

Abstract
The isolable complex [Os(PHMes*)H(PNP)] (Mes*=2,4,6‐tBu3C6H3; PNP=N{CHCHPtBu2}2) exhibits high phosphinyl radical character. This compound offers access to the phosphinidene complex [Os(PMes*)H(PNP)] by P−H proton coupled electron transfer (PCET). The P−H bond dissociation energy (BDE) was determined by isothermal titration calorimetry and supporting DFT computations. The phosphinidene product exhibits electrophilic reactivity as demonstrated by intramolecular C−H activation.
Keywords: osmium, phosphanyl radical, phosphinidene, pincer, proton coupled electron transfer
Electronically unsaturated phosphorous compounds, such as phosphinyl radicals (PR2), are key transient species in P−C bond forming reactions, like olefin phosphination.1, 2 Structural and spectroscopic characterization of free and coordinated phosphinyl radicals facilitated the examination of (electronic) structure/reactivity relationships.3, 4 Free phosphinidenes (PR) could only very recently be sufficiently stabilized.5 While a variety of transition metals form isolable phosphinidene complexes (M=PR) with promising stoichiometric reactivity, such as P−C bond formation,6, 7 catalytic phosphinidene transfer protocols remain rare,8 compared to, for example, nitrene transfer which has emerged as a powerful method for C−N bond formation.9
The scarcity of catalytic PR‐transfer might in part be attributed to a lack of suitable oxidizing PR‐transfer reagents, such as analogs of iminoiodane (ArI=NR) or azide (RN3) nitrene sources. Primary phosphines are attractive precursors from a thermochemical point of view due to lower intrinsic P−H compared to N−H bond dissociation (free) energies (BD(F)Es; Scheme 1).10, 11 The P−H bonds might be further weakened upon coordination to a metal catalyst as was demonstrated, for example, for coordinated H2O and NH3 ligands.12 Amide ligand N−H bond activation by proton coupled electron transfer (PCET) is an active field of research.13 In contrast, phosphide P−H homolysis was only reported by Hillhouse and co‐workers (Scheme 2).14 Inspired by this precedence, we examined P−H PCET as an entry to phosphinidene chemistry. Herein, the formation of a rare phosphinyl radical complex and its transformation to a terminal osmium phosphinidene complex by PCET are reported.
Scheme 1.
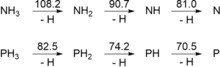
N−H and P−H bond dissociation energies (BDEs) in the gas phase (kcal mol−1).10
Scheme 2.
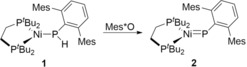
Generation of a nickel phosphinidene complex by PCET reported by Hillhouse and co‐workers. (Mes=2,4,6‐Me3C6H3, Mes*=2,4,6‐tBu3C6H3).14b
We previously utilized the four‐coordinate complex [OsIICl(PNP)] (3, PNP=N{CHCHPtBu2}2; Scheme 3) to stabilize low‐valent imide and nitride complexes.15 Complex 3 readily reacts with PH2Mes* in Et2O to the dark blue P−H oxidative addition product [OsCl(PHMes*)H(PNP)] (4) (Scheme 3). Complex 4 decomposes over the course of a few hours in solution at room temperature but could be fully characterized including single crystal X‐ray diffraction.16 NMR spectra are in agreement with C 1 symmetry at −30 °C. All signals (1H, 13C, 31P) are broadened supporting hindered rotation around the Os−P bond. The phosphide P‐atom exhibits trigonal‐planar coordination in the solid state, indicating P to OsIV π donation.16
Scheme 3.
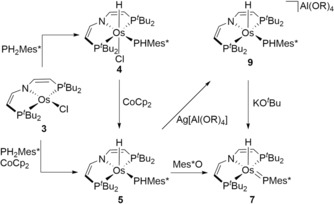
Synthetic access to phosphanide and phosphinidene complexes 4–6 and 9 (R=C(CF3)3).
Reduction of 4 with cobaltocene affords the radical product [OsH(PHMes*)(PNP)] (5) in 80 % isolated yield (Scheme 3). The magnetic moment in solution (μ eff=1.51 μB) derived by Evan's method is in agreement with an S= system with unquenched orbital momentum. Complex 5 can also be directly synthesized from 3 by one pot P−H oxidative addition and subsequent reduction. Characteristic bands for the PHMes* and hydride ligands were detected in the IR spectrum (νPH=2345 cm−1; νOsH=2180 cm−1). In the solid state, complex 5 features distorted square pyramidal coordination geometry with the hydride ligand in the apical position (Figure 1). Upon reduction from 4 to 5 the Os−P bond to the PHMes* ligand is slightly elongated by Δd=0.04 Å.
Figure 1.
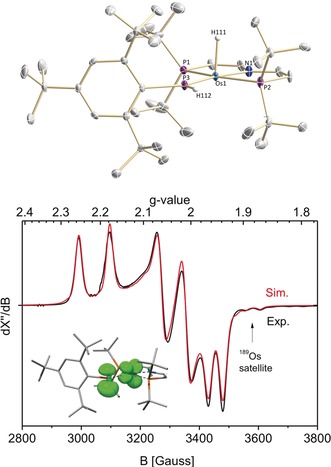
Top: Molecular structure of 5 in the solid state from single‐crystal X‐ray diffraction (thermal ellipsoids set at 50 % probability); solvent molecules, anions, and hydrogen atoms, except the P−H and Os−H hydrogen atoms, are omitted for clarity. Selected bond lengths [Å] and angles [°]: 5 Os1−H111 1.69(6), Os1−N1 2.069(4), Os1−P1 2.3723(12), Os1−P2 2.3537(13), Os1−P3 2.2301(13), P3−H112 1.33(5); P1−Os1−P2 160.20(4); N1−Os1−P3 172.17(11). Bottom: X‐band EPR spectrum of 5 (9.4366 GHz) in toluene at 148 K (black) and simulation (red) with the following parameters (DFT computed values in brackets): gx=1.952 (1.951), gy=2.033 (2.024), gz=2.214 (2.230); Ax(31P)=+57 MHz (+58 MHz), Ay(31P)=+105 MHz (+95 MHz), Az(31P)=+440 MHz (+441 MHz); Euler angles: α=−73 (−74.3), β=+137 (+142.7), γ=−7.5 (−7.5); Ax(189Os)=−240 (−277) MHz. Insert: Computed spin density plot.
The EPR spectrum of 5 in frozen toluene exhibits a rhombic signal that is in agreement with an S= low‐spin configuration (Figure 1). The g‐anisotropy is considerably smaller than that of typical osmium(III) complexes, in line with reduced spin‐orbit interaction due to ligand redox non‐innocence.15a, 17 Accordingly, large and slightly rhombic hyperfine interaction (HFI) with one 31P nucleus is observed in all principal directions of the g‐tensor (Figure 1), supporting considerable spin delocalization to the PHMes* ligand. The free phosphinyl radical PPh2 exhibits axial 31P‐HFI with an isotropic coupling constant (A iso(31P)=260 MHz) close to that of 5 (A iso(31P)=201 MHz).18 The higher isotropic HFI, yet reduced dipolar coupling, found for the transient phosphanyl radical complex [W(PPh2)(CO)5] (A iso(31P)=499 MHz) was attributed to p P→d W spin delocalization (ρ P=75 %) and concomitant polarization of the P lone‐pair.4a For 5, the higher g‐anisotropy and smaller isotropic and anisotropic contributions of the 31P‐HFI tensor are consistent with increased P→M spin delocalization. Comparing the isotropic and anisotropic (Tx=−144 MHz, Ty=−96 MHz, Tz=+239 MHz) contributions to the 31P‐HFI with atomic parameters allows for a rough estimate of phosphorous spin densities (ρ 3s≈2 %; ρ 3p≈42 %) when treating the HFI as approximately axial (T ⊥=(Tx + Ty)/2).18
DFT calculations fully support this interpretation. Both the molecular structure and the EPR parameters (Figure 1) were excellently reproduced computationally. The SOMO of 5 represents an antisymmetric (π*) combination of the metal dyz orbital and a phosphorus p‐orbital. Reduced π‐bonding is expressed by the Os−PHMes* Mayer bond index (1.46). In consequence, the computed spin density (Figure 1) is almost equally distributed between the P (ρ P=47 %) and Os atoms (ρ Os=50 %).
Complex 5 represents an unprecedented, isolable phosphanide complex with large phosphinyl redox non‐innocent character (OsII−.PR2). We therefore examined PCET reactivity of the radical ligand, specifically as an entry to phosphinidene chemistry. Two experimental methods are widely used to estimate E‐H BD(F)Es, that is, a) bracketing based on hydrogen transfer reactions with reference H‐donor/acceptor reagents and b) quantification by the “square‐Scheme” formalism, that is, a thermochemical redox/protonation cycle.19
Complex 5 shows no reactivity with the H‐atom donor TEMPO‐H (TEMPO=2,2,6,6‐tetramethylpiperidinyloxyl; BDFEO‐H=ca. 66 kcal mol−1),19 indicating weak P−H bonds for the hypothetical osmium(II) phosphine complex [Os(PH2Mes*)H(PNP)].20 In turn, 5 readily reacted with the H‐atom acceptors Mes*O and TEMPO. A purple product is obtained in yields around 90 % with low thermal stability at room temperature even in the solid state (see below) but could be characterized by NMR spectroscopy at −30 °C. Retention of the hydride ligand is indicated by a 1H NMR signal at δ H=−15.9 ppm. Formation of the phosphinidene complex [Os(PMes*)H(PNP)] (6) is evidenced by the 31P NMR signal at δ P=825 ppm, that is, assignable to the PMes* group. While suitable crystals for X‐ray diffraction could not be obtained, the structural integrity of the osmium pincer phosphinidene framework is supported by the spectroscopic characterization of the C−H insertion product 7 as a mixture of two diastereomers (Scheme 4). Such electrophilic phosphinidene insertion has previously been reported.6d, 21 Furthermore, addition of CO to 6 gives the five‐coordinate phosphanide complex 8 (Scheme 4) after Os−H reductive elimination.16
Scheme 4.
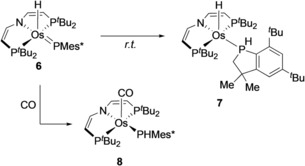
Reactivity of phosphinidene complex 6.
Parent 5 offers two potential sites for H‐atom transfer (HAT). The generation of 6 indicates higher Os−H over P−H bond strength if the reaction proceeds under thermodynamic control. BDE quantification was attempted by stepwise oxidation and deprotonation. The cyclic voltammogram of 5 reveals quasi‐reversible reduction at E p,c=−2.06 V and reversible oxidation E 1/2=−0.88 V (vs. FeCp2 +/0).16 Chemical oxidation with Ag[Al(OC(CF3)3)4] at −35 °C immediately gives the deep blue osmium(IV) phosphide complex 9 (Scheme 3). Complex 9 readily decomposes at room temperate but could be characterized at low temperatures including crystallography.16 Deprotonation of in situ prepared 9 with KOtBu at −80 °C gives phosphinidene 6 almost quantitatively. However, the low thermal stability of 9 hampered reliable pK a determination.
The P−H bond strength of 5 was therefore derived by isothermal titration calorimetry (ITC). Titration of 5 with Mes*O in benzene or THF afforded the reaction enthalpies for HAT (Δr H =−16.5 kcal mol−1, Δr H THF=−17 kcal mol−1) and consequently the BDEP‐H of 5 (BDE = 65.1±1 kcal mol−1, BDETHF=67±1 kcal mol−1).22 DFT analysis of 5 supports the experimental P−H BDE (calibrated BDEP‐H=67.5 kcal mol−1; non‐calibrated value: 64.0 kcal mol−1),23 that is, considerably lower than the Os−H bond (calibrated BDEP‐H=74.2 kcal mol−1; non‐calibrated: 70.1 kcal mol−1), indicating that phosphinidene 6 is the thermodynamic PCET product. The calorimetric and electrochemical data also allows for calculating the pK a of 9 (pK a THF=16) from a thermochemical square‐scheme (Scheme 5).24
Scheme 5.
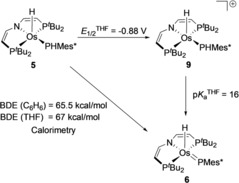
Thermochemical square‐scheme of 5, 6 and 9.
In conclusion, we presented the first spectroscopically and crystallographically characterized phosphide complex with large phosphanyl radical character. The rhombic 31P‐HFI tensor and the DFT model are in line with even spin delocalization over the Os−P core. Versatile access to an electrophilic phosphinidene complex that undergoes intramolecular C−H activation was demonstrated by P−H PCET. Thermochemical analysis by means of ITC was utilized due to thermal instability of 9. The data indicates that concerted or stepwise ET/PT are both viable routes from phosphide to phosphinidene complexes.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was funded by the European Research Council (ERC Grant Agreement 646747), the Fond der Chemischen Industrie (FCI Doktoranden Stipendium for J.A.), the Deutsche Forschungsgemeinschaft (DFG, 389479699/GRK2455) and The Netherlands Organisation for Scientific Research (NWO TOP‐Grant 716.015.001) for financial support. Furthermore, the authors thank Dr. A. C. Stückl and R. Schöne for EPR and NMR measurements, respectively.
J. Abbenseth, D. Delony, M. C. Neben, C. Würtele, B. de Bruin, S. Schneider, Angew. Chem. Int. Ed. 2019, 58, 6338.
This paper is dedicated to Professor Thomas Fässler on the occasion of his 60th birthday
Contributor Information
Prof. Dr. Bas de Bruin, Email: b.debruin@uva.nl.
Prof. Dr. Sven Schneider, Email: sven.schneider@chemie.uni-goettingen.de.
References
- 1. Marque S., Tordo P., Top. Curr. Chem. 2005, 250, 43. [Google Scholar]
- 2.
- 2a. Power P. P., Chem. Rev. 2003, 103, 789; [DOI] [PubMed] [Google Scholar]
- 2b. Armstrong A., Chivers T., Boere R. T., ACS Symp. Ser. 2006, 917, 66; [Google Scholar]
- 2c. Martin C. D., Soleilhavoup M., Bertrand G., Chem. Sci. 2013, 4, 3020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Nesterov V., Reiter D., Bag P., Frisch P., Holzner R., Porzelt A., Inoue S., Chem. Rev. 2018, 118, 9678. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Hinchley S. L., Morrison C. A., Rankin D. W. H., Macdonald C. L. B., Wiacek R. J., Voigt A., Cowley A. H., Lappert M. F., Gundersen G., Clyburne J. A. C., Power P. P., J. Am. Chem. Soc. 2001, 123, 9045; [DOI] [PubMed] [Google Scholar]
- 3b. Bezombes J. P., Borisenko K. B., Hitchcock P. B., Lappert M. F., Nycz J. E., Rankin D. W. H., Robertson H. E., Dalton Trans. 2004, 1980; [DOI] [PubMed] [Google Scholar]
- 3c. Ito S., Kikuchi M., Yoshifuji M., A. J. Arduengo III , Konovalova T. A., Kispert L. D., Angew. Chem. Int. Ed. 2006, 45, 4341; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 4447; [Google Scholar]
- 3d. Kinjo R., Donnadieu B., Bertrand G., Angew. Chem. Int. Ed. 2010, 49, 5930; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6066; [Google Scholar]
- 3e. Back O., Celik M. A., Frenking G., Melaimi M., Donnadieu B., Bertrand G., J. Am. Chem. Soc. 2010, 132, 10262; [DOI] [PubMed] [Google Scholar]
- 3f. Back O., Donnadieu B., von Hopffgarten M., Klein S., Tonner R., Frenking G., Bertrand G., Chem. Sci. 2011, 2, 858; [Google Scholar]
- 3g. Ishida S., Hirakawa F., Iwamoto T., J. Am. Chem. Soc. 2011, 133, 12968; [DOI] [PubMed] [Google Scholar]
- 3h. Pan X., Wang X., Zhao Y., Sui Y., Wang X., J. Am. Chem. Soc. 2014, 136, 9834; [DOI] [PubMed] [Google Scholar]
- 3i. Schwedtmann K., Schulz S., Hennersdorf F., Strassner T., Dmitrieva E., Weigand J. J., Angew. Chem. Int. Ed. 2015, 54, 11054; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11206; [Google Scholar]
- 3j. Schwedtmann K., Zanoni G., Weigand J. J., Chem. Asian J. 2018, 13, 1388. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Ndiaye B., Bhat S., Jouaiti A., Berclaz T., Bernardinelli G., Geoffroy M., J. Chem. Phys A 2006, 110, 9736; [DOI] [PubMed] [Google Scholar]
- 4b. Agarwal P., Piro N. A., Meyer K., Muller P., Cummins C. C., Angew. Chem. Int. Ed. 2007, 46, 3111; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 3171; [Google Scholar]
- 4c. Scheer M., Kuntz C., Stubenhofer M., Linseis M., Winter R. F., Sierka M., Angew. Chem. Int. Ed. 2009, 48, 2600; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2638; [Google Scholar]
- 4d. Tan G., Li J., Zhang L., Chen C., Zhao Y., Wang X., Song Y., Zhang Y.-Q., Driess M., Angew. Chem. Int. Ed. 2017, 56, 12741; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12915; [Google Scholar]
- 4e. Fischbach U., Trincado M., Grützmacher H., Dalton Trans. 2017, 46, 3443; [DOI] [PubMed] [Google Scholar]
- 4f. Kim Y.-E., Lee Y., Angew. Chem. Int. Ed. 2018, 57, 14159; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14355. [Google Scholar]
- 5.
- 5a. Liu L., Ruiz D. A., Munz D., Bertrand G., Chem 2016, 1, 147; [Google Scholar]
- 5b. Hansmann M. M., Jazzar R., Bertrand G., J. Am. Chem. Soc. 2016, 138, 8356. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Lammertsma K., Top. Curr. Chem. 2003, 229, 95; [Google Scholar]
- 6b. Slootweg J. C., Lammertsma K. in Science of Synthesis, Vol. 42 (Eds.: B. M. Trost, F. Mathey), Thieme, Stuttgart, 2009, p. 15; [Google Scholar]
- 6c. Waterman R., Dalton Trans. 2009, 18; [DOI] [PubMed] [Google Scholar]
- 6d. Aktaş H., Slootweg J. C., Lammertsma K., Angew. Chem. Int. Ed. 2010, 49, 2102; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2148. [Google Scholar]
- 7. Amme M. J., Kazi A. B., Cundari T. R., Int. J. Quantum Chem. 2010, 110, 1702. [Google Scholar]
- 8.
- 8a. Pal K., Hemming O. B., Day B. M., Pugh T., Evans D. J., Layfield R. A., Angew. Chem. Int. Ed. 2016, 55, 1690; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1722; [Google Scholar]
- 8b. Pagano J. K., Ackley B. J., Waterman R., Chem. Eur. J. 2018, 24, 2554. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Dequirez G., Pons V., Dauban P., Angew. Chem. Int. Ed. 2012, 51, 7384; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7498; [Google Scholar]
- 9b. Roizen J. L., Harvey M. E., Du Bois J., Acc. Chem. Res. 2012, 45, 911; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Kuijpers P. F., van der Vlugt J. I., Schneider S., de Bruin B., Chem. Eur. J. 2017, 23, 13819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Barkovskii N. V., Tsirel'nikov V. I., Emel'yanov A. M., Khodeev Y. S., Teplofiz. Vys. Temp. 1991, 29, 474; [Google Scholar]
- 10b. Huber K. P., Herzberg G., Molecular Spectra and Molecular Structure of Diatomic Molecules, Van Nostrand, New York, 1979, p. 456; [Google Scholar]
- 10c. Berkowitz J., Curtiss L. A., Gibson S. T., Greene J. P., Hillhouse G. L., Pople J. A., J. Chem. Phys. 1986, 84, 375; [Google Scholar]
- 10d. Cox J. D., Wagman D. D., Medvedev V. A., CODATA Key Values for Thermodynamics, Hemisphere Publishing Corp., New York, 1984, p. 1; [Google Scholar]
- 10e.“NIST-JANAF Themochemical Tables, Fourth Edition”: M. W. Chase, Jr. , J. Phys. Chem. Ref. Data Monogr. 1998, 9, 1. [Google Scholar]
- 11.The BDE(NH2) was calculated using a Hess cycle: BDE(NH2)=Δf H°gas(N) + 2 Δf H°gas(H)−BDE(NH)−Δf H°gas(NH2).
- 12.
- 12a. Kolmar S. S., Mayer J. M., J. Am. Chem. Soc. 2017, 139, 10687; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Bezdek M. J., Guo S., Chirik P. J., Science 2016, 354, 730. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Mindiola D. J., Hillhouse G. L., J. Am. Chem. Soc. 2001, 123, 4623; [DOI] [PubMed] [Google Scholar]
- 13b. Huynh M. H., Meyer T. J., Proc. Natl. Acad. Sci. USA 2004, 101, 13138; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Cowley R. E., Bontchev R. P., Sorrell J., Sarracino O., Feng Y., Wang H., Smith J. M., J. Am. Chem. Soc. 2007, 129, 2424; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d. Nieto I., Ding F., Bontchev R. P., Wang H., Smith J. M., J. Am. Chem. Soc. 2008, 130, 2716; [DOI] [PubMed] [Google Scholar]
- 13e. Cowley R. E., Eckert N. A., Vaddadi S., Figg T. M., Cundari T. R., Holland P. L., J. Am. Chem. Soc. 2011, 133, 9796; [DOI] [PubMed] [Google Scholar]
- 13f. Iluc V. M., Miller A. J. M., Anderson J. S., Monreal M. J., Mehn M. P., Hillhouse G. L., J. Am. Chem. Soc. 2011, 133, 13055; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13g. Wiese S., McAfee J. L., Pahls D. R., McMullin C. L., Cundari T. R., Warren T. H., J. Am. Chem. Soc. 2012, 134, 10114; [DOI] [PubMed] [Google Scholar]
- 13h. Milsmann C., Semproni S. P., Chirik P. J., J. Am. Chem. Soc. 2014, 136, 12099; [DOI] [PubMed] [Google Scholar]
- 13i. Scheibel M. G., Abbenseth J., Kinauer M., Heinemann F. W., Würtele C., de Bruin B., Schneider S., Inorg. Chem. 2015, 54, 9290; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13j. Pappas I., Chirik P. J., J. Am. Chem. Soc. 2016, 138, 13379; [DOI] [PubMed] [Google Scholar]
- 13k. Spasyuk D. M., Carpenter S. H., Kefalidis C. E., Piers W. E., Neidig M. L., Maron L., Chem. Sci. 2016, 7, 5939; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13l. Bezdek M. J., Chirik P. J., Angew. Chem. Int. Ed. 2018, 57, 2224; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2246. [Google Scholar]
- 14.
- 14a. Melenkivitz R., Mindiola D. J., Hillhouse G. L., J. Am. Chem. Soc. 2002, 124, 3846; [DOI] [PubMed] [Google Scholar]
- 14b. Iluc V. M., Hillhouse G. L., J. Am. Chem. Soc. 2010, 132, 15148. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Abbenseth J., Diefenbach M., Bete S. C., Würtele C., Volkmann C., Demeshko S., Holthausen M. C., Schneider S., Chem. Commun. 2017, 53, 5511; [DOI] [PubMed] [Google Scholar]
- 15b. Abbenseth J., Bete S. C., Finger M., Volkmann C., Würtele C., Schneider S., Organometallics 2018, 37, 802. [Google Scholar]
- 16.See the Supporting Information for spectroscopic, electrochemical, crystallographic or computational details.
- 17. McCleverty J. A., Meyer T. J. in Comprehensive Coordination Chemistry II, Vol. 5, Elsevier, Amsterdam, 2003, p. 556. [Google Scholar]
- 18. Geoffroy M., Lucken E. A. C., Mazeline C., Mol. Phys. 1974, 28, 839. [Google Scholar]
- 19. Warren J. J., Tronic T. A., Mayer J. M., Chem. Rev. 2010, 110, 6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steric reasons are unlikely for the lack of reactivity as 5 readily undergoes P−H transfer with TEMPO.
- 21. Masuda J. D., Jantunen K. C., Ozerov O. V., Noonan K. J. T., Gates D. P., Scott B. L., Kiplinger J. L., J. Am. Chem. Soc. 2008, 130, 2408. [DOI] [PubMed] [Google Scholar]
- 22.The BDEO-H of Mes*OH in THF was calculated using the pK a value from (see the Supporting Information): Ding F., Smith J. M., Wang H., J. Org. Chem. 2009, 74, 2679.19275192 [Google Scholar]
- 23.The calculated BDEs were calibrated (taking advantage of internal error cancelation) against the experimentally available BDE of propene, using a Hess cycle (see Supporting Information).
- 24. Cappellani E. P., Drouin S. D., Jia G., Maltby P. A., Morris R. H., Schweitzer C. T., J. Am. Chem. Soc. 1994, 116, 3375. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary