9 and S10). All three compounds demonstrated efficient localization to their target organelles as determined by Pearson's sample correlation coefficients (r=0.95, 0.73, and 0.75 for 11, 12, and 13, respectively) with the respective organelles stains (ER‐tracker Blue, LysoTracker deep‐red, and MitoTracker deep‐red).
Figure 3.
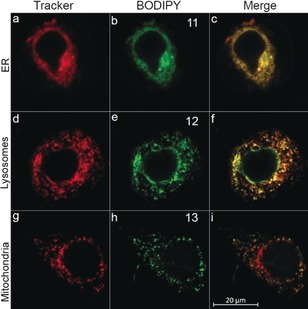
Cellular distribution and co‐localization of compounds 11–13. Confocal fluorescence images of live HeLa cells incubated with a–c) ER‐Tracker Blue (2 μm) and 11 (10 μm, 30 min), d–f) LysoTracker deep‐red (2 μm) and 12 (10 μm, 30 min), g–i) MitoTracker deep‐red (2 μm), and 13 (10 μm, 30 min). Co‐localization appears as yellow/orange (c), (f), (i).
To evaluate the utility of organelle‐targeted BODIPY photocages, we synthesized and tested a caged version of the protonophore 2,4‐dinitrophenol (DNP). DNP is an uncoupler of mitochondrial oxidative phosphorylation, dissipating the proton gradient across the membrane thus decreasing mitochondrial membrane potential13 (Δψ m). Compound 14 was synthesized by ipso‐substitution of 1‐fluoro‐2,4‐dinitrobenzene with meso‐methyl BODIPY and functionalized by employing the above‐described protocol using thioglycolic acid as a nucleophile. A TPP motif was subsequently coupled to obtain the mitochondria‐targeted BODIPY‐DNP 16 (Figures 4 A and Figure S4). Quantum yields, half‐lives (t 1/2), and chemical yields for photorelease of DNP were determined by UV/Vis spectroscopy and confirmed by HPLC‐MS. Compounds 14 and 16 are stable in the dark and release DNP when irradiated with visible light (Figures 4 B and Figures S5 and S6), compound 16 presenting the highest photorelease efficiency (ϵΦ r=43) within this series of compounds (Table S1). Although BODIPY photocages were previously demonstrated to release halides, acids, carbon monoxide, thiols, and xanthates,10 this is first example of direct uncaging of phenols, expanding the palette of functional groups and bioactive molecules amenable for caging by BODIPYs.
Figure 4.
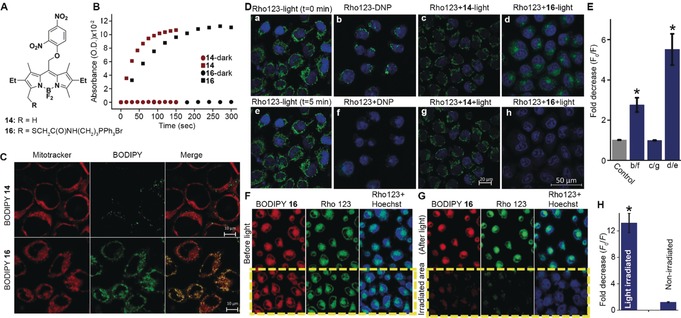
A) Structures of compounds 14 and 16. B) Light‐mediated release of DNP from 14 and 16 (10 μm, ACN/water 7:3) following light irradiation (545/30 nm, 42 mW cm−2) for the indicated times. Absorbance at 367 nm (representing free DNP) vs. time was plotted. C) Distribution and co‐localization of 14 and 16. Confocal images of live HeLa cells treated with Hoechst 33342 (17 μm), MitoTracker deep‐red (2 μm) and 14 (upper) or 16 (lower) (10 μm, 30 min). Areas of co‐localization appear in yellow/orange in Merged. D) Photorelease of DNP in live cells leads to decrease in mitochondrial membrane potential. Confocal images of live HeLa cells stained with Hoechst 33342 (17 μm) and Rhod123 (26 μm, 15 min), a,e) in the absence of light at t=0 and 5 min; b,f) before and after treatment with 200 μm DNP; c,g) compound 14 (25 μm) with and without light (545/25 nm, 1.4 mW cm−2, 15 s); d,h) compound 16 (25 μm) with and without light (545/25 nm, 1.4 mW cm−2, 15 s). E) Decrease in cells fluorescence intensity, where F 0 is fluorescence intensity before either light or DNP treatment and F is fluorescence intensity after either light or DNP treatment. F,G) Localized photoactivation of DNP in live cells. HeLa cells were incubated with Rho123 (26 μm), 16 (25 μm), and Hoechst 33342 (17 μm) for 15 min before (F) and after (G) light irradiation (545/25 nm, 1.4 mW cm−2, 15 s) of a selected region. H) Fold‐decrease in fluorescence intensity of cells in irradiated and non‐irradiated regions, where F 0 and F are defined as in (E). * Statistical significance (one‐way ANOVA with Tukey correction, p<0.05) from control (E) or non‐irradiated cells (H). Error bars show the standard error (SE).
Cellular uptake and mitochondrial localization of 16 were examined and compared to the non‐targeted 14 by live‐cell imaging of HeLa cells (Figures 4 C and Figures S9 and S11). Low Pearson's sample correlation coefficients (r=0.13) confirmed poor targeting of 14 to the mitochondria while 16 showed efficient and specific mitochondrial accumulation (r=0.84).
Next, intracellular photoactivation of 16 was investigated. Changes in Δψ m were assessed using the Δψ m‐sensitive lipophilic cationic dye, rhodamine 123 (Rho123). In intact cells, Rho123 accumulates in mitochondria, leading to a strong localized fluorescence signal.14 Conversely, reduction in Δψ m leads to redistribution of the dye to the cytoplasm, resulting in its dilution and a decrease in fluorescence signal. In HeLa cells treated with Rho123, strong mitochondrial fluorescence could be detected, which was significantly reduced (ca. 3‐fold) by further treatment with 200 μm DNP (Figure 4 D and Figure S12). When similar cells were treated with Rho123 and 16 (25 μm), a mitochondria‐localized fluorescence signal was observed, indicating that 16 by itself does not disrupt Δψ m. However, upon irradiation of the cells (545/25 nm, 1.4 mW cm−2, 15 s), a 6‐fold decrease in Rho123 mitochondrial fluorescence was observed. Importantly, photoactivation of the non‐targeted 14 under similar conditions did not have any effect on Rho123 fluorescence. Cells treated with Rho123 alone and exposed to similar irradiation conditions did not show any change in mitochondrial fluorescence, ruling out direct light effect on Δψ m. In addition, in the absence of light, 16 did not show any sign of toxicity at the applied concentration (Figure S14).
Finally, we tested light‐mediated selective activation of 16 in specific cells. Thus, HeLa cells were treated with Roh123, 16, and the DNA stain Hoechst 33342, and confocal images were acquired at three channels (Figure 4 F and Figure S13 a). Cells were then irradiated for 15 s (545/25 nm, 1.4 mW cm−2) in a selected region (marked by a yellow rectangle) and re‐imaged after 5 min (Figure 4 G and Figure S13 b). Results show a significant (ca. 13‐fold) decrease in fluorescence signal only in cells within the light‐exposed region while cells outside of it remains unaffected. Quantification of the averaged fluorescence intensities of cells within the irradiated area versus those outside of it, before and after light exposure, is shown in Figure 4 H.
To demonstrate the general applicability of BODIPY photocages targeting, we synthesized an ER‐targeted caged version of the protein synthesis inhibitor puromycin15 (19, Figure S15). Compound 19 showed efficient light‐dependent release of puromycin in vitro and in HeLa cells (Figure S15 B) and colocalized efficiently with ER‐tracker blue (r=0.95, Figure S16 C). Following photoactivation of 19 in live cells (20 μm, 545/305 nm, 42 mW cm−2), released puromycin could be detected specifically in the ER, unlike treatment with free puromycin that was detected throughout the cell (ER, cytoplasm, nucleus), as visualized by immunostaining (Figure S16 E).
In summary, we developed a set of BODIPY photocages suitable for visible‐light‐mediated release of bioactive molecules in specific, pre‐designated organelles. We have established a post‐synthetic procedure to straightforwardly introduce conjugatable functional groups onto BODIPY α‐methyl in one synthetic step and without compromising their spectroscopic nor photoreaction properties. This procedure represents a unique post‐synthetic functionalization method applicable to BODIPYs at large, providing a simple and effective solution to the traditional challenge of BODIPY functionalization, usually requiring multi‐step processes.16 Thus, it not only should provide access to conjugation of BODIPY photocages to other small‐ or macro‐molecules but also uniquely represents a simple path to direct activation and further (bio‐)conjugation of BODIPYs when used as fluorescent tags. The developed procedure was applied to generate a set of organelle‐targeted BODIPY photocages in a divergent manner. All organelle‐targeted BODIPY photocages efficiently localized to their pre‐designated sub‐cellular compartments. A mitochondria‐targeted BODIPY was demonstrated to release the protonophore DNP in live cells with exquisite spatio‐temporal control, achieving a much higher effect compared to non‐targeted DNP. Thus, photocaging introduces spatio‐temporal specificity to organelle targeting and leads to higher efficacy of the bioactive molecule, most probably owing to localized and abrupt release. Finally, we expect that our approach could be extended to the selective delivery of a wide range of bioactive molecules to diverse organelles in order to perturb and study their localized processes and functions. The use of BODIPY provides access to photoactivation with biologically benign visible light thus eliminating concerns of phototoxicity associated with traditional UV‐excitable photocages and, potentially, opening the way to organelle‐targeted light‐mediated drug delivery.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
W.R. would like to thank the United States‐Israel Binational Science Foundation (Grant No. 2016060), the Germany‐Israel Foundation (Grant No. I‐2387‐302.5/2015) and the ERC (Grant No. 679189), and D.F. M. to the Israeli Science Foundation (1310/15), for funding this research. D.K. thanks the Planning and Budgeting Committee (PBC) of the Israeli Council for Higher Education for a postdoctoral fellowship. We would like to thank Prof. Evan W. Miller (UC Berkeley) for helpful discussions.
D. Kand, L. Pizarro, I. Angel, A. Avni, D. Friedmann-Morvinski, R. Weinstain, Angew. Chem. Int. Ed. 2019, 58, 4659.
References
- 1.
- 1a. Maly D. J., Papa F. R., Nat. Chem. Biol. 2014, 10, 892–901; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Piao S., Amaravadi R. K., Ann. N. Y. Acad. Sci. 2016, 1371, 45–54; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Sakhrani N. M., Padh H., Drug Des. Dev. Ther. 2013, 7, 585–599; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Weinberg S. E., Chandel N. S., Nat. Chem. Biol. 2015, 11, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaplan J. H., Forbush 3rd B., Hoffman J. F., Biochemistry 1978, 17, 1929–1935. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Riggsbee C. W., Deiters A., Trends Biotechnol. 2010, 28, 468–475; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Shao Q., Xing B., Chem. Soc. Rev. 2010, 39, 2835–2846. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Fournier L., Gauron C., Xu L., Aujard I., Le Saux T., Gagey-Eilstein N., Maurin S., Dubruille S., Baudin J. B., Bensimon D., Volovitch M., Vriz S., Jullien L., ACS Chem. Biol. 2013, 8, 1528–1536; [DOI] [PubMed] [Google Scholar]
- 4b. Nani R. R., Gorka A. P., Nagaya T., Kobayashi H., Schnermann M. J., Angew. Chem. Int. Ed. 2015, 54, 13635–13638; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13839–13842. [Google Scholar]
- 5.
- 5a. Hoye A. T., Davoren J. E., Wipf P., Fink M. P., Kagan V. E., Acc. Chem. Res. 2008, 41, 87–97; [DOI] [PubMed] [Google Scholar]
- 5b. Wisnovsky S., Lei E. K., Jean S. R., Kelley S. O., Cell Chem. Biol. 2016, 23, 917–927. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Xiao H., Li P., Hu X., Shi X., Zhang W., Tang B., Chem. Sci. 2016, 7, 6153–6159; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Xiao H., Liu X., Wu C., Wu Y., Li P., Guo X., Tang B., Biosens. Bioelectron. 2017, 91, 449–455. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Goodman C. A., Pierre P., Hornberger T. A., Proc. Natl. Acad. Sci. USA 2012, 109, E989; author reply E990; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Liu T., Xu Z., Spring D. R., Cui J., Org. Lett. 2013, 15, 2310–2313. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Chalmers S., Caldwell S. T., Quin C., Prime T. A., James A. M., Cairns A. G., Murphy M. P., McCarron J. G., Hartley R. C., J. Am. Chem. Soc. 2012, 134, 758–761; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Leonidova A., Pierroz V., Rubbiani R., Lan Y. J., Schmitz A. G., Kaech A., Sigel R. K. O., Ferrari S., Gasser G., Chem. Sci. 2014, 5, 4044–4056. [Google Scholar]
- 9.
- 9a. Nadler A., Yushchenko D. A., Muller R., Stein F., Feng S., Mulle C., Carta M., Schultz C., Nat. Commun. 2015, 6, 10056; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Wagner N., Stephan M., Hoglinger D., Nadler A., Angew. Chem. Int. Ed. 2018, 57, 13339–13343; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13523–13527; [Google Scholar]
- 9c. Feng S., Harayama T., Montessuit S., David F. P., Winssinger N., Martinou J. C., Riezman H., eLife 2018, 7, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Goswami P. P., Syed A., Beck C. L., Albright T. R., Mahoney K. M., Unash R., Smith E. A., Winter A. H., J. Am. Chem. Soc. 2015, 137, 3783–3786; [DOI] [PubMed] [Google Scholar]
- 10b. Rubinstein N., Liu P., Miller E. W., Weinstain R., Chem. Commun. 2015, 51, 6369–6372; [DOI] [PubMed] [Google Scholar]
- 10c. Sitkowska K., Feringa B. L., Szymanski W., J. Org. Chem. 2018, 83, 1819–1827; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Slanina T., Shrestha P., Palao E., Kand D., Peterson J. A., Dutton A. S., Rubinstein N., Weinstain R., Winter A. H., Klan P., J. Am. Chem. Soc. 2017, 139, 15168–15175. [DOI] [PubMed] [Google Scholar]
- 11. Klán P., Šolomek T., Bochet C. G., Blanc A., Givens R., Rubina M., Popik V., Kostikov A., Wirz J., Chem. Rev. 2013, 113, 119–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ulrich G., Ziessel R., Haefele A., J. Org. Chem. 2012, 77, 4298–4311. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Bestman J. E., Stackley K. D., Rahn J. J., Williamson T. J., Chan S. S., Differentiation 2015, 89, 51–69; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Pinchot G. B., J. Biol. Chem. 1967, 242, 4577–4583. [PubMed] [Google Scholar]
- 14.
- 14a. Annamalai G., Kathiresan S., Kannappan N., Biomed. Pharmacother. 2016, 82, 226–236; [DOI] [PubMed] [Google Scholar]
- 14b. Duan G., Hou S., Ji J., Deng B., Cancer Biomarkers 2018, 22, 29–34; [DOI] [PubMed] [Google Scholar]
- 14c. Lu Y. Y., Chen T. S., Qu J. L., Pan W. L., Sun L., Wei X. B., J. Biomed. Sci. 2009, 16, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Buhr F., Kohl-Landgraf J., tom Dieck S., Hanus C., Chatterjee D., Hegelein A., Schuman E. M., Wachtveitl J., Schwalbe H., Angew. Chem. Int. Ed. 2015, 54, 3717–3721; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3788–3792; [Google Scholar]
- 15b. Elamri I., Heumuller M., Herzig L. M., Stirnal E., Wachtveitl J., Schuman E. M., Schwalbe H., ChemBioChem 2018, 19, 2458–2464; [DOI] [PubMed] [Google Scholar]
- 15c. Herzig L. M., Elamri I., Schwalbe H., Wachtveitl J., Phys. Chem. Chem. Phys. 2017, 19, 14835–14844; [DOI] [PubMed] [Google Scholar]
- 15d. Kohl-Landgraf J., Buhr F., Lefrancois D., Mewes J. M., Schwalbe H., Dreuw A., Wachtveitl J., J. Am. Chem. Soc. 2014, 136, 3430–3438. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Boens N., Verbelen B., Dehaen W., Eur. J. Org. Chem. 2015, 6577–6595; [Google Scholar]
- 16b. Loudet A., Burgess K., Chem. Rev. 2007, 107, 4891–4932; [DOI] [PubMed] [Google Scholar]
- 16c. Wang H., Fronczek F. R., Vicente M. G., Smith K. M., J. Org. Chem. 2014, 79, 10342–10352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary