

Abstract
Objective
Clinical trials of the anti–interleukin‐17A (anti–IL‐17A) antibody secukinumab have demonstrated a crucial role of the cytokine IL‐17A in the pathogenesis of spondyloarthritis (SpA); however, its cellular source in this condition remains a matter of controversy. Group 3 innate lymphoid cells (ILC3s) have been recently identified as potent producers of proinflammatory cytokines, including IL‐17A and IL‐22, in a number of different tissues. This study was undertaken to characterize the presence and composition of ILCs, and investigate whether these cells are an important source of IL‐17A, in the synovial tissue (ST) of patients with SpA.
Methods
Matched ST, synovial fluid, and peripheral blood (PB) samples were obtained from SpA patients with actively inflamed knee joints. ILC subsets were characterized by flow cytometry. Gene expression analysis at the single‐cell level was performed directly ex vivo and after in vitro activation. An IL‐17A enzyme‐linked immunospot assay was used to detect IL‐17A–secreting cells.
Results
ILCs, and particularly NKp44+ ILC3s, were expanded in inflamed arthritic joints. Single‐cell expression analysis demonstrated that ST ILCs were clearly distinguishable from ST T cells and from their PB counterparts. Expression of the Th17 signature transcripts RORC,AHR, and IL23R was detected in a large proportion of ST ILC3s. These cells were capable of inducing expression of IL22 and CSF2, but not IL17A, in response to in vitro restimulation.
Conclusion
Our findings demonstrate that absolute and relative numbers of ILC3s are enriched in the synovial joints of patients with SpA. However, these cells are not a significant source of IL‐17A in this disease.
Introduction
Spondyloarthritis (SpA) is a major form of chronic inflammatory arthritis characterized by inflammation of axial and peripheral joints and extraarticular manifestations such as psoriasis and inflammatory bowel disease (IBD), including Crohn's disease and ulcerative colitis. Interleukin‐17A (IL‐17A) has been demonstrated to be a key driver of inflammation in axial SpA 1, 2 and peripheral SpA 3, 4; however, the cellular source of IL‐17A in this condition remains largely unknown and has been a matter of debate. In the peripheral blood (PB) of patients with ankylosing spondylitis (AS), CD4+ T cells 5, 6, 7 and γδ T cells 8 have been demonstrated to be the major producers of IL‐17A upon ex vivo stimulation. In contrast, direct analysis of synovial membrane, psoriatic skin, and enthesis has identified CD8+ T cells 9, mast cells and neutrophils 10, 11, 12, 13, 14, and CD4−CD8– double‐negative “innate‐like” T cells 15, suggesting that the role of innate immune cells is more pronounced than that of canonical Th17 cells in the inflamed target tissues in SpA.
Innate lymphoid cells (ILCs) are a relatively newly discovered heterogeneous population of innate immune lymphocytes that are strategically positioned at barrier surfaces and regulate immunity, inflammation, and tissue homeostasis 16. ILCs mirror the phenotype and function of T cells and therefore are divided into 3 subsets: the group 1 ILCs express the transcription factor T‐bet and secrete the Th1‐related cytokines interferon‐γ and tumor necrosis factor (TNF). The group 2 ILCs express GATA‐3 and chemoattractant receptor–like molecule expressed on Th2 cells (CRTH2) and produce the Th2‐related cytokines IL‐4, IL‐5, IL‐9, and IL‐13. The group 3 ILCs depend on the transcription factor retinoic acid receptor–related orphan nuclear receptor γt (RORγt) and include lymphoid tissue inducer cells and ILC3s.
In humans, almost all ILC3s express CCR6 and CD117, and can be subdivided into NKp44+ and NKp44− subsets. In adult humans NKp44+ ILC3s reside in tonsil and intestine and represent an important source of IL‐22 17, 18, 19, 20, while IL‐17–producing NKp44− ILC3s have been described only in fetal developing lymph nodes 21, 22. In contrast to basic science studies, recent translational rheumatology studies demonstrated the presence of IL‐17–producing NKp44+ ILC3s in the gut 23 and synovial fluid (SF) 24 of patients with SpA, in the intestine of patients with Crohn's disease 25, and in the normal enthesis 26, but not in the synovial membrane 27, the primary site of inflammation in SpA. The reason for this apparent discrepancy remains to be clarified; however, lack of stringent ILC phenotyping, high functional plasticity of ILCs, and potential contamination with T cells during isolation may play an important role.
In this study we assessed whether ILCs are a significant source of IL‐17A in SpA. Using a combination of 2 unique approaches, rigorous analysis at the individual cell level and examination of directly ex vivo isolated ILCs from SpA ST, we unambiguously demonstrated that ILC3s do not contribute to IL‐17A production, but are a source of IL‐22 and granulocyte–macrophage colony‐stimulating factor (GM‐CSF), in the synovium of patients with SpA.
Patients and Methods
Patients and synovial tissue biopsies
This study included 26 patients with peripheral SpA according to the Assessment of SpondyloArthritis international Society criteria 28 and 11 patients with rheumatoid arthritis (RA) according to the American College of Rheumatology 1987 classification criteria 29. Before enrollment, all patients provided written informed consent to participate in the study as approved by the local medical ethics committee of Academic Medical Center, University of Amsterdam. Patient characteristics are presented in Table 1. ST biopsy specimens were obtained from clinically inflamed knee or ankle joints and processed as described earlier 30.
Table 1.
Demographic and clinical characteristics of the patientsa
| SpA (n = 26) | RA (n = 11) | |
|---|---|---|
| Age, mean ± SD years | 44.9 ± 12.7 | 53.4 ± 15.3 |
| No. male/female | 20/6 | 4/7 |
| Disease duration, median (range) years | 6 (1–16.7) | 3.7 (0.4–11.5) |
| DAS28, mean ± SD | 3.9 ± 1.1 | NA |
| CRP, mean ± SD mg/liter | 33.1 ± 52.4 | 21.9 ± 31.9 |
| ESR, mean ± SD mm/hour | 33.2 ± 34.7 | 18.3 ± 16.6 |
| TJC28 | 3.5 ± 2.8 | 2.8 ± 1.8 |
| SJC28 | 1.9 ± 1.6 | 1.2 ± 0.6 |
| RF positive, no. (%) | NA | 4 (36.4) |
| ACPA positive, no. (%) | NA | 2 (18.2) |
| Psoriatic arthritis, no. (%) | 17 (65.4) | NA |
| Previous DMARD or prednisone therapy, no. (%) | 10 (38.5) | 9 (81,8) |
| Previous biologic therapy, no. (%)b | 10 (38.5) | 2 (18.2) |
SpA = spondyloarthritis; RA = rheumatoid arthritis; DAS28 = Disease Activity Score in 28 joints; NA = data not available; CRP = C‐reactive protein; ESR = erythrocyte sedimentation rate; TJC28 = tender joint count of 28 joints; SJC28 = swollen joint count of 28 joints; RF = rheumatoid factor; ACPA = anti–cyclic citrullinated peptide antibody; DMARD = disease‐modifying antirheumatic drug.
All patients underwent a washout period of at least 3 months before participation in the study.
Isolation of cells
Mononuclear cells were isolated from heparinized PB and from SF by density‐gradient centrifugation using Lymphoprep Ficoll‐Isopaque according to the instructions of the manufacturer (Axis‐Shield). To obtain cell suspensions, ST was cut into small pieces and digested for 60 minutes at 37°C with Liberase TM (125 μg/ml) and DNase I (200 μg/ml) (both from Roche) containing Dulbecco's modified Eagle's medium (Gibco). Cell suspensions were filtered through a 70‐μm nylon mesh, and ST mononuclear cells were directly stained for fluorescence‐activated cell sorting (FACS) analysis.
Flow cytometric analysis
The following antibodies to human proteins were used for flow cytometry: fluorescein isothiocyanate (FITC)–conjugated anti‐CD1a (clone HI149), anti‐CD3 (clone OKT3), anti‐CD94 (clone DX22), anti‐CD123 (clone 6H6), anti‐CD16 (clone 3G8), anti‐CD19 (clone HIB19), anti‐CD4 (clone RPA‐T4), anti–Fcε receptor 1 (clone AER‐37 [CRA‐1]), BV421‐conjugated anti‐CD161 (clone HP‐3G10), phycoerythrin (PE)–conjugated anti‐NKp44 (clone P44‐8) (all from BioLegend); FITC‐conjugated anti‐CD8 (SK1), anti‐CD14 (MϕP9), anti‐CD16 (3G8), anti‐CD34 (clone 8G12), anti–T cell receptor (anti‐TCR) αβ (clone T10B9), anti‐TCRγδ (clone 11F2), Alexa Fluor 647–conjugated anti‐CRTH2 (clone BM16), PE–Cy7–conjugated anti‐CD127 (clone R34.34), PE–Cy5.5–conjugated CD117 (clone 104D2D1) (all from Becton Dickinson); Alexa Fluor 700–conjugated anti‐CD3 (clone UCHT1; eBioscience); and FITC‐conjugated anti‐BDCA2 (clone AC144; Miltenyi Biotec). For phenotypic analysis and sorting by flow cytometry, data were collected with a FACSAria III cell sorter (BD Biosciences) and analyzed with FlowJo software (Tree Star).
Single‐cell gene expression analysis
Single‐cell quantitative polymerase chain reaction (qPCR) analysis was performed on ILCs isolated from synovial joints of 6 patients with SpA (specifically, from ex vivo unstimulated SF [1 patient] and ST [1 patient], phorbol myristate acetate [PMA]/ionomycin–stimulated SF [2 patients], and PMA/ionomycin‐ or IL‐1/IL‐23–stimulated ST [2 patients]). For analysis of ILCs from ST, ILCs from matched PB as well as T cells from both compartments from the same SpA donor were assessed. Single cells were directly sorted into 96‐well PCR plates containing lysis buffer. Index sorting during FACS was used to identify the cell surface marker profiles of sorted individual cells.
Quantitative reverse transcription–PCR was performed using a Two‐Step Protocol with SuperScript Vilo cDNA Synthesis kit (Invitrogen) for reverse transcription reaction and TaqMan PreAmp Master Mix (Applied Biosystems) for specific target amplification. Gene expression profiling was performed using a Biomark 48.48 Dynamic Array (Fluidigm) with EvaGreen Supermix (Bio‐Rad) according to the manufacturers’ protocols. The primers used (from Fluidigm) are listed in Supplementary Table 1, on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40736/abstract. Mean and SD Ct values from housekeeping genes (B2M, ACTB, and GAPDH) for all cells were calculated. Cells with housekeeping gene Ct values exceeding the SD by >2‐fold were eliminated from the analysis.
Freshly isolated single ILCs and single T cells from peripheral blood, synovial fluid, and synovial tissue were analyzed ex vivo or stimulated with PMA (10 ng/ml) and ionomycin (1 μg/ml) (both from Sigma) for 4 hours before analysis. Alternatively, cells were cultured with IL‐1β (50 ng/ml), IL‐23 (50 ng/ml), and IL‐2 (20 units/ml) (all from R&D Systems) for 36 hours and then restimulated with PMA/ionomycin for 4 hours. The frequency of expression of each gene was calculated based on the number of cells with detectable signal (Ct ≤38) from the total number of cells in each group. ILCs with expression of CD3 and T cells without expression of CD3 were omitted from analysis. Expression levels were analyzed with GraphPad Prism version 7. The t‐distributed stochastic neighbor embedding technique for dimensionality reduction 31 was applied to present single‐cell qPCR data in 2‐dimensional space with the Rtsne package and visualized with ggplot2 in Rstudio (www.rstudio.com) for R 3.4.2 (www.r-project.org).
IL‐17A enzyme‐linked immunospot (ELISpot) assay
For IL‐17A ELISpot assay, the PVDF membrane of the culture plates (Millipore) was treated with 70% ethanol and rinsed with phosphate buffered saline. Diluted anti–IL‐17A antibody (eBio64CAP17; eBioscience) was loaded into wells and incubated overnight at 4°C. Lin−CD3−CD127+CD161+ ILCs and CD3+ T cells were sorted from SpA SF, and a total of 2 × 103 cells of each population in 100 μl RPMI medium (Gibco) were seeded into anti–IL‐17A–coated wells and stimulated with PMA/ionomycin overnight at 37°C with 5% CO2 in 96% humidity. After incubation, the cells were washed from the wells and the presence of IL‐17A–producing cells was revealed by incubation of membrane with a biotinylated anti–IL‐17A antibody (eBio64DEC17; eBioscience) for 1 hour at 37°C, followed by incubation with γ‐aminobutyric acid–conjugated streptavidin (U‐Cytech Biosciences) to develop silver spots at places where cells secreted IL‐17A. After the wells were dried, positive reactions (identified as black spots) were analyzed by counting spots on an ELISpot reader (CTL) and read as the number of spots per well.
Statistical analysis
Median and interquartile range (IQR) values for the experimental results were calculated, and the statistical significance of differences between groups was determined by Kruskal‐Wallis test with Dunn's nonparametric post hoc comparison, Wilcoxon's test, or Mann‐Whitney U test. P values less than 0.05 were considered significant. All data were analyzed with GraphPad Prism version 7.
Results
Enrichment of ILC3s in the inflamed ST of patients with SpA
In order to assess whether ILCs are present in the inflamed synovial joint of patients with SpA, we performed a stringent phenotypic analysis of ILC subsets using a previously established gating strategy 32 (Figure 1A). According to the proposed ILC nomenclature 33, all human ILC subsets are lineage marker–negative lymphocytes, which express the cytokine receptor subunit IL‐7Rα (CD127) and the natural killer cell receptor CD161. Analysis of matched PB, SF, and ST samples revealed that the frequency of ILCs was higher in the inflamed ST of patients with SpA (median 0.27% of the lymphocyte population [IQR 0.22–0.62%]) than in corresponding PB (median 0.04% [IQR 0.03–0.08%]) (P = 0.018) (Figure 1B).
Figure 1.
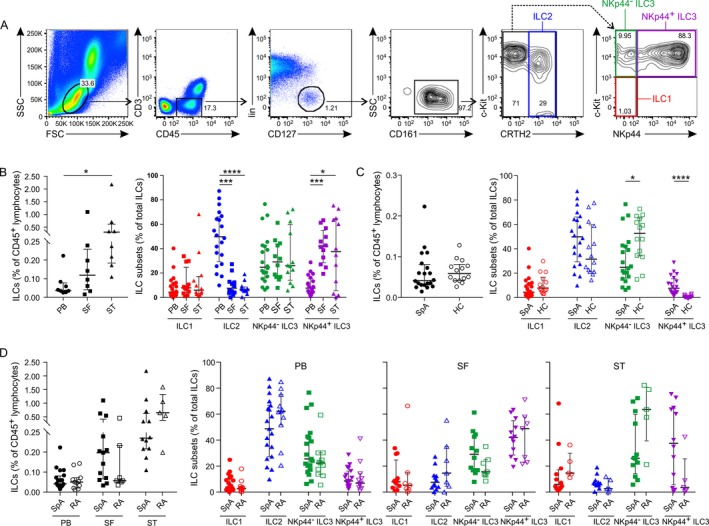
Increased numbers of group 3 innate lymphoid cells (ILC3s) in the inflamed synovial joint. A, Gating strategy for flow cytometric detection of ILCs in spondyloarthritis (SpA) patient synovial fluid (SF), with ILCs defined as negative for lineage markers (CD1a−CD3−CD4−CD8−CD14−CD16−CD19−CD34−CD94−CD123−BDCA‐2−FcεR1α−TCRαβ−TCRγδ−) and CD45+CD127+CD161+. B, Frequencies of ILCs in peripheral blood (PB), SF, and synovial tissue (ST) (paired samples from 8 patients) (left) and ILC subsets in PB (n = 20), SF (n = 12), and ST (n = 12) (right) from patients with SpA. C, Frequencies of ILCs in PB from healthy controls (HC) (n = 14) and patients with SpA (n = 20), shown as total ILCs (left) and by ILC subset (right). D, Frequencies of ILCs in PB (n = 20), SF (n = 12), and ST (n = 12) from patients with SpA and in PB (n = 11), SF (n = 7), and ST (n = 5) from patients with rheumatoid arthritis (RA), shown as total ILCs (left) and by ILC subset (right). Symbols represent individual subjects; bars show the median and interquartile range. * = P < 0.05; *** = P < 0.001; **** = P < 0.0001. CRTH2 = chemoattractant receptor–like molecule expressed on Th2 cells; BDCA‐2 = blood dendritic cell antigen 2; FcεR1α = Fcε receptor 1α; TCRαβ = T cell receptor αβ.
Immunophenotyping of ILC subsets showed a significant increase in the frequency of NKp44+ ILC3s in ST (median 37.5% of the total ILCs [IQR 5.6–64.6%]) and in SF (median 42% [IQR 28.5–55%]) versus PB (median 7.6% [IQR 3.9–15.5%]) (P = 0.015 and P = 0.0002, respectively). This increase was independent of SpA phenotype (psoriatic versus nonpsoriatic) (Supplementary Figure 1, on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40736/abstract). The second most prominent ILC subset in the joint was NKp44− ILC3, composing 26% of the total ILCs, but this population was present in SF and PB compartments at similar frequencies. ILC1 and ILC2 populations were observed in synovium at low frequencies. ILC2 was the predominant population in PB (median 49.8% [IQR 29.3–66.1%]), versus median 7.6% (IQR 3.7–12.8%) in the SF (P = 0.0004) and median 6.6% (IQR 5.2–8.6%) in the ST (P < 0.0001). Frequencies of ILC1 were rather low (<10% of total ILCs), and similar in all compartments (Figure 1B).
As we observed an absolute and relative enrichment of NKp44+ ILC3s in the ST of patients with SpA, we next sought to determine whether these cells were also expanded in the PB of these patients as compared to healthy controls. While the total frequency of ILCs in PB was similar between healthy controls and patients with SpA (Figure 1C), NKp44+ ILC3s were significantly increased in the PB of patients with SpA compared to that of healthy controls (median 7.6% [IQR 3.9–15.5%] versus median 0.5% [IQR 0.2–1.6%]; P < 0.0001) (Figure 1C). NKp44− ILC3s were decreased in PB of SpA patients compared to healthy controls (median 24.6% [IQR 15.9–42.5%] versus 52.7% [IQR 34.2–65.3%]; P = 0.03).
Increase in the NKp44+ ILC3 subset in the synovium and blood of SpA patients is not disease specific
To determine whether the increase in NKp44+ ILC3s in the ST and PB is specific to SpA, we analyzed the frequency and phenotype of ILCs in PB, SF, and ST of patients with RA as an inflammatory arthritis–matched control group. We found that frequencies of total ILCs in the joint and blood were similar between patients with SpA and those with RA (Figure 1D). Furthermore, there were no differences between SpA and RA patients in ILC subset distribution in the PB, SF, or ST (Figure 1D). There was a trend toward a higher percentage of NKp44+ ILC3s in the ST of patients with SpA (median 37.5% [IQR 5.6–64.6%]) compared to the ST of patients with RA (median 3.5% [IQR 0.8–25.6%]); however, this difference did not reach statistical significance (P = 0.13). Also, no difference was detected in the frequencies of NKp44+ ILC3s in the PB of SpA and RA patients, suggesting that the observed increase in NKp44+ ILC3s in the PB of SpA patients as compared to healthy controls is related to chronic inflammation rather than to SpA‐specific pathogenesis.
Presence of IL‐22– and CSF2–expressing, but not IL‐17A–expressing, ILCs in SpA SF
Having identified ILC3 as a predominant ILC population in the SpA synovial joint, we next explored whether these cells are an important source of IL‐17A. To avoid contamination with T cells as much as possible and to assess heterogeneity of ILCs, we sorted 14 single Lin−CD3−CD127+CD161+ ILCs from SpA SF (obtained from 1 patient) and analyzed expression of a selected set of transcripts directly ex vivo, using Biomark arrays. Targets included transcription factors, cytokines, chemokines, cell surface proteins, and cytokine receptors, which characterize the phenotype and function of ILCs (Supplementary Table 1, http://onlinelibrary.wiley.com/doi/10.1002/art.40736/abstract).
Expression analysis (Figure 2A) demonstrated that SF ILCs did not express TBX21, which encodes the transcription factor T‐bet and is known to be expressed by conventional ILC1s. The majority of SF ILCs (79%) expressed GATA3, which encodes the transcription factor GATA‐3 that is essential for development and function of the ILC2 subset. When examining expression of transcription factors important for the ILC3 subset we found that RORC, which encodes the transcription factor RORγt, was present in a subset of SF ILCs (21%). In contrast, expression of AHR, which encodes another transcription factor associated with ILC3 biology, was detected in the majority of SF ILCs (93%) (Supplementary Figure 2A, on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40736/abstract). When analyzing cytokine expression we detected IFNG, TNF, and IL29 transcripts (Figure 2A and Supplementary Figure 2A). Furthermore, we found that freshly isolated SF ILCs expressed genes that encode the chemokine receptors CXCR3, CXCR4, CXCR5, and CCR6 (Supplementary Figure 2A).
Figure 2.
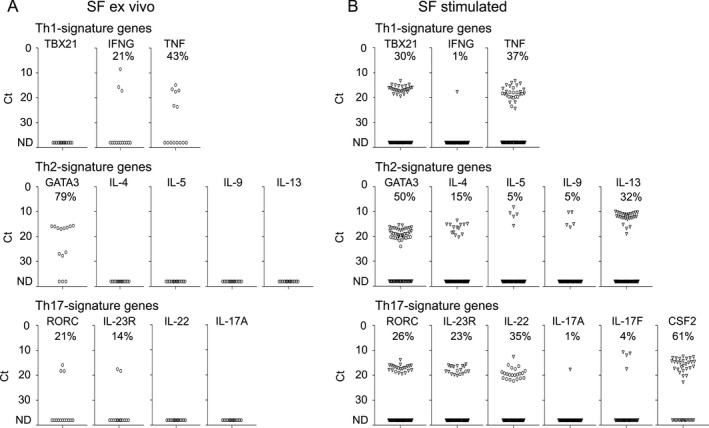
Single‐cell expression profiling for transcripts specific for Th1, Th2, and Th17 signature genes in innate lymphoid cells isolated from spondyloarthritis patient synovial fluid (SF) directly ex vivo (n = 1) (A) or stimulated with phorbol myristate acetate/ionomycin (n = 2) (B). Symbols represent single cells; in B, cells from the first patient are represented by circles, and cells from the second patient by triangles. Frequencies of expression were calculated according to the number of cells with detectable signal (Ct ≤38) from the total cells in each group. ND = not detectable.
As we were not able to detect expression of most of the cytokines assessed, including IL‐17A, directly ex vivo, we examined transcripts of 77 single ILCs in SF from 2 different SpA patients following restimulation with PMA/ionomycin. In general, transcription profiling of stimulated SF ILCs revealed expression patterns very similar to those observed with unstimulated SF ILCs (Figure 2 and Supplementary Figure 2). Again we detected substantial populations of RORC‐expressing, GATA3‐expressing, and AHR‐expressing ILCs (26%, 50%, and 80%, respectively). Similar to the findings with ILCs analyzed ex vivo, we detected expression of TNF, IL29, CXCR3, CXCR4, CXCR5, and CCR6 transcripts after PMA/ionomycin stimulation. Of interest, expression of some transcripts, such as CCL20 and ICOS, was dramatically increased after activation of ILCs (Supplementary Figure 2B). Although we observed expression of TBX21 in 30% of SF ILCs, only 1% of these cells were able to induce expression of IFNG after stimulation with PMA/ionomycin. Expression of Th2 signature cytokines was detected in a fraction of PMA/ionomycin‐activated ILCs (15% for IL4, 5% for IL5, 5% for IL9, and 32% for IL13).
Expression of IL17A and IL17F transcripts was detected in only 1% and 4% of stimulated SF ILCs, respectively. In contrast, we observed expression of IL22 and CSF2 transcripts, encoding IL‐22 and GM‐CSF cytokines, in prominent fractions of restimulated SF ILCs (35% and 61%, respectively) (Figure 2B).
Presence of IL‐22–expressing, but not IL‐17A–expressing, ILC3s in SpA ST
Since we were unable to detect IL17A‐expressing ILCs in SpA SF either directly ex vivo or after in vitro restimulation, we hypothesized that this tissue‐resident cell population could be present exclusively in SpA ST. In order to test this hypothesis, we assessed expression of a selected set of genes at the single‐cell level in 17 ILCs sorted from ST from 1 patient (and as a control, in 13 ILCs sorted from matched PB) directly ex vivo. This analysis revealed that ST ILCs did not express TBX21, 76% expressed GATA3, 23% expressed RORC, and 35% expressed IL23R, while PB ILCs uniformly expressed GATA3 and were negative for IL3R, with expression of TBX21 and RORC detected in minor fractions of these cells (8% and 15%, respectively) (Figure 3A). Expression of TNF, IL29, GRZB, CXCR3, CXCR4, and CXCR5 was detectable in ILCs from both locations. Of note, we detected a small population of IL22‐expressing cells (11.8% of ST ILCs and 7.7% of PB ILCs) (Figure 3). Expression of other cytokines, including IL17A, was not detectable in SpA ST or PB ILCs ex vivo without restimulation (Figure 3 and Supplementary Figure 3, on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40736/abstract).
Figure 3.
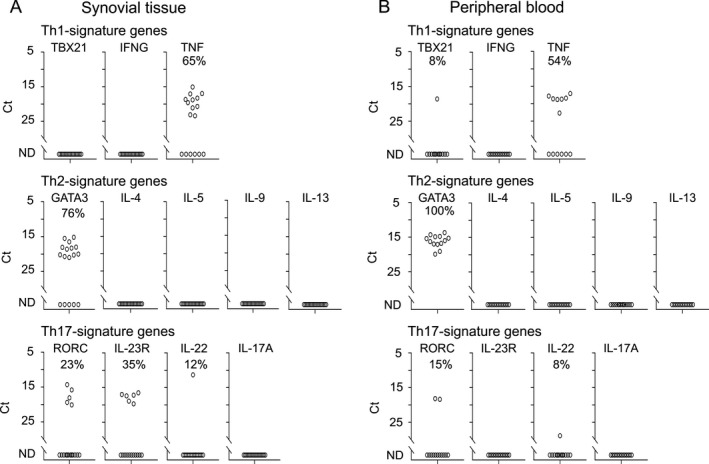
Single‐cell expression profiling for transcripts specific for Th1, Th2, and Th17 signature genes in innate lymphoid cells isolated from spondyloarthritis patient synovial tissue (A) and matched peripheral blood (B) directly ex vivo (n = 1). Symbols represent single cells. ND = not detectable.
Next, to assess the intrinsic capacity of ST ILCs to express IL17A, we sorted single ILC1 cells (0 cells from ST and 4 cells from PB), ILC2 cells (10 cells from ST and 26 cells from PB), NKp44− ILC3 cells (58 cells from ST and 9 cells from PB), and NKp44+ ILC3 cells (3 cells from ST and 0 cells from PB), and, as a control, CD3+ T cells (47 cells from ST and 34 cells from PB) from 2 SpA donors and analyzed their gene expression profile after activation with PMA and ionomycin. Unbiased clustering analysis revealed 4 distinct populations corresponding to ST ILCs, PB ILCs, ST T cells, and PB T cells (Figure 4 and Supplementary Figure 4, http://onlinelibrary.wiley.com/doi/10.1002/art.40736/abstract), demonstrating clear differences in their expression profiles (Figure 5 and Supplementary Figures 5 and 6, http://onlinelibrary.wiley.com/doi/10.1002/art.40736/abstract). No single ILC expressed CD3 transcript, which encodes the pan–T cell marker CD3, whereas all T cells expressed this gene. All ILCs expressed KLRB1, which encodes CD161, the pan marker for all human ILCs (Supplementary Figure 4). Seventy‐one percent of PB‐derived and 36% of ST‐derived T cells expressed TBX21. Accordingly, a significant population of T cells from both locations (67% and 50% from PB and ST, respectively) were able to express IFNG upon stimulation. In contrast, activated ILCs failed to express this Th1 signature cytokine, despite the expression of TBX21 in a substantial proportion of ILC2s (58%) and NKp44− ILC3s (21%) in the ST (Figure 5A).
Figure 4.
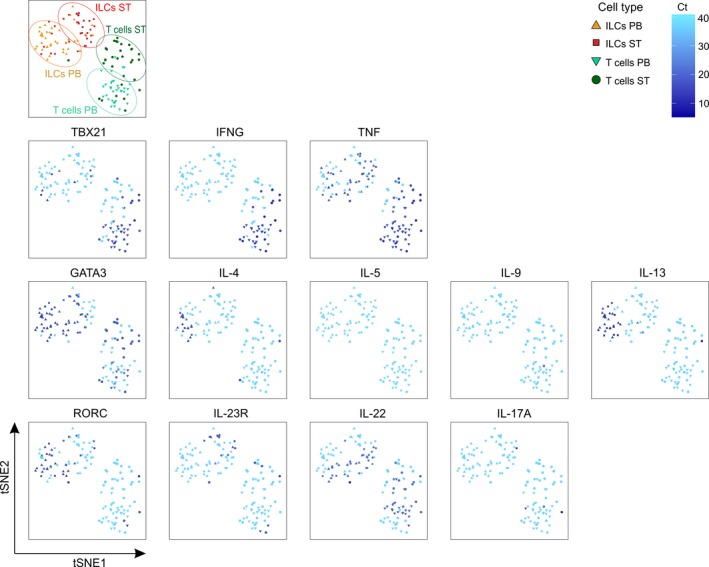
Two‐dimensional t‐distributed stochastic neighbor embedding (tSNE) representation of expression of transcripts specific for Th1, Th2, and Th17 signature genes in phorbol myristate acetate/ionomycin–stimulated innate lymphoid cells (ILCs) and T cells isolated from spondyloarthritis patient peripheral blood (PB) and synovial tissue (ST). Data are representative of 2 independent experiments with 1 donor each. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.40736/abstract.
Figure 5.
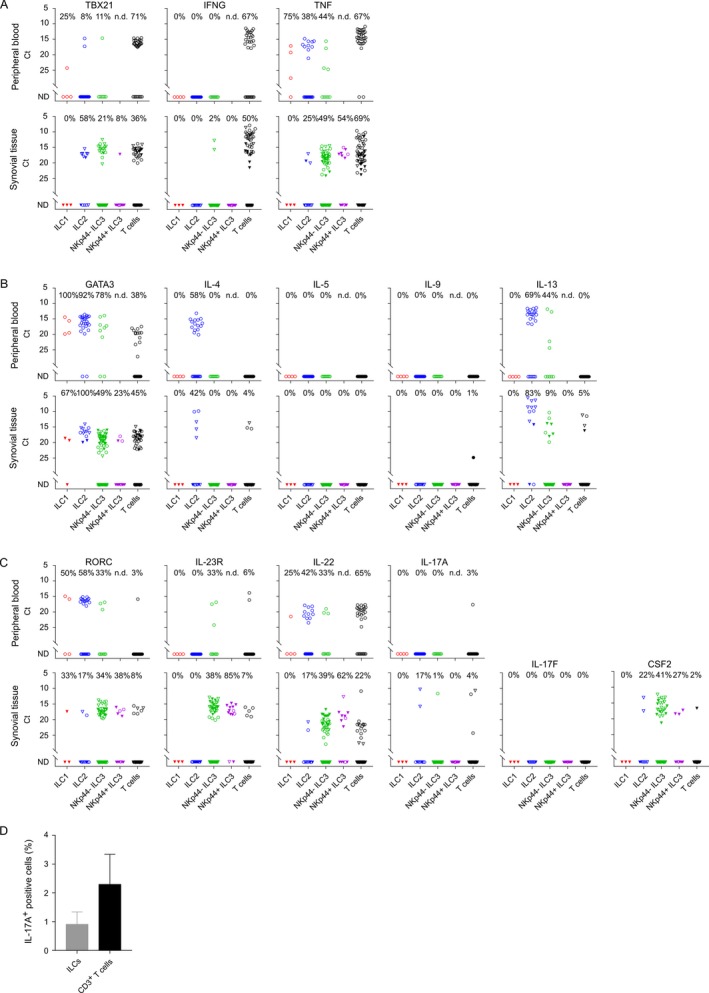
A–C, Single‐cell expression profiling for transcripts specific for Th1 (A), Th2 (B), and Th17 (C) signature genes in phorbol myristate acetate (PMA)/ionomycin–stimulated innate lymphoid cells (ILCs) (ILC1, ILC2, and NKp44− and NKp44+ ILC3) and T cells isolated from spondyloarthritis (SpA) patient peripheral blood (n = 1) and synovial tissue (n = 2). Symbols represent single cells; cells from the first patient are represented by circles, and cells from the second patient by triangles. Open symbols represent cells stimulated with PMA/ionomycin alone; closed symbols represent cells stimulated with an interleukin‐1β (IL‐1β)/IL‐23 cytokine cocktail prior to stimulation with PMA/ionomycin. D, IL‐17A protein produced by PMA/ionomycin–stimulated ILCs and T cells isolated from SpA patient synovial fluid (n = 3) and analyzed by enzyme‐linked immunospot assay. Values are the median and interquartile range. ND = not detectable. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.40736/abstract.
Expression of GATA3 was detected in a majority of PB and ST ILCs (Figure 5B). Correspondingly, a large proportion of stimulated ILC2s were shown to express IL4 (58% of PB cells and 42% of ST cells) and IL13 (69% of PB cells and 83% of ST cells) (Figure 5B). In contrast to ILCs and despite substantial population of GATA3‐expressing cells, PB T cells did not express Th2 signature cytokines in response to activation, and the frequencies of IL4‐ and IL13‐expressing ST T cells were low (4–5%) (Figure 5B).
Analysis of Th17‐associated genes revealed expression of RORC and IL23R in a small fraction of in vitro–stimulated ST T cells (8% and 7%, respectively), which was concordant with a low frequency of IL17A‐expressing ST T cells (4%) (Figure 5C). In contrast, expression of RORC and IL23R was found in 38% and 85% of NKp44+ ILC3s, respectively, and in ~34% and 38% of NKp44− ST ILC3s, respectively. However, strikingly, only 1% of the NKp44− ILC3s and none of NKp44+ ILC3s in ST were able to express IL17A upon stimulation with PMA/ionomycin. We detected IL17A transcript in 17% of ILC2s (2 of 12 cells analyzed); however, these cells did not coexpress RORC or IL23R. No single IL17F‐expressing ILC was detected after PMA/ionomycin stimulation (Figure 5C). In sharp contrast, a marked fraction of ILC3s induced expression of IL22 (39% of NKp44− and 62% NKp44+ cells) and CSF2 (41% of NKp44− and 27% of NKp44+ cells) in response to in vitro stimulation.
With regard to expression of other transcripts, the majority of restimulated ST ILCs, similarly to ex vivo isolated cells, expressed genes that encode subunits of the IL‐1, IL‐2, and IL‐10 receptors, CD161 receptor, the chemokine receptors CXCR3, CXCR4, CXCR5, and CCR6, the transcription factors aryl hydrocarbon receptor and RUNX, and the inducible costimulator and granzyme B proteins. However, expression of some transcripts, such as IL2, CCL20, CD40L, IL12RB2, and PRF1, was detectable in these cells only after in vitro activation (Supplementary Figures 5 and 6, on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40736/abstract).
As we did not detect IL17A expression in a significant proportion of synovial ILCs and as an IL‐1β and IL‐23 cytokine cocktail has been demonstrated to potentiate IL‐17A production from innate cells including ILCs (26, 33, 34), we next examined changes in the expression pattern of IL7A after activation of ILCs with these cytokines. We analyzed 3 ILC1s, 2 ILC2s, 29 NKp44− and 10 NKp44+ ILC3s, and 27 T cells. Single‐cell transcription analysis of IL‐1β/IL‐23–stimulated ST ILCs revealed no IL17A‐ or IL17F‐expressing cells, yet these cells expressed IL23R and RORC and responded to the stimulation by an increase in expression of IL22 transcript (Figure 5C). In order to confirm these data at the protein level, the total number of IL‐17A–producing ILCs and T cells in SpA SF was analyzed by ELISpot assay. This revealed that a median of ~0.9% (IQR 0.8–1.3%) of total ILCs and 2.3% (IQR 1.1–3.3%) of total T cells from SpA SF were able secret IL‐17A in response to PMA/ionomycin stimulation (Figure 5D).
Discussion
ILC3s play an important role in the regulation of tissue homeostasis, repair, and inflammation 20 and, similar to their Th17 cell counterparts, have been proposed to produce IL‐17A, IL‐22, and GM‐CSF 16. Recent studies have suggested that IL‐17–producing ILC3s are amplified in the inflamed tissue of patients with SpA 23, 24, 25. However, whether ILC3s constitute an important source of IL‐17A in SpA synovitis remains an open question. A major concern is the absence of stringent ILC phenotyping criteria. Due to the lack of exclusive markers, during flow cytometric analysis of ILCs a combination of lineage markers is usually used to exclude other immune cells. Different research groups have used nonidentical sets of antibodies to define their lineage, which may have introduced variability in the results. Further, it is plausible to assume that the scarcity of ILCs together with the relatively high number of T cells, which, depending on their activation status, might express different levels of CD3, may yield an ILC population contaminated with T cells. Results obtained from such contaminated ILC populations will be biased since T cells may contribute to the IL‐17A levels detected. Another crucial factor is the analysis of tissues that are not relevant to the disease. Reports from recent translational rheumatology studies described IL‐17–producing ILCs in the gut of patients with AS‐associated IBD 23, in the SF of patients with psoriatic arthritis (PsA) 24, in the skin of patients with psoriasis (35), and in normal enthesis samples obtained from donors without systemic inflammatory disease 26, but not in the inflamed synovium, enthesis, or spine, the key target tissues in SpA.
In the current study we reassessed whether ILC3s contribute significantly to the cellular source of IL‐17A in SpA, attempting to overcome these hurdles. First, we focused our research on peripheral synovitis in SpA as this allowed us to study ILCs directly in the inflamed target tissue. We evaluated the distribution of ILCs in SpA ST using stringent phenotyping criteria according to proposed ILC nomenclature 33. In order to minimize contamination with T cells during phenotypic analysis, we included 2 different anti‐CD3 antibodies in a sequential gating strategy: one in the “dump” channel for lineage markers and the other in the separate fluorescent channel. Finally, using a unique single‐cell approach, which allows profiling of individual cells, we assessed heterogeneity of ST ILCs and their capacity to express Th17 signature cytokines either directly ex vivo or after in vitro restimulation.
We observed an absolute and relative enrichment of NKp44+ ILC3s in the inflamed SpA synovium. A similar increase was described previously in SF 24 and in ST of patients with end‐stage SpA 27, although the latter study did not include an anti‐CRTH2 antibody, crucial for proper definition of the ILC2 subset in the phenotyping panel. Furthermore, analysis of the PB compartment revealed that NKp44+ ILC3s are significantly increased in patients with SpA compared to age‐matched healthy controls, similar to findings reported previously in psoriasis (35, 36), AS 23, and PsA 24. Yet this expansion does not seem to be specific for SpA, as we found comparable frequencies of ILC3s in the PB and ST of patients with RA. We did not detect any IL17A‐expressing ILCs isolated ex vivo from SpA patient SF and ST, despite adequate expression of other proinflammatory mediators such as TNF and IL‐29. Moreover, even after in vitro activation with potent stimuli, IL7A was either absent or expressed by only a miniscule population, even if a notable fraction of synovial ILC3s expressed Th17 signature transcripts RORC, IL23R, AHR, and IL22.
There were some important limitations to this study. Although we did not detect IL17A transcripts in ILCs directly ex vivo or after PMA/ionomycin or IL‐1β/IL‐23 stimulation, we cannot entirely rule out the possibility that these cells would express IL17A in response to other stimuli. Furthermore, detection of rare cell populations can be challenging when the analysis includes a limited number of cells from a limited number of patients. But the unique strength of the present study relies on the single‐cell approach. This high‐resolution technique operates at the level of individual cells, which circumvents the averaging of artifacts associated with traditional bulk population data.
Our study revealed that ILC3s express IL22 and CSF2 but do not contribute significantly to the production of IL‐17A in the inflamed SpA joint. This is consistent with the results of a recently reported study describing GM‐CSF–producing, but not IL‐17–producing, ILCs isolated from synovial tissue of patients with SpA 27.
The pathologic source of IL‐17A in the joint warrants further investigation. The most obvious candidates are T cells since they comprise up to 50% of all immune cells in the inflamed synovial tissue and various T cell subsets are able to produce IL‐17A; however, directly ex vivo, we did not detect any expression of IL‐17A in T cells. The results of RNA sequencing analysis of SpA patient ST confirm that very little IL‐17A is produced locally, in contrast to other proinflammatory cytokines such as TNF, IL‐8, and IL‐6 (Yeremenko NG, et al: unpublished observations). Furthermore, consistent with these data, none of the IL‐17A–producing T cells or ILCs can be detected by immunostaining for IL‐17A protein in the synovium (37). We have previously reported that IL‐17A protein is found exclusively inside mast cells and neutrophils; however, these cells intercept this cytokine rather than producing it (37). Overall, the observation of only low levels of IL‐17A messenger RNA provides evidence against de novo synthesis locally in the synovial tissue. Further work is needed to investigate alternative hypotheses, including production of IL‐17A in other tissues of the joint (e.g., in bone) or at distant sites (e.g., in the gut), and/or abnormal sensitivity of joint cells to IL‐17A.
Finally, our study revealed other interesting features of synovial ILCs, beyond the IL‐17 pathway. For example, we observed that upon activation, these cells have the ability to induce expression of PRF1 and GZMB genes, encoding the lytic enzymes perforin and granzyme B. This finding suggests that investigation of their cytotoxic effector functions in the pathophysiology of SpA is warranted.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Yeremenko had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Blijdorp, Spits, Rogge, Baeten, Yeremenko.
Acquisition of data
Blijdorp, Menegatti, van Mens, van de Sande, Chen, Hreggvidsdottir, Noordenbos, Latuhihin, Bernink, Yeremenko.
Analysis and interpretation of data
Blijdorp, Menegatti, Chen, Noordenbos, Spits, Rogge, Baeten, Yeremenko.
Additional Disclosures
Dr. Spits is an employee of AIMM Therapeutics. Dr. Baeten is an employee of UCB Pharma.
Supporting information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Table 1
Supplementary figures legends
Dr. Baeten's work was supported by the Netherlands Scientific Organization (NWO Vici grant) and the European Research Council (ERC consolidator grant).
Drs. Baeten and Yeremenko contributed equally to this work.
Dr. van de Sande has received speaking fees from AbbVie and Novartis (less than $10,000 each). Dr. Spits owns stock or stock options in and is chief scientific officer of AIMM Therapeutics. Dr. Baeten has received consulting fees, speaking fees, and/or honoraria from AbbVie, Pfizer, MSD, Roche, Bristol‐Myers Squibb, Novartis, Eli Lilly, Janssen, Glenmark, and Boehringer‐Ingelheim (less than $10,000 each) and owns stock or stock options in UCB.
References
- 1. Baeten D, Baraliakos X, Braun J, Sieper J, Emery P, van der Heijde D, et al. Anti‐interleukin‐17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double‐blind, placebo‐controlled trial. Lancet 2013;382:1705–13. [DOI] [PubMed] [Google Scholar]
- 2. Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, et al. Secukinumab, an interleukin‐17A inhibitor, in ankylosing spondylitis. N Engl J Med 2015;373:2534–48. [DOI] [PubMed] [Google Scholar]
- 3. McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, et al. Secukinumab, a human anti‐interleukin‐17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2015;386:1137–46. [DOI] [PubMed] [Google Scholar]
- 4. Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, et al. Secukinumab inhibition of interleukin‐17A in patients with psoriatic arthritis. N Engl J Med 2015;373:1329–39. [DOI] [PubMed] [Google Scholar]
- 5. Bowness P, Ridley A, Shaw J, Chan AT, Wong‐Baeza I, Fleming M, et al. Th17 cells expressing KIR3DL2+ and responsive to HLA‐B27 homodimers are increased in ankylosing spondylitis. J Immunol 2011;186:2672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coffre M, Roumier M, Rybczynska M, Sechet E, Law HK, Gossec L, et al. Combinatorial control of Th17 and Th1 cell functions by genetic variations in genes associated with the interleukin‐23 signaling pathway in spondyloarthritis. Arthritis Rheum 2013;65:1510–21. [DOI] [PubMed] [Google Scholar]
- 7. Jansen DT, Hameetman M, van Bergen J, Huizinga TW, van der Heijde D, Toes RE, et al. IL‐17‐producing CD4+ T cells are increased in early, active axial spondyloarthritis including patients without imaging abnormalities. Rheumatology (Oxford) 2015;54:728–35. [DOI] [PubMed] [Google Scholar]
- 8. Kenna TJ, Davidson SI, Duan R, Bradbury LA, McFarlane J, Smith M, et al. Enrichment of circulating interleukin‐17–secreting interleukin‐23 receptor–positive γ/δ T cells in patients with active ankylosing spondylitis. Arthritis Rheum 2012;64:1420–9. [DOI] [PubMed] [Google Scholar]
- 9. Menon B, Gullick NJ, Walter GJ, Rajasekhar M, Garrood T, Evans HG, et al. Interleukin‐17+CD8+ T cells are enriched in the joints of patients with psoriatic arthritis and correlate with disease activity and joint damage progression. Arthritis Rheumatol 2014;66:1272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Appel H, Maier R, Wu P, Scheer R, Hempfing A, Kayser R, et al. Analysis of IL‐17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17‐mediated adaptive immune response. Arthritis Res Ther 2011;13:R95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast cells and neutrophils release IL‐17 through extracellular trap formation in psoriasis. J Immunol 2011;187:490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Noordenbos T, Yeremenko N, Gofita I, van de Sande M, Tak PP, Canete JD, et al. Interleukin‐17–positive mast cells contribute to synovial inflammation in spondylarthritis. Arthritis Rheum 2012;64:99–109. [DOI] [PubMed] [Google Scholar]
- 13. Keijsers R, Hendriks AG, van Erp PE, van Cranenbroek B, van de Kerkhof PC, Koenen H, et al. In vivo induction of cutaneous inflammation results in the accumulation of extracellular trap‐forming neutrophils expressing RORγt and IL‐17. J Invest Dermatol 2014;134:1276–84. [DOI] [PubMed] [Google Scholar]
- 14. Brembilla NC, Stalder R, Senra L, Boehncke WH. IL‐17A localizes in the exocytic compartment of mast cells in psoriatic skin. Br J Dermatol 2017;177:1458–60. [DOI] [PubMed] [Google Scholar]
- 15. Sherlock JP, Joyce‐Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, et al. IL‐23 induces spondyloarthropathy by acting on ROR‐γt+ CD3+CD4−CD8− entheseal resident T cells. Nat Med 2012;18:1069–76. [DOI] [PubMed] [Google Scholar]
- 16. Artis D, Spits H. The biology of innate lymphoid cells. Nature 2015;517:293–301. [DOI] [PubMed] [Google Scholar]
- 17. Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, et al. A human natural killer cell subset provides an innate source of IL‐22 for mucosal immunity. Nature 2009;457:722–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Glatzer T, Killig M, Meisig J, Ommert I, Luetke‐Eversloh M, Babic M, et al. RORγt(+) innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp44. Immunity 2013;38:1223–35. [DOI] [PubMed] [Google Scholar]
- 19. Sawa S, Lochner M, Satoh‐Takayama N, Dulauroy S, Berard M, Kleinschek M, et al. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol 2011;12:320–6. [DOI] [PubMed] [Google Scholar]
- 20. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol 2016;17:765–74. [DOI] [PubMed] [Google Scholar]
- 21. Hoorweg K, Peters CP, Cornelissen F, Aparicio‐Domingo P, Papazian N, Kazemier G, et al. Functional differences between human NKp44(−) and NKp44(+) RORC(+) innate lymphoid cells. Front Immunol 2012;3:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, et al. Human fetal lymphoid tissue‐inducer cells are interleukin 17‐producing precursors to RORC+ CD127+ natural killer‐like cells. Nat Immunol 2009;10:66–74. [DOI] [PubMed] [Google Scholar]
- 23. Ciccia F, Guggino G, Rizzo A, Saieva L, Peralta S, Giardina A, et al. Type 3 innate lymphoid cells producing IL‐17 and IL‐22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Ann Rheum Dis 2015;74:1739–47. [DOI] [PubMed] [Google Scholar]
- 24. Leijten EF, van Kempen TS, Boes M, Michels‐van Amelsfort JM, Hijnen D, Hartgring SA, et al. Enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol 2015;67:2673–8. [DOI] [PubMed] [Google Scholar]
- 25. Geremia A, Arancibia‐Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL‐23‐responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med 2011;208:1127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cuthbert RJ, Fragkakis EM, Dunsmuir R, Li Z, Coles M, Marzo‐Ortega H, et al. Group 3 innate lymphoid cells in human enthesis. Arthritis Rheumatol 2017;69:1816–22. [DOI] [PubMed] [Google Scholar]
- 27. Al‐Mossawi MH, Chen L, Fang H, Ridley A, de Wit J, Yager N, et al. Unique transcriptome signatures and GM‐CSF expression in lymphocytes from patients with spondyloarthritis. Nat Commun 2017;8:1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rudwaleit M, van der Heijde D, Landewe R, Akkoc N, Brandt J, Chou CT, et al. The assessment of SpondyloArthritis international Society classification criteria for peripheral spondyloarthritis and for spondyloarthritis in general. Ann Rheum Dis 2011;70:25–31. [DOI] [PubMed] [Google Scholar]
- 29. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 30. Baeten D, Van den Bosch F, Elewaut D, Stuer A, Veys EM, De Keyser F. Needle arthroscopy of the knee with synovial biopsy sampling: technical experience in 150 patients. Clin Rheumatol 1999;18:434–41. [DOI] [PubMed] [Google Scholar]
- 31. Van der Maaten LH, Hinton G. Visualizing high‐dimensional data using t‐SNE. J Mach Learn Res 2008;9:2579–605. [Google Scholar]
- 32. Hazenberg MD, Spits H. Human innate lymphoid cells. Blood 2014;124:700–9. [DOI] [PubMed] [Google Scholar]
- 33. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: a proposal for uniform nomenclature. Nat Rev Immunol 2013;13:145–9. [DOI] [PubMed] [Google Scholar]
- 34. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin‐1 and IL‐23 induce innate IL‐17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity 2009;31:321–30. [DOI] [PubMed] [Google Scholar]
- 35. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol 2014;134:984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Teunissen MB, Munneke JM, Bernink JH, Spuls PI, Res PC, Te Velde A, et al. Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol 2014;134:2351–60. [DOI] [PubMed] [Google Scholar]
- 37. Noordenbos T, Blijdorp I, Chen S, Stap J, Mul E, Canete JD, et al. Human mast cells capture, store, and release bioactive, exogenous IL‐17A. J Leukoc Biol 2016;100:453–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Table 1
Supplementary figures legends